
Expanding the thiol–X toolbox: photoinitiation and materials application of the acid-catalyzed thiol–ene (ACT) reaction†
Bryan P.
Sutherland
 a,
Mukund
Kabra
b and
Christopher J.
Kloxin
*ab
a,
Mukund
Kabra
b and
Christopher J.
Kloxin
*ab
aDepartment of Materials Science and Engineering, University of Delaware, 201 DuPont Hall, Newark, DE 19716, USA. E-mail: cjk@udel.edu
bDepartment of Chemical and Biomolecular Engineering, University of Delaware, 150 Academy Street, Newark, DE 19716, USA
First published on 13th January 2021
Abstract
The acid-catalyzed thiol–ene reaction (ACT) is a unique thiol–X conjugation strategy that produces S,X-acetal conjugates. Unlike the well-known radical-mediated thiol–ene and anion-mediated thiol-Michael reactions that produce static thioether bonds, acetals provide unique function for various fields such as drug delivery and protecting group chemistries; however, this reaction is relatively underutilized for creating new and unique materials owing to the unexplored reactivity over a broad set of substrates and potential side reactions. Solution-phase studies using a range of thiol and alkene substrates were conducted to evaluate the ACT reaction as a conjugation strategy. Substrates that efficiently undergo cationic polymerizations, such as those containing vinyl functional groups, were found to be highly reactive to thiols in the presence of catalytic amounts of acid. Additionally, sequential initiation of three separate thiol–X reactions (thiol-Michael, ACT, and thiol–ene) was achieved in a one-pot scheme simply by the addition of the appropriate catalyst demonstrating substrate selectivity. Furthermore, photoinitiation of the ACT reaction was achieved for the first time under 470 nm blue light using a novel photochromic photoacid. Finally, using multifunctional monomers, solid-state polymer networks were formed using the ACT reaction producing acetal crosslinks. The presence of S,X-acetal bonds results in an increased glass transition temperature of 20 °C as compared with the same polymeric film polymerized through the radical thiol–ene mechanism. This investigation demonstrates the broad impact of the ACT reaction and expands upon the diverse thiol–X library of conjugation strategies towards the development of novel materials systems.
Introduction
Thiol–X reactions are a class of robust conjugation reactions between thiols and thiol-conjugate adducts to form carbon sulfur bonds.1–4 The versatility of thiol–X reactions has been demonstrated through the broad range of implementations, from bioconjugations5–10 to polymer synthesis.11–15 A unique aspect to thiol–X reactions is the large functional group library that can react with thiols, including alkenes,16 alkynes,17 epoxides,18 isocyanates,19 and alkyl halogens.20 Reaction control is typically achieved by the type of initiator or catalyst employed enabling near ideal 1-to-1 reactivity of thiols with their desired substrate. Given the precise control in thiol conjugation under mild conditions, thiol–X reactions are often demarcated as potential ‘click’ reactions.21–24The two most prominent thiol–X conjugations, the thiol–ene4 and thiol-Michael reactions,25 have each been hailed as ‘click’ reactions26 for their rapid kinetics, high conversions, and minimal byproduct formation. While both thiol–ene and thiol-Michael reactions result in the formation of a new thioether bond between a thiol and an alkene substrate, they proceed through different reaction pathways.27 The thiol–ene reaction follows a radical-mediated mechanism between a thiol and an electron-rich alkene, such as a vinyl, allyl, or norbornenyl functional group, whereas the thiol-Michael reaction follows an anion-mediated mechanism between a thiol and an electron deficient alkene, such as an acrylate, vinyl sulfone, or maleimide functional group (Scheme 1A and B, respectively). The self-limited, 1-to-1 reactivity combined with distinct mechanisms enables a sequential thiol–ene and thiol-Michael reaction scheme, which has been applied in the design of iterative peptide conjugations28 and sequence-defined polymers.29
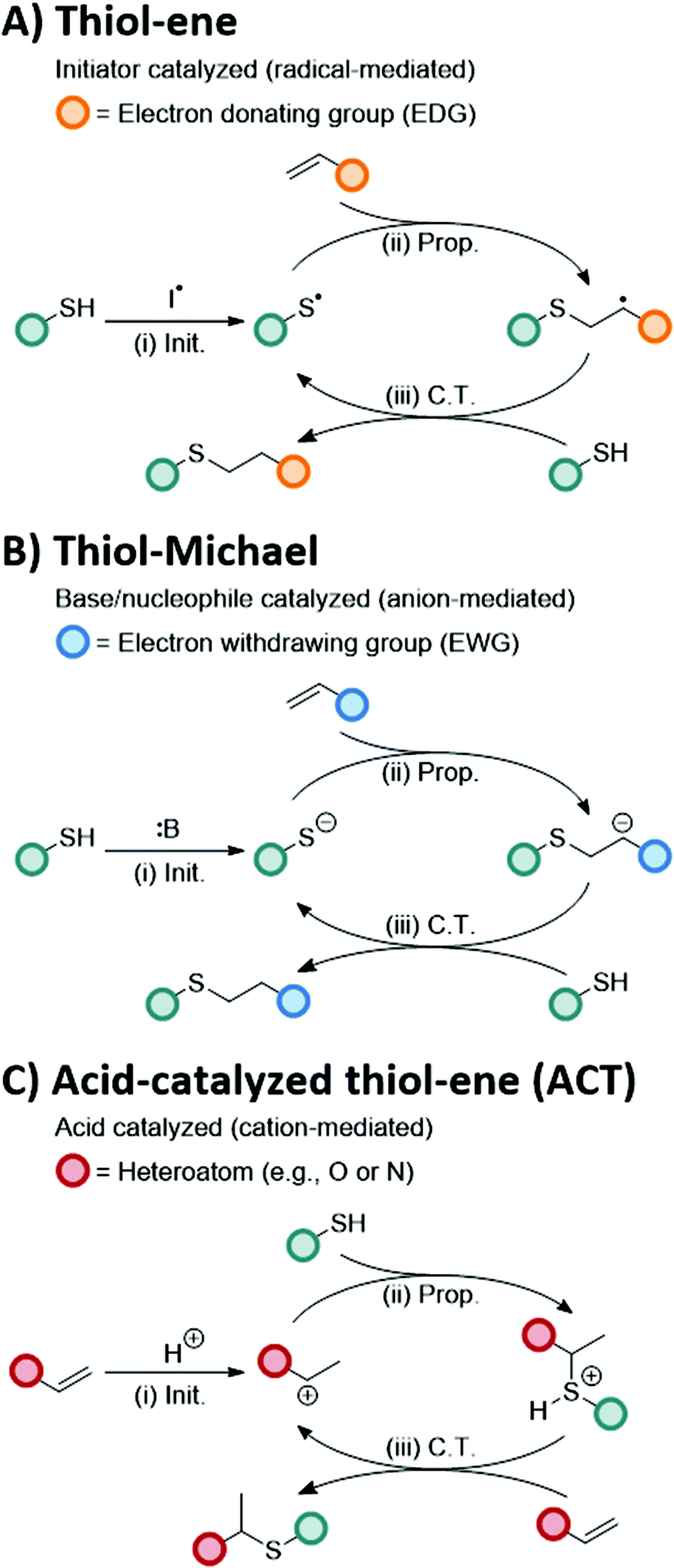 | ||
| Scheme 1 Proposed mechanism of the thiol–ene, thiol-Michael, and ACT reactions, which all undergo (i) an initiation step (Init.) followed by alternating (ii) propagation (Prop.) and (iii) chain transfer (C.T.) steps. (A) The thiol–ene reaction proceeds via a radical-mediated pathway in which an initiator species that generates radicals (e.g., photoinitiator, thermal initiator, etc.) deprotonates a thiol producing a thiyl radical. This thiyl radical reacts with electron-rich alkenes resulting in the formation of thioether bonds. (B) The thiol-Michael reaction proceeds via an anion-mediated pathway in which a base deprotonates a thiol forming a thiolate anion that reacts with electron deficient alkenes forming a thioether bond. (C) The acid-catalyzed thiol–ene (ACT) reaction proceeds via a cation-mediated pathway in which an acid protonates a vinyl substrate forming a carbocation. The carbocation then undergoes electrophilic addition by a thiol generating a Markovnikov-directed thioether bond. | ||
A unique mechanistic pathway distinct from the thiol–ene and thiol-Michael reactions is the cation-mediated reaction between a thiol and a vinyl to produce S,X-acetals in the presence of dilute acid,30,31 referred to here as the acid-catalyzed thiol–ene (ACT) reaction. In the presence of Brønsted acid32 or Lewis acid33 catalysts, vinyl groups become protonated forming a carbenium ion that is quenched through electrophilic addition.30,34–36 The proposed mechanism for the ACT conjugation reaction, shown in Scheme 1C, follows a cyclic process involving (i) carbenium generation (i.e., initiation) in the presence of an acid catalyst, (ii) electrophilic addition (i.e., propagation) of a thiol generating a sulfonium intermediate, and (iii) reformation of a new protonated carbenium substrate through chain transfer.31 Unlike the thiol–ene and thiol-Michael reactions, the ACT reaction undergoes a Markovnikov-directed addition producing an S,X-acetal. Acetal bonds are of particular interest for their broad impact in numerous fields, from amino acid protecting groups37 to chain transfer agents38 to dynamic materials.39–42 Acetals can also undergo acid hydrolysis, making them useful tools for drug delivery43 or antimicrobial agents.44 In work by Du Prez and coworkers,41 the incorporation of acetals into the crosslinks of a polymer network produced a covalent adaptable network.45 Thus, the introduction of acetal bonds into polymeric systems through the ACT reaction provides a new thiol–X route to create novel functional and responsive materials.
