
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRepurposing a supramolecular iridium catalyst via secondary Zn⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) C weak interactions between the ligand and substrate leads to ortho-selective C(sp2)–H borylation of benzamides with unusual kinetics†
C weak interactions between the ligand and substrate leads to ortho-selective C(sp2)–H borylation of benzamides with unusual kinetics†
Jonathan
Trouvé
a,
Vanessa
Delahaye
a,
Michele
Tomasini
 b,
Purushothaman
Rajeshwaran
a,
Thierry
Roisnel
a,
Albert
Poater
*b and
Rafael
Gramage-Doria
*a
b,
Purushothaman
Rajeshwaran
a,
Thierry
Roisnel
a,
Albert
Poater
*b and
Rafael
Gramage-Doria
*a
aUniv Rennes, CNRS, ISCR – UMR 6226, F-35000 Rennes, France. E-mail: rafael.gramage-doria@univ-rennes1.fr
bDepartament de Química, Institut de Química Computacional i Catàlisi, Universitat de Girona, c/Maria Aurèlia Capmany 69, 17003 Girona, Catalonia, Spain. E-mail: albert.poater@udg.edu
First published on 14th June 2024
Abstract
The iridium-catalyzed C–H borylation of benzamides typically leads to meta and para selectivities using state-of-the-art iridium-based N,N-chelating bipyridine ligands. However, reaching ortho selectivity patterns requires extensive trial-and-error screening via molecular design at the ligand first coordination sphere. Herein, we demonstrate that triazolylpyridines are excellent ligands for the selective iridium-catalyzed ortho C–H borylation of tertiary benzamides and, importantly, we demonstrate the almost negligible effect of the first coordination sphere in the selectivity, which is so far unprecedented in iridium C–H bond borylations. Remarkably, the activity is dramatically enhanced by exploiting a remote Zn⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C weak interaction between the substrate and a rationally designed molecular-recognition site in the catalyst. Kinetic studies and DFT calculations indicate that the iridium-catalyzed C–H activation step is not rate-determining, this being unique for remotely controlled C–H functionalizations. Consequently, a previously established supramolecular iridium catalyst designed for meta-borylation of pyridines is now compatible with the ortho-borylation of benzamides, a regioselectivity switch that is counter-intuitive regarding precedents in the literature. In addition, we highlight the role of the cyclohexene additive in avoiding the formation of undesired side-products as well as accelerating the HBpin release event that precedes the catalyst regeneration step, which is highly relevant for the design of powerful and selective iridium borylating catalysts.
C weak interaction between the substrate and a rationally designed molecular-recognition site in the catalyst. Kinetic studies and DFT calculations indicate that the iridium-catalyzed C–H activation step is not rate-determining, this being unique for remotely controlled C–H functionalizations. Consequently, a previously established supramolecular iridium catalyst designed for meta-borylation of pyridines is now compatible with the ortho-borylation of benzamides, a regioselectivity switch that is counter-intuitive regarding precedents in the literature. In addition, we highlight the role of the cyclohexene additive in avoiding the formation of undesired side-products as well as accelerating the HBpin release event that precedes the catalyst regeneration step, which is highly relevant for the design of powerful and selective iridium borylating catalysts.
Introduction
The functionalization of C–H bonds by means of transition metal catalysis is one of the most powerful methodologies in chemical synthesis as it provides access to highly elaborated molecules with step- and atom-economy in a predictive manner.1 Consequently, compounds difficult or impossible to form otherwise are nowadays accessible,2 which is relevant for drug discovery in the application of late-stage functionalization methodologies for upgrading the available chemical space3 as well as in the area of materials sciences to find chemical systems featuring unique photo-, electro- and physicochemical properties.4 Traditionally, the directing group present in the substrate of interest directs the selectivity via metal-coordination in C–H bond functionalizations.5 Consequently, it is highly important to develop selective metal-catalysed C–H bond functionalizations for unbiased substrates, thus reducing the costs devoted to the introduction and further removal of metal-coordinating directing groups.6In this context, C–B bond-forming processes via C–H activation employing iridium catalysts are particularly attractive since they take place without the need of covalently linking directing groups to the substrate of interest7 and well-known methodologies can be applied to further transform the boron-based functional group into more useful carbon- or heteroatom-containing fragments.8 Indeed, the reactivity and the selectivity at iridium are highly affected by the nature of the ligand attached to it, with neutral bipyridine-type ligands being of choice since the pioneering contributions by Ishiyama, Miyaura and Hartwig.9
In the case of aromatic substrates the iridium-catalysed C–H bond borylation generally occurs at the less sterically demanding meta and para positions.10 This regio-selectivity is largely controlled by the rational design of catalysts enabling attractive substrate-to-ligand interactions via hydrogen bonding or ion-pairing in view to place a specific C–H bond at close spatial proximity of the active N,N-chelated iridium site.11 In the case of benzamides, which are important constituents in agrochemicals12 and active pharmaceutical ingredients,13 the selective iridium-catalysed C–H bond borylation at meta and para positions, respectively, were accomplished with unique iridium catalysts developed, independently, by Kuninobu and Kanai, Phipps, Chattopadhyay, and Nakao.14 In the case of iridium-catalysed ortho C–H bond borylations (Fig. 1A), Reek developed a neutral bipyridine ligand featuring a hydrogen bonding site that works specifically for secondary benzamides (Fig. 1B).15 On the other hand, ortho C–H bond borylations of tertiary benzamides were disclosed utilizing anionic hybrid chelating ligands as shown by Maleczka and Smith (P,Si- or N,Si-chelating ligands) and by Chattopadhyay (N,Cthienyl- or N,Cfuryl-chelating ligands) (Fig. 1B).16a–c A series of ligands enabling iridium-catalysed ortho C–H bond borylations of tertiary benzamides with modest activity were reported by Li and co-workers.16d,e Recently, Mascareñas and co-workers reported a neutral CF3-containing bipyridine ligand that led to iridium-catalysed ortho C–H borylated benzamides after extensive trial-and-error ligand screening (Fig. 1B).16f Sawamura also reported ortho C–H borylation of a single example of tertiary benzamide with a heterogeneous phosphine–iridium system supported on silica.16g Iridium-catalysed ortho C–H bond borylations of phenol, aniline and thioanisole derivatives have been achieved exploiting remote weak interactions between the boryl ligands and the functional group in the arene, as well as between Lewis acid boron-containing motifs in the bipyridine ligand and the Lewis base thioether group in the substrate.16h–j In all the above-stated examples, the rather air-sensitive [Ir(COD)(OMe)]2 was used as the catalyst precursor,14–16 and the iridium-catalysed C–H bond activation step was found to be the rate-determining step of the catalytic cycle for the cases in which this was studied.9–16 Note that palladium- and iron-catalysed ortho-selective C–H bond borylations are known.16k,l
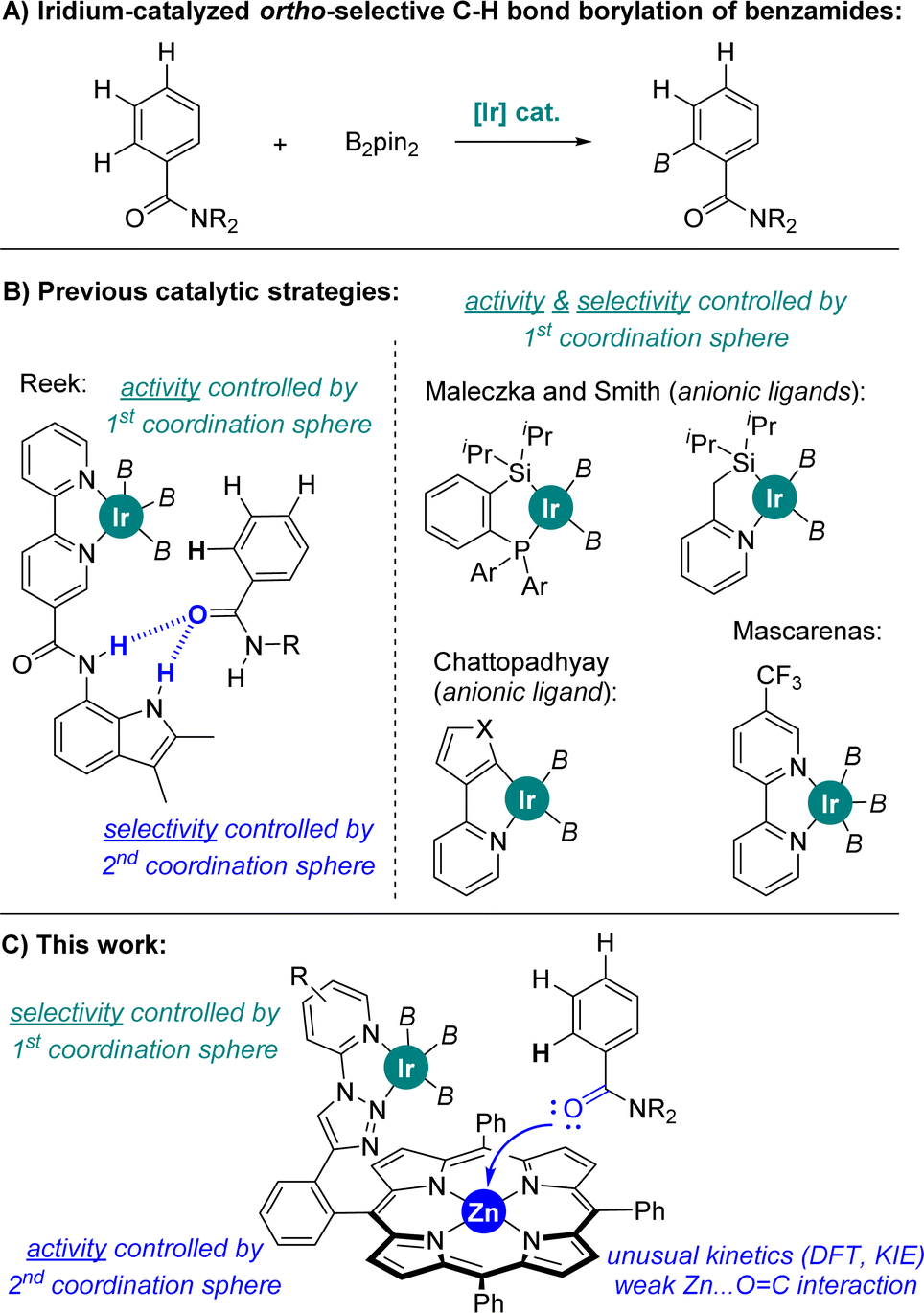 | ||
| Fig. 1 Previous catalysts enabling ortho-selective C–H bond borylation of benzamides (A and B) and the current, repurposed supramolecular approach (C). B2pin2 = bis(pinacolato)diboron, B = (pinacolato)boron, Ar = para-tolyl, X = O, S. | ||
Ultimately, predicting the reactivity of an iridium C–H bond borylating catalyst in a precise manner is a major goal. Herein, we report a predictive catalyst design based on a supramolecular strategy that enables the highly ortho selective C–H bond borylation of tertiary benzamides employing a neutral triazolylpyridine-chelating ligand and a rather air stable [Ir(COD)(Cl)]2 metal precursor. The iridium catalyst is built up around a zinc-porphyrin unit that enables a unique Zn⋯OC weak interaction between the amide group in the substrate and the catalyst in a remote fashion (Fig. 1C). Extensive control experimentation and catalyst evaluation demonstrate that the selectivity is exclusively controlled by the iridium-ligated triazolylpyridine-N,N-chelating site regardless of its chemical nature at the first coordination sphere, whereas the activity is highly enhanced thanks to the crucial substrate pre-organization occurring at the second coordination sphere via a single, remote Zn⋯OC weak interaction. In addition, computational calculations predicted that the iridium-mediated C–H bond activation is not the rate-determining step, but rather the iridium-mediated C–B bond formation, analogous to Chirik's pincer-type cobalt-catalysed borylations.17 This unexpected feature, which explains the observed reactivity of the supramolecular iridium catalyst, was further supported by kinetic isotopic effect studies.
