
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design of poly(N-isopropylacrylamide) coated MnO2 nanoparticles for thermally regulated catalytic decomposition of H2O2†
Anashwara
Babu‡
a,
Samarendra
Maji‡
*ab,
Gomathi
Sivakumar
a and
Richard
Hoogenboom
 *b
*b
aDepartment of Chemistry, Faculty of Engineering and Technology, SRM Institute of Science and Technology (SRMIST), Kattankulathur, Tamil Nadu-603203, India. E-mail: samarenr@srmist.edu.in
bSupramolecular Chemistry Group, Department of Organic and Macromolecular Chemistry, Ghent University, Krijgslaan 281 S4, Ghent, Belgium. E-mail: richard.hoogenboom@ugent.be
First published on 17th June 2024
Abstract
We report the synthesis of thermoresponsive poly(N-isopropylacrylamide) (PNIPAM) coated manganese dioxide nanoparticles (PNIPAM@MnO2 NPs) for the catalytic decomposition of hydrogen peroxide (H2O2), with the goal of developing a temperature-controlled catalytic system. For this purpose, we have developed MnO2 NPs and further modified them with thermoresponsive PNIPAM containing nitrodopamine end groups. The modification of the MnO2 NPs was confirmed using SEM, TEM, XPS, and TGA. Upon varying the temperature above and below the lower critical solution temperature (LCST) of the polymer coating, the NPs undergo spontaneous cyclic swelling–deswelling changes, as confirmed by DLS measurements. Photoluminescence (PL) experiments were performed to explore the catalytic decomposition of H2O2 by PNIPAM@MnO2 NPs, indicating thermal control over the catalytic activity of the MnO2 NPs. The proposed proof-of-concept of these smart PNIPAM@MnO2 NPs for the decomposition of H2O2 may serve as a basis for the development of smart catalytic self-regulating systems in future work.
Introduction
Hydrogen peroxide (H2O2) catalysis plays a crucial role in biological processes as well as various chemical processes including the production of detergents, bleaching agents, and rocket propellants.1,2 H2O2 naturally decomposes into water (H2O) and oxygen (O2) over time, but this process can be expedited and controlled by introducing a catalyst. The catalytic decomposition of H2O2 holds immense importance in laboratory chemical reactions due to its ability to serve as a controlled source of oxygen.3,4 Common catalysts for this purpose include transition metal ions, such as manganese (Mn), iron (Fe), and palladium (Pd).5–8 The controlled release of oxygen in chemical reactions is valuable for enhancing reaction rates and promoting specific pathways, making catalytic decomposition a key aspect of numerous chemical and biochemical processes such as oxidation reactions and the synthesis of organic compounds.9 Understanding the importance of H2O2 catalysis in biological systems provides insights into the delicate balance of cellular processes and may hold implications for therapeutic interventions in diseases associated with oxidative stress.10 Enzymes such as peroxidases and catalases catalyze the decomposition of H2O2 in the biological domain.11–13 The controlled acceleration of this decomposition is crucial for maintaining cellular homeostasis. This process is vital in preventing the accumulation of toxic levels of H2O2 within cells, as high concentrations can lead to oxidative stress and cellular damage.14 Beyond detoxification, H2O2 is also involved in signalling pathways, serving as a secondary messenger that regulates cellular functions. In particular, it is involved in the modulation of gene expression, immune response, and cell proliferation.14–16 Additionally, certain metabolic pathways, such as fatty acid metabolism, utilize H2O2 as a signalling molecule, emphasizing its multifaceted role in various biological activities.17Typically, the primary determinant of catalytic efficiency in heterogeneous catalysis is sufficient crystallinity and expansive surface area, which are achievable through the utilization of either nanoparticles or granular particles exhibiting porous characteristics.18 Hermanek et al.19 conducted an extensive study on the effect of surface area and crystallinity of iron(III) oxides on the catalytic decomposition of H2O2. In a recent study, iron phosphide was loaded onto alumina to create 4–6 mm spheres that can activate H2O2 and facilitate the decomposition of contaminants in wastewater.20 Manganese dioxide nanoparticles (MnO2 NPs) have an attractive combination of stability, high catalytic efficiency, and cost-effectiveness that sets them apart from all other metal-based catalysts for H2O2 decomposition.21,22 Their importance lies not only in their catalytic activity but also in their biocompatibility and ability to operate under mild conditions. The unique structural features of MnO2 NPs, such as their high surface area and reactive sites, make them well-suited for interacting with H2O2 efficiently.23 Owing to the high reactivity of manganese dioxide (MnO2) towards hydrogen peroxide, it is used in fuel cells in space programs for the exothermic catalytic decomposition of H2O2 for the sustained production of O2.24
The decomposition of H2O2 in chemical reactions is highly temperature-dependent, following the Arrhenius equation, which plays a crucial role in determining the reaction rate and pathway.25 Understanding the complicated relationship between temperature and hydrogen peroxide decomposition can be vital for designing efficient and controlled reactions in diverse industrial and laboratory settings. Furthermore, the temperature-sensitive catalytic decomposition of H2O2 provides a means to precisely control the levels of H2O2 in biological environments. The temperature sensitivity ensures that the decomposition rates align with the dynamic nature of cellular processes, allowing for real-time adjustments in response to varying conditions. This temperature-dependent control is particularly relevant in disease states where fluctuations in temperature often accompany physiological changes. Conditions such as inflammation or infections, which are associated with altered temperatures, can benefit from tailored approaches that consider both the catalytic activity and temperature responsiveness of the decomposition process. This understanding opens avenues for developing targeted therapies that leverage temperature-sensitive catalysts to regulate hydrogen peroxide levels in a spatially and temporally controlled manner, which has not been explored much by researchers. A recent study from our group explained the efficient surface modification of MnO2 nanosheets by a nitrodopamine moiety for extracellular and intracellular glutathione detection.26 This work inspired the utilization of a thermoresponsive polymer having a nitrodopamine end group for the surface modification of MnO2 NPs. Polymeric materials, especially thermoresponsive polymers [e.g. poly(N-isopropyl acrylamide) (PNIPAM)] that undergo a temperature-driven reversible phase transition [LCST] in aqueous solution, have been studied for the development of thermoresponsive smart materials.27 Different metal/metal oxide nanoparticle (NP) cores with thermoresponsive PNIPAM have been investigated for the preparation of smart materials for diverse applications, such as sensors and actuators.28,29
These smart NPs have been prepared either via ‘grafting to’ or ‘grafting from’ approaches.30 Polymers synthesized via controlled radical polymerization ‘CRP’ methods (atom transfer radical polymerization (ATRP),31–34 nitroxide mediated polymerization (NMP),35–37 and reversible addition–fragmentation chain transfer (RAFT) polymerization) are extensively used for the modification of inorganic NPs as these methods provide ease of synthesis of polymers with precise molar masses, diverse compositions, and a high degree of chain-end functionality.38–44 Among the CRP methods, RAFT polymerization is very appealing because of its compatibility with a vast array of monomers, mild reaction conditions and because it avoids the use of metal catalysts.43,45 Thermoresponsive LCST polymers show a phase transition due to the change in entropy of solvation of the polymeric chain in aqueous solution as temperature alters.46 PNIPAM is the most widely used thermoresponsive polymer having a sharp phase transition at around 32 °C.47–50 Unlike existing studies that primarily focus on drug delivery or environmental applications by coating polymers on the surface of MnO2 NPs, this study highlights the thermo-responsive catalytic activity. Therefore, inorganic (metal/metal oxide) nanoparticles (NPs) with a temperature-regulating thermoresponsive polymer layer was hypothesized to enable temperature-responsive catalytic decomposition of H2O2.
By coating manganese dioxide nanoparticles with a temperature-responsive PNIPAM (PNIPAM@MnO2 NPs), the decomposition of H2O2 might be controlled. The decomposition of H2O2 takes place when the polymer is hydrated, which is at a temperature below the lower critical solution temperature (LCST) of PNIPAM, allowing H2O2 to reach the manganese oxide catalytic nanoparticles, leading to its decomposition into water and oxygen. Thus, H2O2 is no longer available for the dimerization of the non-fluorescent homovanillic acid as a model substrate into a fluorescent dimer. When the solution temperature rises over the polymer LCST, the polymer dehydrates and collapses onto the MnO2 NPs as a hydrophobic coating. Since in this state, H2O2 cannot reach the catalytic surface due to the presence of the dehydrated polymer layer around the nanoparticles, H2O2 is available for catalyzing the dimerization reaction, forming a fluorescent product, as shown in Fig. 1.
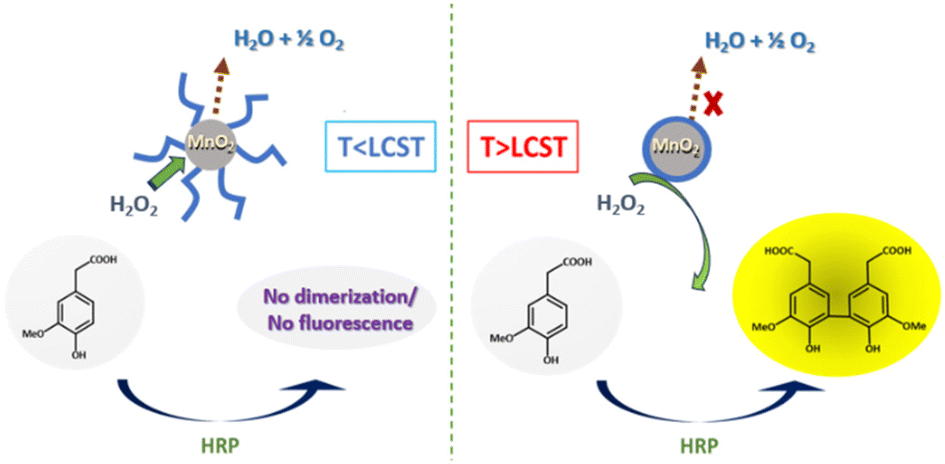 | ||
| Fig. 1 Temperature-driven catalytic decomposition of H2O2. | ||
In this study, we will discuss the synthesis of thermoresponsive PNIPAM-coated MnO2 NPs and their use in the temperature controlled catalytic decomposition of H2O2, which may in the future be used as a basis for the design of smart catalytic self-regulating systems.
Experimental
Materials
N-Isopropylacrylamide (NIPAM) was purchased from Scientific Polymer Products, INC., and was recrystallized twice from n-hexane before use. Potassium permanganate (KMnO4) and sodium nitrite (NaNO2) were obtained from Acros Organics Chemicals. Isoamyl alcohol was purchased from Ferak Berlin GmbH. Azobisisobutyronitrile (AIBN), pentafluorophenol (PFP), N,N′-dicyclohexylcarbodiimide (DCC), 4-dimethylaminopyridine (DMAP), dopamine hydrochloride, and sodium sulfate (Na2SO4) were obtained from Sigma Aldrich. AIBN was recrystallized from MeOH (twice) and stored in a freezer. A 2-(Butylthiocarbonothioylthio)propanoic acid (BTTCP) chain transfer agent was synthesized as previously reported.46 HPLC grade organic solvents dichloromethane (DCM), chloroform (CHCl3), ethylacetate (EtOAc), dimethylformamide (DMF, anhydrous), N,N-dimethylacetamide (DMAc), and diethyl ether (DEE) were obtained from Sigma Aldrich. N,N-Dimethylformamide (DMF) was obtained from Biosolve and n-hexane was from Fischer Scientific. Trimethylamine (TEA) (dry) was purified and collected from the solvent purification system (SPS). Unless specified otherwise, all reagents and solvents were used exactly as purchased from the commercial sources.Instrumentation
1H NMR spectra were recorded on a Bruker Avance 300 and 400 MHz spectrometer at room temperature in chloroform-d (CDCl3) purchased from Euriso-top. The conversion of prepared PNIPAM was analyzed by gas chromatography performed on an Agilent 7890A GC system from Agilent Technologies. Size-exclusion chromatography (SEC) was performed on an Agilent 1260-series HPLC system equipped with a 1260 online degasser, a 1260 ISO-pump, a 1260 automatic liquid sampler (ALS), a thermostatted column compartment (TCC) at 50 °C equipped with two PLgel 5 μm mixed-D columns and a mixed-D guard column in series, a 1260 diode array detector (DAD) and a 1260 refractive index detector (RID). The used eluent was DMA containing 50 mM LiCl at a flow rate of 0.500 mL min−1. The spectra were analyzed using the Agilent Chemstation software with the GPC add-on. The molar mass and dispersity values were calculated against PMMA standards from PSS. The morphologies of MnO2 and PNIPAM@MnO2 nanoparticles were determined by using a scanning electron microscope (FE-SEM, JEOL JSM-7100F) and a high-resolution transmission electron microscope (JEOL TEM 2100 Plus, Japan). Elemental analysis of samples was performed by X-ray photoelectron spectroscopy carried out on a PHI Versa Probe III scanning XPS microprobe (Physical Electronics, USA). The thermal behaviors of samples were measured with a Mettler Toledo TGA-DSC2/LF/1100 thermal gravimetric analyzer. The particle size distributions of products were characterized by a dynamic light scattering (DLS) instrument on a Malvern Zetasizer Nano Series operating a 4 mW He–Ne laser at 633 nm. The analysis was conducted over a range of temperatures, from 10 to 50 °C, with a heating rate of 1.0 °C per minute, at an angle of 173°. Additionally, measurements were taken at a 90° scattering angle using a Zetasizer Malvern/Nano ZS-90 over different temperature range from 20 to 50 °C. UV–vis spectra were measured with a Cary 300 Bio UV-visible spectrophotometer. Temperature-responsive static fluorescence measurements were carried out on a Varian Cary Eclipse fluorescence spectrophotometer. A fluorescence spectrometer Edinburgh FLS 1000 was used to monitor fluorescence at various times in order to analyze the temperature-driven process.Methods
Synthesis of nitrodopamine hydrogensulfate
Nitrodopamine hydrogensulfate was synthesized based on an established procedure.51 A solution of dopamine hydrochloride (5.0 g, 26.4 mmol) and sodium nitrite (6.37 g, 92.3 mmol) in 150 mL distilled water was cooled to 0 °C using an ice bath. Aqueous sulphuric acid (50 mL, 20 v/v%) was slowly added to the reaction mixture. After the addition, the ice bath was removed and the solution was stirred overnight at room temperature. The resulting yellow dispersion was filtered using a sintered funnel and the collected solid was washed with ice-cold water several times and finally dried in a vacuum oven at 50 °C for 2 days to give nitrodopamine hydrogensulfate. 1H NMR spectroscopy was used to confirm the nitrodopamine hydrogen sulfate structure (3.8 g, yield: 49%). A 1H NMR spectrum of the nitrodopamine hydrogen sulfate is shown in Fig. S1.†Synthesis of pentafluorophenol (PFP) containing CTA
BTTCP-PFP was synthesized as described by Stenzel et al.52 BTTCP (1.92 g, 8.1 mmol), PFP (1.60 g, 8.7 mmol) and DMAP (106 mg, 0.87 mmol) were added to a round-bottom flask and dissolved in anhydrous DCM (55 mL). The reaction mixture was cooled to 0 °C in an ice bath and a solution of DCC (1.80 g, 8.7 mmol) in DCM (15 mL) was added drop-wise with fast stirring. The reaction was allowed to stir in an ice bath for two hours and was continued at room temperature overnight. The resulting solution was filtered, and the solvent was evaporated under vacuum. The crude product was purified by column chromatography on silica gel using chloroform as the eluent. The solvent was removed under reduced pressure to obtain dark yellow oil (2.2 g, yield 67%). 1H NMR spectroscopy was used to confirm the BTTCP-PFP structure as shown in Fig. S2.†1H-NMR (300 MHz, CDCl3) δ (ppm): 5.10 (q, J = 7.4, 1H, –CHCH3), 3.38 (t, J = 7.4 Hz, 2H, –SCH2–), 1.76 (d, J = 7.4 Hz, 3H, –CHCH3), 1.69 (tt, J = 7.4 Hz, 2H, –SCH2CH2–), 1.44 (app. sext, J = 7.4 Hz, 2H, –CH2CH3), 0.93 (t, J = 7.4 Hz, 3H, –CH2CH3).Synthesis of nitro DOPA-CTA
Nitro-dopamine (0.72 g, 3.58 mmol) was dispersed in DMF (5 mL) taken in a dry high-pressure test tube kept under an argon atmosphere at room temperature. 1.1 mL of dry TEA was slowly added to the reaction mixture, which was left to stir for 10 minutes. Then BTTCP-PFP (1.02 g, 2.47 mmol) dissolved in DMF (5 mL) was added to the nitro-dopamine solution and left to stir at room temperature overnight. The reaction mixture was then dripped slowly into a phthalate buffer (150 mL, pH 3.0) to obtain a dark yellow dispersion and extracted with CHCl3. The organic layer was dried over Na2SO4 and filtered. Removal of the solvent under reduced pressure gave a crude product as a yellow liquid. (NB: traces of DMF were removed using heptane to make an azeotrope and concentrated at 50 °C under reduced pressure). The crude product was purified by column chromatography on silica gel using chloroform and ethyl acetate (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the eluent. After recrystallization from a toluene:hexane (50:50) mixture, the product was dried under vacuum to yield a solid yellow powder (0.67 g, 65% yield) (LC-MS [M − H]− = 417.0). A 1H NMR spectrum of the nitro DOPA-CTA is shown in Fig. S3.†
:1) as the eluent. After recrystallization from a toluene:hexane (50:50) mixture, the product was dried under vacuum to yield a solid yellow powder (0.67 g, 65% yield) (LC-MS [M − H]− = 417.0). A 1H NMR spectrum of the nitro DOPA-CTA is shown in Fig. S3.†
Synthesis of PNIPAM-nitro DOPA
The RAFT polymerization of NIPAM was performed in a 25 mL Schlenk tube under an argon atmosphere. NIPAM (1.0 g, 8.84 mmol), butyl(1-((4,5-dihydroxy-2-nitrophenethyl)amino)-1-oxopropan-2-yl)carbonotrithioate (BDNAOCT) (37.0 mg, 0.09 mmol), 2,2′-azobisisobutyronitrile (AIBN) (7.3 mg, 0.044 mmol), and DMF (4.4 mL) were charged in the 25 mL Schlenk tube at a molar ratio of 100:1:0.5. The solution was deoxygenated by bubbling argon for 30 min. Then the reaction mixture was placed in a preheated oil bath at 60 °C to initiate the polymerization. After 145 min of polymerization, the Schlenk tube was removed from the oil bath and opened to the air and placed in ice to stop the polymerization. The resulting polymer was precipitated by dropping the polymer solution into 300 mL of cold diethyl ether. After decantation of some of the solvent, the polymer suspended in diethyl ether was centrifuged to isolate it.
Later the polymer was dispersed again in fresh diethyl ether and centrifuged again to isolate the polymer. The resulting powdery light-yellow polymer was dried for 2 days under vacuum at 50 °C. The monomer conversion was determined by GC whereas SEC was used to determine the molecular weight and dispersity (Đ) (Fig. S4†) (conversion = 43%, MnGC = 5.3 kDa, DP = 43; MnSEC(DMA) = 10.9 kDa and Đ = 1.09).
Synthesis of MnO2 NPs
In a typical synthesis, a 20 mM solution of KMnO4 was prepared by dissolving 0.158 g (1 mmol) of KMnO4 in 25 mL of milliQ water, taken in a 250 mL conical flask. A second solution was prepared with 1.1 mL isoamyl alcohol dissolved in 25 mL of milliQ water. The isoamyl alcohol solution was added to the permanganate solution and the resulting reaction mixture was stirred at 700 rpm for 15 h at 25 °C. Then the resulting brown dispersed MnO2 nanoparticles were separated from the reaction mixture by centrifuging the mixture at 8000 rpm for 10 min and the residue was thoroughly washed with milliQ water 2 times, and finally suspended in 8.5 mL of milliQ water. Later suspended MnO2 was dried and used for the next step. The obtained core–shell MnO2 NPs were characterized using UV-Vis, DLS, SEM, TEM, XPS, and TGA.Ligand-exchange with PNIPAM-nitro-DOPA
The MnO2 NPs (14.02 mg) were dispersed in 6 mL of dry DMF and PNIPAM-nitro-DOPA (Mn = 5.3 kg mol−1) (173.0 mg) was added in a 25 mL Schlenk tube. This solution was purged with argon, put in an ultrasonic bath for 30 min, shaken at room temperature for 44 h, ultrasonicated again for 40 min and precipitated in cold diethyl ether (180 mL). After centrifugation at 5000 rpm for 5 min, PNIPAM-modified MnO2 NPs were washed with diethyl ether and centrifuged again. The excess of diethyl ether was air dried and dispersed in milliQ water (180 mL). Later PNIPAM modified MnO2 nanoparticles were separated from the mixture by centrifugation at 8000 rpm for 10 min and the residue was thoroughly washed with milliQ water 3 times, and finally suspended in 8.5 mL of milliQ water. The obtained core–shell PNIPAM@MnO2 NPs were characterized using UV-Vis, DLS, SEM, TEM, XPS and TGA.HRP induced degradation of H2O2
Homovanillic acid/3-methoxy-4-hydroxyphenylacetic acid (HVA) is non-fluorescent; however, in combination with horse radish peroxidase (HRP) and H2O2 a dimer is formed which is fluorescent (Scheme 1). Thus, an addition of HRP and HVA to a sample containing H2O2 can be used to detect the presence of H2O2. A stock solution of HRP in PBS was made (3.41 mg enzyme dissolved in 1 mL buffer = 611 U mL−1), which was combined with a 10 mM stock solution of HVA and H2O2 in PBS.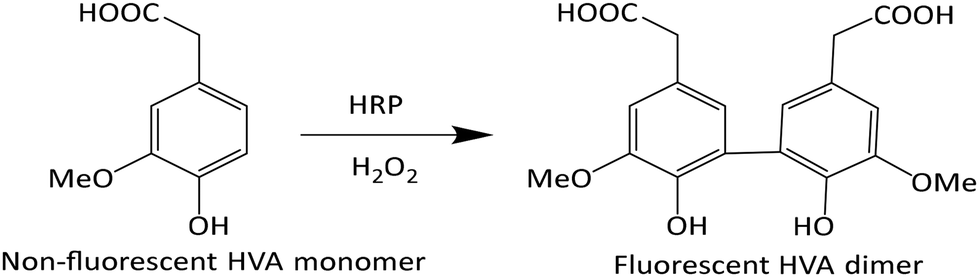 | ||
