DOI:
10.1039/C4RA09541C
(Paper)
RSC Adv., 2014,
4, 61919-61926
The efficient enrichment of U(VI) by graphene oxide-supported chitosan
Received
31st August 2014
, Accepted 12th November 2014
First published on 12th November 2014
Abstract
Graphene oxide-supported chitosan (GO-Ch) composites were synthesized using a covalent method for U(VI) adsorption and were characterized by scanning electron microscopy (SEM), Fourier transform infrared (FT-IR) spectroscopy, thermogravimetric analysis (TGA), differential thermal analysis (DTA) and extended X-ray absorption fine structure (EXAFS). The characteristic results indicated that Ch was successfully grafted onto GO. The adsorption of U(VI) on GO-Ch was investigated under different environmental conditions. The adsorption kinetics showed that the adsorption of U(VI) on GO-Ch followed the pseudo-second-order equation. The maximum adsorption capacity of U(VI) on GO-Ch at pH 4.0 and T = 303 K calculated from the Langmuir model was 225.78 mg g−1. Thermodynamic parameters calculated from temperature-dependent adsorption isotherms suggested that U(VI) adsorption on GO-Ch was an endothermic and spontaneous process. The batch desorption indicated U(VI) cannot be completely desorbed from GO-Ch without intervention, suggesting the irreversible adsorption of U(VI) on GO-Ch. The analysis of FT-IR spectra suggested that the interaction mechanism of U(VI) on GO-Ch was mainly chemical adsorption by –NH2 and –COOH groups. According to EXAFS analysis, the peaks at ∼2.9 Å can be satisfactorily fitted by the U–C/N shell, revealing the formation of inner-sphere surface complexes. It is demonstrated that the GO-Ch nanocomposite can be a promising material for the preconcentration and solidification of U(VI) from large volumes of aqueous solution.
1. Introduction
The release of uranium from mine tailings and the nuclear industry has been of concern due to its radioactivity and biological toxicity.1–3 For the sake of public health and ecosystem stability, it is necessary to eliminate U(VI) from contaminated wastewater prior to releasing it into the natural environment. The adsorption method is considered to be a simple and economical method for the removal of U(VI) from wastewater.4 A great number of researchers have investigated the adsorption of U(VI) on minerals, organic macromolecules and organisms.4–6 In these studies, the impact of environmental factors (e.g., pH, organic substances, ionic strength, and temperature) on the adsorption capacity of adsorbents have been extensively investigated. However, the relatively low adsorption efficacy limited their application to large volumes of aqueous solution. Thus, the development of new artificial adsorbents with high adsorption capacity is of particular importance for the safe treatment and the disposal of U(VI) from aqueous solution.
Graphene oxide (GO) has been evidenced to exhibit great removal of heavy metals,7–9 radionuclides6,10–12 and dye.13–15 Ding et al.16 found the maximum adsorption of U(VI) on GO was 208.33 mg g−1 at pH 3.0 and T = 303 K. Sun et al.12 also demonstrated GO presented high adsorption capacity for Eu(III) (175.44 mg Eu(III) per gram of GO at T = 298 K). However, the irreversible aggregation of GO or the polydisperse in its thickness, lateral size, and shape can hinder the adsorption performance and reduce their adsorption capacity.17 Therefore, the modification of GO by introducing various functional groups has been reported to enhance its dispersity and efficacy.12,18 The functionalization of GO with nanoparticles, organic compounds, biomaterials, and polymers was reviewed by Chen et al.19 As a natural product, chitosan (Ch) is identified to have strong affinity for heavy metals (i.e., Cu(II), Cr(VI), Cd(II), Ag(I)) and organic matter20–23 due to large numbers of amino (–NH2) and hydroxyl (–OH) groups. However, the poor mechanical property and the dissolution of Ch in strong acids greatly affect its experimental study and commercial application.24 It is demonstrated that the modification of Ch by biomaterials not only prevents the dissolution in strong acids, but also increases the porosity of the polymers.25 To the authors' knowledge, few studies on the adsorption of U(VI) on GO-supported chitosan (GO-Ch) composites are reported, and the interaction mechanism between U(VI) and GO-Ch is not well defined.
The objectives of the current study are (1) to synthesize GO- Ch composites and to characterize the microscopic and macroscopic surface properties of GO-Ch composites by using scanning electron microscopy (SEM), thermal gravity analysis and differential thermal analysis (TGA/DTA); (2) to investigate the adsorption of U(VI) on GO-Ch under various environmental conditions (i.e., contact time, pH, ionic strength, initial U(VI) concentration and temperature) by batch techniques; and (3) to discuss the interaction mechanism between U(VI) and GO-Ch composites by using Fourier transformed infrared spectroscopy (FT-IR) and extended X-ray absorption fine structure (EXAFS) spectroscopy.
2. Experimental section
2.1. Materials
All chemicals used in the experiments were purchased in analytical purity and were used without any purification. All reagents were prepared with Milli-Q water. Ch (with a deacetylation degree of 90%) was purchased from Sinopharm Chemical Reagent Co., Ltd, Beijing. The glutaraldehyde (GLA 50 wt% in water, Sigma-Aldrich) was used as the cross-linking agent.
