
Self-assembly of peptides in living cells for disease theranostics
Xiaowei
Mo
a,
Zeyu
Zhang
a,
Jinyan
Song
a,
Yushi
Wang
a and
Zhilin
Yu
 *ab
*ab
aKey Laboratory of Functional Polymer Materials, Ministry of Education, State Key Laboratory of Medicinal Chemical Biology, Institute of Polymer Chemistry, College of Chemistry, Nankai University, 94 Weijin Road, Tianjin 300071, China. E-mail: yzh026@nankai.edu.cn
bHaihe Laboratory of Synthetic Biology, 21 West 15th Avenue, Tianjin 300308, China
First published on 27th March 2024
Abstract
The past few decades have witnessed substantial progress in biomedical materials for addressing health concerns and improving disease therapeutic and diagnostic efficacy. Conventional biomedical materials are typically created through an ex vivo approach and are usually utilized under physiological environments via transfer from preparative media. This transfer potentially gives rise to challenges for the efficient preservation of the bioactivity and implementation of theranostic goals on site. To overcome these issues, the in situ synthesis of biomedical materials on site has attracted great attention in the past few years. Peptides, which exhibit remarkable biocompability and reliable noncovalent interactions, can be tailored via tunable assembly to precisely create biomedical materials. In this review, we summarize the progress in the self-assembly of peptides in living cells for disease diagnosis and therapy. After a brief introduction to the basic design principles of peptide assembly systems in living cells, the applications of peptide assemblies for bioimaging and disease treatment are highlighted. The challenges in the field of peptide self-assembly in living cells and the prospects for novel peptide assembly systems towards next-generation biomaterials are also discussed, which will hopefully help elucidate the great potential of peptide assembly in living cells for future healthcare applications.
 Xiaowei Mo | Xiaowei Mo completed her BSc degree from the College of Chemistry, Nankai University in 2021. Currently, she is pursuing a Master's degree at the Institute of Polymer Chemistry under the supervision of Prof. Zhilin Yu at Nankai University. Her current research is focused on the development of new strategies for the controlled assembly of peptides and the precise construction of peptide nanostructures. |
 Zeyu Zhang | Zeyu Zhang received his MSc degree from Guangxi University in 2022. He is currently a PhD candidate at the Institute of Polymer Chemistry of Nankai University under the supervision of Prof. Zhilin Yu. His research interest is focused on the stimulus-responsive self-assembly of peptides in living cells for disease diagnosis and therapy. |
 Jinyan Song | Jinyan Song received her BSc degree in Polymer Materials and Engineering from Hebei University. Currently, she is pursuing a Master's degree at the Institute of Polymer Chemistry under the supervision of Prof. Zhilin Yu at Nankai University. Her research interests focus on the development of new strategies for the controlled assembly of peptides and the precise creation of peptide nanostructures. |
 Yushi Wang | Yushi Wang received her BSc degree in Materials Science and Engineering from Jilin University, and her MSc degree in Biomedical Engineering from South China University of Technology. Currently, she is perusing a PhD degree in Chemistry at Nankai University, under the supervision of Professor Zhilin Yu. Her research interest is focused on regulating peptide assembly through noncanonical amino acids. |
 Zhilin Yu | Zhilin Yu, a professor at Nankai University, studied chemistry at Tianjin University and was awarded his PhD degree under the supervision of Prof. Stefan Hecht at the Humboldt University of Berlin in Germany in 2013. He conducted his postdoctoral training with Prof. Samuel I. Stupp at Northwestern University. He started his independent career as a principle investigator at Nankai University in 2017. His current research interests focus on the controllable self-assembly of peptides in living systems and creation of dynamic biomaterials, particularly materials with great potential in disease diagnosis and therapy. |
1. Introduction
Biomedical materials serve as the basic platform for most healthcare, ranging from diagnosis to rehabilitation and treatment. Lying at the intersection of biology, chemistry, and materials science, biomedical materials can implement different healthcare processes, primarily including drug delivery, biosensors, and tissue engineering.1,2 Over the past decades, biomaterials have been classified based on their components into different types, such as metals, ceramics, polymers, and hybrids.3,4 In particular, fabricating biomaterials using bottom-up approaches opens a new avenue to improve their performance in healthcare and revolutionize the approaches for improving human health.Conventionally, biomaterials utilized in healthcare are created in an ex vivo manner. In typical processes, guided by healthcare goals, the materials are formulated from individual components with corresponding functions. The bioactivity of the resulting materials is evaluated through in vitro and in vivo studies. Despite the enormous progress achieved in this field, conventional biomaterials prepared under ex vivo conditions still face challenges arising from their preparation under simplified solution environments, whereby their utilization via transfer to the physiological environment can lead to biosafety issues associated with off-targeting or a loss of bioactivity. Therefore, currently available biomaterials consequently exhibit poor treatment outcomes and limited selection between healthy and pathological regions.5,6 To control bioactivity, stimulus-responsive biomedical materials have been developed as second-generation counterparts.7–9 Employing either external or internal stimuli, the bioactivity of stimulus-responsive biomaterials can be precisely regulated at pathological sites. However, preformed biomaterials still suffer from problems in blood circulation and site accumulation. As an extension of stimulus-responsive categories, third-generation biomaterials can be created on site, termed as in situ-formed biomaterials.10,11 Generally, stimulus-responsive materials undergo various responses upon exposure to stimuli, broadly ranging from structural changes to functional implementation. In situ-formed materials are usually created through stimulus-responsive processes. Therefore, in situ-formed materials are, in principle, one category of stimulus-responsive materials. However, the definition of in situ-formed materials emphasizes the formulation on targeting sites and allows for a precise clarification of stimulus-responsive processes. In addition, compared to traditional strategies, biomedical materials prepared in living environments obviously allows for satisfying the demand for healthcare with real-time characterization and detection under physiological conditions.12,13 Accuracy and the precision activation of bioactivity at specific sites could thereby alleviate the possible biosafety issues of such materials.14–16
In situ biomaterials are strongly dependent on stimulus-responsive reactions to regulate their bioactivities. In principle, in situ biomedical materials are typically involve stimulus-formation and stimulus-responsive activation aspects. Stimulus-responsive formation refers to the creation of a biomaterial's individual components, thereby generating the bioactivity.17–21 Stimulus-responsive activation involves the removal of caged functional groups and self-immolative moieties. Considering the advantages of self-assembly for generating nanostructures, bottom-up fabrication has great potential for the creation of in situ biomaterials in living systems.
Peptides consisting of amino acids exhibit great biocompatibility and reliable noncovalent interactions.22,23 The self-assembly of peptides into well-defined nanostructures therefore has been considered as one of the versatile strategies for creating biomedical materials.24–29 Due to their great biocompatibility and reliable noncovalent interactions, peptides have been widely utilized as components for the development of biomedical materials. The resulting peptide-based materials have been broadly utilized in biomedical engineering, ranging from bioimaging, drug delivery, cell culture, and tissue regeneration, to antibacterial therapy.30–34 In addition, expanding knowledge about the peptide assembly mechanism has promoted the development of a considerable number of strategies to manipulate the self-assembly of peptides under various conditions.35–42 While either external or internal stimuli could be employed as the sources to control peptide assembly, stimulus-responsive reactions upon amino acid sequences or individual residues enrich the approaches to efficiently modulate the assembly driving forces for peptides.43,44 Hence, the formation of assembled peptide materials with distinct structural features and bioactivities in living conditions becomes possible. The versatility of peptide-based biomedical materials leads to a vast space for the development of next-generation biomaterials and precise medication.
In situ peptide biomaterials refer to biomaterials formed at targeted sites caused by appropriate stimuli. Only considering the biomaterials utilized for body healthcare, the targeting sites could be defined under different aspects. At the macroscale level, the targeting sites could be on the skin or inside the body; while at the microscale level, the targeting sites could be classified into the surface or interior of tissues; and at the cellular level, the in situ biomaterials could be extracellularly or intracellularly formed. However, peptide-based materials in living cells usually originate from either the internalization of ex vivo-assembled biomaterials or in situ assembly. On the basis of these considerations, in situ peptide biomaterials and peptide-based materials assembled in living cells are different, while exhibiting some overlap between each other. Analogous to ex vivo formed materials, combining an in situ assembling propensity with antimicrobial activity can lead to the in situ formulation of peptide biomaterials with great potential in various areas, such as antibacterial agents.
This review presents an overview of the progress in the self-assembly of peptides in living cells toward biomedical agents that have been achieved within the past few years. Starting with a brief introduction of biomedical materials, we introduce the design principles for peptide assembly in living cells with respect to the stimulus sources, bioactivity generation, and assembly regulation. Afterwards, the corresponding applications of peptide-based biomaterials formed in living environments, predominately including bioimaging, disease diagnosis, and treatment, will be highlighted. Eventually, the challenges faced in the self-assembly of peptides in living cells and the prospects of novel peptide assembly systems toward next-generation biomaterials will be also discussed, thereby helping elucidate their great potential for healthcare in the future.
2. Self-assembly of peptides in living cells
The self-assembly of peptides could be manipulated by external and internal stimuli,45 precisely forming biomedical materials.46 Thus far, various stimulus-responsive strategies, including protonation, cleavage, cross-linking, redox, and isomerization promoted by enzymes, photons, acids, ROS, and glutathione (GSH), have been developed to establish peptide controllable assembly systems.47–49 These responsive reactions provide various options for the formulation of biomedical materials under living conditions.50–52 The in situ formation of peptide-based biomaterials is primarily based on stimulus-assembly (including the formation of less-ordered aggregates) and stimulus-responsive morphological transformation (Scheme 1). | ||
| Scheme 1 Schematic illustration of the self-assembly of peptides in living cells toward biomedical materials with an emphasis on the design principles and applications in bioimaging and disease therapy. | ||
The responsive assembling systems undergo stimulus-responsive assembly prompted by precursor monomers, in which the precursors are converted to assembling monomers and subsequently assemble into bioactive assemblies for disease theranostics. On the contrary, morphological transformation systems are based on biomaterials undergoing a stimulus-responsive morphological transition. Peptide assemblies exhibit morphological diversity and they may be present in diverse forms, including nanoparticles, nanofibers, nanoribbons, and nanotubes. Upon exposure to stimuli, these assemblies can undergo morphological transition, leading to structural changes and thereby affecting the assembly driving forces. Therefore, these assembly systems allow for the development of assembled peptide biomaterials via stimulus-responsive morphological transformation, which is distinct from the case of peptide materials formed through assembly from monomers. Thus far, there are several reference studies that have reported such kinds of peptide materials exhibiting responsive morphological transformation.53–55
The design of peptide assembly systems for biomedical materials is predominately based on the incorporation of stimulus-responsive and functional moieties into peptides. The stimulus-responsive units allow for the formulation of materials at targeting sites, while the functional moieties account for the bioactivity, which many be either simultaneously generated during the self-assembly or activated through specific processes.29,53 The stimulus-induced self-assembly or morphological transformation of peptides is usually promoted by a sequence cleavage or by reactions at the side chains.55 For instance, the selective hydrolysis of amino acid sequences by enzymes allows for regulating the assembly via sequence cleavage, while stimulus-responsive reactions on the side chains of amino acids provide the versatility for the assembly of peptides in living cells. Accompanied with natural amino acids, several noncanonical amino acids have been synthesized and incorporated in peptide sequences.
The incorporation of functional moieties in biomedical materials depends on the healthcare objectives. In most cases, imaging probes, drugs, or epitopes as targeting moieties and growth factors have been covalently attached with peptide sequences and eventually integrated into materials utilized in tissue engineering, bioimaging, and disease therapy. Besides offering possibilities for attaching intrinsically bioactive moieties, stimulus-responsive functional moieties also offer the possibility for the in situ activation of biomaterials, particularly for bioimaging agents. In some special cases, the bioactivity of materials could be enhanced due to formation of ordered nanostructures. In the following sections, we outline some examples of peptide assembly in living cells according to biomedical applications in bioimaging and disease therapy with an emphasis on the design principles for materials formulation and activation of the biomedical functions.
