
Reversal of the enantioselectivity in aldol addition over immobilized di- and tripeptides: studies under continuous flow conditions†
András Gurkaa,
Imre Bucsia,
Lenke Kovácsb,
György Szőllősi*c and
Mihály Bartók*ac
aDepartment of Organic Chemistry, University of Szeged, Dóm tér 8, H-6720 Szeged, Hungary
bInstitute of Pharmaceutical Chemistry, University of Szeged, Eötvös utca 6, H-6720 Szeged, Hungary
cMTA-SZTE Stereochemistry Research Group, Dóm tér 8, H-6720 Szeged, Hungary. E-mail: bartok@chem.u-szeged.hu; szollosi@chem.u-szeged.hu
First published on 10th November 2014
Abstract
Heterogeneous asymmetric direct aldol reactions between aldehydes (2-nitrobenzaldehyde, 2-methylpropanal) and acetone catalyzed by polystyrene resin (PS) supported di- and tripeptides H-Pro-Pro-, H-Pro-Pro-Pro-, H-Pro-Glu(OH)-, H-Pro-Pro-Glu(OH)-, H-Pro-Asp(OH)-, H-Pro-Pro-Asp(OH)-, H-Ser-Glu(OH)-, H-Ser-Ser-Glu(OH)-, H-Val-Glu(OH)-, H-Val-Val-Glu(OH)-MBHA-PS, were studied under identical experimental conditions at room temperature in a continuous-flow fixed-bed reactor (CFBR) system. In the asymmetric aldol reactions reversal of enantioselectivity was observed on H-Pro-Pro-Glu(OH)- and H-Pro-Pro-Asp(OH)-MBHA-PS-supported catalysts (ee 42–67% S) as compared to the H-Pro-Glu(OH)- and H-Pro-Asp(OH)-MBHA-PS-supported catalyst (ee 28–82% R). In the case of H-Pro-Pro- and H-Pro-Pro-Pro-MBHA-PS-supported catalysts reversed enantioselectivity was observed by using the benzoic acid additive (12% S) as compared to the H-Pro-MBHA-PS catalyst (25% R). The stability of the catalysts in the flow system was consistent with the heterogeneous character of the reaction, as was the linear behavior obtained using mixtures of L- and D-enantiomers of the supported H-Pro-MBHA-PS catalyst. The enamine character of the reaction intermediates was supported by ESI-MS measurements. Based on these and the computed structure of the peptides, the conformation of the intermediate adducts is held responsible for chiral induction, therefore for the enantioselectivity inversion observed in these reactions.
1. Introduction
Asymmetric organocatalysis, in general, and stereoselective catalytic formation of C–C bonds, in particular, has become an important method in the production of chiral compounds.1,2 One of the intensively studied reactions utilized for the formation of the C–C bonds is the asymmetric aldol addition. Its significance is emphasized in numerous monographs.1–6 Advantages of the heterogeneous catalytic procedures put the investigation of asymmetric aldol reactions over insoluble solid catalysts at the forefront of catalytic research. Results obtained in such studies have been published continuously.2,7–11 We were among the first to initiate studies on heterogeneous catalytic asymmetric aldol reactions, our first report was published in 2003,12 and we have published on a regular basis ever since.13–16A widely used group of chiral catalysts, especially in asymmetric aldol additions is L-proline (Pro, P) and its synthetic derivatives. In addition, considerable progress has been made in the use of peptide-type organocatalysts. Some short peptides (in particular tripeptides) have proved to act as especially active and enantioselective catalysts.17–19 Asymmetric syntheses using chiral organocatalysts usually lead to the production of important products of high optical purity. An unfortunate property of these catalysts is their low activity requiring reaction times that may range from a few hours to a few days and the relatively high amount of catalysts (up to 30 mol%) needed to obtain reasonable yields. These are among the most important obstacles preventing industrial applications of the numerous organocatalytic methods developed up to now. Moreover, the industry prefers continuous-flow fixed-bed (CFBR) operating methods, which allow process intensification and have several other advantages, such as saving space, time and energy, possibility of increased reactivities and selectivities and easy scale-up of processes.11 These methods have only recently been applied for carrying out asymmetric aldol reactions, so far only 5 publications have reported different variants of such procedures.20–24
Recently we have reported the application of polystyrene resin immobilized proline terminated di- and tripeptides in asymmetric catalytic direct aldol reactions of aldehydes with ketones. Our study revealed that reversal of the enantioselectivity is obtained by increasing the number of the terminal proline units.16 Accordingly, we have shown that both enantiomers of an aldol reaction may be prepared by using polystyrene resin immobilized proline terminated peptide of appropriate length. Moreover, the immobilized peptides may be used in continuous flow systems. In continuation of our research on the use of heterogeneous chiral organocatalysts we have studied the use of immobilized di- and tripeptides in the direct asymmetric aldol reaction of aldehydes and acetone under flow conditions using fixed-bed reactor. The aim of this study was to ascertain that the phenomenon observed in batch system, i.e. the inversion of the enantioselectivity by increasing the peptide length, occurs also in a flow systems where the reactions are carried out under markedly different conditions (i.e. catalyst/reactant ratio can reach high values). Besides the immobilized proline terminated peptides reported in our previous publication, other amino acid terminated supported di- and tripeptides were also prepared and used under identical reaction conditions, to test the role of the terminal proline units in the enantioselectivity inversion. The following resin-supported di- and tripeptides were used as catalysts: P-NH-R, PP-NH-R, PPP-NH-R, PD-NH-R, PPD-NH-R, PE-NH-R, PPE-NH-R, SE-NH-R, SSE-NH-R, VE-NH-R and VVE-NH-R (Fig. 1).