| Scheme 1 Dimerization of HVA by the action of HRP and H2O2. | ||
Effect of temperature on the degradation of H2O2 at 10 and 50 °C
For the investigation of temperature effect on the degradation of H2O2 at 10 and 50 °C, 950 μL of PBS and 50 μL of H2O2 were added to an Eppendorf cuvette and placed in the thermoshaker at 10 °C and 50 °C, respectively. Once the Eppendorf reached those temperatures, 10 μL of the NP solution was added and left to react for 10 min then the Eppendorf was centrifuged at 4 °C (7500 rpm, 10 min) in order to precipitate the NP. 750 μL of the supernatant was taken and added to a cuvette together with 50 μL of HRP, 100 μL of HVA, and 1600 μL of PBS and investigated using a fluorometric method. The same experiment was done in parallel but without the addition of the NP solution, as the reference.Results and discussion
Poly(N-isopropyl acrylamide) (PNIPAM) is a frequently studied thermoresponsive polymer due to its cloud point temperature of about 32 °C, which is quite close to human body temperature. The synthesis of well-defined nitro-dopamine end-functionalized PNIPAM was performed by reversible addition fragmentation chain-transfer (RAFT) polymerization. Therefore, nitrodopamine hydrogensulfate was reacted with pentafluorophenol (PFP) containing CTA to obtain butyl(1-((4,5-dihydroxy-2-nitrophenethyl)amino)-1-oxopropan-2-yl)carbonotrithioate (BDNAOCT) chain transfer agent (CTA). 1H-NMR spectroscopy was used to confirm its structure (Fig. S3†). The polymer was subsequently synthesized in DMF at 60 °C in the presence of AIBN utilizing BDNAOCT as the RAFT chain transfer agent (CTA) (Scheme 2). At around 43% conversion, the polymerization was stopped and the formed polymer was isolated by precipitation in cold diethyl ether. Theoretical molecular weight and the degree of polymerization (DP) were calculated and found to be 5.3 kDa and 43, respectively. SEC results of the synthesized polymer revealed that the polymer is well defined with Mn and dispersity (Đ) at 10.9 kDa and 1.09, respectively (Fig. S4†). MnO2 NPs were prepared by mixing KMnO4 with isoamyl alcohol using the procedure reported in the literature.53 In this method KMnO4 acts as an oxidizing agent that eventually oxidizes the alcohol to the corresponding acid, which caps the spherical MnO2 particles. The purple colour of the KMnO4 solution resulting from the charge transfer (CT) transition turns into a yellowish-brown colour when it transforms into MnO2 NPs (Fig. S5†). The optical properties of the MnO2 NPs in water were investigated and compared with the starting KMnO4. It was observed that the characteristic absorption bands of KMnO4 at λ = 508, 526, 546, and 565 nm resulting from the CT transition vanish and a new broad peak was observed in the range of 300–450 nm, indicating the formation of MnO2 NPs as evident from Fig. 2a. From the DLS measurement of prepared MnO2 NPs, the average particle size was confirmed to be around 278.5 nm measured at 25 °C (Fig. 2b).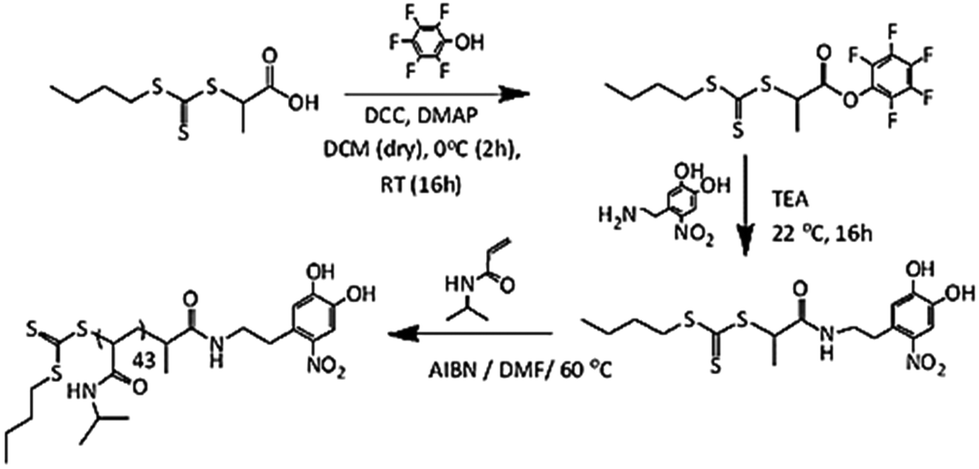 | ||
| Scheme 2 Schematic representation of the synthesis of dopamine-functionalized PNIPAM. | ||
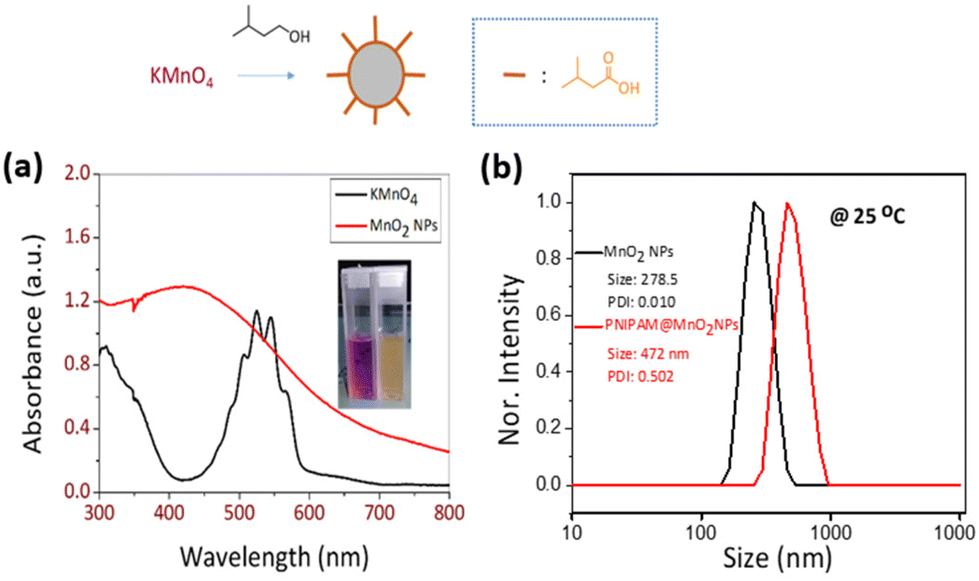 | ||
| Fig. 2 (a) UV-vis spectra of KMnO4 and MnO2 NPs and (b) size distribution obtained from DLS of MnO2 NPs and PNIPAM@MnO2 NPs. It should be noted that the x-axis is on the logarithmic scale. | ||
Next, PNIPAM@MnO2 NPs were prepared by ligand-exchange, which was achieved by shaking the solution of MnO2 NPs and PNIPAM in DMF for 2 days. The resulting PNIPAM@MnO2 NPs was isolated by precipitation in cold DEE followed by centrifugation leading to a solid sample that could be redispersed in water for future investigations (Fig. S6†). The mechanism of surface modification of MnO2 by nitrodopamine is elaborately discussed by Sivakumar et al.,26 as mentioned earlier. Both theoretical and experimental proofs are given to prove the attachment of catechol hydroxyl groups present in nitrodopamine to the MnO2 surface. The strong electron withdrawing nitro group in nitrodopamine makes the catechol hydroxyl more acidic and thus favours the effective binding on the surface of NPs. DLS analysis of the PNIPAM@MnO2 NPs revealed an increase of the size to 472 nm, confirming the presence of the polymer coating, while the PDI was at 0.502 (Fig. 2b). SEM images of the MnO2 NPs and the PNIPAM@MnO2 NPs are shown in Fig. 3. Fig. 3a shows well-dispersed MnO2 NPs with a particle size around the 200–300 nm range as given in Fig. S7a.† The size of the PNIPAM@MnO2 NPs clearly shows an increase in the particle size after polymer modification as shown in Fig. 3b, albeit the image becomes more blurry in the presence of the polymer coating and the resolution of the SEM analysis is insufficient to visualize the polymer coating. Furthermore, it is evident that the MnO2 NPs are coated by the polymer individually or in the agglomerated stage, which is mostly likely due to the dehydration of the polymer during the sample preparation process as DLS confirmed the presence of defined individual particles. In Fig. 3b (right) the particle size was determined to be around 400–600 nm given in Fig. S7b.† Therefore, it is clear that PNIPAM is coated on to the NP surfaces. By using energy-dispersive X-ray (EDX) analysis, we have determined the elemental composition (Fig. 4a). The presence of Mn and oxygen peaks corroborates well with the elemental composition, as shown in Table S1.† Furthermore, the EDX analysis of the PNIPAM@MNO2 NPs shows an enhancement in carbon percentage and the presence of other elements, such as nitrogen and sulphur, in agreement with the polymer coating (Fig. 4b and Table S2†). As the surface of the NPs is modified with the polymer, the Mn composition on the surface is declined, confirming successful polymer coating of the MnO2 NPs.
 | ||
| Fig. 3 (a) SEM image of MnO2 NPs (scale bar is 500 nm; right: scale bar is 1 μm), (b) SEM image of PNIPAM@MnO2 NPs (scale bar is 500 nm; right: scale bar is 1 μm). | ||
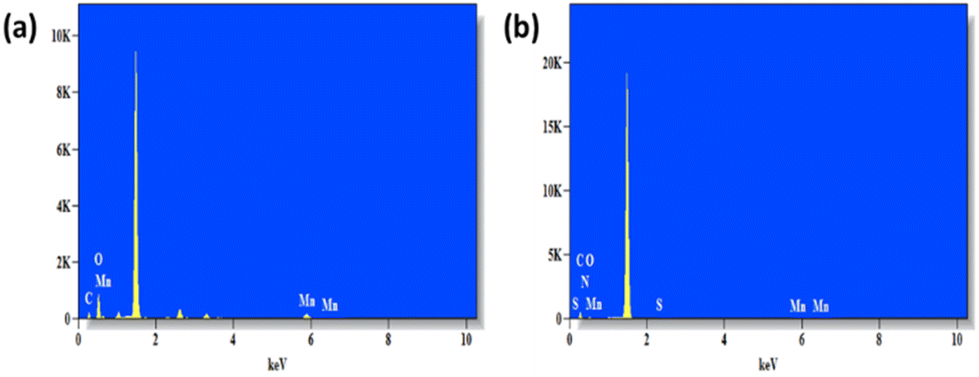 | ||
| Fig. 4 (a) EDX graph of MnO2 NPs and (b) EDX graph of PNIPAM@MnO2 nanoparticles. | ||
TEM images of the synthesized MnO2 NPs showed the flaky structure with microfibrillar morphology. It is evident from a comparison of the images of unmodified and modified nanoparticles (Fig. 5a and b, respectively) that the modified NPs have a visible polymer coating layer.
 | ||
| Fig. 5 (a) TEM micrographs of dispersed MnO2 nanoparticles (scale bar is 20 nm; right: scale bar is 50 nm; the zoomed image shows a MnO2 nanoparticle with a nanofibrillar structure), (b) TEM micrographs of dispersed PNIPAM@MnO2 nanoparticles (scale bar is 20 nm; right: scale bar is 50 nm; the zoomed image shows a MnO2 nanoparticle with a nanofibrillar structure). | ||
X-ray photoelectron spectroscopy was used to perform a basic investigation to identify the corresponding valence states of the materials. Overall the XPS spectra of the MnO2 NPs showed peaks corresponding to all the elements with characteristic valence states as expected in the sample (Fig. 6a). Giving further attention to each element, Mn 2p and O 1s XPS spectra are shown in Fig. 6b and c, respectively. In the deconvoluted Mn 2p peak, the two peaks observed at 654.10 and 642.34 eV with an energy separation of 11.76 eV correspond to the Mn 2p3/2 and Mn 2p1/2 orbitals (Fig. 6b). Similarly, the O 1s peak of MnO2 was curve-fitted as shown in Fig. 6c, and includes three characteristic oxygen bonds, which are associated to the Mn–O–Mn bond, Mn–O–H bond and H–O–H bond with binding energies (BEs) of 529.73 eV, 530.97 eV and 533.08 eV, respectively. Previously reported values of the XPS spectra for the oxidation state of Mn and O in the MnO2 NPs agree well with these findings.54,55
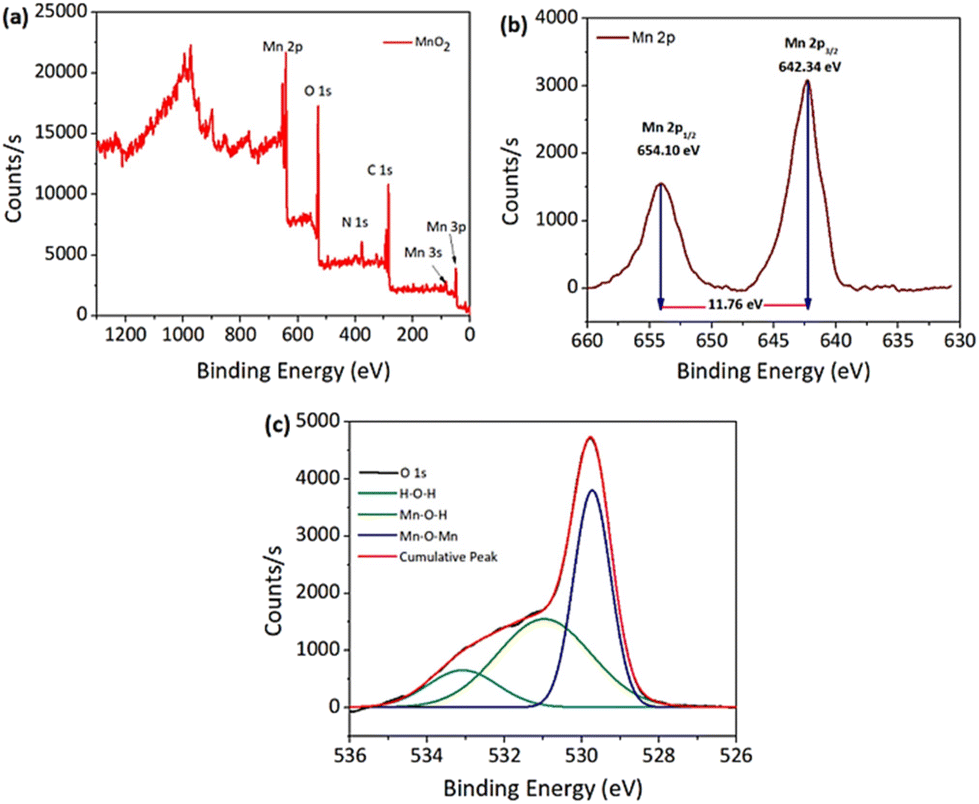 | ||
| Fig. 6 XPS spectra. (a) Overall MnO2 NPs and (b) Mn 2p and (c) O 1s orbitals. | ||
For the PNIPAM@MnO2 NPs, the overall XPS spectrum showed (Fig. S8a†) an increase in the elemental composition of carbon and nitrogen, whereas a drastic decrease in the Mn composition confirms the successful coating of PNIPAM onto the nanoparticles. Fig. S8b and S8c† corresponding to the Mn 2p and O 1s orbitals provide similar deconvoluted peaks as that of the unmodified NPs, but with a lower count of elements due to the enhancement of carbon and nitrogen composition from the polymer.
Nonetheless, the presence of Mn and O is evident from the spectra. Carbon (C 1s) high resolution spectra from PNIPAM were resolved by curve-fitting into five component peaks (Fig. S8d†). The peak with a BE of 284.24 eV corresponds to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds associated with the aromatic end-group of the polymer. A peak with a BE at 285.01 eV corresponds to the C–H/C–C bonds with the aliphatic hydrocarbon part of the polymer. Other peaks with a BE at 286.27, 287.15 and 287.59 eV are attributed to the C–O/C–S, N–CO and CO bonds, respectively. Similar spectra and binding environments (C–H/C–C, CC, C–O/C–S, N–CO and CO) were observed for the C 1s orbital in other literature studies.56–58 Fig. S8e† shows the deconvoluted peaks of the N 1s orbital and the peaks observed at 399 eV and 400 eV are attributed to the C–N and N–O bonds respectively.59,60 Three peaks appeared in the expanded spectra of the S 2p orbital (Fig. S8f†). The first peak can be ascribed to sulphur atoms bound with carbon by CS double bonds at 164 eV; also a small amount of oxidized sulphur was observed at 168.5 eV.61
C bonds associated with the aromatic end-group of the polymer. A peak with a BE at 285.01 eV corresponds to the C–H/C–C bonds with the aliphatic hydrocarbon part of the polymer. Other peaks with a BE at 286.27, 287.15 and 287.59 eV are attributed to the C–O/C–S, N–CO and CO bonds, respectively. Similar spectra and binding environments (C–H/C–C, CC, C–O/C–S, N–CO and CO) were observed for the C 1s orbital in other literature studies.56–58 Fig. S8e† shows the deconvoluted peaks of the N 1s orbital and the peaks observed at 399 eV and 400 eV are attributed to the C–N and N–O bonds respectively.59,60 Three peaks appeared in the expanded spectra of the S 2p orbital (Fig. S8f†). The first peak can be ascribed to sulphur atoms bound with carbon by CS double bonds at 164 eV; also a small amount of oxidized sulphur was observed at 168.5 eV.61
Thermal gravimetric analysis (TGA) also showed the successful coating of PNIPAM onto the MnO2 NP surface (Fig. S9†). The TGA analysis of the PNIPAM@MnO2 NPs showed a two-step weight loss at 190 °C and 400 °C, which is due to the PNIPAM coating on the nanoparticles. The observation of a second decomposition at ∼400 °C of the polymer is in good agreement with that of pure PNIPAM.62 The TGA curve of pure MnO2 possesses only one weight loss step due to the loss of moisture from an MnO2 lattice (adsorbed water molecules).63
Dynamic light scattering (DLS) data indicated that the synthesized MnO2 NPs were stable and well dispersed in an aqueous medium with an average particle size of 278.5 nm and a polydispersity index (PDI) of 0.010 (Fig. S10†). The particle size was remeasured after 15 days and found to be stable at 25 °C. Temperature-dependent aggregation of the MnO2 NPs was also investigated by DLS in an aqueous medium (Fig. S10†). The particle size of the MnO2 NPs remained almost unchanged at 25 °C (278 nm) and 50 °C (287 nm) indicating the absence of temperature-dependent aggregation of unmodified NPs. In contrast with the MnO2 NPs, the PNIPAM@MnO2 NPs showed temperature-responsive behavior when measured at 25 °C, 35 °C, and 50 °C, respectively (Fig. 7a). Upon increasing the temperature from 25 °C to 35 °C, a decrease in the particle size of 127 nm was observed from 472 nm to 345 nm. By further heating above the LCST of PNIPAM at 50 °C, an increment of 157 nm in the particle size was detected. In order to obtain additional information on the temperature-responsive behaviour of the PNIPAM@MnO2 NPs, DLS measurement was analysed in 5 °C intervals from 20 to 50 °C. The change in the diameter of the NPs with temperature can be seen from the graphs plotted in Fig. 7a. At the Tcp of PNIPAM at around 35 °C there is a drop in the diameter when compared to the lower and higher temperatures. At the lower temperature the polymer chains are efficiently hydrated, leading to a larger hydrodynamic diameter. When the temperature approaches the Tcp, the polymer chains are dehydrated and collapse onto the individual NPs, resulting in a decrease in the observed particle size. Upon further heating, the particle size gradually increases as, at temperatures above the Tcp of PNIPAM, the polymer chains are further dehydrated and collapse, inducing hydrophobic interactions that lead to the aggregation of polymer modified NPs. The size reaches a maximum of 579 nm at around 42 °C, followed by a slight decline due to the precipitation of aggregated particles (Fig. 7b).
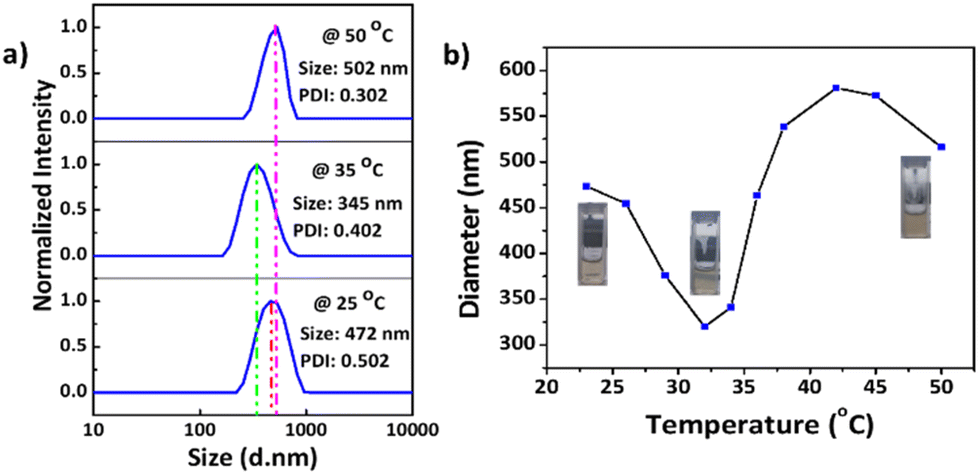 | ||
| Fig. 7 (a) Particle size and PDI of the PNIPAM@MnO2 NPs at three different temperatures: 25 °C, 35 °C and 50 °C, respectively; (b) change in particle sizes with temperature varying from 23 to 50 °C at a concentration of 0.2 mg mL−1 in all cases. | ||
The observed changes in the particle size with temperature suggest that PNIPAM@MnO2 NPs can be used as smart material for the catalytic decomposition of H2O2, based on the dehydration of PNIPAM at increased temperatures.
To investigate the effect of nanoparticle concentration on the temperature-induced aggregation and temperature-induced reversibility of the smart PNIPAM@MnO2 NPs, we lowered the concentration of the nanoparticles to half, leading to an increase in the particle size to 580 nm at 20 °C, indicating more efficient polymer hydration (Fig. S11a†). At this lower concentration, we did not observe NP aggregation above the Tcp of PNIPAM. Instead, the particle size decreased to around 240 nm upon heating to 50 °C, indicating the collapse of the dehydrated polymer chains onto the NPs without any further aggregation. The temperature-induced dehydration of the PNIPAM coating at this lower concentration was also found to be reversible without noticeable deviations during five cycles (Fig. S11b†).
To investigate the temperature effect on the catalytic activity of the developed smart NPs, fluorescence experiments were conducted at three different temperatures, 10, 37, and 50 °C, respectively. As illustrated in Scheme 1, the non-fluorescent HVA monomer is transformed into the fluorescent HVA dimer (2,2′-dihydroxy-3,3′-dimethoxydiphenyl-5,5′-diacetic acid) in the presence of HRP and H2O2, resulting in enhanced fluorescence. The strong reactivity of H2O2 with transition metal-containing heme peroxides, such as HRP, has been reported to result in high-rate constants, in the range of 107–108 M−1 s−1.64 The resulting metal-containing heme peroxide complex catalyzes the dimerization of HVA into the fluorescent dimer. We have successfully utilized this method to investigate the activity of H2O2 in the presence of the smart PNIPAM@MnO2 NPs at different temperatures. Therefore, the change in the fluorescent intensity at 10 and 50 °C in the presence and absence of PNIPAM@MnO2 NPs was analysed as shown in Fig. S12.†
The fluorescence studies were carried out at two different temperatures, 10 °C and 50 °C, respectively, in order to investigate the influence of temperature on the catalytic activity of the PNIPAM@MnO2 NPs for the decomposition of H2O2 (Fig. 8). These two temperatures are taken into account for our analysis since these are below and above the Tcp of PNIPAM (Tcp ∼ 32 °C). Note that we used a reverse assay to monitor the remaining H2O2 through HRP catalysed dimerization of HVA, leading to significant background fluorescence at t = 0 due to the presence of all reactive components before the addition of the PNIPAM@MnO2 NPs.
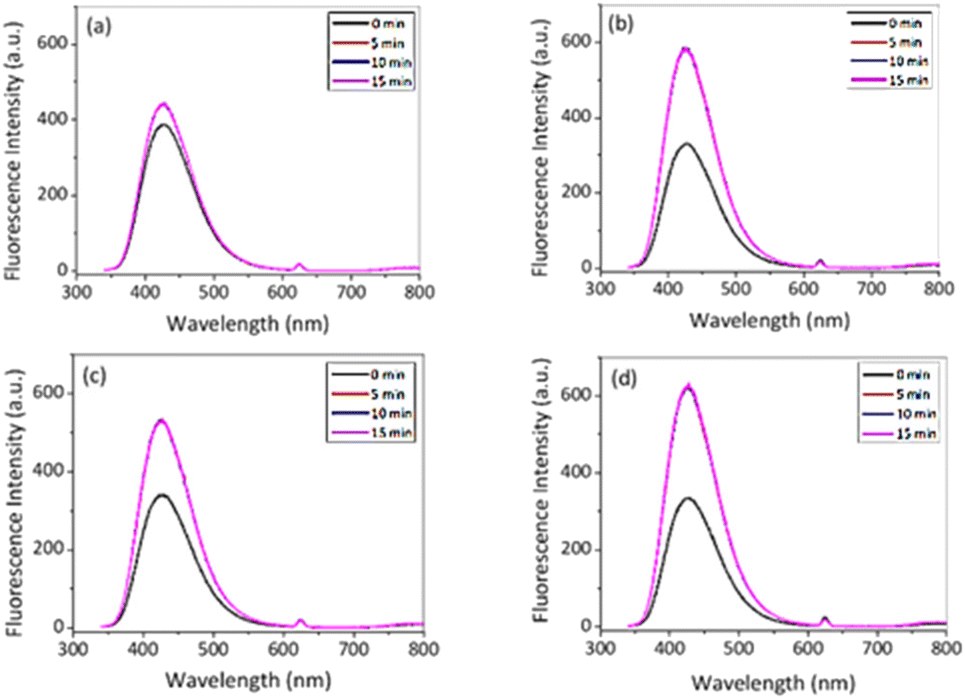 | ||
| Fig. 8 Fluorescence intensity in the presence of HVA, HRP and 50 μL of H2O2 (a) with NPs; (b) without NPs at 10 °C and (c) with NPs; and (d) without NPs at 50 °C in PBS (λex. = 312 nm). | ||
After 15 min in the presence of PNIPAM@MnO2 NPs, the fluorescence intensity only increased from 385 a.u. to 442 a.u. (λem,max@t15/λem,max@t0 = 1.15) at 10 °C, while in the absence of PNIPAM@MnO2 NPs the fluorescence intensity increased from 330 a.u. to 588 a.u. (λem,max@t15/λem,max@t0 = 1.78) during the same time interval. This result clearly demonstrates less dimer formation indicative of a lower H2O2 concentration, implying that H2O2 is catalytically decomposed in the presence of the PNIPAM@MnO2 NPs. In contrast, at 50 °C, the fluorescence intensity increased from 341 to 529 a.u. (λem,max@t15/λem,max@t0 = 1.55) in the presence of the PNIPAM@MnO2 NPs, while in the absence of PNIPAM@MnO2 NPs it increased from 334 a.u. to 632 a.u. (λem,max@t15/λem,max@t0 = 1.89) for the same time interval. As a result, it was evident that the increase in relative fluorescence intensity in the presence of smart NPs is much higher at 50 °C than at 10 °C. As this temperature is above the LCST of PNIPAM (∼32 °C) it induces dehydration and collapse of the PNIPAM chains onto the NPs, preventing the catalyst from coming into contact with the H2O2, resulting in higher retention of the H2O2 and, thus, in greater fluorescence increase due to HVA dimer formation. Control experiments at 37 °C revealed that there is no fluorescence increase without H2O2, but there is a moderate increase in fluorescence in the presence of H2O2, and a strong increase in fluorescence in the presence of both H2O2 and HRP (Fig. S13†).
Furthermore, we have investigated the role of H2O2 towards the dimerization of HVA in the absence of the PNIPAM@MnO2 NPs (Fig. S14†), indicating a strong correlation of fluorescence intensity increase with the H2O2 concentration, further supporting the observed temperature-switchable catalytic activity of the PNIPAM@MnO2 NPs.
Conclusions
In this work we have successfully synthesized and characterized temperature responsive PNIPAM coated MnO2 NPs for the catalytic decomposition of H2O2. Successful coating of the NP surface with nitrodopamine functionalized PNIPAM was confirmed by the analysis of elemental compositions, SEM and XPS. The results were further confirmed by TEM images, as a clear uniform coating of polymers on NPs is visible from the images. The TGA results validated the PNIPAM coating on the surface of MnO2 NPs. The synthesized PNIPAM@MnO2 NPs were proved to be temperature-responsive by DLS and fluorescence spectroscopy. Thermoresponsive swelling and deswelling of PNIPAM@MnO2 NPs were confirmed as the particle size of the nanoparticles altered with the change in temperature. The catalytic decomposition of H2O2 by MnO2 is a renowned exothermic reaction. This reaction is done with the synthesized PNIPAM@MnO2 NPs and a fluorescence assay for HVA dimerization proved the significant catalytic decomposition of H2O2 at a lower temperature (below the Tcp of PNIPAM) by the PNIPAM@MnO2 NPs. At a higher temperature (above the Tcp of PNIPAM), H2O2 decomposition was found to be less significant. These results confirm that the smart nanoparticles can be used for the temperature-dependent catalytic decomposition of H2O2. In the future, we aim to further develop this system towards self-regulated smart catalytic systems.Data availability
The data supporting this article have been included in the main manuscript and as part of the ESI.†Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. B. and G. S. thanks SRMIST for providing the fellowship to support the Ph.D. program. S. M. acknowledges SRMIST for providing the SRMIST seed grant, the Science and Engineering Research Board (SERB), India, for a core research grant (CRG/2021/004203) and FWO. R. H. thanks FWO and Ghent University for financial support. We thank Dr. Vincent Ornelis from UGent, Belgium, for the measurement of PL of PNIPAM@MnO2NP samples and Ms Sushma Kundu from TIFR Mumbai, India, for TGA measurements.References
- N. Di Marzo, E. Chisci and R. Giovannoni, Cells, 2018, 7, 156 CrossRef CAS PubMed.
- L. Zhang, C. Jia, F. Bai, W. Wang, S. An, K. Zhao, Z. Li, J. Li and H. Sun, Fuel, 2024, 355, 129455 CrossRef CAS.
- R. J. Lewis and G. J. Hutchings, Acc. Chem. Res., 2024, 57, 106–119 CrossRef CAS PubMed.
- Z. Wang, L. Peng, P. Zhu, W. Wang, C. Yang, H.-Y. Hu and Q. Wu, ACS Nano, 2024, 18, 2885–2897 CrossRef CAS PubMed.
- B. Kichatov, A. Korshunov, V. Sudakov, A. Golubkov, D. Smovzh, S. Sakhapov and M. Skirda, Phys. Chem. Chem. Phys., 2024, 26, 1612–1615 RSC.
- J. J. Dolhun, J. Chem. Educ., 2014, 91, 760–762 CrossRef CAS.
- T. Zeng, X. Meng, S. Sun, M. Ling, C. Zhang, W. Yuan, D. Cao, M. Niu, L. Y. Zhang and C. M. Li, Small Methods, 2023, 7, 2300791 CrossRef CAS PubMed.
- X. Zhang, G. Zhu, B. Xiao, J. Geng, Y. Yang, D. Wang, J. Li, J. Wang and Y. Zhu, Sep. Purif. Technol., 2024, 330, 125338 CrossRef CAS.
- B. Martin, J. Sedelmeier, A. Bouisseau, P. Fernandez-Rodriguez, J. Haber, F. Kleinbeck, S. Kamptmann, F. Susanne, P. Hoehn, M. Lanz, L. Pellegatti, F. Venturoni, J. Robertson, M. C. Willis and B. Schenkel, Green Chem., 2017, 19, 1439–1448 RSC.
- Z.-L. Wu, C.-K. Li, J.-G. Yu and X.-Q. Chen, Sens. Actuators, B, 2017, 239, 544–552 CrossRef CAS.
- K. Sobańska, P. Pietrzyk and Z. Sojka, ACS Catal., 2017, 7, 2935–2947 CrossRef.
- M. J. Männel, L. P. Kreuzer, C. Goldhahn, J. Schubert, M. J. Hartl and M. Chanana, ACS Catal., 2017, 7, 1664–1672 CrossRef.
- L. P. Kreuzer, M. J. Männel, J. Schubert, R. P. M. Höller and M. Chanana, ACS Omega, 2017, 2, 7305–7312 CrossRef CAS PubMed.
- Y. Wu, P. Balasubramanian, Z. Wang, J. A. S. Coelho, M. Prslja, R. Siebert, M. B. Plenio, F. Jelezko and T. Weil, J. Am. Chem. Soc., 2022, 144, 12642–12651 CrossRef CAS PubMed.
- C. Busquets-Cortés, X. Capó, E. Argelich, M. Ferrer, D. Mateos, C. Bouzas, M. Abbate, J. Tur, A. Sureda and A. Pons, Nutrients, 2018, 10, 1920 CrossRef PubMed.
- M. Zhu, L. Zhu, Y. You, M. Sun, F. Jin, Y. Song, J. Zhang, X. Xu, J. Ji and Y. Du, ACS Nano, 2023, 17, 17285–17298 CrossRef CAS PubMed.
- H. J. Forman and H. Zhang, Nat. Rev. Drug Discovery, 2021, 20, 689–709 CrossRef CAS PubMed.
- S. Mourdikoudis, R. M. Pallares and N. T. K. Thanh, Nanoscale, 2018, 10, 12871–12934 RSC.
- M. Hermanek, R. Zboril, I. Medrik, J. Pechousek and C. Gregor, J. Am. Chem. Soc., 2007, 129, 10929–10936 CrossRef CAS PubMed.
- M. Xu, Y. Wang, J. Yang, J. Wang, D. Liu, L. Zhou and H. Liu, Sep. Purif. Technol., 2024, 330, 125263 CrossRef CAS.
- S. Sisakhtnezhad, M. Rahimi and S. Mohammadi, Biomed. Pharmacother., 2023, 163, 114833 CrossRef CAS PubMed.
- Y. Yang, K. Hu, Z. Zhu, Y. Yao, P. Zhang, P. Zhou, P. Huo, X. Duan, H. Sun and S. Wang, Small Methods, 2023, 7, 2300588 CrossRef CAS PubMed.
- J. P. C. Moura, V. S. Antonin, A. B. Trench and M. C. Santos, Electrochim. Acta, 2023, 463, 142852 CrossRef CAS.
- M. Song, T. Liu, C. Shi, X. Zhang and X. Chen, ACS Nano, 2016, 10, 633–647 CrossRef CAS PubMed.
- A. Matsugi, Combust. Flame, 2021, 225, 444–452 CrossRef CAS.
- G. Sivakumar, A. Gupta, A. Babu, P. K. Sasmal and S. Maji, J. Mater. Chem. B, 2024, 12, 4724–4735 RSC.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- L. Johnson, D. M. Gray, E. Niezabitowska and T. O. McDonald, Nanoscale, 2021, 13, 7879–7896 RSC.
- A. Das, A. Babu, S. Chakraborty, J. F. R. Van Guyse, R. Hoogenboom and S. Maji, Adv. Funct. Mater., 2024, 2402432 CrossRef.
- R. Wang, Q. Wei, W. Sheng, B. Yu, F. Zhou and B. Li, Angew. Chem., Int. Ed., 2023, 62, e202219312 CrossRef CAS PubMed.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS PubMed.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
- G. Vancoillie, S. Pelz, E. Holder and R. Hoogenboom, Polym. Chem., 2012, 3, 1726–1729 RSC.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- G. Moad, Polym. Chem., 2017, 8, 177–219 RSC.
- X. Tian, J. Ding, B. Zhang, F. Qiu, X. Zhuang and Y. Chen, Polymers, 2018, 10, 318 CrossRef PubMed.
- C. Sánchez-Rodríguez, R. Palao-Suay, L. Rodrigáñez, M. Aguilar, S. Martín-Saldaña, J. San Román and R. Sanz-Fernández, Biomolecules, 2018, 8, 97 CrossRef PubMed.
- S. Maji, G. Vancoillie, L. Voorhaar, Q. Zhang and R. Hoogenboom, Macromol. Rapid Commun., 2014, 35, 214–220 CrossRef CAS PubMed.
- S. Maji, Z. Zhang, L. Voorhaar, S. Pieters, B. Stubbe, S. Van Vlierberghe, P. Dubruel, B. G. De Geest and R. Hoogenboom, RSC Adv., 2015, 5, 42388–42398 RSC.
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS PubMed.
- D. A. Gkika, A. K. Tolkou, D. A. Lambropoulou, D. N. Bikiaris, P. Kokkinos, I. K. Kalavrouziotis and G. Z. Kyzas, RSC Appl. Polym., 2024, 2, 127–148 RSC.
- D. Roy, W. L. A. Brooks and B. S. Sumerlin, Chem. Soc. Rev., 2013, 42, 7214 RSC.
- C. Boutris, E. G. Chatzi and C. Kiparissides, Polymer, 1997, 38, 2567–2570 CrossRef CAS.
- S. Maji, B. Cesur, Z. Zhang, B. G. De Geest and R. Hoogenboom, Polym. Chem., 2016, 7, 1705–1710 RSC.
- A. Babu, G. Sivakumar, M. Anandan, P. Adhya, T. Akash, T. Mondal, V. Nutalapati and S. Maji, Eur. Polym. J., 2023, 200, 112527 CrossRef CAS.
- M. A. Cooperstein and H. E. Canavan, Langmuir, 2010, 26, 7695–7707 CrossRef CAS PubMed.
- C. J. Ferguson, R. J. Hughes, D. Nguyen, B. T. T. Pham, R. G. Gilbert, A. K. Serelis, C. H. Such and B. S. Hawkett, Macromolecules, 2005, 38, 2191–2204 CrossRef CAS.
- S. Kurzhals, R. Zirbs and E. Reimhult, ACS Appl. Mater. Interfaces, 2015, 7, 19342–19352 CrossRef CAS PubMed.
- W. Scarano, H. Lu and M. H. Stenzel, Chem. Commun., 2014, 50, 6390–6393 RSC.
- A. K. Sinha, M. Basu, M. Pradhan, S. Sarkar, Y. Negishi and T. Pal, J. Phys. Chem. C, 2010, 114, 21173–21183 CrossRef CAS.
- G. Xie, X. Liu, Q. Li, H. Lin, Y. Li, M. Nie and L. Qin, J. Mater. Sci., 2017, 52, 10915–10926 CrossRef CAS.
- J. M. de O. Cremonezzi, D. Y. Tiba and S. H. Domingues, SN Appl. Sci., 2020, 2, 1689 CrossRef CAS.
- A. Munir, T. ul Haq, A. Qurashi, H. ur Rehman, A. Ul-Hamid and I. Hussain, ACS Appl. Energy Mater., 2019, 2, 363–371 CrossRef CAS.
- P. Shivapooja, L. K. Ista, H. E. Canavan and G. P. Lopez, Biointerphases, 2012, 7, 32 CrossRef CAS PubMed.
- X. Yang, Z. Sun, J. Gao, C. Yang and D. Tang, Polym. Bull., 2020, 77, 963–974 CrossRef CAS.
- A. Agrawal, K. Biswas, S. K. Srivastava and S. Ghosh, J. Solid State Electrochem., 2018, 22, 3443–3455 CrossRef CAS.
- X. Yan, T. Xu, G. Chen, S. Yang, H. Liu and Q. Xue, J. Phys. D: Appl. Phys., 2004, 37, 907–913 CrossRef CAS.
- N. Fechler, T.-P. Fellinger and M. Antonietti, J. Mater. Chem. A, 2013, 1, 14097 RSC.
- L. Wang, Y. Wu, Y. Men, J. Shen and Z. Liu, RSC Adv., 2015, 5, 70758–70765 RSC.
- S. Mallakpour, A. Abdolmaleki and H. Tabebordbar, Polym. Bull., 2017, 74, 2957–2973 CrossRef CAS.
- X. Huang and J. T. Groves, Chem. Rev., 2018, 118, 2491–2553 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H NMR spectra of nitrodopamine hydrogensulfate, PFP containing CTA and nitro DOPA-CTA, SEC analysis for PNIPAM, pictorial representations, particle size, EDX, XPS, TGA and DLS for MnO2 and PNIPAM@MnO2 NPs, and photophysical properties with additional data. See DOI: https://doi.org/10.1039/d4py00595c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |