
Topological analysis and control of post-synthetic metalation sites in Zr-based metal–organic frameworks†
Pol
Gimeno-Fonquernie
 a,
Jorge
Albalad‡
a,
Jason R.
Price
b,
Witold M.
Bloch
c,
Jack D.
Evans
*a,
Christian J.
Doonan
*a and
Christopher J.
Sumby
*a
a,
Jorge
Albalad‡
a,
Jason R.
Price
b,
Witold M.
Bloch
c,
Jack D.
Evans
*a,
Christian J.
Doonan
*a and
Christopher J.
Sumby
*a
aCentre for Advanced Nanomaterials and Department of Chemistry, School of Physics, Chemistry and Earth Sciences, The University of Adelaide Adelaide, SA 5005, Australia. E-mail: j.evans@adelaide.edu.au; christian.doonan@adelaide.edu.au; christopher.sumby@adelaide.edu.au
bANSTO Melbourne, The Australian Synchrotron, 800 Blackburn Rd, Clayton, Vic 3168, Australia
cCollege of Science & Engineering, Flinders University, Sturt Road, Bedford Park, SA 5042, Australia
First published on 22nd December 2023
Abstract
The design criteria needed for the formation of the sqc metal–organic framework (MOF) topology, from an 8-connecting node and a 4-connecting linker, are unclear due to a limited number of reports. After recently reporting the MOF UAM-1000 (UAM = University of Adelaide Material), which has this rare sqc topology, we present a study that explores the effect of flexible tetrapyrazole carboxylate linker structure metrics on the topology of Zirconium-based MOFs (Zr-MOFs). By modifying the linker length and width, three new Zr-MOFs (UAM-10, UAM-11, and UAM-1002) were synthesized and characterized. The study reveals that linker dimensions influence the accessible conformations, and along with fine-tuning of synthetic conditions, allow control over MOF topology. Additionally, linker flexibility plays a crucial role in the formation of the sqc over the more common csq topology. Finally, the presence of free bis-pyrazolyl groups in the reported MOFs allowed us to evaluate the potential for post-synthetic metalation (PSMet). UAM-10 and UAM-11 are too rigid, the pyrazole groups lack the appropriate arrangement and therefore these materials do not undergo PSMet. In contrast, UAM-1002 with its scu topology exhibits the right quanta of flexibility needed for successful PSMet, making it a promising platform for studying the chemistry of anchored organometallic complexes. Moreover, the different topology for UAM-1002 versus UAM-1000 changes the nature of the PSMet site (bidentate versus a tetradentate site) despite these being made from the same node and linker building blocks.
Introduction
The topology of a metal–organic framework (MOF), namely the type of network structure, plays a crucial role in determining its stability, and its adsorption, separation, and catalytic properties.1–4 Different topologies result in distinct pore shapes and sizes that can significantly impact guest adsorption and selectivity of the MOF toward particular adsorbates.5 Similarly, topology can determine substrate and product diffusion rates and thereby catalytic properties.6,7 Despite extensive research on the reticulation of MOFs and the development of the secondary building block (SBU) approach7–9 to MOF design, with the exception of highly connected SBUs,10 predicting MOF structures remains a challenge as multiple net topologies exist for a given combination of metal node and link geometries.11–13 Therefore, understanding topological control in MOFs is crucial to provide phase pure samples and ensure the topology with the most suitable performance characteristics is obtained.14Zirconium (Zr) based MOFs have attracted increasing interest since their 2008 discovery due to the exceptional strength of the Zr–O bond, which enables the formation of highly stable and robust materials.15–17 The typical Zr cluster consists of six Zr atoms forming an octahedron that can coordinate up to 12 carboxylate groups, resulting in a wide variety of topologies with 4, 5, 6, 8, 10, and 12-connected clusters.11 Several studies have provided understanding of the different connectivities of Zr clusters.18 For instance, the formation of the ftw topology, formed by the combination of a 12-connected cluster and a square linker, is only possible with square linkers where the oxygen atoms of the carboxylate groups are in plane with the core of the linker.19 The use of a rectangular linkers, substituents or bulky ligands to induce an out-of-plane arrangement of the carboxylate groups leads to the formation of an 8-connected cluster.20 In some of these cases, different topologies can be possible from the same metal cluster and linker connectivity, and control of Zr MOF topology becomes more challenging. An illustrative example is the reaction of the cubic 8-connected Zr-cluster with a tetratopic linker, which can lead to the formation of four different topologies: flu when the linker adopts a tetrahedral arrangement of the donors and csq, scu, and sqc when the linker adopts a planar conformation (Fig. 1). Several studies have probed the factors, such as the presence of bulky substituents in the organic linker,21,22 synthetic conditions,23,24 modulator rigidity and co-modulator acidity,25 that can influence the formation of different topologies. Furthermore, in certain cases, the addition of a seed of a phase pure MOF has been shown to be required for the synthesis of a particular MOF, while the absence often leads to the formation of a mixture of phases.26
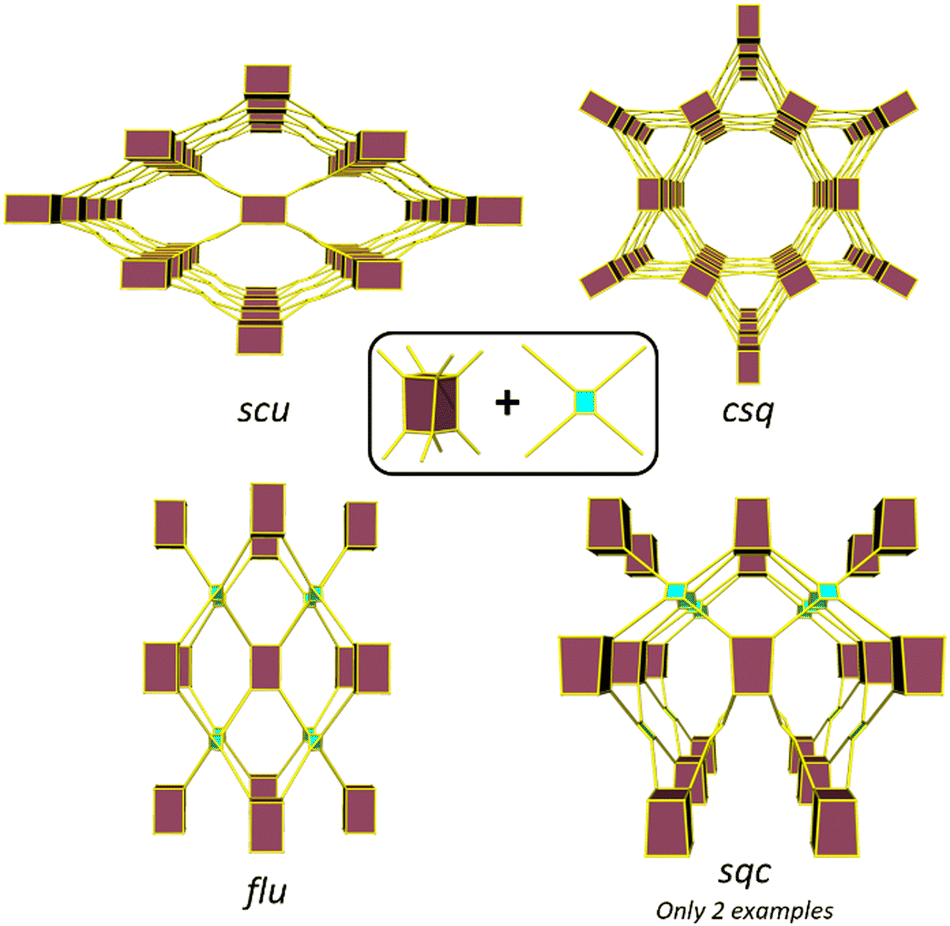 | ||
| Fig. 1 Representations of the four different topologies that can be formed from a cubic 8-connected cluster and a tetratopic linker (with either a tetrahedral or square planar geometry). The maroon polyhedron represents the cluster and the blue shaded rectangle or polygon with yellow lines represents the organic linker. | ||
Despite the existence of at least four possible topologies from reacting a cubic 8-connected Z-cluster with a tetratopic linker, previous studies have mainly focused on differentiating between the formation of the flu, scu and csq topologies.21–24 This is primarily due to the rarity of MOFs with the sqc topology, with only two reported examples to date.27,28 Notably, our group only recently communicated the second synthesis of a MOF with the sqc topology (UAM-1000).28 In this MOF and braced analogues (UAM-1001), a flexible linker containing free bis-pyrazolyl groups allows for post-synthetic metalation (PSMet) with dinuclear complexes. PSMet is a widely applied technique that allows the functionalization of MOFs with secondary, often non-structural metal centres, conferring MOFs with novel physical, optical, sorption and catalytic properties. Given that Zr-MOFs based on tetratopic carboxylate linkers can access different topologies, control over MOF topology can therefore be used to dictate the outcome of PSMet as the spatial disposition of the metalation sites can lead to the isolation of different metalation products.
To enhance our understanding of the synthesis of Zr-MOFs for PSMet, and provide insights into the formation of the sqc topology in UAM-1000, this study investigates the effect of flexible tetrapyrazole carboxylate linker metrics on MOF topology. Starting from the linker 1,1,2,2-tetrakis[4-(4-carboxyphenyl)-1H-pyrazol-1-yl]ethane (TCPE), which was used to form UAM-1000, we modified the length and width of the linker, resulting in two new linkers: 1,1′-methylenebis(1H-pyrazole-4-carboxylic acid) (L1H4) and 1,4-bis(bis(4-carboxy-1H-pyrazol-1-yl)methyl)benzene (L2H4). Each linker, including TCPE, was reacted with ZrCl4 under different conditions to evaluate the formation of different topologies, allowing an assessment of the characteristics needed to form the different MOFs and the effect of MOF topology on PSMet outcome. Overall, the study reports the synthesis and characterization of three new Zr-MOFs (UAM-10, UAM-11, and UAM-1002), each containing non-coordinated bis-pyrazolyl units, and investigates their potential for PSMet, showing the ability to form metalation sites for discrete mononuclear and dinuclear complexes.
Experimental section
Materials and methods
Unless otherwise stated, all chemicals were obtained from commercial sources and used as received. 1,1,2,2-Tetrakis[4-(4-carboxyphenyl)-1H-pyrazol-1-yl]ethane29,30 and 1,4-bis(dibromomethyl)benzene31 were synthesized following an already reported procedure. Acetonitrile (MeCN) was dried from CaH2 under N2 and degassed with Ar prior to use.Powder X-ray diffraction (PXRD) data were collected on a Bruker Advanced D8 diffractometer (capillary stage) using Cu Kα radiation (λ = 1.5456 Å, 40 kW/40 mA, 2θ = 2–52.94°, φ rotation = 20 rotations per min, at 1 s exposure per step with 5001 steps, and using 0.5 mm glass capillaries). Fourier Transform Infrared Spectroscopy (FTIR) spectra were collected on a Shimadzu IR spirit using ATR mode (spectral range: 7800–350 cm−1). Nuclear Magnetic Resonance (NMR) spectra were collected at 25 °C in deuterated solvents on an Agilent DD2 500 MHz NMR with a 5 mm OneNMR probe, using tetramethylsilane (TMS) signals as the internal reference standard. Scanning Electron Microscopy (SEM) images were collected, and Energy Dispersive X-ray (EDX) analysis performed on a FEI Quanta 450 field-emission scanning electron microscope, operating at 10.0–20.0 kV and under ultra-high vacuum (10−7–10−12 hPa) conditions. The samples were prepared for analysis by mounting a mixture of intact and crushed crystals on the SEM stub, measuring the metal composition for areas of the sample, and averaging the measurements. Gas adsorption measurements were performed on a Micromeritics 3-Flex surface area and pore size analyser. Activation conditions for each sample is stated in figure captions (Fig. S10, S15, S21, S30–S32, ESI†). Thermogravimetric analysis data was collected on an STA 449 F3 Jupiter analyser from 45–700 °C at 5 °C min−1 under 21% O2, 79% N2.
Molecular simulations
The ligands L1, L2 and L3 were considered as the corresponding acid and the conformational landscape was assessed using the Conformer-Rotamer Ensemble Sampling Tool (CREST)32 with an energy window of 30 kJ mol−1 and energies computed using GFN2-xTB33 with implicit solvation of DMF considered by the analytical linearized Poisson–Boltzmann approach.34Framework models were produced using the weaver code35 and resulting frameworks first optimised using the UFF36 as implemented by the lammps-interface37 using the lammps simulation engine.38 Subsequently, the geometry and cell parameters for these frameworks were optimised using density functional theory (DFT). These calculations used the PBE functional,39 Goedecker-Teter-Hutter pseudopotential40 and D3 London dispersion corrections,41 together with a double-zeta valence (DZVP) basis set. A grid cutoff of 800 Ry was employed and a relative cutoff of 70 Ry. The DFT approach used the QUICKSTEP42 module of the CP2K program package.43 Pore metrics of these frameworks were computed using the Zeo + + software package.44 Frameworks based on different anti–anti or syn–syn ligand conformations were considered at the classical level of theory (UFF) and the framework was constructed based on the lowest energy conformer observed with anti–anti or syn–syn arrangement.
Single crystal X-ray diffraction
Single crystals were mounted in Paratone-N oil on a MiTeGen micromount. Single-crystal X-ray data of UAM-10, UAM-11 and UAM-1002 were collected at 100 K on the MX1 or MX2 beamlines of the Australian Synchrotron using the Blue-ice software interface,45λ = 0.71073 Å and absorption corrections were applied using multiscan methods using XDS.46,47 UAM-1002[PdCl2] was collected at 100 K on a Rigaku XtaLAB Synergy-s diffractometer equipped with a HyPix-6000HE detector. The structures were solved using SHELXS or SHELXT,48,49 and refined by full-matrix least squares on F2 by SHELXL,50 interfaced through the program X-Seed or OLEX.51,52 In general, all atoms were refined anisotropically and hydrogens atoms were included as invariants at geometrically estimated positions, unless specified otherwise in additional details in ESI.† Where noted, the data was treated with the SQUEEZE routine available in Platon53 or using the solvent masking feature of Olex. Images were produced using the software Diamond Version 4.6.8.54 X-ray experimental data is given in Table S5 (ESI†). CIF data have been deposited with the Cambridge Crystallographic Data Centre, CCDC reference numbers CCDC 2288076–2288079 (2288076 = UAM-10; 2288077 = UAM-11; 2288079 = UAM-1002; 2288078 = UAM-1002[PdCl2]).†Synthetic protocols
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 ethanol/water, and dried in air to give the ester-protected compound. Deprotection was achieved by boiling the white solid in 1 M NaOH overnight. The resulting solution was acidified with 4 M HCl to pH 2 to precipitate H4L1 as a white solid, which was subsequently collected, washed with water and acetone, and dried under vacuum overnight. Yield: 1.8 g (40%). 1H NMR (500 MHz, DMSO-d6): 12.62 (s, 4H, COOH), 8.69 (s, 4H, CH2 pyrazole), 8.52 (s, 2H, N–CH–N), 7.84 (s, 4H, CH2 pyrazole). 13C-NMR spectrum (500 MHz, DMSO-d6): 162.98 (COOH), 141.86 (C pyr), 134.76 (C pyr), 116.03 (C py), 71.98 (C-sp3). MS (ESI†): calcd for C18H13N8O8+ [M]+ 469.08, found 469.08.
:7 ethanol/water, and dried in air to give the ester-protected compound. Deprotection was achieved by boiling the white solid in 1 M NaOH overnight. The resulting solution was acidified with 4 M HCl to pH 2 to precipitate H4L2 as a yellow solid, which was subsequently collected, washed with water and acetone, and dried under vacuum overnight. Yield: 1.08 g (22%).1H NMR (500 MHz, DMSO-d6): 12.62 (s, 4H, COOH), 8.37 (s, 4H, CH2 pyrazole), 8.25 (s, 2H, N–CH–N), 7.98 (s, 4H, CH2 pyrazole), 7.25 (s, 4H, CH phenyl). 13C-NMR spectrum (500 MHz, DMSO-d6): 163.22 (COOH), 141.80 (COOH), 136.21 (C pyr), 134.04 (C pyr), 127.72 (C pheyl), 116.09 (s, 4H, C pyr), 75.93 (C-sp3). MS (ESI†): calcd for C24H17N8O8+ [M]+ 545.12, found 545.12.
:2.0(Pd):4(Cl).
:7 ethanol/water, and dried in air to give the ester-protected compound. Deprotection was achieved by boiling the white solid in 1 M NaOH overnight. The resulting solution was acidified with 4 M HCl to pH 2 to precipitate H4L1 as a white solid, which was subsequently collected, washed with water and acetone, and dried under vacuum overnight. Yield: 1.8 g (40%). 1H NMR (500 MHz, DMSO-d6): 12.62 (s, 4H, COOH), 8.69 (s, 4H, CH2 pyrazole), 8.52 (s, 2H, N–CH–N), 7.84 (s, 4H, CH2 pyrazole). 13C-NMR spectrum (500 MHz, DMSO-d6): 162.98 (COOH), 141.86 (C pyr), 134.76 (C pyr), 116.03 (C py), 71.98 (C-sp3). MS (ESI†): calcd for C18H13N8O8+ [M]+ 469.08, found 469.08.
:7 ethanol/water, and dried in air to give the ester-protected compound. Deprotection was achieved by boiling the white solid in 1 M NaOH overnight. The resulting solution was acidified with 4 M HCl to pH 2 to precipitate H4L2 as a yellow solid, which was subsequently collected, washed with water and acetone, and dried under vacuum overnight. Yield: 1.08 g (22%).1H NMR (500 MHz, DMSO-d6): 12.62 (s, 4H, COOH), 8.37 (s, 4H, CH2 pyrazole), 8.25 (s, 2H, N–CH–N), 7.98 (s, 4H, CH2 pyrazole), 7.25 (s, 4H, CH phenyl). 13C-NMR spectrum (500 MHz, DMSO-d6): 163.22 (COOH), 141.80 (COOH), 136.21 (C pyr), 134.04 (C pyr), 127.72 (C pheyl), 116.09 (s, 4H, C pyr), 75.93 (C-sp3). MS (ESI†): calcd for C24H17N8O8+ [M]+ 545.12, found 545.12.
:2.0(Pd):4(Cl).
Results and discussion
Linker synthesis and preferred conformation
The new flexible tetrapyrazole carboxylate linkers, 1,1′-methylenebis(1H-pyrazole-4-carboxylic acid) (L1H4) and 1,4-bis(bis(4-carboxy-1H-pyrazol-1-yl)methyl)benzene (L2H4), were synthesized using a similar two-step synthesis procedure, where ethyl 4-carboxypyrazole was coupled to 1,1,2,2-tetrabromoethane or 1,4-bis(dibromomethyl)benzene, followed by deprotection of the ester protecting groups. The synthesis of both linkers was confirmed by 1H-NMR (Fig. S1 and S2, ESI†) and 13C-NMR spectroscopy (Fig. S3 and S4, ESI†), and mass spectrometry (Table S1, ESI†).Given that at least three topologies are accessible for a cubic 8-connected cluster and a flexible rectangular tetratopic linker, simulations were employed to explore the conformational flexibility of TCPE and these new ligands, thereby unveiling their preferred conformation (Fig. 2). A series of possible and unique conformers in a relative energy window of 30 kJ mol−1 were computed. The different dihedral angles of the resulting ensembles of conformers were analysed to consider the arrangement of framework forming bonds. Specifically, two sets of NNNN dihedrals were measured, which dictate the syn or anti conformation for each set of pyrazole units, and the HCCH angle that describe the relative positions of the carboxylate groups (planar or tetrahedral) was determined (Fig. 2). Analysis of the NNNN dihedral angle for each of the ligands reveals that L1H4 and TCPE access similar conformational space (Fig. 2(a) and (c)) showing the anti–anti conformation of the bis-pyrazolyl units is the lowest in energy (NNNN ∼ ±180°). The longer arm length (width) of TCPE seems to facilitate the transition from anti–anti to syn–syn conformation as it shows intermediate conformers lower in energy, which suggest an easier transition. A different outcome was observed for L2H4. Analysis of the NNNN dihedral angles for L2H4 reveals a greater range of conformations with low energy but the perfect syn–syn and anti–anti conformations (NNNN ∼ ±180°) are not accessible owing to steric hinderance of the central phenyl ring. Conformations closed to anti–anti conformations (NNNN ∼ ±180°) have the lowest energy. In all cases, we observe the same trends in energy for syn and anti conformations of the pyrazole units as previously reported for a similar bispyrazole carboxylate ligand,55 with an energy difference disfavouring the syn-conformations on the order of 5–10 kJ mol−1.
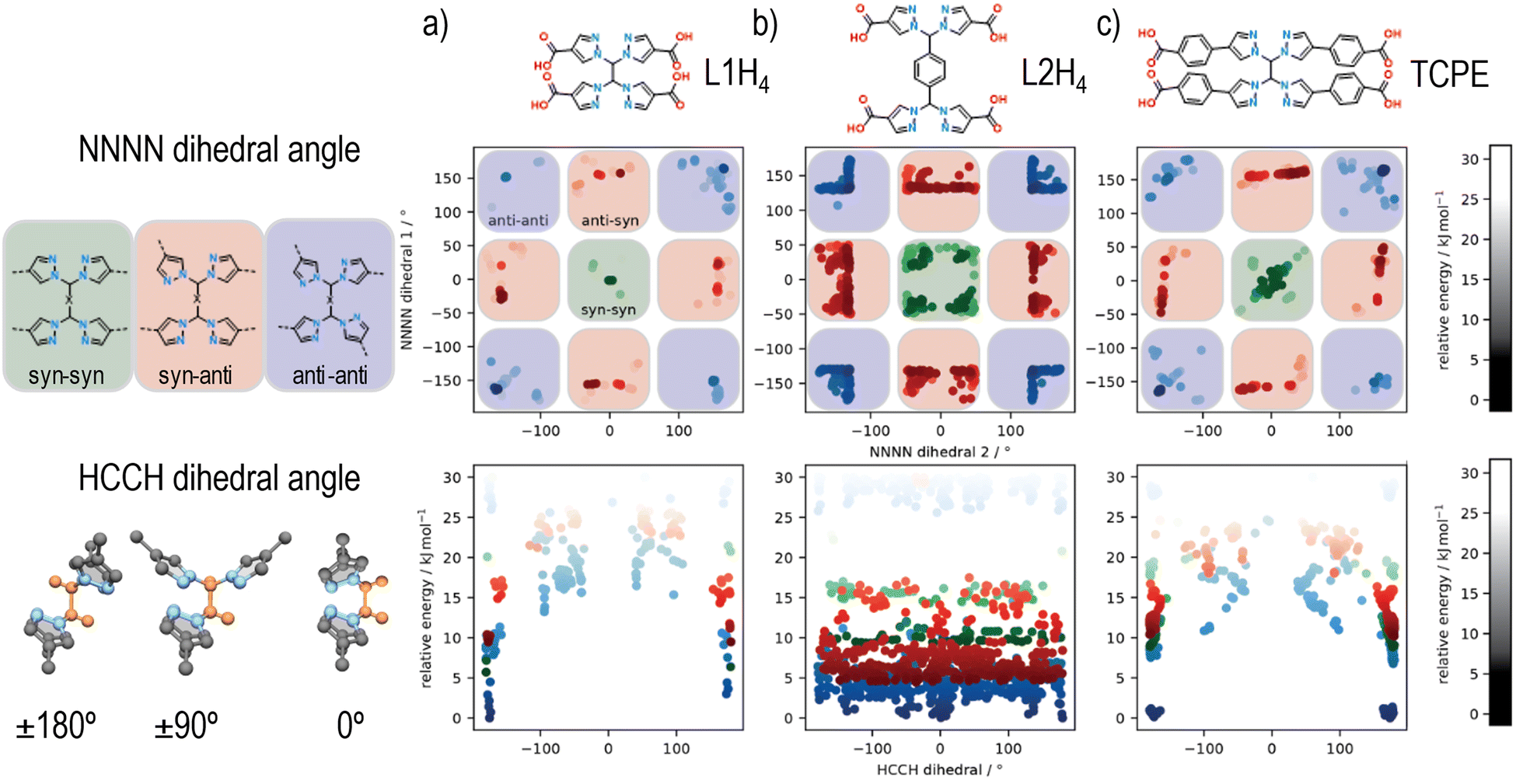 | ||
| Fig. 2 Plots of the simulation output showing the relative energy of different conformations of L1H4, L2H4 and TCPE upon changing the NNNN dihedral angle of the bis-pyrazolyl groups (top row) and the HCCH dihedral angle of the ethylene bridge (bottom row), the latter for each of the three conformations accessible by changing the NNNN dihedral angle. Colour intensity is indicative of the relative energy, with darker colours being lower in energy and lighter colours higher in energy. On the left of the plots are representations of the conformations accessed upon changing each of the dihedral angles. In the representation of the HCCH dihedral angle (bottom-left), the H and C atoms are highlighted in orange. The Y-axis label is shared across plots within the same row. | ||
Analysis of the HCCH angle, reveals the effect of the phenyl unit on L2H4, where the HCCH dihedral of the central ethylene groups, which in turn dictates positions of carboxylates (planar or tetrahedral), can rotate almost without restriction. In comparison, L1H4 and TCPE are restricted to approximately coplanar and anti HCCH conformations. Overall, these simulations demonstrate that a range of different conformations are possible for these flexible tetrapyrazole carboxylate linkers, but the individual bis-pyrazole units typically prefer one of two possible arrangements (anti preferred over syn). In terms of the overall linker geometry, L1H4 and TCPE are restricted to broadly anti HCCH conformations but the phenyl spacer of L2H4 provides even greater conformational flexibility. Based on the restricted flexibility seen in the conformational modelling, it is anticipated that certain MOF topologies should be favoured by the three ligands, with the flu topology likely to be accessible for L2H4, but L1H4 and TPCE likely restricted to the csq, scu, and sqc topologies (e.g., UAM-1000, based on TCPE, adopts the sqc topology).
MOF synthesis and structures
With this information in hand, we investigated the synthesis of MOFs with the two new linkers. Reaction of L1H4 with ZrCl4 using an acetic acid (AcOH) modulator in dimethylformamide (DMF) yielded colourless needle-shaped crystals corresponding to a MOF with a chemical formula of [Zr6(μ3-OH)8(μ2-CH3COO)4(L1)2] (UAM-10) (Fig. S5, ESI†). SCXRD revealed the MOF to have a 3D structure that consists of an 8-connected octahedral Zr6 cluster forming the common cubic SBU (Fig. 3). L1 adopts a rectangular geometry where all four carboxylate groups sit in a plane, as suggested by calculations, connecting four Zr-SBUs. The 3D structure has an scu topology with 17.0 by 15.8 Å rhombic channels that are accessible along the c axis (Fig. 3 and Fig. S6, ESI†). The proximity of the Zr-clusters in the c-axis leads to these being bridged by acetate groups (Fig. S7, ESI†) forming a rod-type MOF (and a different topology when these are considered in the analysis).4 PXRD analysis of a bulk sample of UAM-10 confirmed phase purity (Fig. S8, ESI†). Thermogravimetric analysis (TGA), after activation under vacuum at room temperature, reveals thermal stability up to 350 °C (Fig. S9, ESI†) typical of Zr-MOFs. A 77K N2 adsorption isotherm of UAM-10 after activation shows the material to be porous to nitrogen with a type I isotherm, a Brunauer–Emmett–Teller (BET) surface area of 864 m2 g−1 (Fig. S10, ESI†) and a total pore volume of 0.28 cm3 g−1.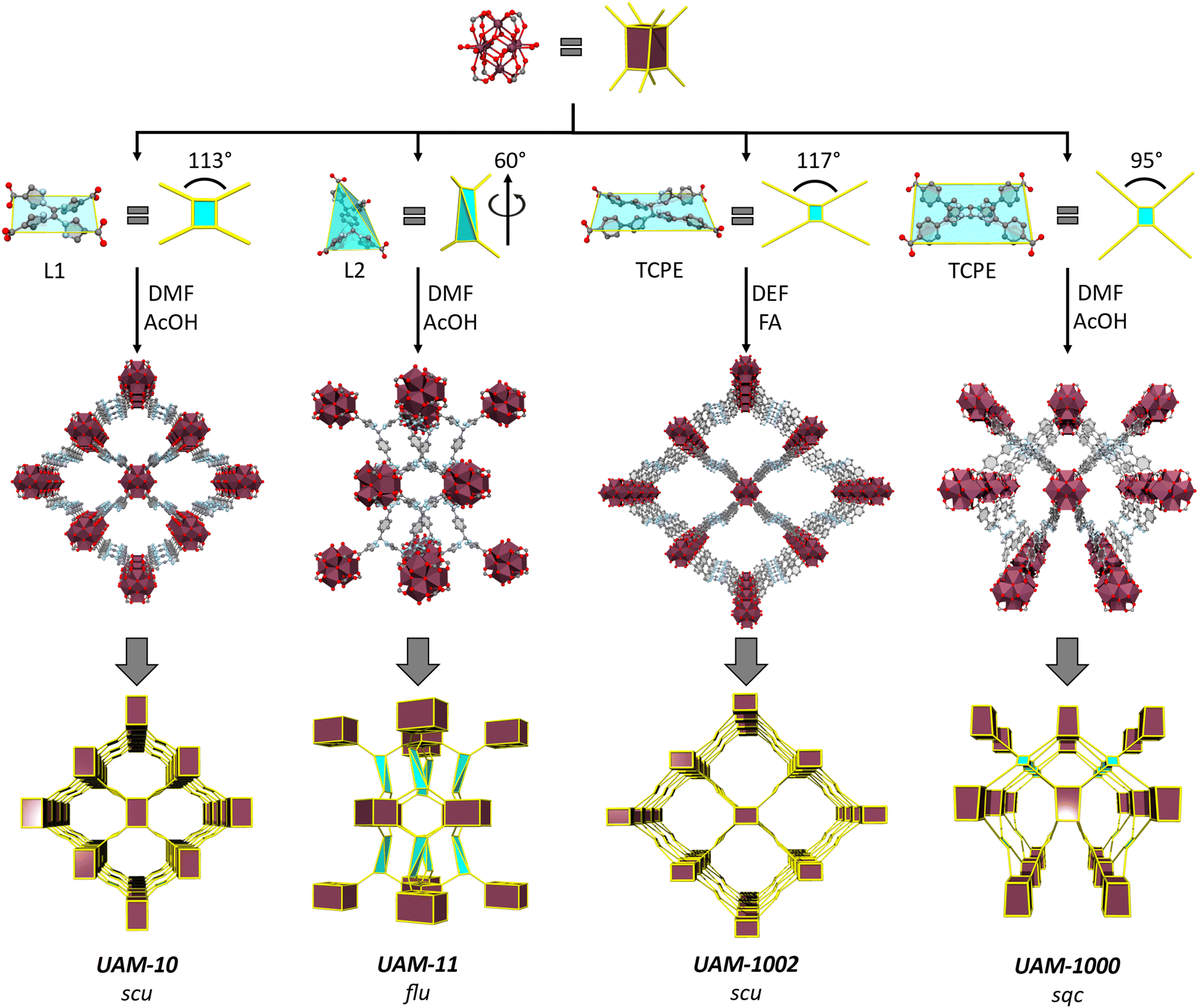 | ||
| Fig. 3 A summary of the synthesis, structure and topologies of UAM-10, UAM-11, UAM-1002 and UAM-1000. All four MOFs are formed from the same 8-connected cubic Zr-cluster but different linkers and linker conformations lead to the formation of different MOF topologies, in part governed by the restrictions on linker flexibility. The maroon polyhedron represents the cluster and the blue shaded rectangle/polyhedron with yellow lines represents the organic linker. | ||
Reaction of L2H4, which possesses the phenyl spacer that leads to decoupling of the bis-pyrazole units, with ZrCl4 and an AcOH modulator in DMF yielded pale yellow crystals corresponding to a MOF with a chemical formula of [Zr6(μ3-OH)8(μ2-CH3COO)4(L2)2] (UAM-11) (Fig. S11, ESI†). UAM-11 also has the cubic 8-connected SBUs with each L2 connecting four Zr SBUs. However, in this case, L2 adopts a tetrahedral geometry to form a flu topology (Fig. 3 and Fig. S12, ESI†) as might be anticipated from the analysis of preferred conformations. Phase purity of the sample was confirmed by PXRD (Fig. S13, ESI†) and TGA also reveals thermal stability up to 350 °C (Fig. S14, ESI†). A 77K N2 adsorption isotherm of UAM-11 after activation shows the material to be porous to nitrogen with a type I isotherm, a BET surface area of 844 m2 g−1 (Fig. S15, ESI†) and a total pore volume of 0.27 cm3 g−1. Despite the ligand length being increased from L1 to L2, the greater flexibility of L2 facilitates access to the more dense flu topology and thereby a similar BET surface area and total pore volume.
The formation of UAM-1000 as the product of the reaction of TCPE with ZrCl4 using trifluoracetic acid (TFA) as modulator in DMF was recently reported by our group.28 The synthesis was reported using TFA as modulator. As the use of different solvents and modulators could lead to different topologies, we reinvestigated the synthesized UAM-1000. Using AcOH as the modulator, UAM-1000AA (acetic acid) was formed and PXRD analysis confirmed the formation of UAM-1000 as the sole product (Fig. S16, ESI†). Briefly, UAM-1000 consists of the cubic Zr-SBU with each linker connecting four Zr-SBUs to form a MOF with sqc topology (Fig. 3, 2nd example reported).8,27 UAM-1000 can also be synthesized in DMF in using benzoic acid (BA) (UAM-1000BA) and TFA (UAM-1000TFA) as confirmed by PXRD (Fig. S16, ESI†). However, a different structure and topology is formed when using both DEF as the solvent and formic acid (FA) as the modulator; using FA in DMF only gives amorphous material. Using the other modulators (AA, BA, and TFA) in DEF also gives amorphous material except in the case of BA which leads to the formation of the sqc topology (UAM-1000). Reaction of TCPE with ZrCl4 in the presence of DEF using FA as modulator provides UAM-1002, which forms as large colourless needle-shaped crystals that could be characterized by SCXRD (Fig. S17, ESI†). Structure solution and refinement by SCXRD revealed the formation of a MOF with an scu topology, like UAM-10, but where the Zr clusters are not bridged by the carboxylate group of the modulator (Fig. 3 and Fig. S18, ESI†).
PXRD analysis of bulk UAM-1002 confirmed phase purity of the samples (Fig. S19, ESI†). Thermogravimetric analysis (TGA) analysis, after activation under vacuum at room temperature, reveals thermal stability up to 475 °C (Fig. S20, ESI†). A 77K N2 adsorption isotherm of UAM-1002 after thermal activation shows a BET surface area of 138 m2g−1 (Fig. S21, ESI†) and a total pore volume of 0.04 cm3 g−1. These values are considerably lower than expected based on calculations conducted on the crystal structure (3400 m2 g−1 and 1.14 cm3 g−1). Activation under supercritical CO2 showed a slight increase in porosity with a BET of 567 m2 g−1 (Fig. S21, ESI†) and a total pore volume of 0.18 cm3 g−1. However, this remained much lower than the calculated values. We hypothesize that the larger pore volume and anticipated flexibility of UAM-1002 is allowing partial collapse of the structure upon activation. PXRD data (Fig. S22, ESI†) for activated UAM-1002 (UAM-1002·activated) shows peaks associated with hkl planes (0,2,0), (0,0,1) and (2,0,0) shift compared to those in as-synthesised UAM-1002. We could not determine the activated structure by SCXRD but the PXRD changes, and the loss of accessible pore volume, is consistent with a transition to a closed phase, as is observed with other similar MOFs (e.g. MIL-53 type MOFs, CuMOF-1).56–58
Topologies of UAM-10, UAM-11, UAM-1002 and UAM-1000
Zr clusters with 4, 5, 6, 8, 10, and 12-connected SBUs have been reported, but the 8 and 12-connected examples are the most common. Thus, we will focus on possible topologies with the 8-connected Zr-SBU (cubic) and the 12-connected SBU (cuboctahedra), and considering that all linkers used are tetratopic linkers, only six topologies are possible. Among the six topologies, ftw is only accessed with a planar linker and a cuboctahedral SBU, ith is the only combination for a tetrahedral linker and a cuboctahedral SBU, and flu is the only possibility of a tetrahedral linker and the cubic SBU. Finally, three possible topologies can be formed with a cubic SBU and a planar linker: csq, scu, and the less common sqc.To understand the linker preferences for different topologies we produced hypothetical models for each framework. We used single conformation of each ligand (based on the lowest energy conformer discovered above) and arranged these to produce likely framework structures for each of the six possible topologies. There are an immense number of combinations of inorganic node and ligand orientations59 but here only a single representative framework was produced to provide a reasonable model for each topology and ligand. We note, the production of framework models of complex topologies, where there a many possible linker orientations and conformations, are challenging and require exhaustive search approaches.35 In this study a single arrangement of the ligands with a single conformation were considered. As a result, these hypothetical frameworks serve as a simple guide of the energy landscape for these topologies.
The ftw topology is edge-transitive and, as noted, based on a cuboctahedral SBU and a square linker. It is the most common topology, when possible, as the Zr6 cluster is fully saturated with the maximum number of carboxylates it can accommodate. However, studies have shown that for this to be possible, the torsion angle between the carboxylates with the backbone (core) of the linker needs to be close to 0°.19 As the linkers used in this work have two sp3 carbons, the pyrazole and phenyl rings are out of the plane formed by the carbon of the carboxylic groups. As the carboxylates are conjugated with the aromatic rings they are attached to, they are going to be out of that plane too, making the ftw topology very unlikely. This is evidenced by analysis of the MOF structures reported in this work; none of the structures have the carboxylates in plane with the core of the linker, and thus ruling out the ftw topology (Fig. S23, ESI†). The hypothetical structure for the ligands arranged in the ftw topology also shows significant strain based on observation of the framework structure. This strain may be partially offset by the fully saturated cluster, but this does not represent a likely framework product. If the linkers adopted a tetrahedral geometry, ith could also be possible. However, this topology is rare, as it is challenging to form a cuboctahedra SBU with a tetrahedral linker; the single example reported was obtained as a byproduct and could not be obtained phase pure.60
Since the ftw and ith topologies are unlikely products, we turned our attention to evaluating the topologies that can be formed with a tetratopic linker and the 8-connected (cubic) Zr-SBU. The combination of a tetratopic linker and a cubic SBU can lead to four different topologies: csq, scu, sqc and flu. The energetics of the framework formation for all different linkers in the possible topologies suggest the csq topology is lowest in energy for L1 and TCPE and the scu topology for L2 (Table S2, ESI†). However, there does not appear to be a large difference in the relative energies for each of the topologies and this suggests all are likely for the three linkers. Notably we find the spread of energies becomes significantly lower in TCPE (∼0.005 eV per atom) compared to L1 (∼0.1 eV per atom) demonstrating that larger more flexible ligands can provide access to a greater range of topologies than smaller, more rigid, ligands.
As noted, all three linkers contain two sp3 carbons, and they can adopt either a tetrahedral or square planar conformation. In the case where the linker adopts a tetrahedral conformation, only one topology is expected: flu. This is observed in UAM-11 due to the conformational flexibility of the central phenyl group, allowing L2 to adopt a “tetrahedral-like” conformation with a torsion angle of 60° between all four carboxylates forming a distorted flu topology. It is not clear why L2 prefers to adopt a tetrahedral conformation over a square planar conformation, but one hypothesis is that the small energy difference between the different HCCH dihedral angles observed in the study of the free ligand means that it does not favour any specific angle within the MOF that is formed. Consequently, we hypothesize that flu could be favoured because, out of the four topologies considered, flu is the only topology that does not form channels, and rather it forms a more rigid and stable structure with large pores accessible only through smaller pore apertures. While other topologies for L2 based MOFs might be accessible based on the simulation data, attempts to induce the formation these by altering the synthetic conditions were unsuccessful, resulting in the production of either amorphous powders or the same flu topology.
When the linker adopts a square/rectangular planar geometry, instead of a tetrahedral geometry, this can lead to the formation of three possible topologies: csq, scu, and sqc, as documented in literature.8 Normally, the csq topology is more commonly observed compared to scu and sqc. Surprisingly, none of the MOFs formed in our study had the csq topology despite this being predicted to be energetically possible (further discussion below); however, our simulation approach did not consider the possibility of coordinating molecules that may act to bridge proximal Zr nodes which is observed in the scu topology, particularly for L1 (Fig. S7, ESI†).
Ligand L1 is observed to form UAM-10, where the framework adopts an scu topology, while TCPE provides MOFs UAM-1000 and UAM-1002, which adopt sqc and scu topologies, respectively. Analysis of structures shows that the adoption of an anti-conformation and thereby a more rectangular linker results in an scu topology, whereas a syn conformation (and more square linker) leads to the sqc topology. This small difference in linker conformation looks to be important, as the scu topology normally forms with rectangular ligands while for the sqc topology appears to prefer a more square arrangement, albeit the latter is based on only two reported examples. Comparing UAM-1000 and UAM-1002 shows that the orientation of the metal clusters and the carboxylates of the ligand are the same in both UAM-1002 and UAM-1000 with the only difference being that in UAM-1000 the ligand has squarer dimensions and in UAM-1002 more rectangular (Fig. 4). Simulations of hypothetical frameworks based on different ligand conformations of L1 and TCPE support these observations (Table S3, ESI†).
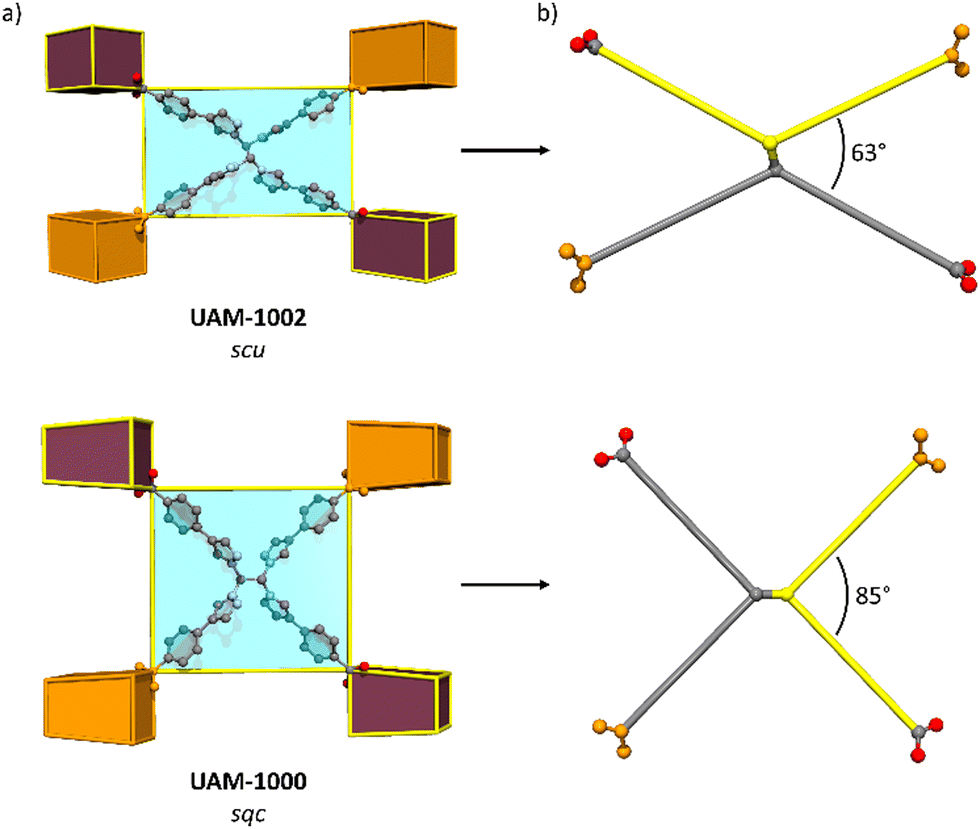 | ||
| Fig. 4 (a) Representations of the scu (top) and sqc (bottom) MOF topologies showing the linker orientation and disposition that allows it to be connected to four cubic clusters that are in the same orientation. (b) Schematic representation of the linkers highlighting in the same colour the two arms that are attached to the same carbon of the central ethylene group. | ||
Finally, as noted, we have observed that the csq topology (e.g., NU-1000 type materials) cannot be obtained with any of the linkers studied. In general, the csq topology tends to be favoured more with square linkers, while scu topology is favoured with more rectangular linkers, which is consistent with our observations in UAM-10 and UAM-1002. We hypothesize that while the linkers L1 and TCPE can adopt a squarer arrangement (seen for the latter in UAM-1000) they may be too flexible to form a csq topology with such large pores, consistent with its absence in this work.
Post-synthetic metalation of the MOFs
To evaluate the ability of the bis-pyrazole units in UAM-10, UAM-11, UAM-1000, and UAM-1002 undergo PSMet, we examined reactions conducted with a relatively inert metal (minimal leaching) that could induce an easily detectable colour change in the crystals. PdCl2 was chosen for this purpose. Metalation of UAM-1000 with PdCl2 was recently investigated by us.28 Partial metalation (ca. 50% PSMet) was achievable at the bis-pyrazole units due to the proximity of the adjacent bis-pyrazole unit, which hindered full metalation for steric reasons. The flexibility of the linker TCPE also posed challenges for determining the structure by SCXRD, but this could be overcome by incorporating a secondary linker to create UAM-1001, where the material was effectively locked.Considering that UAM-10 and UAM-11 have shorter ligands compared to UAM-1000, we anticipated that these compounds would be easier to metalate in a single crystal-to-single crystal manner and characterise the outcome by SCXRD. However, attempts to PSMet UAM-10 and UAM-11 with PdCl2 was unsuccessful. SEM-EDX analysis revealed some Pd present (Table S4, ESI†); however, the metalation was inconsistent across the sample and between samples, and SCXRD analysis revealed the pyrazoles to still be in an anti-conformation. Given that UAM-10 and UAM-11 have linkers with the pyrazoles directly attached to carboxylate groups and exhibit an anti-conformation of the bis-pyrazole units in the as-synthesised form of the MOF, these results suggest there is a significant barrier to convert from anti to syn within the MOF. This is likely due to this anti to syn change requiring a significant geometric and structure metric change in the linker that cannot be tolerated by the MOF topology.
In contrast, heating crystals of UAM-1002 in an acetonitrile solution of PdCl2 resulted in a colour change of the crystals to the typical orange colour of bis-pyrazole coordinated Pd(II) (Fig. S25, ESI†). SEM-EDX analysis confirmed the presence of Pd(II) in the MOF, with a Zr:Pd ratio of 3:2 for UAM-1002 (Table S4, ESI†), consistent with full metalation of the bis-pyrazole sites. This is a notable contrast to UAM-1000, which due to the short 5.3 Å separation between two the two bis-pyrazole groups, only allows 50% metalation with PdCl2 under similar conditions. As UAM-1002 has shown significant structural flexibility (PXRD and gas adsorption data) we also expected the crystals to diffract poorly. To our surprise, the toluene-solvated crystals (formed by solvent exchange) diffracted very well and SCXRD analysis of a crystal of UAM-1002[PdCl2] confirmed that the Pd(II) metal centre was bound to the bis-pyrazole units in a typical square planar geometry, with two chlorides completing the coordination sphere (Fig. 5). Interestingly, compared to as-synthesised UAM-1002, the Zr-clusters where now significantly closer in the c-axis and bridged by formate anions (Fig. 5(a)) transforming its structure from a dot-type to a rod-type of MOF (with a different topology to the parent structure).4 We hypothesize that when chelating the Pd(II) centre, the pyrazoles switch from anti to syn (facilitated by the phenyl spacer in the ligand arms decoupling this motion from the carboxylates, compare with UAM-10 and UAM-11) and the overall ligand changes its donor disposition to a more anisotropic shape, which forces the Zr nodes closer. The proximity of the nodes allow the formate capping ligands to change from chelating the Zr-clusters to bridging them, in turn providing more rigidity to the structure and allowing a better quality diffraction.
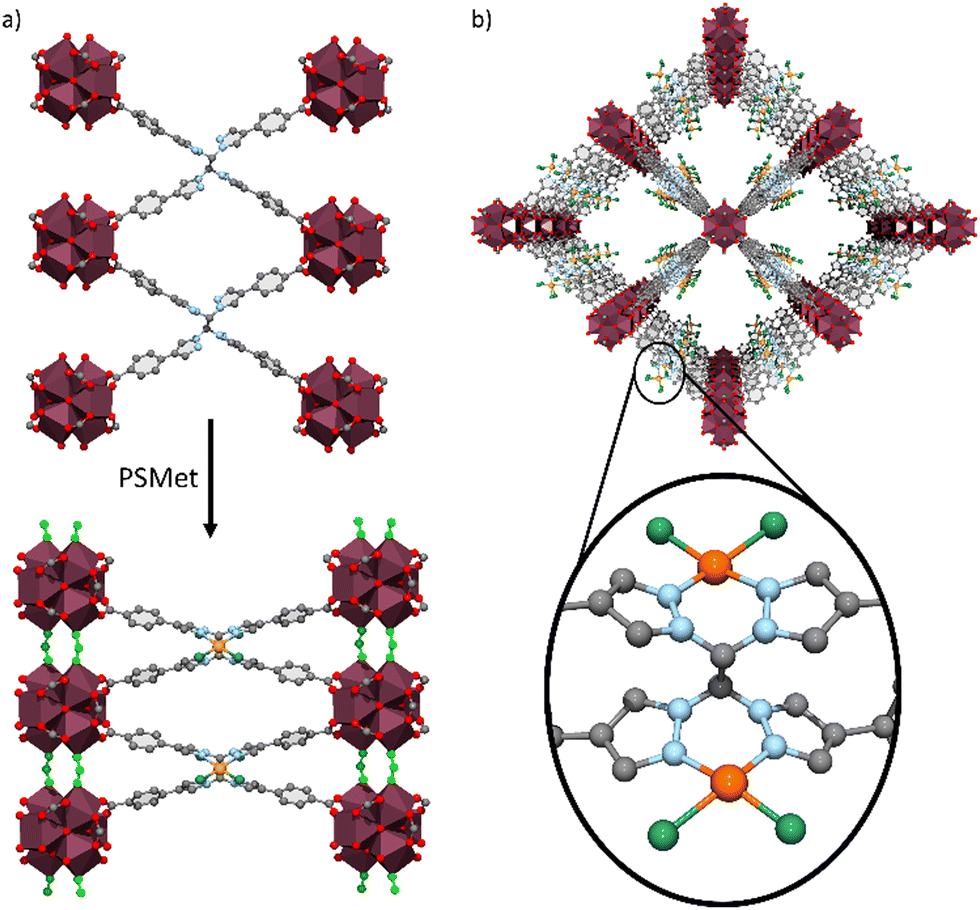 | ||
| Fig. 5 (a) Representations of ligands bound to the Zr clusters in UAM-1002 (top) and UAM-1002[PdCl2] (bottom). The transition from anti-(top) to syn-conformation (bottom) brings the Zr-clusters close enough that they end up being bridged by formate anions (represented in light green). (b) Representation of the structure of UAM-1002[PdCl2] along the c axis, with the enlargement showing the PdCl2 complex bound to the bis-pyrazole groups with a square planar geometry. | ||
Conclusions
In summary, we have reported a study into the formation of Zr-MOFs formed from flexible tetratopic pyrazole carboxylate linkers. Two new linkers have been reported (L1H4 and L2H4) and the formation of three new Zr-MOFs (UAM-10, UAM-11 and UAM-1002) observed. The combination of our experimental observations and simulations reveals that when using flexible pyrazole carboxylate linkers in the synthesis of Zr-MOFs the overall linker length and width (phenyl spacers within the arms) affects the accessible conformations of the linker, thereby limiting or allowing the synthesis of particular MOF topologies. Narrower linkers like L1H4 are more conformationally restricted, allowing access to a single topology while wider linkers like TCPE allow synthesis of different topologies by fine tuning of the synthetic conditions. For example, the reaction of TCPE and ZrCl4 in DMF and TFA form an sqc topology while the reaction in DEF and formic acid forms the scu topology. Interestingly, when TCPE adopts a syn-conformation the sqc topology appears to be preferred and when it adopts an anti-conformation it forms the scu topology. Interestingly, this is the first time that the sqc topology was observed to be the dominant phase among 8,4 connected topologies, whereas typically the csq topology is encountered. L2H4 is somewhat unique in that it has significant conformational freedom yet still forms only one MOF structure preferentially. We hypothesize the formation of the uncommon sqc topology, rather than the scu and csq, is favoured for flexible tetracarboxylate linkers that have a square arrangement of the donors. Alternative topologies, such as csq, may represent unstable frameworks considering the size of the channels in this topology.The length and width of the linker not only affects the topologies that can be formed but also the ability of the bis-pyrazolyl groups to transition from an anti- to a syn-conformation. Shorter and rigid linkers disfavour the transition preventing PSMet, as observed with UAM-10 and UAM-11. Wider linkers like TCPE allow easier transition from an anti to a syn-conformation as the carboxylate and pyrazole core of the ligand is decoupled, allowing PSMet. Consequently, UAM-1002 possesses the right flexibility to allow structural changes upon PSMet and the elucidation of the inserted organometallic complex. These properties, combined with large unidirectional channels make UAM-1002 a great platform that could be exploited as a crystalline sponge for the study of the chemistry that occurs at the anchored organometallic complexes.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. J. S. and C. J. D. gratefully acknowledge the Australian Research Council for funding (DP190101402 and DP220101774). JDE is the recipient of an Australian Research Council Discovery Early Career Award (project number DE220100163) funded by the Australian Government. Phoenix HPC service at the University of Adelaide is thanked for providing high-performance computing resources. This project was undertaken with the assistance of resources and services from the National Computational Infrastructure (NCI), which is supported by the Australian Government. This research was undertaken in part using the MX1 and MX2 beamlines at the Australian Synchrotron, part of ANSTO, and made use of the Australian Cancer Research Foundation (ACRF) detector. P. G.-F. gratefully acknowledges a University of Adelaide International Scholarship and an Australian Institute of Nuclear Science and Engineering Postgraduate Scholarship.Notes and references
- P. M. Bhatt, V. Guillerm, S. J. Datta, A. Shkurenko and M. Eddaoudi, Chem, 2020, 6, 1613–1633 CAS.
- P. Deria, D. A. Gómez-Gualdrón, I. Hod, R. Q. Snurr, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2016, 138, 14449–14457 CrossRef CAS PubMed.
- Z.-J. Li, Y. Ju, X.-L. Wu, X. Li, J. Qiu, Y. Li, Z.-H. Zhang, M.-Y. He, L. Zhang, J.-Q. Wang and J. Lin, Inorg. Chem. Front., 2023, 10, 1721–1730 RSC.
- F. M. Amombo Noa, M. Abrahamsson, E. Ahlberg, O. Cheung, C. R. Göb, C. J. McKenzie and L. Öhrström, Chem, 2021, 7, 2491–2512 CAS.
- I. Kurzydym and I. Czekaj, Tech. Trans., 2020, 117, 1–24 Search PubMed.
- M. C. Wasson, C. T. Buru, Z. J. Chen, T. Islamoglu and O. K. Farha, Appl. Catal., A, 2019, 586, 117214 CrossRef CAS.
- Z. Chen, K. O. Kirlikovali, P. Li and O. K. Farha, Acc. Chem. Res., 2022, 55, 579–591 CrossRef CAS PubMed.
- H. Jiang, D. Alezi and M. Eddaoudi, Nat. Rev. Mater., 2021, 6, 466–487 CrossRef CAS.
- O. M. Yaghi, M. O’Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- Z. Chen, J. Weseliński, K. Adil, Y. Belmabkhout, A. Shkurenko, H. Jiang, P. M. Bhatt, V. Guillerm, E. Dauzon, D.-X. Xue, M. O’Keeffe and M. Eddaoudi, J. Am. Chem. Soc., 2017, 139, 3265–3274 CrossRef CAS PubMed.
- Z. Chen, S. L. Hanna, L. R. Redfern, D. Alezi, T. Islamoglu and O. K. Farha, Coord. Chem. Rev., 2019, 386, 32–49 CrossRef CAS.
- M. J. Kalmutzki, N. Hanikel and O. M. Yaghi, Sci. Adv., 2018, 4, eaat9180 CrossRef CAS PubMed.
- D. Sun, S. Ma, J. M. Simmons, J.-R. Li, D. Yuan and H.-C. Zhou, Chem. Commun., 2010, 46, 1329–1331 RSC.
- E. Biemmi, S. Christian, N. Stock and T. Bein, Microporous Mesoporous Mater., 2009, 117, 111–117 CrossRef CAS.
- G.-Y. Qiao, S. Yuan, J. Pang, H. Rao, C. T. Lollar, D. Dang, J.-S. Qin, H.-C. Zhou and J. Yu, Angew. Chem., Int. Ed., 2020, 59, 18224–18228 CrossRef CAS PubMed.
- D. Feng, Z. Y. Gu, J. R. Li, H. L. Jiang, Z. Wei and H. C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 10307–10310 CrossRef CAS PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- C. Koschnick, M. W. Terban, R. Frison, M. Etter, F. A. Böhm, D. M. Proserpio, S. Krause, R. E. Dinnebier, S. Canossa and B. V. Lotsch, J. Am. Chem. Soc., 2023, 145, 10051–10060 CrossRef CAS PubMed.
- R. Luebke, Y. Belmabkhout, Ł. J. Weseliński, A. J. Cairns, M. Alkordi, G. Norton, Ł. Wojtas, K. Adil and M. Eddaoudi, Chem. Sci., 2015, 6, 4095–4102 RSC.
- J. Ma, L. D. Tran and A. J. Matzger, Cryst. Growth Des., 2016, 16, 4148–4153 CrossRef CAS.
- J. Pang, S. Yuan, J. Qin, C. Liu, C. Lollar, M. Wu, D. Yuan, H.-C. Zhou and M. Hong, J. Am. Chem. Soc., 2017, 139, 16939–16945 CrossRef CAS PubMed.
- J. Lyu, X. Zhang, K.-I. Otake, X. Wang, P. Li, Z. Li, Z. Chen, Y. Zhang, M. C. Wasson, Y. Yang, P. Bai, X. Guo, T. Islamoglu and O. K. Farha, Chem. Sci., 2019, 10, 1186–1192 RSC.
- Y. Chen, X. Zhang, M. R. Mian, F. A. Son, K. Zhang, R. Cao, Z. Chen, S.-J. Lee, K. B. Idrees, T. A. Goetjen, J. Lyu, P. Li, Q. Xia, Z. Li, J. T. Hupp, T. Islamoglu, A. Napolitano, G. W. Peterson and O. K. Farha, J. Am. Chem. Soc., 2020, 142, 21428–21438 CrossRef CAS PubMed.
- H.-L. Xia, K. Zhou, L. Yu, H. Wang, X.-Y. Liu, D. M. Proserpio and J. Li, Inorg. Chem., 2022, 61, 7980–7988 CrossRef CAS PubMed.
- T. Islamoglu, K.-I. Otake, P. Li, C. T. Buru, A. W. Peters, I. Akpinar, S. J. Garibay and O. K. Farha, CrystEngComm, 2018, 20, 5913–5918 RSC.
- H.-Q. Xu, K. Wang, M. Ding, D. Feng, H.-L. Jiang and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 5316–5320 CrossRef CAS PubMed.
- H.-L. Jiang, D. Feng, K. Wang, Z.-Y. Gu, Z. Wei, Y.-P. Chen and H.-C. Zhou, J. Am. Chem. Soc., 2013, 135, 13934–13938 CrossRef CAS PubMed.
- P. Gimeno-Fonquernie, J. Albalad, J. D. Evans, J. R. Price, C. J. Doonan and C. J. Sumby, Submitted 2023.
- C. J. Coghlan, C. J. Sumby and C. J. Doonan, CrystEngComm, 2014, 16, 6364–6371 RSC.
- E. A. Nudnova, A. S. Potapov, A. I. Khlebnikov and V. D. Ogorodnikov, Russ. J. Org. Chem., 2007, 43, 1698–1702 CrossRef CAS.
- H. Cao, Z. Liu and Q. Wang, Polym. Chem., 2017, 8, 3810–3814 RSC.
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- S. Ehlert, M. Stahn, S. Spicher and S. Grimme, J. Chem. Theory Comput., 2021, 17, 4250–4261 CrossRef CAS PubMed.
- J. Keupp and R. Schmid, Faraday Discuss., 2018, 211, 79–101 RSC.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef CAS.
- P. G. Boyd, S. M. Moosavi, M. Witman and B. Smit, J. Phys. Chem. Lett., 2017, 8, 357–363 CrossRef CAS PubMed.
- A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown, P. S. Crozier, P. J. in ’t Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott and S. J. Plimpton, Comput. Phys. Commun., 2022, 271, 108171 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703–1710 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, Chem. Phys., 2010, 132, 154104 Search PubMed.
- J. Vandevondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- T. D. Kühne, M. Iannuzzi, M. Del Ben, V. V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schütt, F. Schiffmann, D. Golze, J. Wilhelm, S. Chulkov, M. H. Bani-Hashemian, V. Weber, U. Borštnik, M. Taillefumier, A. S. Jakobovits, A. Lazzaro, H. Pabst, T. Müller, R. Schade, M. Guidon, S. Andermatt, N. Holmberg, G. K. Schenter, A. Hehn, A. Bussy, F. Belleflamme, G. Tabacchi, A. Glöβ, M. Lass, I. Bethune, C. J. Mundy, C. Plessl, M. Watkins, J. Vandevondele, M. Krack and J. Hutter, Chem. Phys., 2020, 152, 194103 Search PubMed.
- T. F. Willems, C. Rycroft, M. Kazi, J. C. Meza and M. Haranczyk, Microporous Mesoporous Mater., 2012, 149, 134–141 CrossRef CAS.
- T. M. McPhillips, S. E. McPhillips, H. J. Chiu, A. E. Cohen, A. M. Deacon, P. J. Ellis, E. Garman, A. Gonzalez, N. K. Sauter, R. P. Phizackerley, S. M. Soltis and P. Kuhn, J. Synchrotron Radiat., 2002, 9, 401–406 CrossRef CAS PubMed.
- N. P. Cowieson, D. Aragao, M. Clift, D. J. Ericsson, C. Gee, S. J. Harrop, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Williamson and T. Caradoc-Davies, J. Synchrotron Radiat., 2015, 22, 187–190 CrossRef CAS PubMed.
- D. Aragao, J. Aishima, H. Cherukuvada, R. Clarken, M. Clift, N. P. Cowieson, D. J. Ericsson, C. L. Gee, S. Macedo, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Rostan, R. Williamson and T. T. Caradoc-Davies, J. Synchrotron Radiat., 2018, 25, 885–891 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr. Sect., A: Found. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr. Sect., A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr. Sect., C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- L. J. Barbour, Supramol. Chem., 2001, 1, 189–191 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 9–18 CrossRef CAS PubMed.
- K. Dr. H. Putz, Dr. K. Brandenburg GbR, Diamond – Crystal and Molecular Structure Visualization – Crystal Impact, 53227, Bonn, Germany, 1999, 102.
- R. A. Peralta, M. T. Huxley, R. J. Young, O. M. Linder-Patton, J. D. Evans, C. J. Doonan and C. J. Sumby, Faraday Discuss., 2021, 225, 84–99 RSC.
- Y. Liu, J. H. Her, A. Dailly, A. J. Ramirez-Cuesta, D. A. Neumann and C. M. Brown, J. Am. Chem. Soc., 2008, 130, 11813–11818 CrossRef CAS PubMed.
- W. M. Bloch, C. J. Doonan and C. J. Sumby, CrystEngComm, 2013, 15, 9663 RSC.
- L. Chen, J. P. S. Mowat, D. Fairen-Jimenez, C. A. Morrison, S. P. Thompson, P. A. Wright and T. Düren, J. Am. Chem. Soc., 2013, 135, 15763–15773 CrossRef CAS PubMed.
- S. Amirjalayer and R. Schmid, J. Phys. Chem. C, 2008, 112, 14980–14987 CrossRef CAS.
- H. Furukawa, F. Gándara, Y.-B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H-and 13C NMR spectra, IR spectra, SEM-EDX data, PXRD data, adsorption isotherms for all compounds. Additional experimental details, materials, and methods. CCDC 2288076–2288079. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3tc03606e |
| ‡ Present address: Catalan Institute of Nanoscience and Nanotechnology (ICN2)CSIC and The Barcelona Institute of Science and Technology Campus UAB, Bellaterra, Barcelona 08193, Spain. |
| This journal is © The Royal Society of Chemistry 2024 |