
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZinc amidinate-catalysed cyclization reaction of carbodiimides and alkynes. An insight into the mechanism†
Blanca
Parra-Cadenas
a,
Carlos
Ginés
a,
Daniel
García-Vivó
 b,
David
Elorriaga
*b and
Fernando
Carrillo-Hermosilla
*a
b,
David
Elorriaga
*b and
Fernando
Carrillo-Hermosilla
*a
aDepartamento de Química Inorgánica, Orgánica y Bioquímica-Centro de Innovación en Química Avanzada (ORFEO–CINQA), Facultad de Ciencias y Tecnologías Químicas, Universidad de Castilla-La Mancha, 13071 Ciudad Real, Spain. E-mail: Fernando.carrillo@uclm.es
bDepartamento de Química Orgánica e Inorgánica, Instituto Universitario de Química Organometálica “Enrique Moles”, Universidad de Oviedo, Julián Clavería 8, 33006 Oviedo, Spain. E-mail: elorriagadavid@uniovi.es
First published on 28th May 2024
Abstract
A synthesis of iminopyridines based on zinc has been developed. The commercially available ZnEt2 was employed as a precatalyst for this process. A mechanism has been proposed on the basis of Density Functional Theory (DFT) studies and stoichiometric reactions. The zinc amidinato intermediates underscore the critical role of zinc in this synthesis process.
The search for selective, efficient and sustainable processes to obtain organic compounds is the greatest aim to be achieved by chemistry. Among all the known protocols, catalysis is the most efficient, economical and usually greenest strategy to achieve that purpose.1 Commonly, transition metal complexes are the catalysts most extensively employed for organic transformations. However, these elements have potential drawbacks such as limited availability (due to their high cost), high toxicity and frequent metal incorporation in the final product. In order to overcome these limitations, zinc, which is abundant and biologically relevant, is of great interest. Despite its potential,2 the use of zinc derivatives as catalysts has been underdeveloped compared to that of transition metals. In this context, our research group has demonstrated the ability of ZnEt2, an affordable and common organometallic reagent in many synthetic laboratories, to act as a precatalyst in different substrate transformation processes towards more complex organic compounds. In this context, we have shown that this compound is capable of transforming amines and carbodiimides into substituted guanidines3 and alcohols of diverse natures, and carbodiimides into isoureas,4 and, several years ago, we discovered that it could serve as a highly effective precursor for the conversion of terminal alkynes and carbodiimides into propiolamidines (also referred to as propargylamidines) (RN
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(C
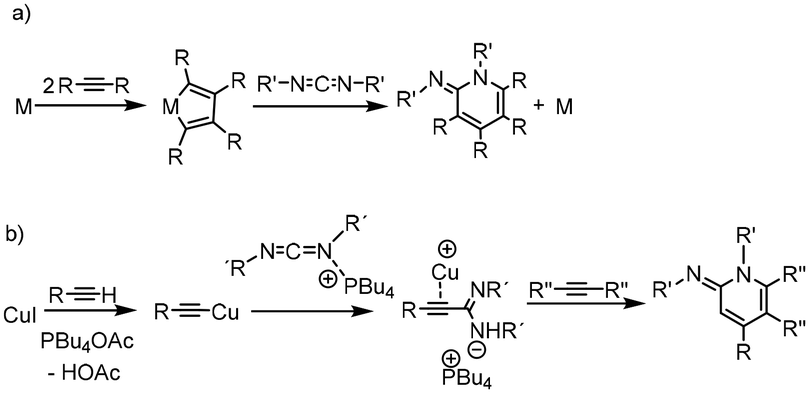
C(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR′)(NHR)). These propargylamidines show the particular feature of a C–C triple bond with a novel factor of potential reactivity.5 The distinctive structural characteristics of the amidines underlie a multitude of applications. In fact, we found that by addition of isocyanates to these propargylamidines, imidazolidinones were synthesized via an intramolecular urea hydroamination process.5 Mechanistic evidence of this transformation showed that the formation of a zinc amidinato complex was not only central to the process of alkyne addition to carbodiimides, but also to the subsequent addition of isocyanates. In this vein, an intramolecular hydroamination process of alkynyl amides to form indoles has also been described using diethylzinc as a precatalyst.6 As a continuation of our interest in the study of simple ZnEt2 as a pre-catalyst, we decided to explore its availability for the catalytic activation and addition of terminal alkynes to carbodiimides, with the aim of obtaining substituted 1,2-dihydropyridines. Typically, these organic compounds are synthesized by a catalytic cyclotrimerization reaction [2 + 2 + 2]7 of alkynes and unsaturated nitrogen-containing molecules such as nitriles,8 isocyanates,9 imines,10 or cyanamides.11 However, carbodiimides are the most attractive alternatives for this transformation because they can produce the 1,2-dihydroiminopyridines in one step (Scheme 1).
