
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExclusive catalytic hydrogenation of nitrobenzene toward p-aminophenol over atomically precise Au36(SR)24 clusters†
Jinzhi Lua,
Kun Tangb,
Guodong Qic,
Chao Juand,
Jun Xu c,
Zhenfeng Caid,
Dan Li*d,
Xiao Caia,
Xu Liua,
Mingyang Chen*b,
Weiping Dinga and
Yan Zhu*a
c,
Zhenfeng Caid,
Dan Li*d,
Xiao Caia,
Xu Liua,
Mingyang Chen*b,
Weiping Dinga and
Yan Zhu*a
aKey Lab of Mesoscopic Chemistry of MOE and Jiangsu Key Lab of Vehicle Emissions Control, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, China. E-mail: zhuyan@nju.edu.cn
bSchool of Materials Science and Engineering, University of Science and Technology Beijing, Beijing 100083, China. E-mail: mychen@ustb.edu.cn
cState Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics, Chinese Academy of Sciences, Wuhan 430071, China
dKey Laboratory of Green Chemistry and Technology of Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, China. E-mail: danli@scu.edu.cn
First published on 10th September 2024
Abstract
Despite the advances in devising green methodologies for selective hydrogenation of nitrobenzene toward p-aminophenol, it is still difficult to realize p-aminophenol as the exclusive product in heterogeneous metal catalysis, as the excessive hydrogenation of nitrobenzene usually results in the aniline byproduct. Herein we report that a metal cluster containing 36 gold atoms capped by 24 thiolate ligands provides a unique pathway for nitrobenzene hydrogenation to achieve a p-aminophenol selectivity of ∼100%. The gold cluster can efficiently suppress the over-hydrogenation of amino groups via hydroxyl rearrangement with the aid of water and sequentially the proton transfer promoted by acid toward p-aminophenol. More notably, remarkable catalytic performances can be extended to clusters with similar structures such as Au28(SR)20 and Au44(SR)28, where only an atomic layer change of 2.1 Å thickness in the Au36(SR)24 cluster can tailor the proton affinity for the amino group of the key intermediate phenylhydroxylamine, thereby altering the activity while the p-aminophenol selectivity remained.
Introduction
p-Aminophenol (PAP) is a primary chemical for the synthesis of medicine, rubber and dyestuffs.1 Currently, the industrial route to PAP production involves the selective hydrogenation of nitrobenzene over heterogeneous catalysts such as Pt/C to the intermediate phenylhydroxylamine (PHA), followed by the Bamberger rearrangement of PHA with the help of 10–15 wt% sulfuric acid toward PAP (Fig. 1a).2–5 However, the excessive hydrogenation of nitrobenzene can lead to the formation of the aniline by-product, which is essentially inevitable. Meanwhile, the catalysts are liable to decompose and deactivate in the sulfuric acid solution. Many strategies have been developed to suppress the continual hydrogenation process of PHA via desorbing from the catalyst surface into the aqueous phase, sequentially to make the rearrangement promoted by Brønsted acids to obtain PAP (Fig. 1b).1,4,6–19 In a series of seminal studies, Wang and colleagues reported that the Pt-enriched surface of PtRh bimetallic nanoparticles can readily inhibit the strong adsorption of the PHA intermediate over the catalyst surface, leading to 95.4% selectivity for PAP.11 Similarly, the toxic behavior of sulfur in the active center of the Pt/C catalyst pretreated with thiourea deliberately sacrificed the dissociation and hydrogen adsorption capacity of the catalyst. This resulted in a relatively low concentration hydrogen environment, thereby reducing the adsorption strength of the intermediates on the catalyst and preventing the over-hydrogenation process, which gave rise to 72.5% selectivity for PAP.18 Extremely high acid concentrations such as >20 wt% sulfuric acid were even used to enhance the selectivity of PAP (>99%), leading to deleterious effects on the catalysts and reactor lifetimes.14 Indeed, a significant breakthrough in the exclusive hydrogenation of nitrobenzene to PAP currently has not been made. Therefore, an alternative approach that would be completely distinct from previously conventional routes is highly desired. | ||
| Fig. 1 (a) Distinct comparison between the traditional route of nitrobenzene hydrogenation and the proposed route shown in this work. The hydrogen atom color codes: black from H2, green from H2SO4, blue from water, and gray from the benzene ring. (b) Comparison of Au36(SR)24 with recently reported catalysts based on the correlation of the PAP selectivity with the molar ratio of H2SO4 to nitrobenzene. (c) Crystal structure of the Au36(SR)24 cluster. Color codes: orange = Au; yellow = S; gray = C. The H atoms are omitted for clarity. (d) Catalytic performances of the Au36(SR)24 catalyst for nitrobenzene hydrogenation with the sulfuric acid concentration. (e) Catalytic performances of the Pt/C catalyst for nitrobenzene hydrogenation with the sulfuric acid concentration. Reaction conditions: 3 mg catalyst, 0.5 mmol nitrobenzene, 10 mg CTAB, 4 MPa H2, 120 °C for 24 h. | ||
Atomically precise metal clusters with exact numbers of atoms and crystallographic structures have been documented to exhibit unexpectedly catalytic performances for many chemical processes, which are quite different from those of typical nanoparticle catalysts.20–23 These metal clusters are of significant interest in catalysis, which can provide novel reaction pathways to control product selectivity, and the changes in the size and composition of clusters such as the addition or removal of a single atom or even the doping of a heteroatom can significantly alter their catalytic properties.24–30 For example, doping of one Pt atom into one core of Au38(SR)24 cluster can elevate the HOMO energy and broaden the HOMO–LUMO gap (HOMO: highest occupied molecular orbital; LUMO: lowest unoccupied molecular orbital), whereas doping of two Pt atoms into the two cores of Au38(SR)24 can narrow the HOMO–LUMO gap, in which the former showed higher activity for CO2 electroreduction reaction than the latter.30
Herein, we report the remarkable performance of a Au36(SR)24 cluster catalyst for the selective hydrogenation of nitrobenzene, in which PAP is yielded as an exclusive product in a low-content sulphuric acid (<1 wt%) and the recyclability of the catalyst is also realized. Notably, a novel reaction pathway for nitrobenzene hydrogenation is navigated over the catalyst surface, which is different from previously reported processes that PHA needs to desorb from the catalyst into the aqueous phase and is further converted to PAP. More interestingly, our study shows that the catalytic performance is not limited to Au36(SR)24, and structurally similar clusters such as Au28(SR)20 and Au44(SR)28 also achieved ∼100% selectivity for PAP from nitrobenzene hydrogenation.