Despite the potential of the ACT reaction as a tool to create new materials, the reaction has been relatively unexplored. The substrate scope and kinetics, especially under ambient conditions, are absent in the literature. Recent work by Uchiyama et al.31 demonstrated that this reaction could be applied towards the formation of linear polymers with thioethers or S,O-acetals simply by switching the initiating species. However, such reactions were performed under sub-freezing temperatures and water-free conditions limiting the utility of the reaction. Additionally, while much work has been done to enable the thiol–ene and thiol-Michael reactions to be triggered using light, the ACT reaction has thus far been limited to traditional, non-photoinitiated cationic species.31,38 The on-demand, spatiotemporal reaction control using photoinitiators is an attractive feature of thiol–ene and thiol-Michael reactions and enables a range of technologies, from advanced coatings to 3D printing.
Herein, the broad substrate scope of the ACT reaction was demonstrated by monitoring the kinetics of the reaction over a range of common thiol and alkene functional groups under ambient conditions. Additionally, photocontrol over the ACT reaction was achieved for the first time using a novel non-radical forming photochromic photoacid for the application of photopolymerizing ACT polymer networks. This work provides fundamental insights into the ACT reaction and demonstrates its broad impact in the fields of both cationic polymerizations and thiol–X material systems.
Results
Model studies of the ACT reaction
Inspired by the broad substrate scope available to thiol–ene and thiol-Michael reactions, vinyl and thiol ACT substrates were investigated to better understand structural effects on the reaction kinetics. Solution-phase kinetic studies were performed using model vinyls and thiols under ambient conditions utilizing nuclear magnetic resonance (NMR) spectroscopy to monitor functional group consumption and product formation. Reactions were performed in deuterated dichloromethane (DCM) and monitored at 30 s intervals using mesitylene as an internal standard. To initiate the reaction, an acid catalyst, methanesulfonic acid (MSA), and a Lewis base, tetramethylurea (TMU), were added. MSA was selected as the initiating species owing to its use as a cationic polymerization initiator46 as well as being a milder alternative to super acid catalysts. A concentration of 1.5 mol% of MSA was utilized in all model reactions. It should be noted that ACT reaction only exhibited a minor rate dependence on acid concentration (i.e., r ∼ [MSA]0.23±0.02, see Fig. S11 in ESI†). In early studies, dimethylformamide (DMF) was used as an internal standard and appeared to influence the overall reaction kinetics and stability of the acetal products. This observation is consistent with prior literature showing that Lewis bases, such as DMF, help mediate the protonation of vinyl functional groups in cationic polymerizations leading to more controlled reactions.47 As such, catalytic amounts of TMU were included to improve the overall control of the reaction and prevent degradation of more acid sensitive acetal products and will be discussed more in detail later.It was observed that the vinyl structure greatly influences the kinetics of the ACT reaction. Commonly used terminal, internal, and cyclic vinyl ethers were evaluated to understand their structural effects on the ACT kinetics. The conversion as a function of time is shown for each vinyl substrate reacted with ethyl mercaptopropionate is shown in Fig. 1A. Ethyl vinyl ether (EVE) proceeded rapidly to roughly 88.6 ± 0.6% conversion after 10 min. The plateauing conversion below 100% is a consequence of vinyl hydrolysis in the presence of water as observed by NMR (see Fig. S1 in the ESI†). Upon forming the cationic vinyl intermediate, water can undergo electrophilic addition to the vinyl group resulting in rapid decomposition to acetaldehyde and alcohol.48 Subsequently, the acetaldehyde that forms can react with newly formed alcohol and residual thiol species to form minor amounts O,O- and S,S-acetal side products. This side reaction is well known to affect typical cationic polymerizations that generally require air- and water-free conditions to avoid degradation and slow reaction kinetics.49 However, in the case of the ACT reaction, acetaldehyde formation is minor, demonstrating the robust nature of the reaction. In the case of 2-methoxypropene (iPVE), the reaction reached a plateauing conversion of 90 ± 1% prior to the first time point (30 s). The highly stable tertiary carbenium ion that forms upon protonation by the acid catalyst leads to the rapid kinetics observed in the reaction.50 The internal vinyl ether substrates, 1-ethyoxypropene (PVE) and dihydropyran (DHP), proceeded slower than the terminal vinyl ether substrates, reaching conversions of 86 ± 1% and 31.2 ± 0.8% after 10 min, respectively. This finding is similar to that of radical-mediated thiol–ene reactions in which internal alkenes typically result in sluggish kinetics owing to steric hinderance of the alkene.4,51,52
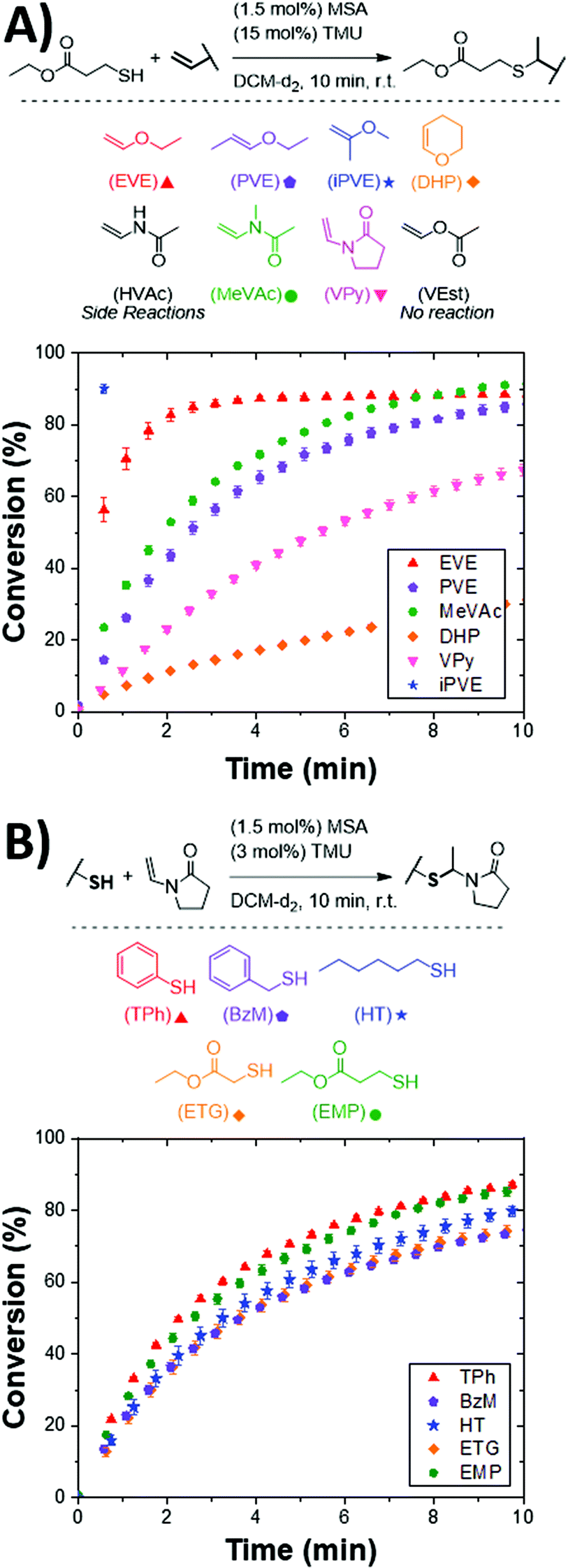 | ||
| Fig. 1 ACT reaction kinetics as a function of vinyl and thiol functional groups. (A) Vinyl substrates (0.2 mmol, 1 eq.) were mixed with EMP (0.2 mmol, 1 eq.) and TMU (15 mol%) in DCM-d2 with mesitylene (0.2 mmol, 1 eq.) as an internal standard. Kinetics were measured via NMR with t = 0 min being prior to adding (1.5 mol%) MSA. Additionally, NMR spectras at t = 16 h were collected to determine final conversions and ensure stability of the final product (see Fig. S1 through S9 in the ESI†). (B) Thiols (0.2 mmol, 1 eq.) were mixed with the VPy (0.2 mmol, 1 eq.) and TMU (3 mol%) in DCM-d2 (calculated to make total final volume 600 μL) with mesitylene (0.2 mmol, 1 eq.) as an internal standard. Kinetics were measured via NMR with t = 0 min being prior to adding MSA (1.5 mol%). | ||