CR′)(NHR)). These propargylamidines show the particular feature of a C–C triple bond with a novel factor of potential reactivity.5 The distinctive structural characteristics of the amidines underlie a multitude of applications. In fact, we found that by addition of isocyanates to these propargylamidines, imidazolidinones were synthesized via an intramolecular urea hydroamination process.5 Mechanistic evidence of this transformation showed that the formation of a zinc amidinato complex was not only central to the process of alkyne addition to carbodiimides, but also to the subsequent addition of isocyanates. In this vein, an intramolecular hydroamination process of alkynyl amides to form indoles has also been described using diethylzinc as a precatalyst.6 As a continuation of our interest in the study of simple ZnEt2 as a pre-catalyst, we decided to explore its availability for the catalytic activation and addition of terminal alkynes to carbodiimides, with the aim of obtaining substituted 1,2-dihydropyridines. Typically, these organic compounds are synthesized by a catalytic cyclotrimerization reaction [2 + 2 + 2]7 of alkynes and unsaturated nitrogen-containing molecules such as nitriles,8 isocyanates,9 imines,10 or cyanamides.11 However, carbodiimides are the most attractive alternatives for this transformation because they can produce the 1,2-dihydroiminopyridines in one step (Scheme 1).
 | ||
| Scheme 1 Transition metal mediated synthesis of 2-iminopyridines. | ||
Although this transformation has been known since the 70's, scarce examples have been reported since then, all of them transition metal-catalysed processes. Those precedents are based on cobalt and nickel complexes as catalysts where evidence suggests that the mechanism goes through a metallacyclopentadiene intermediate formed by coupling of two alkyne molecules (Scheme 1a),12 or by the coupling of an alkyne and a carbodiimide molecule.13 However, those procedures showed troubles with chemoselectivity and low yield. These issues were successfully addressed by employing microwave-mediated solid-supported [CpCo(CO)2] as a catalyst.14 As an alternative to cobalt, Gandon, Aubert and coworkers reported the use of rhodium complexes in this reaction which also provided excellent yields, even under mild reaction conditions.15 Recently, another route was proposed to obtain these compounds, which goes via a carbodiimide activated by tetrabutylphosphonium acetate employing CuI as catalyst.16 This procedure goes through an amidinato intermediate and allows to combine terminal and internal alkynes as shown in Scheme 1b.