3. Peptide assembly in living cells for bioimaging
Self-assembled peptide probes, which can be classified as either “always-on” or “responsive-activated” types, are considered as new bioimaging agents due to their advantages for addressing the need for accumulation and retention at the target, as well as enhancing signal intensity.56 “Always-on” peptide probes generate fluorescent signals through accumulation and retention at the targeting sites.57 For example, Pu's group developed semiconducting polymer nanoprobes for protein sulfonic acids without the influence of fluorescence and PA properties.58 In contrast, “responsive-activated” probes can interact with the detecting targets to activate fluorescence emission. Regardless of the type of self-assembled peptide probes, stimulus-responsive assembly processes can contribute to optimizing the pharmacokinetic parameters and avoid the rapid degradation of peptide probes. Self-assembly into ordered supramolecular structures can also potentially amplify the signal intensity, thereby improving signal-to-noise ratios.59–64 In addition, the stimulated-activation of peptide probes allows for the improvement of imaging sensitivity and for the real-time monitoring of physiological information in response to intrinsic biomarkers at pathological sites.65,66 This section discusses peptide self-assembly in living cells with bioimaging functions based on various imaging models, ranging from fluorescence, to photoacoustic, and magnetic resonance, as well as multi-model systems.3.1. Fluorescence imaging
Fluorescence imaging is one of the most conventional methods for bioimaging due to its affordability, simplicity, and capability for non-invasive and minimally invasive surgeries.67–71 The fluorescence imaging of pathological tissues depends on the precise generation of fluorescence signals at specific sites, for example fluorescence generation in living cells.Recently, our group designed and synthesized an enzyme-activated peptide fluorescence probe (FI-A(2NI)VE) that can undergo morphological transformation in the tumour hypoxic region induced by nitroreductase (NTR) (Fig. 1),72 which is a typical reductase overexpressed in solid tumour hypoxic regions.73 Both the morphological transformation and fluorescence activation were promoted in the living environment by the NTR-reduction of a noncanonical amino acid 2-nitroimidazol-1-yl alanine [A(2NI)]. Conversion of the hydrophobic nitro group into a hydrophilic amino group within the amino acid simultaneously weakened the assembling driving forces and prevented fluorescence quenching. By rationally incorporating A(2NI) into the peptide sequence, the FI-A(2NI)VE probe was created via co-assembling the peptide A(2NI)VE with the dye-labelled derivative IR780-A(2NI)VE. In vivo results demonstrated that the FI-A(2NI)VE probe could efficiently accumulate at the periphery of the solid periphery compared to spherical probes and could also penetrate into the deep region of solid tumours compared to the morphology-persistent fibrous probes. This hypoxic-activated morphological transformable peptide-based fluorescence probe provides a new approach for the efficient imaging of solid tumours, and may potentially be particularly useful for application with tissues with rapid metabolizing characteristics.
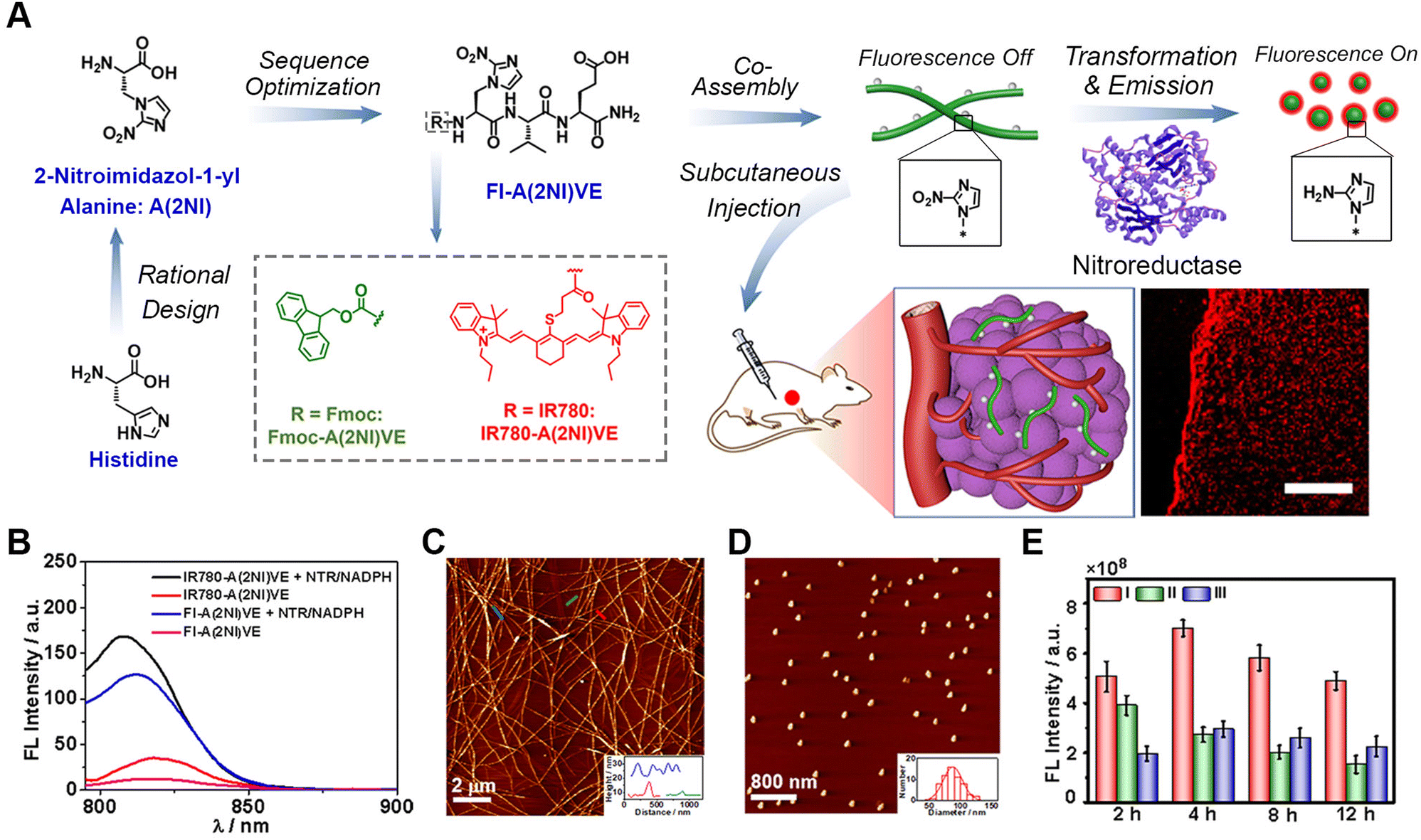 | ||
| Fig. 1 Nitroreductase-induced enzyme-activated transformable supramolecular probes for fluorescence imaging in the tumour hypoxic region. (A) Schematic illustration of the design of NTR-responsive amino acid A(2NI) and the mechanism for efficient hypoxia fluorescent imaging by the morphology-transformable fluorescent supramolecular probe FI-A(2NI)VE. Scale bar: 200 μm. (B) Fluorescence spectra of peptide IR780-A(2NI)VE and probe FI-A(2NI)VE in the presence or absence of NTR (excitation: 780 nm). AFM images of FI-A(2NI)VE (C) and FI-A(2NH2I)VE (D). (E) Quantitative in vivo fluorescence intensity of tumour-bearing mice treated with FI-A(2NI)VE (I), FI-A(2NH2I)VE (II), or FI-A(2NI)VE + dicoumarin (III). Reproduced with permission from ref. 72. Copyright 2021, American Chemical Society. | ||
3.2. Photoacoustic imaging
To overcome the limited tissue penetration and signal interference drawbacks of fluorescence imaging, photoacoustic imaging (PAI) has been developed based on a photoacoustic effect, in which input light energy is converted into heat, subsequently leading to transient thermoelastic expansion and wideband ultrasonic emission.74–76 The resulting ultrasonic waves are then detected by transducers and analysed to generate PA images. Compared to other imaging modes, PAI combines the high contrast of optical imaging with the good tissue penetration of ultrasonic waves, thus allowing it to fill the scale gap between microscopic and macroscopic imaging techniques. Therefore, in the past few years, many PAI contrast agents have been developed for in vivo PAI imaging.77–81 Among these probes, responsive-formed peptide-based PAI probes exhibit real-time, non-invasive, and continuous monitoring features and are therefore considered a promising strategy for designing intelligent in vivo probes.82–84In addition to responsive formation, peptide-based PA probes could also undergo reactive morphological transformation to modulate the bioactivity and signal intensity.85–87 Wang and colleagues designed a photothermal-promoted morphology transformation (PMT) strategy to accelerate the responsive formation of polymer-peptide conjugates into ratiometric PA probes (Fig. 2).87 The polymer-peptide conjugate consisted of a mitochondrial-targeting therapeutic peptide [KLAKLAK]2, an assembly segment KLVFF, a polymer-peptide conjugate PKK-S-PEG, a GSH-responsive unit tailored with hydrophilic PEG, and a photothermal/optoacoustic motif purpurin-18 (P18). While the conjugate formed nanoparticles coated with hydrophilic PEG tails, GSH-induced cleavage of disulphide bonds led to the formation of nanofibers driven by the strong assembling propensity of KLVFF. Upon exposure to light irradiation, the morphological transformation from nanoparticles to nanofibers was accelerated by a photothermal effect associated with P18. The morphological transformation also altered the molecular arrangement of P18 along the nanofibers, thus leading to a red-shift of the UV-vis absorption for P18 and remarkably different ratiometric photoacoustic (RPA) values at 730/685 nm between the nanoparticles and nanofibers. Compared to spontaneous morphology transformation, the responsive PMT-accelerated morphology transformation in living cells or at target sites was also confirmed by PA images of HeLa cells and tumour-bearing mice treated by the conjugates via recording the RPA signals at 730/685 nm. This works demonstrated the potential of photothermal-promoted morphology transformation as an efficient strategy for the responsive formulation of peptide-based nanostructure for biomedical engineering in the future.
 | ||
| Fig. 2 Photothermal-promoted morphological transformation (PMT) of ratiometric probes for PA imaging. (A) Chemical structure of PKK-S-PEG and the process for the photothermal-accelerated responsive transformation of nanofibers monitored by PA imaging. (B) Time-course TEM images of PKK-S-PEG treated with GSH in the dark (left) and upon NIR irradiation (right). (C) RPA difference between NFs and NPs formed by PKK-S-PEG and GSH-reduced PKK-S-PEG at different wavelengths. (D) RPA730/685 images of HeLa cells treated by PKK-S-PEG in either PMT or spontaneous morphology transformation (SMT) process. (E) PA images at 730 or 685 nm in transverse sections of the tumour tissues from the mice treated with PKK-S-PEG for 4 to 12 h. Reproduced with permission from ref. 87. Copyright 2018, American Chemical Society. | ||
3.3. Positron emission tomography
Positron emission tomography (PET) is a powerful non-invasive imaging technique for disease scanning via utilizing radioactive probes and then collecting the annihilating radiation information arising from the probes.88–90 Efficient PET imaging strongly depends on the target accumulation of the probes at the disease sites. However, the limitation of conventional small molecular probes and nanoprobes for target accumulation significantly prevents further contrast improvement. For instance, while small-molecule probes with deep tissue penetration are rapidly metabolized, nanoprobes with high signal-to-background ratios tend to exhibit poor tissue penetration.91,92 Therefore, responsive-formed PET probes have been developed, as exemplified by the hallmark work reported by Rao's group, as well as other research groups.93–95In their early work, Rao et al. reported a strategy of target-enabled in situ ligand aggregation (TESLA) to develop responsive-formed PET tracers through a biorthogonal cyclization between 2-cyano-benzothiazole (CBT) and cysteine (Cys).96,97 The PET tracer was composed of one 18F-labelled tag and one DVED-caged cysteine residue and underwent caspase-3 induced cyclization and subsequent aggregation in doxorubicin-treated tumour cells, which ultimately enhanced retention of the 18F-labelled tracer in cells for imaging caspase-3 activity. A targeted-enabled responsive formulation of PET tracers was also developed by utilizing the intramolecular cyclization between CBT and disulfide-caged cysteine for enabling the imaging activity of different enzymes.98 Recently, Rao and co-authors developed a pre-targeted imaging strategy for the responsive formation of PET tracers based on two biorthogonal reactions, i.e. the condensation between pyrimidinecarbonitrile and cysteine and the inverse-electron demand Diels–Alder (IEDDA) reaction between tetrazine (Tz) and trans-cyclooctene (TCO) (Fig. 3).99 The precursor molecule TCO-C-SNAT4 consisted of a TCO moiety as the handle to rapidly capture imaging probes and could be converted into Cycl-TCO-SNAT4, which underwent responsive aggregation into TCO-labelled nanoparticles. Subsequently, either the fluorescence tag Tz-BDP or PET imaging tag 64Cu-chelated Tz-DOTA was administrated to label the nanoparticles through an IEDDA reaction between Tz and TCO. The caspase-3/7-promoted cleavage of the caged segment and responsive formation of nanoparticles were thoroughly confirmed by a combination of HPLC, dynamic light scattering, and TEM studies. In vivo experiments demonstrated the pre-targeted imaging strategy for both the fluorescence and PET imaging of caspase-3/7 activity, due to the decoupling of enzyme activation and imaging tag immobilization.