In the case of vinyl ethers that are adjacent to an electron deficient carbonyl (vinyl esters), little reactivity is observed towards the desired S,O-acetal product. When performing the same reaction conditions with vinyl acetate (VEst), less than 1% product conversion was observed (see Fig. S5 in the ESI†). The low reactivity of vinyl acetate within the ACT reaction scheme is expected, as it is known to be challenging to polymerize via a cationic mechanism.53 The higher basicity of the ester carbonyl as compared with the vinyl inhibits vinyl protonation, resulting in negligible reaction towards the ACT conjugate.54
Vinyl acetamide (HVAc), N-methyl vinylacetamide (MeVAc), and vinyl pyrrolidone (VPy) were selected as vinyl substrates to examine the reactivity differences in secondary and tertiary vinyl amides. The HVAc substrate resulted in a fast-initial rate, but ultimately resulted in significant homopolymerization55,56 (see Fig. S6 in the ESI†) limiting the conversion of the desired product to 33.8 ± 0.2% after 10 min. However, the tertiary vinyl amide, MeVAc, reached a conversion of 91.5 ± 0.6% after 10 min. The higher conversion of MeVAc as compared to vinyl ethers is likely attributed to the slower hydrolysis rate associated with enamides over vinyl ethers.57 The tertiary cyclic VPy showed slower kinetics, reaching only 64.0 ± 0.2% conversion after 10 min. We identified the cyclic DHP and VPy as model substrates as they showed limited vinyl hydrolysis producing quantitative conversions after 16 h resulting in click-like conjugations. We attribute the near quantitative conversions of DHP and VPy to their slow hydrolysis rates as compared with their noncyclic counterparts.58,59 As such, VPy was used in all subsequent model reactions as an ideal vinyl substrate.
Surprisingly, while the vinyl substrate structure has a significant impact on the ACT reaction kinetics, the thiol substrate structure was observed to have minimal influence. Six commonly employed thiol substrates used in synthesizing and modifying polymeric materials were evaluated. As shown in Fig. 1B, only slight differences in the initial rate and conversion profiles were observed between the six thiol substrates. Such minor differences in kinetics when varying the thiol is in stark contrast to both thiol–ene reactions,60 where glycolate and propionate esters show higher reactivity than alkyl thiols, and thiol-Michael reactions,61 where lower thiol pKa results in faster rates. Interestingly, the minor dependence on thiol structure coincides with kinetic studies showing negligible rate dependence on the ACT reaction for thiol at concentrations utilized in these studies (see Fig. S10 in ESI†).
As noted previously, water has a significant effect on the reaction kinetics, which results in a competing side reaction. An offset in the consumption of the thiol and vinyl substrates was observed during the reaction, suggesting the formation of intermediates (see Fig. S13 and S14 in the ESI† for NMR spectra and conversion versus time plots). It was hypothesized that water influences the resulting offset through the reversible formation of hemiacetals during the reaction process (i.e., see reaction scheme Fig. S12 in ESI†). The reaction between ethyl mercaptopropionate (EMP) to VPy using ‘wet’ deuterated DCM led to the formation of an α-vinyl carbon intermediate during the reaction and larger offsets between the vinyl and thiol conversions during reaction than observed for the reaction using dry deuterated DCM (see Fig. S15 and S16 in the ESI†). Water is known to act as a chain transfer agent in cationic polymerizations and inhibits the electrophilic addition of the thiol to the carbenium ion.62,63 Importantly, the excess water did not hinder the high conversions seen in the 1-to-1 reaction between EMP and VPy, demonstrating its robustness under ambient conditions.
The inclusion of a Lewis base in the ACT reaction reduces the reaction rate but stabilizes the acid-sensitive acetal product. The introduction of ethers,64 thioethers,65 and carbonyls66 have been utilized as Lewis bases to reduce the propagation rate of cationic polymerizations, thus improving control of the overall reaction. In our model system, increasing the amount of the Lewis base, TMU, relative to constant thiol and vinyl concentrations led to a decrease in the product formation rate (see Fig. S17 in the ESI†); however, more acid sensitive products, such as the S,O-acetal, showed increased stability over the course of the reaction (see Fig. S18 in the ESI†). In the absence of TMU, the stoichiometric reaction between BzM and EVE resulted in mixtures of O,O-, S,S-, and S,O-acetals, with conversions after 30 min of 47%, 42%, and 11%, respectively; whereas, in the presence of 3 mol% TMU, conversions after 30 min were <5%, <5%, and 87%, respectively. The high conversion towards the desired S,O-acetal product in the presence of TMU suggests that the Lewis base helps in preventing product degradation at the expense of decreased reaction rate. As an aside, in the extreme case for which the solvent itself can act as a Lewis base, such as for aprotic polar DMSO, the reaction rate became severely inhibited (see Fig. S19 in the ESI†).
Sequential one-pot thiol–X conjugations
Using the unique selectivity of the ACT reaction towards vinyl substrates, its orthogonality to the thiol–ene and thiol-Michael reactions was demonstrated using a model ‘one-pot’ sequential initiation scheme. We hypothesized that a sequential reaction scheme could be accomplished solely based on the catalyst added under ambient conditions. The proposed sequential reaction scheme and the NMR spectra showing alkene conversion upon addition of the appropriate catalyst is shown in Fig. 2. Three equivalents of EMP was dissolved in deuterated DMSO along with three alkene substrates that each undergo different thiol–X reactions. The three chosen alkenes were dimethylacrylamide (DiMA), VPy, and tert-butyl N-allylcarbamate (tBAC) that undergo the thiol-Michael, ACT, and thiol–ene reactions, respectively. It is important to note that TMU was not included in the reaction scheme as the solvent, DMSO, acts as a Lewis base in its absence. To the reaction mixture, a catalytic amount of the nucleophile dimethylphenylphosphine (DMPP) was added to initiate the thiol-Michael reaction.67 After 16 h, the NMR showed no remaining acrylamide double bond chemical shifts (δ = 6.72, 6.12, 5.63 ppm), while the vinyl (δ = 6.97 and 4.40 ppm) and allyl (δ = 5.79 and 5.05 ppm) chemical shifts remained unchanged. To initiate the ACT reaction, a catalytic amount of MSA was added to the same reaction pot. After another 16 h, the vinyl peaks associated with the VPy had completely reacted (δ = 6.98 and 4.40 ppm), and the resulting product chemical shift associated with the tertiary carbon product (δ = 5.43 ppm) appeared showing selective initiation of vinyl substrate. Finally, a catalytic amount of a visible-light sensitive radical photoinitiator (I-819) was added and irradiated with 20 mW cm−2 of 405 nm light for 30 min to initiate the thiol–ene reaction. As shown by the NMR, the final set of alkene peaks (δ = 5.79 and 5.06 ppm) had reacted via the thiol–ene pathway. Excitingly, no appreciable change in the chemical shift integration occurred for the vinyl or allyl when DMPP and MSA were added, respectively. It is important to note that additional acid catalyst (5 mol% of MSA) was needed to neutralize the base catalyst (2.5 mol% of DMPP) in the one-pot scheme to promote the ACT reaction. Although the thiol-Michael reaction was catalyzed prior to the ACT reaction in the presented case, either reaction can be performed first under these sequential conditions barring the respective base or acid catalyst added in the first step is sufficiently neutralized by the subsequent catalyst. The presence of the acid catalyst when attempting a thiol-Michael conjugation is a necessary consideration to make, as acids, such as MSA, are well known to hinder thiol-Michael reaction kinetics.68 However, photoinitiation of the thiol–ene reaction must be performed last as to avoid undesired radical-mediated homopolymerization of the acrylamides or thiol–ene reactions with all available alkenes. The selective initiation of all three reactions demonstrates the orthogonal nature of the three thiol–X reactions and the potential for sequential or simultaneous reactions towards the design of multifunctional or sequenced-defined materials.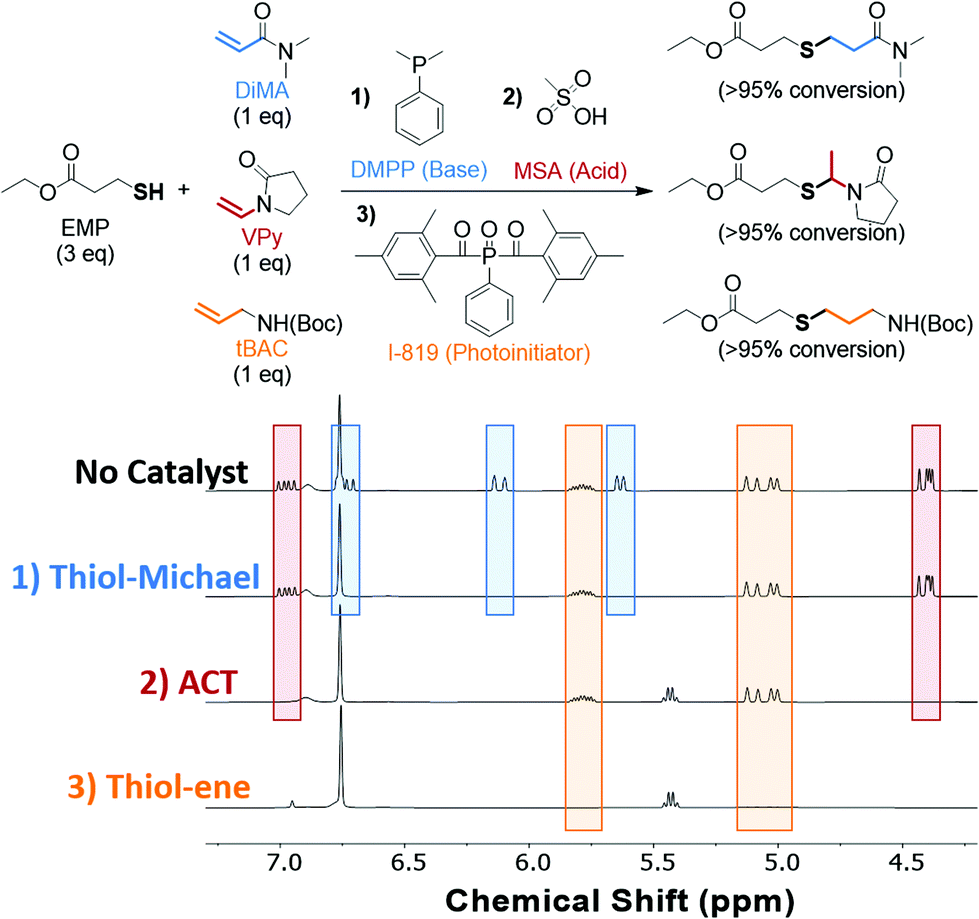 | ||
| Fig. 2 Sequential initiation of thiol-Michael, ACT, and thiol–ene reactions. In a single NMR tube was added EMP (0.6 mmol, 3 eq.), DiMA (0.2 mmol, 1 eq., red), VPy (0.2 mmol, 1 eq., blue), tBAC (0.2 mmol, 1 eq., orange) and DMSO-d6 (600 μL). The ‘no catalyst’ NMR was run prior to the addition of any of the catalysts. To initiate the thiol-Michael reaction, DMPP (2.5 mol%, respective to DiMA amount) was added and allowed to mix for 16 h. Once complete, the NMR showed no remaining alkene peaks associated with DiMA implying that full conversion had been reached. To initiate the ACT reaction, MSA (5 mol%, respective to VPy amount) was added and allowed to mix for 16 h. NMR showed no remaining vinyl functional groups indicating full conversion. Finally, I-819 (5 mol%, respective to tBAC amount) was added to the NMR tube and 20 mW cm−2 of 405 nm light was shined on the sample for 30 min. Post irradiation, the NMR revealed complete conversion of the allyl peaks. | ||
Photoinitiation of the ACT reaction
Although photoinitiation of both thiol–ene71 and thiol-Michael72 reactions have been extensively studied to achieve spatial and temporal control over the reactions, the ACT reaction has been limited by the types of photoacids available and thus far has not been demonstrated to be photoinitiated. Orthogonal photoacid initiation of the ACT reaction is nontrivial, since many commercially available photoacids generate both protons and radicals through homolytic cleavage,73 which can initiate the thiol–ene polymerization.74 An alternative strategy for generating an acidic catalyst is the use of photochromic photoacids.69,75 Under light, terarylenes or spiropyrans can undergo an isomerization reaction producing an acidic proton. A spiropyran-based photoacid was of particular interest for its scalability and simplicity in design (Fig. 3A, SP1).69,70,76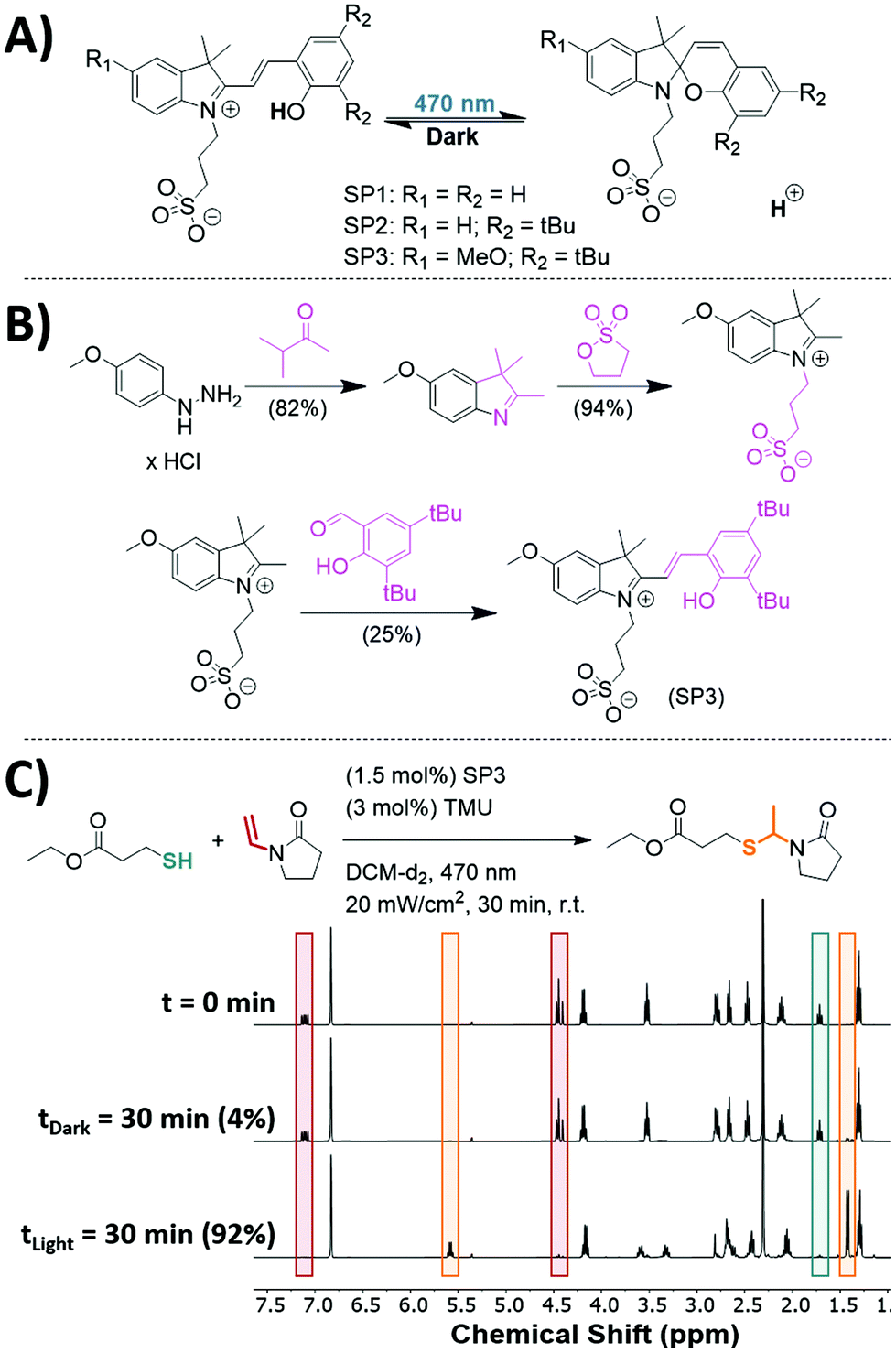 | ||
| Fig. 3 (A) Photoisomerization of three spiropyran-based photoacids (SP1,69 SP2,70 and SP3). (B) Synthetic route to produce SP3, which has reduced dark acidity, improved solubility, and can be produced on a multigram scale. Full synthetic procedures for SP1, SP2, and SP3 can be found in section 10 of the ESI.† (C) Photoinitiation of the ACT reaction was achieved using SP3. EMP (0.2 mmol, 1 eq.), VPy (0.2 mmol, 1 eq.), TMU (3 mol%), and mesitylene (0.2 mmol, 1 eq.) were dissolved in DCM (600 μL). The sample was split into two equal portions in which both received SP3 (1.5 mol%). One sample was left in the dark for 30 min and the other was subjected to 470 nm irradiation (20 mW cm−2, 30 min). | ||
A model system was developed to test SP1 as a photoacid catalyst for the ACT reaction. A stoichiometric amount of EMP and VPy was dissolved in deuterated DCM with TMU and mesitylene as an internal standard. SP1 was synthesized based on previously reported procedures.69 An NMR was taken of the solution (t = 0 min) and subsequently split into two samples where both received 1.5 mol% of SP1. One sample was left in the dark for 30 min and then characterized by NMR while the other sample was irradiated using 470 nm light at 20 mW cm−2 for 30 min and then characterized by NMR spectroscopy. The sample that was left in the dark showed no appreciable conversion, while the sample irradiated with light proceeded to 87% conversion after 30 min (see Fig. S23 in the ESI†). Unfortunately, SP1 was poorly soluble in organic solvents limiting the loading of the catalyst.
A novel modified spiropyran-based photoacid for the ACT reaction was designed to increase solubility in organic solvents while maintaining negligible reaction in the dark. Work by Zayas et al.70 aimed to increase the solubility by adding tert-butyl (tBu) groups on the phenolic ring (Fig. 3A, SP2). Although the SP2 had improved solubility and resulted in high conversions (94%) upon irradiation, the sample stored in the dark exhibited significant conversion (8% conversion after 30 min, see Fig. S24 in the ESI†), owing to the underlying acidity of SP2. Subsequently, Liu et al.76 showed that by introducing an electron donating group to the indole ring, the overall dark acidity of the photoacid was significantly reduced. Encouraged by this result, we designed and synthesized a novel photoacid (SP3), depicted in Fig. 3B. SP3 was facilely synthesized on the gram scale without the need of chromatography. As is shown in Fig. 3C, the model system with SP3 achieved 92% conversion within 30 minutes of irradiation while having significantly less dark acidity (4% conversion after 30 min). These results show the improved effectiveness of SP3 for initiating the ACT reaction as compared with previous spiropyran-based photoacid designs. With the successful redesign of the spiropyran photoacid, photoinitiation of the ACT reaction was achieved for the first time.
Photopolymerization of an ACT polymer network
Utilizing the newly developed photoinitiation scheme, polymer networks were formed using multifunctional thiols and vinyl ethers under blue light irradiation. Resins were formulated at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio of thiols to vinyls using pentaerythritol tetrakis(3-mercaptopropionate) (PETMP) and 1,4-cyclohexanedimethanol divinyl ether (CDDVE) with 0.27 wt% of SP3 as a photoacid generator (structures shown in Fig. 4A). The resin formulation was thoroughly mixed for 2 min and left in the dark for an additional 2 min prior to irradiation with 470 nm light at 20 mW cm−2. The polymerization kinetics were monitored using real-time FTIR, and the conversions are presented in Fig. 4B. Minor dark acidity (less than 6% conversion over 7 min) was observed in the film prior to irradiation, while a control film without any SP3 in the formulation did not completely inhibit the dark reaction, suggesting the resin itself was slightly acidic (see Fig. S25 in the ESI†). The observation of resin instability was expected as the system itself is uninhibited, similar to that of uninhibited thiol–ene resins that exhibit poor shelf-life.77 The acidic nature of the thiol–ene resins, associated with the thiol monomer PETMP to prevent disulfide formation, can lead to unintended ACT polymerizations. It should be noted that the presence of a weak base, such as triphenylphosphine (TPP), resulted in significantly reduced dark acidity; however, polymerization with TPP exhibited reduced conversion (see Fig. S26 in the ESI†). After 80 min of continuous irradiation, the polymerization of the monomer resin plateaued at 99 ± 1% and 90.1 ± 0.3% conversion for the vinyl and thiol, respectively. Based on the solution-phase kinetics, one might speculate that the offset between the vinyl and thiol final conversions is due to vinyl degradation by water. However, no peak changes were observed at 4561 cm−1, associated with acetaldehyde (Fig. S27†);62 thus, it is more likely that the offset owes to minor homopolymerization between vinyls, analogous to that observed in thiol–ene polymerizations formulated with triallyl-1,3,5-triazine-2,4,6-trione (TATATO) or acrylates.78 The resulting polymeric film is a slightly yellow translucent elastomer that turns bright yellow after 24 h, owing to the thermal relaxation of the spiropyran from a ring closed state to a ring opened state (e.g., see Fig. 3A).
:1 stoichiometric ratio of thiols to vinyls using pentaerythritol tetrakis(3-mercaptopropionate) (PETMP) and 1,4-cyclohexanedimethanol divinyl ether (CDDVE) with 0.27 wt% of SP3 as a photoacid generator (structures shown in Fig. 4A). The resin formulation was thoroughly mixed for 2 min and left in the dark for an additional 2 min prior to irradiation with 470 nm light at 20 mW cm−2. The polymerization kinetics were monitored using real-time FTIR, and the conversions are presented in Fig. 4B. Minor dark acidity (less than 6% conversion over 7 min) was observed in the film prior to irradiation, while a control film without any SP3 in the formulation did not completely inhibit the dark reaction, suggesting the resin itself was slightly acidic (see Fig. S25 in the ESI†). The observation of resin instability was expected as the system itself is uninhibited, similar to that of uninhibited thiol–ene resins that exhibit poor shelf-life.77 The acidic nature of the thiol–ene resins, associated with the thiol monomer PETMP to prevent disulfide formation, can lead to unintended ACT polymerizations. It should be noted that the presence of a weak base, such as triphenylphosphine (TPP), resulted in significantly reduced dark acidity; however, polymerization with TPP exhibited reduced conversion (see Fig. S26 in the ESI†). After 80 min of continuous irradiation, the polymerization of the monomer resin plateaued at 99 ± 1% and 90.1 ± 0.3% conversion for the vinyl and thiol, respectively. Based on the solution-phase kinetics, one might speculate that the offset between the vinyl and thiol final conversions is due to vinyl degradation by water. However, no peak changes were observed at 4561 cm−1, associated with acetaldehyde (Fig. S27†);62 thus, it is more likely that the offset owes to minor homopolymerization between vinyls, analogous to that observed in thiol–ene polymerizations formulated with triallyl-1,3,5-triazine-2,4,6-trione (TATATO) or acrylates.78 The resulting polymeric film is a slightly yellow translucent elastomer that turns bright yellow after 24 h, owing to the thermal relaxation of the spiropyran from a ring closed state to a ring opened state (e.g., see Fig. 3A).
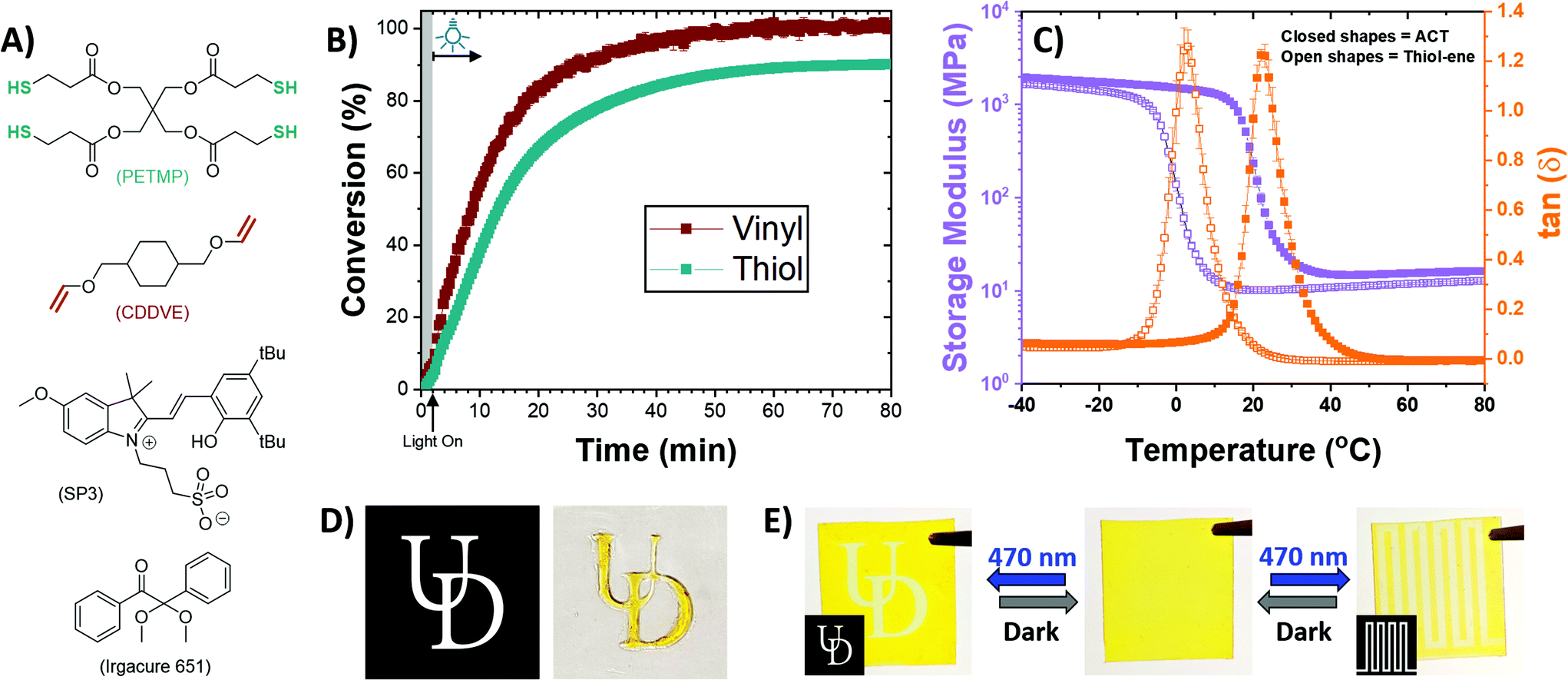 | ||
| Fig. 4 Polymer networks were photopolymerized via the ACT reaction. (A) Stoichiometric ratios of tetrafunctional thiol (PETMP), difunctional vinyl ether (CDDVE), and SP3 (0.27 wt%) were polymerized between two glass slides. (B) Conversion versus time was measured for the ACT reaction using Fourier transform infrared spectroscopy (FTIR) by tracking the thiol (2620–2530 cm−1) and vinyl (6205–6175 cm−1) peaks. The resin was left in the dark for 2 min (shaded region) prior to irradiation of the sample with 470 nm light at 20 mW cm−2. The resultant polymer film (inset) is translucent that turns yellow/orange after a period of 24 h from the relaxation of SP3's closed form to its open form. (C) DMA of the ACT polymerized network compared with a control thiol–ene network polymerized with Irgacure 651. The control thiol–ene resin was formulated with 0.1 wt% of Irgacure 651 and polymerized using 365 nm light at 15 mW cm−2. (D) Spatial temporal control of the ACT reaction was demonstrated by using a photomask with good fidelity (bottom edge = 2 cm). (E) The resulting relaxation of SP3 from its closed form to its open form produces a yellow/orange film that can be subjected to 470 nm irradiation to produce a colorless film. Using photomasks, reversible images can be applied to the polymer film using 15 s of 470 nm irradiation at 20 mW cm−2 and erased after 24 h in the dark (bottom edge = 2 cm). | ||
The thermomechanical properties of the film polymerized via the ACT mechanism produced a homogenous network structure with an increased glass transition temperature (Tg) as compared with a film polymerized via a radical-mediated thiol–ene mechanism. As shown in Fig. 4C, the ACT polymerized film had a Tg of 23 °C as compared with the thiol–ene polymerized film having a Tg of 3 °C. While chemically identical, the two polymer networks are crosslinked via two different isomers resulting in the substantial increase in Tg (i.e., the thioether versus the S,O-acetal crosslink formed via the thiol–ene and ACT reactions, respectively). These results are consistent with higher glass transition temperatures in linear polymers possessing S,O-acetal linkages as compared with their thioether isomers.31 The differences in backbone mobility brought about by the conformational differences in the crosslink and the reduced molecular weight between crosslinks has a significant impact on the Tg. Additionally, the sharp tan delta peak shown in Fig. 4C suggests a homogenous network structure, which is characteristic of a step-growth polymerized network.79 The larger rubbery modulus of the ACT film (16.4 ± 0.4 MPa at 80 °C) as compared with the thiol–ene film (12.8 ± 0.3 MPa at 80 °C) is attributed to the reduced distance between crosslinks produced via the Markovnikov directed conjugation. It should be noted that the relatively minor homopolymerization that is speculated to occur would further enhance the mechanical properties. Additionally, although solution-phase studies suggested the formation of O,O- and S,S-acetal side products for more acid sensitive substrates in the absence of TMU (e.g., with vinyl ethers), one might anticipate the formation of a mixture of O,O-, S,S- and S,O-acetal crosslinks in the polymer network. We speculate that while TMU is absent from the polymer network formulations, the monomer structures themselves contain esters and ethers that may stabilize product formation. Furthermore, only minor amounts of acetaldehyde is formed (see Fig. S27 in the ESI†) and without this key side product forming, O,O- and S,S-acetals cannot form thus suggesting the network crosslinks are primarily S,O-acetal in nature.
The spatiotemporal control of the reaction was demonstrated via photopatterning. By irradiating the resin cast between glass slides through a photomask, the reaction was spatially confined to only the irradiated area, shown in Fig. 4D. Upon completion of the reaction, the clear film turned bright yellow after a period of 24 h as a result of the ring closed spiropyran relaxing to its ring opened state. As such, after polymerization, this reversible photoacid could be spatially ring closed to form images by shining 470 nm light at 20 mW cm−2 for 15 s. As demonstrated in Fig. 4E, multiple photopatterns could be applied to the polymer film with complete reversion back to the ring opened state after 24 h. This reversible writing is a unique feature of the material and could be a useful tool in digital writing on polymeric materials.
Conclusions
In summary, the ACT reaction provides a new thiol–X reaction pathway for solution-phase conjugations and in the design of unique materials systems. The scope of the reaction was evaluated using various thiol and vinyl structures demonstrating a broad range of functional handles. Additionally, selectivity of the cationic-mediated mechanism to both the thiol-Michael and thiol–ene reactions were shown using a sequential one-pot reaction of three different alkene substrates. Furthermore, photoinitiation of the ACT reaction was achieved for the first time using a novel photochromic photoacid. Using the knowledge gained from the solution-phase studies, solid-state polymeric materials were synthesized using the photoinitiated ACT reaction. The polymer networks formed via the cationic pathway resulted in a higher glass transition temperature than a polymer network polymerized via the radical mechanism due to the presence of the S,O-acetal bond as opposed to a thioether linkage. The work presented provides direct insight into utilizing the ACT reaction with the various caveats associated with its successful conjugation. Development of the ACT reaction expands upon a diverse toolbox of thiol conjugation strategies further growing the applicability of thiol–X reactions towards next generation materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
BPS and CJK would like to acknowledge support by the National Institutes of Health (NIH) under Award Number P20GM104316 as well as P30GM110758-02 for additional core instrument support. MK would like to acknowledge support by the National Science Foundation EPSCoR Grant No. 1757353 and the State of Delaware. BPS would like to acknowledge Dr Abhishek U. Shete for his insightful comments and suggestions regarding the written manuscript.References
- T. Posner, Ber. Dtsch. Chem. Ges., 1905, 38, 646–657 CrossRef.
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743–750 CrossRef CAS.
- A. B. Lowe, Polym. Chem., 2014, 5, 4820–4870 RSC.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS.
- N. Gupta, B. F. Lin, L. M. Campos, M. D. Dimitriou, S. T. Hikita, N. D. Treat, M. V. Tirrell, D. O. Clegg, E. J. Kramer and C. J. Hawker, Nat. Chem., 2010, 2, 138–145 CrossRef CAS.
- A. A. Aimetti, R. K. Shoemaker, C. C. Lin and K. S. Anseth, Chem. Commun., 2010, 46, 4061–4063 RSC.
- Y. Jiang, J. Chen, C. Deng, E. J. Suuronen and Z. Zhong, Biomaterials, 2014, 35, 4969–4985 CrossRef CAS.
- J. C. Grim, T. E. Brown, B. A. Aguado, D. A. Chapnick, A. L. Viert, X. Liu and K. S. Anseth, ACS Cent. Sci., 2018, 4, 909–916 CrossRef CAS.
- A. Dondoni and A. Marra, Chem. Soc. Rev., 2012, 41, 573–586 RSC.
- B. P. Sutherland, B. M. El-Zaatari, N. I. Halaszynski, J. M. French, S. Bai and C. J. Kloxin, Bioconjugate Chem., 2018, 29, 3987–3992 CrossRef CAS.
- S. J. Ma, S. J. Mannino, N. J. Wagner and C. J. Kloxin, ACS Macro Lett., 2013, 2, 474–477 CrossRef CAS.
- O. Daglar, U. S. Gunay, G. Hizal, U. Tunca and H. Durmaz, Macromolecules, 2019, 52, 3558–3572 CrossRef CAS.
- G. Chen, Q. Zhang, M. Lu, Y. Liu, S. Wang, K. Wu and M. Lu, Macromol. Chem. Phys., 2019, 220, 1900059 CrossRef.
- W. Xi, S. Pattanayak, C. Wang, B. Fairbanks, T. Gong, J. Wagner, C. J. Kloxin and C. N. Bowman, Angew. Chem., Int. Ed. Engl., 2015, 54, 14462–14467 CrossRef CAS.
- B. P. Sutherland, P. J. LeValley, D. J. Bischoff, A. M. Kloxin and C. J. Kloxin, Chem. Commun., 2020, 56, 11263–11266 RSC.
- D. P. Nair, N. B. Cramer, T. F. Scott, C. N. Bowman and R. Shandas, Polymer, 2010, 51, 4383–4389 CrossRef CAS.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2010, 49, 3415–3417 CrossRef CAS.
- M. C. Stuparu and A. Khan, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3057–3070 CrossRef CAS.
- R. M. Hensarling, S. B. Rahane, A. P. Leblanc, B. J. Sparks, E. M. White, J. Locklin and D. L. Patton, Polym. Chem., 2011, 2, 88–90 RSC.
- X. Y. Wang, Y. G. Li, H. L. Mu, L. Pan and Y. S. Li, Polym. Chem., 2015, 6, 1150–1158 RSC.
- B. Yu, J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3544–3557 CrossRef CAS.
- X. Zhang, W. Xi, S. Huang, K. Long and C. N. Bowman, Macromolecules, 2017, 50, 5652–5660 CrossRef CAS.
- J. Shin, H. Matsushima, C. M. Comer, C. N. Bowman and C. E. Hoyle, Chem. Mater., 2010, 22, 2616–2625 CrossRef CAS.
- S. H. Frayne, R. R. Murthy and B. H. Northrop, J. Org. Chem., 2017, 82, 7946–7956 CrossRef CAS.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- L. T. T. Nguyen, M. T. Gokmen and F. E. Du Prez, Polym. Chem., 2013, 4, 5527–5536 RSC.
- F. Jivan, N. Fabela, Z. Davis and D. L. Alge, J. Mater. Chem. B, 2018, 6, 4929–4936 RSC.
- M. Porel and C. A. Alabi, J. Am. Chem. Soc., 2014, 136, 13162–13165 CrossRef CAS.
- L. A. Oparina, O. V. Vysotskaya, L. N. Parshina, N. K. Gusarova and B. A. Trofimov, Phosphorus, Sulfur Silicon Relat. Elem., 2003, 178, 2087–2093 CrossRef CAS.
- M. Uchiyama, M. Osumi, K. Satoh and M. Kamigaito, Angew. Chem., Int. Ed., 2020, 59, 6832–6838 CrossRef CAS.
- M. Sangermano, I. Roppolo and A. Chiappone, Polymers, 2018, 10(2), 136–144 CrossRef.
- K. Kojima, M. Sawamoto and T. Higashimura, Macromolecules, 1989, 22, 1552–1557 CrossRef CAS.
- V. N. Ipatieff, H. Pines and B. S. Friedman, Reaction of Aliphatic Olefins with Thiophenol, 1938, vol. 60 Search PubMed.
- R. A. Hickner, C. I. Judd and W. W. Bakke, Alkylation of Active Hydrogen Compounds by N-Vinylamides, 1967, vol. 32 Search PubMed.
- M. A. Savolainen and J. Wu, Org. Lett., 2013, 15, 3802–3804 CrossRef.
- I. Ramos-Tomillero, H. Rodriguez and F. Albericio, Org. Lett., 2015, 17, 1680–1683 CrossRef CAS.
- M. Uchiyama, K. Satoh and M. Kamigaito, Macromolecules, 2015, 48, 5533–5542 CrossRef CAS.
- A. G. Orrillo, A. La-Venia, A. M. Escalante and R. L. E. Furlan, Chem. – Eur. J., 2018, 24, 3141–3146 CrossRef CAS.
- A. G. Orrillo, A. M. Escalante and R. L. E. Furlan, Chem. – Eur. J., 2016, 22, 6746–6749 CrossRef CAS.
- N. Van Herck, D. Maes, K. Unal, M. Guerre, J. M. Winne and F. E. Du Prez, Angew. Chemie, 2020, 132, 3637–3646 CrossRef.
- D. N. Amato, D. V. Amato, O. V. Mavrodi, W. B. Martin, S. N. Swilley, K. H. Parsons, D. V. Mavrodi and D. L. Patton, ACS Macro Lett., 2017, 6, 171–175 CrossRef CAS.
- H. Cao, C. Chen, D. Xie, X. Chen, P. Wang, Y. Wang, H. Song and W. Wang, Polym. Chem., 2018, 9, 169–177 RSC.
- D. N. Amato, D. V. Amato, Y. Adewunmi, O. V. Mavrodi, K. H. Parsons, S. N. Swilley, D. A. Braasch, W. D. Walker, D. V. Mavrodi and D. L. Patton, ACS Appl. Bio Mater., 2018, 1, 1983–1991 CrossRef CAS.
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- T. Masuda, M. Sawamoto and T. Higashimura, Die Makromol. Chem., 1976, 177, 2981–2993 CrossRef CAS.
- M. Zsuga and J. P. Kennedy, Electron donors in carbocationic polymerization - IV. Preparation of narrow-dispersity tert.-chlorine-capped polyisobutylene by the trans-2,5-diacetoxy-2,5-dimethyl-3-hexene/BCl3/dimethyl sulfoxide system, 1989, vol. 21 Search PubMed.
- D. M. Jones and N. F. Wood, J. Chem. Soc., 1964, 5391–5392 Search PubMed.
- P. Sigwalt, C. Gobin, P. Nicol, M. Moreau and M. Masure, Makromol. Chem., Macromol. Symp., 1991, 4243, 229–240 CrossRef.
- A. J. Kresge and H. J. Chen, J. Am. Chem. Soc., 1972, 94, 2818–2822 CrossRef CAS.
- M. Claudino, M. Jonsson and M. Johansson, RSC Adv., 2013, 3, 11021–11034 RSC.
- B. H. Northrop and R. N. Coffey, J. Am. Chem. Soc., 2012, 134, 13804–13817 CrossRef CAS.
- G. Wulff and P. Birnbrich, Die Makromol. Chem., 1992, 193, 2405–2411 CrossRef CAS.
- J. E. Puskas and G. Kaszas, in Encyclopedia of Polymer Science and Technology, Wiley, 2016, pp. 1–43 Search PubMed.
- S. N. Zelinskiy, E. N. Danilovtseva, G. Kandasamy, V. A. Pal'Shin, T. A. Shishlyannikova, U. M. Krishnan and V. V. Annenkov, E-Polymers, 2018, 18, 347–357 CAS.
- M. Dréan, P. Guégan, C. Jérôme, J. Rieger and A. Debuigne, Polym. Chem., 2016, 7, 69–78 RSC.
- B. Capon and Z. P. Wu, J. Org. Chem., 1990, 55, 2317–2324 CrossRef CAS.
- J. Ferguson and V. Sundar Rajan, Eur. Polym. J., 1979, 15, 627–630 CrossRef CAS.
- M. F. Frosolono and M. M. Rapport, J. Lipid Res., 1969, 10, 504–506 CrossRef CAS.
- C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301–5338 CrossRef CAS.
- J. W. Chan, C. E. Hoyle, A. B. Lowe and M. Bowman, Macromolecules, 2010, 43, 6381–6388 CrossRef CAS.
- Y. Lin and J. W. Stansbury, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1985–1998 CrossRef CAS.
- R. Huang, B. A. Ficek, S. O. Glover and A. B. Scranton, Effect of water in cationic photopolymerizations: reversible inhibition, 2007, vol. 21 Search PubMed.
- M. Delfour, V. éronique Bennevault-Celton, H. A. Nguyen, A. Macedo and H. Cheradame, Eur. Polym. J., 2004, 40, 1387–1398 CrossRef CAS.
- C. G. Cho, B. A. Feit and O. W. Webster, Macromolecules, 1990, 23, 1918–1923 CrossRef CAS.
- Y. Wu and G. Wu, in Journal of Polymer Science, Part A: Polymer Chemistry, 2002, vol. 40, pp. 2209–2214 Search PubMed.
- J. W. Chan, B. Yu, C. E. Hoyle and A. B. Lowe, Chem. Commun., 2008, 4959–4961 RSC.
- S. Chatani, D. P. Nair and C. N. Bowman, Polym. Chem., 2013, 4, 1048–1055 RSC.
- Z. Shi, P. Peng, D. Strohecker and Y. Liao, J. Am. Chem. Soc., 2011, 133, 14699–14703 CrossRef CAS.
- M. S. Zayas, N. D. Dolinski, J. L. Self, A. Abdilla, C. J. Hawker, C. M. Bates and J. Read de Alaniz, ChemPhotoChem, 2019, 3, 467–472 CrossRef CAS.
- D. Ahn, S. S. Sathe, B. H. Clarkson and T. F. Scott, Dent. Mater., 2015, 31, 1075–1089 CrossRef CAS.
- W. Xi, H. Peng, A. Aguirre-Soto, C. J. Kloxin, J. W. Stansbury and C. N. Bowman, Macromolecules, 2014, 47, 6159–6165 CrossRef CAS.
- N. Zivic, P. K. Kuroishi, F. Dumur, D. Gigmes, A. P. Dove and H. Sardon, Recent Advances and Challenges in the Design of Organic Photoacid and Photobase Generators for Polymerizations, Wiley-VCH Verlag, 2019, vol. 58 Search PubMed.
- H. Wei, Q. Li, M. Ojelade, S. Madbouly, J. U. Otaigbe and C. E. Hoyle, Macromolecules, 2007, 40, 8788–8793 CrossRef CAS.
- T. Nakashima, K. Tsuchie, R. Kanazawa, R. Li, S. Iijima, O. Galangau, H. Nakagawa, K. Mutoh, Y. Kobayashi, J. Abe and T. Kawai, J. Am. Chem. Soc., 2015, 137, 7023–7026 CrossRef CAS.
- J. Liu, W. Tang, L. Sheng, Z. Du, T. Zhang, X. Su and S. X. A. Zhang, Chem. – Asian J., 2019, 14, 438–445 CrossRef CAS.
- P. Esfandiari, S. C. Ligon, J. J. Lagref, R. Frantz, Z. Cherkaoui and R. Liska, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4261–4266 CrossRef CAS.
- N. B. Cramer and C. N. Bowman, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3311–3319 CrossRef CAS.
- A. Bacchi, J. A. Yih, J. Platta, J. Knight and C. S. Pfeifer, J. Mech. Behav. Biomed. Mater., 2018, 78, 235–240 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py01593h |
| This journal is © The Royal Society of Chemistry 2021 |