Results and discussion
The crystal structure of the Au36(SR)24 (SR = 4-tert-butylbenzenethiol) cluster can be disassembled into a twenty-atom gold kernel and eight external staples.31,32 As shown in Fig. 1c, the gold kernel is packed in a face-centered cubic mode, and the surface motifs can be viewed as eight Au2(SR)3 staples, in which four dimeric staples are slightly distorted with the short distance of the gold kernel and another four dimeric staples are co-planar with the large distance of the gold kernel. We next explored the hydrogenation of nitrobenzene catalyzed by this cluster in a four-phase reaction system, where hexadecyl trimethyl ammonium bromide (CTAB) was added in the reaction to effectively increase the solubility of nitrobenzene in the water phase (Fig. S1a†). As shown in Fig. 1d, Au36(SR)24 always exhibited ∼100% selectivity for the PAP product even if sulfuric acid had a much lower concentration (0.97 wt%) that was the lowest value reported (Fig. 1b and Table S1†), which was also impossible for a commercial Pt/C catalyst (Fig. 1e). It was found that the higher the acid concentration, more nitrobenzene was converted into PAP, but the selectivity of PAP remained (Fig. 1d). Regardless of the change of the reaction conditions being temperature, reaction time and pressure, the over-hydrogenation was significantly inhibited and no aniline was detected in the Au36(SR)24 cases (Fig. S1b–d†).It was worth pointing out that the Au36(SR)24 catalyst was conveniently separated from the reaction system by centrifugation and further showed high catalytic performances over multiple cycles (Fig. S2†). The Au36(SR)24 clusters were robust and remained intact under reaction conditions (Fig. S3 and S4†). Interestingly, when the 4-tert-butylbenzenethiol ligand was exchanged into the other thiolate ligand such as the 3,5-dimethylbenzenethiol ligand, the 36-gold-atom cluster with a similar atomic-packing framework still gave ∼100% selectivity for PAP under identical conditions (Fig. S5†). Notably, when the ligands were removed from the surface of the clusters via the calcination pretreatment, the superior catalysis of Au36(SR)24 could not be obtained anymore, in which by-product aniline was produced on the ligand-off gold catalyst (Fig. S5 and S6†). The above observations revealed that the reaction pathway for nitrobenzene hydrogenation over the Au36(SR)24 catalyst should be radically distinct from the traditional route.
To unveil the unique pathway for nitrobenzene hydrogenation on the Au36(SR)24 catalyst, the adsorption behavior of nitrobenzene over the Au36(SR)24 cluster was first studied by Fourier Transform infrared spectroscopy (FTIR). As shown in Fig. 2a, when nitrobenzene was adsorbed onto the Au36(SR)24 cluster at room temperature, the two bands at 1347 and 1523 cm−1 assigned to the symmetric and asymmetric vibrations of the nitro group could be found, respectively.19 Subsequently, after removing potential