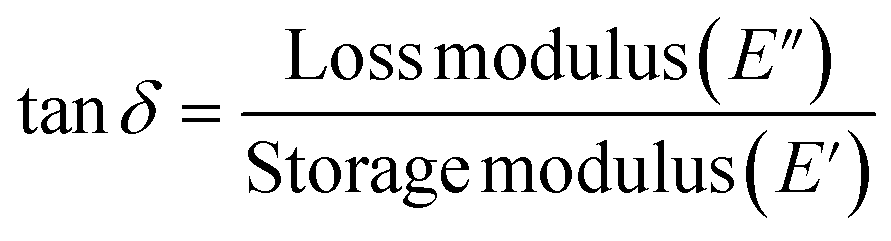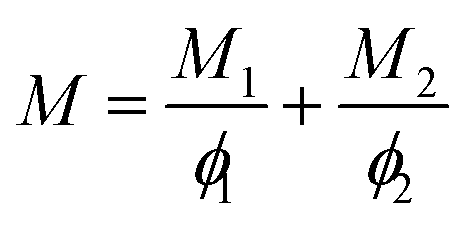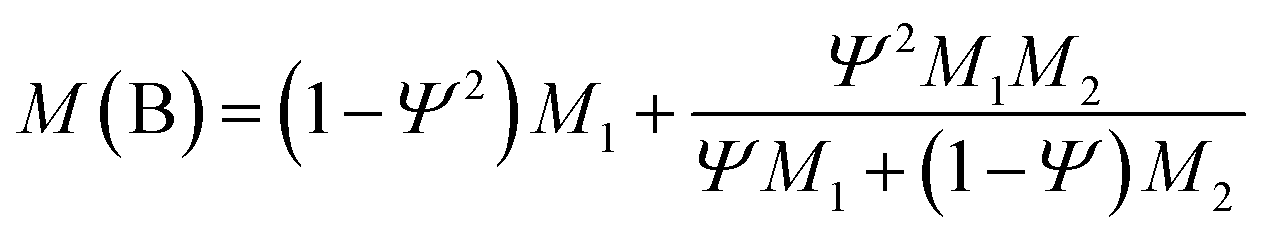DOI:
10.1039/C4RA07823C
(Paper)
RSC Adv., 2014,
4, 60831-60841
Investigation of morphology, mechanical, dynamic mechanical and thermal behaviour of blends based on ethylene vinyl acetate (EVA) and thermoplastic polyurethane (TPU)
Received
30th July 2014
, Accepted 30th October 2014
First published on 30th October 2014
Abstract
Novel blends based on ethylene vinyl acetate/thermoplastic polyurethane (EVA/TPU) at different ratios were prepared via the melt blending technique. Scanning electron microscopy (SEM) studies revealed primarily a two phase morphology in which the minor TPU phase was dispersed in the major continuous EVA matrix. However in the 50/50 blend EVA and TPU appear as a co-continuous phase. The influence of blend ratio on the mechanical properties and hardness was investigated. An 80/20 EVA/TPU blend was observed to exhibit optimum tensile strength and elongation at break. Various models have been applied to predict the change in modulus with blend composition. The Veenstra B model was found the best to correlate the experimental modulus values. All the blends show a single Tg and a shift in Tg with change in blend composition was observed through differential scanning calorimetry (DSC) and dynamic mechanical analysis (DMA), indicating technological compatibility. Thermogravimetric analysis (TGA) was performed to study the thermal stability of the EVA/TPU blends. Incorporation of TPU has also been found to improve the oil resistance properties of the blend system.
Introduction
Blending of two or more structurally and functionally different polymers in suitable proportions has been found to be an important and emerging field for the last few decades to the scientific as well as the industrial community. Blending offers unique, improved and desired properties to a newly developed material that the blending components lack, leading to many and useful applications to revolutionize polymer technology. While synthesizing an entirely new material with all desired properties is a daunting task and demands a high cost, long time and huge effort, effective blending provides a low cost alternative with improved properties and plays a key role in the polymer and elastomer industries, meeting specific purposes. Large numbers of literature reports exist which show that blended polymers provide unusual combinations of mechanical, thermal, chemical, and morphological properties by compensating for the deficiencies of the individual precursors.1,2 Polymer blends are often broadly categorized into miscible, immiscible and compatible blends, among which miscible blends are very few in number. Miscible blends result in a single phase morphology characterized by a single glass transition temperature (Tg). On the other hand, immiscible blends do not satisfy the thermodynamic criteria of miscibility resulting in multiphase morphology causing multiple Tgs. Therefore, the mechanical behavior of immiscible blends becomes more complicated and hence is an intriguing area of research. However, in the case of partially miscible blends, conditional miscibility is exhibited by the corresponding blends depending on the relative proportions of the constituent polymers, mixing temperature and so on.
Ethylene vinyl acetate (EVA) is a random copolymer synthesized from ethylene and vinyl acetate (VA) monomers where the weight percent of VA usually varies from 10 to 45%. The numerous desirable features of EVA, such as high polarity (due to the polar nature of the acetoxy side group), excellent aging resistance, weather resistance, good mechanical properties, resistance to colour change and relatively low cost, have made EVA a candidate of choice for blending with other polymers. In addition, they can provide easier melt processability to the corresponding blends. EVA blends with low density polyethylene (LDPE),3,4 high density polyethylene (HDPE),5 polypropylene (PP),6,7 natural rubber (NR)8 and acrylonitrile butadiene rubber (NBR)9,10 etc. have been extensively studied by several groups of researchers. Blends of EVA/polybutadiene rubber11 have been successfully used for the manufacture of microcellular footwear soling and EVA/PE blends find their application mainly in the cable industry. Naskar et al. studied the miscibility, morphology, crystallization and mechanical properties of EVA/Engage™ blends and successfully used the blend to develop cable insulation and fire resistant low smoke (FRLS) sheathing compounds.12
Thermoplastic polyurethane elastomer (TPU) is one of the most versatile polymers that bridge the gap between rubbers and plastics. They can be used in a wide range of properties, from hard rubbers to soft engineering thermoplastics as they are elastic and melt-processable.13 TPU is a linear segmented block copolymer containing alternating hard (adduct of di-isocyanate and small glycols) and soft (e.g. polyester, polyether etc.) segments connected to each other by the urethane groups (–NH–COO–). By varying the amounts of the hard and soft segments, the properties can be varied over a wide range, and it also influences the properties of the blends. TPUs exhibit a considerable number of superior physical properties like high tensile modulus, resilience, abrasion resistance, wear and tear resistance, good compression set, good chemical and solvent resistance along with low-temperature elasticity.14–17 Such materials can offer flexibility even without the use of plasticizers. However, TPU is a relatively more expensive material. Therefore, attempts have been made to blend TPU with other polar polymers. Maity et al. discussed the effect of blending techniques on the properties of peroxide cured EVA/PU rubber blends.18 Poomalai et al. reported that the impact strength of the PMMA/TPU blends increases significantly with an increase in the percentage of TPU up to 20%, by retaining the tensile strength of PMMA, and a drastic reduction in percentage elongation at break and the tensile modulus of the blends was observed after exposure in different chemical reagents.19 Santra et al. revealed that polyether based TPU and poly(ethylene-co-methyl acrylate) (EMA) blends are miscible throughout the entire composition range via hydrogen bond formation.20 Wang et al. studied the morphology, mechanical properties and rheological behavior of TPU/EPDM blends and improved their compatibility by adding maleic anhydride grafted polyethylene oxide (PEO-g-MA) as a compatibilizer.21 Palanivelu et al. investigated TPU/poly(butylene terephthalate) blends at various compositions and reported that addition of a small amount of TPU to poly(butylene terephthalate) increases the impact strength while the tensile and flexural properties decrease.22
It has already been mentioned that TPUs provide a number of remarkable physical properties like high tensile modulus, excellent abrasion resistance, good compression set, good chemical and solvent resistance etc. On the other hand, EVA copolymer containing 28% VA is widely used as an insulating and sheathing material for medium to high voltage cables, shrinkable films, multilayer packaging and also in the footwear and toy industries due to its high flexibility, good heat resistance and chemical inertness. Since TPU is relatively more expensive than many other polymers, there has been a growing interest in blending TPU with various other polymers in recent years to make it cost effective. This motivated the authors to study the influence of blending TPU with EVA to develop a novel cost effective material. This particular blend system can be used as a new material for the footwear industry and can also find application as a potential candidate for cable sheathing materials with good oil resistance, heat stability and low temperature flexibility. Hence, the objective of this research work is to study the morphology, mechanical, thermal, and dynamic mechanical properties of the blend system in detail and to develop an economically viable useful blend system. However to the best of our knowledge no systematic study has been made so far to develop blends of EVA and TPU. The present investigation explores our observations on the blends of EVA and TPU prepared by the melt blending process in an internal mixer. Attempts have also been made to correlate the mechanical properties with various theoretical models.
Experimental
Materials
Thermoplastic polyurethane (TPU) Desmopan 385 S, composed of 4,4′-diphenylmethane diisocyanate hard segments and polyester based soft segments was procured from Bayer Chemicals, India. The density of TPU is 1.2 g cm−3 with a Shore-A hardness of 84 and melting temperature is around 170 °C. Ethylene vinyl acetate copolymer (EVA) containing 28% vinyl acetate, grade Elvax 265, with a melt flow index (MFI) of 1.7 g 10 min−1 and a density of 0.95 g cm−3, was obtained from Dupont, India.
Preparation of blends
EVA and TPU were mixed in a Haake Rheomix OS (Germany) 600 internal mixer, having a mixing chamber volume of 85 cm3 at 180 °C for 7 min. At first TPU was added and it was allowed to melt for 2 min. After that EVA was added and the mixing was continued for another 5 min. The mixes so obtained were sheeted under hot conditions in an open mill set at 2 mm nip gap. The sheets were compression molded between Teflon sheets for 4 min at 190 °C at a pressure of 5 MPa in an electrically heated hydraulic press (Moore Hydraulic Press, England). The moulded sheets were cooled under pressure to maintain the overall dimensional stability of the moulded articles. Table 1 shows the various blends prepared for this study having different blend ratios of EVA and TPU.
Table 1 Sample designations with composition
| Sample designation |
EVA (wt%) |
TPU (wt%) |
| EVA |
100 |
0 |
| ET 80 |
80 |
20 |
| ET 70 |
70 |
30 |
| ET 60 |
60 |
40 |
| ET 50 |
50 |
50 |
| TPU |
0 |
100 |
Characterization of blends
Mechanical tests
Tensile testing was carried out using a universal testing machine (Hounsfield H10KS) at room temperature at a crosshead speed of 200 mm min−1. Tensile specimens as per ASTM D 412 were punched from the molded sheets using ASTM Die-C. The tensile strength for all of the samples was the maximum stress of the tensile stress–strain curves before they were broken. All results reported were based on the average values of three measurements.
Stress relaxation and tension set
For stress relaxation and tension set measurement, dumbbell specimens were extended up to 100% in the tensile direction at a rate of 200 mm min−1 and kept at that position for 10 min at room temperature and the value of stress was noted after each 30 s interval up to 10 min for stress relaxation measurements. It was then relaxed back to unstressed conditions and the percentage change in dimension in the tensile direction was measured after 15 min and reported as the tension set.| |
 | (1) |
Hardness test
Hardness of the samples was measured on both the Shore A and Shore D scales. Shore D hardness of samples was determined using a Shore D hardness-testing machine (Bowers Mertrology, UK) as per ASTM D2240 standard at room temperature. Hardness reported on the Shore A scale was measured using Rex Durometers (as per ASTM D2240 method) Model 2000, Buffalo Grove, UK. The hardness value was determined by the penetration of the Durometer indenter foot into the sample.
Morphological study
Surface morphology of the blends was examined by using a JEOL JSM 5800 Digital Scanning Electron Microscope (SEM). The accelerating potential 20 kV was used for the analysis of samples. All the blends were cryofractured in liquid nitrogen to avoid any possibility of phase deformation during the cracking process. The cryofractured surface of the blends was etched in tetrahydrofuran (THF) solvent for 4 days in order to remove the TPU phase from the blends. The EVA component of the blend remained unaffected while TPU dissolved in the solvent, appearing as black holes in the SEM photomicrographs. The etched surface after adequate drying for 24 hours at room temperature was gold sputtered and then observed under SEM. From the SEM results particle size determination was done by measuring the domain diameter. The number average (Dn) and weight average (Dw) domain diameter values were calculated using the following equations:| |
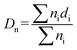 | (2) |
| |
 | (3) |
In eqn (2) and (3), ni is the number of particles with diameter di.
Differential scanning calorimetry (DSC)
Differential scanning calorimetry studies of the pure polymers and the blends were carried out using a DSC Q2000 V24.10 (TA Instruments, USA) at a heating and cooling rate of 10 °C min−1 in an inert atmosphere (N2 atmosphere). Standard aluminum pans were used to analyze the samples. The experiment was conducted from −80 °C to 230 °C for all the samples. Glass transition temperatures (Tg) and melting behaviour were observed from the second heating run of the DSC plot. The data from the second heating cycle were used to eliminate the thermal history.
Thermogravimetric analysis (TGA)
Thermogravimetric analyses (TGA) and derivative thermogravimetry (DTG) of the neat components as well as the EVA/TPU blends were measured by using a thermogravimetric analyzer (TGA Q5000, TA Instruments, USA). The sample weight was 8–10 mg and the heating rate was 10 °C min−1. Tests were performed from ambient temperature to 800 °C under N2 atmosphere.
Dynamic mechanical thermal analysis (DMTA)
Dynamic mechanical thermal analyses of the samples were carried out using an Eplexor 150N DMTA (Gabo Qualimeter, Ahlden, Germany). Tests were carried out at a frequency of 10 Hz under a static strain of 0.50% and a dynamic strain of 0.001%. The samples were first cooled to −100 °C and then subsequently heated at a rate of 2 K min−1. The range of temperature under which the tests were carried out was −100 °C to 80 °C. The sheets were cut into approximately 35 × 10 × 2 mm3 rectangles. The nitrogen flow rate was 200 ml min−1. The temperature corresponding to the peak in the tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ versus temperature plot was taken as the Tg.
δ versus temperature plot was taken as the Tg.
Here tan delta can be defined as the ratio of loss modulus to the storage modulus. It is expressed as:
Oil swelling study
The test specimens were immersed in ASTM 3 oil at room temperature for 7 days. After the required period of time the specimens were removed from the oil, quickly dipped in acetone, and blotted lightly with a clean blotting paper to eliminate the excess oil on the specimen surfaces and the final weight was taken. For every single composition three specimens were tested and their average values are reported. Swelling % was measured as follows:| |
 | (4) |
where M1 = initial mass of specimen in air and M2 = mass of specimen in air after immersion.
Results and discussion
Mechanical properties
Mechanical properties like tensile strength (T.S.), elongation at break (EB), modulus at various strains and hardness are summarized in Table 2. Neat EVA shows a tensile strength of 16.9 MPa and elongation at break of 1061%, whereas TPU exhibits a tensile strength of 35.1 MPa and elongation at break of 805%. However, the tensile strength of an 80/20 EVA/TPU blend is found to be 17.5 MPa with a good elongation at break of 1067%. Thus it is observed that with the addition of only 20 wt% of TPU the tensile strength is improved by 4%. With further addition of TPU in the blend, the tensile strength gradually decreases to 9.3 MPa at 50 wt% of TPU (Fig. 1) Although the value of elongation at break also continuously decreases from 1067% to 494% with increasing TPU in the blend over the entire composition range, ET 80 exhibits a similar elongation at break value to that of neat EVA retaining the excellent elastic properties of the pure polymer.
Table 2 Mechanical properties of the EVA/TPU blendsa
| Sample designation |
TS (MPa) |
EB% |
Mod at 100% (MPa) |
Mod at 200% (MPa) |
Mod at 300% (MPa) |
Shore D Hardness |
| The hardness values in the parentheses indicate the corresponding Shore A hardness. |
| EVA |
16.9 ± 0.4 |
1061 ± 48 |
3.0 ± 0.3 |
3.5 ± 0.3 |
4.0 ± 0.3 |
25.0 (78) |
| ET 80 |
17.6 ± 0.4 |
1067 ± 45 |
3.3 ± 0.2 |
3.9 ± 0.3 |
4.3 ± 0.2 |
25.2 (80) |
| ET 70 |
12.2 ± 0.6 |
832 ± 25 |
3.5 ± 0.3 |
4.0 ± 0.2 |
4.5 ± 0.2 |
25.5 (80) |
| ET 60 |
10.4 ± 0.7 |
722 ± 37 |
3.7 ± 0.3 |
4.1 ± 0.3 |
4.6 ± 0.3 |
29.5 (81) |
| ET 50 |
9.5 ± 0.5 |
494 ± 15 |
3.9 ± 0.2 |
4.7 ± 0.2 |
5.7 ± 0.3 |
31.4 (83) |
| TPU |
35.1 ± 1.1 |
805 ± 32 |
5.9 ± 0.3 |
8.7 ± 0.2 |
11.5 ± 0.4 |
33.5 (86) |
 |
| | Fig. 1 Variation of tensile strength with TPU content. | |
In Fig. 2 the stress–strain curves of EVA and all the blends are shown. It is to be noted that all the EVA/TPU blends have higher moduli than EVA. Fig. 3 clearly shows that the moduli at 100%, 200% and 300% become gradually higher with increasing TPU content in the blend. It is found that in ET 80, ET 70, ET 60 and ET 50 the moduli at 300% show increases of 8%, 15%, 18% and 43% respectively. However, a small variation has been observed at higher strain level (>300% strain) where the modulus of pure EVA becomes slightly higher than those of the 80/20 and 70/30 EVA/TPU blends. It has been observed that after 450% elongation EVA shows crazing phenomena (the sample undergoes yielding and strain hardening due to strain induced crystallization of the amorphous phase). This phenomenon may be attributed to the orientation of the crystalline polyethylene hard segments present in EVA.3,9 In case of ET 60 and ET 50, as the amount of EVA reduces in the blend the crazing phenomenon becomes somewhat restricted. That is why it is observed that the modulus of EVA at higher strain is slightly higher for the 80/20 and 70/30 EVA/TPU blends.
 |
| | Fig. 2 Stress–strain curves of EVA, TPU and their blends. | |
 |
| | Fig. 3 Modulus of the blends as a function of TPU content. | |
The hardness of the EVA/TPU and the blends has been reported on the Shore A and Shore D scale. EVA has a hardness of 25 Shore D and the hardness of the blend varies from 25.2 to 31.4 Shore D. Thus it is found that the hardness of the blend system increases with increasing TPU content.
Mechanical modeling
The mechanical properties of the blend system are found to depend on the relative proportions of the components in the blend. Different theoretical models such as the series, parallel and Veenstra models have been used to describe the change of modulus with composition. The modulus of polymer blends generally ranges between an upper limit given by the parallel model (Reuss model) and a lower limit given by the series model (Voigt model).23,24 The parallel coupling of polymer components means that the strains of all elements are equal and each of them contributes to the final mechanical properties according to the mixing rule. Here the resulting mechanical properties of the blends become independent of interfacial adhesion. The mathematically parallel model is given by the following equation:where M is the mechanical property of the blend under consideration and M1 and M2 are the particular properties of the components 1 and 2 respectively. ϕ1 and ϕ2 indicate the volume fractions of components 1 and 2 respectively.
On the other hand, in the lowest lower bound series model, the contributions of the constituents to the blend system are expressed by the inverted rule of mixing. Here it is assumed that the components are arranged in series with the applied stress. Since in this model all the stress is transmitted through the present interface, interfacial adhesion plays the major role in determining the ultimate properties. The series model is expressed as:
| |
 | (6) |
Veenstra model A and model B can be used for modeling of polymer blends with a droplet/matrix morphology when the dispersed particles are evenly distributed in the matrix. These two models have been mathematically expressed in eqn (7) and (8) respectively:
| |
 | (7) |
| |
 | (8) |
where
Ψ3 = 1 −
ϕ1 =
ϕ2.
M(A) and M(B) indicate the properties obtained from Veenstra model A and model B respectively. Here M1 is taken as the modulus of the matrix phase which in this case is EVA and M2 is the modulus of the dispersed phase, that is TPU.25
These models have been applied by several researchers to characterize the compatibility of various blend systems. Furthermore, although the parallel and series models are valid only for simple and idealized structures, they are often used as limiting models irrespective of morphology. In Fig. 4, the experimental values of 100% moduli of EVA/TPU blends are plotted as a function of wt% of EVA and compared with the theoretical values calculated using the parallel, series, Veenstra A and B models. It is found that the experimental values of the modulus lie very close to the Veenstra B model except for ET 50. It indicates that for the ET 80, ET 70 and ET 60 blends TPU particles are almost evenly dispersed in the EVA matrix which is also further supported by SEM images of the samples.
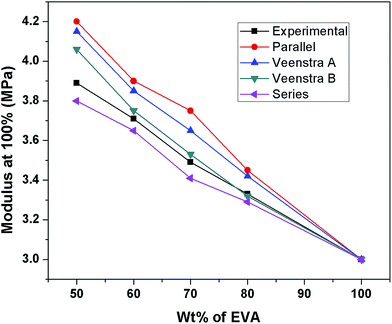 |
| | Fig. 4 Experimental and theoretical modulus at 100% strain for various blends as a function of EVA content. | |
Tension set and stress relaxation
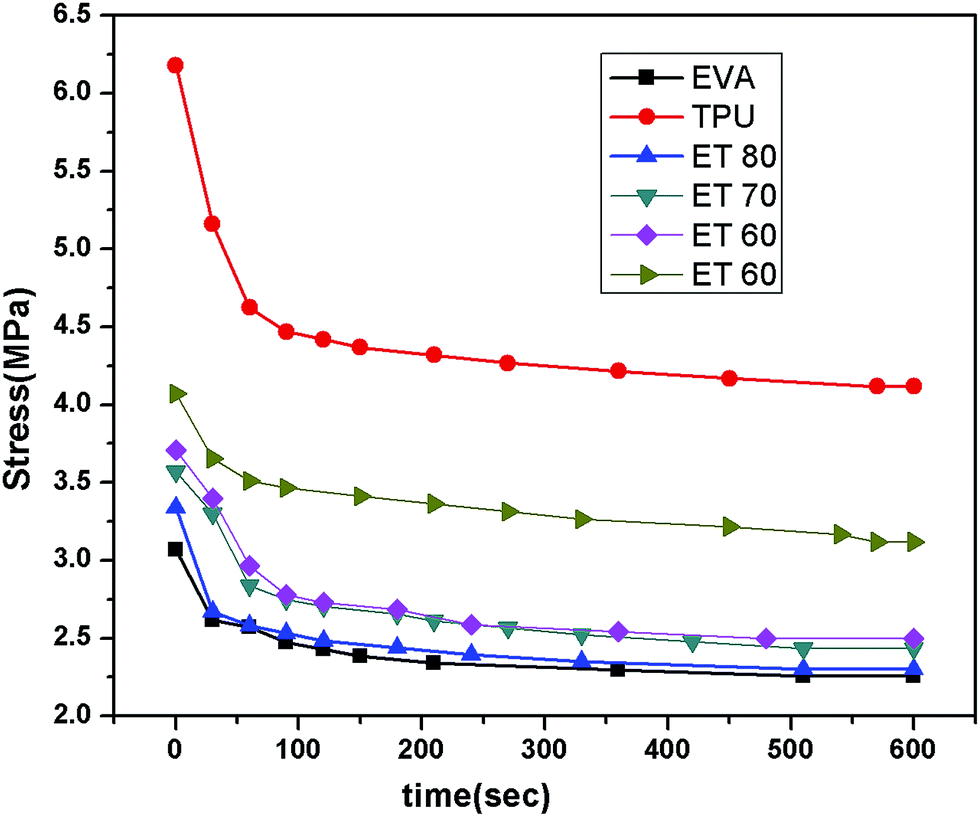
The results of the tension set test are given in Table 3. It clearly shows that pure EVA has somewhat better set properties than TPU. The tension set % for the EVA/TPU blends gradually increase with increasing TPU content in the blends. During this tension set test the change of stress value after each 30 s interval was also noted as the decay of stress up to 10 min. It is a well-known fact that when a constant strain is applied to a rubber sample, the force required for maintaining the strain decreases with time and this behaviour is called ‘stress relaxation’. It has been found from Table 3 and Fig. 5 that the decay of stress (stress relaxation) is relatively lower for EVA and EVA rich blends. As more TPU is added to the blend, the decay of stress slightly increases.
Table 3 Tension set and stress relaxation of different blends
| Sample designation |
Initial stress (MPa) |
Final stress (MPa) |
Stress relaxation (MPa) |
Tension set % |
| EVA |
3.11 |
2.29 |
0.82 |
16 |
| ET 80 |
3.34 |
2.30 |
1.04 |
17 |
| ET 70 |
3.57 |
2.43 |
1.14 |
18 |
| ET 60 |
3.71 |
2.50 |
1.21 |
20 |
| ET 50 |
4.07 |
2.74 |
1.33 |
20 |
| TPU |
6.10 |
4.12 |
1.98 |
21 |
 |
| | Fig. 5 Stress relaxation curves of neat polymers and different blends in tension mode. | |
Morphology
The mechanical properties of the blends are strongly dependent on their morphology (i.e. size, shape and distribution of the blend components) and therefore, the control of morphology plays a significant role in modulating the ultimate performance properties of the blend system.26,27 Factors governing the morphology are composition, interfacial tension, processing conditions and rheological properties of the components. Here THF solvent has been used to preferentially remove TPU, the minor component of the blend, and the black domains indicate the positions of the extracted TPU phase. Fig. 6 shows the morphology of ET 80, ET 70, ET 60, and ET 50 blends. SEM photomicrographs reveal that all the blends, except ET 50, have a two phase morphology. The morphological observation correlates well with the mechanical behavior of the EVA/TPU blends. Fig. 6a shows that in ET 80 the fine, spherical TPU particles are almost homogeneously distributed in continuous EVA matrices leading to good mechanical properties due to better interfacial adhesion and effective stress transfer (from matrix to dispersed phase). The size of dispersed TPU particles becomes coarser in the 70/30 EVA/TPU blend than the 80/20 counterpart. Similarly in the 60/40 blend the particle size of TPU has been found to be even bigger. The number average (Dn) and weight average (Dw) domain diameter values of the blends were calculated for matrix dispersed morphologies and the values are given in Table 4. With increasing TPU content from 20% to 40%, the number average diameter of the dispersed TPU domains increases from 2.95 to 7.15 μm. In case of the 50/50 EVA/TPU blend, both TPU and EVA exist as continuous phases and generate a co-continuous morphology. Fig. 6b and c indicate the presence of larger TPU domains that can be easily removed by THF, whereas in the case of the ET 80/20 blend, extraction of finer TPU particles is somewhat restricted indicating better compatibility and interfacial adhesion. Such composition dependent morphology is related to the mechanical behavior of the blend system and thus ET 80 with finer domain size provides the best combination of physico-mechanical properties with respect to the other blends with coarser domain sizes.
 |
| | Fig. 6 SEM photomicrographs of the THF extracted fractured surfaces of (a) ET 80 (b) ET 70 (c) ET 60 and (d) ET 50. | |
Table 4 Domain diameter of EVA/TPU blends
| Sample designation |
Dn (μm) |
Dw (μm) |

|
| ET 80 |
2.94 |
3.21 |
1.09 |
| ET 70 |
5.60 |
6.18 |
1.10 |
| ET 60 |
7.15 |
7.60 |
1.07 |
Differential scanning calorimetric (DSC) studies
Fig. 7 shows the second heating curve for different blends. TPU shows a glass transition at −37.7 °C whereas the glass transition of EVA is observed at −27.7 °C (Tg). However, for all the EVA/TPU blends a single broad peak appears in the DSC heating curve in between the Tg of the pure polymers. On addition of TPU with EVA, there is a distinct reduction in Tg from −29.12 °C to −32.98 °C as the TPU content in the blend increases from 20 wt% to 50 wt%. Since there is only a very small difference between the Tg of neat EVA and neat TPU, a single Tg does not indicate that EVA and TPU are completely miscible in this composition range. Rather it can be said that there is technological compatibility to some extent between EVA and TPU. Tg values of the various blend systems have been also calculated theoretically using the Fox equation. The equation is mentioned below:| |
 | (9) |
 |
| | Fig. 7 DSC second heating curve for neat polymers and their blends. | |
It is also observed that there is not much difference between the experimental Tg (from DSC) and the Tg calculated using the Fox equation (Table 5). Pure EVA shows a melting endotherm (Tm) at around 78 °C and there is a slight depression of melting point of EVA component in all the blends. However, for different blend compositions the melting temperature of the EVA does not change significantly with variation in TPU content. DSC cooling curves of the samples are shown in Fig. 8. It has been observed that neat EVA exhibits a sharp crystallization peak (Tc) at around 54.9 °C. Although the crystallization peak temperature corresponding to EVA does not change much in the case of the EVA/TPU blends, the crystallization peak height (exothermic peak) corresponding to EVA content gradually decreases with increasing TPU content. Table 5 shows the detailed DSC data of various samples.
Table 5 DSC data of various samples
| Sample designation |
Tg (°C) |
Theoretical Tg (°C) |
Melting temperature, Tm (°C) |
Crystallization temperature, Tc (°C) |
ΔCp (J g−1 K−1) |
| EVA |
−27.70 |
−27.70 |
78.06 |
54.91 |
0.787 |
| ET 80 |
−29.12 |
−29.76 |
77.03 |
55.01 |
0.910 |
| ET 70 |
−31.02 |
−30.78 |
77.64 |
55.03 |
0.875 |
| ET 60 |
−31.83 |
−31.80 |
77.58 |
55.89 |
0.849 |
| ET 50 |
−32.98 |
−32.81 |
77.17 |
56.04 |
0.828 |
| TPU |
−37.76 |
−37.76 |
166.6 |
82.11 |
0.260 |
 |
| | Fig. 8 DSC cooling curves for neat polymers and their blends. | |
Thermogravimetric analysis (TGA)
TGA and DTG plots of EVA, TPU and their blends are shown in Fig. 9 and 10 respectively and the quantitative data are summarized in Table 6. Neat EVA shows a two step degradation; the first decomposition at around 268 °C involves the evolution of acetic acid and the second decomposition which starts at about 379 °C corresponds to the transvinylene formation associated with main chain-scission.28 TPU thermal degradation is also a two stage process. The first step involves the degradation of hard segments (decomposition of urethane group) and the latter is associated with the greatest rate of mass loss of soft segment.29 In case of EVA thermal degradation starts at about 268 °C and about 20.36% weight loss occurs at around 377 °C while for TPU thermal decomposition starts at only 258 °C and % weight loss in the first step is 40.19% up to 380 °C. The results of thermogravimetric analysis show that EVA is thermally somewhat more stable than TPU and the thermal stabilities of all the blends lie in between. From the TGA curve it is observed that the 50% weight loss temperature for TPU is 392 °C whereas for EVA it is rather higher, 457 °C. But in case of ET 80, ET 70 and ET 60, the 50% decomposition temperature has been shifted to 448 °C, 443 °C and 422 °C indicating an increase by 56 °C, 51 °C and 30 °C respectively. For the 50/50 EVA/TPU blend the 50% decomposition temperature is 414 °C which is significantly higher than that of pure TPU. In the case of all the blends with increasing TPU, the 50% decomposition temperatures reduce and the maximum degradation temperatures of the blends are higher indicating better thermal stability of the blends than of TPU.
 |
| | Fig. 9 TGA thermograms of different blends. | |
 |
| | Fig. 10 DTG thermograms of various blends. | |
Similarly from the DTG plots (Fig. 10) it is clearly observed that EVA exhibits two major peaks at 353.4 and 470.2 °C due to deacetylation and main chain degradation respectively. TPU exhibits a first minor peak at 377.6 °C and a second major peak at 409.4 °C associated with its hard and soft segment decomposition respectively. In the case of ET 70, ET 60 and ET 50 initial weight loss occurs in two minor steps at about 350 °C and 450 °C. The first DTG peak arises mainly due to deacetylation and the second one is probably related to the dissociation of TPU into diisocyanate and polyols. However for all the blends the maximum rate of decomposition takes place at around 468 °C due to main chain decomposition of the EVA component. It is also to be noted that the rate of weight loss in the main degradation region gradually decreases with increasing TPU content in the blends.
Table 6 TGA and DTG data of neat polymers and their blends
| Sample designation |
Initial decomposition temperature, Ti (°C) |
Temperature corresponding to 5% decomposition T95 (°C) |
Temperature corresponding to 50% decomposition T50 (°C) |
Maximum decomposition temperature, Tmax (°C) |
| EVA |
267 |
332 |
457 |
470 |
| ET 80 |
262 |
323 |
447 |
467 |
| ET 70 |
262 |
322 |
443 |
466 |
| ET 60 |
260 |
318 |
422 |
466 |
| ET 50 |
258 |
317 |
414 |
465 |
| TPU |
258 |
315 |
392 |
409 |
Dynamic mechanical analysis
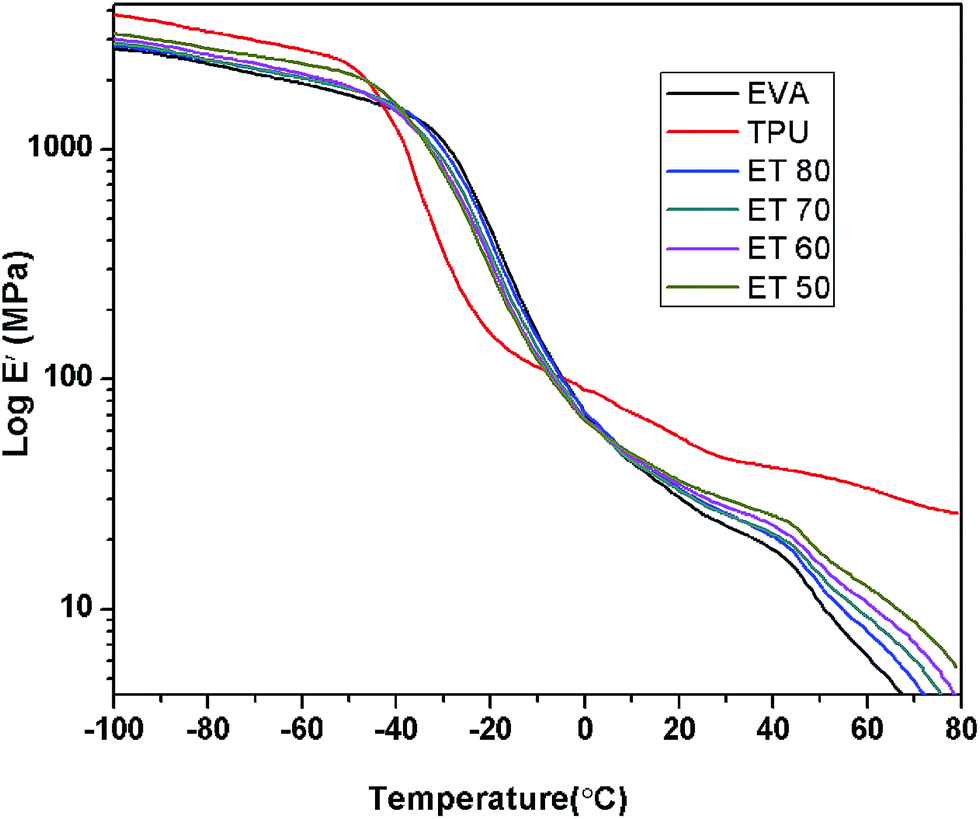
Fig. 11 represents the variation of storage modulus (E′) as a function of temperature in the range −100 °C to 80 °C. Initially the storage modulus of TPU is higher than those of EVA and all the EVA/TPU blends in the low temperature region. The E′ value of the blends gradually goes up with addition of TPU. Then TPU shows a sharp and drastic fall in E′ value at around −48.5 °C due to its glass to rubber transition. However in the case of EVA, the E′ value starts to decrease at the rather higher temperature of −37.8 °C and also the storage modulus drops at a comparatively slower rate than that of TPU. It is clearly seen that in this glass transition region the E′ values of TPU and TPU rich blends become relatively lower than those of EVA and EVA rich blends. Thus at −20 °C the E′ values of ET 80, ET 70, ET 60 and ET 50 are 399.1, 352.1, 330.5 and 297.1 MPa respectively. However at about −9 °C a cross-over region is observed and thereafter again the storage modulus of EVA and EVA rich blends goes down. At ambient temperature TPU shows a much higher storage modulus than EVA and the E′ values of all the blends lie in between those of the pure components. The influence of temperature on the loss modulus of the samples is shown in Fig. 12. From Table 7 it is observed that TPU exhibits a prominent loss peak at about −40.2 °C while for EVA the relaxation corresponding to the loss modulus occurs at −26.2 °C. With increasing TPU content in the blend the maximum loss peak gradually shifts to lower temperature. While ET 80 shows the loss peak at −28.8 °C where the shift is marginal, the other blends show significant shifts to lower temperature as compared to ET 80. The corresponding peaks for ET 70, ET 60, and ET 50 are at −33.3 °C, −34.2 °C and −35.3 °C, respectively. It is also to be noted that the loss peak heights are intermediate between those of the two pure components. Fig. 13 illustrates the effect of temperature on tanδ of the neat polymers and their blends. In the above temperature region TPU shows a single sharp loss tangent peak at −29 °C which is attributed to the glass to rubber transition of the polyester based soft segments of TPU.20 EVA exhibits one major peak at −14.6 °C corresponding to the crystal unconstrained glass–rubber relaxation. This tanδ peak can be assigned as the Tg of EVA and it is higher than the Tg obtained from DSC measurement which is shown by other researchers also. Another tiny peak at around 10 °C is associated with this glass transition which is probably related to the segmental motion in the amorphous zone and corresponds to the crystal constrained glass–rubber transition.30 This α transition peak is mainly prominent in ET 80 and ET 70 due to the presence of higher EVA content. However the Tg values obtained from the E′′ versus temperature plots are always higher than those obtained from tanδ values.9 Tg of all the blends is shifted to lower temperature by a few degrees from the Tg of EVA and a single broad tanδ peak appears in between the tanδ peaks of neat EVA and neat TPU. However, it is to be noted that the glass transition temperatures (Tg) of pure TPU and pure EVA lie very close. Hence, in this case a single tanδ peak does not indicate miscibility in the true sense. It can be said that there is technological compatibility to some extent between EVA and TPU. It is also seen that with increasing TPU content in the blend there is a shifting of the relaxation peak towards the TPU region.
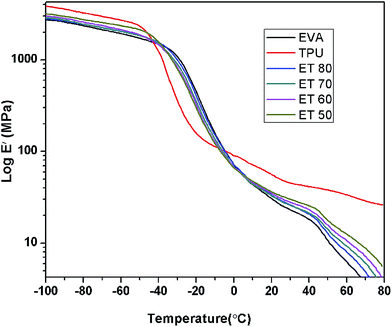 |
| | Fig. 11 Variation of storage modulus with temperature for different blends. | |
 |
| | Fig. 12 Loss modulus versus temperature curves for various blends. | |
Table 7 Dynamic mechanical analysis of different blends
| Sample designation |
tanδ peak (°C) |
Loss modulus peak (°C) |
Storage modulus at −80 °C (MPa) |
Storage modulus at 30 °C (MPa) |
| EVA |
−14.61 |
−26.2 |
2350.12 |
20.67 |
| ET 80 |
−15.80 |
−28.8 |
2450.23 |
23.39 |
| ET 70 |
−16.68 |
−33.3 |
2465.05 |
23.65 |
| ET 60 |
−16.95 |
−34.2 |
2584.69 |
25.68 |
| ET 50 |
−17.45 |
−35.3 |
2759.41 |
27.78 |
| TPU |
−29.02 |
−40.2 |
3251.69 |
43.29 |
 |
| | Fig. 13 tanδ versus temperature plots. | |
Oil swelling study
The oil resistance properties of the blends are measured by immersing the samples in IRM 903 oil for 7 days at room temperature. The results of the oil swelling test are shown in Fig. 14. With increasing TPU content the oil swelling of the blends significantly reduces due to the excellent oil resistance properties of TPU. Neat EVA shows almost 45% swelling after 7 days. However as the amount of TPU increases from 20 wt% to 50 wt% the oil swelling changes from 40% to only 17.6%. It is clearly seen that incorporation of even small amounts of TPU causes a great improvement in the oil resistance properties of EVA.
 |
| | Fig. 14 Oil swelling of different EVA/TPU blends. | |
Conclusions
The mechanical properties of the blends based on EVA and TPU are found to be strongly influenced by the blend ratio and the morphology of the blend system. SEM observation revealed primarily a two phase morphology and the size of dispersed TPU particles increases with increasing TPU content. However, a 50/50 EVA/TPU blend generates a co-continuous morphology. The 80/20 EVA/TPU blend showed the optimum tensile strength and elongation at break which may be due to finer dispersion of the TPU particles in the continuous EVA matrix. With further addition of TPU, the tensile strength and elongation at break gradually reduce but the modulus and hardness continuously improve. The tension set % and the decay of stress at constant strain are lower for EVA rich blends. The experimental moduli have been compared with relevant theoretical models and it was observed that, except for the 50/50 blend, the experimental data fit best with the Veenstra B model suggesting the droplet matrix morphology for rest of the blends which has also been confirmed through morphological analysis. All the blends exhibit single Tg in between the Tg of neat polymers as evidenced from DSC study. With addition of TPU, the Tg of the blends gradually shift towards the Tg of TPU indicating that there is technological compatibility to some extent between the blend components. A similar trend was observed from the loss modulus peak and the loss peak height also takes intermediate values between the pure components. Blends with higher TPU content show higher storage moduli as observed from DMA study throughout the entire temperature range excepting the glass transition region. The oil resistance properties of the blend system significantly improve upon addition of TPU.
Acknowledgements
The authors would like to thank Mr Debdipta Basu (IPF Dresden, Germany) for his kind help in thermal characterization.
References
- J. W. Barlow and D. R. Paul, Polym. Eng. Sci., 1981, 21, 985–996 CAS
 .
. - Polymer alloys and blends—Thermodynamics and rheology, ed. B. D. Favis and L. A. Utracki, Hanser, Munich, 1990 Search PubMed .
- I. Ray and D. Khastgir, Polymer, 1993, 34, 2030–2037 CrossRef CAS .
- M. Faker, M. K. Razavi Aghjeh, M. Ghaffari and S. A. Seyyedi, Eur. Polym. J., 2008, 44, 1834–1842 CrossRef CAS PubMed .
- B. John, K. T. Varughese, Z. Oommen, P. Pötschke and S. Thomas, J. Appl. Polym. Sci., 2003, 87, 2083–2099 CrossRef CAS .
- S. Thomas and A. George, Eur. Polym. J., 1992, 28, 1451–1458 CrossRef CAS .
- D. G. Dikobe and A. S. Luyt, eXPRESS Polym. Lett., 2009, 3, 190–199 CrossRef CAS .
- A. T. Koshy, B. Kuriakose, S. Thomas and S. Varghese, Polymer, 1993, 34, 3428–3436 CrossRef CAS .
- H. Varghese, S. S. Bhagawan, S. S. Rao and S. Thomas, Eur. Polym. J., 1995, 31, 957–967 CrossRef CAS .
- P. Kumari, C. K. Radhakrishnan, S. George and G. Unnikrishnan, J. Polym. Res., 2007, 15, 97–106 CrossRef .
- M. Maiti, R. V. Jasra, S. K. Kusum and T. K. Chaki, Ind. Eng. Chem. Res., 2012, 51, 10607–10612 CrossRef CAS .
- K. Naskar, S. Mohanty and G. B. Nando, J. Appl. Polym. Sci., 2007, 104, 2839–2848 CrossRef CAS .
- Thermoplastic elastomer: A comprehensive review, ed. N. R. Legge, G. Holden and H. E. Schroeders, Hanser, Munich, 1987 Search PubMed .
- Q.-W. Lu and C. W. Macosko, Polymer, 2004, 45, 1981–1991 CrossRef CAS PubMed .
- K. Bagdi, K. Molnár, I. Sajó and B. Pukánszky, eXPRESS Polym. Lett., 2011, 5, 417–427 CrossRef CAS .
- Y. Kim, W.-J. Cho and C.-S. Ha, J. Appl. Polym. Sci., 1999, 71, 415–422 CrossRef CAS .
- F. Feng and L. Ye, J. Appl. Polym. Sci., 2011, 119, 2778–2783 CrossRef CAS .
- M. Maity, C. K. Das, K. N. Pandey and G. N. Mathur, Int. J. Polym. Mater., 2000, 45, 123–133 CrossRef CAS .
- P. Poomalai and Siddaramaiah, J. Macromol. Sci., Part A: Pure Appl.Chem., 2005, 42, 1399–1407, DOI:10.1080/10601320500205764 .
- R. N. Santra, T. K. Chaki, S. Roy and G. B. Nando, Angew. Makromol. Chem., 1993, 213, 7–13 CrossRef CAS .
- X. Wang and X. Luo, Eur. Polym. J., 2004, 40, 2391–2399 CrossRef CAS PubMed .
- K. Palanivelu, P. Sivaraman and M. Dasaratha Reddy, Polym. Test., 2002, 21, 345–351 CrossRef CAS .
- R. C. Willemse, A. Speijer, A. E. Langeraar and A. Posthuma de Boer, Polymer, 1999, 40, 6645–6650 CrossRef CAS .
- O. Martin and L. Avérous, Polymer, 2001, 42, 6209–6219 CrossRef CAS .
- H. Veenstra, P. C. J. Verkooijen, B. J. J. van Lent, J. van Dam, A. P. de Boer and A. P. H. J. Nijhof, Polymer, 2000, 41, 1817–1826 CrossRef CAS .
- M. Baer, J. Appl. Polym. Sci., 1972, 16, 1109–1123 CrossRef CAS .
- L. A. Utracki and Z. H. Shi, Polym. Eng. Sci., 1992, 32, 1824–1833 CAS .
- S. K. Dutta, A. K. Bhowmick, P. G. Mukunda and T. K. Chaki, Polym. Degrad. Stab., 1995, 50, 75–82 CrossRef CAS .
- M. A. Hood, B. Wang, J. M. Sands, J. J. La Scala, F. L. Beyer and C. Y. Li, Polymer, 2010, 51, 2191–2198 CrossRef CAS PubMed .
- B. G. Soares, F. C. F. Alves, M. G. Oliveira and A. C. F. Moreira, J. Appl. Polym. Sci., 2002, 86, 239–249 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.