 | ||
| Fig. 3 Pre-targeted PET imaging of caspase-3/7 activity by the controllable assembly of nanoparticles. (A) Illustration of the pre-target strategy for imaging enzyme activity based on reactive aggregation and the chemical structures of the corresponding ligands. (B) HPLC traces of TCO-C-SNAT4 along with the incubation with caspase-3 or addition of Tz-BDP to cycl-TCO-SNAT4. (C) DLS analysis of the nanoparticle sizes formed by cycl-TCO-SNAT4. (D) TEM image of nanoparticles of cycl-TCO-SNAT4. Scale bar: 500 nm. (E) Decay of the fluorescence signals at the injection sites of cycl-TCO-SNAT4 and TCO-C-SNAT4 over 8 h. (F) Time-course fluorescence imaging of cisplatin-pretreated H460 tumour-bearing mice injected with TCO-C-SNAT4. Reproduced with permission from ref. 99. Copyright 2020, Wiley-VCH GmbH. | ||
3.4. Magnetic resonance imaging
Magnetic resonance imaging (MRI) is based on the principles of magnetic resonance to generate in vivo signals with high resolution and deep tissue penetration.100–103 Due to its mature and robust characteristics, MRI has become a powerful diagnostic tool in clinical medicine. The responsive formulation of MRI contrast agents allows for broadening the difference between pathological and healthy tissues and thus improving the sensitivity of MRI.104–110 Liang's group developed several unique approaches for the responsive formation of gadolinium (Gd)-based MRI contrast agents instructed by enzyme-catalysed reactions.111 For instance, the group designed a γ-glutamultranspeptidase (GGT)-instructed CBT-Cys cyclization for the formulation of Gd-based contrast agents with enhanced T2-weighted MRI under a high magnetic field. Upon GGT cleavage and GSH reduction, the probe, Glu-Cys(StBu)-Lys(DOTA-Gd)-CBT, underwent intermolecular CBT-Cys condensation and formed nanoparticles in GGT-overexpressed tumour cells. Both in vitro and in vivo experiments confirmed the enhanced T2-weighted MRI signal in HeLa cells and tumour-bearing mice, due to the improved cellular uptake and formation of the nanoparticles.In addition, combining MRI with other imaging methods enables further improving the sensitivity for pathological tissue detection.112 In this context, Ye's group established strategies for the responsive formulation of probes with multimodality imaging functions, including NIR fluorescence (FL)/MRI and NIR FL/MRI/PET imaging (Fig. 4).113 The primary challenge for the formulation of such probes lies in integrating the stimulus-responsive self-assembling process with the signal-generating units. Therefore, the authors integrated activatable NIR and MR probes with a dipeptide motif, leading to the bimodal probe P-CyFF-Gd containing a phosphorated merocyanine dye. The probe assembled into nanoparticles at the tumour sites following an ALP-cleavage reaction within the dye, thereby amplifying and accumulating the imaging signals. In particular, the formation of nanoparticles prevented molecular rotation and generated bright T1-weighted MR via increasing the r1 relaxation rate and shortening the water protons′ T1 relaxation. The HeLa cell experiments and tumour-bearing mice studies showed a robust NIR FL signal, a shortened T1 relaxation, and the enhanced fluorescence and MR contrast, thus evidencing the efficient activation of P-CyFF-Gd through ALP dephosphorylation and stimulus-responsive self-assembly.
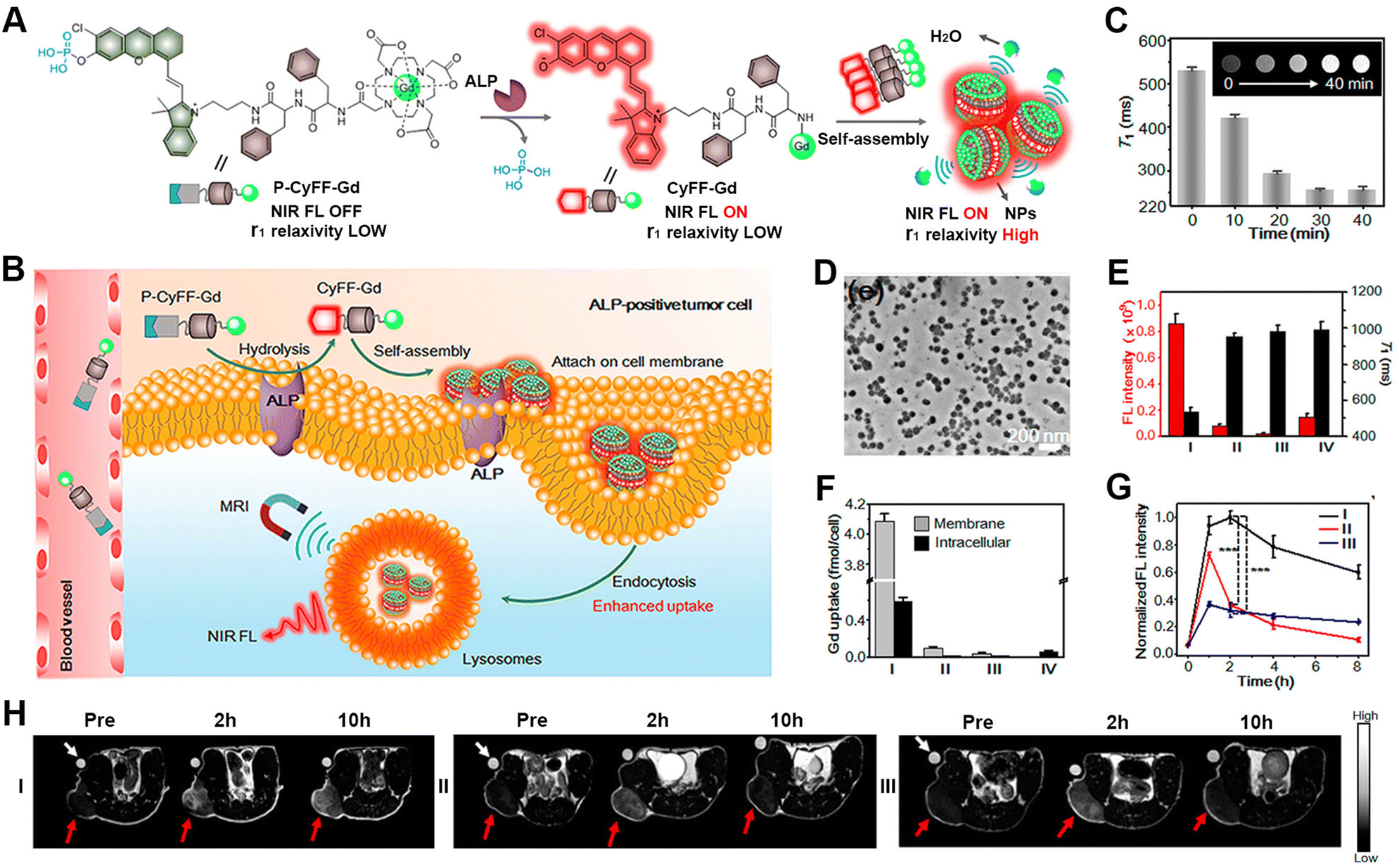 | ||
| Fig. 4 ALP-activatable NIR fluorescence/MRI bimodal probes for in vivo imaging. (A) Chemical structure of P-CyFF-Gd and its ALP-induced assembly into nanoparticles exhibiting enhanced fluorescence and r1 relaxivity. (B) Mechanism of P-CyFF-Gd for the bimodality imaging of ALP-positive tumour cells in vivo. (C) T1-weighted MR images and T1 values for P-CyFF-Gd upon the addition of ALP. (D) TEM image of nanoparticles collected from HeLa cell membranes incubated with P-CyFF-Gd for 1 h. (E) Average FL intensity (red) and T1 value (black) of cell pellets incubated with different treatments. (F) ICP-MS analysis of the Gd contents on the cell membrane or intracellularly after incubation with the indicated treatments. I–IV: HeLa cells incubated with P-CyFF-Gd (I), PCy-Gd (II), P-CyFF-Gd (III), IV: P-Cy-Gd (IV). (G) Normalized FL intensity of HeLa tumour-bearing mice with different treatments. (H) T1-weighted MR images of HeLa tumour-bearing mice with different treatments. In (G) and (H), I–III: HeLa cells incubated with P-CyFF-Gd (I), PCy-Gd (II), or P-CyFF-Gd (III). Reproduced with permission from ref. 113. Copyright 2019, American Chemical Society. | ||
4. Peptide assembly in living cells for disease therapy
The efficient delivery of therapeutic agents to pathological sites is the basis for achieving a perfect curative effect in disease therapy. Conventional delivering vehicles have been demonstrated with limited capability to overcome the physiological barriers present in the delivery pathway, including the blood flow, tumour cell interstitial compartment, and cell membranes.114–117 The application of stimulus-activated peptide assemblies towards drug delivery allows for overcoming the physiological barriers in delivery via the morphological transforming or assembling process.118–121 For example, the assembly process bestows peptide-based nanomedicines with stabilization during blood circulation, whereas the transformation from nanofibers into small nanoparticles facilitates tumour penetration.122 Therefore, due to the precise control of stimulus-responsive self-assembly or morphological transformations, self-assembled peptide nanostructures in living cells have great potential for application in various disease treatments.123,124 This section discusses several recent examples utilizing stimulus-responsive self-assembled peptides in different therapeutic methods, including phototherapy, chemotherapy, immunotherapy, and combination therapy.4.1. Phototherapy
Phototherapy predominately includes photodynamic therapy (PDT) and photothermal therapy (PTT).125–129 With respect to PDT, the exposure of photosensitizers to light leads to the production of ROS, such as hydrogen peroxide (H2O2) and hydroxyl radicals (HO˙) that usually elicit cell death via the oxidization of biomolecules.130 In contrast, the activation of photosensitizers utilized in PTT by light irradiation releases vibrational energy, rather than transferring into ROS species, and subsequently kills cells by local heating. However, high temperature can also cause thermal damage to adjacent healthy tissues. Therefore, mild-temperature PTT strategies that minimize the associated adverse effects have been developed, but suffer from limited therapeutic efficacy or the thermoresistance of cancer cells.131 This can be overcome by combining PTT with other therapeutic modalities, such as immunotherapy or chemotherapy, which is discussed later in the combination therapy section.132–136 It is worth noting that within phototherapy, the integration of photosensitizers with peptide assemblies not only facilitates the target delivery of photosensitizers, it most probably also enhances the production of ROS or heat energy through organization within peptide nanostructures.137–141As an interesting example, our group developed reactive transformable drug delivering vehicles for PDT tumour therapy, termed as tumour microenvironment-adaptable self-assembly (TMAS), based on the reversible acid-responsive self-assembly of the pentapeptide AmpF (Fig. 5).142 While AmpF contains a central 4-aminoproline (Amp) residue and two diphenylalanine fragments on each termini, TMAS is composed of AmpF and its derivative (AmpF-C) modified with the photosensitizer Ce6. The Xaa-Amp amide bond in AmpF adopts a cis- or trans-conformation under neutral and acidic conditions, respectively, thus resulting in the pH-dependent self-assembly of AmpF into superhelices and nanoparticles. Analogous to AmpF, TMAS exhibited a similar reversible morphological transformation between superhelices and nanoparticles triggered by alternating exposure to neutral and acidic environments. This unique pH-responsive property renders the morphology of TMAS adaptable to the pH gradient during delivery pathway and can eventually improve the accumulation of photosensitizers in cancer cells. Both cellular and animal experiments confirmed the enhanced cellular internalization, tumour accumulation, penetration, and retention of photosensitizers due to the adaptable morphology of TMAS, accompanied by improved ROS generation. These results demonstrate the great potential of TMAS for efficient drug delivery to tumour sites for cancer therapy.