Results and discussion
Considerations for catalyst rationale
Previously, we reported on the meta selective C(sp2)–H bond borylation of pyridines using a well-defined supramolecular iridium catalyst equipped with a zinc-porphyrin unit that served for the specific molecular recognition of pyridines via a remote Zn⋯N weak coordination bond (Fig. 2, top, left).18 In order to demonstrate the generality of this approach, we reasoned that other molecules different than nitrogen-containing heterocycles such as benzamides could potentially bind in a reversible manner to the zinc centre of the molecular recognition site, thereby placing potentially reactive C–H sites at close proximity of the catalytically active iridium site. During the course of our studies, we successfully obtained single crystals suitable for X-ray diffraction studies upon slow evaporation of a dichloromethane solution containing a zinc(II)-tetraphenylporphyrin (ZnTPP) and N,N-dimethylformamide (DMF).19 Analogously to the well-known binding of other tertiary amides to zinc-porphyrin derivatives,20 the DMF molecule apically binds to the zinc centre of ZnTPP via the oxygen atom (Fig. 2, top, right). With these considerations, we built a PM3 semi-empirical molecular model combining our supramolecular iridium catalyst with N,N-dimethylbenzamide as the substrate because it could match for C(sp2)–H borylation similar to that for pyridines (Fig. 2). Interestingly, this low-cost prediction suggests that the supramolecular iridium catalyst might be suitable for activating the aromatic ortho C–H bond of the benzamide instead of the anticipated meta one. In fact, the ortho C–H bond of the benzamide is located at a distance of four chemical bonds apart from the molecular recognition site, a distance that is exactly the same for the case of pyridine substrates. As such, a single, remote Zn⋯X (X = N or OC) weak interaction between the substrate and the catalyst could be enough to reverse the regioselectivity between pyridines and benzamides. The fact that the distance and geometry between the active site and the substrate recognition site in this supramolecular approach are key parameters results in a slight influence of the substrate electronic effects on controlling the catalytic outcome. On the other hand, such strategy takes advantage of the difference in the coordinating ability between a cationic iridium that will have more affinity for anionic boryl ligands and a neutral amide that will have more affinity to a neutral zinc-porphyrin unit in agreement with the hard and soft acid and base theory.21 Consequently, this supramolecular approach would lead to an iridium catalyst that will switch the selectivity (from meta to ortho) between pyridines and benzamides, which strikingly contrasts with the pioneering Nakao's observations in which the same iridium, boron-based catalyst led to the same meta regioselectivity for both pyridines and benzamides (Fig. 2, bottom).14f Note that Nakao's group reported Ir/Al-based catalysts that afforded para-borylated products for both pyridines and benzamides.14a
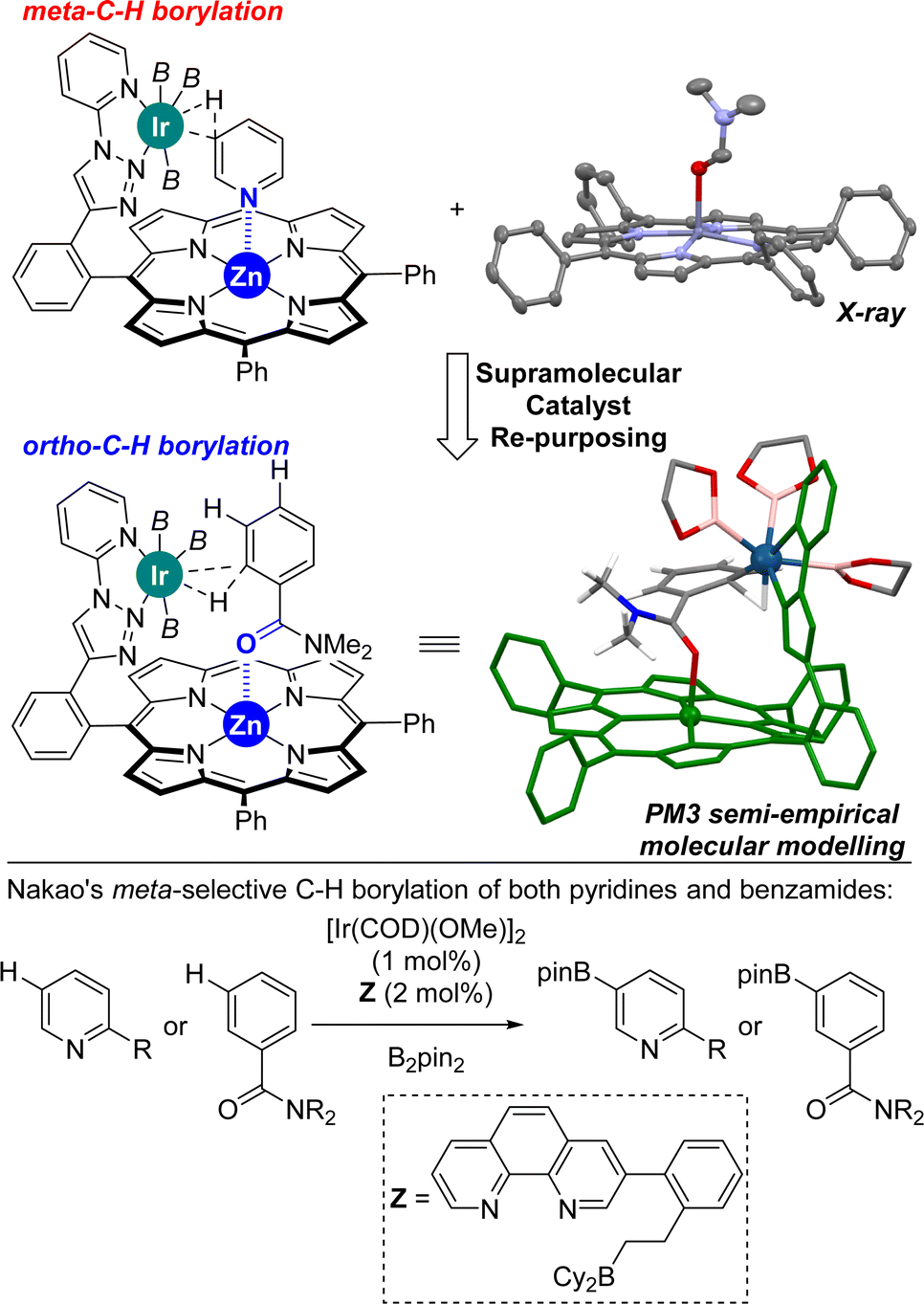 | ||
| Fig. 2 Previous supramolecular iridium-catalysed meta-C–H bond borylation of pyridines (top, left), ORTEP of [ZnTPP ⊂ DMF] determined by single-crystal X-ray diffraction studies (thermal ellipsoids at 50% probability, all hydrogen atoms are omitted, top, right), and current re-purposed supramolecular iridium-catalysed ortho-C–H borylation of benzamides with the PM3-minimized molecular modelling for a plausible intermediate (the methyl groups on the boryl ligand and all hydrogen atoms except those of the amide in the substrate were omitted). Previous iridium-catalysed C–H borylation of both pyridines and benzamides with the same ligand and the same meta-selectivity (bottom). B2pin2 = bis(pinacolato)diboron, B = (pinacolato)boron. | ||
Validation of the supramolecular catalyst action mode: ligand assessment, control experimentation, computational calculations and kinetic studies
Taking the above-stated rationale into consideration, we evaluated the supramolecular ligands L1–L6, which comprise different steric and electronic substituents at the triazolylpyridine site while keeping the same molecular recognition site made of a zinc-porphyrin backbone, in the iridium-catalysed C–H bond borylation of N,N-dimethylbenzamide (1a) as the model substrate (Fig. 3 and Table S1 in the ESI†). For comparison purposes, we also assessed the corresponding triazolylpyridine ligands lacking the zinc-porphyrin substrate recognition site (L1*–L6*, Fig. 3). For the sake of comparison with our previously developed meta-selective C–H borylation of pyridines,18 the reactions were evaluated at 80 °C using para-xylene as the solvent and 1.5 equivalents of the borylating reagent B2pin2 (Fig. 3).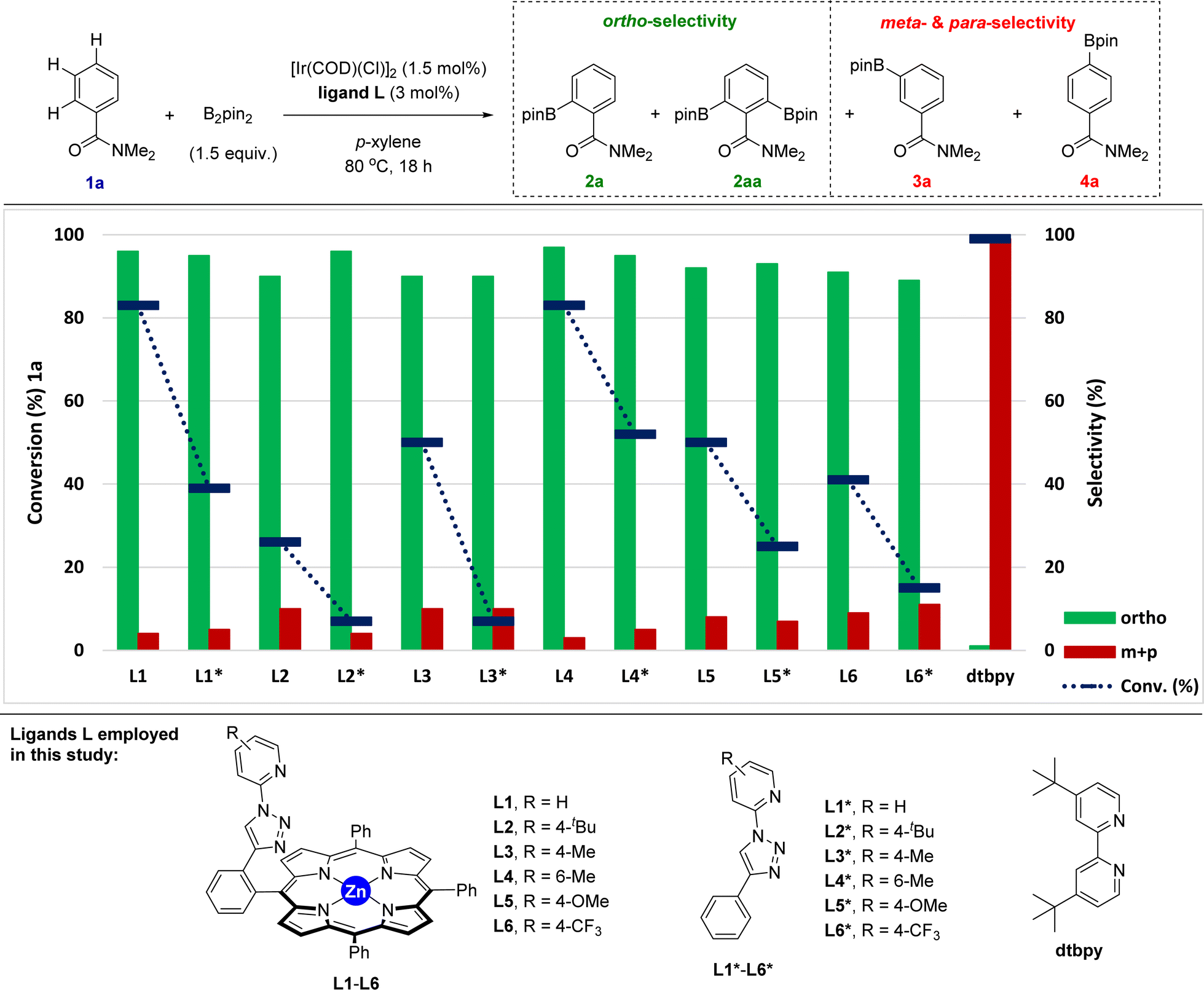 | ||
| Fig. 3 Assessment of the supramolecular and non-supramolecular ligands in the iridium-catalysed C–H bond borylation of N,N-dimethylbenzamide 1a. Reaction conditions: 1a (0.024 g, 0.162 mmol), [Ir(COD)(Cl)]2 (1.7 mg, 2.43 × 10−3 mmol), ligand L (4.86 × 10−3 mmol), B2pin2 (61.7 mg, 0.243 mmol), p-xylene (1 mL), 80 °C, 24 h (conversion of 1 and product selectivity for 2/3/4 were determined by GC analysis using dodecane as internal standard and 1H NMR analysis). | ||
In terms of conversion of 1a, the iridium-catalysed C–H bond borylation employing each supramolecular ligand L outperformed its non-supramolecular counterpart L* by at least a factor of two (blue bars and dashed lines, Fig. 3). On the other hand, the ortho-selectivity was comparable for all supramolecular catalysts derived from L1–L6 being in the range of 89–97% (green colour, Fig. 3). Although this selectivity compares well with previous precedents,16 the major difference of our supramolecular approach is that there is a negligible effect of the first coordination sphere regarding the selectivity outcome of the catalysis. In contrast, and as was expected, using the classical 4,4′-di-tert-butyl-2,2′-bipyridine (dtbpy) ligand afforded a mixture of meta and para borylated products (red colour, Fig. 3) with a complete absence of formation of the ortho isomer. These findings clearly indicate that the selectivity of this transformation when using the supramolecular ligands L1–L6 is exclusively controlled by the trivial iridium-coordinated triazolylpyridine unit in the first coordination sphere and that the activity is enhanced by the presence of the substrate recognition zinc-porphyrin site in the second coordination sphere.