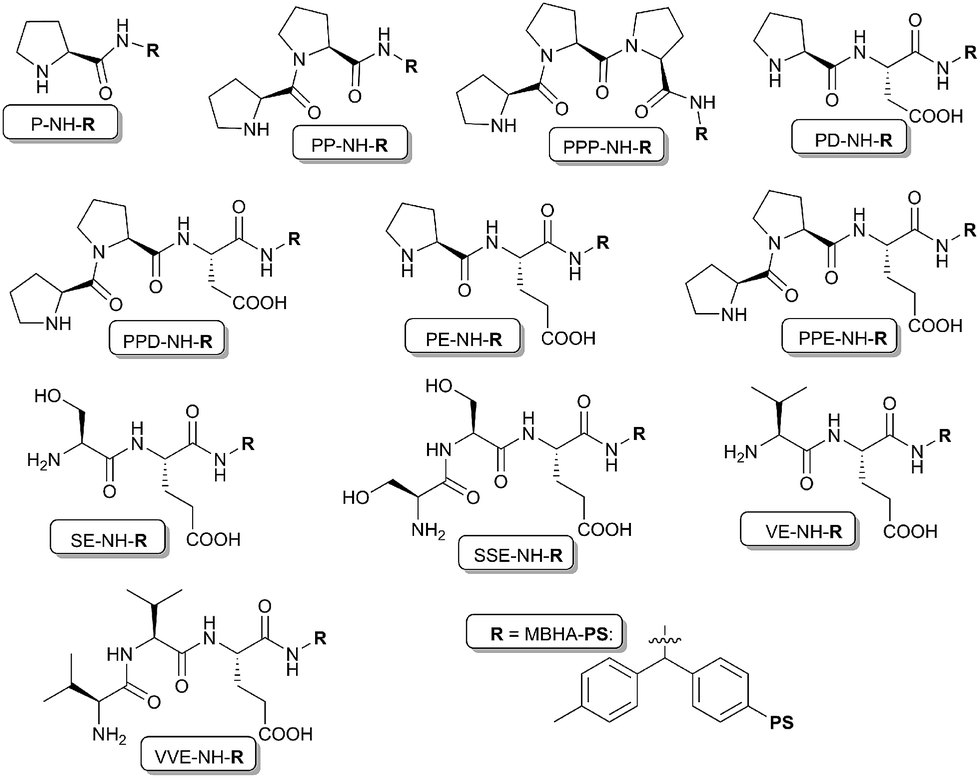 | ||
| Fig. 1 Organocatalysts used in these experiments for aldol reactions (R = MBHA-polystyrene resin). | ||
2. Experimental section
2.1. Preparation and characterization of resin-bonded peptides
The immobilized peptide catalysts were synthesized by a solid-phase technique using Fmoc-strategy.16 Solid-phase synthesis was carried out manually on p-methylbenzhydrylamine polystyrene resin (MBHA-PS resin purchased from Bachem GmbH) with standard methodology. The catalyst loading was calculated from CHN-analysis. IR spectra of the prepared materials were collected on a Bio-Rad Digilab Division FTS-65A/896 FT-IR spectrometer operated in diffuse reflectance mode (DRIFT) between 4000 and 400 cm−1 at 2 cm−1 resolution by averaging 256 scans. The resin-bonded peptides were also characterized by HPLC analysis of the peptides cleaved from the resins and by electrospray ionization mass spectrometry (ESI-MS) according to published methods.162.2. General procedure for the direct aldol reactions
The aldehydes: 2-nitrobenzaldehyde (2-NBA) and 2-methylpropanal (2-MPA) were purchased from Sigma-Aldrich Co. 2-NBA was purified by crystallization, whereas 2-MPA was distilled before use. Acetone of analytical purity was also purchased from Sigma-Aldrich Co. and used as received.The aldol reactions using mixtures of chiral catalysts were conducted over L-Pro-MBHA-PS (P-NH-R) and D-Pro-MBHA-PS catalysts and their mixtures in various ratios. The reactions were carried out in closed glass batch reactors. The given amount of immobilized catalyst (containing 10 mol% of amino acid compared to the aldehyde) was suspended in acetone followed by addition of the given amount of the corresponding aldehyde.