 | ||
| Fig. 5 (A) Schematic illustration of the tumour microenvironment-adaptable self-assembly (TMAS) of pentapeptide AmpF and its co-assembly with Ce6-modified AmpF (AmpF-C) into the nanomedicine TMAS, in which AmpF underwent a reversible morphological transition between nanofibers and nanoparticles due to the acid-sensitive cis/trans-isomerization of Amp amide bonds. (B) 1H NMR spectra of the peptides PF (left) and AmpF (right) under various pH conditions. (C) AFM images of the assemblies formed by AmpF at pH 7.4 (left) and 6.5 (right). (D) Quantification of intracellular ROS generation upon the laser irradiation of TMAS and TMRS by 4T1 cells with time. (E) In vivo fluorescence imaging of 4T1 breast tumour-bearing mice administrated with TMAS, TMRS (formed by PF and its PF-C derivative), and Ce6. (F) Relative tumour volume change in tumour-bearing mice upon different treatments. Reproduced with permission from ref. 142. Copyright 2019, American Chemical Society. | ||
4.2. Chemotherapy
Despite being one of most conventional disease-treatment methods, chemotherapy suffers from drug resistance and adverse effect arising from off-target activity.143–146 Self-assembling peptide-based biomedical materials in living cells represents one promising approach to address these challenges.147–151 Due to the capability of peptide nanostructures, including nanoparticles, nanofibers, and nanofibrous networks, to interfere in physiological processes, stimulus-activatable peptide assemblies have been directly utilized as therapeutic agents.152–157 Xu's group and other colleagues performed pioneering work on rationally controlling the pericellular or intracellular self-assembly of peptides.158–160Dexamethasone (Dex) is a chemical agent commonly used for cancer therapy. Here, we used intracellular peptide materials in combination with Dex for inflammation treatment, broadening the chemotherapy scope for disease treatment. Recently, our group reported the development of an stimulus-activated self-amplifying assembling system, AmpFQB, for inflammation therapy based on the robust relationship between the self-assembly of AmpFQB and the expression of the enzyme NAD(P)H quinone dehydrogenase 1 (NQO1) (Fig. 6).161 AmpFQB consists of quinone propionate (QPA)-modified pentapeptide AmpF (AmpFQ) and its derivative AmpFQ-ETGE, which contains an ETGE domain. NQO1 is a critical cytosolic reductase mediating redox homeostasis in cells, which is regulated by protein nuclear factor erythroid-related factor 2 (Nrf2).162,163 Containing the ETGE sequence, which was derived from Nrf2, AmpFQB coassemblies compete with Nrf2-Keap1 complexes and release Nrf2, thereby promoting NQO1 expression.164 While the modification of AmpFQ with QPA supports its NQO1 responsive conversion to AmpF, the ETGE sequence derived from the Nrf2 domain enables binding with the Keap1 receptor and the release of Nrf2 from Keap1-Nrf2 complexes. The liberated Nrf2 increases the level of NQO1 and thus conversely contributes to the transition from AmpFQB nanoparticles to AmpFB nanofibers. Microscale thermophoresis (MST) revealed a four-fold affinity of AmpFB nanofibers to Keap1 compared to AmpFQB nanoparticles or ETGE. Combining the results from the WB assay of the intracellular protein levels and bio-TEM images of macrophages treated with AmpFQB, the amplifying relationship between the reactive assembly of AmpFQB into nanofibers and the expression of enzyme NQO1. AmpFQB could be utilized to deliver the drug dexamethasone (DEX) to treat acute injured lungs, leading to the nanomedicine DEX@AmpFQB. Both in vitro and in vivo studies confirmed the capability of DEX@AmpFQB to enhance the anti-inflammatory efficacy of DEX via alleviating ROS-associated side effects and downregulating the proinflammatory cytokines. The self-amplifying assembly of peptides in living cells with a regular enzyme level provides a unique strategy for the responsive creation of theranostic agents.
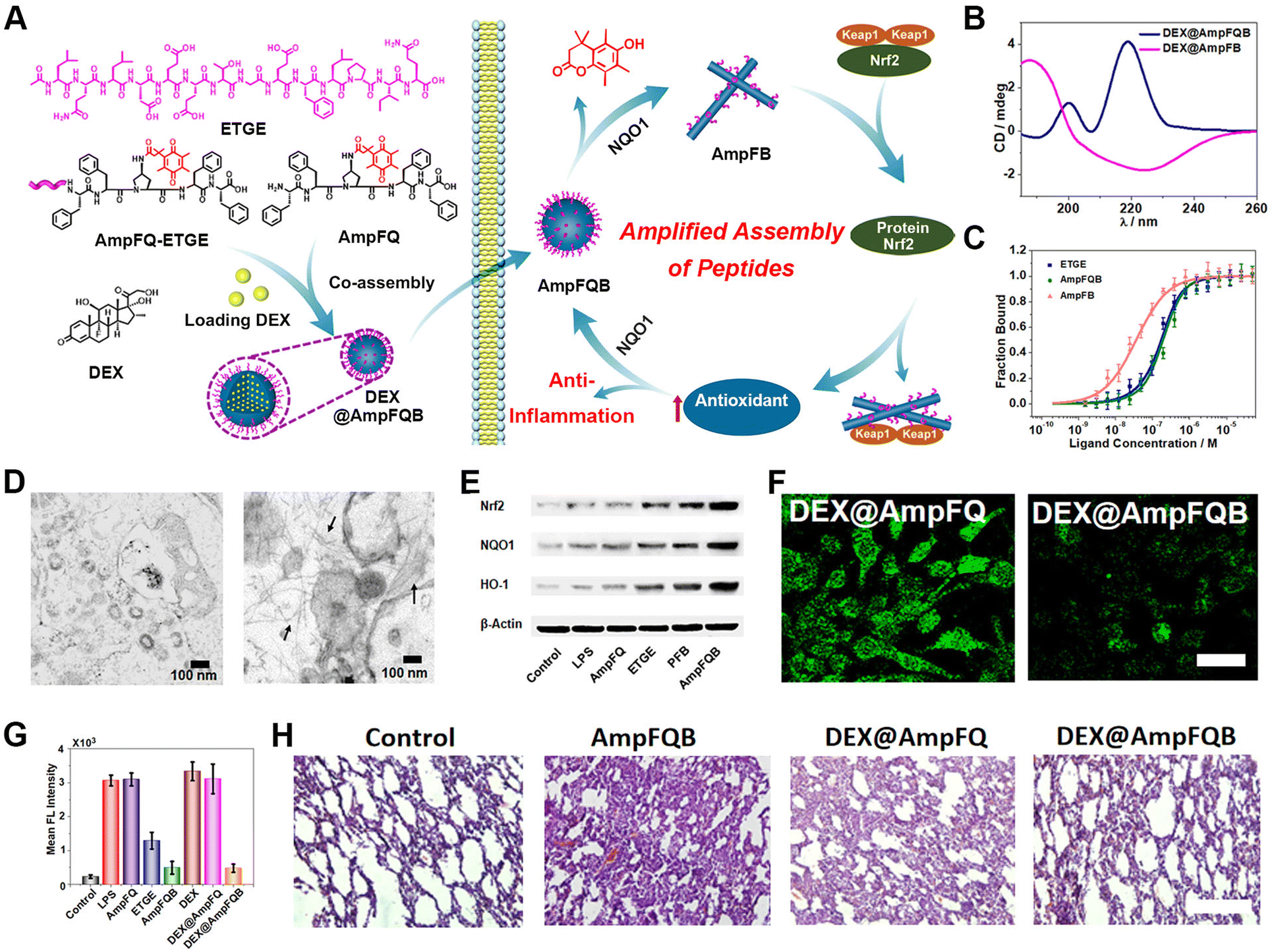 | ||
| Fig. 6 NQO1-activated self-amplifying assembly of peptides in macrophages. (A) Self-amplifying assembly of peptide AmpFQB due to the amplified relationship between peptide assembly and enzyme expression, and its application as a delivery vehicle for dexamethasone (DEX) for improving inflammatory therapy. (B) CD spectra of DEX@AmpFQ before after QPA reduction. (C) MST studies on the free peptide ETGE, AmpFQB, and AmpFB with the protein Keap1. (D) Bio-TEM images of stimulated macrophages incubated with AmpFQ (left) and AmpFQB (right). (E) WB assays of the expression of Nrf2, NQO1, and HO-1 in macrophages treated with AmpFQB and other groups. (F) CLSM images of stimulated macrophages incubated with various treatment stained with DCFH-DA. Scale bar: 10 μm. (G) Quantitative fluorescence intensity associated with DCFH-DA in the treated macrophages. (H) H&E-stained images of lung tissues dissected from injured mice with various treatments. Scale bar: 40 μm. Reproduced with permission from ref. 161. Copyright 2022, American Chemical Society. | ||
4.3. Immunotherapy
Immunotherapy is considered one of the most promising tumour-treatment strategies due to its advanced curative efficacy for both local and remote tumour tissues via eliciting host systemic immune responses.165–167 Combining the advantages of responsive assembled peptides for targeting delivery with immunotherapy, peptide-based cancer immunotherapy represents one of main topics in disease therapy.168–172 Zhang's group designed a tumour protein-engineering system based on the tyrosinase-catalysed polymerization of tripeptide DFY into quinone-rich microfibrils and a reactive collection of immunogenic tumour proteins via a Michael addition between quinone and cysteine (Fig. 7).173 The resulting tumour-protein-captured microfibrils delivered proteins to antigen-presenting cells, which eventually boosted the immune response. Decreases in the levels of tyrosine residues and cysteine residues, as well as an increase in the content of cysteinylcatechol groups, indicated the oxidative polymerization of tyrosine residues and the addition between quinone with protein sulfhydryl groups. In comparison to the lysate from untreated tumour cells, lysate protein-cross-linked pDFY (lysate@pDFY) treatment resulted in a 2.3-fold increase in DC maturation, while tumour-bearing mice subjected to the lysate@pDFY treatment demonstrated a 2.8-fold increase in the maturation of DCs. The co-administration of B16 tumour-bearing mice with DFY and anti-PD1 antibody suppressed tumour growth by 91% compared to monotherapy alone in other treatment groups. Furthermore, a transdermal delivery system incorporating DFY exhibited comparable therapeutic efficacy to that achieved by peritumoural DFY injection. This reactive polymerization of peptides and selective capture of tumour-specific proteins could overcome the systemic inhibition of antigen presentation, providing a promising avenue for modulating the immune system during cancer therapy. | ||
| Fig. 7 Enzyme-catalyzed polymerization of tripeptide DFY for cancer immunotherapy. (A) Schematic illustration of the underlying mechanism for the tyrosinase-induced reactive polymerization of DFY and integration with antigens to form nanofibers. (B) Concentration profiles for tyrosine, cysteine, cysteinylcatechol, and proteins in the polymerizing solution of DFY and tumour lysate. (C) In vitro efficacy of antigen-loaded pDFY in promoting DC maturation. (D) In vivo effect of antigen-loaded pDFY in immunological activation. (E) Tumour volume of B16 tumour-bearing mice from different treated groups. (F) Comparison of therapeutic efficacy between injected DFY and DFY-loaded transdermal hydrogels. Reproduced with permission from ref. 173. Copyright 2021, American Chemical Society. | ||
4.4. Combination therapy
Given the inherent limited therapeutic efficacy and the side effects of individual treating modalities, treating diseases via a combination of different pharmaceutical agents may achieve synergistic therapeutic efficacy while minimizing undesirable adverse effects.174–178 Some examples of such combination therapies include unitary combinations from individual therapeutic modality, binary combinations of phototherapy, chemotherapy, radiotherapy, and immunotherapy, as well as ternary or even more combinations.179–182In terms of binary combination therapy, our group reported an reactive morphological transformation of the peptide scaffold DPEIM for implementing photodynamic and chemodrug combination cancer therapy (Fig. 8).183 The oxidation-responsive peptide scaffold DPEIM was created via the co-assembly of hexapeptide EIMIME with its derivatives Ce6-EIM and CPT-EIM, which were functionalized with the photosensitizer Ce6 and drug CPT, respectively. EIMIME contained two methionine residues, in which the hydrophobic thioether was quantitatively converted to hydrophilic sulfoxide groups upon the addition of H2O2, and thereby promoting nanofibrillar-to-nanoparticle morphological transition for the peptide assemblies. While the GSH-cleavage of disulfide bonds led to the release of the carried drug CPT, the generation of ROS by Ce6 under laser irradiation resulted in an oxidation of methionine residues and also implemented photodynamic cancer therapy. Treating cancer cells by DPEIM led to a higher apoptosis rate compared to individual treatments. In vivo experiments indicated that the laser irradiation-induced morphological transformation of DPEIM improved its tumour accumulation and penetration into the tumour tissue, thereby significantly inhibiting the growth of tumour tissues. Both in vitro and in vivo studies demonstrated the effectiveness of cascade combinatorial photodynamic therapy and chemotherapy arising from CPT and Ce6 moieties within DPEIM.