The best results in terms of selectivity (97% ortho-selectivity) and activity (83% conversion) were encountered for the supramolecular ligands L1 and L4 (Fig. 3), most notably, without the need to employ an [Ir(COD)(OMe)]2 metal precursor, which is in stark contrast with precedents in the literature.7–16 At this stage, the exact action mode of the non-supramolecular catalysts derived from ligands L1*–L6* remains to be addressed without discarding the potential role of the amide unit within the substrate as an ortho-directing group.22 However, for the case of the supramolecular catalysts derived from L1–L6, CPK models indicate that the large porphyrin backbone comprising three bulky phenyl groups in the ligand strongly disfavour the accommodation of the benzamide substrate by ortho-chelation around the catalytically productive iridium-(boryl)n species (n = 2 or 3) due to steric shielding. In other words, the highly sterically congested supramolecular iridium catalysts derived from ligands L1–L6 give no choice to the benzamide substrate than engaging in a Zn⋯OC weak interaction upon approaching the catalyst. In addition, similar to our previous observations using pyridines as substrates in metal catalysis,18,23 such a supramolecular effect was completely lost in the presence of polar coordinating solvents such as THF, DMF or 1,4-dioxane, which disrupt the remote Zn⋯OC weak interaction by solvent-coordination to the zinc centre of the porphyrin backbone via Zn⋯O interaction. Aside from the key substrate-preorganization effects, increasing the effective molarity of the substrate around the catalyst is also a major advantage of the supramolecular iridium catalysts derived from L1–L6.
The only difference between the supramolecular iridium catalysts derived from L1 and L4 was the ratio of mono- versus bis-borylation, i.e.2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2aa (Scheme 1). Using L1 afforded an 81:19 ratio of 2a:2aa (Scheme 1A), whereas the bulkier L4 containing a 6-methyl-substitution pattern in the iridium-coordinated pyridine moiety led to an increased mono-selectivity of 2a:2aa = 90:10 ratio (Scheme 1A). This finding was explained by the significant steric shielding that needs to be overcome in order to get a second borylation in compound 2a within the supramolecular iridium catalyst derived from L4. Additional experiments demonstrate the lack of reactivity when the catalytic reactions were carried out in the absence of any ligand (Scheme 1B). To further understand the unique behaviour of the supramolecular ligand L1 in the ortho-selective C–H bond borylation of benzamide 1a, a reaction was carried out using the molecular-recognition-free ligand L1* and ZnTPP at 3 mol% loading each (Scheme 1C). Whereas 96% ortho-selectivity was obtained, showing again the importance of the metal-coordinated triazolylpyridine ligand backbone, the conversion of 1a significantly dropped to 65% (Scheme 1C) when compared to the supramolecular version L1 (83% conversion of 1a, Scheme 1A). These control experiments demonstrate the requirement of covalently linking the triazolylpyridine fragment to the substrate recognition site to increase the reactivity of the catalyst.
:2aa (Scheme 1). Using L1 afforded an 81:19 ratio of 2a:2aa (Scheme 1A), whereas the bulkier L4 containing a 6-methyl-substitution pattern in the iridium-coordinated pyridine moiety led to an increased mono-selectivity of 2a:2aa = 90:10 ratio (Scheme 1A). This finding was explained by the significant steric shielding that needs to be overcome in order to get a second borylation in compound 2a within the supramolecular iridium catalyst derived from L4. Additional experiments demonstrate the lack of reactivity when the catalytic reactions were carried out in the absence of any ligand (Scheme 1B). To further understand the unique behaviour of the supramolecular ligand L1 in the ortho-selective C–H bond borylation of benzamide 1a, a reaction was carried out using the molecular-recognition-free ligand L1* and ZnTPP at 3 mol% loading each (Scheme 1C). Whereas 96% ortho-selectivity was obtained, showing again the importance of the metal-coordinated triazolylpyridine ligand backbone, the conversion of 1a significantly dropped to 65% (Scheme 1C) when compared to the supramolecular version L1 (83% conversion of 1a, Scheme 1A). These control experiments demonstrate the requirement of covalently linking the triazolylpyridine fragment to the substrate recognition site to increase the reactivity of the catalyst.
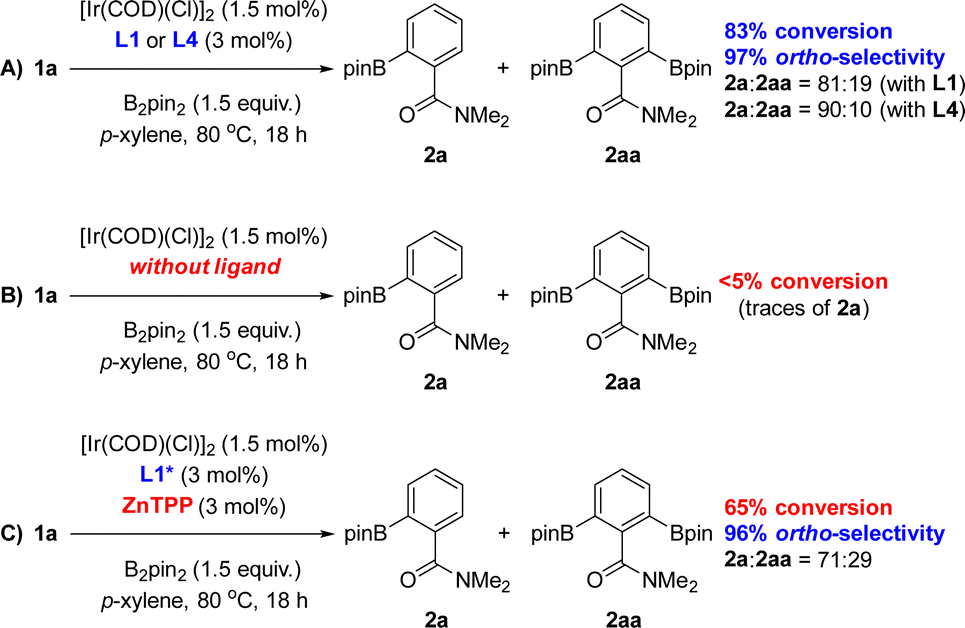 | ||
| Scheme 1 Reaction conditions applied with the optimal supramolecular ligands L1 and L4 (A) and control experimentation (B and C). ZnTPP = zinc(II)-tetraphenylporphyrin. | ||
Interestingly, the amount of bis-functionalized borylated product significantly increased (2a:2aa = 71:29, Scheme 1C) when compared to the reaction carried out in the presence of the supramolecular ligand L1 (2a:2aa = 81:19, Scheme 1A). This observation suggests that the supramolecular ligands are rather bulky and disfavour to some extent the second borylation pathway towards 2aa. Although the binding of amide groups to zinc-porphyrin derivatives is well known including our X-ray data (Fig. 2),20 we did not succeed in detecting direct evidence of the Zn⋯OC weak interaction between the supramolecular ligand L1 and the benzamide substrate 1a in solution by titration NMR studies (1H, 13C and DOSY) because this is known to be a very fast exchange process at the NMR time scale.24 In fact, solvents such as amides (i.e. DMF) and other oxygen-containing derivatives (i.e. acetone, THF, DMSO) are typically used to cleave porphyrin aggregates leading to mono-nuclear and discrete, solvated-zinc-porphyrin species involving Zn⋯O interaction.24 On the other hand, attempts to carry out UV-vis titration studies between the ligand L1 and the benzamide substrate 1a did not provide accurate data for determining the eventual association constant. This is hardly surprising as the association constant between amides and zinc-porphyrins is known to be significantly lower than 102 M−1.20d Nevertheless, the real situation during the catalysis might be different compared to the specific binding between the ligand L1 and the substrate 1a as the iridium site will provide further affinity towards the substrate for activating a C–H bond while simultaneous Zn⋯OC weak interaction occurs at the molecular recognition site. In other words, the supramolecular catalyst may have a stronger affinity for the transition state rather than for the substrate as was observed before for the meta-selective C–H borylation of pyridines18 and previously by Sanders using zinc-porphyrins as organocatalysts.25
The sensitivity to steric shielding associated with the supramolecular iridium catalyst derived from ligand L1 was additionally addressed by evaluating the reactivity of the benzamides 1a–1e bearing different substituents around the nitrogen atom (Fig. 4). For comparison purposes, reaction time of the C–H borylation was kept at 24 hours to ensure the maximum reactivity for each substrate at 80 °C (instead of 18 hours in Fig. 3 and Scheme 1). The supramolecular iridium catalysis was highly affected under these reaction conditions by the steric nature of the tertiary amide group with the conversion decreasing from 90% for 1a to 18% for the bulkiest iso-propyl substituent in 1d (blue colour, Fig. 4). Importantly, the ortho-selectivity was still remarkable ranging from 97% in the best case (1a and 1b) to 87% for 1c and 77% for the challenging 1d (green colour, Fig. 4). The formation of both meta- and para-borylated side-products 3 and 4 increased to 13% for 1c and 23% for 1d, respectively (red colour, Fig. 4), in line with an increase of the steric shielding associated with these substrates that may disfavour the remote Zn⋯OC weak interaction between the substrate and the supramolecular catalyst. In the case that the reaction would be controlled by the classical amide-directed iridium-chelation, little difference of reactivity should have been found, which is not the case for the current case of study with the supramolecular ligand L1.
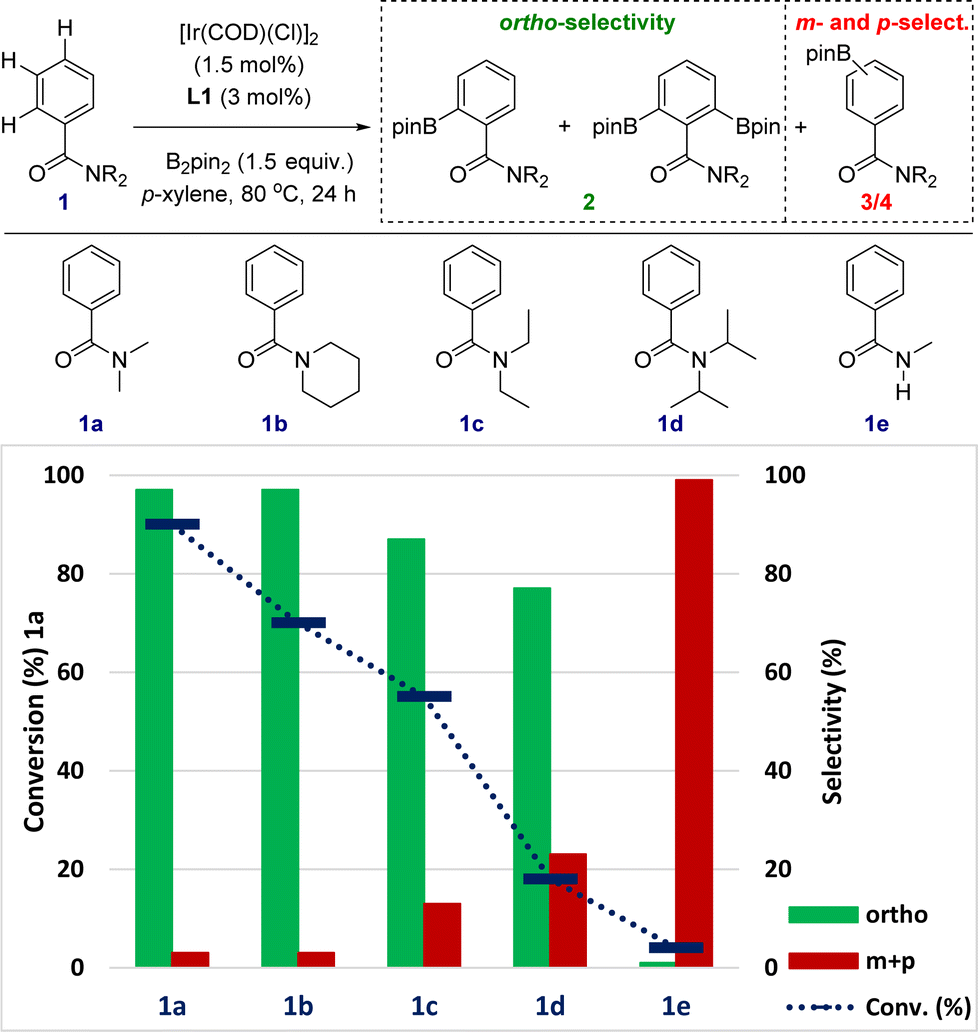 | ||
| Fig. 4 Relevance of the steric parameters of the amide group in benzamides 1a–1e for the supramolecular iridium-catalysed C–H borylation. Reaction conditions: 1 (0.162 mmol), [Ir(COD)(Cl)]2 (1.7 mg, 2.43 × 10−3 mmol), L1 (4.1 mg, 4.86 × 10−3 mmol), B2pin2 (61.7 mg, 0.243 mmol), p-xylene (1 mL), 80 °C, 24 h (conversion of 1 and product selectivity for 2/3/4 were determined by GC analysis using dodecane as internal standard and 1H NMR analysis). | ||
Secondary benzamides such as 1e were not compatible with this supramolecular iridium catalysis (Fig. 4). The almost absence of reactivity encountered in 1e was attributed to (i) the poorer coordinating ability of the carbonyl group due to tautomerization22 and/or (ii) catalyst inhibition due to N-coordination to iridium with HBpin release.26 Overall, the above-stated observations strongly suggest the postulated action mode in which the remote Zn⋯OC weak interaction is beneficial for increasing the reactivity of the catalysis.
In view to further rationalize such findings, Densitiy Functional Theory (DFT) calculations were performed to unravel the mechanism associated with the ortho-C–H borylation of benzamide 1a using the supramolecular ligand L1 at 80 °C. The DFT-computed reaction mechanism is shown in Scheme 2 and the following conclusions can be drawn:
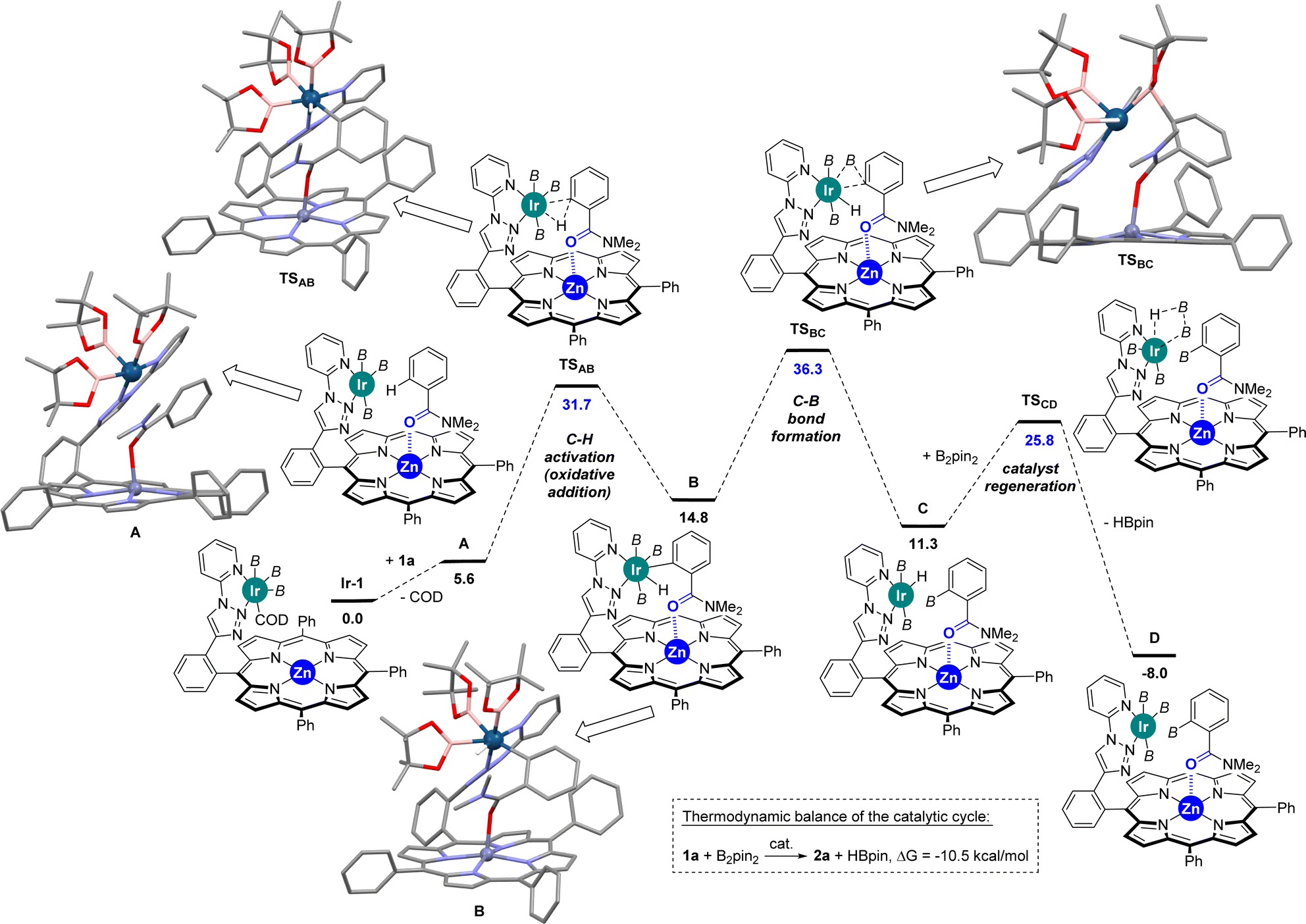 | ||
| Scheme 2 DFT-computed energetic profile of the reaction mechanism for the supramolecular iridium-catalysed ortho-C–H borylation of the benzamide 1a using the supramolecular ligand L1 (Gibbs energies at 353 K in kcal mol−1; in blue the relative energies corresponding to the transition states). B = (pinacolato)boron. | ||
(1) The substrate bound to the catalyst (complex A) via remote Zn⋯OC interaction is less favoured with benzamides 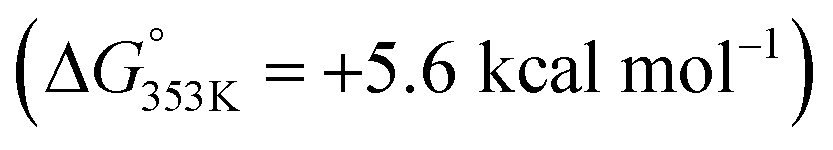 than with pyridines via Zn⋯N interaction
than with pyridines via Zn⋯N interaction 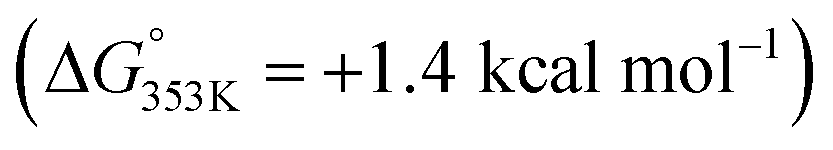 ,18b thereby supporting the previous findings (vide supra) that indicated the relatively low affinity of benzamides to the zinc-porphyrin molecular recognition pocket compared to pyridines, but still being energetically accessible.
,18b thereby supporting the previous findings (vide supra) that indicated the relatively low affinity of benzamides to the zinc-porphyrin molecular recognition pocket compared to pyridines, but still being energetically accessible.
(2) The ortho-C–H activation step (A → B) requires an energetic barrier of ΔG‡ = +31.7 kcal mol−1, which is slightly lower than that observed for the meta-C–H activation of pyridine that was found to be ΔG‡ = +33.7 kcal mol−1.18 Consequently, the iridium-mediated C–H activation and remote Zn⋯OC interaction between the ligand and the substrate contribute simultaneously in a cooperative manner to decreasing the energetic barrier of this key transition state in the benzamide case. Note that DFT calculations considering the cleavage of the Zn⋯OC interaction did not afford any other stable intermediate and that the Zn⋯OC interaction was restored (κ2-N,O-coordination of the substrate to the zinc centre was also ruled out by DFT calculations).
(3) The C–B bond-forming step (B → C), which is ΔG‡ = +36.3 kcal mol−1, is unexpectedly higher in energy compared to the C–H activation step (A → B). Consequently, the C–B bond-forming step appears to be the rate-determining one in the supramolecular iridium-catalysed ortho-C–H borylation of benzamide. For comparison purposes, it is relevant to note that the C–B bond-forming step (B → C) for the meta-C–H borylation of pyridine was computed to be ΔG‡ = +27.1 kcal mol−1,18 a value which is significantly lower than that observed for benzamides.
(4) Catalyst regeneration is accessible with a relatively low barrier of ΔG‡ = +25.8 kcal mol−1 and the release of HBpin is highly favoured while the product is still binding to the molecular recognition pocket.
(5) Although the X-ray data from the [ZnTPP ⊂ DMF] system (Fig. 2) displays a metal coordination of the carbonyl group via an sp2-hybridized oxygen atom (Scheme 3, left), similar to that reported elsewhere for the binding of small DMF to zinc-porphyrin derivatives,20g–o the DFT-computed intermediates and transition states feature a slightly different type of coordination for the Zn⋯OC interaction (Scheme 2). Here, the bulky tertiary benzamide is present in a zwitterionic form with a formal sp3-hybridized oxygen atom engaged in coordination (Scheme 3, right), similarly to that observed by PM3 semi-empirical calculations (Fig. 2) and analogous to what has been reported elsewhere with zinc-porphyrins bound to bulky amides having the formula RCONMe2 (R = alkyl, benzyl).20a–f Details concerning key bond distances and angles are displayed in Table S4 (see the ESI†). As such, the molecular recognition pocket is flexible enough to adopt different coordination geometries concerning the Zn⋯OC interaction,27 which becomes advantageous for this supramolecular catalysis.
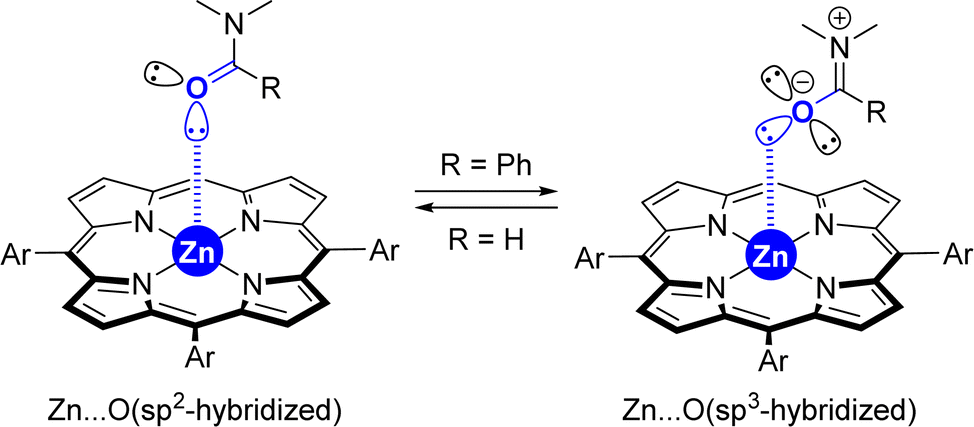 | ||
| Scheme 3 Extreme cases in the difference of the coordination of small amides versus large amides to tetra-aryl-functionalized zinc-porphyrins. | ||
(6) Supramolecular π–π interactions between the bounded benzamide substrate and the peripheral triazolylpyridine ligand are present according to non-covalent interaction NCI plots (Table S2 in the ESI†).
In order to experimentally validate the DFT-computed mechanism, which predicts that the C–H activation step is unexpectedly not rate-determining, kinetic isotope effect (KIE) experiments were carried out.28 In order to have reliable data in a relatively short period of time, the reactions were carried out at 100 °C, which afforded good-quality fitting in the first 10 hours of the reaction. From two parallel experiments, one involving the benzamide starting material 1a and the other one the deuterated 1a-d5, it was found that these reactions do follow a very similar kinetic profile (Fig. 5, top) within the error of the measurements (see the ESI†), thereby indicating an almost negligible kinetic isotope effect. Additionally, from intermolecular competition experiments between 1a and 1a-d5, a very low KIE of 1.6 was observed (Fig. 5, bottom).
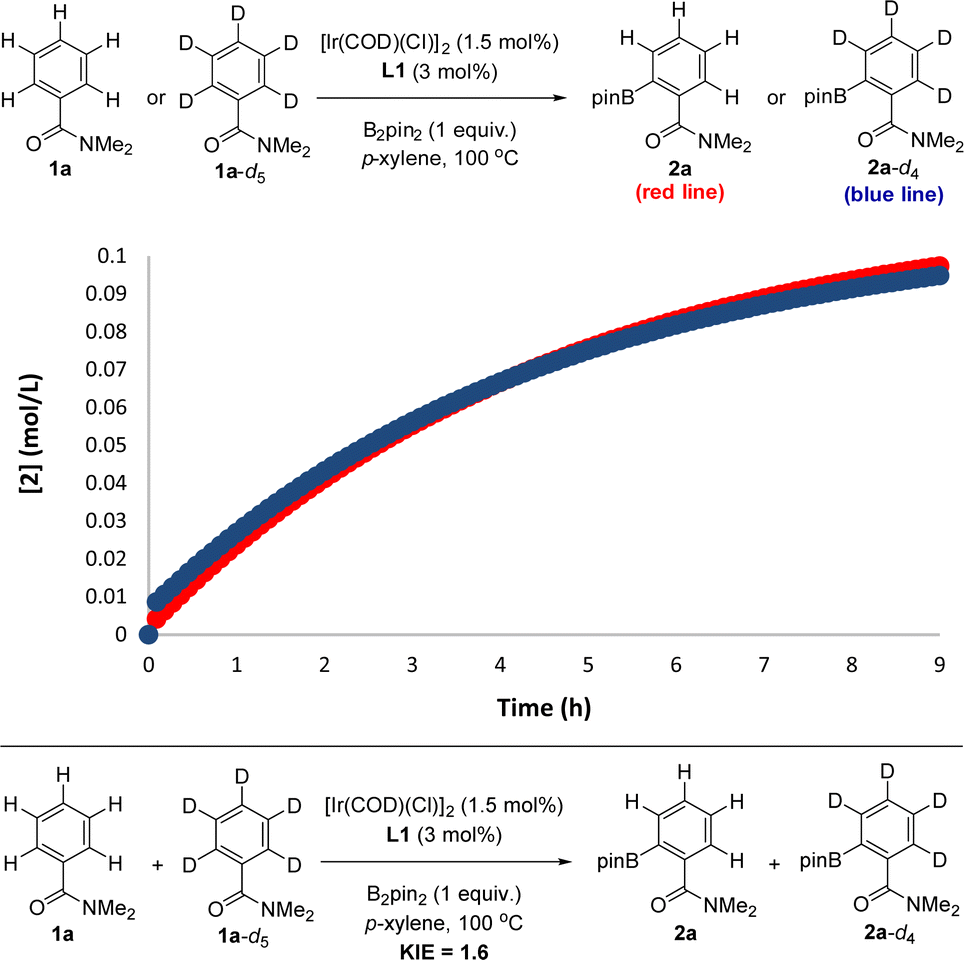 | ||
| Fig. 5 Kinetic isotope effect (KIE) experiments relevant for mechanistic considerations: from two parallel reactions (top) and from an intermolecular competition (bottom). | ||
These experiments clearly indicate that the C–H activation event (A → B, Scheme 2) is not the rate-determining step for the supramolecular ortho C–H borylation of benzamides, which is unprecedented for remote iridium-catalysed C–H borylations so far, although it is known for cobalt-catalysed ones involving rationally designed pincer-type ligands.17 As such, in our supramolecular iridium catalysis there is a formal change in the rate-determining step from pyridines18 to benzamides. Whereas in pyridines the meta-C–H oxidative addition step is turnover limiting,18 the C–B bond-forming step is the rate-determining one for the ortho-C–H borylation of benzamides. Overall, this is a unique case in which a different type of remote weak interaction (Zn⋯OC versus Zn⋯N) in the secondary coordination sphere fundamentally changes the turnover limiting step in a catalytic cycle without altering the overall steric and electronic nature of the catalyst structure. It is relevant to note that ligand-free, directing-group-controlled iridium-catalysed C–H borylations can lead to low KIE with the C–H oxidative addition at iridium still being the rate-determining step,29 which strikingly contrasts with the observations encountered in our supramolecular catalysis.30
Inhibition of side-product formation translates to superior catalysis: scope, limitations and overall reaction mechanism
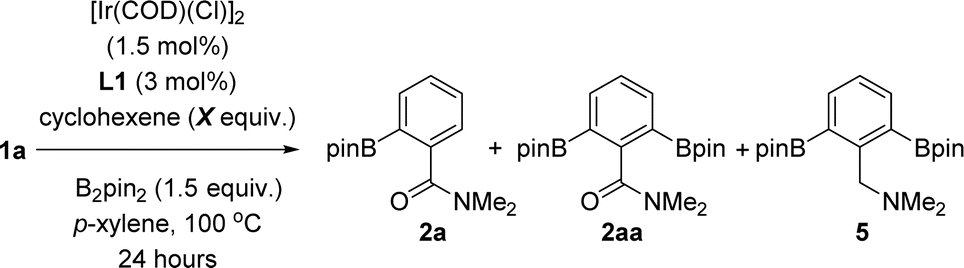
Considering that high temperatures may facilitate overcoming the high reaction barrier (36.3 kcal mol−1) found by DFT calculations (vide supra), we applied a temperature of 100 °C to the C–H borylation of benzamides using the most active and selective supramolecular ligand, namely L1 (Table 1). After 24 hours, although the conversion of the starting material 1a was higher at 100 °C than at 80 °C (90% vs. 83%) and the ortho-borylation reached a superior +99% selectivity (Table 1, entry 1), we observed formation of ca. 20% of deoxygenated borylated product 5 (Table 1, entry 1). We reasoned that the side-product 5 likely resulted from an iridium-catalysed deoxygenative reduction of amides via a hydroboration process with pinacolborane (HBpin) that forms at each turnover during the main iridium-catalysed borylation cycle.31 To overcome this issue, we speculated that the cyclohexene additive could trap the in situ formed HBpin.32 As such, not only could the side-product 5 be reduced but also the reactivity of the supramolecular catalyst could be increased by accelerating the HBpin release event that precedes catalyst regeneration according to the above-described DFT calculations.33 In fact, the iridium-catalysed C–H bond borylation of benzamide 1a performed in the presence of one equivalent of cyclohexene led to a comparable 94% conversion (Table 1, entry 2) as was observed in the absence of cyclohexene (90%, Table 1, entry 1) with an exclusive ortho-selectivity for both cases. Most notably, the side-product borylated amine 5 was reduced from 22% (Table 1, entry 1) to only trace amounts (Table 1, entry 2) when the reaction was carried out in the presence of the cyclohexene additive and the yield of mono-ortho-borylated benzamide 2a increased from 60% (Table 1, entry 1) to 85% (Table 1, entry 2). Leaving the catalysis for longer time durations (30 hours) in the presence of cyclohexene led to full conversion of the starting material and complete ortho-selectivity in an 87% isolated yield of the ortho-mono-borylated benzamide 2a (Table 1, entry 3).|
|
|||||
|---|---|---|---|---|---|
| Entry | X | Conv. 1ab |
Yield 2ab |
Yield 2aab |
Yield 5b |
| a Reaction conditions: 1a (0.024 g, 0.162 mmol), [Ir(COD)(Cl)]2 (1.7 mg, 2.43 × 10−3 mmol), L1 (4.1 mg, 4.86 × 10−3 mmol), B2pin2 (61.7 mg, 0.243 mmol), cyclohexene (13 mg, 16 μL, 0.162 mmol), p-xylene (1 mL), 100 °C, 24 hours. b Conversion of 1 and product selectivity were determined by GC analysis using dodecane as internal standard and 1H NMR analysis. c Reaction performed over 30 hours. d Isolated yield displayed in brackets after purification by column chromatography. | |||||
| 1 | 0 | 90% | 60% | 8% | 22% |
| 2 | 1 | 94% | 85% | 4% | 5% |
| 3c | 1 | 99% | 91% (87%)d | 4% | 5% |
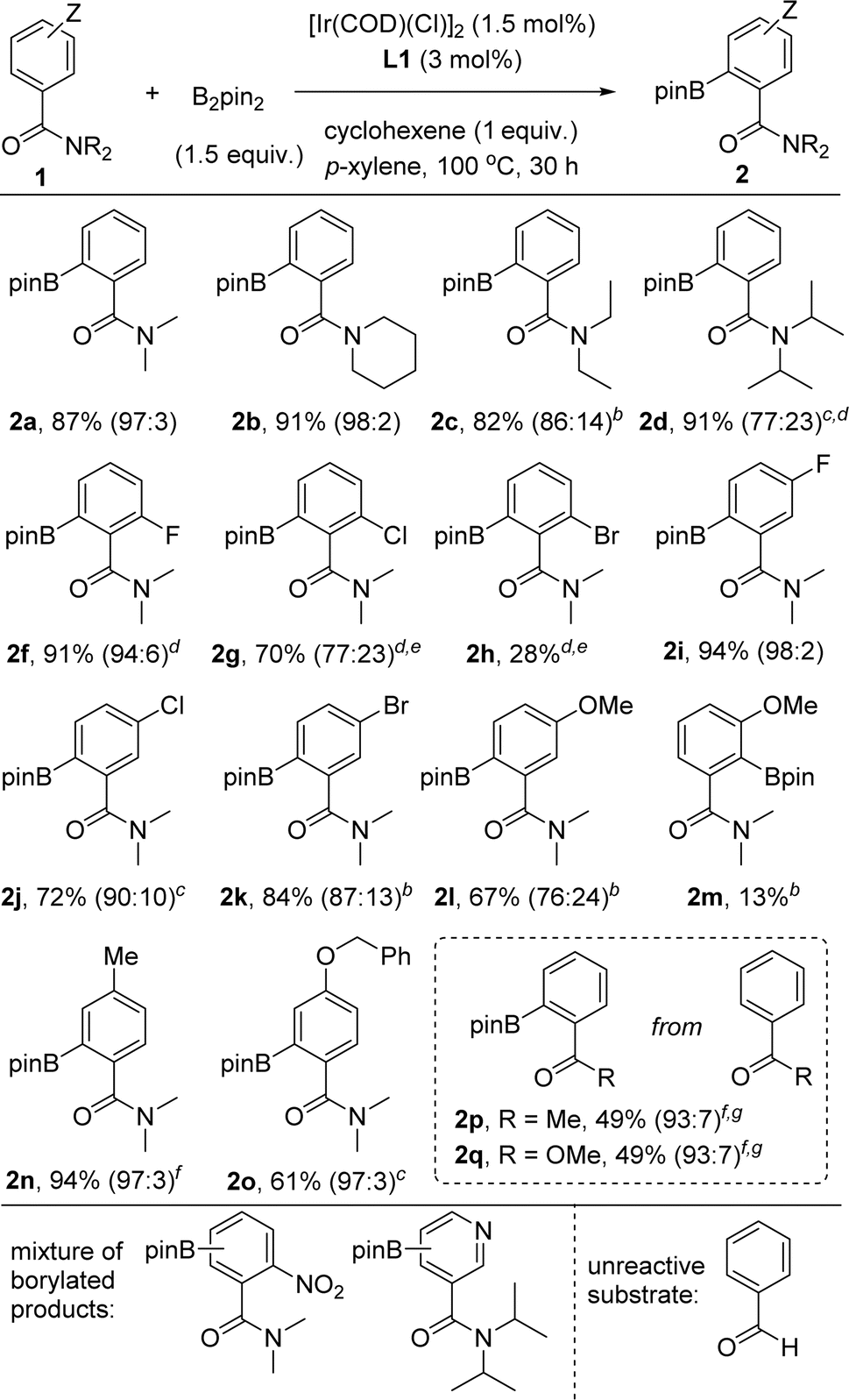
With the optimal conditions in hand using the supramolecular ligand L1 and cyclohexene as additive over 30 hours (Table 1, entry 3), we evaluated the substrate scope for this iridium-catalysed ortho-C–H borylation of tertiary benzamides directed by Zn⋯OC weak interactions between the substrate and the catalyst (Table 2). Different steric patterns at the amide site (methyl, ethyl, iso-propyl) were tolerated including the case in which the amide is part of a piperidine fragment. The corresponding ortho-borylated products 2a–2d were isolated in the range of 82–91% yields with full conversion of the corresponding starting materials 1a–1d and an excellent ortho-selectivity (90–98%). These findings indicate also the benefits of performing the catalysis at higher temperatures for maximizing the dynamic coordination chemistry between the substrate and the catalyst via remote Zn⋯OC weak interaction, as similarly observed for pyridine derivatives.18c As such, substrates that had room for improvement regarding their reactivity at 80 °C (1b–1d, Fig. 4) are efficiently borylated in the ortho position at 100 °C.
| a Reaction conditions: 1 (0.162 mmol), [Ir(COD)(Cl)]2 (1.7 mg, 2.43 × 10−3 mmol), L1 (4.1 mg, 4.86 × 10−3 mmol), B2pin2 (61.7 mg, 0.243 mmol), cyclohexene (13 mg, 16 μL, 0.162 mmol), p-xylene (1 mL), 100 °C, 30 h (isolated yields after purification by column chromatography are reported and values in brackets correspond to the ratio between the ortho-regioisomer and other borylated products). b 40 hours. c 48 hours. d Without cyclohexene additive. e 72 hours. f 24 hours. g 80 °C. |
|---|
|
The catalysis tolerates halides such as fluoride, chloride and bromide in both ortho and meta positions in the aromatic ring as well as alkyl, benzyl and aryl substituents. The reactions appeared sensitive for the case in which the halide substituents are in the other ortho position. For instance, the ortho-borylated product 2f containing a fluoride atom in the other ortho position was obtained in a remarkable 91% isolated yield whereas the yield gradually decreased from chloride (70% for 2g) to bromide (28% for 2h). We reasoned steric shielding between large substituents in the ortho position of the benzamide substrate and the porphyrin macrocycle and/or the meso phenyl substituents being at play regarding the structures of the DFT-computed intermediates and transition states (vide supra). Clearly, the reactivity for halide substituents follows a purely steric effect and not an electronic one, indirectly pointing out that the reactivity occurs when the substrate binds to the molecular recognition site of the supramolecular catalyst via remote Zn⋯OC weak interaction. In the same vein, the ortho-borylated benzamides comprising less sterically demanding meta-functionalized fluoride (2i), chloride (2j) and bromide (2k) were isolated in 94%, 72% and 84% yields, respectively. In these cases, no C–H borylation took place in the ortho C–H bond between the amide and the halide groups. In contrast, a methoxy group placed in the meta position of the benzamide ring led to, besides the expected major borylated compound 2l in 67% yield, 13% of 2m in which the borylation occurred also at the ortho-C–H bond located between the amide and the methoxy group, thus indicating some directing group character for the methoxy group.34 Tertiary benzamides comprising electronically different methyl and ether groups in the para position were compatible for the iridium-catalysed C–H bond ortho-borylation affording the corresponding products 2n and 2o in 94% and 61% isolated yields, respectively. In the case of benzamide 1o, which comprises two aromatic groups, only the benzamide ring was prone to react by affording 2o. A mixture of unidentified, borylated products was obtained when the substrate contained a nitro functional group (Table 2, bottom).
