
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synergy of nanocrystalline carbon nitride with Cu single atom catalyst leads to selective photocatalytic reduction of CO2 to methanol†
Tara M.
LeMercier
 a,
Madasamy
Thangamuthu
*a,
Emerson C.
Kohlrausch
a,
Yifan
Chen
a,
Craig T.
Stoppiello
e,
Michael W.
Fay
b,
Graham A.
Rance
b,
Gazi N.
Aliev
c,
Wolfgang
Theis
c,
Johannes
Biskupek
d,
Ute
Kaiser
d,
Anabel E.
Lanterna
a,
Jesum Alves
Fernandes
a and
Andrei N.
Khlobystov
*a
a,
Madasamy
Thangamuthu
*a,
Emerson C.
Kohlrausch
a,
Yifan
Chen
a,
Craig T.
Stoppiello
e,
Michael W.
Fay
b,
Graham A.
Rance
b,
Gazi N.
Aliev
c,
Wolfgang
Theis
c,
Johannes
Biskupek
d,
Ute
Kaiser
d,
Anabel E.
Lanterna
a,
Jesum Alves
Fernandes
a and
Andrei N.
Khlobystov
*a
aSchool of Chemistry, University of Nottingham, University Park, Nottingham NG7 2RD, UK. E-mail: madasamy.thangamuthu1@nottingham.ac.uk; andrei.khlobystov@nottingham.ac.uk
bNanoscale and Microscale Research Centre (nmRC), University of Nottingham, Nottingham, NG7 2QL, UK
cSchool of Physics and Astronomy, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK
dElectron Microscopy Group of Materials Science, Ulm University, 89081 Ulm, Germany
eCentre for Microscopy and Microanalysis, University of Queensland, St. Lucia, 4072, Australia
First published on 6th March 2024
Abstract
Carbon nitride (C3N4) possesses both a band gap in the visible range and a low-lying conduction band potential, suitable for water splitting and CO2 reduction reactions (CO2RR). Yet, bulk C3N4 (b-C3N4) suffers from structural disorder leading to sluggish reaction kinetics. This can be improved by graphitisation; however, current processes in the literature, lead to a variety of graphitised C3N4 (g-C3N4), making it difficult to link the degrees of graphitisation with the functional properties. Herein, we employ complementary analyses, including electrochemical impedance, photoluminescence, and photocurrent, to elucidate structure–property–function relationships. Guided by the descriptors, we developed a facile two-step annealing method that yields nanocrystalline carbon nitride (nc-C3N4), comprising nanoscale graphitic domains within an amorphous matrix. The nanocrystalline grains of nc-C3N4 allow effective immobilisation of Cu atoms and stabilisation of low oxidation states (Cu(I)). Electron microscopy and energy-dispersive X-ray spectroscopy demonstrate that Cu is atomically dispersed. Importantly, the addition of only 0.11 wt% of copper to nc-C3N4 drastically decreases the charge recombination and resistance to change transfer. The synergy of the Cu single-atom catalyst and nanocrystalline domains in carbon nitride (Cu/nc-C3N4) leads to a remarkable 99% selectivity towards methanol production with a rate of 316 μmol gcat−1 h−1 during the photocatalytic CO2RR, which is absent in Cu/b-C3N4.
1 Introduction
The necessity for effective environmental remediation is only further exacerbated by the current energy crisis, growing the momentum for replacing fossil fuels with sustainable energy sources. Carbon dioxide (CO2) is by far the greatest contributor to global warming. The available mitigation strategies to control future anthropogenic emissions can be divided into two categories: (i) carbon capture and sequestration (CCS) and (ii) carbon valorisation. The inherent difficulties of CCS concern the finiteness of geological sites where emissions can be stored before being removed or utilised, whilst the second incurs the benefits of upcycling CO2 to desirable products thus positively impacting the economy concomitant with reducing global emissions.1 For these reasons, global effort is devoted to stabilising the atmospheric abundance of CO2via carbon valorisation. The conventional thermocatalytic ventures of CO2 hydrogenation have been rapidly overtaken by potentially more sustainable methods, such as photo-, electro- and photo-electrocatalysis, which avoid the use of H2 predominantly sourced from steam reforming, and instead take advantage of the abundance and omnipresence of water.2–4 Photocatalysis in particular offers the added benefit of utilising the inexhaustible solar energy as the external bias for initiating reactions. This technology is being avidly explored as one of the best solutions to pursue a circular economy because it has the potential to transform CO2 into value-added products and chemical feedstocks.2,5,6,7On a practical level, the development of photocatalysts capable of CO2 conversion is still proving to be a challenge due to poor visible light absorption, inadequate adsorption sites, high charge carrier recombination and slow reaction kinetics.8–13 Methods to improve these have been explored extensively; employing defect engineering, the use of co-catalysts and heterojunction formation.14 Though there have been many triumphs in this field, optimisation of materials towards CO2RR is paramount in untangling the complex multielectron transfer processes.15 Currently, most photocatalysts employed for CO2RR require complex syntheses, costly processing, or the use of rare elements, such as silver, yttrium and ruthenium.16–18 To meet the green credentials of CO2RR, sustainable materials, such as carbon-based structures with a small amount of metal, should be used in a facile, effective synthesis with minimal waste and maximal yields of catalyst materials, that must be stable in water and be activated by visible light. Interestingly, carbon nitride (C3N4) meets these credentials, becoming a potentially efficient photocatalyst for CO2RR due to its band gap of 2.7 eV and more importantly its low-lying conduction band (CB), −1.3 eV vs. standard hydrogen electrode (SHE).19–24
Usually, carbon nitride materials are synthesised by the polycondensation of nitrogen-rich precursors, such as urea, cyanamide and melamine. As a solventless reaction this route is very attractive. However, the C3N4 formed in this manner are often poorly structurally defined, consisting of uncondensed polymeric and oligomeric melon structures with a mixture of amorphous, crystalline and quasi-crystalline domains.19 As such, these materials often suffer from slow kinetics, insufficient sunlight absorption and low surface area.25,26 Hence, understanding the role of the material and optimising it for efficient photocatalysis with desired reaction selectivity has been very challenging. Some optimisations include but are not limited to exfoliation, protonation, nitrogen vacancies, defects, and doping.27–30 The most effective methods in addressing these are heterojunction formation and supported single atom catalysts (SACs) or metal nanoclusters (MNCs).20,31–34 The work of Chen, et al. highlights the efficiency of heterojunction formation between C3N4 and WO3 and observed a seven-fold increase in CO2 photoreduction compared with pristine C3N4.35 Incorporating single-atom metal active sites can both enhance reactant adsorption and decrease photogenerated charge carrier recombination.20,36–40 The tri-s-triazine vacancies of C3N4 offer perfect sites for atom binding due to strong interactions with N lone pairs of electrons offering increased stabilisation to support them, thus avoiding agglomeration. In CO2RR it is most common to encounter Cu as SACs, typically chosen for its superiority towards C2+ products.41–44
Despite major advances in this area, there is an inherent lack of correlation between material structure and property and consequently its functionality. C3N4 materials are typically classified into four categories – bulk, graphitic, nanosheets and mesoporous. In reality, C3N4 materials are much more complicated than this simple classification, often existing as a combination or complex mixture of different phases. To better understand the correlation between structure–property–function we must thoroughly understand the catalysts composition at the nanoscale. Building this structure–property-function relationship is crucial for better, more efficient photocatalysis. In this study, we report a method for synthesising Cu/C3N4 SACs where no solvents, reagents or Cu precursor compounds are required. Cu atoms are deposited directly onto carbon nitride materials, which are themselves synthesised by a simple pyrolysis–calcination method from melamine. Firstly, we investigate the composition and structure of C3N4 at the nanoscale, using varying calcination temperatures and conditions that allow controlled formation of graphitised nanoscale domains embedded in an amorphous matrix (nc-C3N4). Low-dose transmission electron microscopy (TEM) imaging allows the visualisation of graphitised domains of carbon nitride in their pristine state without electron beam damage, thanks to a single-electron detector. We utilise photocurrent (PC) response as the key descriptor property for optimisation of the nc-C3N4 structure, which led us to an optimum platform for photocatalysis, as it can simultaneously effectively support Cu atoms and offer optimised physicochemical characteristics, such as decreased charge transfer (CT) resistance and recombination of charge carriers. We demonstrate that it is the synergy between the material components that improve all photophysical properties of the nanocrystalline carbon nitride due to the excellent interface achieved by a solvent-free atomic deposition, translating into enhanced catalytic activity of Cu/nc-C3N4 towards CO2 photoreduction to methanol.
2 Results
2.1 Catalyst preparation and structural characterisation
Carbon nitride is a complex material whose structure and properties are strongly dependent on the method of preparation, often suffering from poor structural definition which results in various environments for metal co-catalyst loading, and concomitantly resulting in poor selectivity towards CO2 reduction products.As post-synthetic annealing plays a crucial role in carbon nitride preparation, we utilised the photocurrent response of carbon nitride as a functional parameter for optimisation of the annealing conditions (Fig. 1a) demonstrating that 2 hours of annealing at 500 °C in air yields the material with maximum photocurrent at 17.6 μA cm−2 (44 times higher than the b-C3N4). Annealing for longer times or under an inert atmosphere have an adverse effect on charge carrier recombination (Fig. 1b).
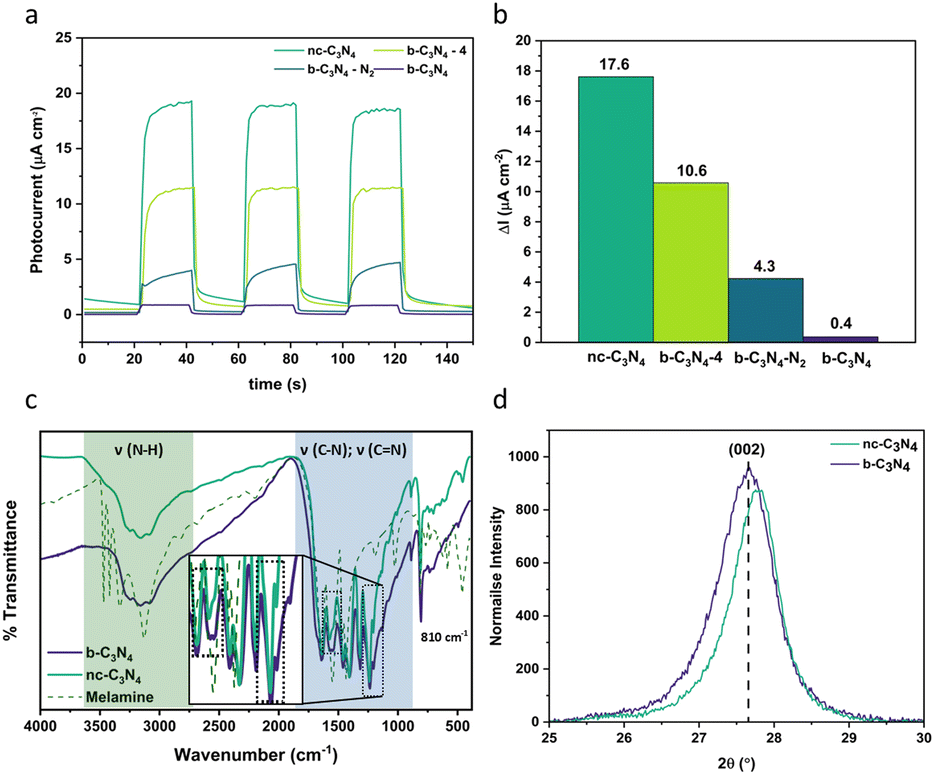 | ||
| Fig. 1 (a) Anodic photocurrent of different forms of carbon nitride prepared under different conditions follows the order nc-C3N4 (dark green) > b-C3N4-4 (light green) > b-C3N4-N2 (dark blue) > b-C3N4 (purple). (b) Change in anodic photocurrent response, ΔI is calculated by taking the difference between photocurrent response of on (75 s) and off (50 s) conditions. (c) Comparison between the FT-IR spectra of melamine precursor (dashed line), b-C3N4 (purple) and nc-C3N4 (dark green). (CN) domains: Aryl breathing mode at 810 cm−1 (sharp peak) and stretching modes between 1200–1600 cm−1 (blue), 2 changes between the relative intensity of samples are observed (black dashed boxes, insert). (NH) stretching modes (3000–3500 cm−1) are less prominent in the C3N4 materials compared to melamine (green). (d) Comparison of the PXRD (002) reflection peak for b-C3N4 (purple) and nc-C3N4 (dark green): a slight shift is observed towards higher 2θ after annealing. | ||
The process of graphitisation was monitored by Fourier transform infra-red (FT-IR) spectroscopy (Fig. 1c) and confirmed with Raman spectroscopy (Fig. S1†) and CHN analysis (Table 1). The typical (CN) bands are observed in the FT-IR spectra (Fig. 1c) for both b-C3N4 and nc-C3N4 with a breathing mode at 810 cm−1 and stretching modes between 1200–1600 cm−1. Differences in the relative intensities of bands at 1567 and 1546 cm−1 are observed. In combination with X-ray photoelectron spectroscopy (XPS) results (Fig. 2), we can attribute these to pyridinic type C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and tertiary amine type C–N bonds, respectively. Upon annealing, the amine-type stretch is reduced in intensity compared to the pyridinic CN stretch. Furthermore, the peaks at 1241 and 1207 cm−1 become narrower after annealing indicating a higher degree of order for C–NH–C stretches. As expected, the (NH) stretching modes (3000–3500 cm−1) are less prominent in the C3N4 materials compared to the melamine precursor.45–48 For both samples, b-C3N4 and nc-C3N4 (where nc stands for nanocrystalline, as shown later), Raman bands are observed on a baseline of increasing fluorescence towards lower Raman shift (Fig. S1†). The extent of fluorescence is reduced for the nc-C3N4 sample, consistent with a change in the degree of polymerisation following annealing. The oxidation onset temperature in air (Fig. S2†) increases significantly upon graphitisation, from 540 to 700 °C, showing that nc-C3N4 has higher thermal stability.
N and tertiary amine type C–N bonds, respectively. Upon annealing, the amine-type stretch is reduced in intensity compared to the pyridinic CN stretch. Furthermore, the peaks at 1241 and 1207 cm−1 become narrower after annealing indicating a higher degree of order for C–NH–C stretches. As expected, the (NH) stretching modes (3000–3500 cm−1) are less prominent in the C3N4 materials compared to the melamine precursor.45–48 For both samples, b-C3N4 and nc-C3N4 (where nc stands for nanocrystalline, as shown later), Raman bands are observed on a baseline of increasing fluorescence towards lower Raman shift (Fig. S1†). The extent of fluorescence is reduced for the nc-C3N4 sample, consistent with a change in the degree of polymerisation following annealing. The oxidation onset temperature in air (Fig. S2†) increases significantly upon graphitisation, from 540 to 700 °C, showing that nc-C3N4 has higher thermal stability.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N = 4:3 confirms the melon structure of carbon nitride materials
:N = 4:3 confirms the melon structure of carbon nitride materials
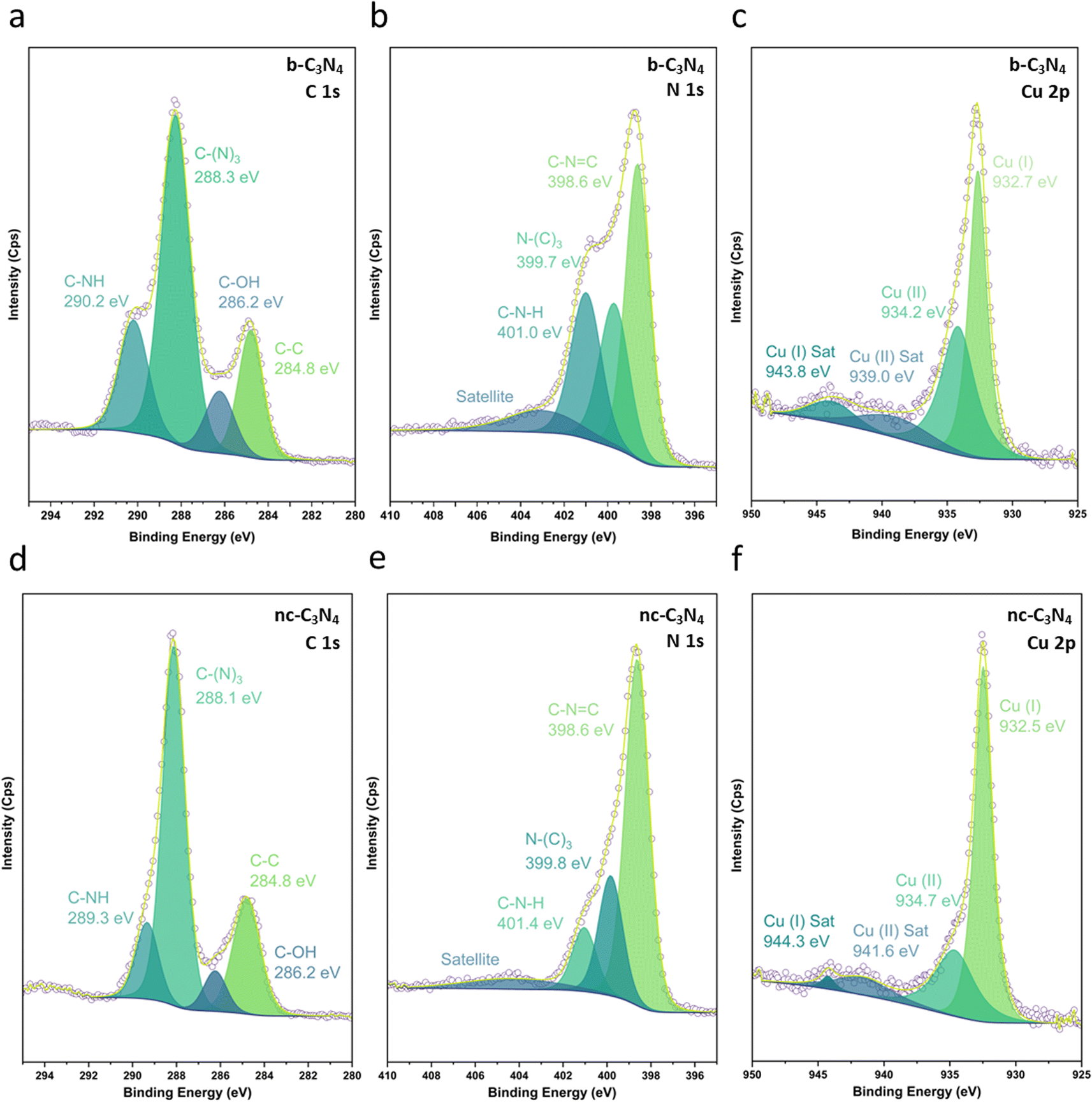 | ||
| Fig. 2 HR XPS spectra of Cu/b-C3N4 in the C 1s (a), N 1s (b) and Cu p3/2 (c) regions and Cu/nc-C3N4 in the C 1s (d), N 1s (e), and Cu p3/2 (f) regions. The raw data (purple circles) is fitted with peaks of oxidation states and satellites in a fitted envelope (yellow). | ||
The crystallinity of carbon nitride phases was assessed by powder X-ray diffraction (PXRD). Both b-C3N4 and nc-C3N4 possess the same diffraction peaks, characteristic of carbon nitride materials (Fig. S3†).48–50 The long-range in-plane structural motif of the tri-s-triazine units is characterised by the (100) reflection and the interlayer stacking by the (002) reflection.48 The width of the (100) peak decreases following thermal treatment, indicating that crystal strain is decreased. Scherrer analysis indicates the crystallite size increases by 1 nm upon annealing. Bragg analysis reveals a characteristic spacing of 0.693–0.695 nm whilst interlayer stacking shows a spacing of 0.320–0.322 nm and Scherrer analysis indicates the average number of layers increases from 35 to 41 under annealing conditions. Emulation of annealing conditions under thermogravimetric analysis (TGA), shows 17% weight loss, consistent with the yield from the original reaction (Fig. S4†). This loss is attributed to absorbed water (2%) and oxidation (15%). During calcination, there are two competing processes: (1) burning off leading to the reduction of stacked layers and (2) annealing of amorphous carbon nitride to the nanocrystalline form, leading to the increase of the number of the layers observed by Scherrer analysis. From this we can assume that the second process outcompetes the first. Furthermore, there is a shift to higher 2θ for the (002) diffraction, evidencing a slight reduction in interlayer distance upon annealing (Fig. 1d). Overall, b-C3N4 and nc-C3N4 are consistent with the melon form of C3N4.49,51 This finding is corroborated with CHN microanalysis whereby the overall atomic ratios for C and N are 3:4 and hydrogen content is decreased compared to the melon structure, confirming successful polymerisation (Table 1). Furthermore, CHN indicates further oxidation of the b-C3N4 occurs upon transforming to nc-C3N4 (1.6%). This is corroborated by TGA, wherein thermal treatment under inert conditions shows 6% less residual weight than for the same conditions in air (Fig. S4†).
XPS was used to evaluate C, N and Cu environments and comparatively evaluate how the material changes with annealing conditions and in turn how this impacts Cu binding. Fig. 2 shows C 1s, N 1s, and Cu 2p XPS for both b-C3N4 (top) and nc-C3N4 (bottom). Notably, upon heating, a significant reduction in the C–N–H peak was observed in both C and N 1s spectra, concomitant with an increase of the C–(N)3 peak (Table S1†). This is characteristic of deamination and successful polycondensation, whereby the N–H bridged melon oligomers convert into N–(C)3 bound tri-s-triazine units, corroborating the melon structure determined by Raman spectroscopy (Fig. S1†).52 The morphology of the materials was characterised by scanning electron microscopy (SEM) and TEM. SEM imaging of b-C3N4 shows a broad distribution of particle sizes, typically between 1 and 10 μm (Fig. S5†). Furthermore, particles are non-uniform and display an irregular morphology (Fig. 3a). In contrast, nc-C3N4 presents more regular domains, in the form of sheets, embedded in remnants of b-C3N4 (Fig. 3b). Bright-field high-resolution TEM (HRTEM) was used to characterise the nanocrystalline domains of nc-C3N4. Carbon nitride is highly susceptible to electron beam damage, which we mitigated by using a K3-IS single-electron detector which enabled us to reduce the electron dose to 5 × 103 e−1 nm−2 and reveal the native crystalline structure of the nc-C3N4 (b-C3N4 was not possible to image even in low dose TEM conditions). Imaging reveals 20 layers of edge-on lattice fringes of 0.32 nm, corresponding to interlayer stacking (002), in agreement with the PXRD data discussed above (Fig. 3c). Complementarily, bright-field AC-HRTEM was used to identify lattice fringes of 0.70 nm within the plane of crystalline domains. These correspond to a lattice parameter of the unit cell of in-plane tri-s-triazine (Fig. 3d), in agreement with previously proposed computational models,53 indicating a high degree of order within the graphitised grains of nc-C3N4. Electron microscopy imaging of b-C3N4 at high magnification is not possible due to the fast decomposition of the material under electron beam irradiation, even under low-dose conditions. This indicates that b-C3N4 has a lower degree of polymerisation than nc-C3N4.
 | ||
| Fig. 3 (a) SEM image of b-C3N4, showing irregular structure (white circles). (b) SEM image of nc-C3N4, showing sheet-like morphology embedded in b-C3N4 (white circles). (c) HRTEM image at 200 kV of interlayer stacking (002), of 0.32 nm between 20 layers (inset: higher magnification of the area in the red box with fast Fourier transform). (d) AC-HRTEM image of in-plane tri-s-triazine motif distance of 0.70 nm corresponding to (100) plane. The area shaded red on the structure depicts the unit cell of a tri-s-triazine lattice with lattice parameter a = 0.70 nm (inset: fast Fourier transform of the area shown in red box). | ||
Overall, structural characterisation confirms that under our annealing conditions, optimised for the photocurrent response, carbon nitride exists in a hybrid form of nanocrystalline domains embedded in a matrix of amorphous carbon nitride, which we term nc-C3N4. Previously, carbon nitride has been demonstrated as an effective support for both metal nanoclusters and single atom catalysts.54,55 With CO2RR in mind, the deposition of Cu was chosen due to its application in methanol synthesis and capabilities of efficiently adsorbing CO2.56–58 To ensure successful incorporation of Cu active centres, without perturbing the structure of carbon nitride by heat or any chemicals, including copper precursor compounds or solvents, deposition of metal atoms was carried out at room temperature from the gas phase using magnetron sputtering (Fig. 7b).55 Cu arrives at the surface of carbon nitride in atomic form, with low energy landing of copper atoms under our conditions.59
Weight loadings were determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) as 0.16 and 0.11 wt% for Cu/b-C3N4 and Cu/nc-C3N4 respectively. The absence of Cu diffractions in the PXRD patterns suggests that Cu exists in the form of atoms or sub-nm clusters (Fig. S3†). HRTEM imaging is consistent with the PXRD patterns, only one area was found to have two particles >1 nm with some smaller clusters distinguishable from the support (Fig. 4a). Aberration-corrected scanning transmission electron microscopy (AC-STEM) imaging shows metal centres more clearly confirming the nature of Cu to be predominantly single atoms (Fig. 4b), comprising 71% of the population of the metal species on carbon nitride, with 29% represented by clusters of atoms smaller than 0.3 nm in diameter (Fig. S6†). Considering the atomic radii of Cu in any oxidation state these clusters are unlikely to contain more than three atoms. HAADF-STEM-EDX corroborates the presence of Cu atoms and their preferential binding sites. Nanocrystalline domains are observed as areas of increased image contrast (Fig. 4c). Encircled (yellow) are areas measured: (i) shows domain with a length of 103 nm and width varying from 2.9 to 12.8 nm, and (ii) presents a length of 48 nm with a uniform width of ca. 1 nm. STEM-EDX mapping detects components of C (red), N (blue) and Cu (green) (Fig. 4d). The proximity expected of C (Fig. 4e) and N (Fig. 4f) within the nc-C3N4 structure is observed by the purple colour depicted. Deconvolution into individual elements allows a clearer evaluation of the nanocrystalline domains. nc-C3N4 is expected to have local areas of higher (g-C3N4) and lower (b-C3N4) density, which results in the areas of higher image contrast observed in both AC-STEM and AC-HRTEM micrographs. This suggests that nanodomains of crystalline carbon nitride are distributed throughout the nc-C3N4 sample and are typically surrounded by an amorphous matrix. As STEM, TEM and PXRD indicate, Cu exists in atomic or near-atomic form, distributed evenly over the whole sample. A brighter Cu signal correlates well with nanocrystalline areas, suggesting there are more binding sites along the nanocrystalline domains (Fig. 4g).
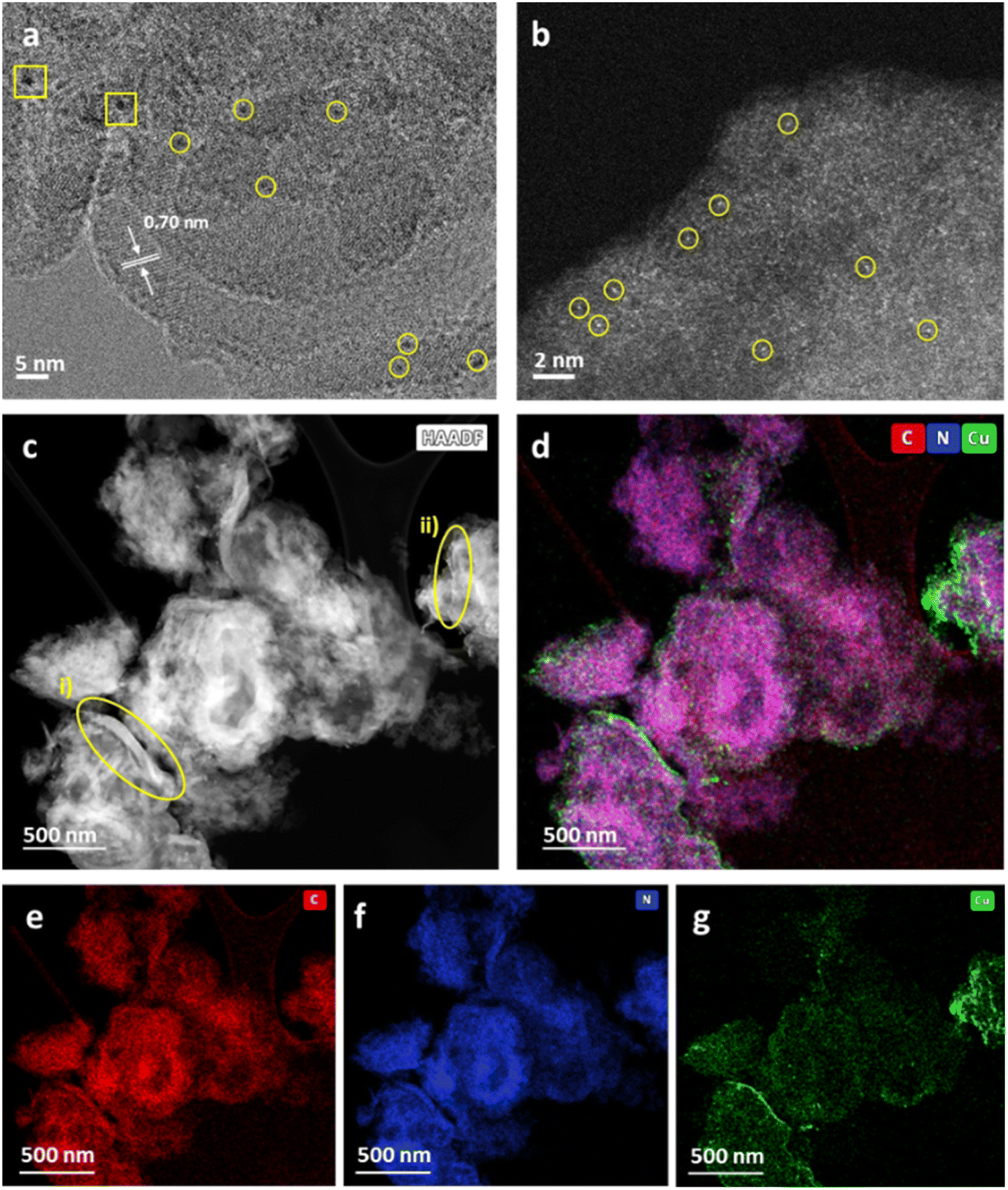 | ||
| Fig. 4 (a) HRTEM image at 200 kV of Cu/nc-C3N4 with a spacing of 0.7 nm between tri-s-triazine units, showing Cu atoms (yellow circles) and CuNPs (yellow squares). (b) AC-STEM image showing Cu atoms (yellow circles). (c) HAADF-STEM image of Cu/nc-C3N4, nanocrystalline domains pertain to brighter areas of the image and the remainder to amorphous carbon nitride domains. Measurable nanocrystalline domains are encircled (yellow). (d) Composed STEM-EDX map of all components overlapping: C (red), N (blue), Cu (green). (e), (f) and (g) Single elemental maps of C, N and Cu respectively. | ||
The Cu1+/Cu2+ ratio was determined by the XPS Cu 2p spectra of b-C3N4 (Fig. 2c) and nc-C3N4 (Fig. 2f), following the methodology outlined by Biesinger and co-works (Table S2 and S3†).60 While this method presumes an overlap in the contribution of Cu0 and Cu1+, in the case where only Cu single atoms are present and bond with C3N4, we assume that the contribution of Cu0 becomes negligible, supported by other works.61,62 Both samples presented Cu1+ and Cu2+ species. However, after thermal treatment, the interaction of Cu single atoms on the surface of nc-C3N4 drive for a notably higher Cu1+ content, around 70%, compared with the b-C3N4, with showed an equivalent 50% contribution for both Cu1+ and Cu2+. This finding implies that a decrease in the C–N–H species on the material's surface, led for a higher proportion of Cu1+ over Cu2+.
2.2 Optical and electrochemical characterisation
The optical absorption properties of the carbon nitride materials were studied using UV-Vis diffuse reflectance spectroscopy (Fig. S7†). All carbon nitride samples show an absorption edge beginning at 450 nm, corresponding to the visible light absorption. Deposition of Cu leads to higher absorption intensities and increased absorption at λ < 450 nm. This phenomenon is characteristic of ligand-to-metal charge transfer (LMCT) and has been noticed to occur in compounds of similar types of nitrogen-bearing ligands and Cu.63,64 The band gap values of the materials were obtained by Tauc plot analysis and are in the range of 2.6–2.67 eV (Fig. S8†).65,66 To explore the band structure of the catalysts, we measured the flat band potential of b-C3N4 using Mott–Schottky analysis. The CB potential is at −0.87 V vs. AgCl/Ag (−1.08 V vs. SHE), which is negative enough to perform photocatalytic CO2RR (Fig. S9†). The valence band (VB) potentials of +1.8 V vs. AgCl/Ag (1.59 V vs. SHE) in b-C3N4 and +1.73 V vs. AgCl/Ag (1.52 V vs. SHE) in nc-C3N4 were derived from the bandgap and CB potential values, as shown in (Fig. 5a).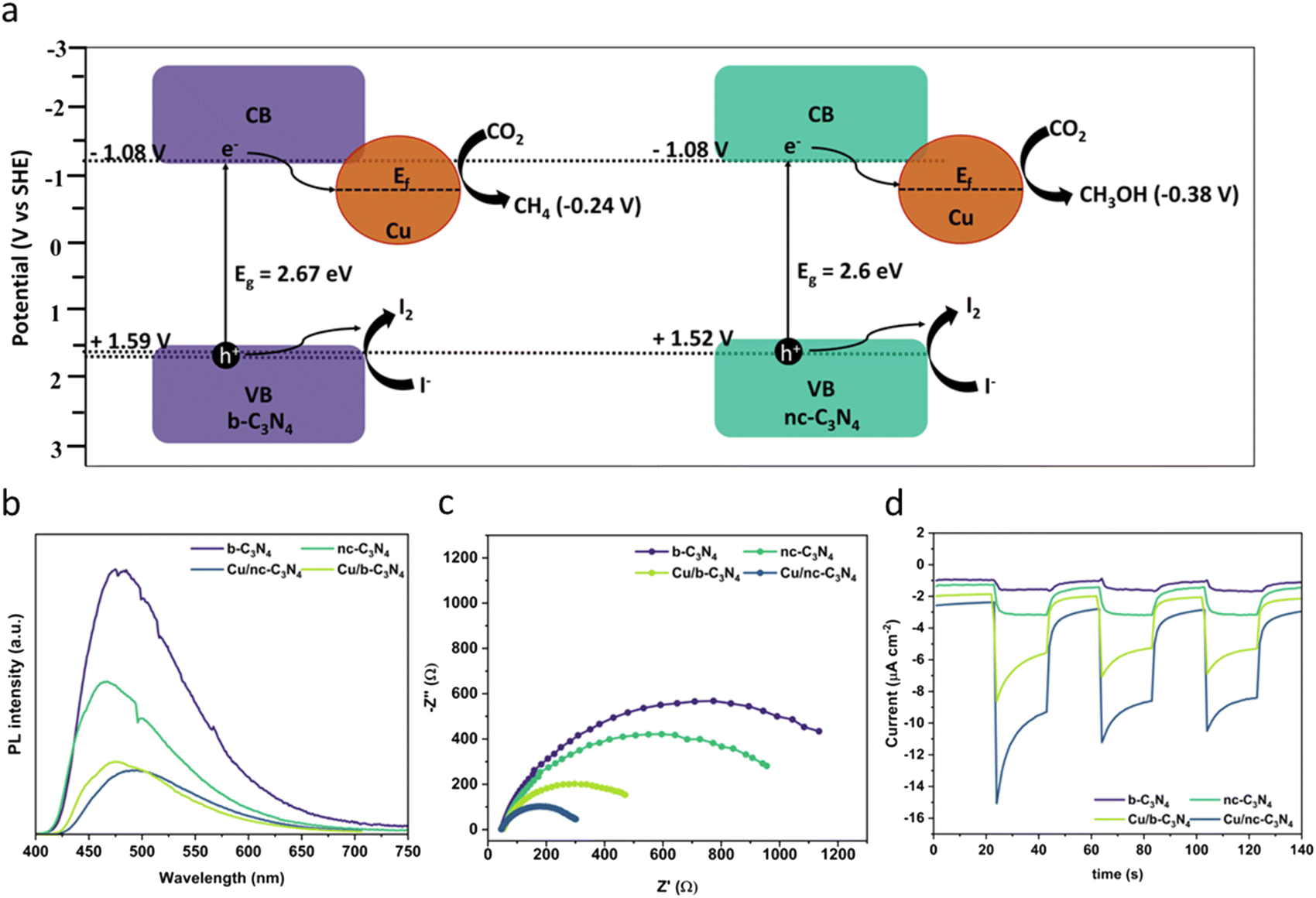 | ||
| Fig. 5 (a) Proposed mechanism for CO2 reduction showing the different band alignments for b-C3N4 and nc-C3N4 derived from Fig. S9–S11† (−0.87 V vs. AgCl/Ag corresponds to −1.08 V vs. SHE). (b) PL spectra (excitation = 445 nm) showing decreasing emission with increased graphitisation and Cu loading. (c) EIS measurements of carbon nitride materials with and without Cu. (d) Photocurrent response showing increasing concentration of charge carriers formed due to light absorption in the following order Cu/nc-C3N4 (dark blue) > Cu/b-C3N4 (light green) > nc-C3N4 (dark green) > b-C3N4 (purple). | ||
The effect of Cu atoms on radiative recombination was investigated by photoluminescence spectroscopy (PL) using 384 nm excitation. A clear trend is observed (Fig. 5b), emission intensity decreases with graphitisation and Cu atom loading, indicating that both decrease the radiative recombination of charge carriers in carbon nitride. Interestingly, graphitisation decreases PL by 42% upon transition from b-C3N4 to nc-C3N4, whereas Cu incorporation decreases PL by 73% for b-C3N4 material and 59% for nc-C3N4. Additionally, we tested the charge transfer property of the catalyst using electrochemical impedance spectroscopy (EIS) and observed that nc-C3N4 shows a small semicircle with an RCT value of 1172 ohms, which is 17% less resistant compared to b-C3N4 (1407 ohms). Incorporation of Cu atoms promotes better charge transfer by 65% and 77% for Cu/b-C3N4 (487 ohms) and Cu/nc-C3N4 (270 ohms), respectively, when compared to the copper-free samples (Fig. 5c). To validate the EIS results and test the photogenerated charge separation, we tested the photocurrent responses of the catalyst before and after loading Cu. Increased photocurrent response correlates with graphitisation and is further increased by Cu loading (Fig. 5d). Change in photocurrent density response is largest for Cu/nc-C3N4 with −7.836 μA cm−2, almost double the response seen for Cu/b-C3N4 with −4.563 μA cm−2 (Fig. S10†). These agree very well with EIS and PL results. The sharp response in Cu-loaded samples indicates a rapid charge accumulation potentially due to the lack of electron scavengers used in this experiment.
2.3 Photocatalytic CO2 reduction
The photocatalytic CO2 reduction activities of b-C3N4, nc-C3N4, Cu/b-C3N4 and Cu/nc-C3N4 were tested in the presence of DI water (pH ∼ 7) containing 2 mM NaI, as a hole scavenger under solar simulated irradiation. All catalysts produce CH4 as the only gas product. Cu/b-C3N4 shows a formation rate two times higher (0.28 μmol gcat−1 h−1) than b-C3N4 (0.13 μmol gcat−1 h−1) and three times higher than the nanocrystalline samples, Cu/nc-C3N4 (0.09 μmol gcat−1 h−1) and nc-C3N4 (0.07 μmol gcat−1 h−1) at 135 min on stream (Fig. 6a). In stark contrast, the addition of only 0.11 wt% of Cu to nc-C3N4 leads to a radical change in selectivity, directing CO2 conversion to liquid product formation. Methanol is produced at a rate of 316 μmol gcat−1 h−1 (Fig. 6b). This radical change is attributed to the nanocrystalline domains improving charge carrier mobility when compared to the bulk material.72 And is related to the fact that Cu atoms help suppress the radiative recombination of excitons in the nanocrystalline form of carbon nitride significantly more than in the amorphous form (Fig. 5b) which leads to a 7-fold increase in photocurrent in Cu/nc-C3N4 (Fig. 5d). This material combination is rarely reported in the literature and to the best of our knowledge the initial formation rate of methanol achieved in this work is 9 times higher than the greatest already reported (Fig. 6b).67–71 Furthermore, we have measured the oxidation products of the reaction using UV-Vis spectroscopy and observed no evidence of IO3− formation. Hence, under the present condition the hole scavenger I− would form I2 or I3− (Fig. 5a). Running the process over several consecutive cycles indicated that methanol may be consumed over the long runs of the catalyst (Fig. S11†), which would necessitate the product separation in future applications of Cu/nc-C3N4. The total apparent quantum yield (AQY) for CH4 and CH3OH was assessed at various monochromatic wavelengths from 420–600 nm (Fig. S12†). The profile matches the light absorption behaviour of nc-C3N4 indicating that the process is photo driven. The AQY at 420 nm is 0.06%, considering the 8 and 6 electron processes involved in CH4 and CH3OH formation from CO2, respectively.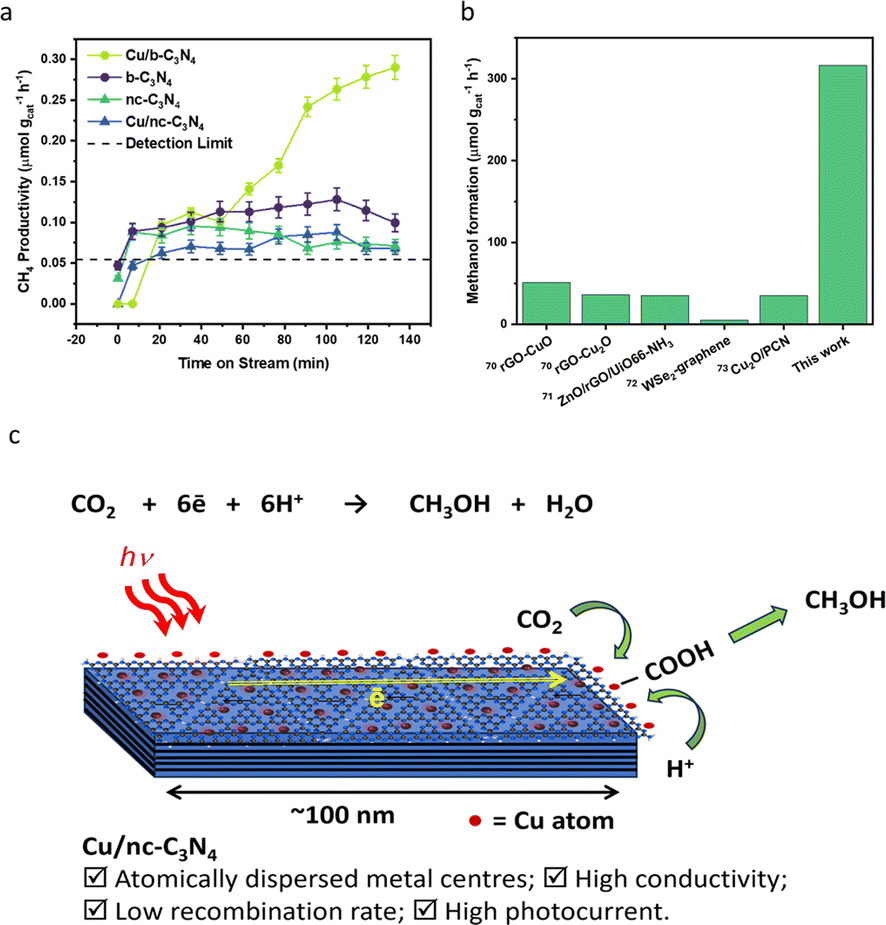 | ||
| Fig. 6 Catalyst performance in photocatalytic reduction of CO2. (a) CH4 productivity over time (μmol gcat−1 h−1) for Cu/b-C3N4 (light green circle), Cu/nc-C3N4 (blue triangle), b-C3N4 (purple circle) and nc-C3N4 (green triangle). (b) Literature comparison of methanol formation rate (μmol gcat−1 h−1) produced over other visible light active catalysts and Cu/nc-C3N4 (this work) catalyst (Fig. S11†).67–71 (c) Schematic representation of CO2 photoreduction on a nanocrystalline domain of carbon nitride decorated with single copper atoms. | ||
3 Discussion
The synergy between the nanocrystalline domains of nc-C3N4 and Cu single atoms has significant implications for the functional properties of this material, with particular relevance to applications in photocatalysis. We adopted an approach where anodic photocurrent response in carbon nitride films deposited on FTO is used as a guide for optimisation of graphitisation conditions. This approach allows the identification of a form of carbon nitride (nc-C3N4) where nanoscale crystalline domains of C3N4 are embedded in a matrix of amorphous b-C3N4 material, as revealed by HRTEM and SEM imaging (Fig. 3). We have extensively investigated this aspect by various methods, demonstrating that some techniques are more appropriate than others for this type of structural evaluation. Whilst IR and Raman spectroscopies are not very sensitive to the changes taking place during the annealing process at 500 °C; PXRD, HRTEM and XPS are highly diagnostic. Carbon nitrides are in general highly sensitive to electron beam damage, and because of this, the structural characterisation of b-C3N4 is challenging due to rapid decomposition under typical electron beam voltages used for TEM analysis (i.e., 80–200 kV). However, the partial graphitisation in nc-C3N4 sufficiently improves the stability of the material under low-dose TEM conditions (single-electron detector) allowing, for example, to measure projections of the (002) planes in HRTEM images with an interlayer spacing of 0.32 nm (Fig. 3c), which is in agreement with the average interplanar distance of 0.321 nm obtained from PXRD analysis and consistent with those reported in literature.73 The in-plane structure of the nanocrystalline domains in nc-C3N4 can also be readily imaged by AC-HRTEM, revealing the tri-s-triazine motif distance of 0.70 nm corresponding to the unit cell lattice parameter (Fig. 3d). Electron microscopy analysis demonstrates that nanocrystalline domains in nc-C3N4 have highly ordered tri-s-triazine sites, ideal for the stabilisation of single metal atoms.The structural and chemical changes caused by annealing in carbon nitride, such as the significant reduction of C–N–H bridge groups shown by XPS (Fig. 2d and e) translate well to the functional properties. Once again, not all methods are sensitive to the formation of nanoscale graphitised carbon nitride domains, as UV-Vis absorption spectra are virtually the same before and after annealing, but there is a dramatic change in PL indicating a significantly faster radiative charge carrier recombination in b-C3N4 compared to nc-C3N4, which can be linked to the larger number of defects acting as recombination centres in the former. Indeed, EIS shows that the nanoscale graphitised domains in carbon nitride decrease resistance to charge transfer by 17%. The most drastic effect of nanoscale graphitisation of carbon nitride is on photocurrent generation, a three-fold increase in the case of nc-C3N4, which can be attributed to a combined effect of better charge mobility and a lower likelihood of radiative recombination due to an increased in-plane electron migration in the nanocrystalline domains.37,72,74,75 In addition, it is important to evaluate the forms of carbon nitride for metal binding, as the metal–semiconductor synergy is important for tuning activity and selectivity of photocatalytic CO2RR. Our method of Cu atoms deposition avoids any artefacts associated with changes in the delicate structure of carbon nitride due to solvents, reagents, or harsh conditions (Fig. 7b) and allows evaluation of the metal–semiconductor interactions in their purest form. Previous studies showed that post-synthesis deposition of metal by wet chemistry methods onto carbon nitride is more likely to lead to nanocluster formation than if the metal is added during the synthesis of carbon nitride.76 Remarkably, in our case, AC-STEM imaging of Cu/nc-C3N4 material reveals that over 70% of the metal loaded is in the form of single-atoms adsorbed on carbon nitride, with the rest forming very small clusters up to ca. 3 atoms of copper (Fig. 4b). STEM-EDX elemental mapping shows that copper atoms are distributed over the surface of nc-C3N4 particles, with a higher concentration along the edges of the nanocrystalline domains (Fig. 4g). Such a fine dispersion of metal means that only 0.11 wt% of Cu vastly enhances the functional properties of carbon nitride. Indeed, radiative recombination is reduced by 59% in nc-C3N4 and 73% in b-C3N4 by the presence of Cu atoms. Both see an improvement, attributed to the Cu centres acting as electron sinks due to Cu reduction potentials being more positive than the bottom of the conduction band of carbon nitride.77 The difference seen by nc-C3N4 suggests that the enhanced electron migration results in electron entrapment at sites rich in b-C3N4 character, thus further decreasing PL intensity. Furthermore, the presence of copper suppresses the resistance of both nc-C3N4 and b-C3N4 by 77% and 65%, respectively, as measured by EIS (Fig. 5c), indicating that Cu atoms help to extract negative charge carriers from carbon nitride. XPS measurements reveal an unexpected influence of the degree of graphitisation of carbon nitride on the oxidation state of copper. nc-C3N4 largely bears Cu(I), which is widely accepted to be the active site for methanol synthesis, both in thermal catalysis and photocatalysis.78–81 The preference for Cu(I) binding is explained by considering the increased number of crystalline domains, which possess the pure melon-type carbon nitride character in nc-C3N4. As tetrahedrally coordinated Cu(I) has a radius of 0.074 nm,82 considering a Cu–N bond length of 0.196 nm (ref. 83) there are three possible coordination sites for each nc-C3N4 heptazine cavity (Fig. S13†) which have the N–N distance of 0.26 nm. It is instructive to compare the effect of metal atoms on carbon nitride activity in CO2 reduction reaction. The addition of Cu atoms to b-C3N4 increases methane production by around 30%, indicating better charge transfer as seen by EIS and photocurrent measurements (Fig. 5c and d). However, the addition of only 0.11 wt% of Cu to nc-C3N4 leads to a radical change in selectivity, directing CO2 conversion to methanol at a rate of 316 μmol gcat−1 h−1 (Fig. 6b). This is related to the fact that Cu atoms are attached directly to nanocrystalline domains, both in-plane (Fig. 4a and b) and at the edges of the plane (Fig. 4g), as evident from electron microscopy. Fig. S14† highlights the Cu binding with nc-C3N4 both in the graphitised and amorphous domains. Such close contact between the Cu atoms and highly ordered planes of semiconductor may allow fast transport of electrons to catalytic centres, where they could be readily utilised for CO2 conversion. Indeed, the addition of Cu atoms suppresses the radiative recombination of excitons in the nanocrystalline form of carbon nitride significantly more than in the amorphous form (Fig. 5b) which leads to a seven-fold increase in photocurrent in Cu/nc-C3N4 (Fig. 5d). This, coupled with the significantly reduced charge transfer resistance of Cu/nc-C3N4 (270 ohms; Fig. 5c), ensures that photo-generated electrons are readily translated to Cu-methanol production sites (Fig. 6c).
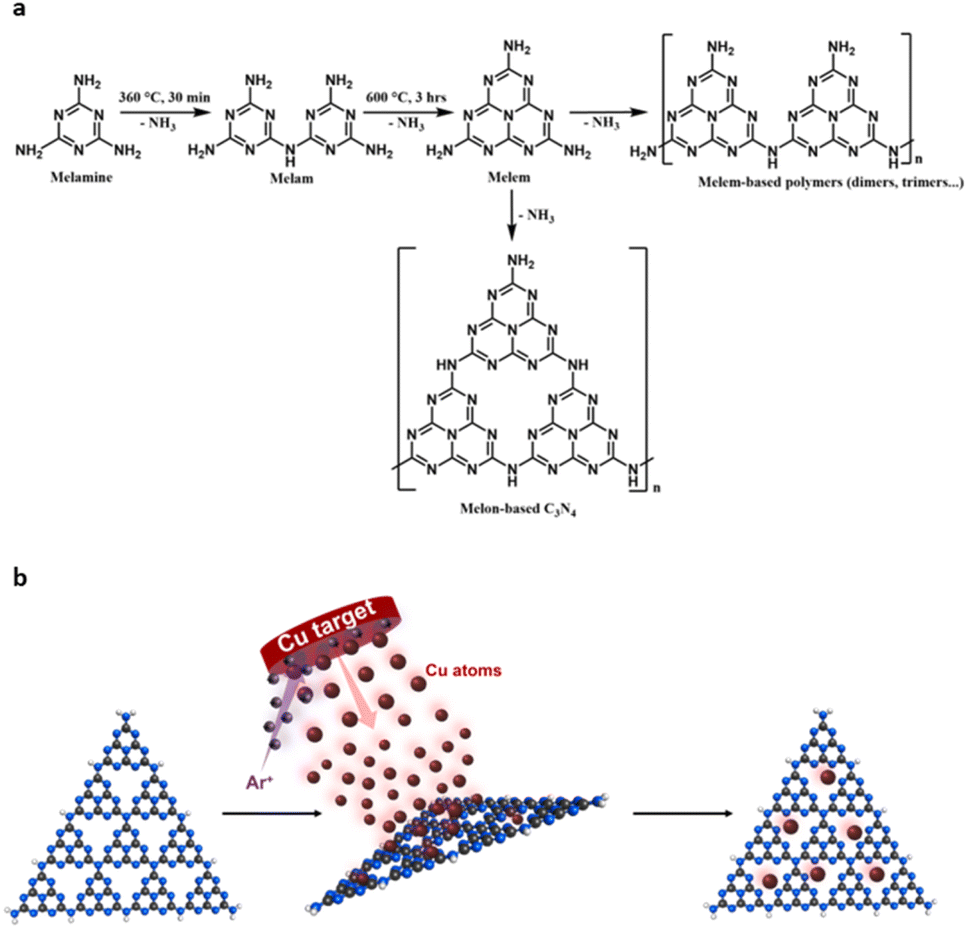 | ||
| Fig. 7 (a) Synthesis of b-C3N4 from melamine via a two-step calcination method. Polycondensation under these conditions can yield melem- and/or melon-based polymers. (b) Schematic representation of Cu atoms deposition by sputtering metal atoms from the solid metal target in vacuum directly onto carbon nitride (copper red, carbon grey, nitrogen blue, hydrogen white). | ||
4 Conclusions
Carbon nitride is a highly promising catalyst for CO2 conversion to high-value products, but there are many open questions regarding the forms of this semiconductor best suited for photocatalysis. In this study, we demonstrate annealing at 500 °C for 2 hours leads to the formation of nanoscale domains of graphitic carbon nitride embedded in a matrix of amorphous carbon nitride – nc-C3N4. Such subtle changes are practically undetectable in bulk methods of characterisation, but they produce drastic changes in the functional properties of carbon nitride decreasing resistance to charge transfer by 17%, recombination of charge carriers measured by PL by 42% and increasing photocurrent three-fold. Importantly, nc-C3N4 enables the stabilisation of single copper atoms both in-plane and at the edges of graphitised nanodomains, as revealed by low-dose HRTEM imaging employing a single-electron detector and STEM-EDX spectral imaging. It is the synergy between the material components that improve all photophysical properties of the nanocrystalline carbon nitride due to the excellent interface achieved by a solvent-free atomic deposition, translating into enhanced catalytic activity of Cu/nc-C3N4 towards CO2 photoreduction to methanol. Importantly, a similarly dispersed atomic Cu on an amorphous form of carbon nitride did not change the catalytic activity of Cu/b-C3N4, emphasising the importance of crystalline domains and Cu atoms both co-existing in close contact for the efficient CO2 reduction catalysis. Our approach links the structure and properties of carbon nitride and provides a framework and methodology for future search for effective photocatalysts using the degree of local graphitisation as a basis for optimisation.5 Experimental
5.1 Materials and methods
Standard reagents, including melamine and sodium iodide, were purchased from Sigma-Aldrich Chemicals and were used as received. Carbon nitrides were synthesized by polycondensation of melamine (Fig. 7a). Post-heat treatments of b-C3N4 were then performed as detailed below.5.2 Preparation of bulk carbon nitride (b-C3N4)
Typically, melamine (8 g) was placed in a quartz crucible with a loose-fitting lid and heated to 360 °C with a heating rate of 5 °C min−1 and held at 360 °C in the air for 30 minutes. Subsequently, the solid was heated to 600 °C at a heating rate of 5 °C min−1 and held at that temperature for 3 hours. This process yields a compressed yellow powder, which was ground to a fine powder and sieved (250 μm), with 52% yield, and denoted as b-C3N4.5.3 Preparation of nanocrystalline carbon nitride (nc-C3N4)
Nanocrystalline carbon nitride (nc-C3N4) was obtained by annealing the b-C3N4 at 500 °C for 2 hours to obtain an 83% yield. Other samples were prepared similarly (Table 2).| Sample | Annealing conditions |
|---|---|
| nc-C3N4 | 500 °C, 2 h, air |
| b-C3N4-4 | 500 °C, 4 h, air |
| b-C3N4-N2 | 500 °C, 2 h, nitrogen |
5.4 Preparation of Cu SACs
Cu SACs were obtained by using an AJA magnetron sputtering system (Fig. 7b). Briefly, a typical sample of carbon nitride (1.5 g) was placed in the glove box and heated under vacuum for 5 hours (120 °C) to remove any moisture. Then, the dried C3N4 analogues were transferred to a custom-built stirring sample holder which was transferred to the load-lock chamber. Once the pressure reached 10−6 torr (15 min), the sample was transferred to the main chamber and left until pressure stabilised (10−7 torr). The Cu depositions were carried out at room temperature with a working pressure of 3 × 10−3 torr Ar plasma applied to a Cu target (99.99%) with a current of 300 mA for 30 minutes.5.5 Materials characterisation
Fourier transform infrared (FT-IR) spectra were recorded at room temperature using a Bruker Tensor 27 spectrometer. All samples were analysed by the KBr pellet method. Typically, a small amount of sample (1–2 wt%) was ground with KBr (125 mg) in an agate mortar and pestle, and thus formed powder was compressed at 8 tonnes for 5 minutes. CHN microanalysis was performed by analytical services at the University of Nottingham. Scanning electron microscopic (SEM) images of the samples were obtained using a JEOL 7000F Field Emission Gun microscope using a 15 kV e-beam and the elemental mapping was performed using an Oxford Instruments ULTIM MAX 100 and Aztec EDX (energy dispersive X-ray) system. Nanocluster size and atomic structure were characterized by a JEOL JEM-2100F aberration-corrected scanning transmission electron microscope (AC-STEM) equipped with a Cs probe corrector (CEOS) at a convergence angle of 19 mrad and annular dark field detector (ADF) operating with an inner angle of 31 mrad and outer angle of 82 mrad at 200 kV. The bright field (BF) detector was also used in parallel. High-resolution transmission electron microscopy (HRTEM) was performed using a JEOL FEG-TEM microscope equipped with a single-electron detector Gatan K3-IS camera operated at 200 kV. Aberration correction (AC-HRTEM) was performed at 80 kV using a chromatic and spherical aberration-corrected TEM (SALVE, http://www.salve-center.de). Additional scanning (S)TEM together with EDX mapping was performed at 200 kV using a Thermofisher Talos 200×. TEM samples were prepared via a drop casting technique, where samples were first dispersed in propan-2-ol using ultrasonication and thus formed suspension was deposited on an Au grid mounted “lacey” carbon films (Agar). All TEM images were processed using Gatan Digital Micrograph. Thermogravimetric analysis (TGA) was executed using a TA Q500 Thermogravimetric Analyzer. All samples were loaded in a platinum pan for analysis under air atmosphere using the following programme: ramp at 10 °C min−1 from 20 to 1000 °C, hold for 10 min at 1000 °C, and air flow at 60 mL min−1, unless otherwise stated. Powder X-ray diffraction (PXRD) measurements were performed using a PANalytical X'Pert Pro diffractometer fitted with a Cu K(α) radiation source (λ = 1.5432 Å, 40 kV 40 mA). Sample adhesion was eased with propan-2-ol addition to a zero background Si plate holder. Typical parameters were start angle: 5°, stop angle: 60°, step size: 0.0525°, time/step: 6080 s, and scan rate: 0.00220° s−1. Inductively coupled plasma-optical emission spectroscopy (ICP-OES) measurements were performed on a PerkinElmer Optima 2000 spectrometer, with Cu/carbon nitride catalysts (10 mg) digested using aqua regia (2 mL) and diluted to 10 mL with a 5% HCl solution. X-ray photoelectron spectroscopy (XPS) was performed using a Kratos AXIS SUPRA PLUS instrument with a monochromatic Al Kα X-ray source (hν = 1486.6 eV) operated at room temperature with 10 mA emission current and 12 kV anode potential. The electron collection spot size was ca. 700 × 300 μm2. A pass energy of 160 eV was used for the survey scans and 20 eV for the high-resolution scans. Spectra were converted into VAMAS format for further analysis. The data was processed using CASA-XPS software. Charge correction in reference to C 1s at 284 eV.84 Ultraviolet-visible spectroscopy (UV-Vis) was performed on an Agilent Cary 5000 UV-Vis NIR Absorption spectrometer using a DRA-900 InGaAs integrating sphere. Photoluminescence (PL) measurements were performed on an Edinburgh Instruments FLS 980 with a 420 nm long pass filter. The excitation wavelength was fixed at 384 nm and the emission range 404–800 nm was scanned with Δλ = 1. Raman spectroscopy was performed using a HORIBA LabRAM HR Raman microscope. Samples were prepared by drop casting onto Si (100) chips from propan-2-ol suspension. Spectra were acquired using a 785 nm laser (at ∼20 mW (100%) power), a 100× objective and a 200 μm confocal pinhole. To simultaneously scan a range of Raman shifts, a 300 lines mm−1 rotatable diffraction grating along a path length of 800 mm was employed. Spectra were detected using a Synapse CCD detector (1024 pixels) thermoelectrically cooled to −60 °C.5.6 Photoelectrochemical characterisation
All the electrochemical and photoelectrochemical measurements were performed in a conventional three-electrode (photocatalyst film deposited on FTO substrate as working electrode, AgCl/Ag reference electrode, and a platinum wire counter electrode) cell using an electrochemical analyser (IVIUM Technologies). A 0.1 M NaHCO3 (pH ∼ 7.6) solution was used as the electrolyte without any hole-scavenger. The photocatalyst film on FTO was prepared by electrophoretic deposition method. Briefly, 1 mg mL−1 photocatalyst suspension in acetone was prepared by ultrasonication for 1 hour and then 10 mg of I2 was added into the suspension as a charging agent. Two identical FTO plates were immersed into the suspension with a distance of 2 cm and 50 V DC bias was applied for 5 min. A film was deposited onto the negative electrode (cathode). Then, the substrates were dried at 150 °C for 1 hour in an oven to remove any residues before electrochemical tests. The area of the photocatalyst thin film was 1 × 1 cm2. A 300 W Xe lamp light source with an AM 1.5 filter was used for photocurrent measurement. The irradiation intensity on the sample was measured to be 30 mW cm−2 (∼0.3 sun) using a spectroradiometer (StellarNet). The charge transfer resistance of the material was estimated using electrochemical impedance spectroscopy (EIS) at a constant bias of 1.1 V vs. AgCl/Ag, with frequency ranging from 100 kHz to 1 Hz with a 10 mV amplitude.5.7 Photocatalytic CO2 reduction measurements
The photocatalytic activity of the catalysts was tested in a custom-built Pyrex continuous flow photoreactor (Fig. S15†), equipped with two mass flow controllers. The reactor was purged with high-purity Ar to eliminate any residual peaks. A 10 mL catalyst suspension (1 mg mL−1 in water) containing 2 mM NaI, which was used as a hole scavenger to improve charge separation, was placed in the reactor. The solution was saturated with CO2 at a rate of 5 sccm for 1 h. Subsequently, the CO2 flow was reduced to 0.5 sccm before the reaction commenced. A control measurement was performed under Ar with a flow rate of 0.5 sccm to confirm the carbon source of all carbonaceous products is CO2 (Fig. S16†). A 300 W xenon arc lamp (Sci-Sun 300 solar simulator) was used to irradiate the catalyst suspension at a working distance of 15 cm. Gas products of the CO2RR were measured by an Agilent 7890B gas chromatographer equipped with a flame ionisation detector (FID), thermal conductivity detector (TCD) and mass spectrometry detector (MSD). All data was processed using MassHunter software and the peak area was converted to the concentration using the calibration curve, which was obtained by a standard gas mixture. The apparent quantum yield (AQY) of the gas products was determined by performing photocatalytic experiments at specific wavelengths by using an appropriate bandpass filter and the AQY value was obtained using eqn (1).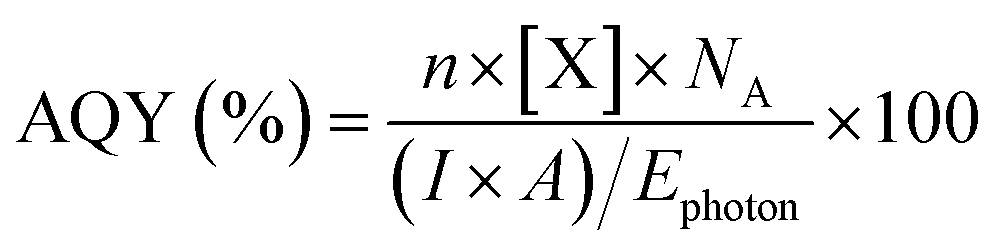 | (1) |
Liquid products were measured by 1H NMR spectroscopy using a Bruker AV(III) 500 with solvent (H2O) suppression (Fig. S17†). An aliquot of the water from the reactor (500 μL) was added to D2O (48 μL) and an aqueous solution of DMSO (4 mM, 40 μL) was added to act as an internal standard, and the concentration was calculated using eqn (2).85
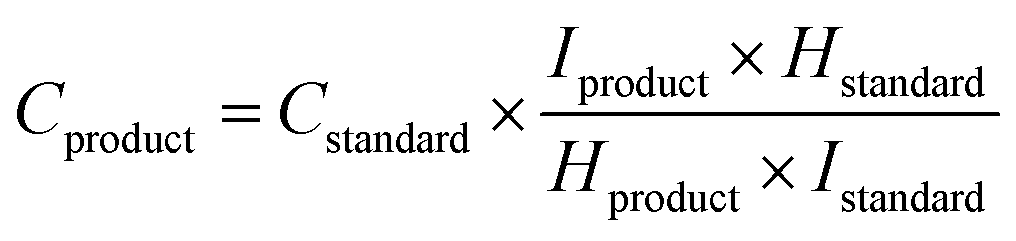 | (2) |
Author contributions
T. M. L., M. T. and A. N. K. developed the methodology. J. B. and U. K. performed AC-HRTEM imaging and STEM-EDS mapping. T. M. L. and E. C. K. synthesised the materials and analysed and interpreted XPS results. M. W. F. and Y. C. aided in acquisition of HRTEM. M. T. performed SEM imaging and all electrochemical characterisations. C. T. S. performed XPS measurements. G. A. R. performed Raman spectroscopy and aided in interpretation. G. N. A., and W. T. performed AC-STEM imaging. T. M. L. performed all other characterisations and analyses; interpreted the data and wrote the manuscript. A. N. K., A. E. L., M. T. and J. A. F. supervised the project and reviewed and edited the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research is supported by the Engineering and Physical Sciences Research Council (Metal Atoms on Surfaces & Interfaces (MASI) for Sustainable Future; EP/V000055/1). J. B. and U. K. acknowledge the financial support of the German Research Foundation (DFG) with the Collaborative Research Center SFB TRR 234 Catalight, (grant # 364549901). FEGTEM work was supported by the Engineering and Physical Sciences Research Council (EPSRC) [under grant EP/W006413/1] and the University of Nottingham Strategic Innovation Fund. C. T. S. acknowledges the facilities, and technical assistance of the Microscopy Australia Facility at the Centre for Microscopy and Microanalysis, The University of Queensland. We are grateful to the Nottingham Nanoscale and Microscale Research Centre (nmRC) for access to equipment.References
- Y. Deng, J. Li, Y. Miao and D. Izikowitz, Energy Rep., 2021, 7, 3506–3516 CrossRef.
- I. Ganesh, Renew. Sustain. Energy Rev., 2014, 31, 221–257 CrossRef CAS.
- N. Muradov, in Compendium of Hydrogen Energy, ed. V. Subramani, A. Basile and T. N. Veziroğlu, Woodhead Publishing, Oxford, 2015, pp. 489–522, DOI:10.1016/B978-1-78242-361-4.00017-0.
- H. Ooka, M. C. Figueiredo and M. T. M. Koper, Langmuir, 2017, 33, 9307–9313 CrossRef CAS PubMed.
- Z. Tong, D. Yang, Z. Li, Y. Nan, F. Ding, Y. Shen and Z. Jiang, ACS Nano, 2017, 11, 1103–1112 CrossRef CAS PubMed.
- R.-P. Ye, J. Ding, W. Gong, M. D. Argyle, Q. Zhong, Y. Wang, C. K. Russell, Z. Xu, A. G. Russell, Q. Li, M. Fan and Y.-G. Yao, Nat. Commun., 2019, 10, 5698 CrossRef CAS PubMed.
- W. Dongliang, M. Wenliang, Z. Huairong, L. Guixian, Y. Yong and L. Hongwei, Energy, 2021, 231, 120970 CrossRef.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC.
- M. Ahmad, E. Ahmed, Z. L. Hong, X. L. Jiao, T. Abbas and N. R. Khalid, Appl. Surf. Sci., 2013, 285, 702–712 CrossRef CAS.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- C. Cheng, A. Amini, C. Zhu, Z. Xu, H. Song and N. Wang, Sci. Rep., 2015, 4, 4181 CrossRef PubMed.
- M. H. A. Azqhandi, B. Vasheghani, F. H. Rajabi and M. Keramati, Results Phys., 2017, 7, 1106–1114 CrossRef.
- K.-L. Bae, J. Kim, C. K. Lim, K. M. Nam and H. Song, Nat. Commun., 2017, 8, 1156 CrossRef PubMed.
- L. Jiang, X. Yuan, Y. Pan, J. Liang, G. Zeng, Z. Wu and H. Wang, Appl. Catal. B Environ., 2017, 217, 388–406 CrossRef CAS.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- J. Deng, C. Zhou, Y. Yang, B. Nan, L. Dong, L. Cai, L. Li, Z.-J. Wang, X. Yang and Z. Chen, Chem. Eng. J., 2023, 462, 142282 CrossRef CAS.
- S. Hu, L. Yang, Y. Tian, X. Wei, J. Ding, J. Zhong and P. Chu, Appl. Catal. B Environ., 2015, 163, 611–622 CrossRef CAS.
- Y. Wang, Y. Li, X. Bai, Q. Cai, C. Liu, Y. Zuo, S. Kang and L. Cui, Catal. Commun., 2016, 84, 179–182 CrossRef CAS.
- C. Fan, J. Miao, G. Xu, J. Liu, J. Lv and Y. Wu, RSC Adv., 2017, 7, 37185–37193 RSC.
- K. Homlamai, T. Maihom, S. Choomwattana, M. Sawangphruk and J. Limtrakul, Appl. Surf. Sci., 2020, 499, 143928 CrossRef CAS.
- A. Kumar, P. Raizada, V. Kumar Thakur, V. Saini, A. Aslam Parwaz Khan, N. Singh and P. Singh, Chem. Eng. Sci., 2021, 230, 116219 CrossRef CAS.
- M. Shen, L. Zhang and J. Shi, Nanotechnology, 2018, 29, 412001 CrossRef PubMed.
- K. Qi, S.-y. Liu and A. Zada, J. Taiwan Inst. Chem. Eng., 2020, 109, 111–123 CrossRef CAS.
- N. D. Shcherban, O. A. Diyuk, V. A. Zazhigalov and D. Y. Murzin, ACS Sustain. Chem. Eng., 2021, 9, 5128–5137 CrossRef CAS.
- G. Zhang, G. Li, Z.-A. Lan, L. Lin, A. Savateev, T. Heil, S. Zafeiratos, X. Wang and M. Antonietti, Angew. Chem., 2017, 129, 13630–13634 CrossRef.
- L. Jiang, X. Yuan, Y. Pan, J. Liang, G. Zeng, Z. Wu and H. Wang, Appl. Catal. B Environ., 2017, 217, 388–406 CrossRef CAS.
- W. Xing, W. Tu, Z. Han, Y. Hu, Q. Meng and G. Chen, ACS Energy Lett., 2018, 3, 514–519 CrossRef CAS.
- Y. Li, X. Feng, Z. Lu, H. Yin, F. Liu and Q. Xiang, J. Colloid Interface Sci., 2018, 513, 866–876 CrossRef CAS PubMed.
- T. Su, Q. Shao, Z. Qin, Z. Guo and Z. Wu, ACS Catal., 2018, 8, 2253–2276 CrossRef CAS.
- J. Liu, W. Fu, Y. Liao, J. Fan and Q. Xiang, J. Mater. Sci. Technol., 2021, 91, 224–240 CrossRef CAS.
- P. Huang, J. Huang, J. Li, T. D. Pham, L. Zhang, J. He, G. W. Brudvig, N. A. Deskins, A. I. Frenkel and G. Li, J. Phys. Chem. C, 2022, 126, 8596–8604 CrossRef CAS.
- Y. Li, B. Li, D. Zhang, L. Cheng and Q. Xiang, ACS Nano, 2020, 14, 10552–10561 CrossRef CAS PubMed.
- W. Shang, W. Liu, X. Cai, J. Hu, J. Guo, C. Xin, Y. Li, N. Zhang, N. Wang, C. Hao and Y. Shi, Adv. Powder Mater., 2023, 2, 100094 CrossRef.
- R. Wang, P. Yang, S. Wang and X. Wang, J. Catal., 2021, 402, 166–176 CrossRef CAS.
- G. Chen, Z. Zhou, B. Li, X. Lin, C. Yang, Y. Fang, W. Lin, Y. Hou, G. Zhang and S. Wang, J. Environ. Sci., 2024, 140, 103–112 CrossRef PubMed.
- D. Zhao, C. L. Dong, B. Wang, C. Chen, Y. C. Huang, Z. Diao, S. Li, L. Guo and S. Shen, Adv. Mater., 2019, 31, 1903545 CrossRef CAS PubMed.
- L. Lin, Z. Yu and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 6164–6175 CrossRef CAS PubMed.
- K. S. Lakhi, D.-H. Park, K. Al-Bahily, W. Cha, B. Viswanathan, J.-H. Choy and A. Vinu, Chem. Soc. Rev., 2017, 46, 72–101 RSC.
- J. Yuan, X. Liu, Y. Tang, Y. Zeng, L. Wang, S. Zhang, T. Cai, Y. Liu, S. Luo, Y. Pei and C. Liu, Appl. Catal. B Environ., 2018, 237, 24–31 CrossRef CAS.
- G. Chen, F. Wei, Z. Zhou, B. Su, C. Yang, X. F. Lu, S. Wang and X. Wang, Sustain. Energy Fuels, 2023, 7, 381–388 RSC.
- C. Cometto, A. Ugolotti, E. Grazietti, A. Moretto, G. Bottaro, L. Armelao, C. Di Valentin, L. Calvillo and G. Granozzi, npj 2D Mater. Appl., 2021, 5, 63 CrossRef CAS.
- B. Wu, R. Yang, L. Shi, T. Lin, X. Yu, M. Huang, K. Gong, F. Sun, Z. Jiang, S. Li, L. Zhong and Y. Sun, Chem. Commun., 2020, 56, 14677–14680 RSC.
- L. Cheng, P. Zhang, Q. Wen, J. Fan and Q. Xiang, Chin. J. Catal., 2022, 43, 451–460 CrossRef CAS.
- W. Huang, H. Ming, X. Bian, C. Yang, Y. Hou, K. Ding and J. Zhang, Chem. Eng. J., 2023, 473, 145230 CrossRef CAS.
- N. D. Shcherban, P. Mäki-Arvela, A. Aho, S. A. Sergiienko, P. S. Yaremov, K. Eränen and D. Y. Murzin, Catal. Sci. Technol., 2018, 8, 2928–2937 RSC.
- C. Huang, J. Wen, Y. Shen, F. He, L. Mi, Z. Gan, J. Ma, S. Liu, H. Ma and Y. Zhang, Chem. Sci., 2018, 9, 7912–7915 RSC.
- B. Jürgens, E. Irran, J. Senker, P. Kroll, H. Müller and W. Schnick, J. Am. Chem. Soc., 2003, 125, 10288–10300 CrossRef PubMed.
- C. Huang, Y. Wen, J. Ma, D. Dong, Y. Shen, S. Liu, H. Ma and Y. Zhang, Nat. Commun., 2021, 12 Search PubMed.
- J. Jiang, RSC Adv., 2016, 6, 47368–47372 RSC.
- X. Li, J. Zhang, L. Shen, Y. Ma, W. Lei, Q. Cui and G. Zou, Appl. Phys. A, 2009, 94, 387–392 CrossRef CAS.
- T. S. Miller, A. B. Jorge, T. M. Suter, A. Sella, F. Corà and P. F. McMillan, Phys. Chem. Chem. Phys., 2017, 19, 15613–15638 RSC.
- Z. Liu, S. Wu, M. Li and J. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 25417–25426 CrossRef CAS PubMed.
- M. Makaremi, S. Grixti, K. T. Butler, G. A. Ozin and C. V. Singh, ACS Appl. Mater. Interfaces, 2018, 10, 11143–11151 CrossRef CAS PubMed.
- I. Cano, A. Weilhard, C. Martin, J. Pinto, R. W. Lodge, A. R. Santos, G. A. Rance, E. H. Åhlgren, E. Jónsson, J. Yuan, Z. Y. Li, P. Licence, A. N. Khlobystov and J. Alves Fernandes, Nat. Commun., 2021, 12 Search PubMed.
- E. C. Kohlrausch, H. A. Centurion, R. W. Lodge, X. Luo, T. Slater, M. J. L. Santos, S. Ling, V. R. Mastelaro, M. J. Cliffe, R. V. Goncalves and J. Alves Fernandes, J. Mater. Chem. A, 2021, 9, 26676–26679 RSC.
- W. N. R. W. Isahak, Z. A. C. Ramli, M. W. Ismail, K. Ismail, R. M. Yusop, M. W. M. Hisham and M. A. Yarmo, J. CO2 Util., 2013, 2, 8–15 CrossRef CAS.
- Y. Ye, H. Yang, J. Qian, H. Su, K.-J. Lee, T. Cheng, H. Xiao, J. Yano, W. A. Goddard and E. J. Crumlin, Nat. Commun., 2019, 10, 1875 CrossRef PubMed.
- K. M. Megha, T. K. Ghanty and A. Banerjee, J. Phys. Chem., 2021, 125, 2558–2572 CrossRef PubMed.
- I. Popov, S. Ghaderzadeh, E. C. Kohlrausch, L. T. Norman, T. J. A. Slater, G. N. Aliev, H. Alhabeadi, A. Kaplan, W. Theis, A. N. Khlobystov, J. A. Fernandes and E. Besley, Nano Lett., 2023, 23, 8006–8012 CrossRef CAS PubMed.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- A. M. Abdel-Mageed, B. Rungtaweevoranit, M. Parlinska-Wojtan, X. Pei, O. M. Yaghi and R. J. Behm, J. Am. Chem. Soc., 2019, 141, 5201–5210 CrossRef CAS PubMed.
- Y. Wang, B. Li, B. Xue, N. Libretto, Z. Xie, H. Shen, C. Wang, D. Raciti, N. Marinkovic, H. Zong, W. Xie, Z. Li, G. Zhou, J. Vitek, J. G. Chen, J. Miller, G. Wang and C. Wang, Sci. Adv., 2023, 9, 30 Search PubMed.
- J. Lv, Y. Lu, J. Wang, F. Zhao, Y. Wang, H. He and Y. Wu, J. Mol. Struct., 2022, 1249, 131638 CrossRef CAS.
- J. Liu, Y. Zou, D. Cruz, A. Savateev, M. Antonietti and G. Vilé, ACS Appl. Mater. Interfaces, 2021, 13, 25858–25867 CrossRef CAS PubMed.
- P. Makuła, M. Pacia and W. Macyk, J. Phys. Chem. Lett., 2018, 9, 6814–6817 CrossRef.
- Y. Xu and S.-P. Gao, Int. J. Hydrogen Energy, 2012, 37, 11072–11080 CrossRef CAS.
- R. Gusain, P. Kumar, O. P. Sharma, S. L. Jain and O. P. Khatri, Appl. Catal. B Environ., 2016, 181, 352–362 CrossRef CAS.
- J. Meng, Q. Chen, J. Lu and H. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 550–562 CrossRef CAS PubMed.
- A. Ali and W.-C. Oh, Sci. Rep., 2017, 7, 1867 CrossRef PubMed.
- K. Wang, M. Cheng, F. Xia, N. Cao, F. Zhang, W. Ni, X. Yue, K. Yan, Y. He, Y. Shi, W. Dai and P. Xie, Small, 2023, 19, 2207581 CrossRef CAS PubMed.
- Z. Y. Tian, L. H. Kong, Y. Wang, H. J. Wang, Y. J. Wang, S. Yao, T. B. Lu and Z. M. Zhang, Small, 2021, 17, 2103558 CrossRef CAS PubMed.
- J. Yuan, Y. Tang, X. Yi, C. Liu, C. Li, Y. Zeng and S. Luo, Appl. Catal. B Environ., 2019, 251, 206–212 CrossRef CAS.
- L. Lin, H. Ou, Y. Zhang and X. Wang, ACS Catal., 2016, 6, 3921–3931 CrossRef CAS.
- L. Lin, H. Ou, Y. Zhang and X. Wang, ACS Catal., 2016, 6, 3921–3931 CrossRef CAS.
- Z. Yu, X. Yue, J. Fan and Q. Xiang, ACS Catal., 2022, 12, 6345–6358 CrossRef CAS.
- Z. Chen, S. Mitchell, E. Vorobyeva, R. K. Leary, R. Hauert, T. Furnival, Q. M. Ramasse, J. M. Thomas, P. A. Midgley, D. Dontsova, M. Antonietti, S. Pogodin, N. López and J. Pérez-Ramírez, Adv. Funct. Mater., 2017, 27, 1605785 CrossRef.
- A. J. Bard, Standard Potentials in Aqueous Solution, CRC Press, 2017 Search PubMed.
- Y. A. Wu, I. McNulty, C. Liu, K. C. Lau, Q. Liu, A. P. Paulikas, C.-J. Sun, Z. Cai, J. R. Guest, Y. Ren, V. Stamenkovic, L. A. Curtiss, Y. Liu and T. Rajh, Nat. Energy, 2019, 4, 957–968 CrossRef CAS.
- B. An, J. Zhang, K. Cheng, P. Ji, C. Wang and W. Lin, J. Am. Chem. Soc., 2017, 139, 3834–3840 CrossRef CAS PubMed.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, Science, 2012, 336, 893–897 CrossRef CAS PubMed.
- S. Kattel, P. J. Ramírez, J. G. Chen, J. A. Rodriguez and P. Liu, Science, 2017, 355, 1296–1299 CrossRef CAS PubMed.
- S. S. Batsanov, Acta Crystallogr. B, 2020, 76, 38–40 CrossRef CAS PubMed.
- A. Sasmal, S. Saha, C. J. Gómez-García, C. Desplanches, E. Garribba, A. Bauzá, A. Frontera, R. Scott, R. J. Butcher and S. Mitra, Chem. Commun., 2013, 49, 7806–7808 RSC.
- M. C. Biesinger, Appl. Surf. Sci., 2022, 597, 153681 CrossRef CAS.
- T. Chatterjee, E. Boutin and M. Robert, Dalton Trans., 2020, 49, 4257–4265 RSC.
- M. Thangamuthu, K. Vankayala, L. Xiong, S. Conroy, X. Zhang and J. Tang, ACS Catal., 2023, 13, 9113–9124 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4se00028e |
| This journal is © The Royal Society of Chemistry 2024 |