
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring enantiopure zinc-scorpionates as catalysts for the preparation of polylactides, cyclic carbonates, and polycarbonates†
Marta
Navarro
 ab,
Sonia
Sobrino
a,
Israel
Fernández
c,
Agustín
Lara-Sánchez
a,
Andrés
Garcés
*b and
Luis F.
Sánchez-Barba
*b
ab,
Sonia
Sobrino
a,
Israel
Fernández
c,
Agustín
Lara-Sánchez
a,
Andrés
Garcés
*b and
Luis F.
Sánchez-Barba
*b
aUniversidad de Castilla-La Mancha, Departamento de Química Inorgánica, Orgánica y Bioquímica-Centro de Innovación en Química Avanzada (ORFEO-CINQA), Campus Universitario, 13071-Ciudad Real, Spain
bUniversidad Rey Juan Carlos, Departamento de Biología y Geología, Física y Química Inorgánica, Móstoles-28933-Madrid, Spain. E-mail: luisfernando.sanchezbarba@urjc.es; andres.garces@urjc.es
cUniversidad Complutense de Madrid. Departamento de Química Orgánica I and Centro de Innovación en Química Avanzada (ORFEO-CINQA), Facultad de Ciencias Químicas, 28040, Madrid, Spain
First published on 29th July 2024
Abstract
New and simple ligand design strategies for the preparation of versatile metal-based catalysts capable of operating under greener and eco-friendly conditions in several industrially attractive processes are in high demand for society development. We present the first nucleophilic addition of an organolithium to ketenimines which incorporates a stereogenic centre in an N-donor atom to prepare new enantiopure NNN-donor scorpionates. We have also verified its potential utility as a valuable scaffold for chirality induction through the preparation of inexpensive, non-toxic and asymmetric zinc complexes. The pro-ligands and the corresponding zinc-based complexes have been characterized by X-ray diffraction studies. DFT studies were carried out to rationalize the different complexation abilities of these pro-ligands. These complexes have proved to act as highly efficient catalysts for a variety of sustainable bioresourced processes that are industrially attractive, with a wide substrate scope. Thus, complex 7 behaves as a highly efficient initiator for the well-behaved living ring-opening polymerization (ROP) of rac-lactide under very mild conditions. The PLA materials produced exhibited enhanced levels of isoselectivity, comparable to the highest value reported so far for zinc-based catalysts (Pi = 0.88). In addition, the combination of 7 with onium halide salts functioned as a very active and selective catalyst for CO2 fixation into five-membered cyclic carbonates through the cycloaddition of CO2 into epoxides under very mild and solvent-free conditions, reaching very good to excellent conversions (TOF = 227 h−1). Furthermore, this bicomponent system exhibits a broad substrate scope and functional group tolerance, including mono- and di-substituted epoxides, as well as the very challenging bio-renewable tri-substituted terpene-derived cis/trans-limonene oxide, whose reaction proceeds with high stereoselectivity. Finally, complex 7 also achieved high activity and selectivity as a one-component initiator for the synthesis of poly(cyclohexene carbonate)s via ring-opening copolymerization (ROCOP) of cyclohexene oxide and CO2 under very soft conditions, affording materials with narrow dispersity values.
Introduction
Complexes containing enantiopure scorpionate ligands, derived from bis(pyrazol-1-yl)methane, have been used for numerous catalytic processes.1 Our research group is a pioneer in the introduction of chirality into the bridging carbon atom of this system,2 and therefore, we have prepared extensive families of complexes with a wide variety of metals supported by NNO-,3 NNS-,4 NNN-,5 and NNCp-6 scorpionate ligands containing an asymmetric centre. However, it is critical in stereocontrolled catalytic processes that after ligand coordination to the metal, the new stereogenic centre in the ligand should remain close to the metal active site.As a result of this continuous motivation for ligand improvement, we have recently reported the first known example of a nucleophilic addition of an organolithium to a ketenimine that was enantiomerically pure, where the asymmetric moiety was attached to the N-donor atom.7 Ketenimines are heterocumulenes with two consecutive carbon atoms, where one of them is in turn linked to an imino group. In addition, these organic compounds have three possible resonance structures,8 where the electrophilic nature of the carbon atom in the α-position justifies their reactivity against organolithiums5b and nucleophiles9 in general (see Chart 1).
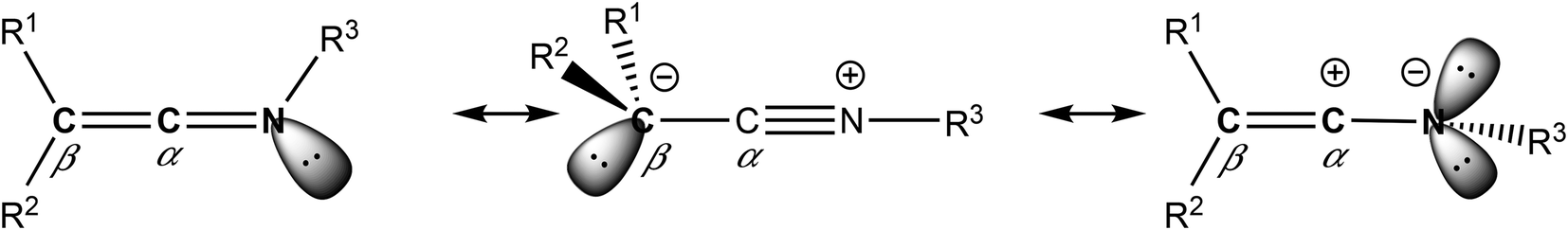 | ||
| Chart 1 Possible resonance structures of ketenimines. | ||
In a further step, a newly prepared7 ketenimine served to design a new enantiopure NNN-scorpionate ligand, which confirmed its potential utility as a valuable scaffold for chirality induction through the preparation of biologically benign zinc-based enantiomerically pure complexes. In addition, these species behaved as highly efficient initiators for the ring-opening polymerization (ROP) of the bio-resourced rac-lactide (rac-LA) and imparted a remarkable influence on the production of highly enriched isotactic poly(lactide)s (PLAs) (Pi = 0.88)7via an enantiomorphic-site control mechanism.6,10 In this context, it is important to highlight that a very small number of zinc-based catalysts bearing enantiopure auxiliary ligands have already succeeded11 (see Fig. 1a) to realize isotactic dyad enchainment from rac-LA in the production of the bioassimilable poly(lactide)s (Pi = 0.84–0.91), which allow an interesting wide array of applications (packaging, microelectronics and biomedical).12
 | ||
| Fig. 1 Representative zinc-based catalysts for the preparation of (a) poly(rac-lactide), (b) cyclic styrene carbonate and (c) poly(cyclohexene carbonate). | ||
Continuing with our previous work, now we take the challenging aim to design a new asymmetric ketenimine to prepare another enantiomerically pure scorpionate ligand with higher chirality induction, confirming the reliability of the method, and to explore new opportunities for the prepared zinc initiators in additional sustainable catalytic procedures. For instance, CO2 valorisation processes13a such as the production of the 100% atom-economical cyclic carbonates (CCs)13b,c or polycarbonates (PCs)14 are currently two scientific areas in high demand.
In this regard, recently our research group has also successfully developed efficient zinc-based scorpionate catalysts for the production of 5-membered cyclic carbonates15 through the cycloaddition of CO2 with a wide range of epoxides, including terminal, internal and bio-based-derived substrates. Very active and selective zinc-based16 catalysts have also been reported for the production of cyclic carbonates, with the assistance of a nucleophile as a co-catalyst (see Fig. 1b).
In parallel, very recently we have also succeeded in the production of poly(cyclohexene carbonate) (PCHC) via ring-opening co-polymerization (ROCOP) of cyclohexene oxide and CO2 by employing analog zinc-based scorpionate initiators.17 Alternatively, highly efficient zinc-based18 initiators have been developed for the selective ROCOP of carbon dioxide and epoxides, frequently with the assistance of a cocatalyst (see Fig. 1c).
Nevertheless, the search for versatile single zinc-based catalysts with a wider substrate scope capable of operating efficiently on each of these sustainable processes mentioned above, i.e., the ROP of rac-LA as well as CO2 fixation processes such as the cycloaddition and the ROCOP of CO2 with epoxides, remains poorly explored (see Fig. 1).
Herein, we report an unprecedented method for the design of two new ketenimines as asymmetric reagents for the synthesis of NNN-scorpionate ligands, and their transfer to prepare enantiomerically pure zinc complexes capable of behaving as single-component living initiators for the highly isoselective production of poly(rac-lactide)s. These complexes have also been investigated in detail as catalysts for CO2 fixation into both five-membered cyclic carbonates in combination with TBAB, exhibiting a very broad substrate scope, as well as one-component initiators for the synthesis of poly(cyclohexene carbonate), under very mild and solvent-free conditions.
Results and discussion
Synthesis and structural characterization of enantiopure scorpionate ligands
In order to prepare the enantiomerically pure ligands, two new enantiopure ketenimines have been obtained by the aza-Wittig synthesis method.19 For this purpose, firstly, two new phosphanimines 1 and 2 have been prepared from the reaction of triphenylphosphane dibromide20 and the commercially enantiopure amines (−)-cis-myrtanylamine or (R)-(+)-bornylamine (see Scheme 1), and were obtained as a yellow oil and a white solid, respectively, in very good yields (approx. 90%). Subsequently, the reaction of the phosphanimines 1 and 2 with diphenylketene and their subsequent treatment gave the enantiomerically pure ketenimines 3, as a yellow oil, and 4 as a white solid, both with good performance (approx. 85%) (see Scheme 2). It is also worth noting to highlight that the ketenimine 4 will allow us to prepare an enantiomerically pure scorpionate ligand with higher chirality induction given the absence of the CH2 spacer group bound to the N atom. | ||
| Scheme 1 Synthesis of phosphanimines 1 and 2. | ||
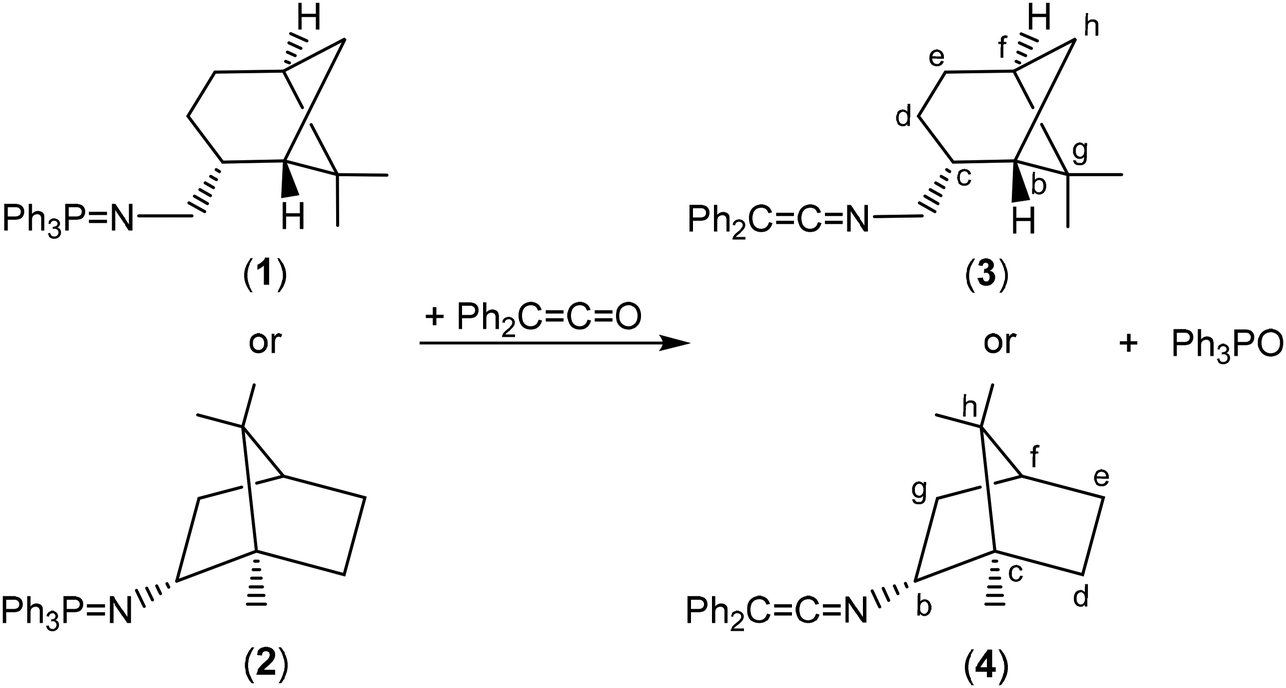 | ||
| Scheme 2 Synthesis of enantiomerically pure ketenimines 3 and 4. | ||
The preparation of enantiopure scorpionate ligands using the novel reaction based on the nucleophilic addition of organolithium reagents to ketenimines7 was carried out by the reaction of lithium bis(3,5-dimethylpyrazol-1-yl)methanide, prepared in situ from nBuLi and bis(3,5-dimethylpyrazol-1-yl)methane at −70 °C, with the new ketenimines 3 and 4, as shown in Scheme 3. The reaction was complete after 1 h and with the appropriate work-up, the enantiomerically pure heteroscorpionate ligands 5 and 6 were obtained in the form of their corresponding tautomers. Ligand 5 gave a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the two enantiopure scorpionate tautomers (enamine or imine derivative), (−)-cis-bpmyH (5a) {bpmyH = 3,3-bis(3,5-dimethyl-1H-pyrazol-1-yl)-N-(((1S,2R,5S)-6,6-dimethylbicyclo[3.1.1]heptan-2-yl)methyl)-1,1-diphenylprop-1-en-2-amine} and (−)-cis-bpmy′H (5b) {bpmy′H = 1,1-bis(3,5-dimethyl-1H-pyrazol-1-yl)-N-(((1S,2R,5S)-6,6-dimethylbicyclo[3.1.1]heptan-2-yl)methyl)-3,3-diphenylpropan-2-imine} as yellow solids, in 68% yield (see Scheme 3a). Both tautomers can be separated by crystallization (see the ESI†). However, the ligand (R)-(+)-bpmbnH (6c) {bpmbnH = (1R,2S,4R,E)-N-(1,1-bis(3,5-dimethyl-1H-pyrazol-1-yl)-3,3-diphenylpropan-2-ylidene)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-imine} was isolated as a single tautomer, corresponding to an alternative enamine form, as a white solid in a 70% yield (see Scheme 3b).