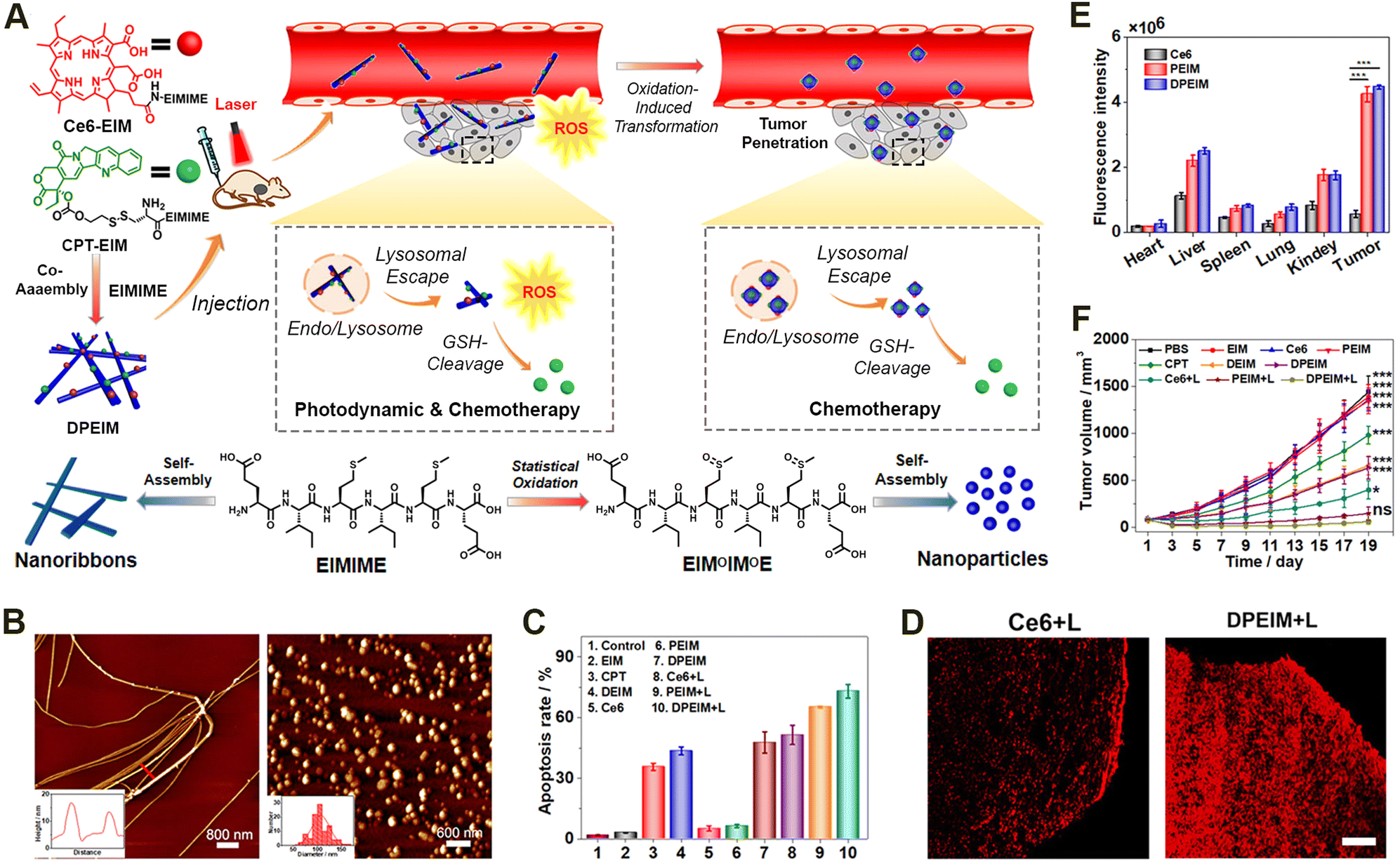 | ||
| Fig. 8 Oxidation-regulated transformation of peptide nanomedicines. (A) Graphic illustration of the mechanism for the creation of the oxidation-regulated reactive transformable peptide nanomedicine DPEIM and its facilitated tumour penetration and cascade combinatorial cancer therapy. (B) AFM images of DPEIM before (left) and after (right) methionine oxidation. (C) Cellular apoptosis studies of 4T1 cells induced with different treatments estimated by flow cytometric analysis. (D) CLSM images of the transverse sections of the tumour tissues from mice administrated with free Ce6 or DPEIM in the presence of laser irradiation. Scale bar: 250 μm. (E) Quantitative fluorescence intensity of ex vivo signals at tumour sites or major organs from mice administrated with free Ce6, PEIM, and DPEIM. (F) Growth profiles for the tumour tissues of mice administrated with different treatments with or without laser irradiation. Reproduced with permission from ref. 183. Copyright 2021, Elsevier. | ||
5. Conclusions and outlook
In this review, we summarized the progress in the stimulus-responsive self-assembly of peptides into biomedical agents in living cells that have been achieved during the past decades. After a brief introduction to the research background, we discussed the design principles of stimulus-responsive peptide materials. We highlighted the achievements of stimulus-responsive peptide biomaterials for implementing disease diagnosis and therapy, providing several examples reported recently to demonstrate their great potential towards precise and real-time healthcare.Due to the remarkable biocompatibility and robust controllable assembling propensity of peptides, the stimulus-responsive self-assembly of peptides in living cells presents a versatile strategy for the controllable formulation of biomaterials at the target site. A simultaneous incorporation of bioactive moieties into peptides usually leads to responsive-formed biomaterials for implementing healthcare objectives.46,184 The bioactivity of the resulting stimulus-reactive biomaterials not only depends on the incorporated functional moieties, but is also associated with the organization and dynamic natures of the peptide assemblies.185–187 In addition, the accumulation and retention of the bioactive moieties at targeting sites could be optimized by the stimulus-responsive assembly processes of peptides.
In terms of peptide assembly-based biomedical materials, the gap between the few number of stimulus-responsive peptide assembling strategies and the complex biological conditions significantly limits their biomedical applications. Hence, the development of stimulus-responsive self-assembling systems is critical for its applications in different fields. In this context, our group intensively engaged in developing noncanonical amino acids containing stimulus-responsive moieties through organic synthesis.188 The rational incorporation of these amino acids into peptides allows for establishing stimulus-responsive assembling systems for the formulation of peptide-based materials in living cells or the body. In addition, exploiting new enzymes as internal stimulus sources for regulating peptide assembly is another direction to expand researchers and designers toolkits.44 This strongly depends on understanding the pathological mechanism, which reveals the biological pathways and involved enzymes corresponding to abnormal physiological behaviour. The finding of new stimulus sources usually exponentially increases the stimulus-responsive units for regulating peptide assembly. Simultaneously, the development of in situ characterizing approaches also facilitates a direct understanding of the assembly process in living systems, which thus far suffers from a lack of appropriate approaches to confirm the assembling process compared to solution conditions.
Despite the achieved progress in stimulus-responsive biomaterials, including peptide assembly-based systems, several critical concerns remain to be addressed with respect to the development of reactive synthetic approaches, the understanding of the underlying mechanism as well as the need for preclinical studies to pave the way towards their clinical usage. Thus far, the approaches available for obtaining stimulus-responsive materials are confined to a few number of versatile methods that are broadly utilized under solution conditions. The utilization of solution methods in responsive biomedical materials studies has been significantly hindered by the limited accessibility of the internal biological structures and signals or the low biocompatibility of the conditions. For instance, mass spectroscopy and scattering techniques could potentially facilitate elucidating the biological structures from the molecular and microscopic levels, respectively.189–194 The stimulus-responsive formulation of biomaterials in living environments might benefit from the development of synthetic approaches involving novel physical phenomena. For example, liquid droplet formation in living cells based on liquid–liquid phase transition provides a new approach for the responsive formulation of biomaterials at targeting sites.195,196 In addition, due to the versatility of machine learning in scanning functional monomers, designing reactive biomaterials based on machine learning is a potentially powerful method to enrich experimental findings.197,198
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Haihe Laboratory of Synthetic Biology (22HHSWSS00010), the Beijing-Tianjin-Hebei Basic Research Cooperation Project (22JCZXJC00010), and the National Natural Science Foundation of China (52073148 & 52273130).Notes and references
- Y. Fang, L. Meng, A. Prominski, E. N. Schaumann, M. Seebald and B. Tian, Chem. Soc. Rev., 2020, 49, 7978–8035 RSC.
- O. Valenzuele, M. Cannataro, I. Rusur, J. Wang, Z. Zhao and I. Rojas, MBC Bioinformatics, 2023, 24, 361 Search PubMed.
- N. Huebsch and D. J. Mooney, Nature, 2009, 462, 426–432 CrossRef CAS.
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2016, 15, 13–26 CrossRef CAS.
- F. Ajalloueian, G. Lemon, J. Hilborn, I. S. Chronakis and M. Fossum, Nat. Rev. Urol., 2018, 15, 155–174 CrossRef.
- D. J. Lipomi, M. Vosgueritchian, B. C. K. Tee, S. L. Hellstrom, J. A. Lee, C. H. Fox and Z. N. Bao, Nat. Nanotechnol., 2011, 6, 788–792 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS.
- M. Karimi, A. Ghasemi, P. Sahandi Zangabad, R. Rahighi, S. M. Moosavi Basri, H. Mirshekari, M. Amiri, Z. Shafaei Pishabad, A. Aslani, M. Bozorgomid, D. Ghosh, A. Beyzavi, A. Vaseghi, A. R. Aref, L. Haghani, S. Bahrami and M. R. Hamblin, Chem. Soc. Rev., 2016, 45, 1457–1501 RSC.
- P. Yang, F. Zhu, Z. Zhang, Y. Cheng, Z. Wang and Y. Li, Chem. Soc. Rev., 2021, 50, 8319–8343 RSC.
- H. J. He, W. Y. Tan, J. Q. Guo, M. H. Yi, A. N. Shy and B. Xu, Chem. Rev., 2020, 120, 9994–10078 CrossRef CAS PubMed.
- J. Kim, S. Lee, Y. Kim, M. Choi, I. Lee, E. Kim, C. G. Yoon, K. Pu, H. Kang and J. S. Kim, Nat. Rev. Mater., 2023, 8, 710–725 CrossRef.