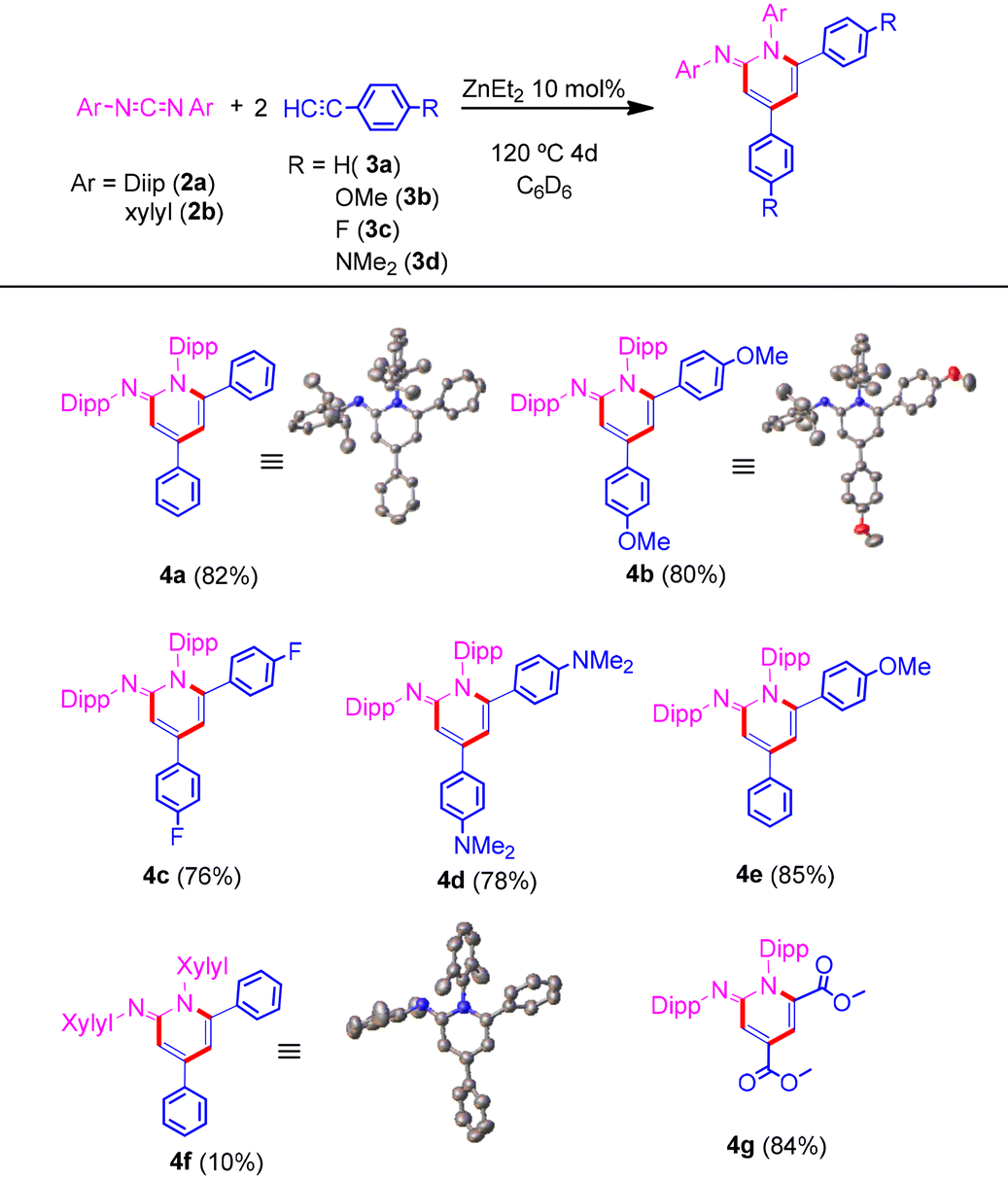
Based on our previous experience, and to begin our study, we firstly focused on our previously reported reaction using ZnEt21a as an outstanding pre-catalyst for the synthesis of propiolamidines, with good yields (∼80%) at 70 °C in 24 h.5 In a further study of this reaction, we found that by increasing the temperature up to 120 °C the reaction took place with yields up to almost 100%. Thus, reacting one equivalent of N,N′-bis(2,6-diisopropylphenyl)carbodiimide 2a with two equivalents of phenylacetylene 3a, using 5 mol% of 1a as precatalyst, after 36 h of reaction at 120 °C, provided a new compound, 4a, in 47% yield. When the precatalyst loading was increased to 10 mol%, the yield improved up to 70%. Upon further increasing the time, we were pleased to observe an 82% yield, after 4 days of reaction at 120 °C (Table 1). Exactly the same results were obtained when dimethyl zinc (1b) was used as the precatalyst. On the other hand, increasing the temperature up to 150 °C did not improve the performance of the reaction. The 1H-NMR spectrum of 4a shows, in addition to the characteristic signals of four inequivalent isopropyl groups and the signals of the aromatic protons of the phenyl groups, two novel doublets at 6.75 and 6.25 ppm, respectively, which are attributed to the protons within the heterocyclic core.
|
Besides, we examined a range of terminal alkynes to assess the diversity of our protocol (Table 1). All reactions went smoothly providing the corresponding iminopyridines. Similar yields were obtained regardless of the electronic character of the substituents in the terminal aromatic alkyne. Moreover, when the aromatic alkyne was changed to a methyl propiolate, the reaction gave rise to the formation of 4g in excellent yield. At this point, it is worth noting that this reaction tolerates different functional groups in the alkyne substrate, such as halogen or ester groups, remaining unaltered. The molecular structures of compounds 4a, 4b and 4f were obtained by single crystal X-ray diffraction, confirming the proposed structure.
As described above, ZnEt2 is known to be an excellent catalyst for the transformation of terminal alkynes and carbodiimides into propiolamidines.5 Also, it was assumed that the cycloaddition process would begin with the amidine formation. Based on this hypothesis, in a sealed NMR tube, the reaction of one equivalent of 2a with an equimolar quantity of 3a in the presence of 10 mol% of 1a was carried out. After 24 h at 120 °C full conversion into the corresponding amidine was confirmed. Next and without any separation or purification step, one equivalent of 4-ethynylanisole 3b was added. Heating at 120 °C for 3 days provided iminopyridine 4e, which shows different aromatic substituents on the heterocyclic core, in excellent yield (85%). However, when the reaction was carried out in one step starting from an equimolar mix of alkynes, selective formation towards one of the possible products was not observed. Additionally, when an internal alkyne was added after the propiolamidine formation step, no cyclotrimerization product was detected at all. The study was also extended to other aromatic substituted carbodiimides, such as N,N′-diphenyl-, N,N′-bis(2,6-dimethylphenyl)-, and N,N′-di-para-tolylcarbodiimide, and aliphatic ones like N,N′-diisopropyl- and N,N′-dicyclohexylcarbodiimide. Surprisingly, these less bulky substrates gave rise to a complex mixture of compounds where in the case of N,N′-bis(2,6-dimethylphenyl)carbodiimide (2b), the corresponding iminopyridine 4f was obtained in 10% yield after just 6 h of reaction at 120 °C. Although better behavior would be expected in these cases, the influence of both the aromatic character and the appropriate steric volume of the substituents is clear.
It is noteworthy that when the catalytic reaction was carried out using PhCCD (3e) in the second step of the process, after having used the non-deuterated alkyne 3a in the amidine formation step, the heterocycle obtained appears as a mixture, in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, of the product substituted with deuterium at position 3 and the alternative product substituted at position 5. This fact indicates the possibility of an alkyne exchange process between the previously formed amidine and the deuterated alkyne. The deuterium derivative substituted at both olefinic positions 4a-D2 can be easily obtained by carrying out the catalytic reaction between two equivalents of 3e and one equivalent of the carbodiimide 2a.
:1 ratio, of the product substituted with deuterium at position 3 and the alternative product substituted at position 5. This fact indicates the possibility of an alkyne exchange process between the previously formed amidine and the deuterated alkyne. The deuterium derivative substituted at both olefinic positions 4a-D2 can be easily obtained by carrying out the catalytic reaction between two equivalents of 3e and one equivalent of the carbodiimide 2a.
To gain mechanistic insights into this process, a series of stoichiometric and catalytic reactions were performed. The involvement of zinc amidinato intermediates in the synthesis of propiolamidines using ZnEt2 as a precatalyst has been demonstrated previously.5 Since it seems reasonable to assume a similar formation of the amidine as a first step in this new process, through amidinato complexes, the synthesis of a bisamidinato complex was carried out by reaction between preformed amidine 5a (N,N′-bis(2,6-diisopropylphenyl)-3-phenylpropiolimidamide) and diethylzinc in 1:2 metal:amidine ratio. The complete conversion of the starting reagents had taken place after 16 h at room temperature. The 1H NMR spectrum in C6D6 of the new compound shows one broad peak and a doublet with the corresponding septuplet for the isopropyl groups, a pattern that fits with the expected symmetric bis(amidinato) complex [Zn{κ2-C(CCPh)(N(Dipp))2}2] 6 (Dipp = 2,6-iPr2C6H3). To obtain more information about the real structure of this new complex, suitable X-ray quality crystals were obtained from a saturated solution in hexane. Diffraction studies (Fig. 1) reveal a mononuclear compound with the amidinato ligands coordinated in a chelated κ2-fashion, which produces a very distorted pseudotetrahedral coordination around the zinc atom, with a bite angle for the chelating ligand (N1–Zn1–N2) of 67.25(13)° and a structural parameter τ4 of 0.65.17 Complex 6 shows delocalization at the amidinato core (C–N ∼1.32 Å, sum of bond angles at C1 ∼360°).
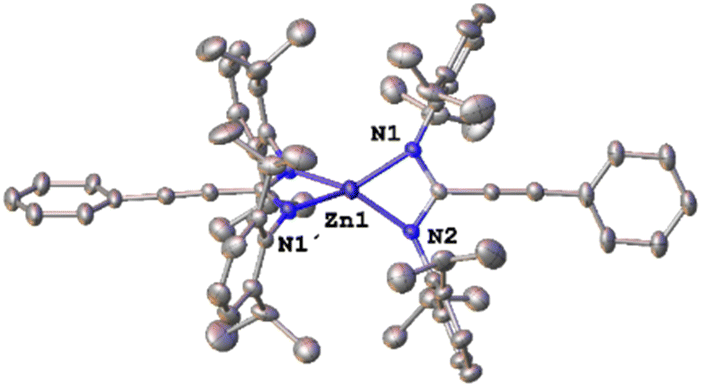 | ||
| Fig. 1 Molecular structure of 6 (H atoms are omitted for clarity). | ||
The structure of compound 6 contrasts with that found for the dinuclear complex [Zn{κ2-C(CCPh)(NiPr)2}{μ-κ1-κ1-C(CCPh)(NiPr)2}]2,16 where the amidinato ligands are in both chelated and bridging form, which was in equilibrium with a mononuclear form similar to that found in compound 6. This difference is probably explained by the larger steric bulk of the nitrogen atom substituents in compound 6, which inhibits the formation of bridging ligands.
Following by 1H NMR the reaction between complex 6 and two equivalents of PhCCH, at room temperature, it was found that a fast transformation takes place to obtain a complex mixture, one of the main features of which being a broad peak near δ 11 ppm, along with broad peaks assigned to phenyl and isopropyl groups, and free alkyne (see the ESI†). Surprisingly, when PhCCD was used, that peak disappears, the rest of the spectrum remaining unaltered (see the ESI†). A similar result was reported previously using [Zn{κ2-C(CCPh)(NiPr)2}{μ-κ1-κ1-C(CCPh)(NiPr)2}]2 as the starting reagent. At that time, it was shown that the low-field peak could be assigned to the formation of a zinc complex in which the C–H bond of the alkyne had been activated to form an alkynyl compound with neutral amidines coordinated to the metal.5 In contrast, if the reaction between complex 6 and two equivalents of alkyne 3a is carried out for 20 hours at 120 °C, it results in the formation of 4a as the major product. This provides us with the possibility of the presence of amidinato derivatives similar to 6 as intermediates in the catalytic cyclization process. Indeed, compound 6 in a 10 mol% ratio was then used as a catalyst for the cyclization process of alkyne 3a and compound 2a giving rise to heterocycle 4a in an 80% yield after 3 days at 120 °C.
To shed light upon the plausible mechanism for the cycloaddition of alkynes and carbodiimides using ZnEt2 as a precatalyst, and zinc bis(amidinates) as the real active species, DFT calculations were conducted (see the ESI†). In agreement with experimental results, the coupling of carbodiimide 2a with two equivalents of the alkyne 3a to form the iminopyridine 4a is a highly exergonic process (ca. −86 kcal mol−1), for which we have been able to model a complete catalytic cycle starting from the bis(amidinato) complex 6 (Scheme 2). Thus, the first step would involve the coordination of the alkyne 3a to the Zn atom in 6, this resulting in a modest energy increase (I1, ca. +3 kcal mol−1). Following the coordination of the alkyne, a nucleophilic attack of one of the amidinato N atoms on the C(Ph) atom of the Zn-bound alkyne would take place, this forming a new C–N bond, and configuring a six-membered metallacylic ring as found in intermediate I2. This step takes place through a high energy transition state TS1 (ca. +27 kcal mol−1), in which the Zn atom is further stabilized via C–H⋯Zn agostic interactions involving one of the Dipp groups (dZnH = 2.24 Å). It is postulated that the presence of a sterically demanding group on the nitrogen atoms of the amidinato ligand could facilitate the requisite chelate opening, thereby enabling the proposed nucleophilic attack. Then, an exchange of Zn-bound groups in the newly formed ligand takes place, this involving the decoordination of the imine N(Dipp) atom and the loose coordination of the amidinato alkyne moiety (I3; dZnC = 3.22 and 3.71 Å), while the Zn center retains significant agostic interactions with one of the Dipp groups (shortest dZnH = 2.44 Å), the latter surely alleviating the coordinative unsaturation of the metal created upon N decoordination. This rearrangement has a modest energy penalty of ca. 5 kcal mol−1 from I2 and places the amidinato alkyne group in a perfect position to allow for the formation of the C–C bond which configures the final six-membered heterocyclic ring found in 4a. Thus, this C–C formation step takes place next through an energetic transition state TS2 (ca. +34 kcal mol−1), in which the metal atom is also strongly involved facilitating the process. Thus, the alkyne carbon atoms are more tightly bound to the Zn atom in TS2 (dZnC = 2.92 and 3.43 Å) when compared to the looser coordination found in the preceding intermediate I3, while the new C–C bond has been substantially formed (dCC = 2.15 Å). As corroborated by IRC calculations, this transition state leads to the formation of a highly stable aryl amidinato zinc complex (I4, ca. −32 kcal mol−1), whereby the Zn(amidinato) group is attached to one of the C atoms of the newly formed heterocyclic ring. To complete the process, a quite exergonic reaction with an additional equivalent of alkyne would free up the final product 4a also forming an amidinato alkynyl complex I5, this latter then regenerating the starting bis(amidinato) complex 6 upon reaction with a carbodiimide molecule.
