
Recent advances in developing nanoscale electro-/photocatalysts for hydrogen production: modification strategies, charge-carrier characterizations, and applications
Mohammed-Ibrahim
Jamesh†
a,
Haihang
Tong†
ao,
Shella Permatasari
Santoso
 b,
Wenxin
Niu
c,
Ji-Jung
Kai
d,
Chang-Wei
Hsieh
ef,
Kuan-Chen
Cheng
*ghij,
Fang-Fang
Li
k,
Bin
Han
*l,
Juan Carlos
Colmenares
mn and
Hsien-Yi
Hsu
*ao
b,
Wenxin
Niu
c,
Ji-Jung
Kai
d,
Chang-Wei
Hsieh
ef,
Kuan-Chen
Cheng
*ghij,
Fang-Fang
Li
k,
Bin
Han
*l,
Juan Carlos
Colmenares
mn and
Hsien-Yi
Hsu
*ao
aSchool of Energy and Environment, Department of Materials Science and Engineering, Centre for Functional Photonics (CFP), City University of Hong Kong, Kowloon Tong, Hong Kong, China. E-mail: sam.hyhsu@cityu.edu.hk
bDepartment of Chemical Engineering, Faculty of Engineering, Widya Mandala Surabaya Catholic University, Kalijudan No. 37, Surabaya 60114, East Java, Indonesia
cState Key Laboratory of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, 5625 Renmin Street, Changchun, Jilin 130022, P. R. China
dDepartment of Mechanical Engineering, City University of Hong Kong, Kowloon Tong, Hong Kong, China
eDepartment of Food Science and Biotechnology, National Chung Hsing University, 145 Xingda Rd., South Dist., Taichung City, Taiwan
fDepartment of Food Science, National Ilan University, Shennong Road, Yilan City 26047, Taiwan
gGraduate Institute of Food Science Technology, National Taiwan University, Taipei 10617, Taiwan. E-mail: kccheng@ntu.edu.tw
hInstitute of Biotechnology, National Taiwan University, Taipei 10617, Taiwan
iDepartment of Medical Research, China Medical University Hospital, China Medical University, Taichung, Taiwan
jDepartment of Optometry, Asia University, 500 Lioufeng Rd., Wufeng, Taichung, Taiwan 41354
kSchool of Materials Science and Engineering, Huazhong University of Science and Technology, 1037 Luoyu Road, Wuhan 430074, China
lMaterials Institute of Atomic and Molecular Science, Shaanxi University of Science and Technology, Xi’an 710021, China. E-mail: binhan@sust.edu.cn
mInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01224, Warsaw, Poland
nEngineering Research Institute (In3), Universidad Cooperativa de Colombia, Medellín 50031, Colombia
oShenzhen Research Institute of City University of Hong Kong, Shenzhen 518057, P. R. China
First published on 23rd August 2024
Abstract
For clean hydrogen (H2) production, electrocatalysis and photocatalysis are widely regarded as promising technologies to counter the increasing energy crisis. However, developing applicable catalysts with high H2 production performances still poses a challenge. In this review, state-of-the-art nanoscale electrocatalysts for water electrolysis and photocatalysts for water splitting, tailored for different reaction environments, including acidic electrolytes, alkaline electrolytes, pure water, seawater, and hydrohalic acids, are systematically presented. In particular, modification approaches such as doping, morphology control, heterojunction/homojunction construction, as well as the integration of cocatalysts and single atoms for efficient charge transfer and separation are examined. Furthermore, the unique properties of these upgraded catalysts and the mechanisms of promoted H2 production are also analyzed by elucidating the charge carrier dynamics revealed by photophysical and photoelectrochemical characterization methods. Finally, perspectives and outlooks on future developments for H2 production using advanced electrocatalysts and photocatalysts are proposed.
1. Introduction
The production of hydrogen fuel through water electrolysis, photocatalytic water splitting, and photocatalytic hydrohalic acid (HX) splitting offers a promising green synthesis approach for the development of renewable energy.5–13 Nevertheless, approximately 95% of global hydrogen production relies on fossil fuels, resulting in carbon emissions, whereas approximately only 4% of global hydrogen production depends on water electrolysis.19,20 Current research efforts are focused on water electrolysis and photocatalysis to mitigate the reliance on fossil fuels for hydrogen energy production.5,7–9,22–24Electrochemical water splitting comprises two half-cell reactions: the hydrogen evolution reaction (HER) at the cathode, which involves the transfer of two electrons, and the oxygen evolution reaction (OER) at the anode, which necessitates the participation of four electrons. These reactions occur in acidic or alkaline media. Presently, noble metal electrocatalysts such as Ir/Ru-based compounds for the OER and Pt for the HER are considered to be promising electrocatalysts for water electrolysis.27 Furthermore, the OER electrocatalysts currently utilized for water electrolysis in acidic media are both costly and scarce.27,29 In contrast, utilizing electrocatalysts in an alkaline environment can significantly lower the overall expense of water electrolyzer systems.12,30,31 Consequently, it is crucial to focus on the development of electrocatalysts specifically designed for water electrolysis under alkaline conditions.
Nevertheless, the use of bifunctional catalysts for water electrolysis may lead to the mixing of gases (H2 and O2) during collection, introducing additional costs for gas separation, which represents a potential drawback. In this context, proton exchange membrane (PEM) water electrolysis in acidic environments could mitigate the issue of gas mixing to a certain extent. Nonetheless, this approach necessitates the development of acid-stable HER and OER catalysts for efficient PEM water electrolysis.33 Therefore, developing electrocatalysts for water electrolysis in an acidic environment is essential.
Water electrolysis technologies, including proton exchange membranes (PEMs) and alkaline electrolyzers, traditionally rely on high-purity water as a feedstock.34,35 Given that Earth-abundant seawater constitutes approximately 96.5% of the world's water reserves, its utilization as a feedstock for water electrolysis could significantly reduce the reliance on scarce freshwater resources. This shift is particularly critical for regions with high freshwater demands, such as coastal countries, arid zones, and islands.23,36 However, the adoption of seawater electrolysis for industrial applications is currently limited due to the complex composition of seawater electrolytes.37,38 Consequently, there is a pressing need to develop electrocatalysts tailored to the seawater electrolysis environment to overcome these challenges. Photocatalytic H2 production via water splitting using photocatalysts is a promising route to convert solar energy into clean chemical fuels.11 Designing photocatalysts with suitable bandgaps for the efficient absorption of sunlight, excellent charge separation and transfer efficiency, and satisfactory surface reactions is essential. Besides, photocatalytic H2 production using hydrohalic acid (HX) is a promising strategy, where value-added chemicals (such as X2/X3−) can also be generated along with clean, renewable H2 fuel.5,9,10,49 Developing photocatalysts that are both resistant to strong acids and active under visible light, especially eco-friendly, lead-free options for hydrogen production via hydrohalic acid (HX) splitting, presents a significant challenge. Thus, advancing the creation of such materials is crucial for efficient photocatalytic H2 generation.
The Nyquist plot, from electrochemical impedance spectroscopy (EIS) data, is a simple and useful tool for developing electrocatalysts for hydrogen production, where comparing the quantified charge transfer resistance of the electrocatalyst with its counterparts can provide valuable information about the conductivity of the electrocatalysts. Generally, a wide range of electrocatalysts demonstrate high activity and low charge transfer resistance for the hydrogen evolution reaction (HER) and/or the oxygen evolution reaction (OER) across various environments, including alkaline, acidic, and seawater.1,4,40,50 However, exceptions exist,39,51 as the catalytic activity for the HER or the OER is not solely determined by conductivity. It also relies on several critical factors: the availability of abundant active sites, optimal adsorption energies for HER/OER intermediates, a suitable electronic structure, a large electrochemically active surface area, and the ability to facilitate gas evolution.30,33,36,52,53 Xi et al.4 observed that Cu RuO2activated exhibited a lower charge transfer resistance (Rct1: 0.27 W cm−2; Rct2: 0.09 W cm−2) for the HER compared to RuO2activated (Rct1: 4.49 W cm−2; Rct2: 0.44 W cm−2), and it also demonstrated a higher activity (η of −19 mV at −10 mA cm−2) and larger electrochemical surface area for the HER in 1 M KOH compared to RuO2activated. Thus, doping Cu into RuO2 facilitates the partial formation of metallic Ru as active sites for the HER during electrochemical activation, leading to enhanced conductivity and activity for the HER.
Liu et al.40 reported that Ru–Ni3N/NiO exhibited a lower charge transfer resistance (Rct: 1.52 Ω or Ω cm−2) and higher activity (η of −190 mV at a huge current density of −1000 mA cm−2) for the HER than those of Ru–Ni3N and Ru–NiO, and it also exhibited a much higher activity for the OER (η of 385 mV at a huge current density of 1000 mA cm−2) and overall water splitting (potential of 1.74 V at a huge current density of 1000 mA cm−2) in 1 M KOH. Theoretical calculations suggested that the structural stability, orbital coupling, and electronic distributions of Ru–Ni3N/NiO could be enhanced by the Ni3N/NiO heterostructures when compared to those of Ru–Ni3N and Ru–NiO; this enhancement could lower the energy barriers and increase the activity for the HER and the OER. In addition, Zhao et al.1 reported that strained Mn0.98Ir0.02O2 exhibited a higher activity (η of 105 mV at 10 mA cm−2; mass activity: 300 mV at 5681 A g−1) and lower charge transfer resistance than those of unstrained Mn0.98Ir0.02O2 for the OER in 0.5 M H2SO4. The incorporation of Ir into MnO2 can induce strain, leading to an elongated Mn–O bond length and pushing the d-band center nearer to the Fermi level, while the holes in the O 2p orbitals can more readily transfer to the catalyst surface, leading to acceleration in charge transfer, and thereby modify the electronic structure, leading to improved activity for the OER. Moreover, Wang et al.50 observed that Co–N,P-HCS (HCS: hollow carbon spheres) exhibited a higher activity and lower charge transfer resistance than those of HCS, Co–HCS, and Co–N-HCS for the HER and the OER in seawater with 1 M KOH, and it exhibited high stability for 1000 h. Co–N,P-HCS is composed of an asymmetric Co–N3P1 structure, which is implanted onto hollow carbon spheres. DFT calculations reveal that asymmetric Co–N3P1 sites with symmetry-breaking electronic structures can be formed due to P-doping of Co–N–C, which enhances the affinity of strong oxygen-containing intermediates with weak Cl− adsorption and moderate H adsorption. On the other hand, Seenivasan et al.39 observed that Nb4N5 exhibited a significantly higher activity (η of −7 mV at −10 mA cm−2; η of −281 mV at a huge current density of −1000 mA cm−2) and lower charge transfer resistance (Rct: 0.892 Ω; Table 1) than those of Fe4N/Nb4N5 (Rct: 2.165 Ω) for the HER in 1 M KOH, while Fe4N/Nb4N5 exhibited a significantly higher activity (η of 438 mV at a huge current density of 1000 mA cm−2) and lower charge transfer resistance (Rct: 0.206 Ω) than those of Nb4N5 (Rct: 1.369 Ω) for the OER in 1 M KOH. This obviously demonstrates the role of conductivity on the activity of the HER or the OER, where the conductivity and activity of Nb4N5 for the HER are higher than those of Fe4N/Nb4N5, whereas the conductivity and activity of Fe4N/Nb4N5 for the OER are higher than those of Nb4N5. The electrochemical reconstruction processes in Nb4N5 and Fe4N/Nb4N5 are the underlying reason. Specifically, Nb4N5 remains stable under HER conditions. However, under OER conditions, the electrochemical reconstruction of Nb4N5 leads to the formation of inactive KNbO3 species. Additionally, the presence of Fe ions in Fe4N/Nb4N5 alters the electrochemical reconstruction process, resulting in the generation of highly active FeOOH species that contain high-valence Fe3+ ions.
| HER electrocatalysts | η at −10 mA cm−2 (mV) | R ct or Rct1 | R ct2 | Stability | Ref. |
|---|---|---|---|---|---|
| HER: hydrogen evolution reaction; η: overpotential; Rct or Rct1 or Rct2: charge transfer resistance; NA: not applicable. | |||||
| Nb4N5 | −7 | 0.892 Ω | NA | NA | 39 |
| Ru–Ni3N/NiO | −15 | 1.52 Ω or Ω cm−2 | NA | 1000 h (500 mA cm−2) | 40 |
| Cu–RuO2activated | −19 | 0.27 Ω cm−2 | 0.09 Ω cm−2 | 50 h | 4 |
| FeCoCrCuOx | −40 | NA | NA | 100 h (500 mA cm−2) | 41 |
| Zr0.1Ni1.9P | −68 | 5 Ω | NA | 100 h | 42 |
| P/Fe–NiSe2 | −74 | 4.78 Ω | NA | Less than 5% current density loss over 24 h (100 mA cm−2) | 43 |
| Fe@Co/Se2 | −78 | 10.80 Ω | NA | 0.3% loss of over potential after 20 h (10 mA cm−2) | 14 |
| DH–CuCo–P@NC/CC | −85 | 2.4 Ω | NA | 12 h | 44 |
| cNiFe/aNiFeOOH@NiMo | −91.9 at −20 mA cm−2 | 0.946 Ω | NA | 100 h (100 mA cm−2) | 45 |
| 1T-Mn–VS2@Co2P | −98 | 2 Ω | NA | 100 h (500 mA cm−2) | 46 |
| CoP3–Nb2P/PCC | −111 | 3.5 Ω | NA | 6.4% loss of current density after 50 h (100 mA cm−2) | 47 |
| NiCoSe2/MoSe2 | −244 at −50 mA cm−2 | 13.1 Ω | NA | 72 h | 48 |
A comparison of charge transfer resistance across various photocatalysts provided by EIS reveals crucial insights into their charge separation efficiencies.5,7,54 High activity in hydrogen evolution depends on a range of factors beyond just charge separation resistance.17,55,56 These include effective charge separation and transfer, minimized recombination of photogenerated charges, rapid diffusion rates of charges, prolonged lifetimes of excitons, efficient use of visible light, appropriate bandgaps, and the optimization of charge distributions at interfaces. Additionally, ensuring efficient electron transport for proton reduction and providing active sites with low energy barriers for hydrogen production are essential.5,8,9,18,32,54 Moon et al.7 found that the Pt/g-C3N4/TiO2/IrOx (PCTI) hollow sphere photocatalyst demonstrated a lower charge transfer resistance (Rct = 1.52 kΩ) compared to that of other co-catalysts on g-C3N4/TiO2, indicating more rapid charge transfer in PCTI. This configuration significantly enhances the transient photocurrent density, which is attributed to efficient surface charge separation where photogenerated electrons are trapped by Pt on the CT's external surface and photoexcited holes are trapped by IrOx internally. This arrangement boosts the water redox reaction performance, illustrating that strategic spatial separation of co-catalysts on a Z-scheme heterostructure can accelerate surface reaction rates, ensuring effective charge separation and transfer, and thus, improving photocatalytic H2 evolution. Tang et al.5 observed that the bismuth-based perovskite heterojunction MA3Bi2I9/DMA3BiI6 (BBP-5; MA: methylammonium; DMA: dimethylammonium) achieved a remarkable H2 evolution rate of 198.2 μmol h−1 g−1 and superior apparent quantum efficiency, outperforming its counterparts without noble metal co-catalysts under visible light (λ ≥ 420 nm). BBP-5's lower charge transfer resistance (Rct: 1.15 kΩ cm2) underscores its enhanced charge separation capabilities. This discovery suggests that the in situ formation of such heterojunctions can not only facilitate charge separation and reduce charge recombination but also extend exciton lifetimes and promote efficient electron transport for H+ ion reduction, culminating in heightened photocatalytic H2 evolution via HI splitting. Furthermore, Guo et al.17 noted that the NiCo2S4/ZnIn2S4 hollow nanocages (NCS/ZIS-HNCs) showcased a lower charge transfer resistance and a higher transient photocurrent density than those of ZIS-HNCs alone, indicating effective photogenerated electron migration at the NCS/ZIS-HNCs interface. Despite NCS exhibiting a lower charge resistance than NCS/ZIS-HNCs, its photocatalytic activity is negligible, whereas NCS/ZIS-HNCs exhibit a significantly higher H2 production activity. This highlights that optimal photocatalytic performance relies on more than just low charge resistance, including the photothermal effect, which is markedly enhanced in NiCo2S4/ZnIn2S4 without cooling water, achieving a H2 evolution rate 5.98 times greater than that with cooling. This enhancement is attributed to improved electron dynamics facilitated by interfacial S atoms with vacancies, indicating that efficient carrier transfer and the localized surface plasmon resonance (LSPR) effect, leading to the generation of hot electrons, are critical for maximizing photocatalytic H2 evolution.
This review embarks on an in-depth exploration of nanostructured electrocatalysts and photocatalysts, showcasing their pivotal roles in the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER) across varied environments including alkaline, acidic, and seawater media. It particularly emphasizes diverse material compositions such as oxides, hydroxides, sulfides, phosphides, nitrides, and innovative carbon-based, alloy, and metal–organic frameworks (MOFs), alongside the strategic implementations of heterojunctions, co-catalyst loading, defect engineering, and elemental doping. The narrative extends to evaluating photocatalytic strategies for hydrogen production, highlighting advancements in photocatalyst design for both water splitting and hydrohalic acid (HX) splitting, with a focus on the integration of lead-free and perovskite-based materials. Through this review, we aim to encapsulate the latest developments, draw comparisons among various catalytic systems, and identify promising directions for future research in the quest for efficient and sustainable hydrogen production technologies.
2. Water electrolysis in an alkaline environment
2.1. Oxide/hydroxide/layered double hydroxide based nano-electrocatalysts
Cu doping of RuO2 has been shown to facilitate the formation of metallic Ru active sites for the hydrogen evolution reaction (HER) during electrochemical activation, enhancing both conductivity and HER activity. Xi et al.4 observed that Cu-doped RuO2, prepared through a hydrolysis method followed by calcination at 300 °C for 2 h and electrochemically activated with five cycles of cyclic voltammetry (CV) from 0 to 1.03 V versus RHE for 7.5 min, exhibited significantly higher HER activity. The presence of Cu2+ sites, due to their lower valence state than that of Ru4+, leads to oxygen vacancies and the disruption of Ru–O octahedra, thereby reducing adjacent Ru4+ under certain voltages during CV activation. This process generates Ru0 sites on the surface, as evidenced by the extended X-ray absorption fine structure (EXAFS) spectrum at the Ru K-edge, showing distinct interaction peaks for Ru–O, Ru–Ru, and Ru–Cu in Cu-doped RuO2, indicative of altered Ru–O octahedral structures (Fig. 1(a)). Similarly, as shown in Fig. 1(b), the EXAFS spectrum at the Cu K-edge revealed changes in Cu–O and the presence of Cu–Ru metallic coordination, confirming the activation effect of Cu doping. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images further confirmed the formation of Ru0 and highlighted lattice distortions and defects post-activation (Fig. 1(c)). Density functional theory (DFT) calculations supported the activation mechanism, showing that Cu2+ doping activated adjacent Ru4+. Notably, Cu-doped RuO2 displayed a significantly lower charge transfer resistance and higher electrochemical surface area for the HER compared to undoped RuO2, underlining its superior activity and conductivity (Fig. 1(d)). In 1 M KOH, this catalyst achieved an overpotential of −19 mV at −10 mA cm−2, showcasing exceptional activity, and maintained negligible decay over 93 h, indicating remarkable stability. For overall water splitting in the same medium, the HER performance of Cu–RuO2//Cu–RuO2 demonstrated very high activity, requiring potentials of 1.66 V at 100 mA cm−2 and 1.78 V at 200 mA cm−2. This comprehensive analysis underscores the significant impact of Cu doping on enhancing the electrocatalytic performance of RuO2 for efficient hydrogen production.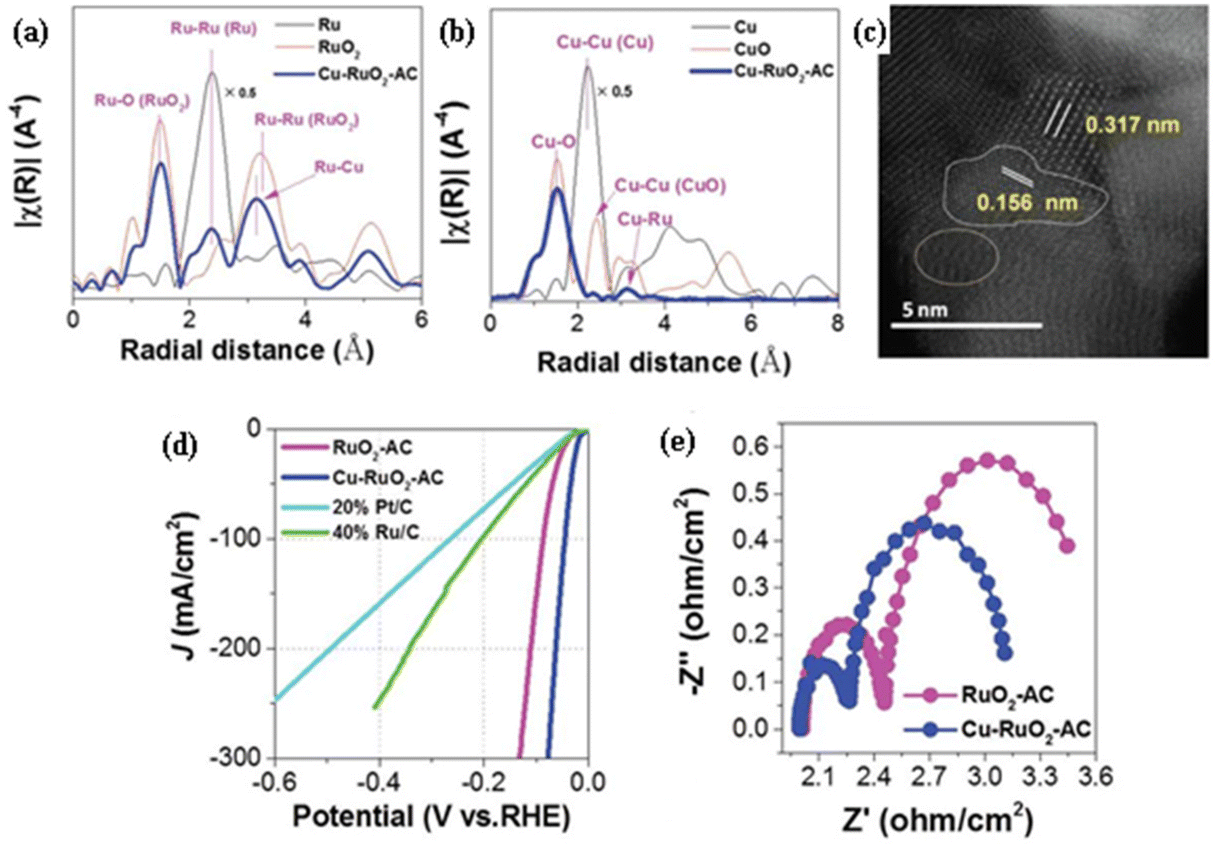 | ||
| Fig. 1 (a) EXAFS spectrum obtained at the Ru K-edge of Cu–RuO2activated in comparison with those of Ru and RuO2; (b) EXAFS spectrum obtained at the Cu K-edge of Cu–RuO2activated in comparison with those of Cu and CuO; (c) HADDF-STEM image of Cu–RuO2activated, where the oval pink area reveals the obvious lattice distortion; (d) LSV curves for the HER of Cu–RuO2activated in comparison with those of RuO2–AC, 20% Pt/C and 40% Ru/C; (e) EIS plot for the HER of Cu–RuO2activated in comparison with that of RuO2–AC obtained at −0.1 V versus RHE (reproduced with permission from ref. 4. Copyright 2023, Wiley-VCH GmbH). | ||