2.2. Preparation of GO-Ch composites
GO was synthesized by the modified Hummers method.26 Briefly, graphite powder (2.0 g) and sodium nitrate (1.5 g) were added into concentrated sulfuric acid (0 °C) under stirring conditions. Then, potassium permanganate (9.0 g) was slowly added to the suspension under ice bath conditions. The mixtures were reacted at room temperature for 5 days under vigorous stirring conditions. After that, 230 mL of the diluted H2SO4 (5 wt%) was injected when the aforementioned suspension was heated to 98 °C. The residual MnO4− was removed by adding 100 mL of hydrogen peroxide (30 wt%) at 60 °C. The GO nanosheets were obtained by centrifuging it at 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 60 min after ultrasonic treatment several times.
000 rpm for 60 min after ultrasonic treatment several times.
The GO-Ch composites were synthesized by chemical method. Typically, 2.0 g of Ch power was dissolved in a 100 mL of 4 wt% acetic acid solution under ultrasonic treatment (100 w) for 30 min. Follow that, 15 mL GLA and 0.5 g GO was added into the aforementioned suspension under vigorous stirring conditions at room temperature for 90 min. Then the mixture was adjusted to pH 9–10 and was stirred at 80 °C for 1 h. The precipitate was washed with ethanol and followed distilled water until the pH neutral.
2.3. Characterization of GO-Ch composites
The morphology of GO-Ch composites was conducted by SEM (JSM-6320F FE-SEM electron microscope), TGA/DTA (TGA Q5000 V3.15 Build 263TA Instruments) with heating rate of 10 K min−1 and a flow rate of 75 mL min−1. The potentiometric acid–base titrations were conducted under argon using a DL50 Automatic Titrator (Mettler Toledo) in 0.01 M NaClO4 as a background electrolyte. FT-IR (Perkin-Elmer Spectrum 100, USA) in the range of 4000–400 cm−1 was performed using KBr pellets. Uranium LIII-edge EXAFS of samples was conducted at the Shanghai Synchrotron Radiation Facility (SSRF, Shanghai, China). All of wet samples were measured in the fluorescence mode using Ge detector (Canberra Ultra-LE Ge) due to the low uranium concentration. EXAFS data fitting was performed using Athena and Artemis interfaces to IFFEFIT 7.0 software. The EXAFS threshold energies (E0) were defined as 17174 eV for the U samples, indicating that uranium in these systems was U(VI) species. The incident X-ray intensity I0 is measured with an Ar-filled ionization chamber at ambient pressure. 25 scans were recorded for each individual sample. The Autobk algorithm was applied for background removal and the frequency cutoff parameter Rbkg was set to 1.20. In all cases, the models are fitted to the k3-weighted raw EXAFS spectra.
2.4. Adsorption and desorption Experiments
Batch adsorption experiments were conducted in the polycarbonate tubes with 0.75 g L−1 GO-Ch and 1.7 × 10−4 mol L−1 U(VI) in the presence of 0.01 mol L−1 NaClO4. The bulk suspensions of GO-Ch and NaClO4 were pre-equilibrated for 24 h, and then U(VI) solution was spiked into the bulk suspension. The breakthrough experiment was conducted according to the method of previous paper.27 Briefly, 0.5 g dried GO-Ch was packed into the glass column with an inner-radius of 0.4 cm, then the column was injected with constant water. Before U(VI) injection, the column was percolated with a 0.01 mol L−1 NaClO4 solution (pH 4.0) at a flow rate of 0.25 mL min−1. The adsorbent volume has an average of 0.87 mL (V0). The solution containing 3 × 10−3 mol L−1 U(VI) (C0) was injected into the GO-Ch column. The pH of suspension was adjusted from pH 2.5 to 10.0 by the drop wise addition of the negligible volume of 0.1 mol L−1 HClO4 or NaOH solutions. The isothermal adsorptions of U(VI) on GO and GO-Ch were investigated at pH 4.0 with the initial concentration in the range of 10–60 mg L−1. The method of synthetic wastewater used in this study is similar to that of Yan et al.28 The suspensions were shaken at T = 303 K for 24 h, which was demonstrated to be adequate for the suspension to obtain equilibrium by the preliminary experiments. The solid phase was then separated from the solution phase by the centrifugation it at 18000 rpm for 30 min. The adsorption percentage of U(VI) (adsorption (%)) and adsorption capacity (Qs, mg g−1) can be expressed as eqn (1) and (2), respectively:| | |
adsorption (%) = (C0 − Ceq)/C0 × 100%
| (1) |
| | |
Qs = V × (C0 − Ceq)/m
| (2) |
where C0 (mg L−1) and Ceq (mg L−1) are initial concentration and concentration after adsorption, respectively. m (g) and V (mL) are the mass of adsorbents and the volume of the suspension, respectively.
The desorption experiments were conducted by lowering U(VI) concentration. After adsorption equilibrium, half volume of the supernatant was displaced by the equivalent volume of U(VI)-free background electrolyte solution (0.01 M NaClO4) with the same pH values. The amount of desorbed U(VI) was measured in terms of the residual concentrations of U(VI) on GO-Ch after desorption. The concentration of U(VI) was measured by a kinetic phosphorescence analyzer (KPA-11, Richland, USA). All experimental data were the average of triplicate determinations and the relative errors were within ±5%.