:1 mixture of the two enantiopure scorpionate tautomers (enamine or imine derivative), (−)-cis-bpmyH (5a) {bpmyH = 3,3-bis(3,5-dimethyl-1H-pyrazol-1-yl)-N-(((1S,2R,5S)-6,6-dimethylbicyclo[3.1.1]heptan-2-yl)methyl)-1,1-diphenylprop-1-en-2-amine} and (−)-cis-bpmy′H (5b) {bpmy′H = 1,1-bis(3,5-dimethyl-1H-pyrazol-1-yl)-N-(((1S,2R,5S)-6,6-dimethylbicyclo[3.1.1]heptan-2-yl)methyl)-3,3-diphenylpropan-2-imine} as yellow solids, in 68% yield (see Scheme 3a). Both tautomers can be separated by crystallization (see the ESI†). However, the ligand (R)-(+)-bpmbnH (6c) {bpmbnH = (1R,2S,4R,E)-N-(1,1-bis(3,5-dimethyl-1H-pyrazol-1-yl)-3,3-diphenylpropan-2-ylidene)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-imine} was isolated as a single tautomer, corresponding to an alternative enamine form, as a white solid in a 70% yield (see Scheme 3b).
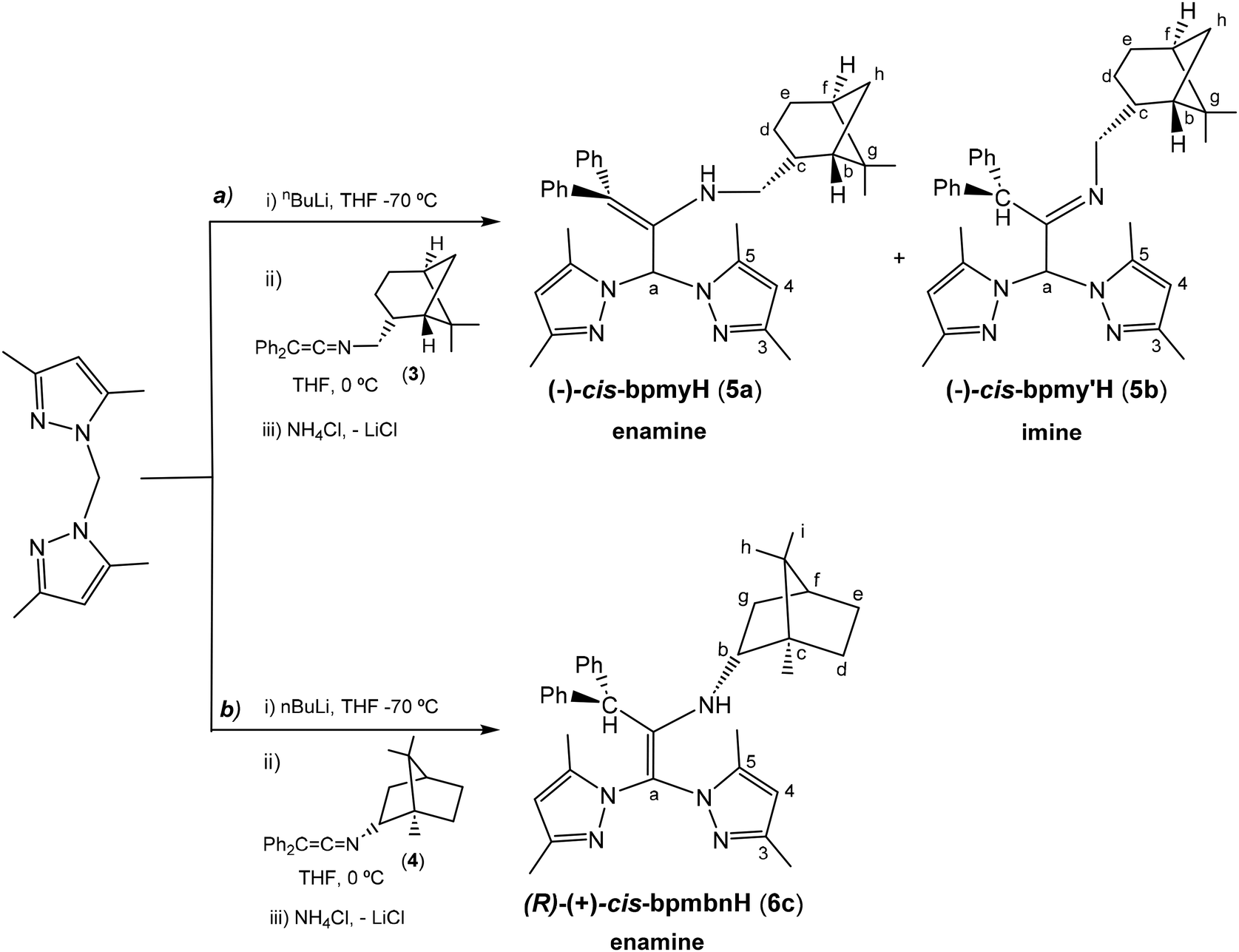 | ||
| Scheme 3 Synthesis of enantiomerically pure scorpionate ligands (−)-cis-bpmyH (5a), (−)-cis-bpmy′H (5b) and (R)-(+)-bpmbnH (6c). | ||
The different compounds were characterized spectroscopically. The 1H NMR spectra of the phosphanimines 1 and 2 as well as the ketenimines 3 and 4 (see Fig. S1 and S2 in the ESI†) show the corresponding signals for the phenyl groups and a multiplet for the methylene group in compounds 1 and 3. In addition, the spectra exhibit the signals corresponding to the bicyclic fragment (six sets of resonances for Hb, Hc, Hd, He, Hf, Hg or Hh and two singlets for the methyl groups). The 1H and 13C {1H} NMR spectra of scorpionate ligand 5 (see Fig. S3 in the ESI†) exhibit four singlets for the H4, Me3 and Me5 pyrazole protons, indicating the presence of two tautomers and the inequivalence of two pyrazole rings. The 1H NMR spectrum also contains two singlets for the CHa bridge to the two pyrazole rings and two multiplets for the methylene group. Two sets of signals for the phenyl groups and the bicycle moiety were also observed related to both tautomers. In compound 5a the signal corresponding to the N–H group can be observed, while in 5b it disappears, and a new signal corresponding to the proton CHPh2 is observed. However, the 1H and 13C {1H} NMR spectra of the ligand 6c (see Fig. S4 in the ESI†) show two singlets for the H4, Me3 and Me5 pyrazole protons, indicating the presence of only one tautomer, and the inequivalence of the two pyrazole rings. Furthermore, the signal corresponding to the N–H group can be observed.
Suitable crystals of the ligand 6c were studied by X-ray diffraction analysis and the ORTEP diagram is depicted in Fig. 2. The molecular structure of 6c consists of a monomeric arrangement in the solid state. Selected bond lengths and angles are listed in Table 1 (see Crystallographic details in Table S1 in the ESI†). The three stereogenic centres are maintained in compound 6c as in the starting bornylamine, since the nucleophilic addition of the organolithium to the ketenimine 4 cannot modify the original stereogenic centres, providing therefore its enantiomerically pure character. The most important feature of the molecule is the C(11)–C(12) or C(51)–C(52) bond length of 1.351(3)–1.344(3) Å, in agreement with a double C–C bond (∼1.33 Å). The sp2 and planar nature of the carbon atoms C(11) or C(51) and C(12) or C(52), respectively, is further confirmed by the summations of the angles (∼120°) around them (360°). It is also worth noting that the distance C(12)–C(13) or C(52)–C(53) of 1.534(3)–1.531(3) Å parallels that of a C–C single bond (∼1.54 Å). Similarly, the distance of the C(12)–N(5) or C(52)–N(15) bond of 1.372(3)–1.358(3) Å is typical of a C–N single bond, and slightly shorter than that of C(11)–N(2) or C(51)–N(12) (1.414(3)–1.423(3) Å) in the pyrazole rings. The hydrogen atom is located on the N(5) atom. These selected bond distances and angles confirm that only the enamine tautomer observed in solution was also present in the solid state.
 | ||
| Fig. 2 ORTEP view of scorpionate ligand (R)-(+)-bpmbnH (6c). Hydrogen atoms have been omitted for clarity. Thermal ellipsoids are drawn at the 30% probability. | ||
| (R)-(+)-bpmbnH ( 6c )a | |||||
| Bond lengths (Å) | |||||
| C(11)–C(12) | 1.351(3) | C(12)–C(13) | 1.534(3) | C(11)–N(4) | 1.427(4) |
| C(12)–N(5) | 1.372(3) | C(11)–N(2) | 1.414(3) | ||
| Angles (°) | |||||
| N(2)–C(11)–N(4) | 115.3(2) | C(12)–C(11)–N(4) | 122.7(2) | C(11)–C(12)–C(13) | 119.3(2) |
| C(12)–C(11)–N(2) | 122.1(2) | C(11)–C(12)–N(5) | 127.1(2) | C(11)–C(12)–N(5) | 127.1(2) |
| (R)-(+)-bpmbnH ( 6c )b | |||||
| Bond lengths (Å) | |||||
| C(51)–C(52) | 1.344(3) | C(52)–C(53) | 1.531(3) | C(51)–N(14) | 1.417(4) |
| C(52)–N(15) | 1.358(3) | C(51)–N(12) | 1.423(3) | ||
| Angles (°) | |||||
| N(12)–C(51)–N(14) | 115.1(2) | C(52)–C(51)–N(14) | 123.7(2) | C(51)–C(52)–C(53) | 119.6(2) |
| C(52)–C(51)–N(12) | 121.2(2) | C(51)–C(52)–N(15) | 128.3(2) | C(51)–C(52)–N(15) | 128.3(2) |
| [ZnMe(κ 3 -cis-bpmyO)] ( 9 ) | |||||
| Bond lengths (Å) | |||||
| Zn(1)–C(23) | 1.962(13) | N(4)–C(6) | 1.350(15) | C(7)–C(8) | 1.397(19) |
| Zn(1)–N(5) | 1.986(10) | N(4)–C(11) | 1.468(15) | C(8)–C(10) | 1.492(16) |
| Zn(1)–N(1) | 2.117(9) | N(5)–C(12) | 1.325(15) | C(11)–C(12) | 1.572(16) |
| Zn(1)–N(3) | 2.122(10) | N(5)–C(13) | 1.483(13) | C(13)–C(14) | 1.512(14) |
| Zn(1)–C(23) | 1.962(13) | O(1)–C(12) | 1.235(14) | C(14)–C(19) | 1.548(13) |
| N(1)–C(3) | 1.350(14) | C(1)–C(2) | 1.362(18) | C(14)–C(15) | 1.549(13) |
| N(1)–N(2) | 1.368(13) | C(1)–C(4) | 1.496(18) | C(15)–C(16) | 1.555(15) |
| N(2)–C(1) | 1.363(14) | C(2)–C(3) | 1.408(17) | C(16)–C(17) | 1.520(15) |
| N(2)–C(11) | 1.459(14) | C(3)–C(5) | 1.455(18) | C(17)–C(18) | 1.496(16) |
| N(3)–C(8) | 1.339(16) | C(6)–C(7) | 1.385(18) | C(17)–C(20) | 1.565(15) |
| N(3)–N(4) | 1.376(13) | C(6)–C(9) | 1.488(17) | C(18)–C(19) | 1.540(15) |
| Angles (°) | |||||
| C(23)–Zn(1)–N(5) | 134.1(5) | C(8)–N(3)–N(4) | 105.5(9) | N(1)–C(3)–C(2) | 107.6(11) |
| C(23)–Zn(1)–N(1) | 124.3(5) | C(8)–N(3)–Zn(1) | 139.4(9) | N(1)–C(3)–C(5) | 120.7(11) |
| N(5)–Zn(1)–N(1) | 87.4(4) | N(4)–N(3)–Zn(1) | 115.0(7) | C(2)–C(3)–C(5) | 131.8(11) |
| C(23)–Zn(1)–N(3) | 120.0(5) | C(6)–N(4)–N(3) | 111.7(10) | N(4)–C(6)–C(7) | 106.0(12) |
| N(5)–Zn(1)–N(3) | 91.6(4) | C(6)–N(4)–C(11) | 129.9(11) | N(4)–C(6)–C(9) | 122.7(12) |
| N(1)–Zn(1)–N(3) | 85.7(4) | N(3)–N(4)–C(11) | 118.0(9) | C(7)–C(6)–C(9) | 131.3(12) |
| C(3)–N(1)–N(2) | 107.1(10) | C(12)–N(5)–C(13) | 112.7(10) | C(8)–C(7)–C(6) | 106.8(13) |
| C(3)–N(1)–Zn(1) | 138.9(9) | C(12)–N(5)–Zn(1) | 119.4(8) | N(3)–C(8)–C(7) | 109.9(11) |
| N(2)–N(1)–Zn(1) | 113.8(7) | C(13)–N(5)–Zn(1) | 127.8(8) | N(3)–C(8)–C(10) | 121.0(12) |
| C(1)–N(2)–N(1) | 110.8(10) | N(2)–C(1)–C(2) | 106.1(11) | C(7)–C(8)–C(10) | 129.0(13) |
| C(1)–N(2)–C(11) | 129.2(10) | N(2)–C(1)–C(4) | 121.7(12) | N(2)–C(11)–N(4) | 108.3(9) |
Synthesis and structural characterization of the scorpionate complexes [Zn(Me)(κ3-cis-bpmy)] (7), [Zn(CH2SiMe3)(κ3-cis-bpmy)] (8) and [ZnMe(κ3-cis-bpmyO)] (9)
The deprotonation of 5 (as a mixture of tautomers 5a and 5b) with ZnR2 (R = Me, CH2SiMe3), in a 1:1 molar ratio in toluene, after the appropriate work-up, gave the complexes [Zn(R)(κ3-cis-bpmy)],7 R = Me (7); R = CH2SiMe3 (8), which were isolated as yellow solids in good yield (ca. 85%) (see Scheme 4). Surprisingly, all attempts to react the pro-ligand 6c with various zinc dialkyls unfortunately resulted fruitless after testing different reaction conditions.