- X. Wu, R. Wang, N. Kwon, H. Ma and J. Yoon, Chem. Soc. Rev., 2022, 51, 450–463 RSC.
- J. R. Sempionatto, J. A. Lasalde-Ramírez, K. Mahato, J. Wang and W. Gao, Nat. Rev. Chem., 2022, 6, 899–915 CrossRef.
- A. K. Gaharwar, I. Singh and A. Khademhosseini, Nat. Rev. Mater., 2020, 5, 686–705 CrossRef CAS.
- D. Wu, J. Lei, Z. Zhang, F. Huang, M. Buljan and G. Yu, Chem. Soc. Rev., 2023, 52, 2911–2945 RSC.
- H. Qin, T. Zhang, N. Li, H.-P. Cong and S.-H. Yu, Nat. Commun., 2019, 10, 2202 CrossRef.
- Z. Ge and S. Liu, Chem. Soc. Rev., 2013, 42, 7289–7325 RSC.
- Y. Lu, A. A. Aimetti, R. Langer and Z. Gu, Nat. Rev. Mater., 2016, 2, 16075 CrossRef.
- L. Cui, S. Vivona, B. R. Smith, S.-R. Kothapalli, J. Liu, X. Ma, Z. Chen, M. Taylor, P. H. Kierstead, J. M. J. Fréchet, S. S. Gambhir and J. Rao, J. Am. Chem. Soc., 2020, 142, 15575–15584 CrossRef CAS PubMed.
- E. Fuentes, M. Gerth, J. A. Berrocal, C. Matera, P. Gorostiza, I. K. Voets, S. Pujals and L. Albertazzi, J. Am. Chem. Soc., 2020, 142, 10069–10078 CrossRef CAS PubMed.
- H. Yan, Y. Wang, F. Huo and C. Yin, J. Am. Chem. Soc., 2023, 145, 3229–3237 CrossRef CAS.
- C. J. Bowerman and B. L. Nilsson, Biopolymers, 2012, 98, 169–184 CrossRef CAS.
- S. Zhang, Interface Focus, 2017, 7, 20170028 CrossRef PubMed.
- J. Zhou, X. Du, C. Berciu, H. He, J. Shi, D. Nicastro and B. Xu, Chem, 2016, 1, 246–263 CAS.
- S. Eskandari, T. Guerin, I. Toth and R. J. Stephenson, Adv. Drug Delivery Rev., 2017, 110–111, 169–187 CrossRef CAS.
- G. Wei, Z. Su, N. P. Reynolds, P. Arosio, I. W. Hamley, E. Gazit and R. Mezzenga, Chem. Soc. Rev., 2017, 46, 4661–4708 RSC.
- F. Sheehan, D. Sementa, A. Jain, M. Kumar, M. Tayarani-Najjaran, D. Kroiss and R. V. Ulijn, Chem. Rev., 2021, 121, 13869–13914 CrossRef CAS PubMed.
- J. A. Raskatov, J. P. Schneider and B. L. Nilsson, Acc. Chem. Res., 2021, 54, 2488–2501 CrossRef CAS PubMed.
- S. Panja and D. J. Adams, Chem. Soc. Rev., 2021, 50, 5165–5200 RSC.
- R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664–675 RSC.
- C. Yan and D. J. Pochan, Chem. Soc. Rev., 2010, 39, 3528–3540 RSC.
- N. Svensen, J. G. A. Walton and M. Bradley, Trends Pharmacol. Sci., 2012, 33, 186–192 CrossRef CAS PubMed.
- T. Lehto, K. Ezzat, M. J. A. Wood and S. E. Andaloussi, Adv. Drug Delivery Rev., 2016, 106, 172–182 CrossRef CAS.
- R. Chang, L. Zhao, R. Xing, J. Li and X. Yan, Chem. Soc. Rev., 2023, 52, 2688–2712 RSC.
- J. Wang, K. Liu, R. Xing and X. Yan, Chem. Soc. Rev., 2016, 45, 5589–5604 RSC.
- M. P. Hendricks, K. Sato, L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2017, 50, 2440–2448 CrossRef CAS.
- E. G. Baker, G. J. Bartlett, K. L. Porter Goff and D. N. Woolfson, Acc. Chem. Res., 2017, 50, 2085–2092 CrossRef CAS.
- C. Yuan, W. Ji, R. Xing, J. Li, E. Gazit and X. Yan, Nat. Rev. Chem., 2019, 3, 567–588 CrossRef CAS.
- M. Hebel, A. Riegger, M. M. Zegota, G. Kizilsavas, J. Gačanin, M. Pieszka, T. Lückerath, J. A. S. Coelho, M. Wagner, P. M. P. Gois, D. Y. W. Ng and T. Weil, J. Am. Chem. Soc., 2019, 141, 14026–14031 CrossRef CAS PubMed.
- Q. Song, Z. Cheng, M. Kariuki, S. C. L. Hall, S. K. Hill, J. Y. Rho and S. Perrier, Chem. Rev., 2021, 121, 13936–13995 CrossRef CAS PubMed.
- J. Zhu, N. Avakyan, A. Kakkis, A. M. Hoffnagle, K. Han, Y. Li, Z. Zhang, T. S. Choi, Y. Na, C.-J. Yu and F. A. Tezcan, Chem. Rev., 2021, 121, 13701–13796 CrossRef CAS PubMed.
- C. Wu, H. Zhang, N. Kong, B. Wu, X. Lin and H. Wang, Angew. Chem., Int. Ed., 2023, 62, e202303455 CrossRef CAS PubMed.
- Y. Cong, Z. Qiao and H. Wang, Adv. Thermoelectr., 2018, 1, 1800067 Search PubMed.
- J. Gao, J. Zhan and Z. Yang, Adv. Mater., 2020, 32, e1805798 CrossRef PubMed.
- Y. Marciano, V. del Solar, N. Nayeem, D. Dave, J. Son, M. Contel and R. V. Ulijn, J. Am. Chem. Soc., 2023, 145, 234–246 CrossRef CAS PubMed.
- D. W. P. M. Löwik, E. H. P. Leunissen, M. V. D. Heuvel, M. B. Hansen and J. C. M. V. Hest, Chem. Soc. Rev., 2010, 39, 3394–3412 RSC.
- Y. Shen, X. Fu, W. Fu and Z. Li, Chem. Soc. Rev., 2015, 44, 612–622 RSC.
- R. Zou, Q. Wang, J. Wu, J. Wu, C. Schmuck and H. Tian, Chem. Soc. Rev., 2015, 44, 5200–5219 RSC.
- N. Habibi, N. Kamaly, A. Memic and H. Shafiee, Nano Today, 2016, 11, 41–60 CrossRef CAS PubMed.
- S. Lee, J. Xie and X. Chen, Chem. Rev., 2010, 110, 3087–3111 CrossRef CAS PubMed.
- M. Zelzer and R. V. Ulijn, Chem. Soc. Rev., 2010, 39, 3351–3357 RSC.
- K. Tao, A. Levin, L. Adler-Abramovich and E. Gazit, Chem. Soc. Rev., 2016, 45, 3935–3953 RSC.
- I. W. Hamley, Chem. Rev., 2017, 117, 14015–14041 CrossRef CAS PubMed.
- L. L. Li, Z. Y. Qiao, L. Wang and H. Wang, Adv. Mater., 2019, 31, e1804971 CrossRef PubMed.
- S. Chagri, D. Y. W. Ng and T. Weil, Nat. Rev. Chem., 2022, 6, 320–338 CrossRef PubMed.
- Z. Liu, G. Liang and W. Zhan, Chem. Res. Chin. Univ., 2021, 37, 889–899 CrossRef CAS.
- Y. Lyu and K. Pu, Adv. Sci., 2017, 4, 1600481 CrossRef PubMed.
- Y. Lyu, X. Zhen, Y. Miao and K. Pu, ACS Nano, 2017, 11, 358–367 CrossRef CAS PubMed.
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2016, 116, 2775–2825 CrossRef CAS PubMed.
- P. Zhang, Y. Cui, C. F. Anderson, C. Zhang, Y. Li, R. Wang and H. Cui, Chem. Soc. Rev., 2018, 47, 3490–3529 RSC.
- X. X. Zhao, L. L. Li, Y. Zhao, H. W. An, Q. Cai, J. Y. Lang, X. X. Han, B. Peng, Y. Fei, H. Liu, H. Qin, G. Nie and H. Wang, Angew. Chem., Int. Ed., 2019, 58, 15287–15294 CrossRef CAS PubMed.
- B. Sun, K. Tao, Y. Jia, X. Yan, Q. Zou, E. Gazit and J. Li, Chem. Soc. Rev., 2019, 48, 4387–4400 RSC.
- G. Kwek, T. C. Do, X. Lu, J. Lin and B. Xing, ACS Appl. Bio Mater., 2021, 4, 2192–2216 CrossRef CAS PubMed.
- Y. Zhong, J. Zhan, G. Xu, Y. Chen, Q. Qin, X. Liao, S. Ma, Z. Yang and Y. Cai, Angew. Chem., Int. Ed., 2021, 60, 8121–8129 CrossRef CAS PubMed.
- D. J. Ye, A. J. Shuhendler, L. N. Cui, L. Tong, S. S. Tee, G. Tikhomirov, D. W. Felsher and J. H. Rao, Nat. Chem., 2014, 6, 519–526 CrossRef CAS PubMed.
- Q. C. Jiang, X. Y. Liu, G. L. Liang and X. B. Sun, Nanoscale, 2021, 13, 15142–15150 RSC.
- S. He, J. Song, J. Qu and Z. Cheng, Chem. Soc. Rev., 2018, 47, 4258–4278 RSC.
- S. A. Hilderbrand and R. Weissleder, Curr. Opin. Chem. Biol., 2010, 14, 71–79 CrossRef CAS PubMed.
- H. Kobayashi and P. L. Choyke, Acc. Chem. Res., 2011, 44, 83–90 CrossRef CAS PubMed.
- P. Sarder, D. Maji and S. Achilefu, Bioconjugate Chem., 2015, 26, 963–974 CrossRef CAS PubMed.
- L. Yuan, W. Y. Lin, K. B. Zheng, L. W. He and W. M. Huang, Chem. Soc. Rev., 2013, 42, 622–661 RSC.
- B. Hu, N. Song, Y. Cao, M. Li, X. Liu, Z. Zhou, L. Shi and Z. Yu, J. Am. Chem. Soc., 2021, 143, 13854–13864 CrossRef CAS PubMed.
- M. Ikeda, T. Tanida, T. Yoshii, K. Kurotani, S. Onogi, K. Urayama and I. Hamachi, Nat. Chem., 2014, 6, 511–518 CrossRef CAS PubMed.
- R. E. Borg and J. Rochford, Photochem. Photobiol., 2018, 94, 1175–1209 CrossRef CAS PubMed.
- J. G. Huang and K. Y. Pu, Angew. Chem., Int. Ed., 2020, 59, 11717–11731 CrossRef CAS PubMed.
- Y. Liu, L. Teng, B. Yin, H. Meng, X. Yin, S. Huan, G. Song and X. B. Zhang, Chem. Rev., 2022, 122, 6850–6918 CrossRef CAS PubMed.
- X. L. Dean-Ben, S. Gottschalk, B. Mc Larney, S. Shoham and D. Razansky, Chem. Soc. Rev., 2017, 46, 2158–2198 RSC.
- P. K. Upputuri and M. Pramanik, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2020, 12, e1618 Search PubMed.
- W. T. Yang, W. S. Guo, W. J. Le, G. X. Lv, F. H. Zhang, L. Shi, X. L. Wang, J. Wang, S. Wang, J. Chang and B. B. Zhang, ACS Nano, 2016, 10, 10245–10257 CrossRef CAS PubMed.
- Q. Chen, C. Liang, X. Q. Sun, J. W. Chen, Z. J. Yang, H. Zhao, L. Z. Feng and Z. Liu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5343–5348 CrossRef CAS PubMed.
- X. J. Cheng, R. Sun, L. Yin, Z. F. Chai, H. B. Shi and M. Y. Gao, Adv. Mater., 2017, 29, 1604894 CrossRef PubMed.
- Y. Wang, X. Hu, J. Weng, J. Li, Q. Fan, Y. Zhang and D. Ye, Angew. Chem., Int. Ed., 2019, 58, 4886–4890 CrossRef CAS PubMed.
- C. Wang, W. Du, C. Wu, S. Dan, M. Sun, T. Zhang, B. Wang, Y. Yuan and G. Liang, Angew. Chem., Int. Ed., 2022, 61, e202114766 CrossRef CAS PubMed.
- X. Wen, R. Zhang, Y. Hu, L. Wu, H. Bai, D. Song, Y. Wang, R. An, J. Weng, S. Zhang, R. Wang, L. Qiu, J. Lin, G. Gao, H. Liu, Z. Guo and D. Ye, Nat. Commun., 2023, 14, 800 CrossRef CAS PubMed.
- C. Wu, R. Zhang, W. Du, L. Cheng and G. Liang, Nano Lett., 2018, 18, 7749–7754 CrossRef CAS PubMed.
- Y. Sun, Y. Liang, W. Dai, B. He, H. Zhang, X. Wang, J. Wang, S. Huang and Q. Zhang, Nano Lett., 2019, 19, 3229–3237 CrossRef CAS PubMed.
- X. H. Zhang, D. B. Cheng, L. Ji, H. W. An, D. Wang, Z. X. Yang, H. Chen, Z. Y. Qiao and H. Wang, Nano Lett., 2020, 20, 1286–1295 CrossRef CAS PubMed.
- B. Foster, U. Bagci, A. Mansoor, Z. Y. Xu and D. J. Mollura, Comput. Biol. Med., 2014, 50, 76–96 CrossRef PubMed.
- K. Lameka, M. D. Farwell and M. Ichise, Handb. Clin. Neurol., 2016, 135, 209–227 Search PubMed.
- S. Preshlock, M. Tredwell and V. Gouverneur, Chem. Rev., 2016, 116, 719–766 CrossRef CAS PubMed.
- Y. Geng, P. Dalhaimer, S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS PubMed.
- L. L. Lock, C. D. Reyes, P. Zhang and H. Cui, J. Am. Chem. Soc., 2016, 138, 3533–3540 CrossRef CAS PubMed.
- B. Shen, J. Jeon, M. Palner, D. Ye, A. Shuhendler, F. T. Chin and J. Rao, Angew. Chem., Int. Ed., 2013, 52, 10511–10514 CrossRef CAS PubMed.
- L. Qiu, W. Wang, K. Li, Y. Peng, G. C. Lv, Q. Z. Liu, F. Gao, Y. Seimbille, M. H. Xie and J. G. Lin, Theranostics, 2019, 9, 6962–6975 CrossRef CAS PubMed.
- Y. Wang, H. Bai, Y. Miao, J. Weng, Z. Huang, J. Fu, Y. Zhang, J. Lin and D. Ye, Angew. Chem., Int. Ed., 2022, 61, e202200369 CrossRef CAS PubMed.
- T. H. Witney, A. Hoehne, R. E. Reeves, O. Ilovich, M. Namavari, B. Shen, F. T. Chin, J. Rao and S. S. Gambhir, Clin. Cancer Res., 2015, 21, 3896–3905 CrossRef CAS PubMed.
- M. Palner, B. Shen, J. H. Jeon, J. G. Lin, F. T. Chin and J. H. Rao, J. Nucl. Med., 2015, 56, 1415–1421 CrossRef CAS PubMed.
- J. Lin, D. Gao, S. Wang, G. Lv, X. Wang, C. Lu, Y. Peng and L. Qiu, J. Am. Chem. Soc., 2022, 144, 7667–7675 CrossRef CAS PubMed.
- Z. Chen, M. Chen, K. Zhou and J. Rao, Angew. Chem., Int. Ed., 2020, 59, 7864–7870 CrossRef CAS PubMed.
- E. T. Ahrens and J. W. M. Bulte, Nat. Rev. Immunol., 2013, 13, 755–763 CrossRef CAS PubMed.
- J. Estelrich, M. J. Sanchez-Martin and M. A. Busquets, Int. J. Nanomed., 2015, 10, 1727–1741 CAS.
- J. J. Futterer, A. Briganti, P. De Visschere, M. Emberton, G. Giannarini, A. Kirkham, S. S. Taneja, H. Thoeny, G. Villeirs and A. Villers, Eur. Urol., 2015, 68, 1045–1053 CrossRef PubMed.
- D. Ni, W. Bu, E. B. Ehlerding, W. Cai and J. Shi, Chem. Soc. Rev., 2017, 46, 7438–7468 RSC.
- G. Liang, J. Ronald, Y. Chen, D. Ye, P. Pandit, M. L. Ma, B. Rutt and J. Rao, Angew. Chem., Int. Ed., 2011, 50, 6283–6286 CrossRef CAS PubMed.
- D. Ye, A. J. Shuhendler, P. Pandit, K. D. Brewer, S. S. Tee, L. Cui, G. Tikhomirov, B. Rutt and J. Rao, Chem. Sci., 2014, 5, 3845–3852 RSC.
- C. Diaferia, E. Gianolio, P. Palladino, F. Arena, C. Boffa, G. Morelli and A. Accardo, Adv. Funct. Mater., 2015, 25, 7003–7016 CrossRef CAS.
- H. Nejadnik, D. J. Ye, O. D. Lenkov, J. S. Donig, J. E. Martin, R. Castillo, N. Derugin, B. Sennino, J. H. Rao and H. Daldrup-Link, ACS Nano, 2015, 9, 1150–1160 CrossRef CAS PubMed.
- Y. Yuan, J. Zhang, X. Qi, S. Li, G. Liu, S. Siddhanta, I. Barman, X. Song, M. T. McMahon and J. W. M. Bulte, Nat. Mater., 2019, 18, 1376–1383 CrossRef CAS PubMed.
- G. Gambino, T. Gambino, L. Connah, F. La Cava, H. Evrard and G. Angelovski, J. Med. Chem., 2021, 64, 7565–7574 CrossRef CAS PubMed.
- L. J. Luo, X. M. Liu, X. Zhang, J. Liu, Y. Gao, T. Y. Sun and L. L. Li, Nano Lett., 2022, 22, 1694–1702 CrossRef CAS PubMed.
- Z. Hai, Y. Ni, D. Saimi, H. Yang, H. Tong, K. Zhong and G. Liang, Nano Lett., 2019, 19, 2428–2433 CrossRef CAS PubMed.
- Y. Hu, J. Zhang, Y. Miao, X. Wen, J. Wang, Y. Sun, Y. Chen, J. Lin, L. Qiu, K. Guo, H. Y. Chen and D. Ye, Angew. Chem., Int. Ed., 2021, 60, 18082–18093 CrossRef CAS PubMed.
- R. Yan, Y. Hu, F. Liu, S. Wei, D. Fang, A. J. Shuhendler, H. Liu, H. Y. Chen and D. Ye, J. Am. Chem. Soc., 2019, 141, 10331–10341 CrossRef CAS PubMed.
- Q. Sun, Z. Zhou, N. Qiu and Y. Shen, Adv. Mater., 2017, 29, 1606628 CrossRef PubMed.
- M. T. Manzari, Y. Shamay, H. Kiguchi, N. Rosen, M. Scaltriti and D. A. Heller, Nat. Rev. Mater., 2021, 6, 351–370 CrossRef CAS PubMed.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- Z. X. Zhou, M. Vazquez-Gonzalez and I. Willner, Chem. Soc. Rev., 2021, 50, 4541–4563 RSC.
- K. Li, C. J. Liu and X. Z. Zhang, Adv. Drug Delivery Rev., 2020, 160, 36–51 CrossRef CAS PubMed.
- G. Li, T. Sasaki, S. Asahina, M. C. Roy, T. Mochizuki, K. Koizumi and Y. Zhang, Chem, 2017, 2, 283–298 CAS.
- M. T. Jeena, L. Palanikumar, E. M. Go, I. Kim, M. G. Kang, S. Lee, S. Park, H. Choi, C. Kim, S.-M. Jin, S. C. Bae, H. W. Rhee, E. Lee, S. K. Kwak and J.-H. Ryu, Nat. Commun., 2017, 8, 26 CrossRef CAS PubMed.
- G. Li, X. Hu, X. Wu and Y. Zhang, Nano Lett., 2021, 21, 3052–3059 CrossRef CAS PubMed.
- M. Li, Z. Wang, X. Liu, N. Song, Y. Song, X. Shi, J. Liu, J. Liu and Z. Yu, J. Controlled Release, 2021, 340, 35–47 CrossRef CAS PubMed.
- C. Zhang, W. Wu, R. Q. Li, W. X. Qiu, Z. N. Zhuang, S. X. Cheng and X. Z. Zhang, Adv. Funct. Mater., 2018, 28, 1804492 CrossRef.
- G.-B. Qi, Y.-J. Gao, L. Wang and H. Wang, Adv. Mater., 2018, 30, e1703444 CrossRef PubMed.
- Y. J. Liu, P. Bhattarai, Z. F. Dai and X. Y. Chen, Chem. Soc. Rev., 2019, 48, 2053–2108 RSC.
- J. C. Li and K. Y. Pu, Chem. Soc. Rev., 2019, 48, 38–71 RSC.
- T. C. Pham, V. N. Nguyen, Y. Choi, S. Lee and J. Yoon, Chem. Rev., 2021, 121, 13454–13619 CrossRef CAS PubMed.
- Q. Y. Zheng, X. M. Liu, Y. F. Zheng, K. W. K. Yeung, Z. D. Cui, Y. Q. Liang, Z. Y. Li, S. L. Zhu, X. B. Wang and S. L. Wu, Chem. Soc. Rev., 2021, 50, 5086–5125 RSC.
- B. Sun, J. Y. Teo, J. Wu and Y. Zhang, Acc. Chem. Res., 2023, 56, 1143–1155 CrossRef CAS PubMed.
- Z. J. Zhou, J. B. Song, L. M. Nie and X. Y. Chen, Chem. Soc. Rev., 2016, 45, 6597–6626 RSC.
- G. Gao, X. Sun and G. Liang, Adv. Funct. Mater., 2021, 31, 2100738 CrossRef CAS.
- Y. W. Chen, Y. L. Su, S. H. Hu and S. Y. Chen, Adv. Drug Delivery Rev., 2016, 105, 190–204 CrossRef CAS PubMed.
- D. de Melo-Diogo, C. Pais-Silva, D. R. Dias, A. F. Moreira and I. J. Correia, Adv. Healthcare Mater., 2017, 6, 1700073 CrossRef PubMed.
- X. Y. Huang, Y. Lu, M. X. Guo, S. Y. Du and N. Han, Theranostics, 2021, 11, 7546–7569 CrossRef CAS PubMed.
- M. Y. Chang, Z. Y. Hou, M. Wang, C. X. Li and J. Lin, Adv. Mater., 2021, 33, 2004788 CrossRef CAS PubMed.
- J. Qin, X. Wang, G. Fan, Y. Lv and J. Ma, Adv. Thermoelectr., 2023, 6, 2200218 CAS.
- M. Abbas, Q. Zou, S. Li and X. Yan, Adv. Mater., 2017, 29, 1605021 CrossRef PubMed.
- W. Ma, S. N. Sha, P. L. Chen, M. Yu, J. J. Chen, C. B. Huang, B. Yu, Y. Liu, L. H. Liu and Z. Q. Yu, Adv. Healthcare Mater., 2020, 9, e1901100 CrossRef PubMed.
- L. Zhang, Y. Wu, X. Yin, Z. Zhu, T. Rojalin, W. Xiao, D. Zhang, Y. Huang, L. Li, C. M. Baehr, X. Yu, Y. Ajena, Y. Li, L. Wang and K. S. Lam, ACS Nano, 2021, 15, 468–479 CrossRef CAS PubMed.
- T. Ma, R. Chen, N. Lv, Y. Li, Z. R. Yang, H. Qin, Z. Li, H. Jiang and J. Zhu, Small, 2022, 18, e2204759 CrossRef PubMed.