physiosorbed species via purging argon at 120 °C for 30 min, the two vibration bands of the nitro group respectively shifted to 1342 and 1517 cm−1 (Fig. 2a). In the case of the Pt/C, as shown in Fig. 2b, the IR bands of adsorbed nitrobenzene onto the catalyst red shifted to 1345 and 1520 cm−1 after treatment of argon purging. This observation suggested that the nitro group of nitrobenzene can be absorbed onto the catalysts, whereas its adsorption strength on the Au36(SR)24 cluster might be weaker than that on Pt/C.18
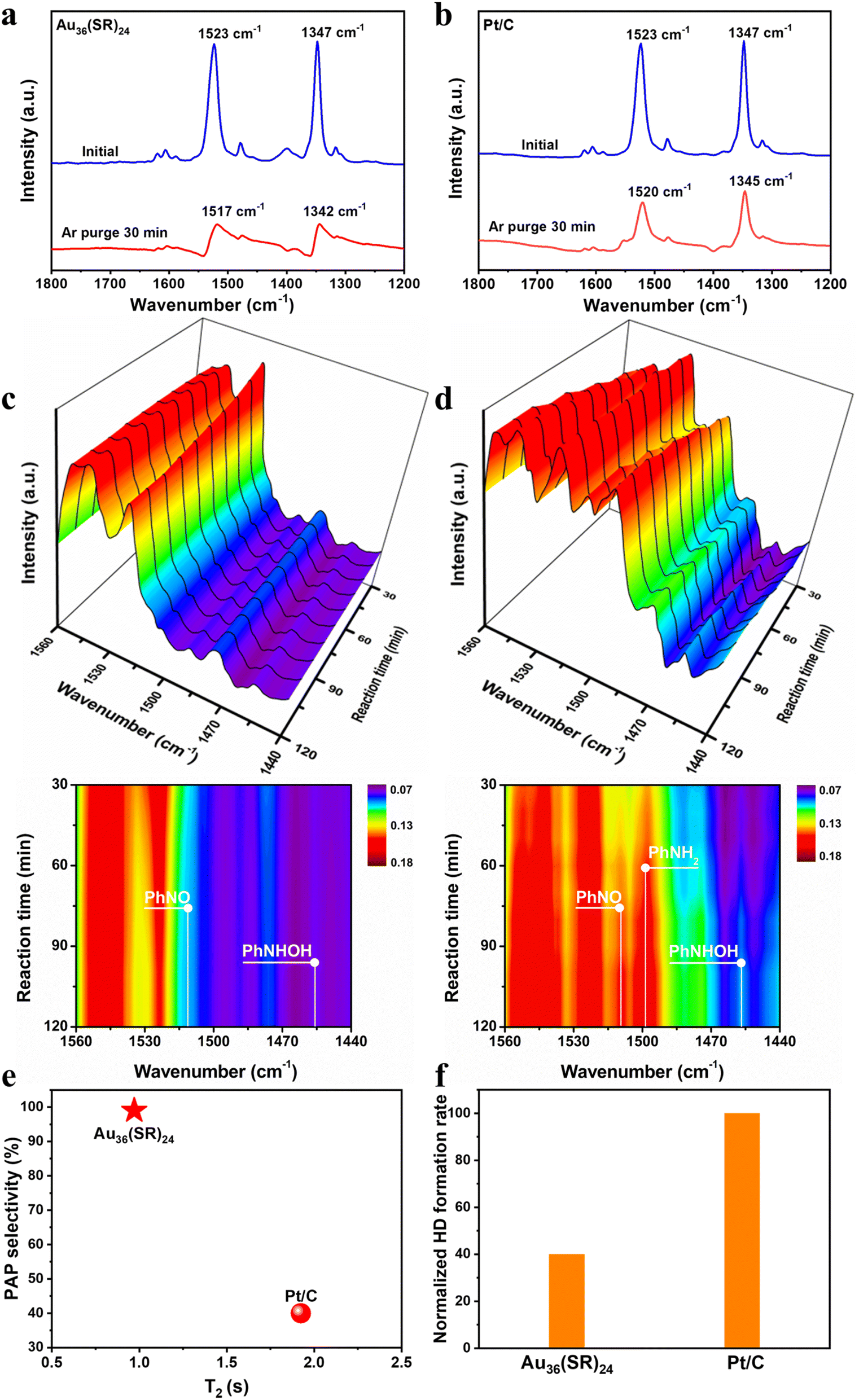 | ||
| Fig. 2 In situ FTIR spectra of adsorbed nitrobenzene onto (a) Au36(SR)24 and (b) Pt/C. The adsorption of nitrobenzene for 30 min at room temperature and then Ar was purged for 30 min at 120 °C. Time-resolved in situ FTIR spectra of the reaction intermediates formed on (c) Au36(SR)24 and (d) Pt/C for nitrobenzene hydrogenation with 4 MPa H2 at 120 °C. (e) PAP selectivity versus T2 value measured by NMR. (f) Normalized HD formation rate during the H2–D2 exchange reaction over Au36(SR)24 and Pt/C, respectively. | ||
Next, the reaction intermediates formed on the two catalysts were detected by time-resolved in situ FTIR spectroscopy with a gas–solid IR cell. The two intermediate species such as PhNO (∼1509 cm−1) and PHA (PhNHOH, ∼1457 cm−1) were observed on the Au36(SR)24 surface (Fig. 2c), but the IR signals of the two species were much weaker than those on the Pt/C system (Fig. 2c and S7†), indicating that the hydrogenation capacity of Au36(SR)24 was weaker than that of Pt/C.19,33,34 Meanwhile, aniline (PhNH2, ∼1497 cm−1) was only observed over the Pt/C system (Fig. 2d), implying that the over-hydrogenation of nitrobenzene only occurred on the Pt/C catalyst. This pointed out that PAP was not observed on the two IR systems, mainly as the IR cell could not be introduced using water and sulphuric acid, which also revealed that both H2O and acid significantly impacted the subsequent conversion of the PHA intermediate to the PAP product.
Furthermore, as seen from in situ Raman spectra obtained from the nitrobenzene hydrogenation over the Au36(SR)24 system (Fig. S8†), characteristic bands at 1345 and 1043 cm−1 occurred after 30 min of the catalytic reaction, which were attributed to vNO2 and vring from nitrobenzene, respectively.35 With the reaction, the peak intensity of vNO2 decreased as compared to that of vring, suggesting the gradual consumption of nitrobenzene; meanwhile a new band appeared at 462 cm−1. Density functional theory (DFT) calculations were performed to simulate the Raman spectra of PHA, PAP and aniline. As shown in Fig. S8,† the characteristic O–H vibration mode from PHA and PAP molecules can be observed at 425 and 455 cm−1, respectively. The observations further suggested that the hydrogenation process on the Au36(SR)24 catalyst followed the pathway from nitrobenzene to PHA and then to PAP, in which aniline was not produced on the Au36(SR)24 surface.