As could be expected considering our previous contributions,18 using a pyridine derivative containing a tertiary amide group in the meta position afforded a mixture of borylated products as the nitrogen atom from pyridine and the carbonyl group from the amide are both engaged in binding to the zinc-porphyrin molecular recognition site. Next, we aimed at exploring other carbonyl-containing substrates different than amides 1 in the supramolecular iridium-catalysed ortho-C–H borylation (Table 2, framed). Acetophenone and methylbenzoate afforded the corresponding ortho-borylated products 2p and 2q, respectively, in promising 49% isolated yield in both cases. This observation also indirectly indicates the key role of the remote, weak Zn⋯OC interaction in increasing the reactivity of the iridium catalyst. No reactivity was observed when using the very challenging benzaldehyde as the substrate.35 It is relevant to note that the formation of deoxygenated side-products such as amine 5 (Table 1) was substrate-dependent, being not necessarily the use of the cyclohexene additive in several cases. As such, addition of cyclohexene might be regarded as a new tool for obtaining selective C–H borylations in challenging substrates.
Considering the many specificities presented by this supramolecular iridium catalysis as well as previous iridium-catalysed C–H bond borylations,7–18 a reaction mechanism is proposed in Scheme 4. Initially, Ir-1 species, which is a tris-boryl-iridium(COD) (COD = 1,5-cyclooctadiene) species coordinated to the triazolylpyridine site of L1, formed and further reacted with the benzamide substrate 1 liberating the COD ligand to afford intermediate A, in which the carbonyl group from the tertiary benzamide binds to the zinc centre of the porphyrin molecular recognition site. After selective ortho-C–H activation (B) and C–B bond formation at iridium (C), which is rate-determining (vide supra), the catalyst was regenerated with B2pin2 delivering one equivalent of HBpin. This catalyst regeneration may take place with the product still bound to the molecular recognition site as it could stabilize the transition state via additional Ir⋯π interactions,18b as supported by DFT calculations (vide supra). Final product release and substrate binding enables the catalytic cycle (D → A) to be pursued. The generated HBpin at each turnover in the catalyst regeneration step C → D may partially serve to deoxygenate product 2 forming 5 (red frame, Scheme 4). However, the presence of stoichiometric amounts of cyclohexene in the reaction mixture suppresses this reaction pathway since the HBpin trapping event (blue frame, Scheme 4) is faster than the deoxygenative reduction (red frame, Scheme 4).
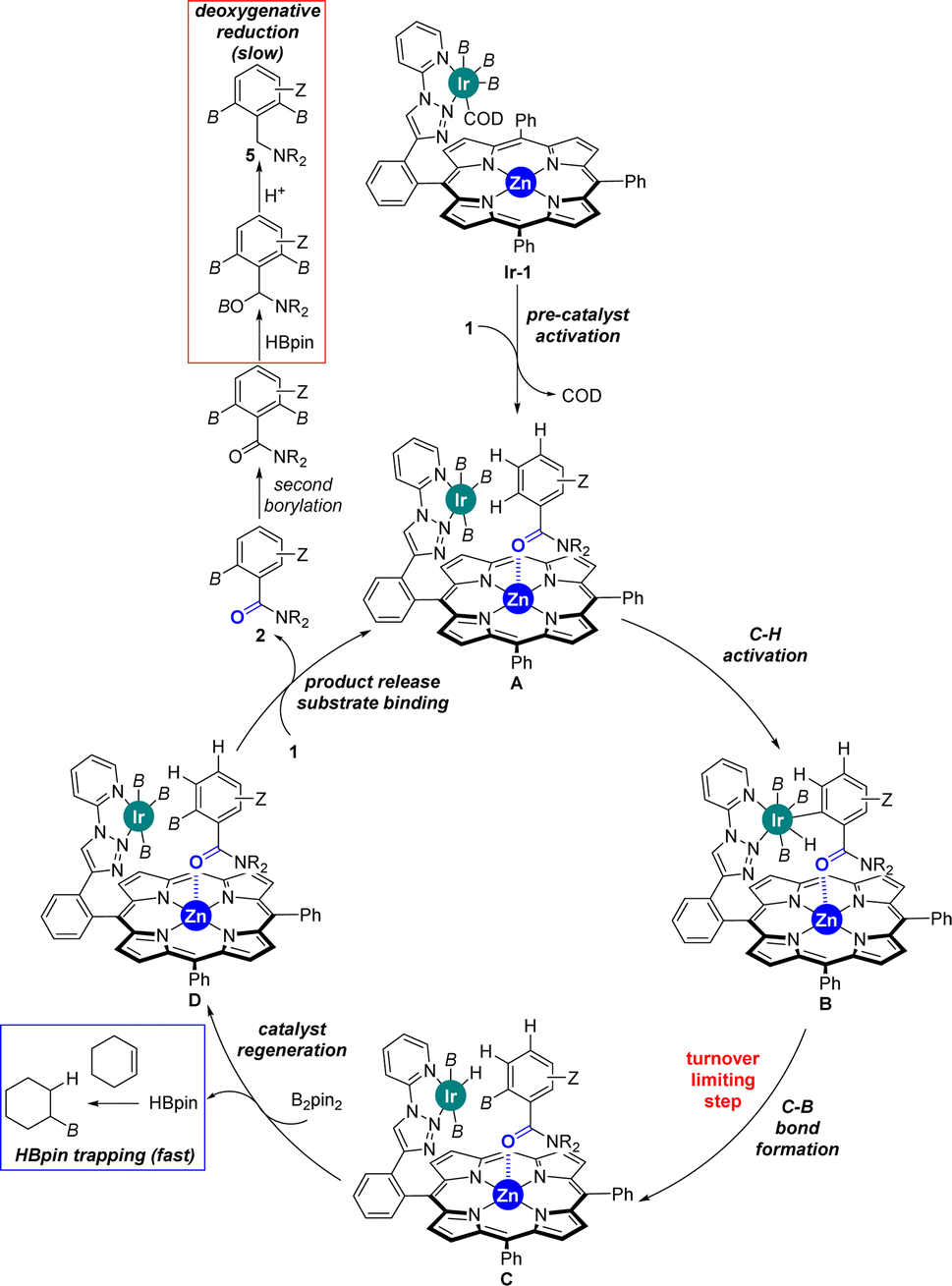 | ||
| Scheme 4 Postulated reaction mechanism for the supramolecular iridium-catalysed ortho-selective C–H borylation of tertiary benzamides 1 using L1. B = (pinacolato)boron. | ||
Conclusions
In summary, we have reported the use of Zn⋯OC weak interactions to exert control on the reactivity of metal catalysts. In particular, we have demonstrated that the iridium-catalysed C–H bond borylations of tertiary benzamides occur in the ortho position when using a triazolylpyridine ligand equipped with a zinc-porphyrin molecular recognition site. The selectivity is exclusively dominated by the first coordination sphere of the catalyst since fine-tuning the triazolylpyridine fragment results in negligible changes regarding the regioselectivity ratio of the borylated products. Interestingly, the activity was controlled by the presence of the zinc-porphyrin backbone that brings the substrate close to the active site as demonstrated by a number of control experiments. Such behaviour is reminiscent of the Michaelis complex formed between a substrate and an enzyme in biological catalysis.36 The optimization of the reaction conditions enabled the identification of [Ir(COD)(Cl)]2 as a suitable precursor, which is rare for iridium-catalysed C–H borylations as most of the studies employed the more air sensitive [Ir(COD)(OMe)]2.18,37 Careful analysis of reaction conditions by mass balance analysis revealed the unexpected formation, in some cases, of a deoxygenated borylated side-product (5) resulting from the reaction of the HBpin formed during the catalyst regeneration with the borylated benzamide product 2. This issue was circumvented by utilizing one equivalent of cyclohexene during the catalysis as a HBpin scavenger. In this way, ortho-borylated tertiary benzamides with a wide variety of functional groups at different positions were obtained according to a substrate evaluation. Preliminary studies indicate that enlarging the substrate scope to other oxygen-containing substrates is feasible. Importantly, detailed kinetic studies and DFT calculations shed light on the reaction mechanism showing that the C–H activation step is not the turnover limiting one in the catalytic cycle, which may explain the similar selectivity obtained for all supramolecular ligands (L1–L6). Overall, this contribution represents a unique case in which (i) not the selectivity but the activity is controlled by weak interactions at the secondary coordination sphere in transition metal-catalysed C–H functionalizations, (ii) a formal change in the rate-determining step for unbiased substrates is evidenced compared to state-of-the-art catalysts thanks to a supra-molecular catalyst equipped with a well-defined substrate-recognition site, and (iii) a catalytic system previously known for meta-borylation switches its selectivity towards the ortho-regioisomer.
Data availability
The data supporting the findings of this study are available within the article and its ESI.†Author contributions
JT: conceptualization, data curation, formal analysis, investigation, methodology, visualization, writing – original draft. VD: data curation, formal analysis, investigation, validation. MT: data curation, formal analysis, investigation, methodology, visualization. PR: data curation, formal analysis, investigation, validation. TR: data curation, formal analysis, investigation. AP: data curation, formal analysis, investigation, funding acquisition, methodology, project administration, supervision, validation, visualization. RGD: conceptualization, data curation, funding acquisition, methodology, project administration, supervision, visualization, writing – original draft, review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The CNRS, University of Rennes, Fondation Rennes (MSc grant to P. R.), Agence Nationale de la Recherche (ANR-19-CE07-0039, PhD grant to J. T.) and Ministère de l'Enseignement supérieur et de la Recherche (PhD grant to V. D.) are acknowledged for financial support. A. P. is a Serra Húnter Fellow and recipient of ICREA Academia Prize 2019. A. P. thanks the Spanish Ministerio de Ciencia e Innovación for project PID2021-127423NB-I00 and the Generalitat de Catalunya for project 2021SGR623.Notes and references
- (a) M. C. White, Science, 2012, 335, 807 CrossRef CAS PubMed; (b) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900 RSC; (c) S. K. Sinha, S. Guin, S. Maiti, J. P. Biswas, S. Porey and D. Maiti, Chem. Rev., 2022, 122, 5682 CrossRef CAS PubMed; (d) T. Dalton, T. Faber and F. Glorius, ACS Cent. Sci., 2021, 7, 245 CrossRef CAS PubMed; (e) J. A. Labinger, Chem. Rev., 2017, 117, 8483 CrossRef CAS PubMed; (f) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS PubMed; (g) M. M. Diaz-Requejo and P. J. Perez, Chem. Rev., 2008, 108, 3379 CrossRef CAS PubMed.
- (a) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546 RSC; (b) P. S. Fier and J. F. Hartwig, Science, 2013, 342, 956 CrossRef CAS PubMed; (c) W. Liu and J. T. Groves, Acc. Chem. Res., 2015, 48, 1727 CrossRef CAS PubMed; (d) M. Simonetti, D. M. Cannas, X. Just-Baringo, I. J. Vitorica-Yrezabal and I. Larrosa, Nat. Chem., 2018, 10, 724 CrossRef CAS PubMed; (e) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed; (f) Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787 CrossRef CAS PubMed; (g) J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 2 CrossRef CAS PubMed; (h) Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333 CrossRef CAS PubMed.
- (a) D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383 CrossRef CAS PubMed; (b) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (c) L. Guillemard, N. Kaplaneris, L. Ackermann and M. J. Johansson, Nat. Rev. Chem, 2021, 5, 522 CrossRef CAS PubMed; (d) R. Jana, H. M. Begam and E. Dinda, Chem. Commun., 2021, 57, 10842 RSC; (e) R. R. Karimov and J. F. Hartwig, Angew. Chem., Int. Ed., 2018, 57, 4234 CrossRef CAS PubMed; (f) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC.
- (a) D. J. Schipper and K. Fagnou, Chem. Mater., 2011, 23, 1594 CrossRef CAS; (b) H. Bohra and M. Wang, J. Mater. Chem. A, 2017, 5, 11550 RSC; (c) L. G. Mercier and M. Leclerc, Acc. Chem. Res., 2013, 46, 1597 CrossRef CAS PubMed; (d) L. Xing and C. K. Luscombe, J. Mater. Chem. C, 2021, 9, 16391 RSC; (e) Y. Segawa, T. Maekawa and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 66 CrossRef CAS; (f) I. A. Stepek and K. Itami, ACS Mater. Lett., 2020, 2, 951 CrossRef CAS.
- (a) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (b) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS; (c) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (d) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (e) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603 RSC; (f) S. De Sarkar, W. Liu, S. I. Kozhushkov and L. Ackermann, Adv. Synth. Catal., 2014, 356, 1461 CrossRef CAS; (g) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (h) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (i) G. Meng, N. Y. S. Lam, E. L. Lucas, T. G. Saint-Denis, P. Verma, N. Chekshin and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142, 10571 CrossRef CAS PubMed; (j) R. Shang, L. Ilies and E. Nakamura, Chem. Rev., 2017, 117, 9086 CrossRef CAS PubMed; (k) S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788 CrossRef CAS PubMed; (l) R.-Y. Zhu, M. E. Farmer, Y.-Q. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 10578 CrossRef CAS PubMed; (m) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236 CrossRef CAS PubMed; (n) R. Gramage-Doria, Chem.–Eur. J., 2020, 26, 9688 CrossRef CAS PubMed; (o) N. Y. S. Lam, Z. Fan, K. Wu, H. S. Park, S. Y. Shim, D. A. Strassfeld and J.-Q. Yu, J. Am. Chem. Soc., 2022, 144, 2793 CrossRef CAS PubMed.
- (a) K. Liao, S. Negretti, D. G. Musaev, J. Bacsa and H. M. L. Davies, Nature, 2016, 533, 230 CrossRef CAS PubMed; (b) Z. Zhang, K. Tanaka and J.-Q. Yu, Nature, 2017, 543, 538 CrossRef CAS PubMed; (c) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed; (d) P. Wang, P. Verma, G. Xia, J. Shi, J. X. Qiao, S. Tao, P. T. W. Cheng, M. A. Poss, M. E. Farmer, K.-S. Yeung and J.-Q. Yu, Nature, 2017, 551, 489 CrossRef CAS PubMed; (e) N. Della Ca, M. Fontana, E. Motti and M. Catellani, Acc. Chem. Res., 2016, 49, 1389 CrossRef CAS PubMed; (f) D. Lichosyt, Y. Zhang, K. Hurej and P. Dydio, Nat. Catal., 2019, 2, 114 CrossRef CAS; (g) R. Gramage-Doria and C. Bruneau, Coord. Chem. Rev., 2021, 428, 21362 CrossRef; (h) J. F. Hartwig and M. A. Larsen, ACS Cent. Sci., 2016, 2, 281 CrossRef CAS PubMed; (i) M. T. Mihai, G. R. Genova and R. J. Phipps, Chem. Soc. Rev., 2018, 47, 149 RSC.
- (a) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed; (b) R. Bisht, C. Haldar, M. M. M. Hassan, M. E. Hoque, J. Chaturvedi and B. Chattopadhyay, Chem. Soc. Rev., 2022, 51, 5042 RSC; (c) L. Xu, G. Wang, S. Zhang, H. Wang, L. Wang, L. Liu, J. Jiao and P. Li, Tetrahedron, 2017, 73, 7123 CrossRef CAS; (d) J. S. Wright, P. J. H. Scott and P. G. Steel, Angew. Chem., Int. Ed., 2021, 60, 2796 CrossRef CAS PubMed; (e) Y. Kuroda, K. Park, Y. Shimazaki, R.-L. Zhong, S. Sakaki and Y. Nakao, Angew. Chem., Int. Ed., 2023, 62, e202300704 CrossRef CAS PubMed; (f) M. M. M. Hassan, S. Guria, S. Dey, J. Das and B. Chattopadhyay, Sci. Adv., 2023, 9, eadg3311 CrossRef CAS PubMed; (g) M. R. Smith III, R. Bisht, C. Haldar, G. Pandey, J. E. Dannatt, B. Ghaffari, R. E. Maleczka Jr and B. Chattopadhyay, ACS Catal., 2018, 8, 6216 CrossRef PubMed; (h) S. Guria, M. M. M. Hassan and B. Chattopadhyay, Org. Chem. Front., 2024, 11, 929 RSC.
- (a) D. G. Hall, Boronic Acids. Preparation, Applications in Organic Synthesis, Wiley-VCH, Weinheim, 2011 CrossRef; (b) E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091 CrossRef CAS PubMed.
- (a) J. Takagi, K. Sato, J. F. Hartwig, T. Ishiyama and N. Miyaura, Tetrahedron Lett., 2002, 43, 5649 CrossRef CAS; (b) T. Ishiyama, Y. Nobuta, J. F. Hartwig and N. Miyaura, Chem. Commun., 2003, 2924 RSC; (c) M. A. Larsen, R. J. Oeschger and J. F. Hartwig, ACS Catal., 2020, 10, 3415 CrossRef CAS PubMed; (d) M. A. Larsen, C. V. Wilson and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 8633 CrossRef CAS PubMed; (e) R. Oeschger, B. Su, Y. Yu, C. Ehinger, E. Romero, S. He and J. Hartwig, Science, 2020, 368, 736 CrossRef CAS PubMed; (f) O. Kuleshova, S. Asako and L. Ilies, ACS Catal., 2021, 11, 5968 CrossRef CAS; (g) C. Haldar, M. E. Hoque, J. Chaturvedi, M. M. M. Hassan and B. Chattopadhyay, Chem. Commun., 2021, 57, 13059 RSC; (h) M. E. Hoque, S. Dey, M. M. M. Hassan, J. Chaturvedi, S. Guria, J. Das, B. Roy and B. Chattopadhyay, Tetrahedron Chem, 2022, 3, 100028 CrossRef CAS.
- (a) E. Fernandez, Top. Organomet. Chem., 2020, 69, 207 CrossRef; (b) T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390 CrossRef CAS PubMed; (c) T. Ishiyama, J. Takagi, J. F. Hartwig and N. Miyaura, Angew. Chem., Int. Ed., 2002, 41, 3056 CrossRef CAS; (d) C. W. Liskey, C. S. Wei, D. R. Pahlsa and J. F. Hartwig, Chem. Commun., 2009, 5603 RSC; (e) R. J. Oeschger, M. A. Larsen, A. Bismuto and J. F. Hartwig, J. Am. Chem. Soc., 2019, 141, 16479 CrossRef CAS PubMed; (f) A. Ros, R. Fernandez and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229 RSC; (g) J. Chaturvedi, C. Haldar, R. Bisht, G. Pandey and B. Chattopadhyay, J. Am. Chem. Soc., 2021, 143, 7604 CrossRef CAS PubMed; (h) C. Haldar, R. Bisht, J. Chaturvedi, S. Guria, M. M. M. Hassan, B. Ram and B. Chattopadhyay, Org. Lett., 2022, 24, 8147 CrossRef CAS PubMed; (i) S. Guria, M. M. M. Hassan, J. Ma, S. Dey, Y. Liang and B. Chattopadhyay, Nat. Commun., 2023, 14, 6906 CrossRef CAS PubMed.
- (a) W. Chang, Y. Chen, S. Lu, H. Jiao, Y. Wang, T. Zheng, Z. Shi, Y. Han, Y. Lu, Y. Wang, Y. Pan, J.-Q. Yu, K. N. Houk, F. Liu and Y. Liang, Chem, 2022, 8, 1775 CrossRef CAS; (b) S. Pandit, S. Maiti and D. Maiti, Org. Chem. Front., 2021, 8, 4349 RSC; (c) X. Zou and S. Xu, Chin. J. Org. Chem., 2021, 41, 2610 CrossRef CAS; (d) J. Wang, T. Torigoe and Y. Kuninobu, Org. Lett., 2019, 21, 1342 CrossRef CAS PubMed; (e) X. Lu, Y. Yoshigoe, H. Ida, M. Nishi, M. Kanai and Y. Kuninobu, ACS Catal., 2019, 9, 1705 CrossRef CAS; (f) H. J. Davis, M. T. Mihai and R. J. Phipps, J. Am. Chem. Soc., 2016, 138, 12759 CrossRef CAS PubMed; (g) B. Lee, M. T. Mihai, V. Stojalnikova and R. J. Phipps, J. Org. Chem., 2019, 84, 13124 CrossRef CAS PubMed; (h) M. T. Mihai, H. J. Davis, G. R. Genov and R. J. Phipps, ACS Catal., 2018, 8, 3764 CrossRef CAS; (i) M. E. Hoque, R. Bisht, C. Haldar and B. Chattopadhyay, J. Am. Chem. Soc., 2017, 139, 7745 CrossRef CAS PubMed; (j) Y. Wang, W. Chang, S. Qin, H. Ang, J. Ma, S. Lu and Y. Liang, Angew. Chem., Int. Ed., 2022, 61, e202206797 CrossRef CAS PubMed; (k) J. L. Douthwaite and R. J. Phipps, Tetrahedron, 2022, 117–118, 132831 CrossRef CAS; (l) R. Bisht and B. Chattopadhyay, J. Am. Chem. Soc., 2016, 138, 84 CrossRef CAS PubMed; (m) R. Bisht, J. Chaturvedi, G. Pandey and B. Chattopadhyay, Org. Lett., 2019, 21, 6476 CrossRef CAS PubMed.
- M. Ditzen, M. Pellegrino and L. B. Vosshall, Science, 2008, 319, 1838 CrossRef CAS PubMed.
- (a) G. A. Bakken and P. C. Jurs, J. Med. Chem., 2000, 43, 4534 CrossRef CAS PubMed; (b) X. He, A. Alianc and P. R. Ortiz de Montellano, Bioorg. Med. Chem., 2007, 15, 6649 CrossRef CAS PubMed; (c) M. A. Letavic, L. Aluisio, R. Apodaca, M. Bajpai, A. J. Barbier, A. Bonneville, P. Bonaventure, N. I. Carruthers, C. Dugovic, I. C. Fraser, M. L. Kramer, B. Lord, T. W. Lovenberg, L. Y. Li, K. S. Ly, H. Mcallister, N. S. Mani, K. L. Morton, A. Ndifor, S. D. Nepomuceno, C. R. Pandit, S. B. Sands, C. R. Shah, J. E. Shelton, S. S. Snook, D. M. Swanson and W. Xiao, ACS Med. Chem. Lett., 2015, 6, 450 CrossRef CAS PubMed; (d) C. J. Gerry and S. L. Schreiber, Nat. Rev. Drug Discovery, 2018, 17, 333 CrossRef CAS PubMed.
- (a) L. Yang, K. Semba and Y. Nakao, Angew. Chem., Int. Ed., 2017, 56, 4853 CrossRef CAS PubMed; (b) M. E. Hoque, R. Bisht, A. Unnikrishnan, S. Dey, M. M. M. Hassan, S. Guria, R. N. Rai, R. B. Sunoj and B. Chattopadhyay, Angew. Chem., Int. Ed., 2022, 61, e202203539 CrossRef CAS PubMed; (c) R. Bisht, M. E. Hoque and B. Chattopadhyay, Angew. Chem., Int. Ed., 2018, 57, 15762 CrossRef CAS PubMed; (d) Y. Kuninobu, H. Ida, M. Nishi and M. Kanai, Nat. Chem., 2015, 7, 712 CrossRef CAS PubMed; (e) S. Lu, T. Zheng, J. Ma, Z. Deng, S. Qin, Y. Chen and Y. Liang, Angew. Chem., Int. Ed., 2022, 61, e202201285 CrossRef CAS PubMed; (f) L. Yang, N. Uemura and Y. Nakao, J. Am. Chem. Soc., 2019, 141, 7972 CrossRef CAS PubMed.
- (a) S.-T. Bai, C. B. Bheeter and J. N. H. Reek, Angew. Chem., Int. Ed., 2019, 58, 13039 CrossRef CAS PubMed; (b) C. Liu, L. Zhang, L. Li and M. Lei, J. Org. Chem., 2021, 86, 16858 CrossRef CAS PubMed.