The reaction mixture was stirred at rt. After the specified reaction time the catalyst was removed by filtration and products were analyzed by GC.
Isolated yields were determined following purification of the aldol products by flash chromatography on silica gel using hexane/ethyl acetate 7/3 (2-NBA) or hexane/ethyl acetate 3/1 (2-MPA) mixtures as eluent. The purified products were characterized by their 1H and 13C NMR spectra recorded on a Bruker AVANCE DRX 400 NMR instrument (for spectra see ESI†). 4-Hydroxy-4-(2-nitrophenyl)-butan-2-one: pale yellow oil, 1H NMR (400 MHz, CDCl3) δ (ppm): 7.94 (1H, d, J = 8.1 Hz, ArH), 7.88 (1H, d, J = 8.1, ArH), 7.65 (1H, tr, J = 7.6 Hz, ArH), 7.42 (1H, tr, J = 7.1 Hz, ArH), 5.66 (1H, dd, J = 9.6, 2.0 Hz), 3.10 (1H, dd, J = 17.6, 2.0 Hz), 2.72 (1H, dd, J = 17.6, 9.6 Hz), 2.22 (3H, s, CH3); 13C NMR (100 MHz, CDCl3) δ (ppm): 208.6, 147.2, 138.4, 133.7, 128.2, 124.4, 65.6, 51.0, 30.4. 4-Hydroxy-5-methylhexan-2-one: pale yellow oil, 1H NMR (400 MHz, CDCl3) δ (ppm): 3.77 (1H, m, J = 9.6, 5.5, 3.0 Hz), 2.45–2.60 (2H, m), 2.15 (3H, s, CH3CO), 1.64 (1H, m), 0.85–0.90 (6H, 2 d, J = 9.6 Hz, C(CH3)2); 13C NMR (100 MHz, CDCl3) δ (ppm): 210.1, 72.2, 46.9, 33.0, 30.7, 18.2, 17.6 (where s singlet, d doublet, tr triplet, dd double doublet, m multiplet).
The transformations of the peptide catalysts were followed by ESI-ion-trap-MS measurements on a AGILENT 1100 LC-MSD TRAP SL ion-trap MS instrument operated under positive ion and auto MS-MS mode using the following parameters: ESI: capillary (needle) voltage = 3.5 kV, capillary exit voltage = 136 V, drying gas (N2) = 9 L min−1, drying gas temperature = 623 K, nebulizer gas = 40 psi; ion-trap: scan range = 80–350 m/z, max. accumulation time = 300 ms, fragmentation amplitude = 1.5 V, fragmentation time = 40 ms. Solvent: methanol/0.1% AcOH; flow rate: 0.5 mL min−1; concentration of sample: 0.1 μmol L−1; injected volume 1.5 μL.
3. Results and discussion
Catalysts having been used to date in asymmetric aldol reactions using the CFBR method are shown in Fig. 3. The main objective of these studies, worthy of being described as pioneering, was to confirm the applicability of catalysts shown in Fig. 3 earlier proven to be active in the batch system for the purpose of efficient synthesis of chiral β-hydroxy ketones. Using catalysts with the necessary activity and stability, enantioselectivities exceeding even 90% could be occasionally achieved in the reactions of various aldehydes and ketones.20–24 Among the catalysts having outstanding activities are Pro-Pro-Asp-based tripeptide organocatalysts developed by Wennemers et al.17,19,25 The remarkable novelty of our research lies in the recognition and its experimental confirmation that (S)-amino acids, easily and economically prepared from proteins, and their derivatives can act as catalysts of chiral building blocks not only of (R)- but also of (S)-configuration. On this basis, making use of the justified and attested successful combination of enamine catalysis with the flow technique26 – in accordance with batch measurements16 – we performed experiments using Pro terminated di- and tripeptides followed by testing immobilized peptides containing other amino acids. The catalysts studied are presented in Fig. 1. | ||
| Fig. 2 Continuous-flow aldol reaction in fixed-bed microreactor. | ||
 | ||
| Fig. 3 Organocatalysts studied under continuous flow conditions in direct asymmetric aldol reaction20–24 (PS = polystyrene, TG = TentaGel). | ||
3.1. Aldol reactions in CFBR condition
The MBHA-PS-immobilized peptide catalysts were synthesized according to published method.12,16 Immobilization of the amino acids was confirmed by IR spectroscopy and by HPLC and ESI-MS analysis of the peptides obtained by cleavage from the resins. The IR spectra of the prepared materials were compared with the spectra of the HCl × MBHA-PS resin, as described earlier.16 The appearance of vibrational bands characteristic of amino acids showed the immobilization of these over the resin. Furthermore, the cleavage of the peptides from the resins using HF and analysis of the obtained compounds by HPLC and ESI-MS confirmed the presence of the desired peptides on the surface in 80–95% purity.Results of aldol additions using the immobilized peptides are summarized in Table 1. It was expedient to start our studies on aldol reactions with P-, PP- and PPP-NH-R catalysts, since proline had been the basic chiral component of the catalysts used in the already published experiments. Furthermore, no flow-type experiments with such catalysts had been described prior to our present study. According to these data, the catalysts containing only proline units exhibited satisfactory activities in the reaction of 2-NBA under the given experimental conditions. Their enantioselectivities, however, proved to be moderate. Most important in the reaction carried out over all three catalysts, the reaction yielded the aldol addition product of (R)-configuration in excess (entries 1–3).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Aldehydeb | Conversion % | Selectivityc % | Eed % | ||
| mg | mmol | ||||||
| a CFBR = continuous-flow fixed-bed reactor, immobilized catalyst/SiO2: 1/1.b 2-NBA = 2-nitrobenzaldehyde, 2-MPA = 2-methylpropanal.c Dehydration of the aldol addition product also occurred.d Enantiomeric excess and the configuration of the excess enantiomer.e Isolated yields of the aldol product following experiments prolonged to 5 h time on stream under otherwise identical conditions. | |||||||
| 1 | P-NH-R | 250 | 0.22 | 2-NBA | 31 | 89 | 13R |
| 2 | PP-NH-R | 250 | 0.19 | 2-NBA | 24 | 63 | 13R |
| 3 | PPP-NH-R | 240 | 0.14 | 2-NBA | 37 | 83 | 7R |
| 4 | PE-NH-R | 250 | 0.095 | 2-NBA | 76 | 95 | 28R |
| 5 | PPE-NH-R | 250 | 0.085 | 2-NBA | 70; 63e | 95 | 28S |
| 6 | PD-NH-R | 250 | 0.1 | 2-NBA | 87; 82e | 96 | 24R |
| 7 | PPD-NH-R | 250 | 0.07 | 2-NBA | 78 | 96 | 42S |
| 8 | SE-NH-R | 300 | 0.125 | 2-NBA | 10 | 97 | 52R |
| 9 | SSE-NH-R | 300 | 0.12 | 2-NBA | 2 | 94 | 60R |
| 10 | VE-NH-R | 300 | 0.125 | 2-NBA | 14 | 96 | 54R |
| 11 | VVE-NH-R | 300 | 0.11 | 2-NBA | 2 | 94 | 66R |
| 12 | PE-NH-R | 250 | 0.095 | 2-MPA | 7 | 99 | 82R |
| 13 | PPE-NH-R | 250 | 0.085 | 2-MPA | 3 | 99 | 35S |
| 14 | PD-NH-R | 250 | 0.1 | 2-MPA | 18; 10e | 98 | 80R |
| 15 | PPD-NH-R | 250 | 0.07 | 2-MPA | 13 | 98 | 67S |

Next, the investigation of di- and tripeptide catalysts containing proline, aspartic acid and glutamic acid was undertaken. Experiments using PE-, PPE-, PD- and PPD-NH-R catalysts were performed under completely identical experimental conditions. However, due to the solvent-dependent, varied swellability of the resins, the peptide catalysts immobilized on the resins would occasionally yield non-reproducible experimental data. Consequently, immobilized peptide catalysts mixed with silica gel were used for experiments on aldol reactions. The catalysts exhibited moderate ee and satisfactory activities even without optimization (entries 4–7). The most interesting observation is, however, the reversal of the ee in the reactions of tripeptide containing catalysts as compared to the reactions of dipeptide catalysts. Namely in case of tripeptide catalysts (S)-product formed, while in the presence of immobilized dipeptides (R)-product was obtained in excess. Accordingly, these results showed that the phenomenon observed earlier in batch system was also detected in continuous flow system, in which the catalyst/aldehyde ratio in the reactor is significantly different. To the best of our knowledge, results obtained using immobilized di- and tripeptide catalysts under identical continuous flow conditions, have not been described in the literature. Optimization of the individual reactions was not among our goals; nevertheless, the reversal of enantioselection was significant.
Further experiments were carried out using di- and tripeptide catalysts lacking proline. As shown in Table 1, SE-, SSE-, VE- and VVE-NH-R catalysts, which were not examined until now, were less active in the aldol reaction of 2-NBA, but at the same time afforded good ee values (52–66%) and produced the (R)-configuration product in excess (entries 8–11).
This observation is in agreement with results obtained in experiments using immobilized chiral primary amine catalysts, a group of material studied scarcely before.21,27–30 In addition to the aromatic aldehyde 2-NBA, the experimental results of the aldol reaction of an aliphatic aldehyde, 2-MPA over PD-, PPD-, PE- and PPE-NH-R catalysts are also presented in Table 1 (entries 12–15). These catalysts exhibited lower activities in the transformation of 2-MPA as compared to 2-NBA, but gave significantly higher and also reversed enantioselectivities (80–82% (R), 35–67% (S)).
The positive effect of acids – among them, benzoic acid (BA) – on the efficiency of the aldol reaction has been reported.31,32 These observations led us to examine the behavior of the less active and enantioselective P-, PP- and PPP-NH-R catalysts in the presence of BA. To this end, the reaction between 2-NBA and acetone was studied in the presence of each of the three catalysts, and the effect of BA on conversion and enantioselection at various temperatures was investigated. The experimental data shown in Fig. 4 allow the following conclusions: (i) naturally, conversion increases with temperature on all three catalysts; (ii) it is remarkable, however, that conversion in the presence of BA is significantly higher and its increase by the effect of temperature is faster than in the reactions without BA; (iii) the most prominent novelty is the difference in ee brought about by the presence of BA: on P-NH-R catalyst the (R)-product is formed in excess both in the presence and absence of BA, whereas on the di- and tripeptide catalysts the product of reverse configuration, i.e. the (S)-product is formed in higher amount in the presence of BA.
 | ||
Fig. 4 Effect of the temperature on the conversion (open symbols) and enantioselectivity (closed symbols) of the aldol addition of 2-NBA and acetone in the absence ( , ,  ) and presence of benzoic acid (BA) ( ) and presence of benzoic acid (BA) ( , ,  ) using (a) P-, (b) PP-, (c) PPP-NH-R supported catalysts under flow conditions. Reagents and conditions: 2-NBA (0.026 mmol mL−1), acetone (25 mL), BA (0.04 mmol mL−1), catalyst (250 mg, 0.9 mmol g−1), flow rate 0.1 mL min−1, Conv. = conversion of 2-NBA. ) using (a) P-, (b) PP-, (c) PPP-NH-R supported catalysts under flow conditions. Reagents and conditions: 2-NBA (0.026 mmol mL−1), acetone (25 mL), BA (0.04 mmol mL−1), catalyst (250 mg, 0.9 mmol g−1), flow rate 0.1 mL min−1, Conv. = conversion of 2-NBA. | ||
To our best knowledge, no similar observation has been published in the field of asymmetric direct aldol reactions and studies on this interesting phenomenon are carried out presently in our laboratory and will be reported in due course. One may assume a conformational change of the flexible di- and tripeptide due to protonation with benzoic acid.
3.2. ESI-MS measurements
ESI-MS and MS2 spectra of the filtrates of aldol reactions over Pro-Glu(OH)-NH2 and Pro-Pro-Glu(OH)-NH2 peptides on the one hand and over derivatives of the same peptides immobilized on MBHA-PS resin (PE-NH-R and PPE-NH-R) on the other hand were recorded. The experimental data obtained in these studies allowed to propose the reaction pathways outlined in Scheme 1, which are consistent with certain part-steps of the reaction mechanism suggested in earlier mechanistic studies.33–35 | ||
| Scheme 1 Hypothetic intermediates in asymmetric aldol reaction and species consistent with the results of the ESI-MS measurements (underlined). | ||
According to results of these measurements we concluded: (i) the aldol reaction takes place on surface active centers of immobilized peptide catalysts, because neither the peptides, nor their fragmented products (leached amino acids) can be detected in the solution; (ii) the experimentally identified intermediates suggested the validity of the so-called enamine mechanism for these catalysts as well; (iii) regrettably, due to their less stable nature, the presence of intermediates that also include the aldehyde cannot be convincingly verified under the experimental conditions applied.