Moreover, the adsorption strength of PHA molecules on the Au36(SR)24 cluster and Pt/C catalyst was compared via measuring the relaxation time of adsorbed PHA using 1H NMR (nuclear magnetic resonance) spectroscopy described by T1 (longitudinal relaxation time) and T2 (transverse relaxation time), while T2 alone explained the problem of locomotion and a lower T2 value corresponded to a stronger interaction with the surface.36 The transverse relaxation time for the Au36(SR)24 cluster and Pt/C catalyst was respectively shown in Fig. S9 and S10,† and the T2 that was clearly correlated with the PAP selectivity is shown in Fig. 2e. The Au36(SR)24 cluster showed a lower T2 value than Pt/C, indicating that the former had higher affinity to PHA than the latter. The result was very surprising and distinct from that in previous reports, that is, higher affinity for PHA onto the Pt/C catalyst resulting in lower PAP selectivity, due to the PHA over-hydrogenation.
H2–D2 exchange experiments were also performed to gain in-depth insights into the ability for H2 activation on the two catalysts, where the normalized rate of HD formation was used to measure H2 activation behavior for Au36(SR)24 and Pt/C.37 Fig. 2f shows that the rate of HD formation was much lower on the Au36(SR)24 cluster than that on the Pt/C catalyst, revealing that Au36(SR)24 had much weaker ability to activate hydrogen than Pt/C, which might lead to no excessive hydrogenation of PHA on the Au36(SR)24 surface.
In order to further study the reaction mechanism of nitrobenzene hydrogenation in the Au36(SR)24 cluster, the DFT calculations were performed. The reaction pathways without the catalyst are first predicted (Fig. 3a). The PhNO2 precursor is reduced to form PhNO via an exergonic deoxygenation reaction (as the combination of hydrogenation and dehydration). PhNO then undergoes hydrogenation to form a perpetual intermediate, PHA, with an exergonicity of 26.3 kcal mol−1. Dehydration of PHA yields a phenylnitrene (PhN:) triplet intermediate, which is endergonic by 26.6 kcal mol−1 at 298 K. PhN:, as a diradical, is expected to be highly reactive and possibly act as a transient intermediate for further reactions. The relevance of PhN: in the reaction of PHA is evinced by the experiment results. Without any catalyst, H2, or acid, PHA can degrade to yield azoxybenzene,38 which can be viewed as the combination of PhN: and PhNO. In the presence of the Au36(SR)24 catalyst, without H2 or acid, PHA degrades mainly into azobenzene and azoxybenzene (>90% selectivity); the former is a dimer of PhN:. The degradation of PHA into azobenzene or azoxybenzene is effectively a dimerization process involving PhN:, which is highly exergonic (purple paths in Fig. 3a). Due to the nature of dimerization, such pathways are contingent on the diffusion of the monomers and thus could be inhibited when competing reactions with favorable thermodynamics and fast kinetics are ongoing, e.g., the potential reactions of PhN: with H2 or acid.