- (a) J. E. Dannatt, A. Yadav, M. R. Smith III and R. E. Maleczka Jr, Tetrahedron Lett., 2022, 109, 132578 CrossRef CAS PubMed; (b) B. Ghaffari, S. M. Preshlock, D. L. Plattner, R. J. Staples, P. E. Maligres, S. W. Krska, R. E. Maleczka Jr and M. R. Smith III, J. Am. Chem. Soc., 2014, 136, 14345 CrossRef CAS PubMed; (c) M. E. Hoque, M. M. M. Hassan and B. Chattopadhyay, J. Am. Chem. Soc., 2021, 143, 5022 CrossRef CAS PubMed; (d) G. Wang, L. Liu, H. Wang, Y.-S. Ding, J. Zhou, S. Mao and P. Li, J. Am. Chem. Soc., 2017, 139, 91 CrossRef CAS PubMed; (e) J. Jiao, W. Nie, P. Song and P. Li, Org. Biomol. Chem., 2021, 19, 355 RSC; (f) D. Marcos-Atanes, C. Vidal, C. D. Navo, F. Peccati, G. Jiménez-Osés and J. L. Mascareñas, Angew. Chem., Int. Ed., 2023, 62, e202214510 CrossRef CAS PubMed; (g) S. Kawamorita, H. Ohmiya, K. Hara, A. Fukuoka and M. Sawamura, J. Am. Chem. Soc., 2009, 131, 5058 CrossRef CAS PubMed; (h) H. L. Li, Y. Kuninobu and M. Kanai, Angew. Chem., Int. Ed., 2017, 56, 1495 CrossRef CAS PubMed; (i) H.-L. Li, M. Kanai and Y. Kuninobu, Org. Lett., 2017, 19, 5944 CrossRef CAS PubMed; (j) J. Zeng, M. Naito, T. Torigoe, M. Yamanaka and Y. Kuninobu, Org. Lett., 2020, 22, 3485 CrossRef CAS PubMed; (k) Y. Kuninobu, T. Iwanaga, T. Omura and K. Takai, Angew. Chem., Int. Ed., 2013, 52, 4431 CrossRef CAS PubMed; (l) Y. Yoshigoe and Y. Kuninobu, Org. Lett., 2017, 19, 3450 CrossRef CAS PubMed; (m) J. M. Zakis, A. M. Messinis, L. Ackermann, T. Smejkal and J. Wencel-Delord, Adv. Synth. Catal., 2024, 366, 2292 CrossRef CAS.
- (a) J. V. Obligacion, S. P. Semproni, I. Pappas and P. J. Chirik, J. Am. Chem. Soc., 2016, 138, 10645 CrossRef CAS PubMed; (b) J. V. Obligacion and P. J. Chirik, ACS Catal., 2017, 7, 4366 CrossRef CAS PubMed; (c) H. Li, J. V. Obligacion, P. J. Chirik and M. B. Hall, ACS Catal., 2018, 8, 10606 CrossRef CAS PubMed; (d) T. P. Pabst, J. V. Obligacion, E. Rochette, I. Pappas and P. J. Chirik, J. Am. Chem. Soc., 2019, 141, 15378 CrossRef CAS PubMed; (e) R. Arevalo, T. P. Pabst and P. J. Chirik, Organometallics, 2020, 39, 2763 CrossRef CAS PubMed; (f) T. P. Pabst, L. Quach, K. T. MacMillan and P. J. Chirik, Chem, 2021, 7, 237 CrossRef CAS PubMed.
- (a) J. Trouvé, P. Zardi, S. Al-Shehimy, T. Roisnel and R. Gramage-Doria, Angew. Chem., Int. Ed., 2021, 60, 18006 CrossRef PubMed; (b) M. Tomasini, L. Caporaso, J. Trouvé, J. Poater, R. Gramage-Doria and A. Poater, Chem.–Eur. J., 2022, 28, e202201970 CrossRef CAS PubMed; (c) J. Trouvé, P. Rajeshwaran, M. Tomasini, A. Perennes, T. Roisnel, A. Poater and R. Gramage-Doria, ACS Catal., 2023, 13, 7715 CrossRef.
- Deposition Number 2216009 (ZnTPP ⊂ DMF) contains the supplementary crystallographic data for this paper. These data are provided free of charge by the Cambridge Crystallographic Data Centre†.
- (a) M. M. Williamson, C. M. Prosser-McCartha, S. Mukundan Jr and C. L. Hill, Inorg. Chem., 1988, 27, 1061 CrossRef CAS; (b) S. Lipstman, S. Muniappan and I. Goldberg, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, m2330 CrossRef CAS; (c) B. Boitrel, V. Baveux-Chambenoît and P. Richard, Eur. J. Org Chem., 2001, 4213 CrossRef CAS; (d) L. H. Tong, P. Pengo, W. Clegg, J. P. Lowe, P. R. Raithby, J. K. M. Sanders and S. I. Pascu, Dalton Trans., 2011, 40, 10833 RSC; (e) L. Carlucci, G. Ciani, D. M. Proserpio and F. Porta, CrystEngComm, 2005, 7, 78 RSC; (f) G. Huang, C.-K. Tsang, Z. Xu, K. Li, M. Zeller, A. D. Hunter, S. S.-Y. Chui and C.-M. Che, Cryst. Growth Des., 2009, 9, 1444 CrossRef CAS; (g) S. J. Rodgers, C. A. Koch, J. R. Tate, C. A. Reed, C. W. Eigenbrot and W. R. Scheidt, Inorg. Chem., 1987, 26, 3647 CrossRef CAS; (h) C. A. Swamy and P. Thilagar, Chem.–Eur. J., 2015, 21, 8874 CrossRef PubMed; (i) Y. Zhu, Y. Huang, Q. Li, D. Zang, J. Gu, Y. Tang and Y. Wei, Inorg. Chem., 2020, 59, 2575 CrossRef CAS PubMed; (j) R. Rubbiani, W. Wu, A. Naik, M. Larocca, L. Schneider, R. Padrutt, V. Babu, C. Konig, D. Hinger, C. Maake, S. Ferrari, G. Gasser and B. Spingler, Chem. Commun., 2020, 56, 14373 RSC; (k) I. Goldberg and A. Karmakar, CrystEngComm, 2010, 12, 4095 RSC; (l) T. Nakamura, H. Ube, M. Shiro and M. Shionoya, Angew. Chem., Int. Ed., 2013, 52, 720 CrossRef CAS PubMed; (m) E. Nichols, J. S. Derrick, S. K. Nistanaki, P. T. Smith and C. J. Chang, Chem. Sci., 2018, 9, 2952 RSC; (n) H. Huang, H. Sato, J. Pirillo, Y. Hijikata, Y. S. Zhao, S. Z. D. Cheng and T. Aida, J. Am. Chem. Soc., 2021, 143, 15319 CrossRef CAS PubMed; (o) D. K. Kumar, A. Das and P. Dastidar, Inorg. Chem., 2007, 46, 7351 CrossRef CAS PubMed.
- (a) R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533 CrossRef CAS; (b) R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512 CrossRef CAS; (c) R. G. Pearson, Coord. Chem. Rev., 1990, 100, 403 CrossRef CAS.
- J.-Y. Cho, C. N. Iverson and M. R. Smith, J. Am. Chem. Soc., 2000, 122, 12868 CrossRef CAS.
- (a) M. Kadri, J. Hou, V. Dorcet, T. Roisnel, L. Bechki, A. Miloudi, C. Bruneau and R. Gramage-Doria, Chem.–Eur. J., 2017, 23, 5033 CrossRef CAS PubMed; (b) P. Zardi, T. Roisnel and R. Gramage-Doria, Chem.–Eur. J., 2019, 25, 627 CrossRef CAS PubMed; (c) N. Abuhafez and R. Gramage-Doria, Faraday Discuss., 2023, 244, 186 RSC.
- (a) K. M. Kadish, K. M. Smith and R. Guilard, Handbook of Porphyrin Science, World Scientific, Singapore, vol. 1–44, 2010 Search PubMed; (b) I. Beletskaya, V. S. Tyurin, A. Y. Tsivadze, R. Guilard and C. Stern, Chem. Rev., 2009, 109, 1659 CrossRef CAS PubMed.
- (a) R. P. Bonar-Law, L. G. Mackay, C. J. Walter, V. Marvaud and J. K. M. Sanders, Pure Appl. Chem., 1994, 66, 803 CrossRef CAS; (b) J. K. M. Sanders, Pure Appl. Chem., 2000, 72, 2265 CrossRef CAS.
- D. W. Robbins and J. F. Hartwig, Angew. Chem., Int. Ed., 2013, 52, 933 CrossRef CAS PubMed.
-
(a) H. Sigel and R. B. Martin, Chem. Rev., 1982, 82, 385 CrossRef CAS;
(b) S. Adachi, N. Kumagai and M. Shibasaki, Chem. Sci., 2017, 8, 85 RSC;
(c) The calculated MBO values in the solvent phase of the Zn⋯OC interaction are 0.138 for benzamide substrate and 0.122 of the Zn⋯N interaction for pyridine substrate..
- (a) W. D. Jones, Acc. Chem. Res., 2003, 36, 140 CrossRef CAS PubMed; (b) S. A. Blum, K. L. Tan and R. G. Bergman, J. Org. Chem., 2003, 68, 4127 CrossRef CAS PubMed; (c) G. Parkin, Acc. Chem. Res., 2009, 42, 315 CrossRef CAS PubMed; (d) M. Gómez-Gallego and M. A. Sierra, Chem. Rev., 2011, 111, 4857 CrossRef PubMed; (e) E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066 CrossRef CAS PubMed; (f) Z. Mao and C. T. Campbell, ACS Catal., 2020, 10, 4181 CrossRef CAS; (g) X. Gao, X.-Y. Yua and C.-R. Chang, Phys. Chem. Chem. Phys., 2022, 24, 15182 RSC.
- M. M. M. Hassan, B. Mondal, S. Singh, C. Haldar, J. Chaturvedi, R. Bisht, R. B. Sunoj and B. Chattopadhyay, J. Org. Chem., 2022, 87, 4360 CrossRef PubMed.
- A low KIE effect has recently been reported for directing-group-controlled iridium-catalyzed C–H borylation in which the rate-determining step is the isomerization of the iridium(III) C–H-activated complex, see: N. Le, N. L. Chuang, C. M. Oliver, A. V. Samoshin, J. T. Hemphill, K. C. Morris, S. N. Hyland, H. Gua, Ch. E. Webster and T. B. Clark, ACS Catal., 2023, 13, 12877 CrossRef CAS.
- (a) C. J. Barger, R. D. Dicken, V. L. Weidner, A. Motta, T. L. Lohr and T. J. Marks, J. Am. Chem. Soc., 2020, 142, 8019 CrossRef CAS PubMed; (b) M. K. Bisai, K. Gour, T. Das, K. Vanka and S. S. Sen, Dalton Trans., 2021, 50, 2354 RSC; (c) P. Ghosh and J. A. von Wangelin, Angew. Chem., Int. Ed., 2021, 60, 16035 CrossRef CAS PubMed; (d) M.-L. Yuan, J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, ACS Catal., 2016, 6, 3665 CrossRef CAS.
- (a) D. J. Pasto and J. Hickman, J. Am. Chem. Soc., 1968, 90, 4445 CrossRef CAS; (b) D. J. Pasto, J. Chow and S. K. Arora, Tetrahedron, 1969, 25, 1557 CrossRef CAS; (c) J. Niziol and T. Ruman, Lett. Org. Chem., 2012, 9, 257 CrossRef CAS; (d) Y. Yamamoto, R. Fujikawa, T. Umemoto and N. Miyaura, Tetrahedron, 2004, 60, 10695 CrossRef CAS; (e) L. P. Press, A. J. Kosanovich, B. J. McCulloch and O. V. Ozerov, J. Am. Chem. Soc., 2016, 138, 9487 CrossRef CAS PubMed.
- H. Tamura, H. Yamazaki, H. Sato and S. Sakaki, J. Am. Chem. Soc., 2003, 125, 16114 CrossRef CAS PubMed.
- (a) B. A. Vanchura II, S. M. Preshlock, P. C. Roosen, V. A. Kallepalli, R. J. Staples, R. E. Maleczka Jr, D. A. Singleton and M. R. Smith III, Chem. Commun., 2010, 46, 7724 RSC; (b) K. Yamazaki, S. Kawamorita, H. Ohmiya and M. Sawamura, Org. Lett., 2010, 12, 3978 CrossRef CAS PubMed; (c) B. Chattopadhyay, J. E. Dannatt, I. L. Andujar-De Sanctis, K. A. Gore, R. E. Maleczka Jr, D. A. Singleton and M. R. Smith III, J. Am. Chem. Soc., 2017, 139, 7864 CrossRef CAS PubMed.
- Replacing the amide group in the substrate 1 by a sulfone, namely methyphenylsulfone, provided a mono-borylated product in a very modest 27% yield, being not possible to isolate it for further characterization unfortunately.
- (a) V. L. Schramm, Acc. Chem. Res., 2015, 48, 1032 CrossRef CAS PubMed; (b) B. Ma, S. Kumar, C. J. Tsai, Z. Hu and R. Nussinov, J. Theor. Biol., 2000, 203, 383 CrossRef CAS PubMed; (c) Y. Peng, A. L. Hansen, L. Bruschweiler-Li, O. Davulcu, J. J. Skalicky, M. S. Chapman and R. Brüschweiler, J. Am. Chem. Soc., 2017, 139, 4846 CrossRef CAS PubMed.
- (a) C. C. C. Johansson Seechurn, V. Sivakumar, D. Satoskar and T. J. Colacot, Organometallics, 2014, 33, 3514 CrossRef CAS; (b) E. D. Slack and T. J. Colac ot, Org. Lett., 2021, 23, 1561 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2216009. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01515k |
| This journal is © The Royal Society of Chemistry 2024 |