3.3. Stability of the catalysts and reactions with mixtures of L- and D-Pro-NH-R catalysts
One of the most important characteristic of heterogeneous catalysts which may be studied easily in continuous flow system is the stability of the catalysts during reaction. Deactivation may be caused either by leaching of the catalytically active species or by poisoning of the active centers by irreversible reaction with one of the reactants or products. The stability of the heterogeneous catalysts developed for asymmetric aldol additions was seldom tested in a continuous flow system.20–22 Results obtained during reactions of 8 h in stream using catalysts PD-NH-R and PPD-NH-R are presented in Fig. 5. Both catalysts were found to be stable following 8 h use, though the tripeptide containing material lost some activity during the first few hours of use. | ||
| Fig. 5 Stability of PD- and PPD-NH-R catalysts during time on stream in the asymmetric aldol addition of acetone to 2-NBA. Reagents and conditions: 300 mg catalyst + SiO2 (1/1), 2-NBA (0.0326 mmol mL−1) in acetone, flow rate 0.1 mL min−1, TON (turn over number) number of transformations over one active site during the given time. | ||
Both materials provided the same conversions at 8 h, which confirmed the successful immobilization of the corresponding amino acids in each preparation step. The enantioselectivity was also found constant during time on stream following a short induction period. The turn over number obtained in 8 h was similar (∼7.5) as obtained in batch reactor considering the product obtained in two reactions of 4 h. Thus these catalysts may be used in continuous flow system providing the similar productivity as in batch mode and sparing the work-up procedure necessary in the latter case.
One of the methods for studying the reaction mechanism of asymmetric reactions is to determine the relationship between the chirality of the catalyst and the ee of the product formed. Non-linear relationship may indicate the involvement of diastereomeric intermediates with different reactivity of the hetero- and homochiral complexes.36 There has been significant progress in studying the phenomenon in amino acid catalyzed direct aldol reactions,37,38 to our best knowledge, however, data on supported peptide catalysts have not been published. Thus, in our efforts to obtain further information on the mechanism of the asymmetric aldol reaction taking place on the supported peptide catalysts experiments using mixtures of resins having bonded L- and D-proline on their surface (L-P-NH-R + D-P-NH-R) were carried out. Data obtained in these experiments are summarized in Fig. 6, which showed a linear behavior in the reaction of 2-NBA and acetone as a function of the composition of the catalyst mixture.
 | ||
| Fig. 6 Effect of L-P-NH-R and D-P-NH-R catalyst mixture composition on the ee obtained in the aldol reaction of 2-NBA with acetone (reaction condition: batch reactor, 2-NBA 0.2 mmol, acetone 1 mL, catalyst 0.02 mmol, rt, reaction time 22 h). | ||
This clearly linear behavior is consistent with the heterogeneous nature of the reaction, as in homogeneous catalytic systems low solubility and aggregation of the amino acid used as catalyst cause deviations from the linearity.15,36
4. Interpretation and conclusion
Our studies on the direct asymmetric aldol reaction between 2-NBA or 2-MPA and acetone over peptide-type catalysts performed using a CFBR procedure led to the following conclusions: (i) under the given experimental conditions, in the presence of dipeptide-NH-R-type catalysts (PP-, PE-, PD-, SE-, VE-NH-R) and P-NH-R β-hydroxy ketones of (R) absolute configuration are formed; (ii) (R)-products are also formed on tripeptide catalysts containing only proline (PPP-NH-R) and on those containing no proline (SSE- and VVE-NH-R); (iii) catalysts containing proline and either aspartic acid or glutamic acid (PPD-NH-R, PPE-NH-R) promote the formation of (S)-products in excess; (iv) according to additional new experimental observation, in the presence of benzoic acid reversal of enantioselection takes place on catalysts containing only proline di- and tripeptides. Namely, on P-NH-R the (R)-product, whereas on PP-NH-R and PPP-NH-R catalysts the (S)-product is formed in larger amount; (v) prolonged experiments over two catalysts showed good stability of the immobilized peptides both as concerns their activity and the enantioselectivity of the products; (vi) the results of ESI-MS as well as those of studies on mixtures of the L- and D-P-NH-R catalysts supported the enamine mechanism and the heterogeneous character of the asymmetric direct aldol reaction occurring on the peptides bonded to the resin support.The stereochemistry of organocatalyzed asymmetric reactions is usually determined by the absolute configuration of the chiral catalyst.1–5 According to numerous experimental observations, however, other factors may also play decisive roles in determining the sense of enantioselection, i.e. the absolute configuration of the product.14 Results so far obtained in asymmetric aldol reactions catalyzed by peptide catalysts highlight the importance of the three-dimensional position of the functional groups of catalysts and the conformation of intermediates leading to chiral induction.17,19
Based on the hypotheses, computation and experimental data reported to date, the interpretation of the above can be summarized as follows. The conformations of the di- and tripeptide catalysts studied, as optimized according to our calculations, are shown in Fig. 7.16 In accordance with results earlier published,17,19 as well as with our own calculations,16 the PPD- and PPE-NH-R tripeptides have a turn-like structure, in which the secondary amine of proline is in close proximity of the carboxylic acid group of aspartic acid17 and glutamic acid. The higher activity and the stronger effect on ee of PPD as compared to PPE can be explained by the difference between the “close proximities” in the two tripeptides. The P-, PP- and PPP-NH-R catalysts contain no carboxylic group, and in the case of the SSE- and VVE-NH-R catalysts the distance between the primary amine and the carboxyl groups is not optimal. Therefore, not only activity and ee will be lower, but the sense of enantioselection may also be unchanged as compared with the immobilized dipeptides.