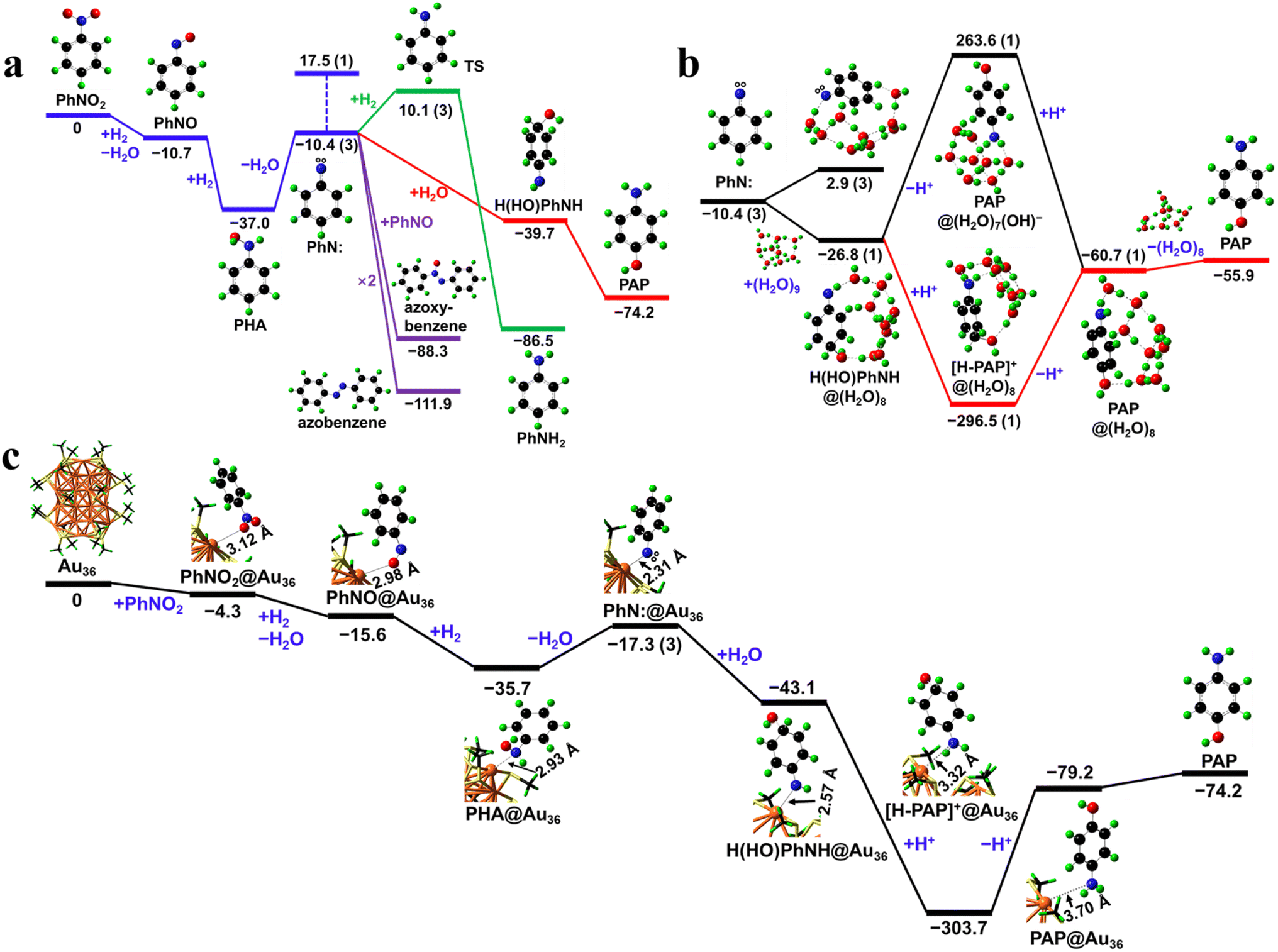 | ||
| Fig. 3 (a) Reaction pathways for catalyst-free reactions of nitrobenzene (PhNO2), including the proton-transfer pathway toward PAP (red), the hydrogenation pathway toward aniline (green), and the dimerization pathways toward azobenzene and azoxybenzene (purple). (b) The proton-transfer pathway in aqueous solution. (c) The nitrobenzene reaction pathways for the Au36(SR)24 catalyst. The Gibbs free energies at 298 K are given in kcal mol−1; the spin multiplicities are given in the parenthesis (singlet if omitted). Color code: O = red, N = blue, C = black, H = green, Au = orange, and S = yellow. The unpaired electrons are indicated by the white circles. The distances between the active Au site and the organic species are labelled with texts. | ||
When H2 is present, PhN: can be reduced to PhNH2 (green path in Fig. 3a) with high exergonicity (76.1 kcal mol−1) and low activation energy (20.5 kcal mol−1). When acid is present, PAP can be formed through an intramolecular proton transfer process (Fig. 3a). To understand this homogeneous process, solvent effects must be considered. DFT calculations using a (H2O)9 cluster as the explicit solvent show that the (H2O)9 cluster dissociates and reconstructs spontaneously into a (H2O)8 cluster, –H adsorbing at N, and –OH adsorbing at the p-position of phenyl, with a solvation energy of −16.4 kcal mol−1. Therefore, the PhN: diradical exists, in effect, as the H(HO)PhNH singlet in the aqueous solution (Fig. 3b). Calculations show that the amino group of the gas-phase H(HO)PhNH has a proton affinity of 237.2 kcal mol−1 and the hydride at the p-position of phenyl has a deprotonation energy of 340.7 kcal mol−1 (Fig. 4c). H(HO)PhNH is hence amphiprotic with its amino and p-hydride serving as a strong base (with proton affinity comparable to that of Li(OH), 239.0 kcal mol−1) and weak acid (comparable to acetic acid with a deprotonation energy of 341.1 kcal mol−1),39,40 respectively. The amino and p-hydride of the same H(HO)PhNH molecule can act as the proton acceptor and donor, respectively; when mediated by solvent or solvated molecules, exergonic amino protonation and endergonic p-hydride deprotonation may occur synergistically, leading to an intramolecular proton transfer process with much lower energy barrier than the otherwise intermolecular proton transfer process. Solvated acid can donate protons to the amino group while the acid anion can accept protons from p-hydride. Hence, acid promotion is conducive to the intramolecular proton transfer of H(HO)PhNH in solution, which can thus be broken down into amino protonation and subsequent p-hydride deprotonation steps (red path in Fig. 3b).
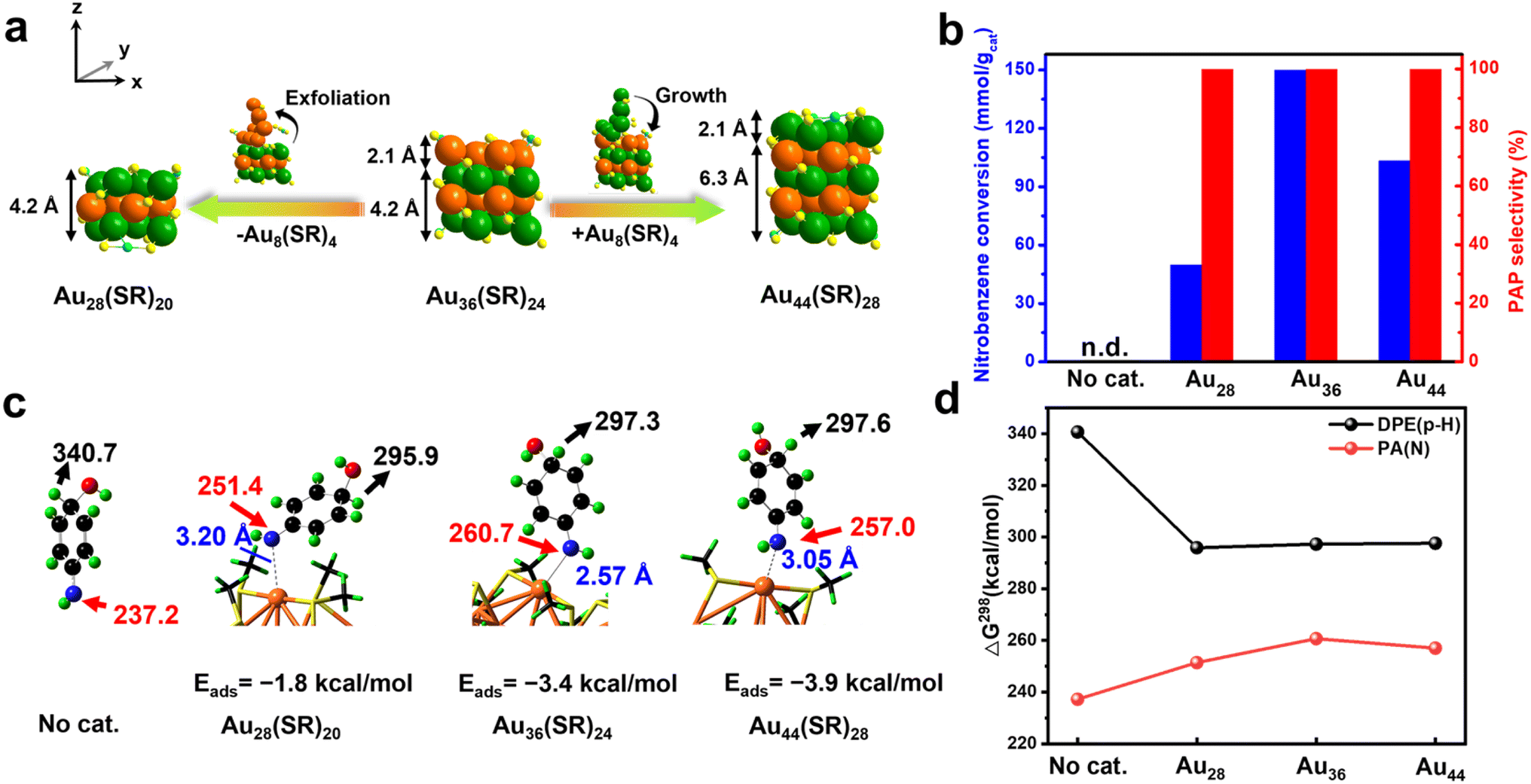 | ||
| Fig. 4 (a) Structural evolution of Au28(SR)20 and Au44(SR)28 from Au36(SR)24 along the z-direction. Color codes in the clusters: green/orange/blue = Au and yellow = S. (b) Catalytic performances of Au28(SR)20, Au36(SR)24 and Au44(SR)28 for nitrobenzene hydrogenation. (c) Gas-phase deprotonation energy (DPE, black) for the para-H and proton affinity (PA, red) for the amino group of H(OH)PhNH with no catalyst, and Au28(SR)20, Au36(SR)24 and Au44(SR)28 at 298 K at the DFT level. Color codes in the H(OH)PhNH: black = C, green = H, red = O, and dark blue = N. (d) Corresponding PA and DPE comparisons. The adsorption energy (Eads), PA and DPE at 298 K are given in kcal mol−1. | ||
Since H2 is required for the reduction of PhNO2 into PHA, the coexistence of H2 and acid is mostly inevitable for the subsequent reactions. The desired catalyst for the PAP synthesis via the PHA intermediate must be capable of suppressing the hydrogenation or promoting the proton transfer in an acidic solution, in order to reduce the yield of the aniline byproduct. Our calculations show that both PhN: and H(HO)PhNH, i.e. active species resulting from PHA, can adsorb on the Au36(SR)24 catalyst (Fig. 3c). The adsorption of PhN:, leading directly towards catalyst-adsorbed H(HO)PhNH in the solution, may cause further reactions to take place at the catalyst surfaces and inhibit the hydrogenation of the amino group, owing to the steric effects of the cluster ligands.
Excitedly, the excellent hydrogenation of nitrobenzene to PAP can be applied to structurally similar Au28(SR)20 and Au44(SR)28 clusters. Au28(SR)20 was obtained through subsequently exfoliating an Au8(SR)4 layer with 2.1 Å thickness from Au36(SR)24 along the z-direction, while Au44(SR)28 was formed through bandaging an additional Au8(SR)4 layer on the Au36(SR)24 cluster along the z-direction, with the x- and y-directions being constant (Fig. 4a).31 As shown in Fig. 4b, the three clusters exhibited an excellent selectivity for PAP, although they had different activities, which might be due to the structural flexibility of the clusters during the reactions (Fig. S11 and S12†). The mechanisms of nitrobenzene hydrogenation over Au28(SR)20 and Au44(SR)28 clusters were also explored by DFT calculations. It also shows that both PhN: and H(HO)PhNH can adsorb on Au28(SR)20 and Au44(SR)28 clusters (Fig. S13†), thereby causing further reactions to take place at the catalyst surfaces and inhibiting the hydrogenation of the amino group. The adsorption of H(HO)PhNH over the Au clusters substantially increases the basicity of its amino group and the acidity of its p-hydride, according to the predicted proton affinities and deprotonation energies (Fig. 4c and d); such a change in the electronic properties will undoubtedly promote intramolecular/intermolecular proton transfer. In addition, when an Au cluster catalyst is used, the endergonicity for the dehydration of PHA into PhN: is reduced as compared to no catalyst case (Fig. S14†), implying an improved kinetics to form the active species. Overall, the whole reaction process of nitrobenzene hydrogenation occurred completely on the gold cluster surface: nitrobenzene hydrogenation generated PHA and then PHA absorbed onto the gold cluster followed by removing a fraction of H2O; then PHA reacted with the nucleophile H2O to form H(OH)PhNH; finally, with the help of acid, the proton transfer occurred to produce the target product PAP. Thus, the novel reaction pathway can effectively inhibit the over-hydrogenation of PHA on the catalyst surface.