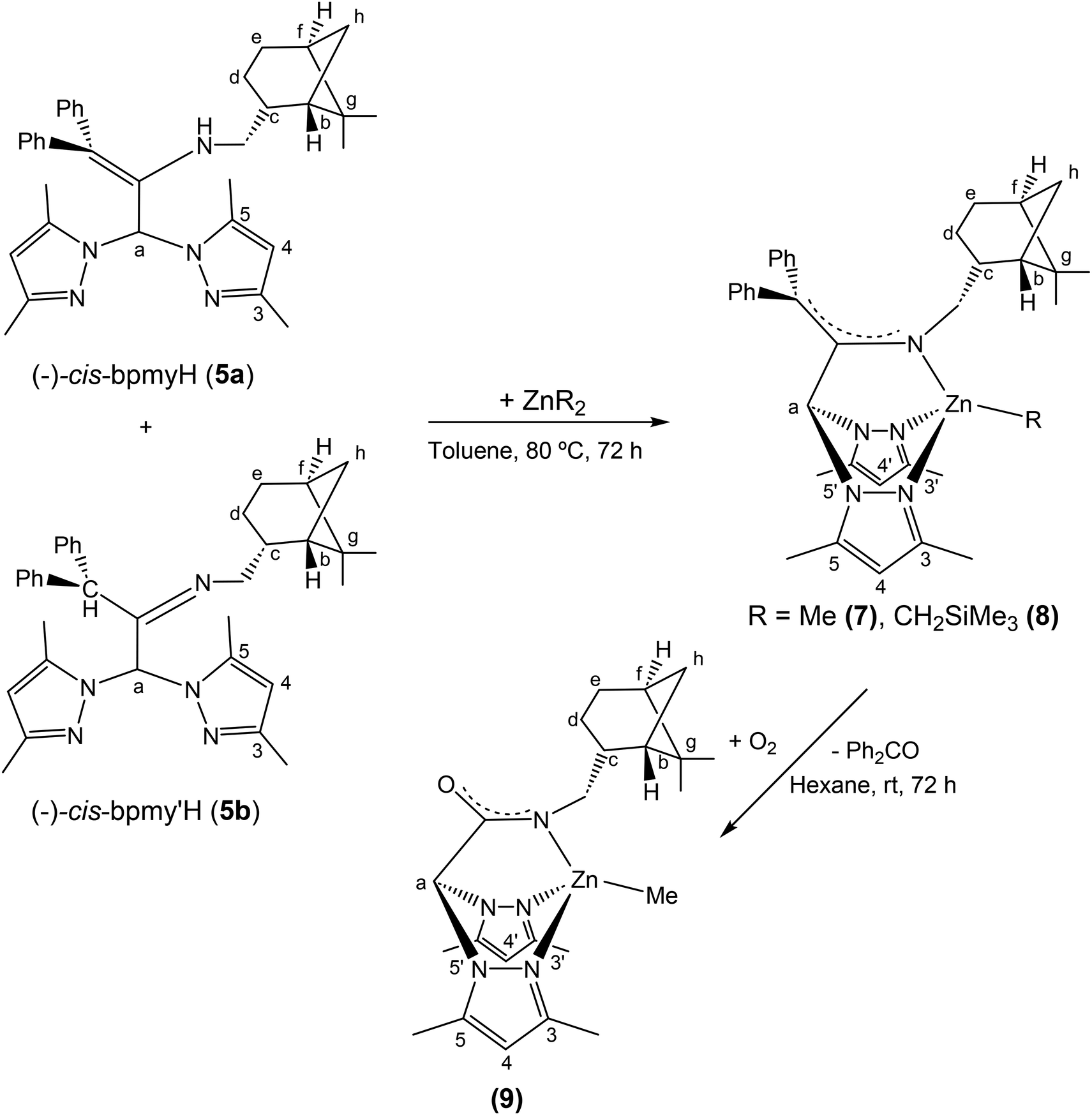 | ||
| Scheme 4 Synthesis of enantiomerically pure scorpionate complexes [Zn(Me)(κ3-cis-bpmy)] (7), [Zn(CH2SiMe3)(κ3-cis-bpmy)] (8) and [ZnMe(κ3-cis-bpmyO)] (9). | ||
Density functional theory (DFT) calculations at the dispersion-corrected PCM-(toluene)-B3LYP-D3/def2-SVP level were carried out to investigate the different reactivities of these species in their ZnMe2-mediated deprotonation/coordination. As depicted in Fig. 3, the process begins with the coordination of the transition metal complex to the pyrazole moiety leading to the initial 5a/6c-ZnMe2 complexes. From these species, the deprotonation reaction takes place through the corresponding transition states TS, a saddle point associated with the release of CH4 and formation of the corresponding bidentate complex. From the data in Fig. 3 it becomes evident that whereas this step proceeds with an accessible barrier of 19.7 kcal mol−1 for 5c (which is compatible with the experimental reaction conditions), the analogous process involving 6c requires a rather high barrier of ca. 37 kcal mol−1, rendering this transformation unfeasible, as observed experimentally.
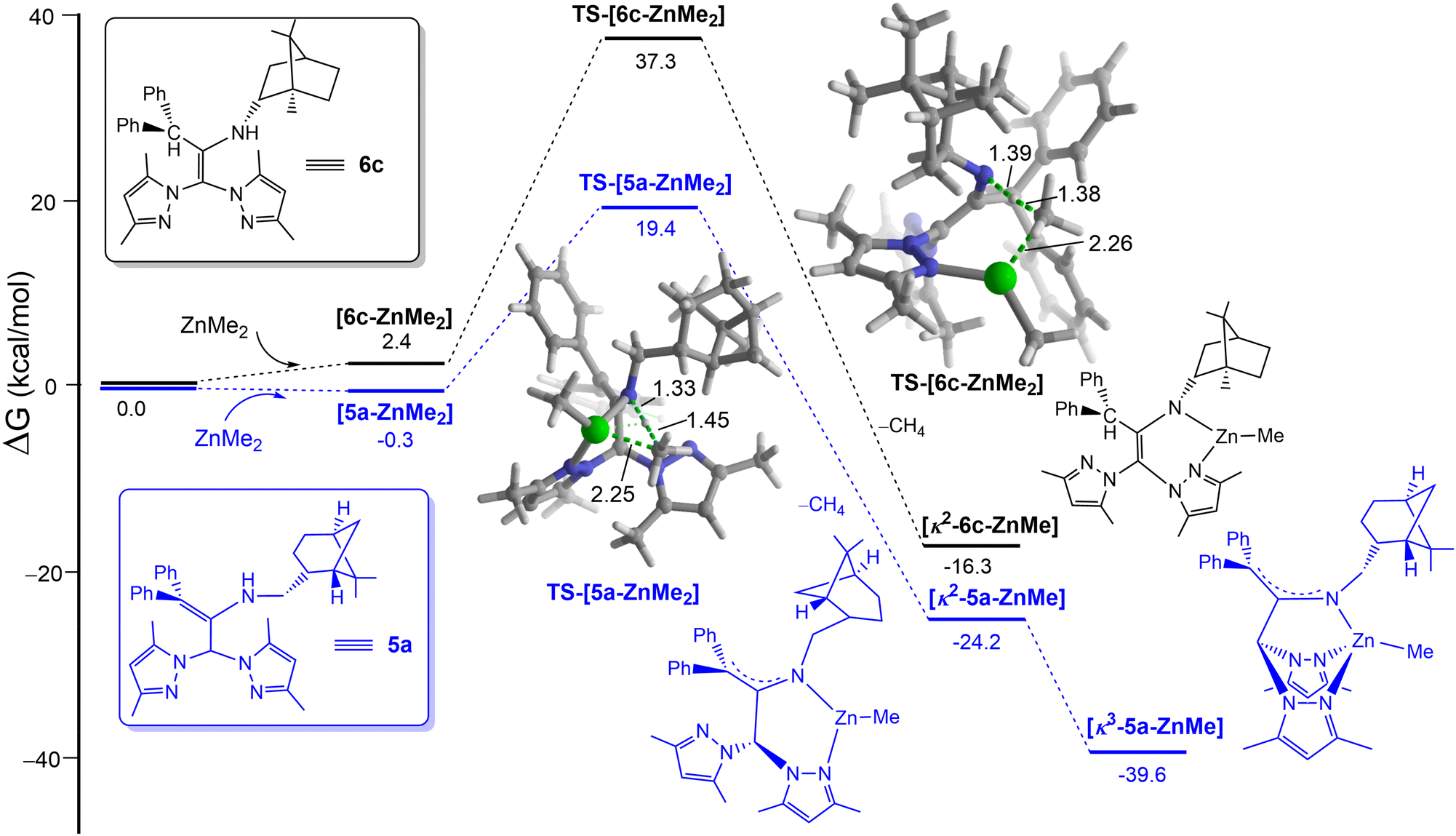 | ||
| Fig. 3 Computed reaction profile for the deprotonation reaction of 5a and 6c. Relative free energies and bond distances are given in kcal mol−1 and angstroms, respectively. All data were computed at the PCM-(toluene)-B3LYP-D3/def2-SVP level. | ||
In addition, subsequent reorganization of bidentante species [κ2-5a-ZnMe] into a scorpionate-arranged complex [κ3-5a-ZnMe] results in an additional thermodynamic stabilization (−39.6 kcal mol−1) in comparison with [κ2-6c-ZnMe] (−16.3 kcal mol−1), which cannot further rearrange into the corresponding κ3-NNN disposition due to the restrictive sp2-hybridation of the carbons in the enamine 6c.
These results are therefore fully consistent with the experimental difference of the reactivity observed in the enamine pro-ligands 6cversus5a with ZnMe2 under otherwise identical conditions.
Finally, to obtain suitable crystals of these complexes for X-ray diffraction analysis, a hexane solution of 7 was slowly evaporated at room temperature in air, and pale-orange crystals were collected from the solution after 72 h, which were identified as the enantiopure zinc complex [ZnMe(κ3-(−)-cis-bpmyO)]7 (9) [bpmyO = 2,2-bis(3,5-dimethyl-1H-pyrazol-1-yl)-N-(((1S,2R,5S)-6,6-dimethylbicyclo[3.1.1]heptan-2-yl)methyl)acetamide] (see Scheme 4).
The evolution of complex 7 to 9 can be envisaged as the final result of the oxidative cleavage of the enamine double bond. We believe that the oxidation that takes place here is probably catalysed by the presence of the metal centre (zinc), since this process was not observed in compound 5 under otherwise identical conditions. In the mother liquor of the crystallization solution, we have been able to observe by 1H NMR the presence of diphenyl ketone. All attempts to crystallize complexes 7 and 8 under an inert atmosphere and using different solvent mixtures were unsuccessful.
The 1H and 13C NMR spectra of complexes 7–9 (see Fig. S5 and S6 in the ESI†) show two sets of signals for each of the H4, Me3 and Me5 pyrazole protons, indicating the inequivalence of the two pyrazole rings. Furthermore, the 1H NMR spectra present one singlet for the CHa bridge, one multiplet (ABX system by coupling with the Hc of by bicycle) for the methylene group, and one set of signals for the bicycle and the phenyl groups for the complexes 7 and 8. However, complex 9 does not present the set of signals corresponding to the phenyl group, while in the 13C NMR spectrum, a signal corresponding to carbonyl carbon is observed at 166.0 ppm. In complexes 7 and 9 we can observe a singlet corresponding to the methyl group coordinated to zinc, while complex 8 shows two singlets corresponding to the CH2SiMe3 group, around zero ppm. 1H NOESY-1D and 1H–13C heteronuclear correlation (g-HSQC) experiments allowed the unequivocal assignment of all 1H resonances and the 13C{1H} NMR spectra, respectively. In addition, the specific rotation of all compounds was examined using optical polarimetry.
The structure of 9 was verified by single crystal X-ray diffraction analysis and is depicted in Fig. 4. Selected bond lengths and angles are listed in Table 1 (see Crystallographic details in Table S1 in the ESI†). The scorpionate ligand retains the enantiopure configuration in the complex. The zinc atom has a distorted tetrahedral geometry in which the scorpionate ligand acts in a tridentate fashion and is coordinated by the two nitrogens of the pyrazole rings and the nitrogen atom of the amide group, with the Zn-methyl group occupying the fourth vacancy of the tetrahedron. The N(1)–Zn and N(3)–Zn bond lengths [2.117(9) Å and 2.122(10) Å] compare well with that observed in other NNO-scorpionate zinc alkyls,21 but are considerably longer than the N(5)–Zn bond length [1.986(10) Å]. The Zn–C(23) bond length [1.962(13) Å] can be regarded as normal considering the distances reported for related zinc alkyls.21 Finally, partial delocalization in the acetamidate O(1)–C(12)–N(5) fragment is also observed [O(1)–C(12) = 1.235(14) Å, C(12)–N(5) = 1.325(15) Å], a common behaviour in related families of mononuclear organoderivatives.22
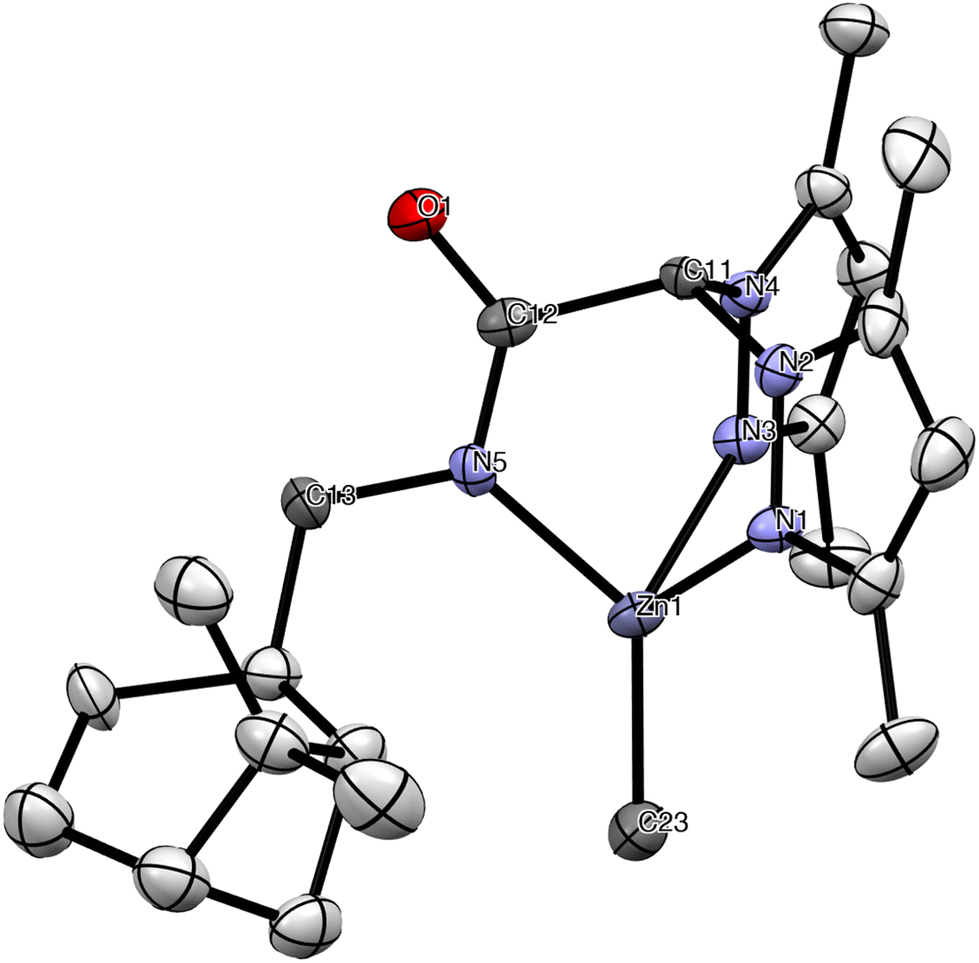 | ||
| Fig. 4 ORTEP view of complex [Zn(Me)(κ3-cis-bpmyO)] (9). Hydrogen atoms have been omitted for clarity. Thermal ellipsoids are drawn at the 30% probability. | ||
Catalytic studies for industrially demanding processes using complexes [ZnMe(κ3-cis-bpmy)] (7), [Zn(CH2SiMe3)(κ3-cis-bpmy)] (8) and [ZnMe(κ3-cis-bpmyO)] (9)
 | ||
| Scheme 5 Ring-opening polymerization of rac-lactide catalysed by complexes [ZnMe(κ3-cis-bpmy)] (7), [Zn(CH2SiMe3)(κ3-cis-bpmy)] (8) and [ZnMe(κ3-cis-bpmyO)] (9). | ||
The experimental Mn values of the PLAs produced showed good agreement with the expected theoretical values [Mn(calcd)-PLA100 = 14400 g mol−1] (see Table 2). Inspection of the size exclusion chromatography (SEC) data for the resulting polyesters showed a monomodal weight distribution, with dispersity values ranging from 1.07 to 1.22 (see Fig. S7 in the ESI†), suggesting well-controlled living propagations and the existence of a single type of reaction site.
a
| Entry | Catalyst | Time (h) | Yield (g) | Conv.b (%) |
M
n (theor.)c (Da) |
M
nd (Da) |
M
w/Mnd |
P
ie |
|---|---|---|---|---|---|---|---|---|
| a Polymerization conditions: [initiator]0 = 90 μmol of catalyst, 10 mL of tetrahydrofuran as solvent, [rac-lactide]0/[initiator]0 = 100, at 50 °C. b Percentage conversion of the monomer [(weight of polymer recovered/weight of monomer) × 100]. c Theoretical Mn = (monomer/catalyst) × (% conversion) × (Mw of lactide). d Determined by size exclusion chromatography relative to polystyrene standards in tetrahydrofuran. Experimental Mn was calculated considering the Mark–Houwink corrections23 for Mn [Mn(obsd) = 0.58 × Mn(GPC)]. e The parameter Pi (i = isotactic) is the probability of forming a new i-dyad. The Pi and the Ps (s = syndiotactic) values were calculated from the following tetrad probabilities based on enantiomorphic site control statistics10a in the polymerization of rac-lactide: sis, sii, iis = [Pi2(1 − Pi) + Pi(1 − Pi)2]/2; iii = [Pi2(1 − Pi)2 + Pi3 + (1 − Pi)3]/2; isi = [Pi(1 − Pi) + Pi(1 − Pi)]/2. f 20 mL of toluene as solvent. g Experiment at 35 °C. | ||||||||
| 1 | 7 | 2.5 | 0.48 | 37 | 5300 | 4900 | 1.07 | 0.88 |
| 2 |
7f |
5 | Traces | — | — | — | — | — |
| 3 |
7g |
5 | Traces | — | — | — | — | — |
| 4 | 7 | 6 | 0.84 | 65 | 9400 | 9700 | 1.07 | — |
| 5 | 7 | 10 | 1.19 | 92 | 13200 |
13500 |
1.09 | — |
| 6 | 8 | 2.5 | 0.42 | 32 | 4600 | 4900 | 1.20 | 0.85 |
| 7 | 9 | 2.5 | 0.44 | 34 | 4900 | 4700 | 1.22 | 0.87 |
| 8 | 8 | 10 | 1.06 | 82 | 11800 |
12100 |
1.19 | — |
| 9 | 9 | 10 | 1.10 | 85 | 12200 |
12000 |
1.18 | — |
Thus, 7 acted as an efficient single-component initiator and 37% of rac-LA was transformed at 50 °C after 2.5 h in tetrahydrofuran, with low molecular weight PLA materials and narrow polydispersity values (Table 2, entry 1; Mn = 4900, Mw/Mn = 1.07). The analog alkyls 8 and 9 were similarly active, with monomer conversions of 32 and 34%, respectively, under otherwise identical conditions, with similar features in the materials produced and a slight increase in the dispersity values (Table 2, entries 1, 6 and 7). After 10 h, almost complete conversion was reached for 7, whereas 8 and 9 showed slightly lower conversions (Table 2, entries 5, 8 and 9). The use of toluene as a solvent led to a drastic decrease in the catalytic activity, as only traces were obtained (Table 2, entry 2). This sharply contrasts with the process in tetrahydrofuran, which, at variance, is able to coordinate to the zinc ions and likely increases the nucleophilicity of the alkyl initiating group and the alkoxide propagating chains. A similar effect was observed with a decrease in the reaction temperature up to 35 °C (Table 2, entry 3). MALDI-ToF MS (see Fig. S8 in the ESI†) of low molecular weight materials evidenced an initial addition of the methyl fragment to the monomer. More interestingly, microstructural analysis of the poly(rac-lactide) revealed that initiator 7 exerts a remarkable preference for isotactic dyad enchainment, reaching a Pi value of 0.88 (see Fig. 5 and Fig. S9, Table S2 in the ESI†), paralleling the highest achieved for the very scarce zinc catalysts bearing chiral auxiliary ligands, which have already succeeded in the isoselective ROP of rac-LA (see Fig. 1a; Pi = 0.93,11a 0.92,11b and 0.8411c,d), and much higher than our previously reported NNCp-scorpionate alkyl zinc initiator (Pi = 0.77).6
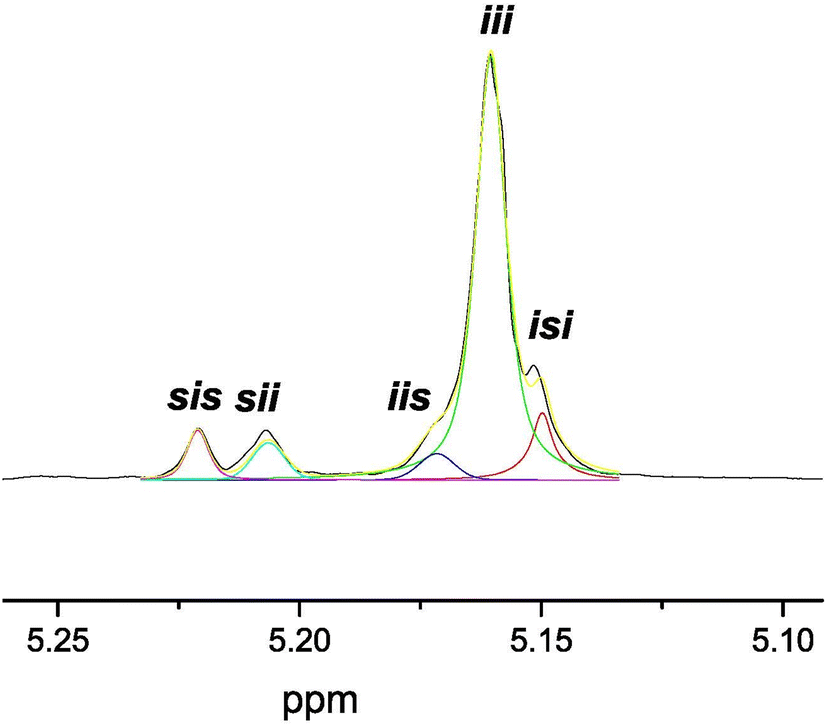 | ||
| Fig. 5 Deconvoluted 1H NMR spectra (500 MHz, 298 K, CDCl3) of the homodecoupled CH resonance of poly(rac-lactide) prepared using [ZnMe(κ3-(−)-cis-bpmy)] (7) as the initiator in tetrahydrofuran at 50 °C for 2.5 h (Table 2, entry 1). | ||
The origin of the isoselectivity observed is probably due to the high level of restrictive rotation of the myrtanyl bicyclic fragment in the C13–N5 bond. Additionally, inspection of the tetrads resulting from stereoerrors (i.e., tetrads other than iii) suggested that an enantiomorphic site control mechanism10 is dominant ([sis]/[sii]/[iis]/[isi] = 1/1/1/2 ratio, see Fig. S9 and Table S2 in the ESI†). This behaviour for catalyst 7 is most probably the result of the high level of homosteric control caused by the accurate architecture of this novel enantiomerically pure NNN′-scorpionate ligand.
:1 molar ratio, using a catalyst loading of 5 mol% in the presence of tetrabutylammonium bromide (Bu4NBr, TBAB). The results are presented in Table 3.
 | ||
| Scheme 6 Cyclic carbonate synthesis catalysed by complexes [ZnMe(κ3-cis-bpmy)] (7), [Zn(CH2SiMe3)(κ3-cis-bpmy)] (8) and [ZnMe(κ3-cis-bpmyO)] (9). | ||
a
| Entry | Catalyst | [Cat]:[co-cat] [mol%] |
Conversion [%] | |
|---|---|---|---|---|
| 25 °C, 1 bar,b 24 h | 50 °C, 10 bar,b 16 h (TOF, h−1)c | |||
| a Reactions carried out at 1–10 bar CO2 pressure during 16–24 h, using 0.25–5 mol% of complexes 7–9/0.25–5 mol% of TBAB as co-catalyst unless specified otherwise (see the ESI†). b Determined by 1H NMR spectroscopy of the crude reaction mixture. c TOF (turnover frequency) = number of moles of styrene oxide consumed/(moles of catalyst × time of reaction). d Reaction carried out during 8 h. e Reaction carried out at 100 °C during 1.5 h. f Not determined. | ||||
| 1 | 7 | 5.0:5.0 |
71 | ndf |
| 2 | 8 | 5.0:5.0 |
55 | nd |
| 3 | 9 | 5.0:5.0 |
47 | nd |
| 4 | 7 | 5.0:5.0 (TBAF) |
7 | nd |
| 5 | 7 | 5.0:5.0 (TBAC) |
54 | nd |
| 6 | 7 | 5.0:5.0 (TBAI) |
59 | nd |
| 7 | 7 | 5.0:5.0 (NMI) |
17 | nd |
| 8 | 7 | 5.0:5.0 (DMAP) |
6 | nd |
| 9 | 7 | 1.0:1.0 |
30 | 100 |
| 10 | 7 | 0.25:0.25 |
nd | 100 (25) |
| 11 |
7d |
0.25:0.25 |
nd | 86 (43) |
| 12 | 7 | 0.25:0 |
nd | 0 |
| 13 | — | 0:0.25 |
nd | 6 |
| 14 | Ligand 5 | 0.25:0.25 |
nd | 5 |
| 15 |
7e |
0.25:0.25 |
nd | 85 (227) |
Styrene oxide 10a conversion into the styrene carbonate 11a was determined by 1H NMR without any further purification (see Fig. S10 in the ESI†). Not surprisingly, the formation of styrene polycarbonate was not detected under the aforementioned conditions (selectivity >99%).
Complex 7 exhibited high catalytic activity (71%) for the synthesis of 11a, while derivatives 8 and 9 showed lower conversion values under identical experimental conditions, possibly due to both the lower steric hindrance of the methyl group and the stabilizing electronic effect of the enamine fragment in complex 7. Both beneficial effects promote the initial epoxide coordination and further catalytic performance in 7 (Table 3, entries 1–3).
Moreover, the effect of halide counter ions in the catalyst system was next inspected for complex 7 by employing different onium salts at 25 °C and 1 bar CO2 pressure for 24 hours employing this catalyst:co-catalyst loading (5 mol%). Interestingly, whereas the fluoride counter ion led to a lower catalytic activity than the chloride and iodide counterions, the bromide anion exhibited the highest activity (Table 1, entries 1 and 4–6), evidencing that this counterion performs as both a good nucleophile to ring-open the epoxide and a good leaving group for cyclic carbonate formation. Furthermore, 1-methylimidazol (NMI) and 4-dimethylaminopyridine (DMAP) were also examined as co-catalysts, resulting in poorly active systems (Table 1, entries 7 and 8, respectively). Therefore, we identified Bu4NBr as the most efficient co-catalyst for complex 7 under these conditions.
Catalyst and co-catalyst loadings were also inspected for the binary system 7/TBAB at 50 °C and 10 bar, and they could be reduced down to 0.25 mol% to reach complete conversion in 16 hours (Table 1, entries 9 and 10); therefore, it was identified as the optimal loading for the bicomponent 7/Bu4NBr under these experimental conditions for further catalytic studies. Interestingly, near complete full conversion (86%) was reached at 50 °C employing a combination of 0.25 mol% of complex 7 and 0.25 mol% of TBAB at 10 bar CO2 pressure after only 8 hours (Table 1, entry 11). Consistently, control experiments under these reaction conditions reveal no catalytic activity for 7 in the absence of TBAB, while the use of TBAB without the presence of 7 produced minimal conversion (6%), confirming the necessity of both catalyst components to perform successfully in this cycloaddition process. Also, the corresponding ancillary enantiopure pro-ligand 5, as a mixture of the tautomers (−)-cis-bpmyH and (−)-cis-bpmy′H, displayed very poor conversion in the presence of TBAB (5%) under otherwise identical conditions (Table 1, entries 12–14), supporting the catalytic performance of complex 7 in the process.
It is also worth noting that under these conditions, the bicomponent system 7/Bu4NBr parallels the catalytic performance observed (Table 1, entry 11, TOF = 43 h−1, 50 °C, 10 bar) for the recently reported efficient binary systems that include mononuclear zinc complexes supported by sterically demanding amidinate-based scorpionates with different electron-withdrawing groups, under identical conditions (TOF = 47 h−1, 50 °C, 10 bar).15a7/Bu4NBr was also much more active than the NNO′-scorpionate zinc-based bicomponent mononuclear (TOF = 2.33 h−1), the dinuclear (TOF = 3.0 h−1) and the bifunctional (TOF = 2.9 h−1) analogs also under mild conditions (50 °C), previously reported by our group.15b In addition, 7/Bu4NBr can operate very efficiently for the production of styrene carbonate 11a under softer and comparable experimental conditions (Table 1, entry 11, TOF = 43 h−1, 50 °C and entry 15, TOF = 227 h−1, 100 °C, respectively) than the alternative very well-performed zinc-based catalysts previously reported (see Fig. 1b).16
In view of these encouraging results, we evaluated a variety of terminal substrates including alkyl, aryl and functionalized terminal epoxides 10b–10i, using the 7/TBAB bicomponent system (see Scheme 6), at 50 °C and 10 bar of CO2 pressure, with 0.25 mol% of catalyst/co-catalyst loading in a 1:1 molar ratio in the absence of a solvent (see Fig. S11–S18 in the ESI†). Interestingly, excellent conversions were achieved under these conditions in 16 hours, including those substrates bearing aryl or alkyl substituents as well as alcohol or ether functionalities (see Fig. 6).
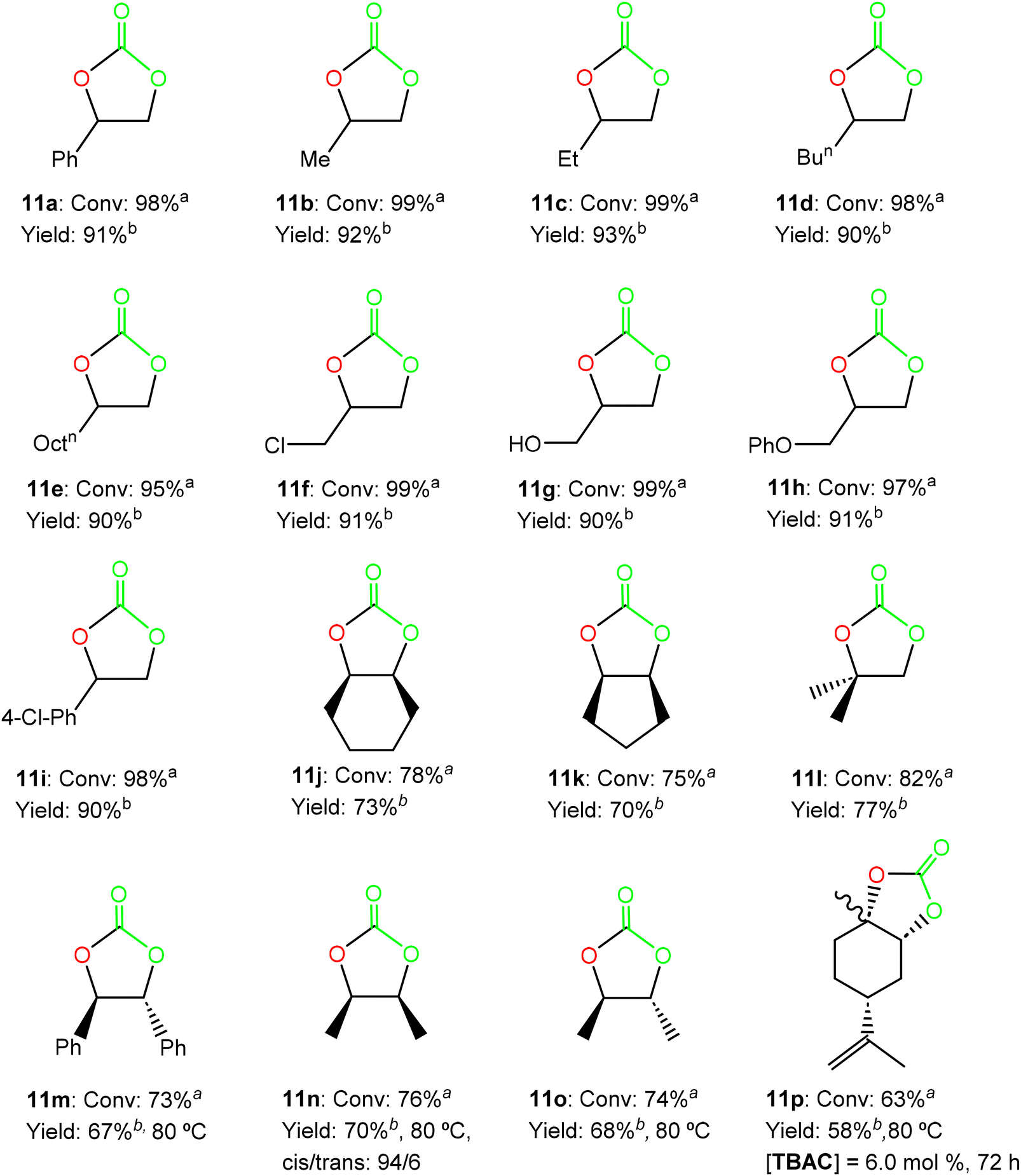 | ||
| Fig. 6 Synthesis of cyclic carbonates 11a–11i from epoxides 10a–10i using 0.25 mol% of the bicomponent system formed by 7/TBAB at 50 °C and 10 bar CO2 pressure for 16 hours, and cyclic carbonates 11j–11p from epoxides 10j–10p, using 2.0 mol% of the binary system 7/TBAB at 50 °C and 20 bar CO2 pressure for 24 hours, unless specified otherwise (see the ESI†). aConversion and selectivity were determined using 1H NMR. bIsolated yield after column chromatography. | ||
Considering the high activity displayed by the bicomponent system 7/TBAB, we decided to extend the substrate scope for catalyst 7 and evaluate the conversion of internal and disubstituted epoxides 10j–10o into the corresponding cyclic carbonates 11j–11o (see Fig. 6).
Thus, we increased CO2 pressure to 20 bar and catalyst:co-catalyst loadings up to 2.0 mol% while maintaining the temperature (50 °C) for the cyclohexene oxide (CHO) 10j, cyclopentene oxide (CPO) 10k and 1,2-isobutylene oxide 10l (see Scheme 6). To our delight, a 1:1 ratio of the binary system 7/TBAB displayed very good activities (conversion >75%) for these internal and disubstituted substrates in 24 hours, with retention in the epoxide stereochemistry. In addition, only an increase in temperature of up to 80 °C was needed for the transformation of the disubstituted trans-stilbene oxide 10m, cis-2,3-epoxybutane 10n and trans-2,3-epoxybutane 10o to reach very good conversions (conv. >73%) under these mild and solvent-free conditions (see Fig. 6), showing the efficiency of this bicomponent system (see Fig. S19–S24 in the ESI†). We have witnessed a significant progress in recent years employing catalysts based on metals such as Fe(II)-,24 Ca(II)-25 and Al(III)-,26 considering the lower reactivity of these disubstituted epoxides.27 However, few examples of Zn(II)-based complexes15,28 have been reported for the efficient and selective synthesis of the cyclic carbonates 11j–11o (5 mol%, 20 bar of CO2, 80 °C, 24 h),15b but not under the current lower catalyst/cocatalyst loadings15a (see Fig. 6).
Encouraged by these results, we finally decided to extend this study to produce a very challenging bio-renewable bicyclic carbonate such as limonene carbonate 11p obtained from limonene oxide 10p (see Scheme 6), a highly substituted monocyclic unsaturated terpene derived from biomass29 (extracted from the peel of citrus fruits).
We were also pleased to find that the bicyclic limonene carbonate 11p was obtained in high yield (conv. >60%) from the commercially available limonene oxide 10p (as a mixture of cis/trans isomers, 43:57), employing a combination of 2.0 mol% of complex 7 and 6.0 mol% of TBAC at 20 bar of CO2 pressure, 80 °C of temperature after 72 h of reaction (see Fig. 6), in accordance with the lower reactivity and the higher steric hindrance of this tri-substituted epoxide, and in agreement with our previous expertise with this challenging substrate (see Fig. S25 in the ESI†).15a,26a,30,32c
As far as we are aware, very few examples of well-defined zinc-based catalysts31 have been reported to date that can operate under mild conditions for the successful cycloaddition reaction of CO2 to limonene oxide.26a,c,27,30,32
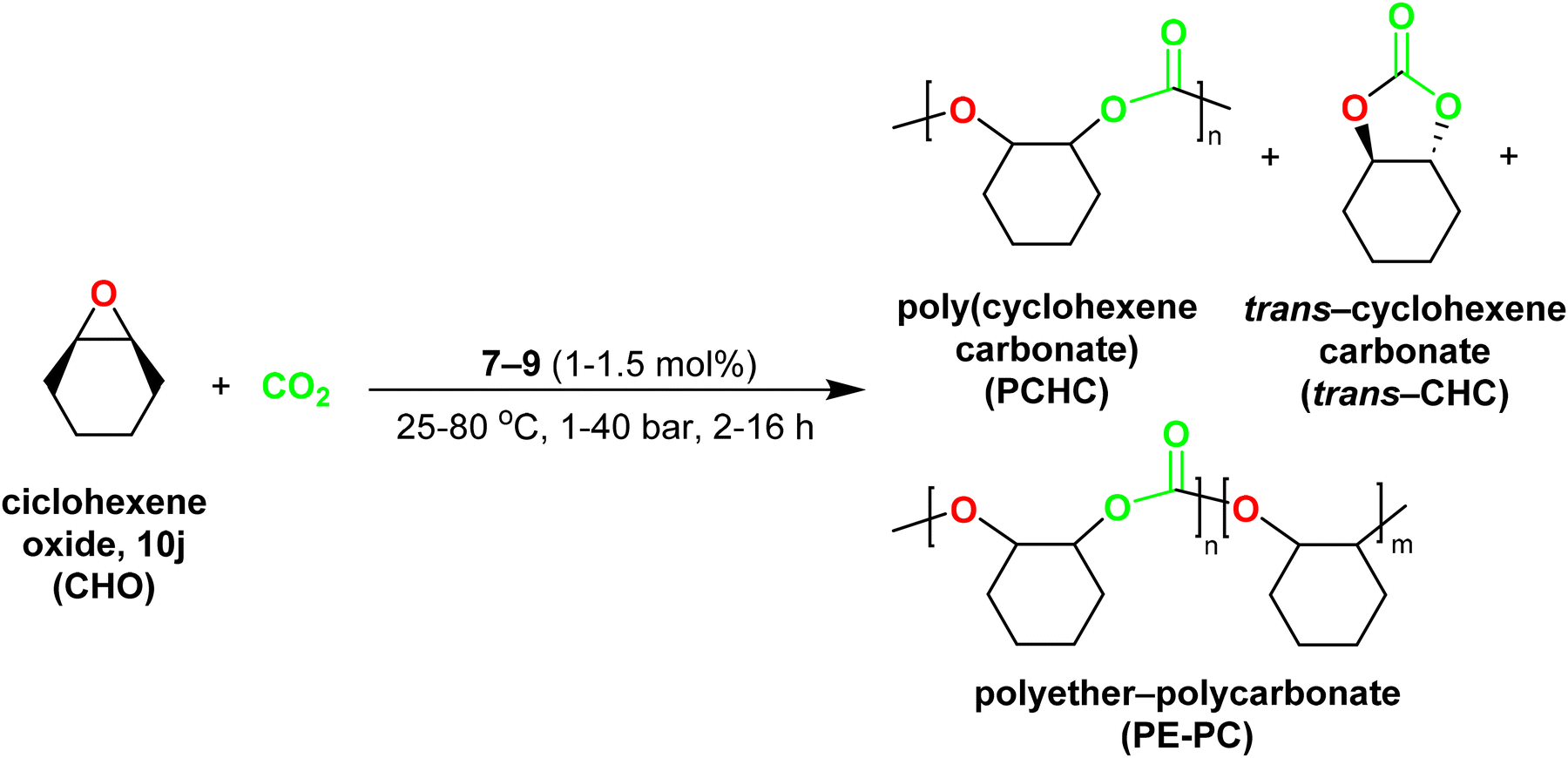 | ||
| Scheme 7 Synthesis of poly(cyclohexenecarbonate) catalysed by complexes [ZnMe(κ3-cis-bpmy)] (7), [Zn(CH2SiMe3)(κ3-cis-bpmy)] (8) and [ZnMe(κ3-cis-bpmyO)] (9). | ||
1H NMR spectroscopy was employed to analyse each reaction without further purification and to determine the conversion of CHO into PCHC, trans-cyclohexenecarbonate (trans-CHC) or polyether-poly(cyclohexenecarbonate) (PE-PCHC) (see Scheme 7). The results are presented in Table 4. Interestingly, complexes 7–9 showed good to excellent conversions of cyclohexene oxide, in the absence of a co-catalyst, thus indicating that these complexes can initiate copolymerization by themselves.33
a
| Entry | Temp (°C) | Pres. (bar) | Time (h) | Conv.b (%) | %CHCb | %Copolymer (%carbonate linkage)b | TOFc (h−1) | M n(exp) (Da)d (Đ)d |
|---|---|---|---|---|---|---|---|---|
| a Reactions carried out using 1 mol% of catalyst 7 unless specified otherwise (see the ESI†). b Conversion, % of trans-CHC, % of PCHC and % of carbonate linkages determined using 1H NMR spectroscopy of the crude reaction mixture. c TOF = (moles of product)/(moles of catalyst × time (h−1)). d Determined by GPC relative to polystyrene standards in tetrahydrofuran. e Experiment using 1 mol% of catalyst 8. f Experiment using 1 mol% of catalyst 9. g Experiment using 1.5 mol% of catalyst 7. h Experiment using 0.5 mol% of catalyst 7. | ||||||||
| 1 | 80 | 40 | 16 | 68 | 11 | 89 (>99) | 3.78 | 8500 (1.33) |
| 2 | 80e | 40 | 16 | 47 (8) | 17 | 83 (>82) | 2.43 | 6000 (1.27) |
| 3 | 80f | 40 | 16 | 40 (9) | 27 | 73 (>74) | 1.82 | 4800 (1.21) |
| 4 | 80g | 40 | 16 | 72 (1.5 mol%) | 12 | 88 (>99) | — | — |
| 5 | 80h | 40 | 16 | 26 (0.5 mol%) | 10 | 90 (>99) | — | — |
| 6 | 70 | 40 | 16 | 59 | 10 | 90 (>91) | 3.31 | 7100 (1.07) |
| 7 | 60 | 40 | 16 | 60 | 9 | 91 (>93) | 3.41 | 8000 (1.15) |
| 8 | 50 | 40 | 16 | 35 | 6 | 94 (>99) | 2.06 | 5100 (1.03) |
| 9 | 25 | 40 | 16 | Traces | — | — | — | — |
| 10 | 50 | 30 | 16 | 57 | 2 | 98 (>98) | 3.49 | 7600 (1.32) |
| 11 | 50 | 20 | 16 | 73 | 1 | 99 (>99) | 4.51 | 12200 (1.06) |
| 12 | 50 | 10 | 16 | 37 | 7 | 93 (>98) | 2.15 | 5200 (1.03) |
| 13 | 50 | 1 | 16 | 20 | 13 | 87 (>98) | 1.08 | 2700 (1.09) |
| 14 | 50 | 20 | 2 | 15 | 2 | 98 (>99) | 7.12 | 2900 (1.26) |
| 15 | 50 | 20 | 5 | 29 | 1 | 99 (>99) | 5.10 | 4800 (1.17) |
| 16 | 50 | 20 | 8 | 51 | 3 | 97 (>99) | 5.86 | 8000 (1.14) |
| 17 | 50 | 20 | 10 | 59 | 2 | 98 (>99) | 5.25 | 9100 (1.09) |
| 18 | 50 | 20 | 15 | 69 | 1 | 99 (>99) | 4.23 | 11500 (1.04) |
In the case of the methyl derivative 7, the only polymeric species identified by 1H NMR spectroscopy was PCHC, in conjunction with trans-CHC, as a result of backbiting reactions (Table 4, entry 1). Contrarily, the trimethylsilylmethyl analog 8 and the acetamidate-derivative 9 exhibited lower conversion and selectivity for the formation of PCHC (Table 4, entries 2 and 3) with higher PE-PCHC production than complex 7 (Table 4, entries 1–3). Complex 7 exhibited an outperformed catalytic activity and carbonate selectivity for the ROCOP of CHO and CO2 (Table 4, entry 1), affording 68% conversion with a high content of carbonate linkages (>99) and a high PCHC/CHC ratio (89/11), possibly due to the stabilizing electronic effect of the enamine fragment in complex 7.
Since complex 7 was more active and selective than 8 and 9 for the synthesis of PCHC, we inspected the effect of varying catalyst loading, temperature, pressure, and time using this complex for further optimisation of the reaction (see Table 4).
Under the present reaction conditions (80 °C, 40 bar of CO2 pressure), the catalyst loading was finally optimized to 1 mol% (Table 4, entries 1, 4 and 5).
Furthermore, the catalytic activity of complex 7 was clearly dependent on the reaction temperature (see Table 4). Thus, decreasing the temperature from 80 °C to 50 °C afforded a significant decrease in the conversion of CHO into PCHC (35%), while, interestingly, it led to an increase in selectivity to a PCHC/CHC ratio of 94/6. A reduction of up to 25 °C resulted in a dramatic reduction of the conversion up to traces (Table 4, entries 1 and 6–9). Therefore, the optimised temperature for further experiments was 50 °C.
Interestingly, reduction of the pressure from 40 to 20 bar provoked an increase in conversion and selectivity of complex 7 (73%, PCHC/CHC = 99/1). Expectedly, a decrease of up to 10–1 bar produced a significant drop in the conversion value (37–20%) in conjunction with a lack of PCHC/CHC selectivity (93/7–87/13). It was also noteworthy that the carbonate linkage (>98%) remained essentially constant in the range of 1 to 40 bar (Table 4, entries 8 and 10–13). Therefore, we selected 20 bar as the optimal CO2 pressure.
Finally, the influence of reaction time on catalytic activity was also investigated for complex 7. As expected, after reduction from 16 to 2 h of reaction the conversion decreased from 73% to 15% (Table 4, entries 11 and 14–18), with near maintenance of polymer selectivity (99% to 97%). Importantly, an increase in the reaction time produced PCHC with higher molecular weight, with low monodispersity (Đ) values ranging from 1.26 to 1.03 (see Fig. S26 in the ESI†), evidencing living propagations (see Fig. 7) and the absence of back-biting reactions during the ROCOP event.
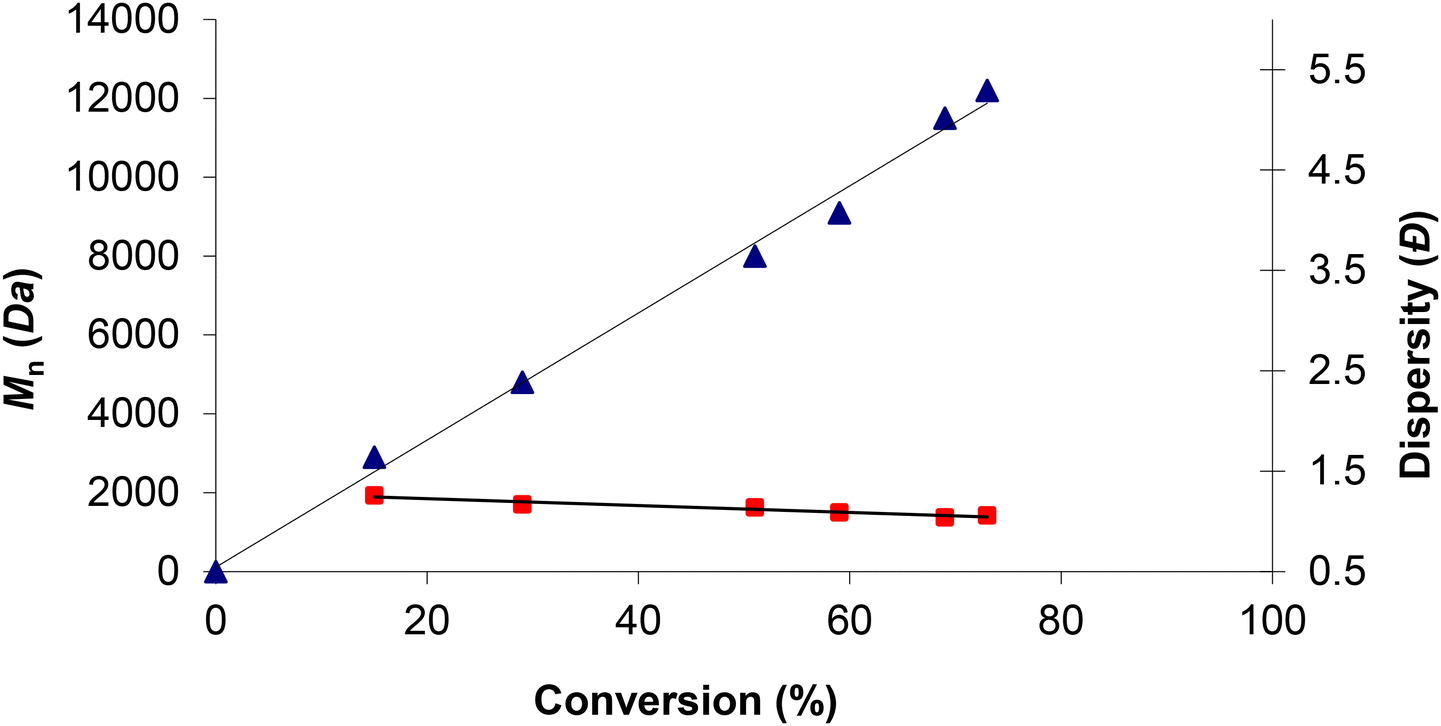 | ||
Fig. 7 Plot of Mnversus CHO conversion ( ) and Mw/Mn ( ) and Mw/Mn ( ) versus CHO conversion for compound 7 (1 mol%) at 50 °C and 20 bar CO2. ) versus CHO conversion for compound 7 (1 mol%) at 50 °C and 20 bar CO2. | ||
In this context, these activity values exhibited by complex 7 are considerably lower than that reported for alternative very efficient and selective multinuclear zinc catalysts for the production of PCHC (see Fig. 1c)18 However, a very scarce number of examples for zinc-based catalysts have been reported to succeed under these mild conditions (50 °C, 20 bar of CO2), displaying a high level of PCHC selectivity.17a,18b,34
In addition, the polymer microstructure produced by 7 was inspected by 1H and 13C-NMR spectroscopy (see Fig. S27 in the ESI†), and MALDI-TOF MS (matrix-assisted laser desorption time-of-flight mass spectrometry) to determine the end-group in these materials (see Fig. 8). Thus, the analysis of the carbonyl region by 13C NMR spectroscopy allowed us to examine the tacticity of these poly(carbonate)s. Regrettably, the isotactic (153.7 ppm) and syndiotactic (153.7–153.0 ppm) dyads were identified (see Fig. S27b in the ESI†), indicating that atactic polymers were obtained.
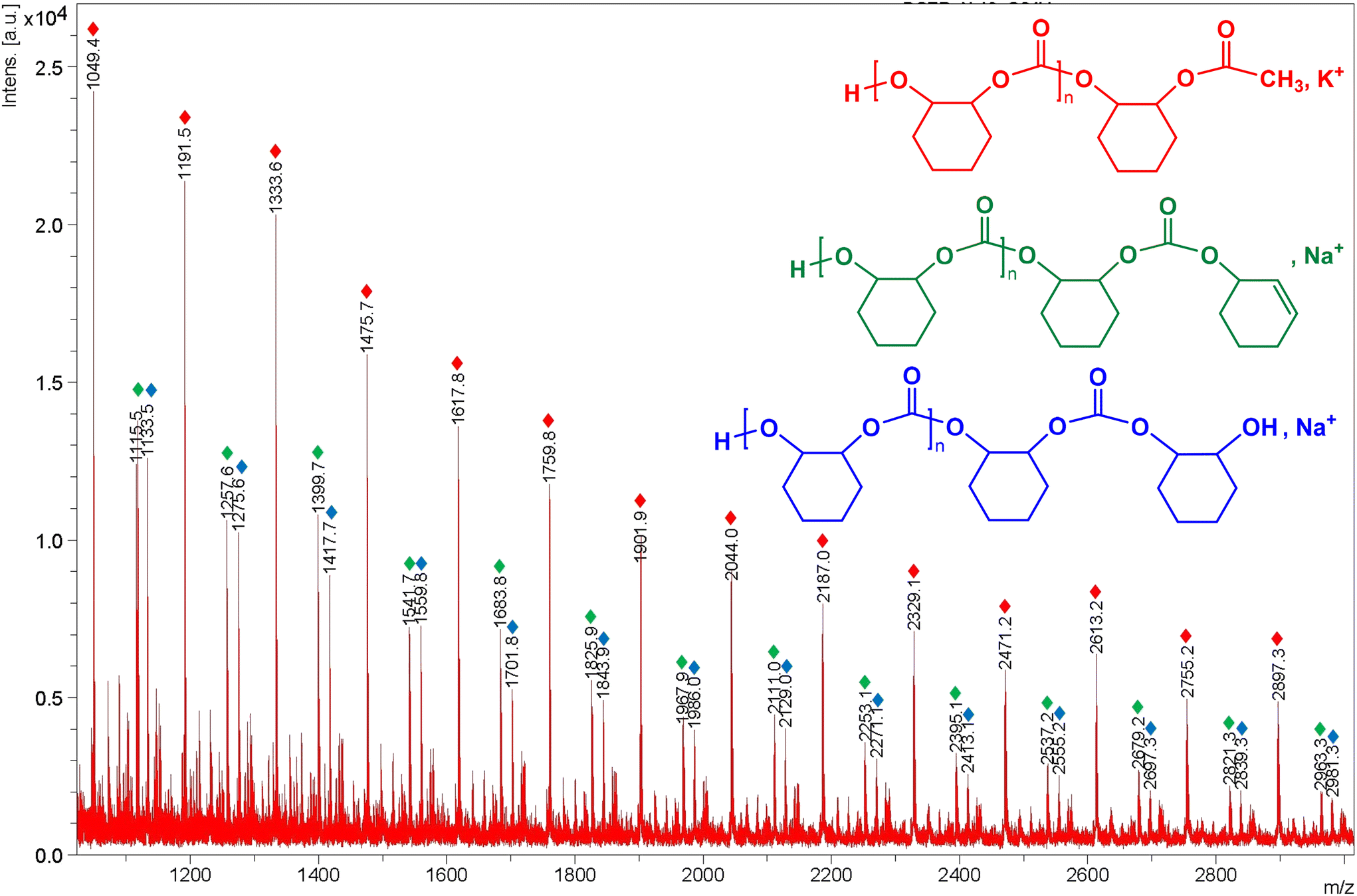 | ||
Fig. 8 Expansion of the MALDI-ToF mass spectrum of poly(cyclohexene carbonate) sample from Table 4 (entry 16) using zinc complex 7 as a catalyst at 50 °C and 20 bar CO2 (Fig. S28 in the ESI†). Assignment of peaks: series  has a repeat unit m/z = 1.01 + (142.07 × DPn+1) + 15.02 + 39.09, where n = 6–19. Series has a repeat unit m/z = 1.01 + (142.07 × DPn+1) + 15.02 + 39.09, where n = 6–19. Series  has a repeat unit m/z = 1.01 + (142.07 × DPn+1) + 115.08 + 22.99, where n = 6–19. Series has a repeat unit m/z = 1.01 + (142.07 × DPn+1) + 115.08 + 22.99, where n = 6–19. Series  has a repeat unit m/z = 1.01 + (142.07 × DPn+1) + 97.07 + 22.99, where n = 6–19. has a repeat unit m/z = 1.01 + (142.07 × DPn+1) + 97.07 + 22.99, where n = 6–19. | ||
Moreover, MALDI-TOF MS spectrum was acquired using trans-2-[3-(4-tert-butyl-phenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) as matrix and NaI as a cationization agent. Different initiation possibilities for complex 7 were observed in the spectrum since the PCHC distribution included three end-group series of peaks separated by a molecular mass of 142 Da, with the loss of a cyclohexene carbonate unit (C7H10O3) in all polymer chains (see Fig. 8 and Fig. S28 in the ESI†). In particular, a major series possessing one hydroxyl end group and one methyl end group in the polycarbonate ( ) can be assigned, in addition to another series bearing one cyclohexenyl group and one hydroxyl end-group (
) can be assigned, in addition to another series bearing one cyclohexenyl group and one hydroxyl end-group ( ) and a third sequence containing one cyclohexanol group and one hydroxyl end-group (
) and a third sequence containing one cyclohexanol group and one hydroxyl end-group ( ). This behaviour suggests that chain transfer reactions operate due to the presence of either adventitious water and/or cyclohexenediol through the polymerisation event, as previously reported.17d,35
). This behaviour suggests that chain transfer reactions operate due to the presence of either adventitious water and/or cyclohexenediol through the polymerisation event, as previously reported.17d,35
Finally, the thermal properties of the polycarbonates were also evaluated and determined by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). TGA analysis showed that PCHC was stable from 0 to 200 °C (see Fig. 29 in the ESI†). A single Tg value of 116.5 °C was observed by DSC analysis (see Fig. 30 in the ESI†), which is similar to values previously reported for atactic poly(cyclohexene) carbonate.36
Conclusions
In conclusion, we present herein a successful ligand design strategy to introduce a chiral substrate in a N-donor atom, through the first example reported of a nucleophilic addition of an organolithium to an enatiomerically pure ketenimine. In this case, we report the preparation of new enantiopure NNN-donor heteroscorpionates. In addition, one of these compounds has proved to be an excellent reagent to introduce chirality in metal complexes, a fact confirmed by the preparation of sustainable, inexpensive and low-toxicity zinc complexes. X-ray diffraction studies confirmed ligand coordination in a κ3-arrangement as well as a stereogenic substrate proximity to the metal centre.These enantiopure zinc complexes have successfully confirmed to behave as highly efficient catalysts for a variety of sustainable bioresourced processes, with a wide range of substrates. Thus, complex 7 can act as an efficient initiator for the living ROP of rac-LA under very mild conditions after a few hours, exerting a significant homosteric control in the production of highly enriched isotactic poly(lactide)s, reaching a Pi value of up to 0.88, which parallels the highest value reported so far using these species. The PLA materials produced exhibited narrow dispersity values and good agreement between the experimental Mn values and the monomer ratio.
Importantly, complex 7, in combination with tetra-n-butylammonium bromide/chloride, behaves as a highly efficient and selective system for the cycloaddition of CO2 to epoxides into five-membered cyclic carbonates under very mild and solvent-free conditions, achieving TOF values of up to 227 h−1. Furthermore, this bicomponent system exhibits a broad substrate scope and functional group tolerance, including not only terminal and internal epoxides but also the bio-renewable tri-substituted terpene-derived cis/trans-limonene oxide, with high stereoselectivity in the formation of the bicyclic trans-limonene carbonate. Moreover, complex 7 also shows good catalytic performance and high selectivity as a one-component initiator for the synthesis of poly(cyclohexene carbonate) via ring-opening copolymerization of cyclohexene oxide and CO2 under very soft conditions (50 °C and 20 bar), affording materials with narrow dispersity values.
Although several highly active zinc-based catalytic systems have been independently reported for sustainable processes in recent years7,11,15–18 (see Fig. 1), the preparation of versatile and efficient single zinc-based catalysts with a wider substrate scope for the processes mentioned above remains poorly investigated. We consider that these results represent an important further step forward in the search for more sustainable, inexpensive, and low-toxicity metal-based catalysts capable of operating efficiently under mild conditions in these industrially demanding processes.
Author contributions
M. N. and S. S. carried out the synthesis and characterisation of the complexes and performed the catalytic studies. A. G. and I. F. accomplished the DFT studies. L. F. S.-B. wrote the manuscript. L. F. S.-B. and A. G. supervised all results. L. F. S.-B. and A. L.-S. ensured financial support. All authors helped in the conceptualization and critical reading of the manuscript.Data availability
Data for this article, including ROP of LA catalysed by 7–9, cycloaddition of styrene oxide and carbon dioxide catalyzed by 7–9, ROCoP of cyclohexene oxide and carbon dioxide catalyzed by 7–9, bond_and_angles_comp._6c and 9 are available at ecienciaDATOS at https://doi.org/10.21950/P3HODS.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support: grants PID2020-117788RB-I00 and PID2022-139318NB-I00 funded by MCIN/AEI/10.13039/501100011033, grant RED2022-134287-T funded by MCIN/AEI, grants SBPLY/21/180501/000132 and SBPLY/22/180502/000059 funded by Junta de Comunidades de Castilla-La Mancha and by the EU through “Fondo Europeo de Desarollo Regional” (FEDER), and grant 2021-GRIN-31240 funded by Universidad de Castilla-La Mancha. L.F.S.-B. thanks Dr Juan Fernández Baeza for his scientific contribution to this work.References
- (a) L. M. D. R. S. Martins, Inorg. Chim. Acta, 2022, 541, 121069 CrossRef CAS; (b) K. H. C. Pettinari, Eur. J. Inorg. Chem., 2016, 2202–2657 Search PubMed; (c) A. Otero, J. Fernández-Baeza, A. Lara-Sánchez and L. F. Sánchez-Barba, Coord. Chem. Rev., 2013, 257, 1806–1868 CrossRef CAS.
- A. Otero, J. Fernández-Baeza, J. Tejeda, A. Antiñolo, F. Carrillo-Hermosilla, E. Díez-Barra, A. Lara-Sánchez, M. Fernández-López, M. Lanfranchi and M. A. Pellinghelli, J. Chem. Soc., Dalton Trans., 1999, 3537–3539 RSC.
- M. Martínez de Sarasa Buchaca, F. de la Cruz-Martínez, L. F. Sánchez-Barba, J. Tejeda, A. M. Rodríguez, J. A. Castro-Osma and A. Lara-Sánchez, Dalton Trans., 2023, 52, 3482–3492 RSC.
- L. F. Sánchez-Barba, A. Garcés, J. Fernández-Baeza, A. Otero, M. Honrado, A. Lara-Sánchez, A. M. Rodríguez and I. López-Solera, Eur. J. Inorg. Chem., 2014, 11, 1922–1928 CrossRef.
- (a) A. Otero, J. Fernández-Baeza, A. Garcés, L. F. Sánchez-Barba, A. Lara-Sánchez, J. Martínez-Ferrer, M. P. Carrión and A. M. Rodríguez, Dalton Trans., 2017, 46, 6654–6662 RSC; (b) A. Otero, J. Fernández-Baeza, J. Tejeda, A. Lara-Sánchez, S. Franco, J. Martínez-Ferrer, M. P. Carrión, I. López-Solera, A. M. Rodríguez and L. F. Sánchez-Barba, Inorg. Chem., 2011, 50, 1826–1839 CrossRef CAS PubMed.
- M. Honrado, A. Otero, J. Fernández-Baeza, L. F. Sánchez-Barba, A. Garcés, A. Lara-Sánchez, J. Martínez-Ferrer, S. Sobrino and A. M. Rodríguez, Organometallics, 2015, 34, 3196–3208 CrossRef CAS.
- M. Honrado, S. Sobrino, J. Fernández-Baeza, L. F. Sánchez-Barba, A. Garcés, A. Lara-Sánchez and A. M. Rodríguez, Chem. Commun., 2019, 55, 8947–8950 RSC.
- P. Lu and Y. Wang, Chem. Soc. Rev., 2012, 41, 5687–5705 RSC.
- (a) A. D. Allen and T. T. Tidwell, Chem. Rev., 2013, 113, 7287–7342 CrossRef CAS PubMed; (b) M. Alajarin, M. Marin-Luna and A. Vidal, Eur. J. Org. Chem., 2012, 5637–5653 CrossRef CAS.
- (a) C. Bakewell, G. Fateh-Iravani, D. W. Beh, D. Myers, S. Tabthong, P. Hormnirun, A. J. P. White, N. Long and C. K. Williams, Dalton Trans., 2015, 44, 12326–12337 RSC; (b) T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1316–1326 CrossRef CAS PubMed.
- (a) C. Kan, J. Hu, Y. Huang, H. Wang and H. Ma, Macromolecules, 2017, 50, 7911–7919 CrossRef CAS; (b) S. Abbina and G. Du, ACS Macro Lett., 2014, 3, 689–692 CrossRef CAS; (c) H. Wang, Y. Yang and H. Ma, Macromolecules, 2014, 47, 7750–7764 CrossRef CAS; (d) H. Wang and H. Ma, Chem. Commun., 2013, 49, 8686–8688 RSC.
- (a) B. Tyler, D. Gullotti, A. Mangraviti, T. Utsuki and H. Brem, Adv. Drug Delivery Rev., 2016, 107, 163–175 CrossRef CAS PubMed; (b) S. Inkinen, M. Hakkarainen, A.-C. Albertsson and A. Södergård, Biomacromolecules, 2011, 12, 523–532 CrossRef CAS PubMed; (c) A. P. Pego, B. Siebum, M. J. A. Van Luyn, X. Van Seijen, A. A. Poot, D. W. Grijpma and J. Feijen, Tissue Eng., 2003, 9, 981–994 CrossRef CAS PubMed.
- (a) F. Vidal, E. R. van der Marel, R. W. F. Kerr, C. McElroy, N. Schroeder, C. Mitchell, G. Rosetto, T. T. D. Chen, R. M. Bailey, C. Hepburn, C. Redgwell and C. K. Williams, Nature, 2024, 626, 45–57 CrossRef CAS PubMed; (b) L. Guo, K. J. Lamb and M. North, Green Chem., 2021, 23, 77–118 RSC; (c) V. Aomchad, À. Cristòfol, F. Della Monica, B. Limburg, V. D'Elia and A. W. Kleij, Green Chem., 2021, 23, 1077–1113 RSC.
- (a) G. Rosetto, F. Vidal, T. M. McGuire, R. W. F. Kerr and C. K. Williams, ChemRxiv, 2024, Version 2, DOI: 10.26434/chemrxiv-2024-7sdx8-v2 Search PubMed; (b) C. M. Kozak, K. Ambrose and T. S. Anderson, Coord. Chem. Rev., 2018, 376, 565–587 CrossRef CAS; (c) G. Trott, P. K. Saini and C. K. Williams, Philos. Trans. R. Soc., A, 2016, 374, 20150085 CrossRef PubMed.
- (a) M. Navarro, A. Garcés, L. F. Sánchez-Barba, D. González-Lizana and A. Lara-Sánchez, Dalton Trans., 2023, 52, 6105–6116 RSC; (b) S. Sobrino, M. Navarro, J. Fernández-Baeza, L. F. Sánchez-Barba, A. Garcés, A. Lara-Sánchez and J. A. Castro-Osma, Dalton Trans., 2019, 48, 10733–10742 RSC.
- (a) L. Cuesta-Aluja, A. Campos-Carrasco, J. Castilla, M. Reguero, A. M. Masdeu-Bultó and A. Aghmiz, J. CO2 Util., 2016, 14, 10–22 CrossRef CAS; (b) C. Maeda, J. Shimonishi, R. Miyazaki, J.-Y. Hasegawa and T. Ema, Chem. – Eur. J., 2016, 22, 6556–6563 CrossRef CAS; (c) R. Ma, L.-N. He and Y.-B. Zhou, Green Chem., 2016, 18, 226–231 RSC; (d) X.-D. Lang, Y.-C. Yu and L.-N. He, J. Mol. Catal. A: Chem., 2016, 420, 208–215 CrossRef CAS.
- (a) F. de la Cruz-Martínez, M. Martínez de Sarasa Buchaca, A. del Campo-Balguerias, J. Fernández-Baeza, L. F. Sánchez-Barba, A. Garcés, C. Alonso-Moreno, J. A. Castro-Osma and A. Lara-Sánchez, Polymers, 2021, 13, 1651 CrossRef PubMed; (b) S. Sobrino, M. Navarro, J. Fernández-Baeza, L. F. Sánchez-Barba, A. Lara-Sánchez, A. Garcés, J. A. Castro-Osma and A. M. Rodríguez, Polymers, 2020, 12, 2148 CrossRef CAS PubMed; (c) F. de la Cruz-Martínez, M. Martínez de Sarasa Buchaca, J. Martínez, J. Tejeda, J. Fernández-Baeza, C. Alonso-Moreno, A. M. Rodríguez, J. A. Castro-Osma and A. Lara-Sánchez, Inorg. Chem., 2020, 59, 8412–8423 CrossRef; (d) J. Martínez, J. A. Castro-Osma, A. Lara-Sánchez, A. Otero, J. Fernández-Baeza, J. Tejeda, L. F. Sánchez-Barba and A. Rodríguez-Diéguez, Polym. Chem., 2016, 7, 6475–6484 RSC.
- (a) J. R. Pankhurst, S. Paul, Y. Zhu, C. K. Williams and J. B. Love, Dalton Trans., 2019, 48, 4887–4893 RSC; (b) M. Reiter, S. Vagin, A. Kronast, C. Jandl and B. Rieger, Chem. Sci., 2017, 8, 1876–1882 RSC; (c) S. Kernbichl, M. Reiter, F. Adams, S. Vagin and B. Rieger, J. Am. Chem. Soc., 2017, 139, 6787–6790 CrossRef CAS; (d) M. Schütze, S. Dechert and F. Meyer, Chem. – Eur. J., 2017, 23, 16472–16475 CrossRef PubMed; (e) S. Kissling, P. T. Altenbuchner, M. W. Lehenmeier, E. Herdtweck, P. Deglmann, U. B. Seemann and B. Rieger, Chem. – Eur. J., 2015, 21, 8148–8157 CrossRef CAS PubMed; (f) S. Kissling, M. W. Lehenmeier, P. T. Altenbuchner, A. Kronast, M. Reiter, P. Deglmann, U. B. Seemann and B. Rieger, Chem. Commun., 2015, 51, 4579–4582 RSC; (g) D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, Angew. Chem., Int. Ed., 2002, 41, 2599–2602 CrossRef CAS.
- F. Palacios, C. Alonso, D. Aparicio, G. Rubiales and J. M. de los Santos, Tetrahedron, 2007, 63, 523–575 CrossRef CAS.
- (a) K.-W. Lee and L. A. Singer, J. Org. Chem., 1974, 39, 3780–3781 CrossRef CAS; (b) K. Hiroi and S. Sato, Chem. Pharm. Bull., 1985, 33, 2331–2338 CrossRef CAS.
- A. Otero, J. Fernández-Baeza, L. F. Sánchez-Barba, S. Sobrino, A. Garcés, A. Lara-Sánchez and A. M. Rodríguez, Dalton Trans., 2017, 46, 15107–15117 RSC.
- J. A. Castro-Osma, C. Alonso-Moreno, I. Márquez-Segovia, A. Otero, A. Lara-Sánchez, J. Fernández-Baeza, A. M. Rodríguez, L. F. Sánchez-Barba and J. C. García-Martínez, Dalton Trans., 2013, 42, 9325–9337 RSC.
- (a) J. Baran, A. Duda, A. Kowalski, R. Szymanski and S. Penczek, Macromol. Rapid Commun., 1997, 18, 325–333 CrossRef CAS; (b) I. Barakat, P. Dubois, R. Jérôme and P. Teyssié, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 505–514 CrossRef CAS.
- (a) F. Chen, Q.-C. Zhang, D. Wei, Q. Bu, B. Dai and N. Liu, J. Org. Chem., 2019, 84, 11407–11416 CrossRef CAS PubMed; (b) H. Büttner, C. Grimmer, J. Steinbauer and T. Werner, ACS Sustainable Chem. Eng., 2016, 4, 4805–4814 CrossRef.
- L. Longwitz, J. Steinbauer, A. Spannenberg and T. Werner, ACS Catal., 2018, 8, 665–672 CrossRef CAS.
- (a) M. Navarro, L. F. Sánchez-Barba, A. Garcés, J. Fernández-Baeza, I. Fernández, A. Lara-Sánchez and A. M. Rodríguez, Catal. Sci. Technol., 2020, 10, 3265–3278 RSC; (b) L. Peña Carrodeguas, À. Cristòfol, J. M. Fraile, J. A. Mayoral, V. Dorado, C. I. Herrerías and A. W. Kleij, Green Chem., 2017, 19, 3535–3541 RSC; (c) G. Fiorani, M. Stuck, C. Martín, M. M. Belmonte, E. Martin, E. C. Escudero-Adán and A. W. Kleij, ChemSusChem, 2016, 9, 1304–1311 CrossRef CAS PubMed.
- (a) C. J. Whiteoak, E. Martin, M. M. Belmonte, J. Benet-Buchholz and A. W. Kleij, Adv. Synth. Catal., 2012, 354, 469–476 CrossRef CAS; (b) D. J. Darensbourg, A. Horn Jr and A. I. Moncada, Green Chem., 2010, 12, 1376–1379 RSC.
- C.-Y. Li, Y.-C. Su, C.-H. Lin, H.-Y. Huang, C.-Y. Tsai, T.-Y. Lee and B.-T. Ko, Dalton Trans., 2017, 46, 15399–15406 RSC.
- (a) K. A. Maltby, M. Hutchby, P. Plucinski, M. G. Davidson and U. Hintermair, Chem. – Eur. J., 2020, 26, 7405–7415 CrossRef CAS PubMed; (b) A. Rehman, A. M. López Fernández, M. F. M. Gunam Resul and A. Harvey, J. CO2 Util., 2019, 29, 126–133 CrossRef CAS.
- F. de la Cruz-Martínez, M. Martínez de Sarasa Buchaca, J. Martínez, J. Fernández-Baeza, L. F. Sánchez-Barba, A. Rodríguez-Diéguez, J. A. Castro-Osma and A. Lara-Sánchez, ACS Sustainable Chem. Eng., 2019, 7, 20126–20138 CrossRef.
- G. N. Bondarenko, O. G. Ganina, A. A. Lysova, V. P. Fedin and I. P. Beletskaya, J. CO2 Util., 2021, 53, 101718 CrossRef CAS.
- (a) A. Rehman, F. Saleem, F. Javed, A. Ikhlaq, S. W. Ahmad and A. Harvey, J. Environ. Chem. Eng., 2021, 9, 105113 CrossRef CAS; (b) J. Chen, X. Wu, H. Ding, N. Liu, B. Liu and L. He, ACS Sustainable Chem. Eng., 2021, 9, 16210–16219 CrossRef CAS; (c) J. Fernández-Baeza, L. F. Sánchez-Barba, A. Lara-Sánchez, S. Sobrino, J. Martínez-Ferrer, A. Garcés, M. Navarro and A. M. Rodríguez, Inorg. Chem., 2020, 59, 12422–12430 CrossRef PubMed.
- P. P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2, 2169–2187 RSC.
- V. Paradiso, V. Capaccio, D. H. Lamparelli and C. Capacchione, Catalysts, 2020, 10, 825 CrossRef CAS.
- (a) M. R. Kember and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 15676–15679 CrossRef CAS PubMed; (b) Y. Xiao, Z. Wang and K. Ding, Macromolecules, 2006, 39, 128–137 CrossRef CAS.
- P. K. Saini, C. Romain, Y. Zhu and C. K. Williams, Polym. Chem., 2014, 5, 6068–6075 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Description of materials and methods employed, preparation and spectroscopy details of compounds 1–6 and complexes 7–9, details for crystallographic studies and structural refinement for 6 and 9 as well as details for the ring-opening polymerization of rac-lactide, the synthesis of cyclic carbonates and the synthesis of poly(cyclohexene carbonate)s using complex 7. CCDC 2350072. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01526f |
| This journal is © The Royal Society of Chemistry 2024 |