3. Results and discussion
3.1. Characterization of GO-Ch
As shown by SEM image in Fig. 1A, the nanosheet-like structure of GO with large thickness, smooth surface, and wrinkled edge is observed. The GO-Ch (Fig. 1B) presents the aggregation and much rougher surface, revealing that Ch has been assembled on the surface of the GO layers with a high density. Additionally, the formation of some holes is also observed on the GO-Ch surface. The TGA curves of GO, Ch and GO-Ch are shown in Fig. 1C, in the first decomposition phase (0–100 °C), approximately 7%, 10% and 15% of weight loss are registered for GO, Ch and GO-Ch, respectively, which are assigned to the evaporation of adsorbed water. In the 150–300 °C region, the Ch and GO-Ch lose about 31% of their weight, whereas the GO loses about 50% of its weight. The weight loss of GO can be due to the decomposition of massive oxygenated functional groups. The GO-Ch exhibits significant weight loss at temperatures about 450 °C, whereas little weight loss is registered for the GO and Ch. The rate of weight loss of GO-Ch at 0–250 °C is significantly lower than GO and Ch, it is apparent that GO-Ch has the different decomposition patterns as compared to pure GO and Ch, indicating the Ch is successfully grafted on the surface GO. Information about the differences in the chemistry of the surfaces can be seen in the DTA curves (Fig. 1D). The first peak centered at about 80–90 °C for all samples is due to the evaporation of physically adsorbed water. For the GO sample, the second peak presented between 250 and 300 °C is related to the decomposition of massive oxygenated functional groups such as epoxy and carboxyl groups.29 It is observed that Ch displays an exothermic peak around 250 °C, which can be attributed to the decomposition of the hydroxyl and amino groups of chitosan.30 The broad peak at about 500–600 °C for Ch can be assigned to the further decomposition of chitosan residues. Two sharp exothermic peaks of GO-Ch at around 200 and 450 °C represent the degradation of GO and Ch, respectively. According to Fig. 1E, the pHpzc of GO and GO-Ch is 4.12 and 5.45, respectively.
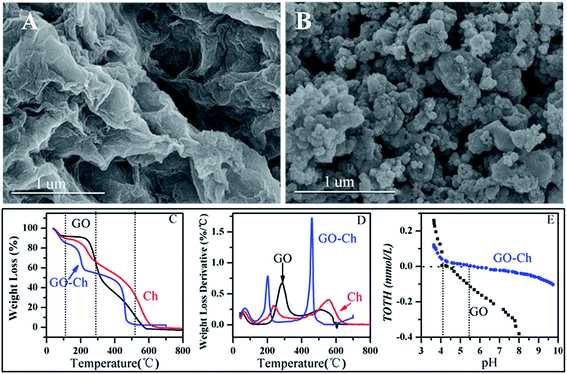 |
| | Fig. 1 Characterization of GO and GO-Ch composites. A and B: SEM images of GO and GO-Ch composites; C and D: TGA and DTA analysis; E: potentiometric acid-base titrations, respectively. | |
The FTIR spectra of GO, Ch, GO-Ch before and after adsorption are showed in Fig. 2. It is observed that GO nanosheets show characteristic bands at 1057.76 (epoxy groups C–O), 1620.87 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1727.91 (CO bonds from carboxyl groups) and the broad band at 2200–3800 cm−1 (C–OH stretching from adsorbed water).12 The FTIR spectrum of Ch exhibits the stretching vibration mode of –OH group at 3200–3400 cm−1, –CO stretching vibration of –NHCO (amide I) at 1647.88 cm−1, the –NH– deformation of –NH2 at 1597.74 cm−1, and the amino group (−NH2) at 1420.32 cm−1. It should be noted that the peak of GO-Ch at 1727.91 cm−1 (carboxyl groups) disappears as compared with GO, moreover the peak density of –NH2 groups at 1420.32 cm−1 on GO-Ch decreases while the increase of characteristic peaks of –NHCO– and –NH– (at 1647.88 and 1597.74 cm−1) are observed. This is evidence that the –NH2 groups on the macromolecular chain of Ch can react with carboxyl groups of GO under cross-linking agent conditions. The GO-Ch composites are synthesized by the interaction between carboxyl groups of GO and amino groups of Ch.
C), 1727.91 (CO bonds from carboxyl groups) and the broad band at 2200–3800 cm−1 (C–OH stretching from adsorbed water).12 The FTIR spectrum of Ch exhibits the stretching vibration mode of –OH group at 3200–3400 cm−1, –CO stretching vibration of –NHCO (amide I) at 1647.88 cm−1, the –NH– deformation of –NH2 at 1597.74 cm−1, and the amino group (−NH2) at 1420.32 cm−1. It should be noted that the peak of GO-Ch at 1727.91 cm−1 (carboxyl groups) disappears as compared with GO, moreover the peak density of –NH2 groups at 1420.32 cm−1 on GO-Ch decreases while the increase of characteristic peaks of –NHCO– and –NH– (at 1647.88 and 1597.74 cm−1) are observed. This is evidence that the –NH2 groups on the macromolecular chain of Ch can react with carboxyl groups of GO under cross-linking agent conditions. The GO-Ch composites are synthesized by the interaction between carboxyl groups of GO and amino groups of Ch.
 |
| | Fig. 2 FTIR spectra of the controls including GO, Ch and GO-Ch, and U(VI)-adsorbed samples including GO and GO-Ch at different pH values. | |
3.2. Adsorption kinetics
Fig. 3A shows the effect of contact time on U(VI) adsorption on GO-Ch and GO. The adsorption rates of U(VI) sharply increase during the initial 30 min, then increase slowly to reach equilibrium at 2 h, which is consistent with previous studies.11,13 The initially rapid adsorption may be attributed to the relatively larger available sites on the adsorbent. It is obvious that GO-Ch exhibits greater adsorption capacity for U(VI) than GO. Approximately 75% and 65% of U(VI) are adsorbed on GO-Ch and GO at 24 h, respectively. The pseudo-first-order and pseudo-second-order kinetic equations are applied to describe the experimental data. Their linear forms are given in eqn (3)31 and (4),12 respectively:| | |
ln(qe − qt) = lnqe − kf × t
| (3) |
| | |
t/qt = 1/(ks × qe2) + t/qe
| (4) |
where qe and qt (mg g−1) are the amount of U(VI) adsorbed at equilibrium and at time t, respectively. kf and ks are the pseudo-first order and pseudo-second order kinetic rate constant, respectively. As shown in Fig. 3A, the adsorption of U(VI) on GO-Ch and GO can be satisfactorily fitted by pseudo-second-order kinetic equation (R2 > 0.99), which indicates that the rate-limiting step of U(VI) adsorption on GO-Ch and GO are chemisorptions rather than interparticle diffusion.31 Table 1 shows the equilibrium time of U(VI) adsorption on different adsorbents, GO-Ch has a shorter time to achieve equilibrium.
 |
| | Fig. 3 A: Adsorption kinetics of U(VI) on GO and GO-Ch (A) pH = 4.0, T = 303 K, C0 = 1.7 × 10−4 mol L−1, I = 0.01 mol L−1 NaClO4. The inserted curves: the fitting of pseudo-second order kinetic model; B: breakthrough curve of U(VI) on GO-Ch, pH = 4.0, T = 303 K, C0 = 3 × 10−3 mol L−1, I = 0.01 mol L−1 NaClO4. | |
Table 1 Comparison of the U(VI) sorption capacity of GO-Ch with other sorbents
| Material |
Experimental conditions |
Qmax (mg g−1) |
Equilibration time (h) |
Reference |
| Nanoporous alumina |
pH 6.8, T = 298 K |
11.6 |
4 |
37 |
| Multi-walled carbon nanotubes |
pH 5.0, T = 298 K |
26.18 |
6 |
38 |
| Akaganéite |
pH 6.0, T = 303 K |
90.44 |
8 |
39 |
| GO |
pH 4.0, T = 303 K |
208.33 |
2 |
16 |
| GO-Ch |
pH 4.0, T = 303 K |
225.78 |
2 |
This work |
| PANI@GO |
pH 3.0, T = 298 K |
242.52 |
2 |
12 |
The breakthrough curve of U(VI) on the GO-Ch is depicted in Fig. 3B. The breakthrough is shown as the ratio of the outlet to the inlet concentrations (C/C0) as a function of the ratio of eluted pore to adsorbent volume (V/V0). As shown from Fig. 3B, the C/C0 of influent samples was zero before the pore volume was 50. Even the pore volume of influent reached 170, the C/C0 of influent samples was less than 0.93. These results indicate that the GO-Ch composites have a high kinetic adsorption capacity.
3.3. Effect of pH and ionic strength
The effect of pH on the adsorption of U(VI) on GO and GO-Ch is shown in Fig. 4(A and B). As shown in Fig. 4A, U(VI) adsorption on GO increases with increasing pH from 3.0 to 5.0, then maintains this high level at pH 5.0-7.0. The decreased adsorption is observed at pH > 7.0, which is agreement with our previous work.16 The deprotonation of GO increases with increasing pH, especially above the pHpzc = 4.12, which favors the electrostatic attraction between negative charged GO and positive charged U(VI). Ding et al.16 has determined that the increasingly U(VI) hydrolyzed species (e.g., UO2OH+, (UO2)3(OH)5+, (UO2)4(OH)7+ and UO2(OH)2 species) are formed at pH > 4.0. However, at pH > 7.0, the negatively charged U(VI) species (i.e., UO2(OH)3− and (UO2)3(OH)7− species) increase, which probably explains the suppressed adsorption at pH > 7.0 because of the electrostatic repulsion between negative U(VI) species and negative charged GO. It should be noted that the decreased adsorption at pH > 7.0 is not observed for GO-Ch (Fig. 4B) due to the different functional groups contributed to U(VI) adsorption. It is worth noting that the pHpzc of GO-Ch (about 5.45) is higher than GO (Fig. 1E) due to the introduction of –NH2 groups. Sun et al.12 has demonstrated that the enriched amino and amido groups on PANI@GO have a high affinity for U(VI) at alkaline pH because of the formation of strong complexes with cations. Therefore, residual –NH2 groups can also react with the negative charged U(VI), thus, keeping the adsorption in plateau at pH > 7.0. It is also worth noting that U(VI) adsorption on GO-Ch is higher than that on GO at variable pH due to the presence of oxygen and nitrogen-containing functional groups of GO-Ch composites.
 |
| | Fig. 4 The effect of pH and ionic strength on U(VI) adsorption onto GO and GO-Ch, T = 303 K, C0 = 1.7 × 10−4 mol L−1. | |
The effect of ionic strength on U(VI) adsorption on GO and GO-Ch are also shown in Fig. 4A and B. Ion strength effect is performed by using NaClO4 since ClO4− has little capacity to coordinate with metal ions.32 Therefore, NaClO4 would not affect U(VI) species with varied pH and would contribute to the mechanistic description of U(VI) adsorption on GO-Ch. The increasing ionic strength has little effect on the adsorption of U(VI) on GO and GO-Ch throughout a wide pH range, which indicates that inner-sphere surface complexation dominates U(VI) adsorption on GO and GO-Ch.11,12 The inner-sphere surface complexes are formed by the complexation of adsorbate with amphoteric groups by chemical bonds, which are less ionic strength-dependent.33,34 The oxygen/nitrogen-containing functional groups contribute to the inner-sphere adsorption of U(VI) and GO or GO-Ch by chemical bonding.
3.4. Adsorption isotherms and thermodynamics
Adsorption isotherms of U(VI) on GO and GO-Ch are displayed in Fig. 5A and B. The adsorption of U(VI) on GO and GO-Ch increases with increasing solution concentration. It can be observed that the adsorption amount of U(VI) on GO-Ch is higher than GO, indicating that GO-Ch composites present greater adsorption capacity for U(VI) compared to GO. The adsorption isotherms are fitted by Langmuir, Freundlich35 and D-R models,36 their linear formations can be given in eqn (5)–(7), respectively:| | |
Ceq/Qs = 1/(b × Qmax) + Ceq/Qmax
| (5) |
| | |
logQs = logKF +1/nlogCeq
| (6) |
| | |
lnQs = lnQmax − βξ2
| (7) |
where Qmax (mg g−1) is the maximum adsorption capacity of adsorbent at complete monolayer coverage. b (L mg−1) is a Langmuir constant, 1/n is the heterogeneity of the adsorption sites, KF represents equilibrium coefficient, β is the activity coefficient related to the mean adsorption energy (mol2 kJ−2), and ξ is the Polanyi potential, which is equal to eqn (8):| | |
ξ = RTln(1 + 1/Ceq)
| (8) |
where R is ideal gas constant (8.314 J (mol−1 K−1)), and T is the absolute temperature in Kelvin (K).
 |
| | Fig. 5 A and B: adsorption isotherm of U(VI) onto GO and GO-Ch, respectively; C and D: adsorption isotherm of U(VI) onto GO and GO-Ch in simulating wastewater; E and F: desorption of U(VI) from GO and GO-Ch, respectively; pH = 4.0, C0 = 1.7 × 10−4 mol L−1, I = 0.01 mol L−1 NaClO4. | |
The corresponding parameters of Langmuir, Freundlich and D–R models are tabulated in Table 2. The fitting results reveal that the adsorption of U(VI) on GO and GO-Ch can be well described by Langmuir model (R2 > 0.99), which indicates U(VI) adsorption on GO and GO-Ch are monolayer adsorption. The maximum adsorption capacities of GO and GO-Ch calculated from Langmuir model at pH 4.0 and T = 303 K are 190.10 and 225.78 mg g−1 for U(VI), respectively. GO-Ch exhibits superior performance for U(VI) removal as compared with other materials (Table 1) reported in previous researches. The effect of temperature on the adsorption of U(VI) on the two adsorbents is also shown in Fig. 5A and B. For GO and GO-Ch, the adsorption increases with increasing temperature from 303 K to 323 K, suggesting U(VI) adsorption on GO and GO-Ch are promoted at higher temperature. A thermodynamic analysis is realized based on the equilibrium data for isotherms at three different temperatures. The Gibbs free energy change (ΔG0, kJ mol−1) of the adsorption process is expressed by the van't Hoff equation (eqn (9)):
| | |
ΔG0 = −RTln(K0).
| (9) |
Table 2 Equilibrium parameters for the adsorption of U(VI) onto GO and GO-Ch composites at 303, 313, and 323 K
| |
T (K) |
Langmuir equation |
Freundlich equation |
D–R equation |
| Qmax (mg g−1) |
b (L mg−1) |
R2 |
kF (mg1−n Ln g−1) |
1/n |
R2 |
Qmax (mg g−1) |
B (mg2 kJ−2) |
R2 |
| GO |
303 |
190.10 |
0.213 |
0.989 |
2.22 |
3.679 |
0.829 |
136.83 |
28.98 |
0.872 |
| 313 |
205.70 |
0.394 |
0.996 |
5.75 |
2.403 |
0.994 |
135.33 |
12.24 |
0.991 |
| 323 |
222.53 |
0.435 |
0.997 |
40.55 |
0.908 |
0.946 |
165.89 |
3.22 |
0.941 |
| GO-Ch |
303 |
225.78 |
0.129 |
0.974 |
81.28 |
0.458 |
0.970 |
165.14 |
1.69 |
0.847 |
| 313 |
231.72 |
0.130 |
0.956 |
87.10 |
0.446 |
0.928 |
174.94 |
1.16 |
0.870 |
| 323 |
240.72 |
0.151 |
0.940 |
103.51 |
0.414 |
0.934 |
188.21 |
0.85 |
0.904 |
The sorption equilibrium constant lnK0 can be calculated by plotting lnKd versus Ce and extrapolating Ce to zero. The adsorption coefficient (Kd) can be calculated by eqn (10):
where
Qs (mg L
−1) is the amount adsorbed on the solid at equilibrium.
The change in entropy [ΔS0, kJ (mol−1 K−1)] and the enthalpy of adsorption (ΔH0, kJ mol−1) at a constant temperature T (K) can be calculated from the eqn (11):
Based on eqn (9)–(11), one can write
| | |
lnK0 = ΔS0/R − ΔH0/RT.
| (12) |
The values of ΔH0 and ΔS0 are calculated from a plot of ln(Kd) versus 1/T. The negative values of ΔG0 (−11.49 kJ mol−1 at 303 K) testify that U(VI) adsorption on GO-Ch is spontaneous process. The value of ΔG0 of U(VI) on GO-Ch is lower than that on GO, suggesting that U(VI) is more prone to be adsorbed on GO-Ch. The positive values of ΔH0 (25.20 kJ mol−1) suggest the endothermic nature of the adsorption process, which are consistent with the increased adsorption capacities with the rise of temperature. The positive values of ΔS0 (117.14 J mol−1 K) express the increased randomness at the solid–liquid interface, which also reveals U(VI) adsorption on GO-Ch and GO are a spontaneous process. Thermodynamic parameters calculated from temperature-dependent adsorption isotherms suggests that U(VI) adsorption on GO-Ch is an endothermic and spontaneous process.
The adsorption isotherms of U(VI) in simulated wastewater are shown in Fig. 5C and D. In simulated wastewater, the maximum adsorption of U(VI) on GO and GO-Ch at 303 K and pH 4.0 are ∼150 and 180 mg g−1, respectively. Although both adsorbents exhibit decreased adsorption capacity in simulated wastewater, the superior performance of GO-Ch for U(VI) removal is still observed compared with GO. The poor U(VI) removal in synthetic groundwater was also evidenced by previous studies.40,41 The synthetic wastewater contains a range of cationic and anionic chemical components: K+, Mg2+, Na+, Ca2+, CO32−, HCO3− and NO3− etc., which cause the competing reactions. It was reported that carbonate can form complexes with U(VI) (carbonato-U(VI) complexes) which is more stable in solutions, thus decreasing U(VI) adsorption.42
3.5. Desorption isotherms and regeneration
The desorption isotherms (desorption induced by replacing radionuclide supernatant) of U(VI) from GO and GO-Ch are shown in Fig. 5E and F. As shown in Fig. 5E and F, the separation of the lines is observed between desorption and adsorption curves, indicating the irreversible desorption hysteresis.43 The irreversible hysteresis indicates that desorption can be not completely achieved without intervention under same pH value.
From the viewpoints of low economic cost and environmental renewable, it is needful to take advantage of renewable material. On this account, the recycle use of GO-Ch is checked to assess its application potential in the actual environment for purifying of U(VI)-bearing outlet water. Acid wash could be a simple and feasible way for the recycling of U(VI)-loaded GO-Ch. Our results show that 0.5 mol L−1 HCl solution can completely desorbed U(VI) from GO-Ch. The results (Fig. 6) reveal that the adsorption ability of GO-Ch slightly reduces from 220 mg g−1 to 200 mg g−1 after 8 recycles. the relationship between number of regeneration cycles and the after 8 cycles. The outstanding power of regeneration and the excellent performance of repeated use indicated that GO-Ch could endure long term use of wastewater treatment.
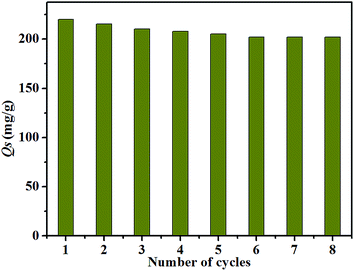 |
| | Fig. 6 Recycling of GO-Ch in the removal of U(VI), pH = 4.0, T = 303 K, C0 = 1.7 × 10−4 mol L−1, I = 0.01 mol L−1 NaClO4. | |
3.6. Interaction mechanism
The interaction mechanism between U(VI) and GO-Ch is demonstrated by FTIR analysis. As shown in Fig. 2, the spectrum of U-adsorbed GO presents a decrease in the peak at 1727.91 cm−1 (CO bonds from carboxyl groups) versus GO, indicating the carboxyl groups on GO is responsible for the adsorption of U(VI). The decrease of –NHCO– peaks (at ∼1630 and ∼1566 cm−1) and –NH2 peaks (at ∼1420 cm−1) are also observed for U-adsorbed GO-Ch at pH 4.0 and 8.0 as compared with GO-Ch, indicating that the nitrogen containing functional groups are responsible for U(VI) adsorption on GO-Ch. The analysis of FITR reveals that the adsorption of U(VI) on GO and GO-Ch depend on different functional groups, and the higher adsorption on GO-Ch is attributed to the introduction of amine groups.
Fig. 7 shows the uranium LIII-edge EXAFS spectra Fourier transforms (FT) of references (UO22+) and adsorption samples at different pH conditions. The fitted results are summarized in Table 3. The similar spectra of GO and GO-Ch samples at pH 4.0 and 8.0 are observed. The FT peaks centered at ∼1.4 and 1.9 Å can be satisfactorily fitted by 2 axial O atoms (at 1.8 Å) and 4–5 equatorial O atoms (at ∼2.5 Å), respectively, which was consistent with the structural results obtained previously for the fully hydrated uranyl ion.43 The FT peaks at 2.9 Å can be fitted by U–C/N shell,44,45 revealing the formation of stable inner-sphere complexes of UO22+ with GO and GO-Ch. This is consistent with the results of FT-IR. The FT feature of GO and GO-Ch (pH 8.0) at 3.5 Å can be fitted by U–U shell at 3.94 Å very well (Table 3), indicating surface precipitation of U(VI) onto the GO and GO-Ch at high pH conditions.46 This result can mainly explain the high adsorption of U(VI) on GO and GO-Ch between pH 6–8. Therefore, the results from EXAFS analysis indicate that the adsorption of U(VI) at pH 4.0 and 8.0 could be attributed to the inner-sphere surface complexation and surface co-precipitation, respectively.
 |
| | Fig. 7 U LIII-edge Fourier transforms (FT) EXAFS data, T = 303 K, I = 0.01 mol L−1 NaClO4. | |
Table 3 EXAFS parameters of U(VI) LIII-edge, T = 303 K, I = 0.01 mol L−1 NaClO4
| Samples |
Shell |
Ra (Å) |
CNb |
σ2c (Å2) |
| R is the bond distance. CN is coordination numbers of neighbors. σ2 is the Debye–Waller factor. |
| UO22+ |
U–Oax |
1.74(8) |
1.8(7) |
0.00466 |
| U–Oeq |
2.25(9) |
5.5(5) |
0.00573 |
| GO pH 4.0 |
U–Oax |
1.81(1) |
2.0(0) |
0.00425 |
| U–Oeq |
2.45(8) |
4.6(9) |
0.00666 |
| U–C/N |
2.93(2) |
3.2(5) |
0.00482 |
| GO pH 8.0 |
U–Oax |
1.80(1) |
2.0(0) |
0.00224 |
| U–Oeq |
2.39(1) |
4.1(3) |
0.00923 |
| U–C/N |
2.91(6) |
3.0(7) |
0.00226 |
| U–U |
3.95(8) |
0.5(7) |
0.03274 |
| GO-Ch pH 4.0 |
U–Oax |
1.75(8) |
2.0(0) |
0.00250 |
| U–Oeq |
2.41(7) |
4.5(5) |
0.00522 |
| U–C/N |
2.95(8) |
1.4(2) |
0.01464 |
| GO-Ch pH 8.0 |
U–Oax |
1.79(1) |
2.0(0) |
0.00081 |
| U–Oeq |
2.35(4) |
4.5(2) |
0.00987 |
| U–C/N |
2.94(8) |
1.3(0) |
0.01903 |
| U–U |
3.94(7) |
0.5(8) |
0.02285 |
4. Conclusions
As prepared GO-Ch was achieved by grafting Ch on the surface of GO via covalent method. The adsorption kinetics showed that the adsorption of U(VI) on GO and GO-Ch can be followed by pseudo-second-order kinetic model very well. The maximum sorption capacity of U(VI) on GO-Ch composites calculated from the Langmuir models was 225.78 mg g−1 at pH 4.0 and T = 303 K, which is slightly higher than that of GO (∼190 mg g−1). The thermodynamic parameters suggested that adsorption of U(VI) on GO-Ch was an endothermic and spontaneous process. The batch desorption revealed the irreversible adsorption of U(VI) on GO-Ch. Based on the analysis of FTIR and EXAFS, the interaction mechanism between U(VI) and GO-Ch was inner-sphere surface complexation. It is determined that GO-Ch composite exhibits higher adsorption capacities than any other natural materials in the preconcentration of U(VI) ions from large volumes of aqueous solutions, which will further enhance the application of GO-based composite in environmental pollution management.
Acknowledgements
Financial support from National Natural Science Foundation of China (21207135, 21207136 and 91126020), National “863” Plan (no. 2011AA10A10401) and Hefei Center for Physical Science and Technology (2012FXZY005) are acknowledged.
References
- C. Basset, O. Averseng, P.-J. Ferron, N. Richaud, A. s. Hagège, O. Pible and C. Vidaud, Chem. Res. Toxicol., 2013, 26, 645–653 CrossRef CAS PubMed.
- M. Carrière, C. Thiebault, S. Milgram, L. Avoscan, O. Proux and B. Gouget, Chem. Res. Toxicol., 2006, 19, 1637–1642 CrossRef PubMed.
- L. He, M. Neu and L. Vanderberg, Environ. Sci. Technol., 2000, 34, 1694–1701 CrossRef CAS.
- Q. Y. Wu, J. H. Lan, C. Z. Wang, C. L. Xiao and Y. L. Zhao, J. Phys. Chem. A, 2014, 118, 2149–2158 CrossRef CAS PubMed.
- Z. Li, F. Chen and L. Yuan, Chem. Eng. J., 2012, 210, 539–546 CrossRef CAS PubMed.
- Y. Sun, J. Li and X. Wang, Geochim. Cosmochim. Acta, 2014, 140, 621–643 CrossRef CAS PubMed.
- G. Zhao, J. Li, X. Ren, C. Chen and X. Wang, Environ. Sci. Technol., 2011, 45, 10454–10462 CrossRef CAS PubMed.
- G. Sheng, Y. Li, X. Yang, X. Ren, S. Yang, J. Hu and X. Wang, RSC Adv., 2012, 2, 12400–12407 RSC.
- X. Yang, C. Chen, J. Li, G. Zhao, X. Ren and X. Wang, RSC Adv., 2012, 2, 8821–8826 RSC.
- A. Y. Romanchuk, A. S. Slesarev, S. N. Kalmykov, D. V. Kosynkin and J. M. Tour, Phys. Chem. Chem. Phys., 2013, 15, 2321–2327 RSC.
- Y. Sun, Q. Wang, C. Chen, X. Tan and X. Wang, Environ. Sci. Technol., 2012, 46, 6020–6027 CrossRef CAS PubMed.
- Y. Sun, D. Shao, C. Chen, S. Yang and X. Wang, Environ. Sci. Technol., 2013, 47, 9904–9910 CrossRef CAS PubMed.
- Q. J. Du, J. K. Sun, Y. H. Li, X. X. Yang, X. H. Wang, Z. H. Wang and L. H. Xia, Chem. Eng. J., 2014, 245, 99–106 CrossRef CAS PubMed.
- T. Yang and H. Chen, RSC Adv., 2014, 4, 29357–29364 RSC.
- C. Yong, H. Yong, L. Yan and G. Jia, RSC Adv., 2013, 3, 7254–7258 RSC.
- C. Ding, W. Cheng, Y. Sun and X. Wang, Dalton Trans., 2014, 43, 3888–3896 RSC.
- A. Green and M. C. Hersam, J. Phys. Chem. Lett., 2009, 1, 544–549 CrossRef PubMed.
- G. L. Li, G. Liu, M. Li, D. Wan, K. Neoh and E. Kang, J. Phys. Chem., 2010, 114, 12742–12748 CAS.
- D. Chen, H. Feng and J. Li, Chem. Rev., 2012, 112, 6027–6053 CrossRef CAS PubMed.
- S. Hasan, A. Krishnaiah, T. K. Ghosh, D. S. Viswanath, V. M. Boddu and E. D. Smith, Ind. Eng. Chem. Res., 2006, 45, 5066–5077 CrossRef CAS.
- Y. Zhang, C. Gao, W. Zhao, Z. Zhou, W. Yan, X. Li, Y. Liu, Z. Sun, G. Zhao and J. Gao, RSC Adv., 2014, 4, 33840–33847 RSC.
- S. Peng, H. Meng, Y. Ouyang and J. Chang, Ind. Eng. Chem. Res., 2014, 53, 2106–2113 CrossRef CAS.
- H. Liu and C. Wang, RSC Adv., 2014, 4, 3864–3872 RSC.
- W. Wan Ngah, L. Teong and M. Hanafiah, Carbohydr. Polym., 2011, 83, 1446–1456 CrossRef CAS PubMed.
- G. Crini and P.-M. Badot, Prog. Polym. Sci., 2008, 33, 399–447 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- R. Artinger, W. Schuessler, T. Schaefer and J. Kim, Environ. Sci. Technol., 2002, 36(20), 4358–4363 CrossRef CAS.
- S. Yan, B. Hua, Z. Bao, J. Yang, C. Liu and B. Deng, Environ. Sci. Technol., 2010, 20, 7783–7789 CrossRef PubMed.
- M. Seredych, R. Pietrzak and T. J. Bandosz, Ind. Eng. Chem. Res., 2007, 46, 6925–6935 CrossRef CAS.
- N. Travlou, G. Kyzas, N. Lazaridis and E. Deliyanni, Langmuir, 2013, 29, 1657–1668 CrossRef CAS PubMed.
- S. Lagergren, K. Sven. Vetenskapsakad. Handl., 1898, 24, 1–39 Search PubMed.
- E. Tertre, S. Castet, G. Berger, M. Loubet and E. Giffaut, Geochim. Cosmochim. Acta, 2006, 70, 4579–4599 CrossRef CAS PubMed.
- T. Rabung, M. Pierret, A. Bauer, H. Geckeis, M. Bradbury and B. Baeyens, Geochim. Cosmochim. Acta, 2005, 69, 5393–5402 CrossRef CAS PubMed.
- V. Sinitsyn, S. Aja, D. Kulik and S. Wood, Geochim. Cosmochim. Acta, 2000, 64, 185–194 CrossRef CAS.
- M. LeVan and T. Vermeulen, J. Phys. Chem., 1981, 85, 3247–3250 CrossRef CAS.
- G. Wood, Carbon, 2002, 40, 231–239 CrossRef CAS.
- Y. Sun, S. Yang, G. Sheng, Z. Guo, X. Tan, J. Xu and X. Wang, Sep. Purif. Technol., 2011, 83, 196–203 CrossRef PubMed.
- D. Shao, Z. Jiang, X. Wang, J. Li and Y. Meng, J. Phys. Chem. B, 2009, 113, 860–864 CrossRef CAS PubMed.
- S. Yusan and S. Akyil, J. Hazard. Mater., 2008, 160, 388–395 CrossRef CAS PubMed.
- R. Crane, M. Dickinson, I. Popescu and T. Scott, Water Res., 2011, 45(9), 2931–2942 CrossRef CAS PubMed.
- T. Scott, I. Popescu, R. Crane and C. Noubactep, J. Hazard. Mater., 2011, 186(1), 280–287 CrossRef CAS PubMed.
- Y. Sun, J. Li and X. Wang, Geochim. Cosmochim. Acta, 2014, 140, 621–643 CrossRef CAS PubMed.
- T. Undabeytia, S. Nir, G. Rytwo, C. Serban, E. Morillo and C. Maqueda, Environ. Sci. Technol., 2002, 36, 2677–2683 CrossRef CAS.
- C. Gaillard, A. Chaumont, I. Billard, C. Hennig, A. Ouadi and G. Wipff, Inorg. Chem., 2007, 46, 4815–4826 CrossRef CAS PubMed.
- C. Lucks, A. Rossberg, S. Tsushima, H. Foerstendorf, A. C. Scheinost and G. Bernhard, Inorg. Chem., 2012, 51, 12288–12300 CrossRef CAS PubMed.
- G. Vazquez, C. Dodge and A. Francis, Inorg. Chem., 2008, 47, 10739–10743 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 