Conclusions
In summary, our study shows that atomically precise metal clusters indeed provide access to inaccessible issues of traditional catalysts. A Au36(SR)24 cluster exhibits a remarkable performance in the exclusive conversion of nitrobenzene to p-aminophenol via a distinguishable reaction route from typical industrial routes. The strong affinity of the key intermediate phenylhydroxylamine with the cluster enables the whole reaction process composed of nitrobenzene hydrogenation, nucleophilic rearrangement and intramolecular/intermolecular proton transfer carried out on the cluster surface, while the over-hydrogenation and Bamberger rearrangement that are well-known and recognized by traditional routes did not occur on the cluster catalyst. Additionally, excellent performances for this reaction can be extended to structurally similar catalysts with Au36(SR)24, which holds great promise in the design of high-performance catalysts for important chemical processes. Looking into the future of heterogeneous catalysts, the studies of atomically precise metal cluster catalysts will lead to an unexpected help in advances in catalysis mechanisms and practical application.Data availability
The data shown in this work can be available from the ESI† or can be obtained from the authors.Author contributions
Y. Z. conceived the project. J. L. synthesized the clusters and conducted the catalytic reactions. K. T. and M. C. carried out the DFT calculations. G. Q. and J. X. conducted the NMR measurements. C. J., Z. C. and D. L. conducted the Raman measurements. X. C. and X. L. analyzed the crystal data. W. D. discussed the results. All authors wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (22125202, 92361201 and 21932004), Natural Science Foundation of Jiangsu Province (BK20220033), Programs for high-level entrepreneurial and innovative talents of Jiangsu Province, and China Postdoctoral Science Foundation (2024T170400 and 2022M721551).References
- L. Zou, Y. Cui and W. Dai, Chin. J. Chem., 2014, 32, 257–262 CrossRef CAS.
- H. E. Heller, E. D. Hughes and C. K. Ingold, Nature, 1951, 168, 909–910 CrossRef CAS.
- T. Zhang, J. Jiang and Y. Wang, Chin. Chem. Lett., 2017, 28, 307–311 CrossRef CAS.
- K. I. Min, J. S. Choi, Y. M. Chung, W. S. Ahn, R. Ryoo and P. K. Lim, Appl. Catal., A, 2008, 337, 97–104 CrossRef CAS.
- S. Yamabe, G. Zeng, W. Guan and S. Sakaki, Beilstein J. Org. Chem., 2013, 9, 1073–1082 CrossRef CAS PubMed.
- A. Zoran, O. Khodzhaev and Y. J. Sasson, J. Chem. Soc., Chem. Commun., 1994, 19, 2239–2240 RSC.
- C. V. Rode, M. J. Vaidya and R. V. Chaudhari, Org. Process Res. Dev., 1999, 3, 465–470 CrossRef CAS.
- R. Joncour, A. Ferreira, N. Duguet and M. Lemaire, Org. Process Res. Dev., 2018, 22, 312–320 CrossRef CAS.
- S. K. Tanielyan, J. J. Nair, N. Marin, G. Alvez, R. J. McNair, D. Wang and R. L. Augustine, Org. Process Res. Dev., 2007, 11, 681–688 CrossRef CAS.
- Y. Liu, Y. Sheng, Y. Yin, J. Ren, X. Lin, X. Zou, X. Wang and X. Lu, ACS Omega, 2022, 7, 11217–11225 CrossRef CAS PubMed.
- Y. Sheng, Y. Liu, Y. Yin, X. Zou, J. Ren, B. Wu, X. Wang and X. Lu, Chem. Eng. J., 2023, 452, 139448 CrossRef CAS.
- J. M. Nadgeri, N. S. Biradar, P. B. Patil, S. T. Jadkar, A. Garade and C. V. Rode, Ind. Eng. Chem. Res., 2011, 50, 5478–5484 CrossRef CAS.
- F. Zhang, Y. Jiang, S. Dai, X. Wei, Y. Ma, H. Liao, Y. Qin, Q. Peng, X. Zhao and Z. Hou, Ind. Eng. Chem. Res., 2023, 62, 5814–5825 CrossRef CAS.
- T. Wang, Z. Dong, T. Fu, Y. Zhao, T. Wang, Y. Wang, Y. Chen, B. Han and W. Ding, Chem. Commun., 2015, 51, 17712–17715 RSC.
- C. V. Rode, M. J. Vaidya, R. Jaganathan and R. V. Chaudhari, Chem. Eng. Sci., 2001, 56, 1299–1304 CrossRef CAS.
- T. Wang, Z. Dong, W. Cai, Y. Wang, T. Fu, B. Zhao, L. Peng, W. Ding and Y. Chen, Chem. Commun., 2016, 52, 10672–10675 RSC.
- J. Luo, C. Zhang, C. Yao, D. Ma, Y. Chen, M. Tian, H. Xie, L. Pan, Y. Zhen, R. Chen, J. Wu, C. Lu, F. Feng, X. Xu, Q. Wang, Q. Zhang and X. Li, Mol. Catal., 2023, 545, 113216 CrossRef CAS.