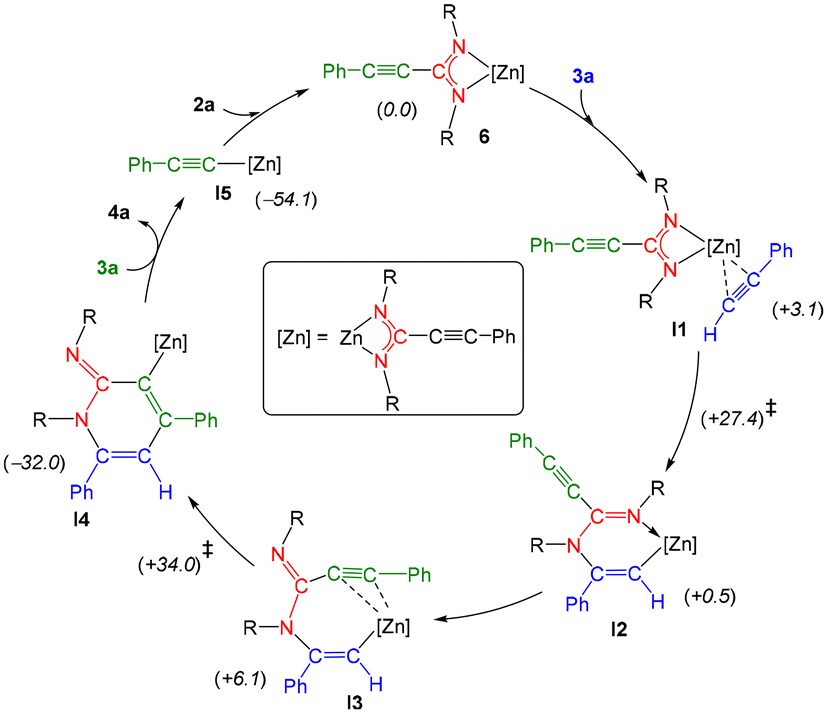 | ||
| Scheme 2 DFT calculated catalytic cycle for the formation of compound 4a catalysed by bis(amidinato) complex 6 (R = Dipp). The solvation and entropy-corrected relative free energies (in parentheses) are given in kcal mol−1 (see the ESI† for full computational details). | ||
Conclusions
In conclusion, we have presented here a zinc-catalysed reaction involving bulky carbodiimides and appropriately substituted terminal alkynes, which results in the synthesis of cyclic dihydropyridine-type derivatives. DFT studies and stoichiometric experiments allowed us to propose zinc amidinates as a crucial component of the catalytic cycle. To the best of our knowledge, this represents the first [2 + 2 + 2] annulation process of this type facilitated by a non-transition metal catalyst, specifically zinc. Our laboratory is currently conducting additional investigations into zinc-catalysed multicomponent cycloaddition reactions, with a particular focus on novel unsaturated substrates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from MCIN/AEI/10.13039/501100011033 (Project Numbers PID2020-117353GB-I00, PID2021-123964NB-I00 and RED2022-134287-T) and the Universidad de Castilla-La Mancha (Project Number 2022-GRIN-34031) and the SCBI (Supercomputer and Bioinformatics) Center of the University of Málaga (Spain) for access to computing facilities.References
- P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686–694 CrossRef CAS PubMed.
