DOI:
10.1039/C4RA08715A
(Paper)
RSC Adv., 2014,
4, 55529-55538
Reversible-deactivation radical polymerization of chloroprene and the synthesis of novel polychloroprene-based block copolymers by the RAFT approach†
Received
15th August 2014
, Accepted 1st October 2014
First published on 2nd October 2014
Abstract
Reversible addition–fragmentation chain transfer (RAFT) polymerization of the reactive monomer chloroprene (2-chloro-1,3-butadiene) mediated by ethyl 2-(ethoxycarbonyl)prop-2-yl dithiobenzoate (EPDTB), 4-cyano-4-(phenylcarbonothioylthio) pentanoic acid (CPDTB) and dibenzyl trithiocarbonate (DBTTC) was investigated in benzene using 2,2′-azobis(isobutyronitrile) (AIBN) as initiator. Polychloroprene (PCP) chains with predetermined molecular weights and low molar mass dispersities were synthesized by RAFT polymerization using EPDTB and CPDTB. The work described here also showed for the first time that well-defined polystyrene-block-polychloroprene (PSt-b-PCP) and poly(methyl methacrylate)-block-polychloroprene (PMMA-b-PCP) with controlled number averaged molecular weights and molecular weight distributions can be prepared in solution polymerization, employing EPDTB and 2-cyanoprop-2-yl dithiobenzoate (CPDB), respectively, as the initial RAFT agent. The success of the block copolymerization was showed by the shift toward higher molar mass of the size exclusion chromatography (SEC) chromatograms recorded before and after block copolymerization. Structural confirmation of the diblock copolymers was accomplished by 1H NMR measurements. The results obtained from SEC analysis together with 1H NMR spectroscopy demonstrate the possibility to design and prepare well-defined PCP-based block copolymers.
Introduction
Since the development of polychloroprenes (PCPs) by DuPont in 1931,1 they have been an extremely important class of synthetic polymers and widely used in the tire and rubber industry and in the production of adhesive materials. PCP, compared to other elastomers, exhibits outstanding physical toughness, a wider operating temperature range than general purpose hydrocarbon elastomers, and excellent resistance to ozone, sun, and general weather conditions. Articles made with this rubber include electrical insulating and sheathing materials, hoses, conveyor belts, flexible bellows, transmission belts, sealing materials, diving suits and other protective suits. Adhesive grades of PCP are used mainly in the footwear industry. PCP latexes have been used for dipped goods (balloons, gloves), latex foam, fibre binders, adhesives and rug backing. So far, many attempts are being made to modify PCP either through the blend formation or by graft copolymerization.2–7 For example, methyl acrylate, acrylonitrile, alkyl methacrylates (e.g., methyl, octyl, lauryl), fumaronitrile, methacrylic acid, and dichlorobutadiene have been employed in solution and emulsion graft polymerization with PCP. By these processes many new materials have been formed which differ completely in both physical and chemical properties from that of PCP.
Well-defined complex macromolecular architectures with controlled molecular weights are of particular interest when they can lead to new material properties. Recent progress in reversible-deactivation radical polymerization (RDRP),8 especially nitroxide-mediated radical polymerization (NMRP),9,10 atom transfer radical polymerization (ATRP),11,12 and reversible addition–fragmentation chain transfer (RAFT)13 polymerization create a new way to prepare well-defined polymers. In recent years, block copolymers have gained increasing attention. There can be a number of advantages in block copolymer has well-defined microstructure. RDRP is the process of choice to obtain block copolymers. RAFT polymerization, the most universal technique, has a great tolerance for functional groups of monomers and reaction conditions, and has been successfully applied to the preparation of a wide range of new functional polymeric architectures.14–17 Moreover, RAFT polymerization provides a tool to produce block copolymers with controlled molecular weight distribution (MWD).18–20 There is now a growing body of work that uses the RAFT process to make block copolymers consisting of a wide range of polymer compositions.21–25
Owing to the numerous potential applications of rubber materials (tyres, adhesives, etc.), the RDRP of conjugated dienes, including butadiene, isoprene, and chloroprene, has received more and more attention.26–30 Among RDRPs, nitroxide-mediated free radical polymerization was first implemented to isoprene and butadiene, but branching reactions of dienes becomes significant when the monomer conversion was high.26 Hua et al. prepared polybutadiene with predictable molecular weights and low molar mass dispersities (Đ = Mw/Mn < 1.5) by reverse atom transfer radical polymerization.27 Dürr et al. synthesized nitrile-butadiene rubber (NBR)-based block and miktoarm copolymers by RAFT solution polymerization.28 The RAFT-mediated emulsion polymerization of conjugated dienes can be conducted under either seeded or ab initio conditions.29 Hlalele et al. recently reported ab RAFT-mediated ab initio emulsion copolymerization of acrylonitrile with 1,3-butadiene employing 2-(((dodecylsulfanyl)carbonothioyl)sulfanyl)propanoic acid (DoPAT) as chain transfer agent.30
The electron-rich vinyl group and electronegative chlorine atom facilitates the high reactivity of chloroprene (2-chloro-1,3-butadiene, CP) monomer. Industrially, practically all the PCP producers use free-radical emulsion polymerization for its synthesis via uncontrolled fashion with sulfur or mercaptans used as chain-terminating agents.31 RDRP of CP represents a special challenge because of its high reactivity. Only scant reference can be found in the open literature involved in the reversible-deactivation radical polymerization behavior of CP. Ajellal et al. illustrated the copolymerization of dimethyl 1,3-butadiene-1-phosphonate (BPMe) with CP using NMRP. The results showed that P(CP-co-BPMe) copolymers were synthesized successfully by solution polymerization. Unfortunately, the GPC chromatograms showed high Đ of the final product (Đ >1.5).32 The appropriate RAFT agent for a particular monomer has been explored by many researchers. The choice of the substituents R and Z is of particular importance in synthesis of end-functional, block and star copolymers. Indeed, as far as we know, the only example of chloroprene monomer polymerized by RAFT has been 2-cyanoprop-2-yl dithiobenzoate (CPDB) as the RAFT chain transfer agent (RAFT-CTA) in which Z is phenyl and R is C(CH3)2CN.33 In this regard, more efforts should be concentrated on the preparation of PCP with appropriate RAFT agents via the RAFT technique. And surprisingly no published data are available on the preparation of well-defined block copolymers composed of CP and other monomers by the RAFT technique.
In the work reported here, homopolymerization of CP was investigated by using 2-(ethoxycarbonyl)prop-2-yl dithiobenzoate (EPDTB) and 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (CPDTB) as the RAFT-CTAs in which Z is phenyl and R is C(CH3)2COOC2H5/C(CH3) (CN)CH2COOH. Aiming at preparing polychloroprene-based block copolymers, we also demonstrate the possibility to synthesize polystyrene-block-polychloroprene (PSt-b-PCP) and poly(methyl methacrylate)-block-polychloroprene (PMMA-b-PCP) diblock copolymers in a controlled fashion, employing EPDTB and CPDB, respectively, as the initial RAFT agent. This type of diblock copolymers may prove useful as a blend compatibilizer or as an adhesion promoter for chloroprene rubber or coatings on more polar substrates such as metals. Therefore, PCP-based block copolymers can significantly expand the range of applications.