- X. Liu, W. Zhan, G. Gao, Q. Jiang, X. Zhang, H. Zhang, X. Sun, W. Han, F. G. Wu and G. Liang, J. Am. Chem. Soc., 2023, 145, 7918–7930 CrossRef CAS PubMed.
- M. Li, Y. Ning, J. Chen, X. Duan, N. Song, D. Ding, X. Su and Z. Yu, Nano Lett., 2019, 19, 7965–7976 CrossRef CAS PubMed.
- A. Overbeek, M. H. van den Berg, F. E. van Leeuwen, G. J. L. Kaspers, C. B. Lambalk and E. van Dulmen-den Broeder, Cancer Treat. Rev., 2017, 53, 10–24 CrossRef CAS PubMed.
- R. Oun, Y. E. Moussa and N. J. Wheate, Dalton Trans., 2018, 47, 6645–6653 RSC.
- X. Wang, H. Y. Zhang and X. Z. Chen, Cancer Drug Resist., 2019, 2, 141–160 Search PubMed.
- T. T. Hu, H. L. Gong, J. Y. Xu, Y. Huang, F. B. Wu and Z. Y. He, Pharmaceutics, 2022, 14, 1606 CrossRef CAS PubMed.
- E. De Santis and M. G. Ryadnov, Chem. Soc. Rev., 2015, 44, 8288–8300 RSC.
- M. Pieszka, S. Han, C. Volkmann, R. Graf, I. Lieberwirth, K. Landfester, D. Y. W. Ng and T. Weil, J. Am. Chem. Soc., 2020, 142, 15780–15789 CrossRef CAS PubMed.
- N. J. Sinha, M. G. Langenstein, D. J. Pochan, C. J. Kloxin and J. G. Saven, Chem. Rev., 2021, 121, 13915–13935 CrossRef CAS PubMed.
- J. Wang, Y. Li and G. Nie, Nat. Rev. Mater., 2021, 6, 766–783 CrossRef CAS PubMed.
- X.-J. Wang, J. Cheng, L.-Y. Zhang and J.-G. Zhang, Drug Delivery, 2022, 29, 1184–1200 CrossRef CAS PubMed.
- J. Zhan, Y. Cai, S. He, L. Wang and Z. Yang, Angew. Chem., Int. Ed., 2018, 57, 1813–1816 CrossRef CAS PubMed.
- L. Zhang, D. Jing, N. Jiang, T. Rojalin, C. M. Baehr, D. Zhang, W. Xiao, Y. Wu, Z. Cong, J. J. Li, Y. Li, L. Wang and K. S. Lam, Nat. Nanotechnol., 2020, 15, 145–153 CrossRef CAS PubMed.
- F. Wang, D. Xu, H. Su, W. Zhang, X. Sun, M. K. Monroe, R. W. Chakroun, Z. Wang, W. Dai, R. Oh, H. Wang, Q. Fan, F. Wan and H. Cui, Sci. Adv., 2020, 6, eaaz8985 CrossRef PubMed.
- C. Keum, J. Hong, D. Kim, S.-Y. Lee and H. Kim, ACS Appl. Mater. Interfaces, 2021, 13, 14866–14874 CrossRef CAS PubMed.
- L. Xie, Y. Ding, Y. Wang, X. Li, Z. Yang and Z.-W. Hu, Adv. Funct. Mater., 2022, 32, 2206969 CrossRef CAS.
- L. Hu, Y. Li, X. Lin, Y. Huo, H. Zhang and H. Wang, Angew. Chem., Int. Ed., 2021, 60, 21807–21816 CrossRef CAS PubMed.
- R. A. Pires, Y. M. Abul-Haija, D. S. Costa, R. Novoa-Carballal, R. L. Reis, R. V. Ulijn and I. Pashkuleva, J. Am. Chem. Soc., 2015, 137, 576–579 CrossRef CAS PubMed.
- S. Liu, Q. Zhang, H. He, M. Yi, W. Tan, J. Guo and B. Xu, Angew. Chem., Int. Ed., 2022, 61, e202210568 CrossRef CAS PubMed.
- W. Tan, Q. Zhang, M. C. Quiñones-Frías, A. Y. Hsu, Y. Zhang, A. Rodal, P. Hong, H. R. Luo and B. Xu, J. Am. Chem. Soc., 2022, 144, 6709–6713 CrossRef CAS PubMed.
- Y. Song, M. Li, N. Song, X. Liu, G. Wu, H. Zhou, J. Long, L. Shi and Z. Yu, J. Am. Chem. Soc., 2022, 144, 6907–6917 CrossRef CAS PubMed.
- D. Ross and D. Siegel, Redox Biol., 2021, 41, 101950 CrossRef CAS PubMed.
- J. Chang, W. Cai, C. Liang, Q. Tang, X. Chen, Y. Jiang, L. Mao and M. Wang, J. Am. Chem. Soc., 2019, 141, 18136–18141 CrossRef CAS PubMed.
- Z.-Y. Jiang, M.-C. Lu and Q.-D. You, J. Med. Chem., 2016, 59, 10837–10858 CrossRef CAS PubMed.
- D. J. Irvine, M. C. Hanson, K. Rakhra and T. Tokatlian, Chem. Rev., 2015, 115, 11109–11146 CrossRef CAS PubMed.
- A. Ribas and J. D. Wolchok, Science, 2018, 359, 1350–1355 CrossRef CAS PubMed.
- A. D. Waldman, J. M. Fritz and M. J. Lenardo, Nat. Rev. Immunol., 2020, 20, 651–668 CrossRef CAS PubMed.
- M. Li, X. Zhao, J. Dai and Z. Yu, Sci. China Mater., 2019, 62, 1759–1781 CrossRef.
- R. Chang and X. Yan, Small Struct., 2020, 1, 2000068 CrossRef.
- M. D. Wang, G. T. Lv, H. W. An, N. Y. Zhang and H. Wang, Angew. Chem., Int. Ed., 2022, 61, e202113649 CrossRef CAS PubMed.
- Y. Ding, D. Zheng, L. Xie, X. Zhang, Z. Zhang, L. Wang, Z. W. Hu and Z. Yang, J. Am. Chem. Soc., 2023, 145, 4366–4371 CrossRef CAS.
- L. Zhang, Z. Jiang, X. Yang, Y. Qian, M. Wang, S. Wu, L. Li, F. Jia, Z. Wang, Z. Hu, M. Zhao, X. Tang, G. Li, H. Shang, X. Chen and W. Wang, Adv. Mater., 2023, 35, 2207330 CrossRef CAS PubMed.
- Q. L. Zhang, D. Zheng, X. Dong, P. Pan, S. M. Zeng, F. Gao, S. X. Cheng and X. Z. Zhang, J. Am. Chem. Soc., 2021, 143, 5127–5140 CrossRef CAS PubMed.
- W. P. Fan, B. Yung, P. Huang and X. Y. Chen, Chem. Rev., 2017, 117, 13566–13638 CrossRef CAS PubMed.
- S. A. Patel and A. J. Minn, Immunity, 2018, 48, 417–433 CrossRef CAS PubMed.
- X. W. Wang, X. Y. Zhong, Z. Liu and L. Cheng, Nano Today, 2020, 35, 100946 CrossRef CAS.
- J. C. Dawson, A. Serrels, D. G. Stupack, D. D. Schlaepfer and M. C. Frame, Nat. Rev. Cancer, 2021, 21, 313–324 CrossRef CAS PubMed.
- S. G. Lim, T. F. Baumert, C. Boni, E. Gane, M. Levrero, A. S. Lok, M. K. Maini, N. A. Terrault and F. Zoulim, Nat. Rev. Gastroenterol. Hepatol., 2023, 20, 238–253 CrossRef CAS PubMed.
- G. Gao, Y. W. Jiang, W. Zhan, X. Liu, R. Tang, X. Sun, Y. Deng, L. Xu and G. Liang, J. Am. Chem. Soc., 2022, 144, 11897–11910 CrossRef CAS PubMed.
- Z. Zhang, K. Wang, M. Liu, P. Hu, Y. Xu, D. Yin, Y. Yang, X. Dong, C. Qu, L. Zhang, J. Ni and X. Yin, Drug Delivery, 2022, 29, 1608–1619 CrossRef CAS PubMed.
- W. Jiang, C. Cheng, X. Qiu, L. Chen, X. Guo, Y. Luo, J. Wang, J. Wang, Z. Xie, P. Li, Z. Wang, H. Ran, Z. Zhou and J. Ren, Adv. Sci., 2023, 10, e2204989 CrossRef PubMed.
- Y. Sun, B. Lyu, C. Yang, B. He, H. Zhang, X. Wang, Q. Zhang and W. Dai, Bioact. Mater., 2023, 22, 47–59 CAS.
- N. Song, Z. Zhou, Y. Song, M. Li, X. Yu, B. Hu and Z. Yu, Nano Today, 2021, 38, 101198 CrossRef CAS.
- Y. Li, G. Yang, L. Gerstweiler, S. H. Thang and C. X. Zhao, Adv. Funct. Mater., 2022, 33, 2210387 CrossRef.
- S. Lou, X. Wang, Z. Yu and L. Shi, Adv. Sci., 2019, 6, 1802043 CrossRef PubMed.
- P. Makam and E. Gazit, Chem. Soc. Rev., 2018, 47, 3406–3420 RSC.
- D. M. Raymond and B. L. Nilsson, Chem. Soc. Rev., 2018, 47, 3659–3720 RSC.
- Y. Song, Z. Zhang, Y. Cao and Z. Yu, ChemBioChem, 2023, 24, e202200497 CrossRef CAS PubMed.
- R. Kubota, W. Tanaka and I. Hamachi, Chem. Rev., 2021, 121, 14281–14347 CrossRef CAS PubMed.
- J. Hofmann, H. S. Hahm, P. H. Seeberger and K. Pagel, Nature, 2015, 526, 241–244 CrossRef CAS PubMed.
- M. A. Walti, F. Ravotti, H. Arai, C. G. Glabe, J. S. Wall, A. Bockmann, P. Guntert, B. H. Meier and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E4976–E4984 CrossRef CAS PubMed.
- L. Wei, Z. X. Chen, L. X. Shi, R. Long, A. V. Anzalone, L. Y. Zhang, F. H. Hu, R. Yuste, V. W. Cornish and W. Min, Nature, 2017, 544, 465–470 CrossRef CAS PubMed.
- R. Hervas, M. J. Rau, Y. Park, W. Zhang, A. G. Murzin, J. A. J. Fitzpatrick, S. H. W. Scheres and K. Si, Science, 2020, 367, 1230–1234 CrossRef CAS PubMed.
- Y. Yang, D. Arseni, W. Zhang, M. Huang, S. Lövestam, M. Schweighauser, A. Kotecha, A. G. Murzin, S. Y. Peak-Chew, J. Macdonald, I. Lavenir, H. J. Garringer, E. Gelpi, K. L. Newell, G. G. Kovacs, R. Vidal, B. Ghetti, B. Ryskeldi-Falcon, S. H. W. Scheres and M. Goedert, Science, 2022, 375, 167–172 CrossRef CAS PubMed.
- Y. Sun, S. Y. Lau, Z. W. Lim, S. C. Chang, F. Ghadessy, A. Partridge and A. Miserez, Nat. Chem., 2022, 14, 274–283 CrossRef CAS PubMed.
- J. Liu, E. Spruijt, A. Miserez and R. Langer, Nat. Rev. Mater., 2023, 8, 139–141 CrossRef CAS.
- J. Huang, Y. Xu, Y. Xue, Y. Huang, X. Li, X. Chen, Y. Xu, D. Zhang, P. Zhang, J. Zhao and J. Ji, Nat. Biomed. Eng., 2023, 7, 797–810 CrossRef CAS PubMed.
- M. Zhu, J. Zhuang, Z. Li, Q. Liu, R. Zhao, Z. Gao, A. C. Midgley, T. Qi, J. Tian, Z. Zhang, D. Kong, J. Tian, X. Yan and X. Huang, Nat. Nanotechnol., 2023, 18, 657–666 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |