
A one-pot approach towards polycarbonate-b-polyester block copolymers via chemoselective polymerization catalyzed by a one-component phosphonium borane Lewis pair†
Yutao
Li
a,
Xinhui
Kou
 b,
Xiaowu
Wang
*c,
Lin
Xia
*a and
Zhibo
Li
*ac
b,
Xiaowu
Wang
*c,
Lin
Xia
*a and
Zhibo
Li
*ac
aCollege of Polymer Science and Engineering, Qingdao University of Science and Technology, 53 Zhengzhou Road, Qingdao 266042, China. E-mail: xia28lin@126.com
bAnalyses and Testing Center Qingdao University of Science and Technology, 53 Zhengzhou Road, Qingdao, 266042, China
cCollege of Chemical Engineering, Qingdao University of Science and Technology, 53 Zhengzhou Road, Qingdao 266042, China. E-mail: wangxw@qust.edu.cn; zbli@qust.edu.cn
First published on 24th May 2024
Abstract
Chemoselective polymerization from mixtures of monomers to control the polymer sequence is a challenging task. In this contribution, we describe a unique chemoselective polymerization affording predictable (co)polymer compositions from a mixture of monomers. The use of a one-component phosphonium borane Lewis pair and a mixture of L-lactide (L-LA), cyclohexene oxide (CHO) and CO2 enables the selective synthesis of either a polyester, polycarbonate or poly(carbonate-b-ester). The selectivity depends on the nature of the borane–oxygen functionality at the growing polymer chain end, and can be controlled by the addition of CO2 as an exogeneous switch reagent.
Introduction
Some of the key challenges in polymer chemistry are the controllable realization of polymerization from a mixed monomer feedstock and the chemoselective control of the incorporation of monomers to regulate the polymer microstructure and consequently the thermal and mechanical properties.1–4 The realization of chemoselective polymerization is highly dependent on the thermodynamic parameters of monomers, polymerization methods and catalysts. Epoxides, oxetanes, cyclic esters and anhydrides are important classes of monomers due to the promising applications of the resulting products. Chemoselective polymerization of these monomers via ring-opening polymerization (ROP) and ring-opening copolymerization (ROCOP) offers an atom-economical way to prepare low carbon polymers, namely, polyethers, polyesters as well as their block copolymers. These constitute a large variety of polymer materials. For example, polyester or polycarbonate based block polyols are versatile, potentially bio-based and/or degradable materials widely applied in the manufacture of coatings, resins, polyurethanes and other products.5 Introducing a degradable component would endow the aforementioned low carbon polymers with degradable property and cater the sustainability of our society.Thanks to the intense development of well-defined organometallic complexes2,6–20 and recent progress of organocatalysis,21–29 several catalysts have shown their capability in the chemoselective polymerization of the above-mentioned monomer mixtures. In particular, the catalyst functions in two different catalytic cycles in a polymerization reaction which can be termed switchable polymerization.2 The merits of switchable polymerization offer the possibility of synthesizing block or multi-block polymers and open the option of overcoming adverse properties of the respective homopolymers, thus resulting in an enhanced performance of the copolymer.
In recent years, borane-based organocatalysts have played an indispensable role in the ROP of epoxides, ROCOP of epoxides and anhydrides, and ROCOP of epoxides and CO2, thus offering a pathway for the preparation of polyether, polyester, and polycarbonate based block copolymers (Fig. S1†).24,30–42Although borane-based Lewis pairs offer a promising alternative for the generation of low carbon polymers, the success of the ROP of lactone or lactide is rare, especially an extra hydrogen-bonding additive needs to be introduced.25,43 We previously reported a one-component phosphonium bisborane Lewis pair, PBB-Br, enabling living/controlled polymerization of PO.44,45 Moreover, it has been disclosed that tetraalkylammonium possesses the hydrogen bonding characteristic of α, β carbon bonds by Reetz et al. in 1993.46 Hydrogen bonding existing in other ammonium or phosphonium functionalized boranes is also well recognized.47–49 In particular, the larger phosphorus radius leads to greater dispersive-type interactions and more positively charged H atoms on α- and β-carbons. The charge delocalization therefore makes Hα and Hβ better H-bond donors.50 This would facilitate the activation of carbonyl compounds, like lactone or lactide, which is unexplored with one-component borane Lewis pairs. Preliminary results indicated that PBB-Br was capable of catalyzing CHO/CO2 copolymerization. Herein, we employed PBB-Br for the ring-opening polymerization of lactide and sought the possibility of selectively copolymerizing CHO/CO2 and lactide using PBB-Br to synthesize the polycarbonate-b-polyester copolymer.
Herein, we report the selective synthesis of either a polyester or polycarbonate or poly(carbonate-b-ester), which were prepared using the PBB-Br Lewis pair and a mixture of L-lactide (L-LA), CHO and CO2 (Fig. 1). The factors that differentiate polymerization pathways were investigated and insights into the observed selectivity are provided.
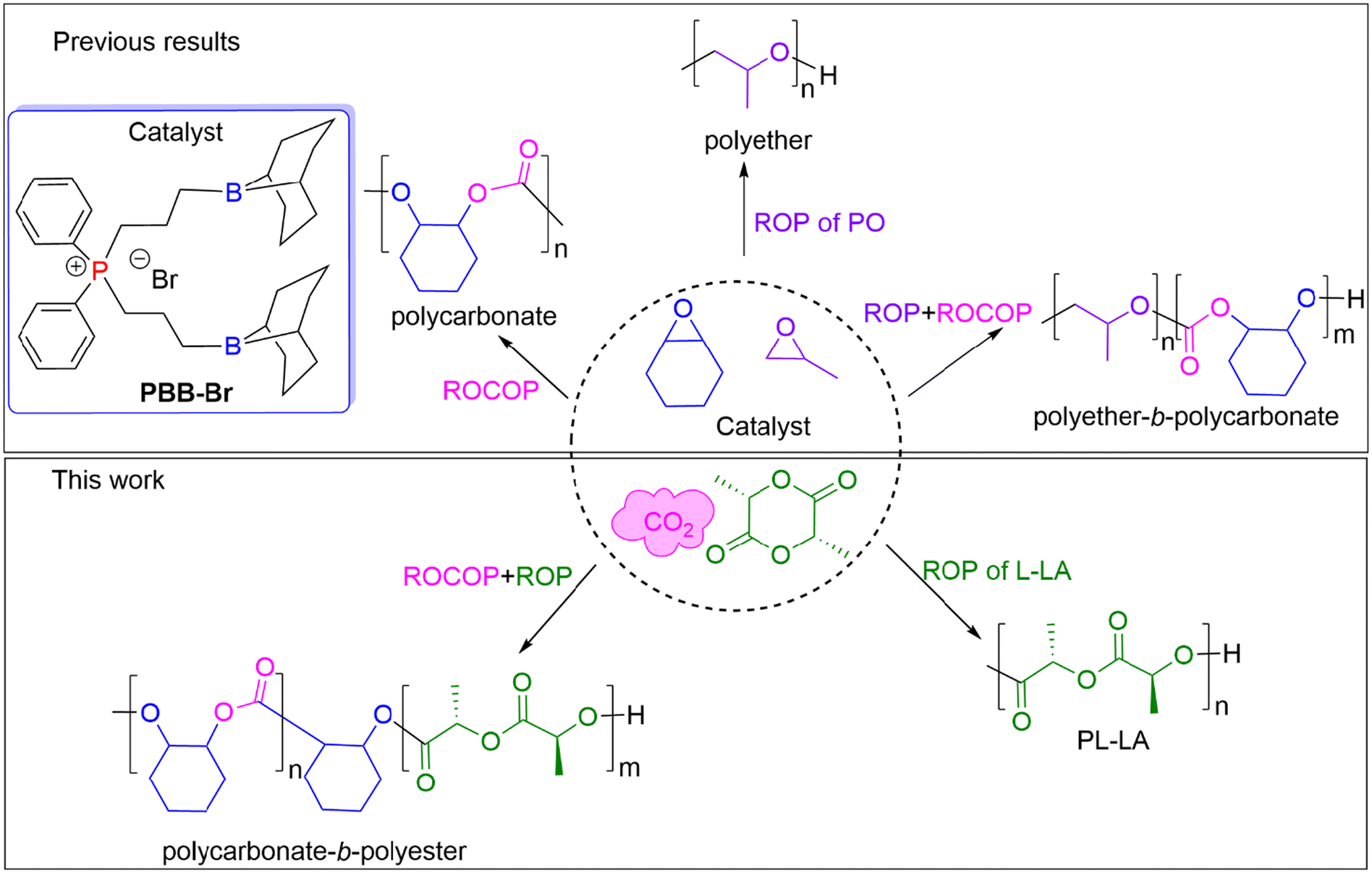 | ||
| Fig. 1 Three selected monomers and the possible range of the produced polymer products using chemoselective catalysis by PBB-Br. | ||
Results and discussion
Previously, we demonstrated that PBB-Br was highly active in catalyzing the ROP of PO and showed excellent living/controlled and immortal polymerization characteristics in the presence of a chain transfer agent (CTA).44,45 Its synthesis and characterization were previously reported.44 The following work in our group indicated that the ROCOP of PO and CO2 catalyzed by PBB-Br only led to the formation of 99% polypropylene oxide (PPO). The exclusive PPO formation was due to the fast and selective PO homopolymerization mediated by PBB-Br. Subsequently, we employed it for the ROCOP of CHO and CO2. Under [CHO]0/[PBB-Br]0 = 100/1 molar ratio and 20 bar CO2 conditions, the conversion of CHO is up to 93% in 2 h at 45 °C (Table 1, run 1). The generated poly(cyclohexene oxide-alt-CO2) (PCHC) showed narrow dispersity (Đ) and possessed >99% carbonate linkages. 1H NMR characterization suggested that almost no ether linkages were observed (Fig. S2,† the 1H NMR spectrum of the generated PCHC, e.g. 4.65 ppm for the PCHC; ∼3.45 ppm for ether linkages). Note that PBB-Br surprisingly showed a very high selectivity for PCHC formation, without detection of any formation of cyclic carbonate byproducts. Otherwise, the characteristic signal from cyclic carbonate should be observed at ∼4.01 ppm. Moreover, the number average molecular weights (Mns) of the generated PCHCs were controllable and showed narrow dispersities. The Mns increased linearly with the conversion of CHO, but were much lower than the theoretical values (Table S1 and Fig. S3†). Such discrepancies are commonly observed in epoxide/CO2 copolymerization and have been attributed to the presence of contaminating water or other protic compounds.51,52 These results indicated that PBB-Br showed controlled polymerization behaviour in the ROCOP of CHO and CO2. Matrix-assisted laser desorption/ionization time of flight mass spectroscopy (MALDI-TOF-MS) of the generated PCHC revealed that both α-Br, ω-OH PCHC and α-OH, ω-OH PCHC were produced (Fig. S4†).| Runa | [CHO]/[L-LA]/[Cat.] | Press. (bar) | Temp. (°C) | Time (h) | Conv.b (%) |
M
Theo.n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c (kg mol−1) c (kg mol−1) |
M
GPCnd (kg mol−1) |
Đ | |
|---|---|---|---|---|---|---|---|---|---|
| CHO | L-LA | ||||||||
| a The polymerizations were performed using PBB-Br as a catalyst (18.6 μmol, 11 mg). 200 μL THF was added as the solvent. b Conv. (%) is the conversion of the CHO or L-LA, which was determined by 1H NMR spectroscopy. c M Theo.n = 142.15 × [CHO + CO2] × Conv.% + 144 × [L-LA] × Conv.%. d Determined by GPC in THF, calibrated with standard polystyrene. e M Theo.n(PL-LA) = 144 × [L-LA] × Conv.%. | |||||||||
| 1 | 100/—/1 | 20 | 45 | 2 | 93 | — | 13.2 | 8.6 | 1.21 |
| 2e | 5/100/1 | 0 | 45 | 24 | — | 93 | 13.4 | 16.1 | 1.40 |
| 3 | 100/50/1 | 20 | 45 | 0.5 | 20 | — | 2.8 | N/D | N/D |
| 0 | 45 | 24 | 20 | — | 2.8 | N/D | N/D | ||
| 4 | 100/50/1 | 5 | 45 | 2 | 70 | — | 9.9 | 3.9 | 1.40 |
| 5 | 50/50/1 | 5 | 45 | 1 | 99 | 0 | 7.0 | 2.0 | 1.25 |
| 0 | 45 | 24 | 99 | 47 | 10.4 | 8.6 | 1.40 | ||
| 6 | 100/50/1 | 5 | 45 | 0.5 | 38 | 0 | 5.4 | 1.2 | 1.20 |
| 0 | 45 | 24 | 38 | 69 | 10.3 | 3.9 | 1.40 | ||
Ring-opening polymerization of L-LA
Although one-component bi(multi)-functional borane systems have shown great promise in the ROP of epoxides,44,45,53 ROCOP of epoxides and anhydrides,54 and ROCOP of epoxides and CO2,52 the capability of these systems to achieve the ROP of cyclic esters: ε-caprolactone (CL), δ-valerolactone (VL) and L-LA, is rare.30 In order to broaden the application range of PBB-Br, we first investigated the ROP of the representative cyclic ester L-LA using PBB-Br. As stated in the Introduction part and given the strong evidence of the hydrogen bonding characteristic of α,β-carbons of PBB-Br, the Hα and Hβ serving as H-bond donors would facilitate the activation of the carbonyl group of L-LA and its subsequent ROP. It is known that a halide is not nucleophilic enough to realize the ROP of L-LA.9 This was also reflected by a control experiment where PBB-Br alone is inactive for the ROP of L-LA (Table S2†). Alkoxide active species usually enable the successful anionic ROP of L-LA.3,55 Therefore, we attempted to perform the ROP of L-LA in the presence of CHO. Strikingly, adding 5 mol% CHO resulted in 93% L-LA conversion within 24 h (45 °C, Table 1, run 2, Fig. S5†). The GPC curve of the generated poly(L-lactide) (PL-LA) featured a bimodal characteristic and the Mn of PL-LA (16.1 kg mol−1) was slightly higher than the theoretical Mn (13.4 kg mol−1) (Fig. S6†), probably due to the initiation by 1,2-cyclohexanediol or water impurity. To further provide evidence for the alkoxide active species being responsible for the ROP of L-LA, a series of control experiments were performed. Equimolar amounts of KOMe and PBB-Br were mixed in THF, and L-LA was added subsequently to allow the reaction mixture to react at 45 °C for 12 h, which resulted in 85.4% L-LA conversion (Table S3, run 1†). The 1H NMR spectrum of the obtained PL-LA showed the typical signal at 3.32 ppm, indicating that OMe initiated the ROP of L-LA. Moreover, 0.5 equiv. of KOMe also worked well when excess benzyl alcohol (5 equiv.) was introduced. The ROP of L-LA ran smoothly at 45 °C for 24 h and gave 65.4% L-LA conversion (Table S3, run 2†). The 1H NMR spectrum of the obtained PL-LA showed typical signals at 7.32 and 4.32 ppm, indicating that benzyl alcohol initiated the ROP of L-LA. This result revealed that benzyl alcohol could be activated by KOMe to form the benzyloxy active species, thus generating benzyloxyl initiated PL-LA.To gain insight into the ROP of L-LA, we performed an in situ NMR study. Therefore, equimolar amounts of PBB-Br and L-LA were mixed in 0.5 mL of CDCl3. As expected, the chemical shift of the αH of PBB-Br shifted slightly from 3.39 ppm to 3.29 ppm (Fig. 2a). The up-field shift of the αH indicated the activation of the carbonyl group of L-LA by hydrogen bonding. The chemical resonance of the carbonyl carbon of L-LA in 13C{1H} NMR shifted from 167.6 ppm to 168.0 ppm (Fig. S9†), which provided further evidence for the activation of L-LA. A minor peak of 11B{1H} NMR appeared at 58.6 ppm indicating the possible coordination of L-LA with the boron center (Fig. 2b). These results indicated that L-LA could be activated by PBB-Brvia hydrogen bonding.
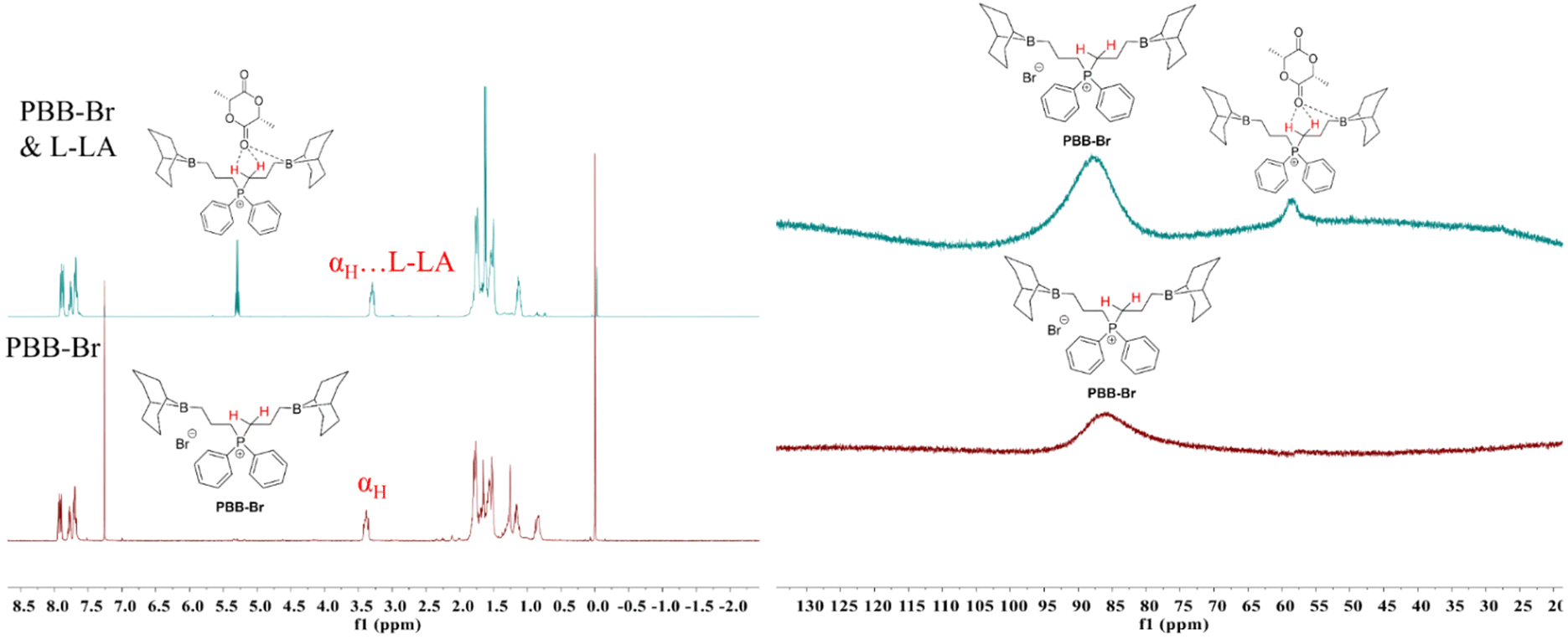 | ||
| Fig. 2 (a) In situ1H NMR (400 MHz, CDCl3, 298 K) spectrum of equimolar amounts of PBB-Br and L-LA (left) (bromide is omitted for clarity). (b) In situ11B{1H} NMR (128 MHz, CDCl3, 298 K) spectrum of equimolar amounts of PBB-Br and L-LA (right) (bromide is omitted for clarity). | ||
Overall, the above experiments revealed that PBB-Br can serve as a catalyst in the ROP of L-LA albeit requiring an anioic activator such as alkoxide. The unique structure of PBB-Br having multiple functional sites, e.g. a basic site, Lewis acidic borane sites, and hydrogen-bonding sites, played a different role in mediating the ROP of L-LA. Specifically, Lewis acidic borane sites accounted for the activation of CHO while the Lewis basic site attacked CHO to generate the alkoxide active species. Such active species was sufficient for the ROP of L-LA due to the activation of L-LA assisted by hydrogen-bonding interaction. Such a scenario afforded the possible chemoselecitive polymerization of CHO, CO2 and L-LA as proposed in Fig. S12.†
Chemoselective polymerization of CHO, CO2 and L-LA
Since PBB-Br was effective for both the ROCOP of CHO/CO2 and ROP of L-LA, we hypothesized that it might be able to serve as a switchable catalyst for the terpolymerization of CHO, CO2 and L-LA. To prove our hypothesis, the terpolymerization of CHO/CO2/L-LA was investigated. Under [CHO]0/[L-LA]0/[PBB-Br]0 = 100/50/1 molar ratio with 20 bar CO2 pressure at 45 °C conditions, homopolymerization of L-LA was completely inhibited and only PCHC was generated in a 20% conversion within 0.5 h (Table 1, run 3). The release of CO2 pressure to 1 bar and prolonging the reaction time to 24 h did not further increase the conversion of CHO and L-LA. This was reasonably explained by the fact that the active species under higher CO2 pressure was carboxylate, which was inactive for the ROP of L-LA. Such a phenomenon was also observed in organo-metal catalyzed L-LA polymerization.56 Although CO2 was released, the residual CO2 dissolved in the reaction mixture inhibited the transformation of the carboxylate active species to alkoxide species. This was reflected by a control experiment that 1 bar CO2 pressure was not high enough to facilitate the ROCOP of CHO and CO2 (Table S1, run 4†). Interestingly, PBB-Br was effective for the ROCOP of CHO and CO2 at 5 bar CO2 while the formation of PCHC was up to 70% within 2 h (Table 1, run 4). To minimize the CO2 effect on the ROP of L-LA, we performed the terpolymerization of CHO, CO2, and L-LA under low CO2 pressure (5 bar CO2). CO2 was released when the copolymerization of CHO and CO2 was run for 1 h (Table 1, run 5). The conversion of CHO was up to 99%. After the release of CO2, L-LA started to polymerize and PCHC-b-PL-LA was obtained. The successful synthesis of the PCHC-b-PL-LA copolymer was further proved by GPC analyses and the DOSY NMR spectrum, in which all the signals had the same diffusion coefficients (D = 1.738 × 10−10 m2 s−1, Fig. 3). Interestingly, the Mn of the generated block copolymer could be adjusted using the feeding ratio of CHO and L-LA (Table 1, run 6). The discrepancy between MGPCn and MTheo.n was ascribed to the presence of water and 1,2-cyclohexanediol diol as a chain transfer agent, which is quite common in copolymerization.52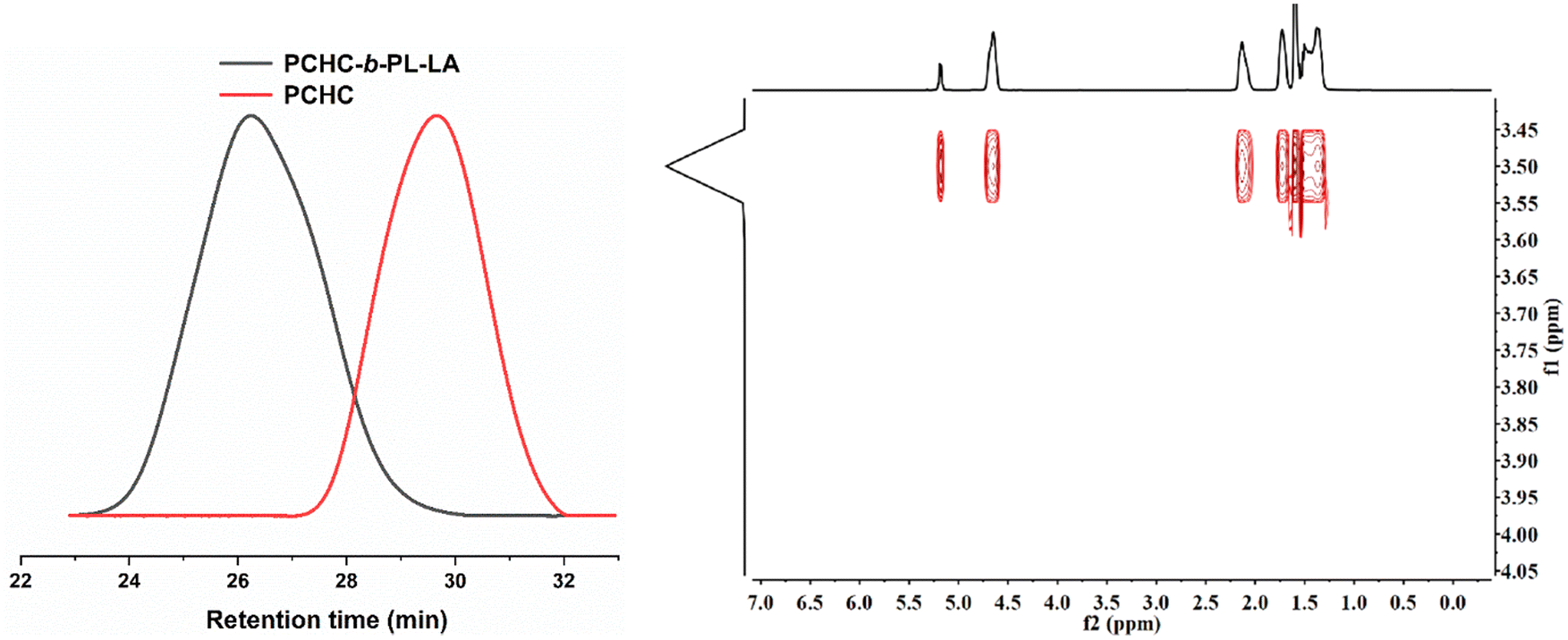 | ||
| Fig. 3 (a) Expanded overlaid GPC traces of PCHC and PCHC-b-PL-LA under [CHO]0/[L-LA]0/PBB-Br = 50/50/1 (Table 1, run 5) obtained using THF as an eluent (left). (b) DOSY NMR spectrum of the PCHC-b-PL-LA block copolymer (right). | ||
A plausible mechanism was proposed for PBB-Br catalyzed chemoselective polymerization of CHO/CO2/L-LA as shown in Fig. 4. In the presence of CO2, it proceeded first due to rapid CO2 insertion (k1 is fast, ROCOP is zero order in carbon dioxide). The formed borane–carbonate intermediate would undergo a ring-opening reaction with the CHO monomer (k2 is slow) but is not nucleophilic enough for the ROP of L-LA. Thus, when CO2 was present, only PCHC homopolymers were produced. After CO2 removal, a borane–alkoxide species was generated in situ, which started the ROP of L-LA (k3) to form borane–LA–alkoxide which subsequently afforded PCHC-b-PL-LA (Fig. 4).
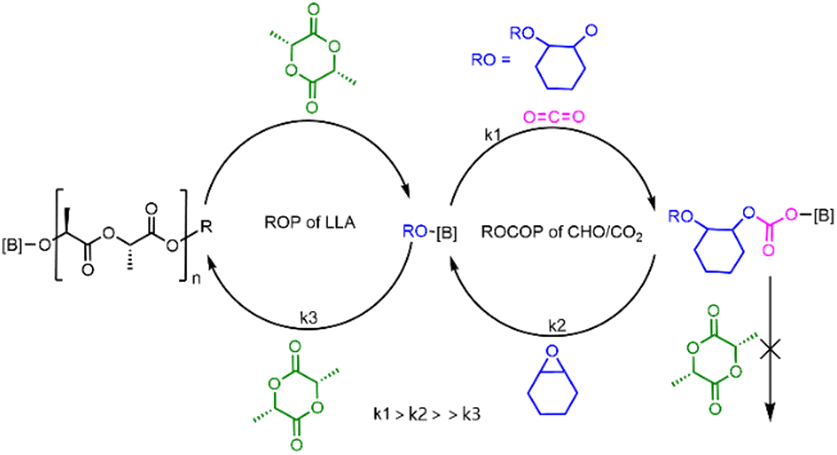 | ||
| Fig. 4 Proposed mechanism of the chemoselective polymerization of CHO, CO2 and L-LA. | ||
Conclusions
In conclusion, chemoselective polymerization using mixtures of CHO, CO2, and L-LA was investigated. The PBB-Br Lewis pair enables the selective synthesis of either a polycarbonate or polyester. The selectivity depends on the nature of the borane–oxygen functionality at the growing polymer chain end, and can be controlled by the addition of an exogeneous switch reagent, CO2. The synthesis takes advantage of the ability of PBB-Br switching between the ROCOP of CHO and CO2 and the ROP of L-LA. CO2 serves as an inhibitor of the ROP of L-LA, thus guaranteeing the selective copolymerization of CHO and CO2 in the presence of L-LA. Release of CO2 switches the ROCOP of CHO and CO2 to the ROP of L-LA. Finally, a straightforward one-pot synthesis of low carbon block polymers featuring polycarbonate-b-polyester is achieved. The access to more complex but controlled low carbon polymers is expected to furnish materials with properties matched to high-value emerging applications such as degradable low carbon thermoplastic elastomers and chemically recyclable polymers.Author contributions
Yutao Li: investigation, visualization and data curation. Xiaowu Wang: investigation, methodology, conceptualization, supervision, writing – original draft and writing – review & editing. Lin Xia: supervision. Zhibo Li: methodology, conceptualization, supervision, and writing – review &editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The National Key R&D Program of China (2021YFA1501600) and the National Natural Science Foundation of China (no. 22175105 and 22031005) are gracefully acknowledged.References
- A. B. Biernesser, K. R. D. Chiaie, J. B. Curley and J. A. Byers, Angew. Chem., Int. Ed., 2016, 55, 5251–5254 CrossRef CAS PubMed.
- A. C. Deacy, G. L. Gregory, G. S. Sulley, T. T. D. Chen and C. K. Williams, J. Am. Chem. Soc., 2021, 143, 10021–10040 CrossRef CAS PubMed.
- M. Kröger, C. Folli, O. Walter and M. Döring, Adv. Synth. Catal., 2006, 348, 1908–1918 CrossRef.
- M. J. Supej, B. M. Peterson and B. P. Fors, Chem, 2020, 6, 1794–1803 CAS.
- C. J. Zhang, H. Y. Duan, L. F. Hu, C. H. Zhang and X. H. Zhang, ChemSusChem, 2018, 11, 4209–4213 CrossRef CAS PubMed.
- A. B. Biernesser, K. R. D. Chiaie, J. B. Curley and J. A. Byers, Angew. Chem., Int. Ed., 2016, 55, 5251–5254 CrossRef CAS PubMed.
- G. Chen, L. Xia, F. Wang, Z. Zhang and Y.-Z. You, Polym. Chem., 2021, 12, 3740–3752 RSC.
- O. Coulembier, S. Moins, R. Todd and P. Dubois, Macromolecules, 2014, 47, 486–491 CrossRef CAS.
- Z. Gao, B. Gao, S. Meng, Z. Yang, Z. Liang, Z. Sun, Y. Zhou and X. Pang, Macromolecules, 2023, 56, 2062–2069 CrossRef CAS.
- G. L. Gregory and C. K. Williams, Macromolecules, 2022, 55, 2290–2299 CrossRef CAS PubMed.
- C. Hu, X. Pang and X. Chen, Macromolecules, 2022, 55, 1879–1893 CrossRef CAS.
- R. C. Jeske, J. M. Rowley and G. W. Coates, Angew. Chem., Int. Ed., 2008, 47, 6041–6044 CrossRef CAS PubMed.
- S. Kernbichl, M. Reiter, F. Adams, S. Vagin and B. Rieger, J. Am. Chem. Soc., 2017, 139, 6787–6790 CrossRef CAS PubMed.
- S. Paul, C. Romain, J. Shaw and C. K. Williams, Macromolecules, 2015, 48, 6047–6056 CrossRef CAS.
- C. Romain and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 1607–1610 CrossRef CAS PubMed.
- C. Romain, Y. Zhu, P. Dingwall, S. Paul, H. S. Rzepa, A. Buchard and C. K. Williams, J. Am. Chem. Soc., 2016, 138, 4120–4131 CrossRef CAS PubMed.
- T. Stößer, G. S. Sulley, G. L. Gregory and C. K. Williams, Nat. Commun., 2019, 10, 2668–2676 CrossRef PubMed.
- G. S. Sulley, G. L. Gregory, T. T. D. Chen, L. P. Carrodeguas, G. Trott, A. Santmarti, K.-Y. Lee, N. J. Terrill and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 4367–4378 CrossRef CAS PubMed.
- Z. Yang, C. Hu, Z. Gao, R. Duan, Z. Sun, Y. Zhou, X. Pang and X. Chen, Macromolecules, 2023, 56, 2370–2378 CrossRef CAS.
- X. Zheng, J. Yang, Z. Xu, Q. Wang, J. Wu, E. Zhang, S. Dou, W. Sun, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202205946 CrossRef CAS PubMed.
- M. Zhao, S. Zhu, G. Zhang, Y. Wang, Y. Liao, J. Xu, X. Zhou and X. Xie, Macromolecules, 2023, 56, 2379–2387 CrossRef CAS.
- Y. Ma, X. You, J. Zhang, X. Wang, X. Kou, S. Liu, R. Zhong and Z. Li, Angew. Chem., Int. Ed., 2023, 62, e202303315 CrossRef CAS PubMed.
- J. Liu, M. Jia, Y. Gnanou and X. Feng, Macromolecules, 2023, 56, 1615–1624 CrossRef CAS.
- Y.-Y. Zhang, G.-W. Yang, R. Xie, X.-F. Zhu and G.-P. Wu, J. Am. Chem. Soc., 2022, 144, 19896–19909 CrossRef CAS PubMed.
- J. Liu, Y. Gnanou and X. Feng, Macromolecules, 2022, 55, 1800–1810 CrossRef CAS.
- V. K. Chidara, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, Macromolecules, 2021, 54, 2711–2719 CrossRef CAS.
- S. Zhu, Y. Zhao, M. Ni, J. Xu, X. Zhou, Y. Liao, Y. Wang and X. Xie, ACS Macro Lett., 2020, 9, 204–209 CrossRef CAS PubMed.
- Y. B. Ivanova, A. S. Semeikin, T. V. Lyubimova and N. Z. Mamardashvili, Russ. J. Org. Chem., 2020, 56, 1691–1695 CrossRef CAS.
- H. Li, G. He, Y. Chen, J. Zhao and G. Zhang, ACS Macro Lett., 2019, 8, 973–978 CrossRef CAS PubMed.
- C. Zhang, X. Geng, X. Zhang, Y. Gnanou and X. Feng, Prog. Polym. Sci., 2023, 136, 101644–101666 CrossRef CAS.
- J. Tang, M. Li, X. Wang and Y. Tao, Angew. Chem., 2022, 134, e202115465 CrossRef.
- N. Patil, Y. Gnanou and X. Feng, Polym. Chem., 2022, 13, 2988–2998 RSC.
- J. Zhang, L. Wang, S. Liu, X. Kang and Z. Li, Macromolecules, 2021, 54, 763–772 CrossRef CAS.
- Y. Xia, G. M. Scheutz, C. P. Easterling, J. Zhao and B. S. Sumerlin, Angew. Chem., Int. Ed., 2021, 60, 18537–18541 CrossRef CAS PubMed.
- S. Zhu, Y. Wang, W. Ding, X. Zhou, Y. Liao and X. Xie, Polym. Chem., 2020, 11, 1691–1695 RSC.
- C.-J. Zhang, X. Zhang and X.-H. Zhang, Sci. China: Chem., 2020, 63, 1807–1814 CrossRef CAS.
- S. Ye, W. Wang, J. Liang, S. Wang, M. Xiao and Y. Meng, ACS Sustainable Chem. Eng., 2020, 8, 17860–17867 CrossRef CAS.
- D. Augustine, N. Hadjichristidis, Y. Gnanou and X. Feng, Macromolecules, 2020, 53, 895–904 CrossRef CAS.
- S. Liu, T. Bai, K. Ni, Y. Chen, J. Zhao, J. Ling, X. Ye and G. Zhang, Angew. Chem., Int. Ed., 2019, 58, 15478–15487 CrossRef CAS PubMed.
- H.-Y. Ji, D.-P. Song, B. Wang, L. Pan and Y.-S. Li, Green Chem., 2019, 21, 6123–6132 RSC.
- K. A. Andrea and F. M. Kerton, ACS Catal., 2019, 9, 1799–1809 CrossRef CAS.
- K. A. Andrea, H. Plommer and F. M. Kerton, Eur. Polym. J., 2019, 120, 109202 CrossRef CAS.
- J. Liu, M. Jia, Y. Gnanou and X. Feng, Macromolecules, 2023, 56, 1615–1624 CrossRef CAS.
- X. Wang, M. Shi, J. Hui, X. Kou, J. Wang, R. Zhong and Z. Li, Macromolecules, 2023, 56, 4030–4040 CrossRef CAS.
- X. Wang, J. Hui, M. Shi, X. Kou, X. Li, R. Zhong and Z. Li, ACS Catal., 2022, 12, 8434–8443 CrossRef CAS.
- M. T. Reetz, S. Huette and R. Goddard, J. Am. Chem. Soc., 1993, 115, 9339–9340 CrossRef CAS.
- G.-W. Yang, Y.-Y. Zhang and G.-P. Wu, Acc. Chem. Res., 2021, 54, 4434–4448 CrossRef CAS PubMed.
- G.-W. Yang, Y.-Y. Zhang, R. Xie and G.-P. Wu, J. Am. Chem. Soc., 2020, 142, 12245–12255 CrossRef CAS PubMed.
- Y.-Y. Zhang, C. Lu, G.-W. Yang, R. Xie, Y.-B. Fang, Y. Wang and G.-P. Wu, Macromolecules, 2022, 55, 6443–6452 CrossRef CAS.
- J. Schaefer, H. Zhou, E. Lee, N. S. Lambic, G. Culcu, M. W. Holtcamp, F. C. Rix and T.-P. Lin, ACS Catal., 2022, 12, 11870–11885 CrossRef CAS.
- F. Jutz, A. Buchard, M. R. Kember, S. B. Fredriksen and C. K. Williams, J. Am. Chem. Soc., 2011, 133, 17395–17405 CrossRef CAS PubMed.
- X.-W. Wang, J.-W. Hui, Y.-T. Li, Y.-R. Gu and Z.-B. Li, Chin. J. Polym. Sci., 2023, 41, 735–744 CrossRef CAS.
- Y. Gu, X. Kou, X. Wang and Z. Li, Angew. Chem., Int. Ed., 2024, 63, e202318645 CrossRef CAS PubMed.
- J. Hui, X. Wang, X. Yao and Z. Li, Polym. Chem., 2022, 13, 6551–6563 RSC.
- L. Mezzasalma, A. P. Dove and O. Coulembier, Eur. Polym. J., 2017, 95, 628–634 CrossRef CAS.
- Z. Yang, C. Hu, F. Cui, X. Pang, Y. Huang, Y. Zhou and X. Chen, Angew. Chem., Int. Ed., 2022, 61, e202117533 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00329b |
| This journal is © The Royal Society of Chemistry 2024 |