Experimental section
Materials
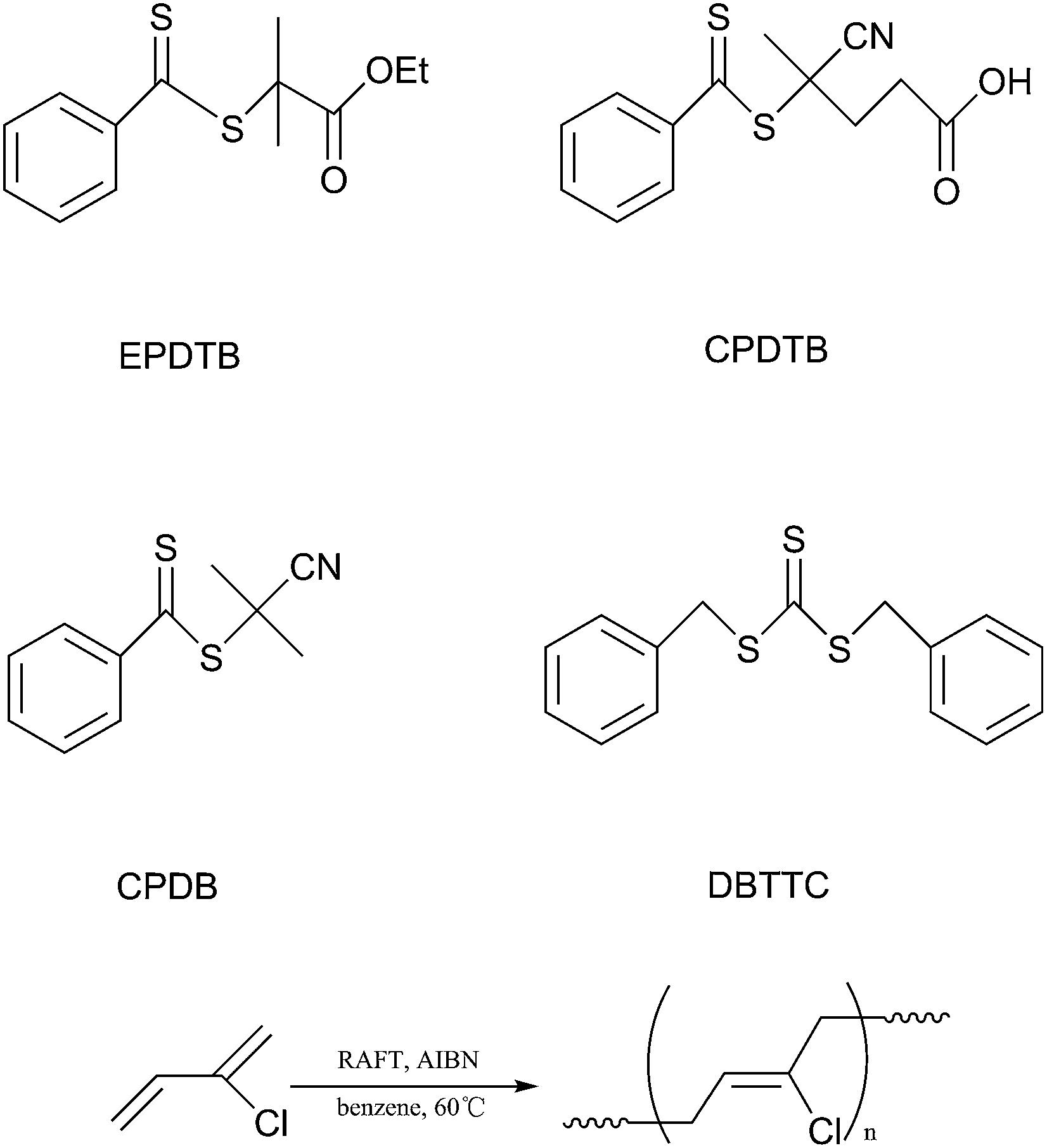
Chloroprene was provided by Shanxi Synthetic Rubber Group Co., Ltd. (Datong, China) and used without further purification. Styrene (St) and methyl methacrylate (MMA) (99%, Tianjin fuchen chemical plant) were distilled under reduced pressure and stored in a freezer before use. 2,2′-Azobisisobutyronitrile (AIBN) was recrystallized from methanol. Benzene and tetrahydrofuran (THF) were dried over CaH2, distilled before use. 2-(Ethoxycarbonyl)prop-2-yl dithiobenzoate (EPDTB) and dibenzyl trithiocarbonate (DBTTC) were synthesized according to literature.34,35 4-Cyano-4-(phenylcarbonothioylthio)pentanoic acid (CPDTB, >97%) and 2-cyanoprop-2-yl dithiobenzoate (CPDB, >97%) were purchased from Sigma-Aldrich and used as received.
Synthesis of polychloroprene
The typical procedures for the RAFT polymerization are as follows: CP, RAFT (EPDTB/CPDTB/DBTTC), AIBN with predetermined molar ratio and benzene were added to a dry round bottomed flask equipped with a magnetic bar. Keeping the molar ratio of the initiator to the RAFT agents constant (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) and the monomer concentration at 5.64 M, the reactivity of different RAFT agents to CP was investigated. The solution was bubbled with argon for approximately 10 minutes to eliminate the oxygen. Afterwards, the flask was degassed by five freeze–thaw–pump cycles, kept under argon, and placed in an oil bath thermostated at 60 °C. At the designed time, about 1.5 mL of reaction mixture was withdrawn from the flask using degassed syringes to determine the monomer conversion and the molecular weight of the polymers.
:4) and the monomer concentration at 5.64 M, the reactivity of different RAFT agents to CP was investigated. The solution was bubbled with argon for approximately 10 minutes to eliminate the oxygen. Afterwards, the flask was degassed by five freeze–thaw–pump cycles, kept under argon, and placed in an oil bath thermostated at 60 °C. At the designed time, about 1.5 mL of reaction mixture was withdrawn from the flask using degassed syringes to determine the monomer conversion and the molecular weight of the polymers.
Synthesis of the block copolymers
The general procedure for synthesizing polystyrene (PSt) macro-RAFT agent was as follows. In a round bottom flask, PSt macro-RAFT agent was synthesized as described previously with EPDTB (1.072 g, 4 mmol), St (20.8 g, 200 mmol), AIBN (0.168 g, 1 mmol), and THF (40 g), heated to 70 °C for 20 h.34 The polymer was purified via precipitations into methanol (Mn = 2900 g mol−1, Mw/Mn = 1.23). Then 2 g of PSt-macro RAFT agent from the preceding synthesis, 12.21 g of CP (138 mmol), 0.0283 g of AIBN (0.17 mmol) were dissolved in 18.3 g of benzene and purged with argon for 10 min. After five freeze–thaw–pump cycles, the flask was filled with argon and immersed in a preheated oil bath at 60 °C. During the polymerizations, a trace amount of sample was taken from reactor at suitable time intervals, and then the polymer was finally dried under vacuum at room temperature. The monomer conversions were calculated by gravimetric analysis. 1H NMR and SEC analyses were performed on the dry solid of the diblock copolymer.
PMMA macro-RAFT agent was prepared according to the literature.34 Block polymerization of CP from the resulted PMMA macro-RAFT agent by RAFT polymerization was carried out as follows. In a round bottom flask, PMMA (Mn = 1800 g mol−1, Mw/Mn = 1.33) (2 g, 1.11 mmol), CP (10 g, 113 mmol), AIBN (0.0966 g, 0.588 mmol), and benzene (15 g) were introduced. After five freeze–thaw–pump cycles, the flask was filled with argon and immersed in a preheated oil bath at 60 °C. During the polymerizations, a trace amount of sample was taken from reactor at suitable time intervals for analysis. PMMA-b-PCP copolymers prepared in this work were analyzed by 1H NMR and SEC to determine their composition, molecular weight, and molar mass dispersity.
Characterization techniques
The monomer conversion was determined by the gravimetric method. The molecular weight and the Đ of the synthesized polymers were measured by gel permeation chromatography using a TOSOH HLC-8320 instrument, which consisted of a solvent delivery system, a column set with two TSK gel Super Multipore HZ-M columns, and a differential refractometer index (RI) detector. The eluent was THF at a flow rate of 0.35 mL min−1 at 40 °C. Polystyrene standards were used to generate the calibration curve. 1H NMR spectra were obtained on a Bruker AV400-MHz spectrometer in CDCl3 at room temperature. 1H chemical shifts were referenced to tetramethylsilane (TMS) via the residual non-deuterated solvent signal at δ = 7.26 ppm.
Results and discussion
RAFT homopolymerization of chloroprene
The nature of RAFT agents strongly influences the control over the polymerization of different vinyl monomers. Generally, the RAFT agents for controlled polymerization of “more-activated” monomers (MAMs), such as, styrene, methyl acrylate will inhibit or retard polymerizations of “less-activated” monomers (LAMs). As few reports were associated with controlled RAFT polymerization of conjugated diene monomer, the choice of suitable RAFT agents for CP polymerization needs to be explored. In this article, three different RAFT agents (Scheme 1), a trithiocarbonate (DBTTC) and two dithiobenzoates (EPDTB and CPDTB), were investigated in the RAFT polymerization of CP at 60 °C. Keeping the molar ratio of the initiator to the RAFT agents constant (1:4) and the monomer concentration at 5.64 M, the reactivity of different RAFT agents to CP was investigated.
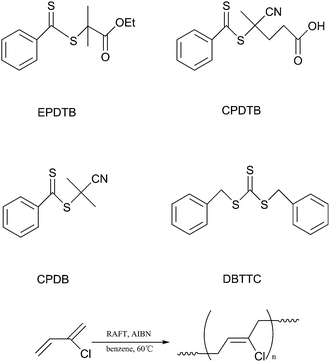 |
| | Scheme 1 Chemical formulas of the RAFT agents and RAFT polymerization of CP. | |
Radical polymerization of chloroprene in the presence of EPDTB
RAFT polymerizations to form the PCP homopolymers were carried out with EPDTB as RAFT agent and AIBN as initiator at 60 °C. The results of polymerization at different [CP]0/[EPDTB]0 molar ratios are listed in Table 1.
Table 1 The synthetic parameters and results of PCP by RAFT polymerization with EPDTB
| No. |
[CP]0:[EPDTB]0:[AIBN]0 |
Time (h) |
Conversion (%) |
Mn,tha |
Mn,SEC |
Mw/Mn |
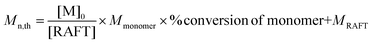 ; [M]0 = initial concentration of the monomer; [RAFT] = concentration of the RAFT agent; Mmonomer = molecular weight of the monomer, in this case, MCP = 88.54; and MRAFT = molecular weight of the RAFT agent. ; [M]0 = initial concentration of the monomer; [RAFT] = concentration of the RAFT agent; Mmonomer = molecular weight of the monomer, in this case, MCP = 88.54; and MRAFT = molecular weight of the RAFT agent. |
| 1 |
200:1:0.25 |
5 |
15.3 |
3000 |
2900 |
1.29 |
| 10 |
21.5 |
4100 |
3900 |
1.29 |
| 20 |
33.3 |
6200 |
6400 |
1.34 |
| 25 |
37.8 |
7000 |
7300 |
1.36 |
| 30 |
42.9 |
7900 |
8600 |
1.38 |
| 2 |
400:1:0.25 |
5 |
14.6 |
5500 |
6000 |
1.24 |
| 9 |
25.8 |
9400 |
10400 |
1.21 |
| 17 |
29.3 |
10700 |
12100 |
1.33 |
| 24 |
32.8 |
11900 |
13000 |
1.38 |
| 67 |
59.1 |
21200 |
22200 |
1.52 |
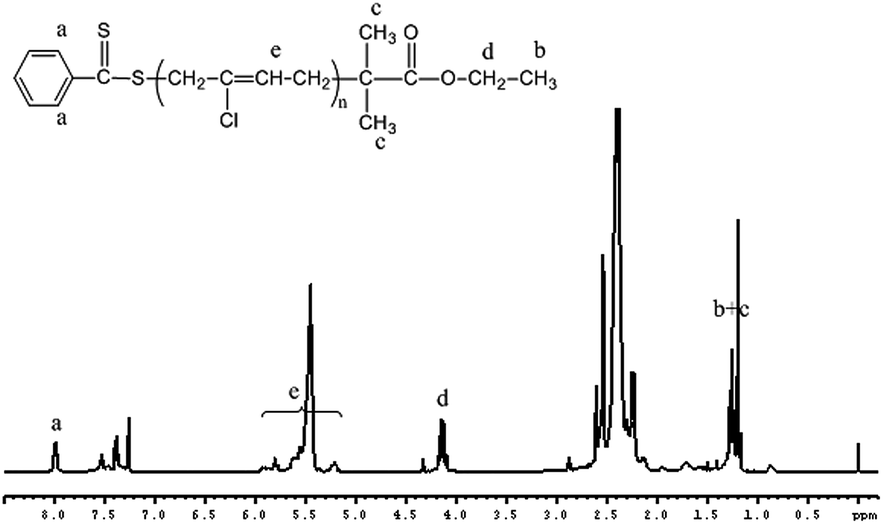
To examine the effectiveness of EPDTB as RAFT-agent, we first maintain a constant 200/1 [CP]0/[EPDTB]0 ratio (entry 1 in Table 1). Fig. 1(A) shows the SEC curves of the PCP samples prepared at different polymerization times. A continuous shift of the curves toward shorter elution times is observed, indicating continuously increasing molar masses. Furthermore, the plots of Mn and Mw/Mn vs. monomer conversion in Fig. 1(B) show that the molecular weight is developed linearly with monomer conversion and close to the theoretical values. The dispersities increased with conversion, showing a leveling trend above ca. 40% conversion. The linear increase in molecular weight and low dispersity verify that the polymerization of CP with EPDTB as RAFT agent is a reversible deactivation radical polymerization. Also, a linear increase of ln([M]0/[M]) with reaction time in Fig. 1(C) was observed for about 30 h (conversion is 42.9%), confirming that the concentration of radical species remained relatively constant. Additionally, the 1H NMR spectrum of PCP is shown in Fig. 2. All the characteristic protons signals of the RAFT terminals and chloroprene units are clearly observed. The peaks at δ = 7.92 ppm, originating from RAFT agent-EPDTB, appeared in the obtained polymers. The signals d (4.15) are attributed to the methylene protons of –COO–CH2–CH3. The signals b and c (1.20) are attributed to the methyl protons of –C(CH3)2–COO–CH2–CH3. The signals e (5.2–5.9) are attributed to the methylidyne protons of PCP.
 |
| | Fig. 1 (A) SEC traces for PCP showing the evolution of molecular mass with time. (B) Plots of Mn and Mw/Mn vs. monomer conversion. (C) The corresponding rate plot for PCP in benzene at 60 °C (entry 1 in Table 1). | |
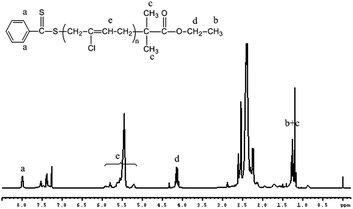 |
| | Fig. 2 1H NMR spectrum of PCP with EPDTB as the CTA in benzene at 60 °C ([CP] = 5.64 M, [CP]0/[EPDTB]0/[AIBN]0 = 200:1:0.25). | |
The number averaged molecular weights based on 1H NMR spectrum (Mn,NMR) can be calculated according to eqn (1):
| |
 | (1) |
where
Ie and
Ia are the integral values of the peaks e and a, respectively. The calculated
Mn,NMR was 2700, which was close to the theoretical value (3000). These demonstrate that the polymerization of chloroprene was controlled by the RAFT process.
The polymerization of CP was investigated in the presence of EPDTB at 400/1 [CP]0/[EPDTB]0 ratio (entry 2 in Table 1). Molecular weight increases with monomer conversion. At low conversion, the molecular weight is in good agreement with the theoretical prediction. However, at high conversion, molecular weight becomes larger than the theoretical prediction. Dispersities gradually increase from 1.24 at 14.6% to 1.52 at 59.1% conversion. The increase in Đ might be ascribed to the vinyl bonds (pendant and internal) in the backbone of the polymer that can act as branching points in the polymer, and further chemical cross-linking of CP might occur when the target molecular weight is much high in RAFT polymerization, which are well accepted in the radical polymerization of conjugated dienes.36,37 Taking into account the high reactivity of CP monomer, RAFT polymerization of CP are much better than the other conjugated dienes.27,29,38–41 It is clear that EPDTB is a good mediator for CP reversible deactivation radical polymerization.
Radical polymerization of chloroprene in the presence of CPDTB
CP was also polymerized in benzene solvent using AIBN as an initiator and CPDTB as a CTA (Scheme 1). As shown in Table 2, molecular weight increases with monomer conversion as predicted, and be controlled up to 14500 with dispersities typically being less than 1.30. The SEC traces, the pseudo first-order kinetic plot and Mn and Đ versus monomer conversion for the polymerization with CPDTB are shown in Fig. 3 ([CP]0/[CPDTB]0/[AIBN]0 = 200:1:0.25, entry 3 in Table 2). First, Mn increases linearly with monomer conversion (Fig. 3(B)) and agrees well with the theoretical Mn. Second, the linearity of the pseudo first order kinetic plot (Fig. 3(C)) implies a constant radical concentration during the polymerization. Third, a relatively narrow molar-mass dispersity (Fig. 3(B)) is observed throughout the polymerization. These plots are as expected for a controlled RAFT polymerization. Fig. 4 displays the 1H NMR of PCP prepared via RAFT polymerization mediated by CPDTB. The resonances at δ = 5.2–5.9 (b) are attributed to the methylidyne protons of PCP backbone. The resonances at δ = 7.8–8.0 (a) are due to the aromatic protons of RAFT end group, as shown in Fig. 4.
Table 2 The synthetic parameters and results of PCP by RAFT polymerization with CPDTB
| No. |
[CP]0:[CPDTB]0:[AIBN]0 |
Time (h) |
Conversion (%) |
Mn,th |
Mn,SEC |
Mw/Mn |
| 3 |
200:1:0.25 |
5 |
9.4 |
1900 |
1800 |
1.21 |
| 10 |
19.4 |
3700 |
4200 |
1.17 |
| 20 |
39.5 |
7300 |
8100 |
1.21 |
| 25 |
48.4 |
8900 |
9500 |
1.24 |
| 30 |
56.7 |
10300 |
10400 |
1.28 |
| 4 |
400:1:0.25 |
5 |
4.2 |
1800 |
1600 |
1.29 |
| 10 |
11.1 |
4200 |
4600 |
1.23 |
| 20 |
28.5 |
10400 |
10700 |
1.23 |
| 25 |
31.5 |
11400 |
12800 |
1.24 |
| 30 |
39.8 |
14400 |
14500 |
1.27 |
 |
| | Fig. 3 (A) SEC traces for PCP showing the evolution of molecular mass with time. (B) Plots of Mn and Mw/Mn vs. monomer conversion. (C) The corresponding rate plot for PCP in benzene at 60 °C (entry 3 in Table 2). | |
 |
| | Fig. 4 1H NMR spectrum of PCP with CPDTB as the CTA in benzene at 60 °C ([CP] = 5.64 M, [CP]0/[CPDTB]0/[AIBN]0 = 200:1:0.25). | |
The number averaged molecular weights based on 1H NMR spectrum (Mn,NMR) can be calculated according to eqn (2):
| |
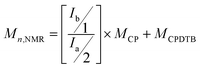 | (2) |
where
Ib and
Ia are the integral values of the peaks b and a, respectively. The calculated
Mn,NMR was 2000, which was close to the theoretical value (1900). All these indicate that RAFT polymerization of CP employing CPDTB as CTA was a controlled process. In other words, CPDTB was an excellent RAFT agent for mediating the reversible deactivation radical polymerization of CP.
Radical polymerization of chloroprene in the presence of DBTTC
The conditions of the polymerization of CP and the results (conversions, molar masses and the molar mass dispersities) in the presence of symmetrical trithiocarbonate RAFT agent DBTTC are listed in Table 3. The kinetic plots, SEC traces and plots of Mn and Mw/Mn vs. monomer conversion can be found in the ESI, Fig. S1.† The kinetic plots show that there is no marked retardation employing DBTTC as the RAFT agent. A continuous shift of the SEC curves toward shorter elution times is observed, indicating continuously increasing molar masses. Molecular weight increases with monomer conversion. As can be seen, the number-averaged molecular weights of the polymers are larger than those theoretical values (Mth) at low monomer conversion (conversion < 25%), while the Mns are slightly lower than Mth at high monomer conversion (conversion > 25%). Higher than predicted molecular weights for low monomer conversions indicates that DBTTC is less active and has a low transfer constant. The data listed in Table 3 also show that the dispersities of the polymers are broad using DBTTC as RAFT agent. In our three RAFT agents, Z group (benzyl) of DBTTC is less active than Z group (phenyl) of EPDTB and CPDTB; and the homolytic leaving ability of R group (benzyl) of DBTTC is poor with respect to that of EPDTB and CPDTB (R: C(CH3)2COOC2H5, C(CH3)(CN)CH2COOH, respectively). This fact implies that the efficiency of DBTTC as CTA was poor in the RAFT polymerization of CP.
Table 3 The synthetic parameters and results of PCP by RAFT polymerization with DBTTC
| No. |
[CP]0:[DBTTC]0:[AIBN]0 |
Time (h) |
Conversion (%) |
Mn,th |
Mn,SEC |
Mw/Mn |
| 5 |
200:1:0.25 |
5 |
14.3 |
2800 |
4000 |
1.96 |
| 10 |
28.8 |
5400 |
4400 |
1.92 |
| 17 |
32.8 |
6100 |
4700 |
1.93 |
| 20 |
36.7 |
6800 |
5100 |
1.89 |
| 25 |
43.4 |
8000 |
6000 |
1.79 |
| 6 |
400:1:0.25 |
5 |
14.7 |
5500 |
9500 |
1.91 |
| 10 |
21.5 |
7900 |
9700 |
2.02 |
| 16 |
29.4 |
10700 |
10000 |
2.00 |
| 20 |
34.2 |
12400 |
11200 |
1.90 |
| 25 |
38.9 |
14100 |
11800 |
1.83 |
The above results of CP polymerization in the presence of different RAFT agents lead us to conclude that the two dithiobenzoates both carrying tertiary R-groups and electron-withdrawing groups, EPDTB and CPDTB, proved to be very efficient to polymerize CP, while the polymerization of CP with DBTTC showed poor controlled characters. This is in full agreement with the results in literature that the addition–fragmentation ability of the RAFT agents increases in the series: DBTTC ≪ EPDTB < CPDTB.42
Synthesis of the block copolymers
The RAFT polymerization can provide not only homopolymers mediated by small molecule RAFT agent but also block copolymers with controlled structures through the chain growth of the second monomer in the presence of appropriate macro-RAFT agent. On the basis of the controlled CP homopolymerization, we try to synthesize PCP-based diblock copolymers with the formed PCP capped with dithioester group as the macro-RAFT agent and St/MMA as the second monomer. However, all of the SEC elution profiles were bimodal, and the peaks of the lower molecular fraction were identical to that of the PCP. These results showed that the transfer of the radical to the PCP macro-RAFT agent is not sufficient and rapid enough. This may be attributed to the fragmentation of the poly(styrene/methyl methacrylate) radical from the intermediate poly(chloroprene)-SC(Z)S-poly(styrene/methyl methacrylate) is faster than that of the poly(chloroprene) radical.20,43 In other words, due to low Ctr of PCP macro-RAFT to styrene/methyl methacrylate monomer, only part of the PCP chain was grown by PSt/PMMA block. This indicates that the synthesis of the PCP-b-P(St/MMA) block copolymer facing difficulties when the PCP is synthesized first by the RAFT techniques (see ESI Fig. S3 and S5, Tables S1 and S2†). Therefore, we try to prepare the block copolymers employing the reverse routes.
Synthesis of polystyrene-block-polychloroprene
In this section, EPDTB was used as the initial RAFT agent, which was an excellent RAFT agent mediating the reversible deactivation radical polymerization of styrene.34 Based on the RAFT polymerization mechanism, the polymer obtained from the RAFT polymerization of styrene contains dithiobenzoate group at one end of polymer chain, which can be used as macro-RAFT agent for synthesis of polystyrene-block-polychloroprene diblock copolymers.
To obtain a PSt block of short chain length, a monomer/CTA/AIBN ratio of 50/1/0.25 was chosen. A PSt macro-RAFT agent with a narrow molecular weight distribution (Mn = 2900 g mol−1, Mw/Mn = 1.23, see the Experimental section) was prepared and precipitated twice in cold CH3OH to remove traces of monomer and initiator. The polymer was subsequently used as a macro-RAFT agent for the growth of the PCP second block, see Scheme 2. The growth of the second block was conducted by re-initiation with AIBN in benzene at 60 °C. As shown in Fig. 5(A), starting with a PSt macro-RAFT agent with a molecular weight of 29 00 g mol−1, the SEC traces of the four samples show a monomodal distribution, and chain extension of PSt resulted in a clear shift of the SEC peaks toward higher molecular weights with time, indicating the growth of a PCP block. The number-average molecular weights and the dispersities of PSt macro-RAFT and corresponding copolymers are showed in Fig. 5(B). With increasing reaction time, the molecular weight of the resulting block copolymer increases linearly with conversion. The linear dependence showed the control over the polymerization as well. The dispersity indicates the narrow molecular weight distribution of the resulting block copolymers ranging between 1.24 and 1.44. The narrow molecular weight distribution suggests that the transfer of the radical to the macro-RAFT agent is successful. The kinetic curve (Fig. 5(C)) indicated that the polymerization was also a first-order reaction. For further confirmation of the formation of diblock copolymers, PSt-b-PCP was further characterized by 1H NMR to acquire the composition (Fig. 6). It can be seen that the signals at 6.3–7.2 ppm and 5.2–5.9 ppm correspond to St and CP units respectively, while the signals at 7.53–8.01 ppm correspond to the aromatic protons originated from EPDTB moieties, confirming the formation of PSt-b-PCP. Combining 1H NMR results as well as the controlled experiment we considered the formation of block copolymers composed of polystyrene and polychloroprene to be successful.
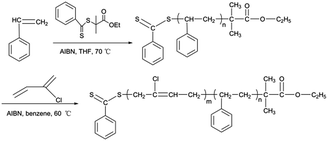 |
| | Scheme 2 Synthetic pathways for the preparation of polystyrene-block-polychloroprene diblock copolymers via RAFT polymerization. | |
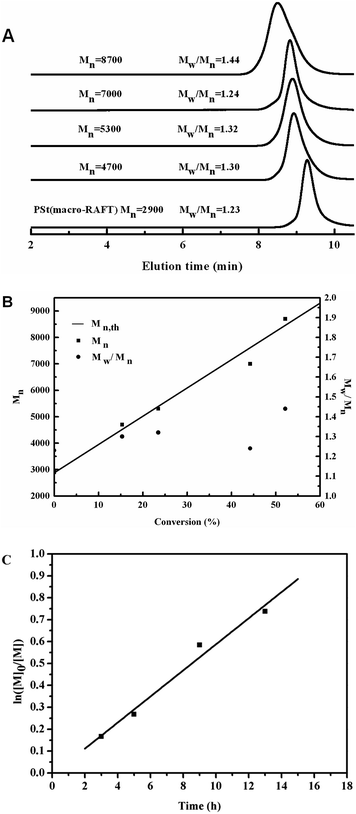 |
| | Fig. 5 (A) SEC traces, (B) Plots of Mn and Mw/Mn vs. conversion, and (C) relationships of ln([M]0/[M]) with polymerization time in the RAFT polymerization of CP using PSt (Mn = 2900 g mol−1, Mw/Mn = 1.23) as macro-chain-transfer agent. | |
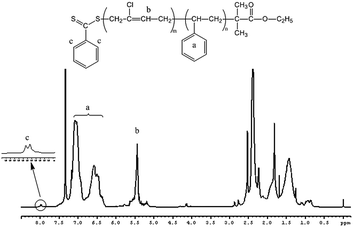 |
| | Fig. 6 1H NMR spectrum recorded in CDCl3 of PSt-b-PCP diblock copolymer (time = 9 h, and conversion = 44.2%). | |
Synthesis of poly(methyl methacrylate)-block-polychloroprene
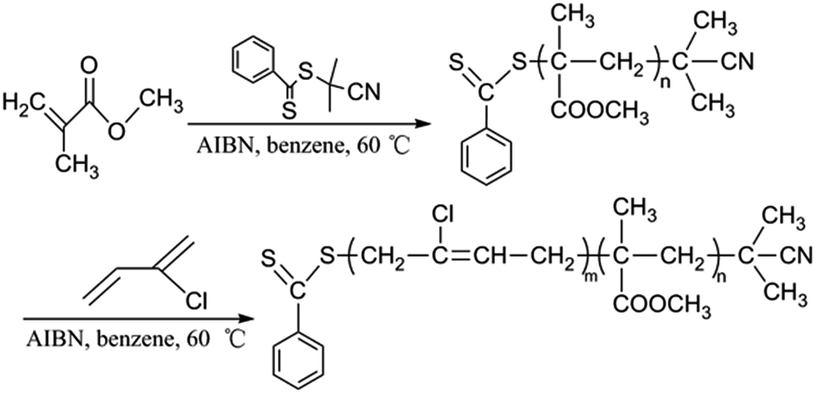
It is widely known that, when preparing a block copolymer, for which one block is based on a methacrylate monomer and the other on a styrene or an acrylate monomer, the methacrylate block should be prepared first.20,43,44 Methacrylate macro-RAFT agent could easily be chain extended via acrylates, styrene or acrylamide. Similarly in our case, the polymethacrylate propagating radicals are relatively good homolytic leaving groups; therefore, the polymethacrylate block was synthesized first, and the polychloroprene block second. PMMA with controlled, narrow molecular weight distribution were successfully synthesized by the RAFT process using CPDB as a CTA.34 Therefore, PMMA-b-PCP diblock polymers were synthesized using CPDB as the initial RAFT agent. The synthetic pathway is shown in Scheme 3. The diblock copolymer was prepared in two steps: synthesis of the first block PMMA, block copolymerization of CP to build the second block of PMMA-b-PCP. The RAFT polymerization of MMA was performed in benzene at 60 °C using CPDB as RAFT-agent and AIBN as initiator. The monomer/CPDB/AIBN molar ratio was lowered to 30/1/0.5 to produce shorter polymeric chains. Precipitation of the final PMMA in cold petroleum ether gave a clean polymer exhibiting all the characteristics required (Mn = 1800 g mol−1, Đ = 1.33 obtained by SEC using a PSt calibration) to be used as the macro-RAFT agent in the polymerization of chloroprene. The second block was synthesized via RAFT polymerization of CP, using the obtained PMMA homopolymer as the macro-RAFT agent, by re-initiation with AIBN in benzene at 60 °C. During the polymerizations, samples were taken from reactor at suitable time intervals for gravimetric conversion measurement, SEC analysis (using PSt standards) and 1H NMR analysis.
 |
| | Scheme 3 Synthetic outline for the preparation of poly(methyl methacrylate)-block-polychloroprene diblock copolymers via RAFT polymerization. | |
The kinetic plots obtained from the samples are displayed in Fig. 7(A). A linear relationship between ln([M]0/[M]) versus time was observed, indicating the first-order kinetics of the polymerization with respect to the concentration of CP. Fig. 7(B) shows the molecular weight distribution of the PMMA macro-CTA and those of the resulting PMMA-b-PCP block copolymers. The overlay of SEC chromatograms showed obvious increase of the molecular weight, with a high molecular weight shoulder. The controlled character of the polymerization was confirmed by the chain extension of the macro transfer agent which led to PMMA-b-PCP of Mn = 6200 g mol−1 (Đ = 1.40) at 50.4% monomer conversion starting from PMMA of Mn = 1800 g mol−1 (Đ = 1.33). However, a small shoulder peak at a high molecular weight was found in the SEC spectrum of the diblock polymer, and this resulted in an increase in Đ of the diblock polymers. A shoulder peak at a high molecular weight could be due to the bimolecular termination and the branching reactions in the polymerization reaction.14,45,46 In a long time under the reaction conditions, the side reaction is obvious. The values of the number averaged molecular weight and Đ is depicted in Fig. 7(C). The number-average molar masses of samples at different conversions clearly show the growth of the PMMA-b-PCP chains. Due to the differences in hydrodynamic volumes of PMMA-b-PCP block copolymers and polystyrene standards, experimental molecular weights of the diblock copolymers evaluated by SEC were slightly lower than the theoretical molecular weights at low and moderate conversions, whereas at 85.5% conversion, the experimental molecular weights appeared to be above the theoretical molecular weights. Branching reactions of conjugated dienes becomes significant when the monomer conversion was high, and the molecular weights slightly deviate from the calculated values towards higher molecular weights.32 Dispersities are low but gradually increase from 1.38 at 34.9% conversion to 1.49 at 72.4% conversion. At conversion 85.5%, Đ of 1.53 were obtained, and this shows that the control is imperfect. For these reactions, the increase in Đ might be ascribed to the irreversible termination due to the quite high initiator level (RAFT to initiator ratio of 2:1) and the possible branching reactions.46
 |
| | Fig. 7 (A) Relationships of ln([M]0/[M]) with polymerization time, (B) SEC traces, and (C) plots of Mn and Mw/Mn vs. conversion in the RAFT polymerization of CP using PMMA (Mn = 1800 g mol−1, Mw/Mn = 1.33) as macro-chain-transfer agent. | |
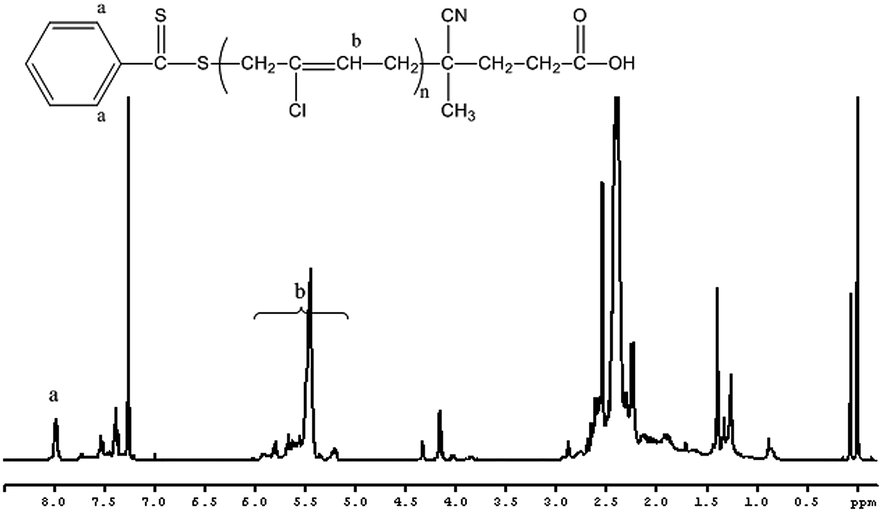
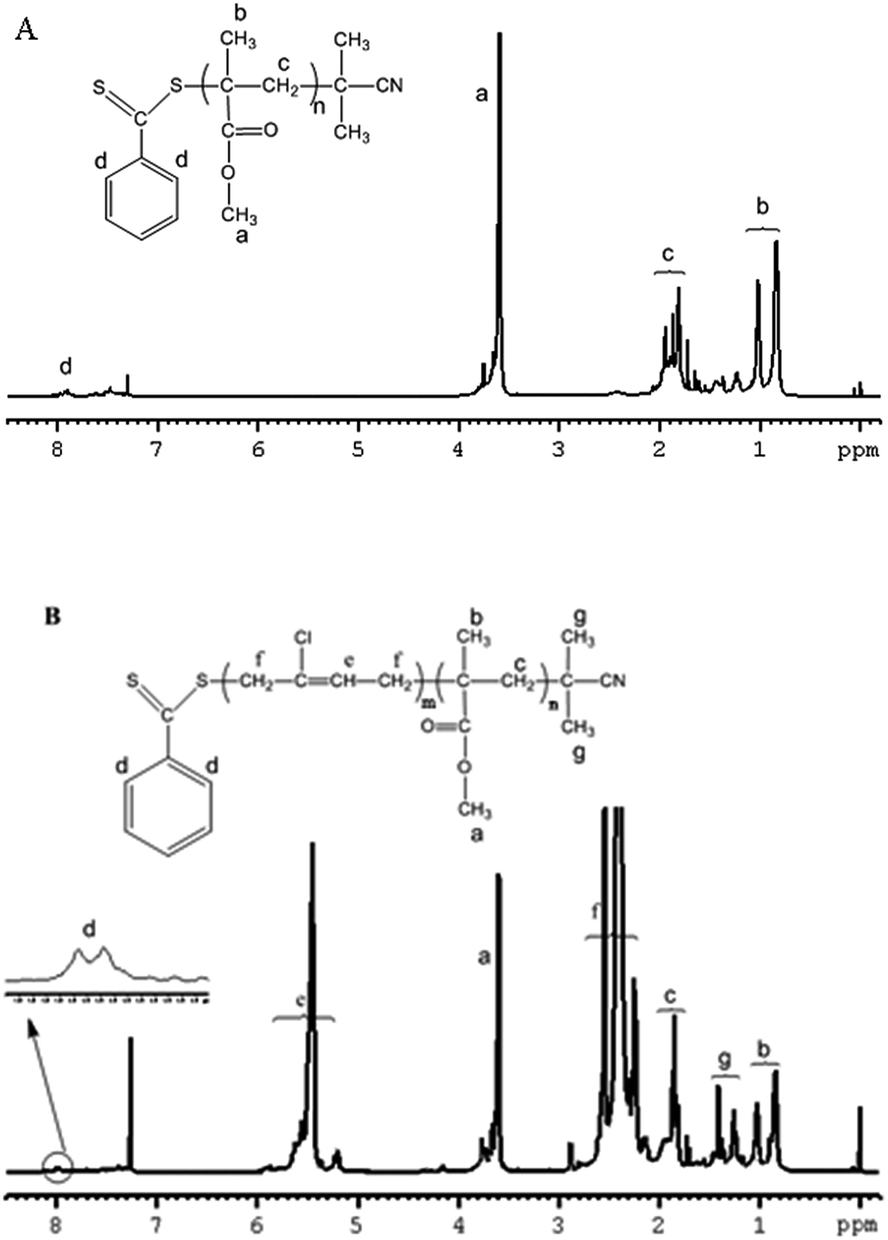
1H NMR spectra were used to characterize the structure of the copolymer. As shown in Fig. 8, the large absorptions of the methyl ester group, –CH2C(CH3)(COOCH3) of the MMA repeating units at 3.6 ppm (a) were observed. The signals b are attributed to the methyl protons of –CH2C(CH3)(COOCH3). Another observation was that the characteristic signals of methylidyne protons of chloroprene appeared at 5.2–6.0 ppm (e) and the characteristic signals of methylene protons of chloroprene appeared at 2.1–3.0 ppm (f), while the signals at 7.92–8.01 ppm (d) correspond to the aromatic protons of CPDB units. H NMR spectra of the copolymers indicate that CP polymerized in the presence of PMMA macro-CTA. Additionally, a clear shift of the SEC curves toward higher molecular weights is observed. These confirm the incorporation of the PCP onto the PMMA chains.
 |
| | Fig. 8 (A) 1H NMR spectrum recorded in CDCl3 of PMMA by RAFT Polymerization with CPDB; (B) 1H NMR spectrum recorded in CDCl3 of PMMA-b-PCP diblock copolymer (time = 39 h, and conversion = 85.5%). | |
Conclusions
The well-defined PCP homopolymers were synthesized, employing ethyl 2-(ethoxycarbonyl)prop-2-yl dithiobenzoate (EPDTB) and 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (CPDTB) as the RAFT-CTAs. However, dibenzyl trithiocarbonate (DBTTC) was shown to be ineffective in RAFT polymerization of CP. We have demonstrated that the successful synthesis of PCP-based block copolymers by RAFT polymerization depends strongly on the order in which the monomers are polymerized to form the blocks. It has proven to be very difficult if the CP block is grown first, followed by the St/MMA block. However, the reverse strategy succeeds. The dithioester end-capped polystyrene was employed as a macro-CTA for the synthesis of diblock copolymer, PSt-b-PCP, employing EPDTB as the initial RAFT agent. Also, well-defined diblock copolymer PMMA-b-PCP has been successfully prepared through the RAFT polymerization using 2-cyano-2-propylbenzodithioate (CPDB) as the initial RAFT agent.
Acknowledgements
The authors acknowledge the financial support from the China National Natural Science Funds (no. 51073010, 51373021).
References
- W. H. Carothers, I. Williams, A. M. Collins and J. E. Kirby, J. Am. Chem. Soc., 1931, 53, 4203–4225 CrossRef CAS.
- K. S. V. Srinivasan, N. Radhakrishnan and M. K. Pillai, J. Appl. Polym. Sci., 1989, 37, 1551–1558 CrossRef CAS.
- S. H. Botros and S. Y. Tawfik, Polym.-Plast. Technol. Eng., 2006, 45, 829–837 CrossRef CAS.
- M. S. Khan, D. Lehmann and G. Heinrich, Acta. Mater., 2009, 57, 4882–4890 CrossRef PubMed.
- M. Cui, L. Liu and J. Kim, J. Appl. Polym. Sci., 2012, 125, 3000–3005 CrossRef CAS.
- K. Zukiene, V. Jankauskaite and S. Petraitiene, Appl. Surf. Sci., 2014, 292, 506–513 CrossRef CAS PubMed.
- M. J. Azizli, G. Naderi, G. R. Bakhshandeh, S. Soltani, F. Askari and E. Esmizadeh, Rubber Chem. Technol., 2014, 87, 10–20 CrossRef CAS.
- A. D. Jenkins, R. G. Jones and G. Moad, Pure Appl. Chem., 2010, 82, 483–491 CAS.
- M. K. Georges, R. P. N. Veregin, P. M. Kazmaier and G. K. Hamer, Macromolecules, 1993, 26, 2987–2988 CrossRef CAS.
- V. Delplace, S. Harrisson, A. Tardy, D. Gigmes, Y. Guillaneuf and J. Nicolas, Macromol. Rapid Commun., 2014, 35, 484–491 CrossRef CAS PubMed.
- K. Matyjaszewski and N. V. Tsarevsky, J. Am. Chem. Soc., 2014, 136, 6513–6533 CrossRef CAS PubMed.
- C. Scholz and K. Matyjaszewski, Polym. Int., 2014, 63, 801–802 CrossRef CAS.
- J. Chiefari, Y. K. B. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- A. Goto, K. Sato, Y. Tsujii, T. Fukuda, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2001, 34, 402–408 CrossRef CAS.
- D. J. Keddie, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2012, 45, 5321–5342 CrossRef CAS.
- Y. Luo and X. Liu, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 6248–6258 CrossRef CAS.
- J. Zhu, D. Zhou, X. Zhu and G. Chen, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2558–2565 CrossRef CAS.
- J. T. Sun, C. Y. Hong and C. Y. Pan, Polym. Chem., 2013, 4, 873–881 RSC.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS.
- D. J. Keddie, Chem. Soc. Rev., 2014, 43, 496–505 RSC.
- S. I. Yusa, K. Fukuda, T. Yamamoto, K. Ishihara and Y. Morishima, Biomacromolecules, 2005, 6, 663–670 CrossRef CAS PubMed.
- G. H. Zheng and C. Y. Pan, Polymer, 2005, 46, 2802–2810 CrossRef CAS PubMed.
- Z. P. Cheng, X. L. Zhu, E. T. Kang and K. G. Neoh, Langmuir, 2005, 21, 7180–7185 CrossRef CAS PubMed.
- R. Bussels, C. Bergman-Gottgens, J. Meuldijk and C. Koning, Polymer, 2005, 46, 8546–8554 CrossRef CAS PubMed.
- J. C. Chen, M. Z. Liu, C. M. Gao, S. Y. Lu, X. Y. Zhang and Z. Liu, RSC Adv., 2013, 3, 15085–15093 RSC.
- D. Benoit, E. Harth, P. Fox, R. M. Waymouth and C. J. Hawker, Macromolecules, 2000, 33, 363–370 CrossRef CAS.
- J. Hua, X. Li, Y. S. Li, L. Xu and Y. X. Li, J. Appl. Polym. Sci., 2007, 104, 3517–3522 CrossRef CAS.
- C. J. Dürr, L. Hlalele, A. Kaiser, S. Brandau and C. Barner-Kowollik, Macromolecules, 2013, 46, 49–62 CrossRef.
- R. Z. Wei, Y. W. Luo and Z. S. Li, Polymer, 2010, 51, 3879–3886 CrossRef CAS PubMed.
- L. Hlalele, D. R. D Hooge, C. J. Dürr, A. Kaiser, S. Brandau and C. Barner-Kowollik, Macromolecules, 2014, 47, 2820–2829 CrossRef CAS.
- W. E. Mochel and J. H. Peterson, J. Am. Chem. Soc., 1949, 71, 1426–1432 CrossRef CAS.
- N. Ajellal, C. M. Thomas and J. F. Carpentier, Polymer, 2008, 49, 4344–4349 CrossRef CAS PubMed.
- N. Pullan, M. Liu and P. D. Topham, Polym. Chem., 2013, 4, 2272–2277 RSC.
- Y. K. Chong, J. Krstina, T. P. T. Le, G. Moad, A. Postma, E. Rizzardo and S. H. Thang, Macromolecules, 2003, 36, 2256–2272 CrossRef CAS.
- S. Fréal-Saison, M. Save, C. Bui, B. Charleux and S. Magnet, Macromolecules, 2006, 39, 8632–8638 CrossRef.
- R. Wang, Y. Luo, B. Li and S. Zhu, Macromolecules, 2008, 42, 85–94 CrossRef.
- A. Kaiser, S. Brandau, M. Klimpel and C. Barner-Kowollik, Macromol. Rapid Commun., 2010, 31, 1616–1621 CrossRef CAS PubMed.
- J. L. Pradel, B. Boutevin and B. Ameduri, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3293–3302 CrossRef CAS.
- R. Wei, Y. Luo, W. Zeng, F. Wang and S. Xu, Ind. Eng. Chem. Res., 2012, 51, 15530–15535 CrossRef CAS.
- I. W. Cheong, C. M. Fellows and R. G. Gilbert, Polymer, 2004, 45, 769–781 CrossRef CAS PubMed.
- C. J. Durr, S. Emmerling, P. Lederhose, A. Kaiser, S. Brandau, M. Klimpel and C. Barner-Kowollik, Polym. Chem., 2012, 3, 1048–1060 RSC.
- P. Lebreton, B. Ameduri, B. Boutevin and J. M. Corpart, Macromol. Chem. Phys., 2002, 203, 522–537 CrossRef CAS.
- Y. K. Chong, T. P. T. Le, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1999, 32, 2071–2074 CrossRef CAS.
- C. Barner-Kowollik, T. P. Davis, J. P. A. Heuts, M. H. Stenzel, P. Vana and M. Whittaker, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 365–375 CrossRef CAS.
- A. Goto and T. Fukuda, Macromolecules, 1997, 30, 5183–5186 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra08715a |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ; [M]0 = initial concentration of the monomer; [RAFT] = concentration of the RAFT agent; Mmonomer = molecular weight of the monomer, in this case, MCP = 88.54; and MRAFT = molecular weight of the RAFT agent.
; [M]0 = initial concentration of the monomer; [RAFT] = concentration of the RAFT agent; Mmonomer = molecular weight of the monomer, in this case, MCP = 88.54; and MRAFT = molecular weight of the RAFT agent.