- (a) S. Enthaler, ACS Catal., 2013, 3, 150–158 CrossRef CAS; (b) K. K. Krishnan, S. M. Ujwaldev, S. Saranya, G. Anilkumar and M. Beller, Adv. Synth. Catal., 2019, 361, 382–404 CrossRef; (c) X.-F. Wua and H. Neumann, Adv. Synth. Catal., 2012, 354, 3141–3160 CrossRef.
- C. Alonso-Moreno, F. Carrillo-Hermosilla, A. Garcés, A. Otero, I. López-Solera, A. M. Rodríguez and A. Antiñolo, Organometallics, 2010, 29, 2789–2795 CrossRef CAS.
- A. Ramos, F. Carrillo-Hermosilla, R. Fernández-Galán, D. Elorriaga, J. Naranjo, A. Antiñolo and D. García-Vivó, Organometallics, 2022, 41, 2949–2957 CrossRef CAS PubMed.
- (a) A. Martínez, S. Moreno-Blázquez, A. Rodríguez-Diéguez, A. Ramos, R. Fernández-Galán, A. Antiñolo and F. Carrillo-Hermosilla, Dalton Trans., 2017, 46, 12923–12934 RSC; (b) R. K. Sahoo, A. G. Patro, N. Sarkar and S. Nembenna, Organometallics, 2023, 42, 1746–1758 CrossRef CAS.
- Y. Yin, W. Ma, Z. Chai and G. Zhao, J. Org. Chem., 2007, 72, 5731–5736 CrossRef CAS PubMed.
- M. O. Faruk Khan, M. S. Levi, C. R. Clark, S. Y. Ablordeppey, S.-L. Law, N. H. Wilson and R. F. Borne, Stud. Nat. Prod. Chem., 2008, 34, 753–787 Search PubMed.
- P. Matton, S. Huvelle, M. Haddad, P. Phansavath and V. Ratovelomanana-Vidal, Synthesis, 2022, 4–32 CAS.
- Y. Xie, C. Wu, C. Jia, C.-H. Tung and W. Wang, Org. Chem. Front., 2020, 7, 2196–2201 RSC.
- (a) D. D. Young and A. Deiters, Angew. Chem., Int. Ed., 2007, 46, 5187–5190 CrossRef CAS PubMed; (b) K. M. Oberg, E. E. Lee and T. Rovis, Tetrahedron, 2009, 65, 5056–5061 CrossRef CAS PubMed.
- M. Ohashi, Y. Hoshimoto and S. Ogoshi, Dalton Trans., 2015, 44, 12060–12073 RSC.
- (a) L. V. R. Boñaga, H.-C. Zhang and B. E. Maryanoff, Chem. Commun., 2004, 2394–2395 RSC; (b) P. Hong and H. Yamazaki, Tetrahedron Lett., 1977, 15, 1333–1336 CrossRef; (c) H. Hoberg and G. Burkhart, Synthesis, 1979, 525 CrossRef CAS; (d) H. Hoberg and W. Richter, J. Organomet. Chem., 1980, 195, 355–362 CrossRef CAS; (e) P. Diversi, G. Ingrosso, A. Lucherini and S. Malquori, J. Mol. Catal., 1987, 40, 359–377 CrossRef CAS.
- T. Takahashi, F.-Y. Tsai, Y. Li, H. Wang, Y. Kondo, M. Yamanaka, K. Nakajima and M. Kotora, J. Am. Chem. Soc., 2002, 124, 5059–5067 CrossRef CAS PubMed.
- D. D. Young and A. Deiters, Angew. Chem., Int. Ed., 2007, 46, 5187–5190 CrossRef CAS PubMed.
- (a) M. Amatore, D. Leboeuf, M. Malacria, V. Gandon and C. Aubert, J. Am. Chem. Soc., 2013, 135, 4576–4579 CrossRef CAS PubMed; (b) M. Ishii, F. Mori and K. Tanaka, Chem. – Eur. J., 2014, 20, 2169–2174 CrossRef CAS PubMed.
- M. S. Jalali, M. Manafi, S. S. Homami, B. Gorji and A. Monzavi, Monatsh. Chem., 2020, 151, 1173–1181 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2346809–2346812. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01265h |
| This journal is © The Royal Society of Chemistry 2024 |