- J. Luo, C. Yao, D. Ma, Y. Chen, M. Tian, H. Xie, R. Chen, J. Wu, Y. Zhen, L. Pan, C. Lu, F. Feng, X. Xu, Q. Wang, Q. Zhang and X. Li, Appl. Catal., A, 2023, 660, 119198 CrossRef CAS.
- C. Yin, Z. Xiang, Y. Yao, X. Li, C. Ma, X. Liu, Y. Zhou, W. He, C. Zhou, F. Feng, Q. Zhang, J. Lyu, Y. Liu, C. Lu and X. Li, ACS Catal., 2023, 13, 13756–13767 CrossRef CAS.
- R. Jin, G. Li, S. Sharma, Y. Li and X. Du, Chem. Rev., 2021, 121, 567–648 CrossRef CAS PubMed.
- X. Liu, X. Cai and Y. Zhu, Acc. Chem. Res., 2023, 56, 1528–1538 CrossRef CAS PubMed.
- J. Yan, B. K. Teo and N. Zheng, Acc. Chem. Res., 2018, 51, 3084–3093 CrossRef CAS PubMed.
- M. R. Narouz, K. M. Osten, P. J. Unsworth, R. W. Y. Man, K. Salorinne, S. Takano, R. Tomihara, S. Kaappa, S. Malola, C. T. Dinh, J. D. Padmos, K. Ayoo, P. J. Garrett, M. Nambo, J. H. Horton, E. H. Sargent, H. Hakkinen, T. Tsukuda and C. M. Crudden, Nat. Chem., 2019, 11, 419–425 CrossRef CAS PubMed.
- K. Kwak, W. Choi, Q. Tang, M. Kim, Y. Lee, D. E. Jiang and D. A. Lee, Nat. Commun., 2017, 8, 14723 CrossRef PubMed.
- S. Li, D. Alfonso, A. V. Nagarajan, S. D. House, J. C. Yang, D. R. Kauffman, G. Mpourmpakis and R. Jin, ACS Catal., 2020, 10, 12011–12016 CrossRef CAS.
- X. Cai, Y. Liu, G. Li, W. Hu, X. Liu, M. Chen, W. Ding and Y. Zhu, Adv. Mater., 2023, 35, 2301466 CrossRef CAS PubMed.
- J. Zhao, Q. Li, S. Zhuang, Y. Song, D. J. Morris, M. Zhou, Z. Wu, P. Zhang and R. Jin, J. Phys. Chem. Lett., 2018, 9, 7173–7179 CrossRef CAS PubMed.
- Y. Tan, X. Y. Liu, L. Zhang, A. Wang, L. Li, X. Pan, S. Miao, M. Haruta, H. Wei, H. Wang, F. Wang, X. Wang and T. Zhang, Angew. Chem., Int. Ed., 2017, 56, 2709–2713 CrossRef CAS PubMed.
- X. Cai, H. Wang, Y. Tian, W. Ding and Y. Zhu, ACS Catal., 2024, 14, 11918–11930 CrossRef CAS.
- X. Liu, E. Wang, M. Zhou, Y. Wan, Y. Zhang, H. Liu, Y. Zhao, J. Li, Y. Gao and Y. Zhu, Angew. Chem., Int. Ed., 2022, 61, e202207685 CrossRef CAS PubMed.
- C. Zeng, Y. Chen, K. Iida, K. Nobusada, K. Kirschbaum, K. J. Lambright and R. Jin, J. Am. Chem. Soc., 2016, 138, 3950–3953 CrossRef CAS PubMed.
- X. Liu, W. W. Xu, X. Huang, E. Wang, X. Cai, Y. Zhao, J. Li, M. Xiao, C. Zhang, Y. Gao, W. Ding and Y. Zhu, Nat. Commun., 2020, 11, 3349 CrossRef CAS PubMed.
- Y. Wei, S. Wang, Y. Zhang, M. Li, J. Hu, Y. Liu, J. Li, L. Yu, R. Huang and D. Deng, ACS Catal., 2023, 13, 15824–15832 CrossRef CAS.
- R. Gilles, A. Jeroen, B. Van, Y. M. Neuhold, M. Makosch and H. Konrad, Phys. Chem. Chem. Phys., 2011, 13, 12463–12471 RSC.
- J. Ma, Z. Wang, T. Majima and G. Zhao, ACS Catal., 2022, 12, 14062–14071 CrossRef CAS.
- W. Wang, W. Zhou, Y. Tang, W. Cao, S. R. Docherty, F. Wu, K. Cheng, Q. Zhang, C. Coperet and Y. Wang, J. Am. Chem. Soc., 2023, 145, 12928–12934 CrossRef CAS PubMed.
- Y. Wang, J. Yang, R. Gu, L. Peng, X. Guo, N. Xue, Y. Zhu and W. Ding, ACS Catal., 2018, 8, 6419–6425 CrossRef CAS.
- A. R. Becker and L. A. Sternson, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 2003–2007 CrossRef CAS PubMed.
- E. P. L. Hunter and S. G. Lias, J. Phys. Chem. Ref. Data, 1998, 27, 413–656 CrossRef CAS.
- R. W. Taft and R. D. Topsom, Prog. Phys. Org. Chem., 1987, 16, 1–83 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05018e |
| This journal is © The Royal Society of Chemistry 2024 |