 | ||
| Fig. 7 Computed conformations of immobilized peptides. | ||
Based on these new experimental data, it is reasonable to assume that the presence of the enamine-type intermediate plays a determinant role on supported catalysts under continuous-flow conditions as well. The probable intermediates detected by our measurements suggest similar mechanisms for the formation of (S) and (R) products, because the formation of hydroxylamine and the pertinent enamine-type intermediates in the presence of acetone can be assumed on both di- and tripeptide catalysts (Scheme 1). Taking into consideration these new experimental results, the cause of the reversal of enantioselectivity observed on catalysts PPD-NH-R and PPE-NH-R as compared the other catalysts used in this study is the divergent structures of the intermediate adducts formed between the enamine of the chiral catalysts and the aldehyde. Consequently, catalysts containing Glu and Asp exhibited similar behavior as regards the inversion of the enantioselectivities.
To sum up, in the asymmetric aldol addition catalyzed by polystyrene-supported di- and tripeptides containing proline and aspartic or glutamic acid reversal of enantioselectivity was observed in the CFBR conditions, too. The determinant role of the conformation of the intermediate adducts is held responsible for chiral induction. These results point our attention to further tasks such as (i) optimizations (including the preparation of the catalysts) in order to increase the activities and enantioselectivities of these catalysts in flow conditions, (ii) studying further chiral catalysts in the aldol reaction and other asymmetric reactions in flow conditions, too. The experimental data reported above highlight the practical significance of the possibility that further research in this promising direction may permit the development of procedures to synthesize chiral building blocks with (R) or (S) absolute configuration using the same cheap chiral source, i.e. (S)-amino acids.
Acknowledgements
Financial support by the Hungarian National Science Foundation (OTKA Grant K 109278) is highly appreciated. The research was supported in the framework of TÁMOP 4.2.4. A/2-11-1-2012-0001 “National Excellence Program – Elaborating and operating an inland student and researcher personal support system” key project. The project was subsidized by the European Union and co-financed by the European Social Fund.References
- Asymmetric Organocatalysis, ed. A. Berkessel and H. Gröger, Wiley-VCH, 2005 Search PubMed.
- Comprehensive Enantioselective Organocatalysis, ed. P. I. Dalko, Wiley-VCH, 2013 Search PubMed.
- S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471–5569 CrossRef CAS PubMed.
- D. E. A. Colby, S. M. Mennen, Y. Xu and S. J. Miller, Chem. Rev., 2007, 107, 5759–5812 CrossRef PubMed.
- B. M. Trost and C. S. Brindle, Chem. Soc. Rev., 2010, 39, 1600–1632 RSC.
- Modern Methods in Stereoselective Aldol Reactions, ed. R. Mahrwald, Wiley-VCH, 2013 Search PubMed.
- Handbook of Asymmetric Heterogeneous Catalysis, ed. K. Ding and Y. Uozumi, Wiley-VCH, 2008 Search PubMed.
- M. Gruttadauria, F. Giacalone and R. Noto, Chem. Soc. Rev., 2008, 37, 1666–1668 RSC.
- A. F. Trindade, P. M. P. Gois and C. A. M. Afonso, Chem. Rev., 2009, 109, 418–517 CrossRef CAS PubMed.
- M. Benaglia, Recoverable and Recyclable Catalysts, Wiley & Sons, UK, 2009 Search PubMed.
- T. Tsubogo, T. Ishiwata and S. Kobayashi, Angew. Chem., Int. Ed., 2013, 52, 6590–6604 CrossRef CAS PubMed.
- G. Szőllősi, G. London, L. Baláspiri, C. Somlai and M. Bartók, Chirality, 2003, 15, S90–S96 CrossRef PubMed.
- G. London, G. Szőllősi and M. Bartók, J. Mol. Catal. A: Chem., 2007, 267, 98–101 CrossRef CAS PubMed.
- M. Bartók, Chem. Rev., 2010, 110, 1663–1705 CrossRef PubMed.
- G. Szőllősi, M. Fekete, A. A. Gurka and M. Bartók, Catal. Lett., 2014, 144, 478–486 CrossRef PubMed.
- G. Szőllősi, A. Csámpai, C. Somlai, M. Fekete and M. Bartók, J. Mol. Catal. A: Chem., 2014, 382, 86–92 CrossRef PubMed.
- P. Krattiger, R. Kovasy, J. D. Revell, S. Ivan and H. Wennemers, Org. Lett., 2005, 7, 1101–1103 CrossRef CAS PubMed.
- M. R. M. Andreae and A. P. Davis, Tetrahedron: Asymmetry, 2005, 16, 2487–2492 CrossRef CAS PubMed.
- J. D. Revell and H. Wennemers, Adv. Synth. Catal., 2008, 350, 1046–1052 CrossRef CAS.
- A. Massi, A. Cavazzini, L. Del Zoppo, O. Pandoli, V. Costa, L. Pasti and P. P. Giovannini, Tetrahedron Lett., 2011, 52, 619–622 CrossRef CAS PubMed.
- A. L. W. Demuynck, L. Peng, F. de Clippel, J. Vanderleyden, P. A. Jacobs and B. F. Selsa, Adv. Synth. Catal., 2011, 353, 725–732 CrossRef CAS.
- C. Ayats, A. H. Henseler and M. A. Pericas, ChemSusChem, 2012, 5, 320–325 CrossRef CAS.
- S. B. Ötvös, I. M. Mándity and F. Fülöp, J. Catal., 2012, 295, 179–185 CrossRef PubMed.
- G. Rulli, K. A. Freriksen, N. Duangdee, T. Bonge-Hansen, A. Berkessel and H. Gröger, Synthesis, 2013, 45, 2512–2519 CrossRef CAS PubMed.
- J. D. Revell, D. Gantenbein, P. Krattiger and H. Wennemers, Biopolymers, 2006, 84, 105–113 CrossRef CAS PubMed.
- Y. Arakawa and H. Wennemers, ChemSusChem, 2013, 6, 212 CrossRef CAS.
- X. Shao, C. Li, S. W. Chen, K. Yao and M. Yao, Colloids Surf., A, 2013, 424, 66–73 CrossRef CAS PubMed.
- S. Luo, X. Zheng and J.-P. Cheng, Chem. Commun., 2008, 5719–5721 RSC.
- W. Wang, X. B. Ma, J. W. Wan, J. Cao and Q. Tang, Dalton Trans., 2012, 5715–5726 RSC.
- I. Muylaert, A. Verberckmoes, J. Spileers, A. Demuynck, L. Peng, F. de Clippel, B. Sels and P. Van Der Voort, Mater. Chem. Phys., 2013, 138, 131–139 CrossRef CAS PubMed.
- J. G. Hernandez and E. Juaristi, Tetrahedron, 2011, 67, 6953–6959 CrossRef CAS PubMed.
- B. Wang, X.-W. Liu, L.-Y. Liu, W.-X. Chang and J. Li, Eur. J. Org. Chem., 2010, 5951–5954 CrossRef CAS.
- B. List, L. Hoang and H. J. Martin, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5839–5842 CrossRef CAS PubMed.
- C. Marquez and J. O. Metzger, Chem. Commun., 2006, 1539–1541 RSC.
- D. A. Bock, C. W. Lehmann and B. List, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20636–20641 CrossRef CAS PubMed.
- C. Girard and H. B. Kagan, Angew. Chem., Int. Ed., 1998, 37, 2922–2959 CrossRef.
- K. Sakthivel, W. Notz, T. Bui and C. F. Barbas III, J. Am. Chem. Soc., 2001, 123, 5260–5267 CrossRef CAS PubMed.
- M. Klussmann, H. Iwamura, S. P. Mathew, D. H. Wells Jr, U. Pandya, A. Armstrong and D. G. Blackmond, Nature, 2006, 441, 621–623 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra07188c |
| This journal is © The Royal Society of Chemistry 2014 |