Besides, Triolo et al.57 found that the high-entropy spinel oxide (Cr1/5Mn1/5Fe1/5Co1/5Ni1/5)3O4 nanofibers exhibited higher activity and lower charge transfer resistance for the OER than those of (Cr1/5Mn1/5Fe1/5Co1/5Zn1/5)3O4 and (Cr1/5Mn1/5Fe1/5Ni1/5Zn1/5)3O4. This could be attributed to the eg filling with nearer to its optimal value, lower outer 3d-electron number, and greater occupation of the 16d sites by the most redox-active species. Moreover, Liu et al.41 observed that FeCoCrCuOx exhibited higher activity and lower charge transfer resistance for the HER than those of FeCoOx, FeCoCrOx, and FeCoCuOx, and it exhibited high activity and stability for overall water splitting in 1 M KOH. In addition, it only requires 3.18 V to achieve >30 A (∼1.84 A cm−2) on a large-sized electrode (16.5 cm2) for alkaline water electrolysis. The superior performance of FeCoCrCuOx is attributed to the synergistic interaction between multiple metals. Additionally, surface activation and robust Cr–OH interactions upon activation also play a crucial role in enhancing the HER performance.
Integration of the heterostructured nanosheet array with Ru nanoparticles could provide optimal metal–support interactions, enhance conductivity, improve orbital coupling, provide optimal adsorption energy with intermediates, and facilitate gas evolution, which could enhance the performance for the HER and OER. Liu et al.40 fabricated a Ru–Ni3N/NiO catalyst with an optimal metal–support interaction, where Ru nanoparticles were uniformly dispersed on a 2D Ni3N/NiO heterostructured nanosheet array, and it exhibited significantly higher activity and stability for the HER and OER. Theoretical calculations suggested that the structural stability, orbital coupling, and electronic distributions of Ru–Ni3N/NiO could be enhanced by the Ni3N/NiO heterostructures when compared to those of Ru–Ni3N and Ru–NiO, leading to lower energy barriers and increased activity for the HER and OER. Ru–Ni3N/NiO demonstrates enhanced electrochemical performance, exhibiting lower charge transfer resistance (Rct: 1.52 Ω cm−2) and superior hydrogen evolution reaction (HER) activity compared to both Ru–Ni3N and Ru–NiO individually. In 1 M KOH, this catalyst achieves an impressive overpotential (η) of 385 mV at a substantial current density of 1000 mA cm−2 for the oxygen evolution reaction (OER), highlighting its remarkable activity. Similarly, for the HER under the same conditions, it reaches an overpotential of −190 mV at a current density of −1000 mA cm−2, further underscoring its exceptional performance. When applied to overall water splitting in 1 M KOH, Ru–Ni3N/NiO maintains a potential of 1.74 V at a current density of 1000 mA cm−2, reflecting its high activity. Moreover, it shows outstanding stability with negligible decay over 1000 h at a current density of 500 mA cm−2, demonstrating its robustness for long-term applications.
The activation of Ni(OH)2 followed by electrolysis in 1 M KOH with the addition of Fe(III) could enable the formation of highly active species for the OER, which could enhance the kinetics for the OER in 1 M KOH with Fe(III) compared to that in 1 M KOH. Huang et al.16 observed that Ni(OH)2 exhibited a lower charge transfer resistance (Fig. 2(d) and (e)) and higher activity (Fig. 2(b)) for the OER in 1 M KOH with 500 μM of Fe(III) than that in 1 M KOH. The Ni(OH)2 nanosheet arrays were obtained on Ni foam through a one-step ultrasonic process, where Ni foam was sonicated for 30 min in 5 wt% HCl, while Ni(OH)2 was activated in 1 M KOH or 1 M KOH with 500 μM of Fe(III). Moreover, Fig. 2(a) depicts a schematic diagram of the synthesis process for the Ni(OH)2 nanosheet arrays and subsequent structural evolution during the surface activation process by a chronopotentiometry test in 1 M KOH in the presence of 500 μM Fe(III) at different time intervals. The morphology, structure, and in/ex situ electrochemical investigations suggest that Fe(III) acts as the active site and enhances the kinetics for the OER in the following two ways (Fig. 2(c)): (i) the formation of several intermediates in a cyclic process on the electrode/electrolyte interface such as Fe(III), (Ni)Fex+, (Ni)Fe–OH, (Ni)Fe–O, (Ni)Fe–OOH, followed by Fe(III) (+O2); (ii) growth of the (oxy)hydroxide interface (Ni–FeOOH/Fe–NiOOH) on the surface of Ni(OH)2. In 1 M KOH, Ni(OH)2 exhibits an impressive overpotential (η) of 616 mV at a current density of 1000 mA cm−2, indicating its outstanding activity for the oxygen evolution reaction (OER). When supplemented with 500 μM of Fe(III), the performance of Ni(OH)2 is further improved, achieving an overpotential of 367 mV at the same current density, which not only highlights its enhanced efficiency but also its high stability under these conditions.
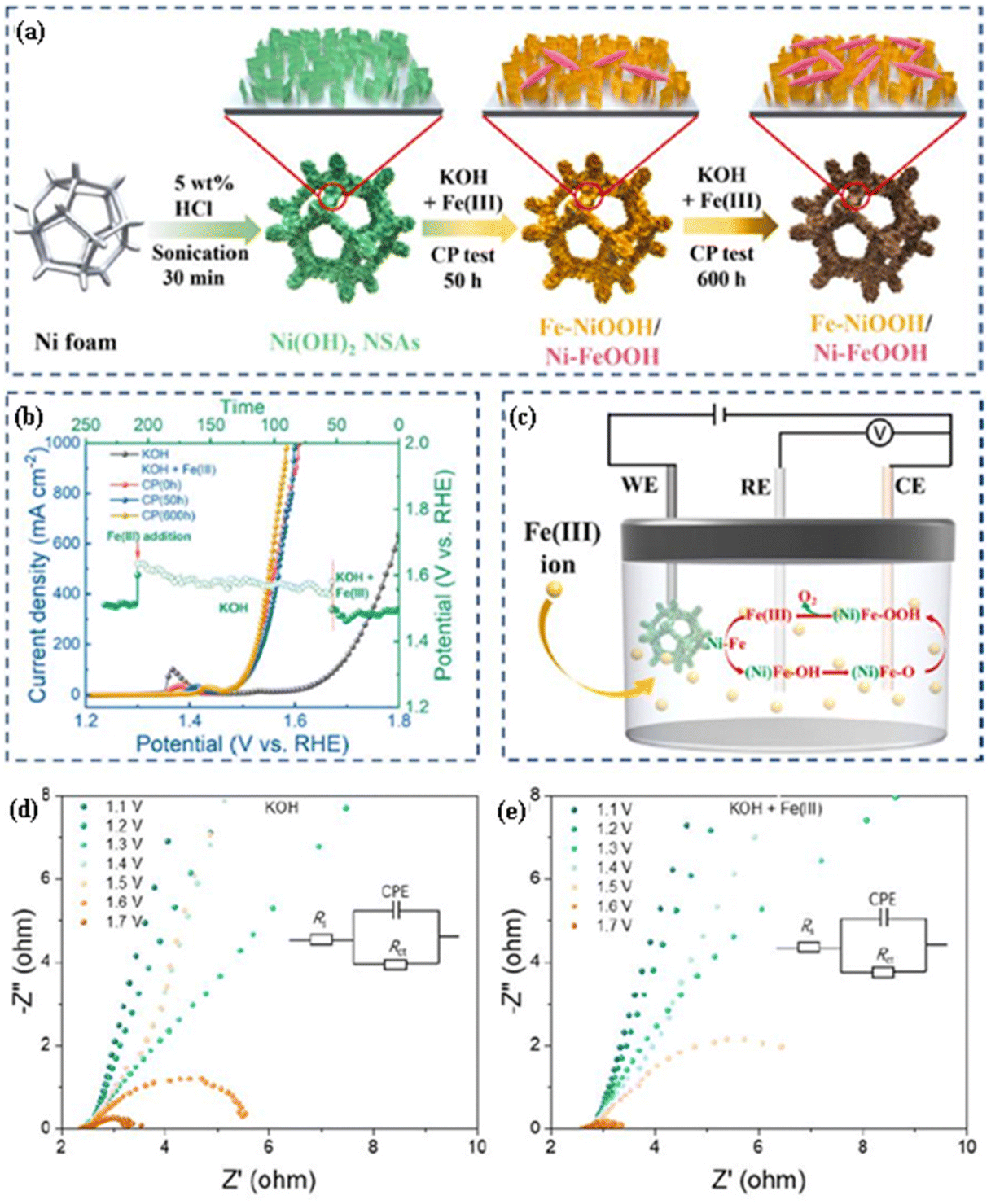 | ||
| Fig. 2 (a) Schematic diagram representing the synthesis process for Ni(OH)2 nanosheet arrays and their subsequent structural evolution during a chronopotentiometry test in 1 M KOH in the presence of 500 μM Fe(III) at different time intervals. (b) The LSV and chronopotentiometry plot for the OER of Ni(OH)2 in KOH with Fe(III) in comparison with those of Ni(OH)2 in KOH. (c) Proposed mechanism for the OER on Ni(OH)2 in alkaline electrolyte with Fe(III); CE, RE, and WE are the counter electrode, reference electrode, and working electrode, respectively. Nyquist plot for the OER of (d) Ni(OH)2 in KOH with Fe(III) in comparison with that of (e) Ni(OH)2 in KOH (reproduced with permission from ref. 16. Copyright 2023, Elsevier B.V.). | ||
The loading of Ce single atoms on NiV LDH (LDH: layered double hydroxide) can tune the band gap from semiconductor to metallic behavior, to afford an optimal adsorption energy with intermediates, and this could enhance the performance for the OER. Zeng et al.58 observed that Ce/NiV LDH exhibited a lower charge transfer resistance (Rct: 0.6 Ω), higher electrochemically active surface area, and higher activity than that of NiV LDH for the OER in 1 M KOH. The Ce/NiV LDH is composed of Ce single atoms, which are anchored on monolayer nickel–vanadium layered double hydroxide. Theoretical calculations and in situ electrochemical impedance spectroscopy investigations reveal that the immobilization of monatomic Ce on NiV LDH narrows the band gap, transforming its semiconducting properties into metallic behavior. This transformation strengthens the density of states near the Fermi level, providing an optimal adsorption energy with intermediates and reducing the charge transfer barrier. Consequently, this enhances the performance for the OER.
2.2. Sulfide/selenide based nano-electrocatalysts
The doping of Ni3S2 with Co could enhance the conductivity and optimize the adsorption energy with intermediates, thereby improving the performance for the OER. Additionally, this doping can reduce the cell voltage for hydrogen production by substituting the OER with sulfion oxidation or urea oxidation, further enhancing the overall efficiency of the system. Li et al.59 reported that Co-doped Ni3S2 demonstrated superior activity and reduced charge transfer resistance (Rct: 33.12 W or W cm−2) for the OER compared to pure Ni3S2. This enhanced performance is achieved through the preparation of Co–Ni3S2 on Ni mesh, involving an electrodeposition step followed by a hydrothermal-sulfurization reaction. Furthermore, the utilization of a sulfion-containing electrolyte has enabled a significant reduction in the cell voltage to 0.80 V at 50 mA cm−2, which is about 1.20 and 0.94 V lower than that of water splitting and urea electrolysis, respectively. The enhanced sulfion oxidation is attributed to direct electron transfer to the surface active sites, while the improved urea oxidation is ascribed to the formation of a suitable oxidation state of the NiIII on the surface, which facilitates a shortened oxidation pathway.The construction of bimetallic sulfide nanoarray surfaces has been shown to promote gas evolution, improve conductivity, and subsequently boost the HER and OER performance. Findings from He et al.60 showed that the FeNiS nanoarray exhibited higher activity and lower charge transfer resistance (Rct: 17.1 Ω cm−2) for the OER than those of NiS, while it exhibited high activity and stability for overall water electrolysis in 1 M KOH. The FeNiS nanoarray is composed of FeS nanosheets, which are anchored on Ni9S8 nanorods. Their phase-field simulations revealed that the flow field of the arrayed electrode surface focused the shear force directly onto the evolved bubbles, leading to enhancement in gas evolution, which could reduce the shielding effect by passage of flow through channels and voids. Moreover, the flow field exerts a more significant influence on the arrayed surface compared to a nanoscale smooth surface, effectively reducing the size of the evolving bubbles.
Integration of a transition metal phosphide with sulfide could enhance the conductivity, to afford an optimal adsorption energy with intermediates, and this could enhance the performance for the HER and OER. It was demonstrated by Dhakal et al.46 that 1T-Mn–VS2@Co2P exhibited higher activity and lower charge transfer resistance (Rct: 2 Ω) for the HER than those of 1T-VS2@Co2P, while it exhibited remarkable activity and stability for overall water electrolysis in 1 M KOH. It is composed of a HER-active hierarchical 2D–2D tangled 3D heterostructure of Mn-doped 1T VS2, which is assembled with OER-active cobalt phosphide.
Incorporation of transition metal multi-cation species into nanostructured selenides could modify the electronic structure and enhance the conductivity, which could boost the performance for the HER and OER. Ibraheem et al.14 observed that the Fe@Co/Se2 nanorods exhibited higher activity and lower charge transfer resistance (for the HER, Rct: 10.80 Ω (Fig. 3j); for the OER, Rct: 15.04 Ω (Fig. 3g)) than those of Fe–Se2 and Co–Se2 for the HER and OER in 1 M KOH. Fe@Co/Se2 contains FeSe2 and CoSe2 phases (Fig. 3(c)), while the HRTEM image (Fig. 3(a)) and the corresponding SAED pattern (Fig. 3(b)) depict inter-planar distances of about 2.6 Å, 1.40 Å, 1.86 Å, and 3.07 Å consistent with the (1 1 1), (3 1 1), (2 2 0) and (0 1 1) lattice planes of the FeSe2 and CoSe2 phases. It contains Fe, Co, and Se, which are homogenously distributed (Fig. 3(d)). By means of a Fe-coordinated Co-bridged bond, the Fe2+/3+/Co2+ species was highly coupled with selenide, suggesting a modified electronic structure.
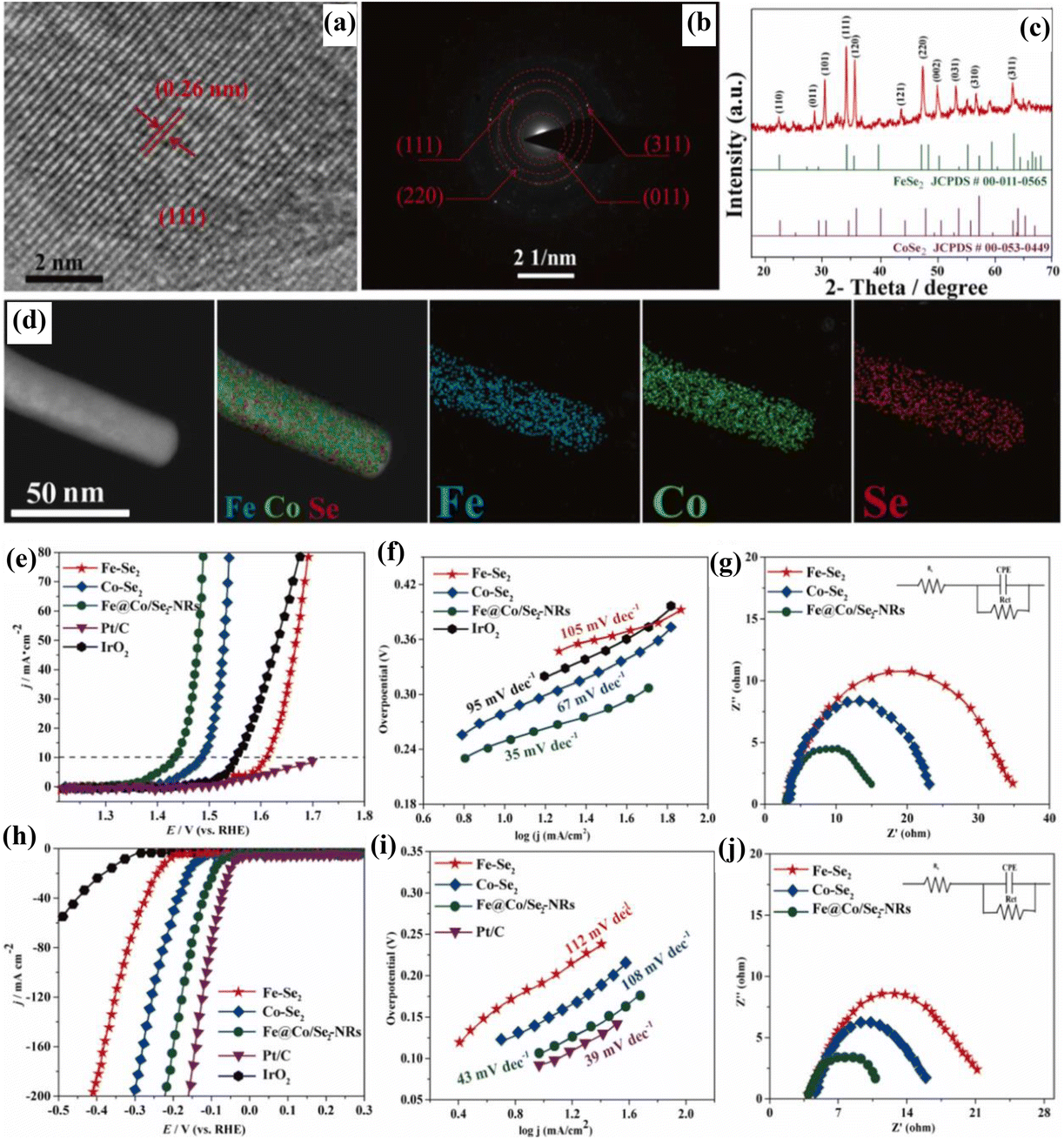 | ||
| Fig. 3 (a) HRTEM image, (b) the corresponding SAED pattern, (c) XRD pattern, and (d) HAADF-STEM image (with elemental mapping images of Fe, Co and Se) of Fe@Co/Se2; (e) LSV curve, (f) Tafel slope, and (g) Nyquist plot for the OER of Fe@Co/Se2 in comparison with those of Fe–Se2 and Co–Se2 in 1 M KOH; (h) LSV curve, (i) Tafel slope, and (j) Nyquist plot for the HER of Fe@Co/Se2 in comparison with those of Fe–Se2 and Co–Se2 in 1 M KOH (reproduced with permission from ref. 14. Copyright 2021, Elsevier B.V.). | ||
Creating nanostructured heterojunctions from transition metal selenides can alter their electronic structures and improve conductivity, thereby enhancing their efficiency in both the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER). Meshesha et al.48 observed that NiCoSe2/MoSe2 exhibited higher activity and lower charge transfer resistance (Rct: 13.1 Ω) than those of NiCoSe2 and MoSe2 for the HER in 1 M KOH, while it exhibited high activity and stability for overall water electrolysis. The enhanced performance is attributed to the formation of a heterojunction of nickel–cobalt selenide with metal phase MoSe2 nanospheres.
Wang et al.43 observed that ultrathin P/Fe–NiSe2 nanosheets exhibited higher activity and lower charge transfer resistance (Rct: 4.78 Ω) than those of Fe–NiSe2 for the HER in 1 M KOH, while they exhibited high efficiency for hydrazine-assisted water splitting and a Zn-hydrazine battery, achieving a remarkable current density of 100 mA cm−2 at potentials of −168 and 200 mV for the HER and HzOR, respectively. P/Fe–NiSe2 has a much lower apparent activation energy value (38.9 kJ mol−1) than those of Fe–NiSe2 (68.3 kJ mol−1) and NiSe2 (74.9 kJ mol−1), indicating that the introduction of P atoms can significantly reduce the kinetic barrier and facilitate the HER process. This superior performance is credited to the ultrathin nanosheet structure and the synergistic effect of P/Fe co-doping in NiSe2, which collectively enhance the electronic structure, conductivity, and interactions with intermediates.
2.3. Phosphide/nitride based electrocatalysts
The incorporation of high-valence Zr into nickel phosphide can optimize the adsorption energy with intermediates and enhance conductivity, improving the performance for the HER and OER. Fan et al.42 observed that Zr0.1Ni1.9P exhibited higher activity and lower charge transfer resistance (Rct: 5 Ω) than those of Ni2P for the HER in 1 M KOH, while it exhibited high activity and stability for overall water electrolysis. Their DFT calculations reveal that the incorporation of high-valence Zr can reduce the free energy change for the potential-determining step (O* → OOH*) to enhance the alkaline OER, and it can improve the dissociation and adsorption processes of H2O to facilitate the HER process.The synergistic effect of bimetallic phosphide could enhance the conductivity and provide an optimal adsorption energy with intermediates, which could enhance the performance for the HER. Xiang et al.47 observed that CoP3–Nb2P/PCC exhibited a significantly higher activity (η of −375 mV at a huge current density of −1000 mA cm−2) and lower charge transfer resistance (Rct: 3.5 Ω) than those of CoO–NbO/PCC for the HER in 1 M KOH (PCC: plasma-defect-engineered carbon cloth). Their in situ Raman spectroscopy and DFT calculations reveal that the outstanding performance for the HER is attributed to the synergistic interaction between Co and Nb phosphide. Furthermore, the Nb–Nb vibration weakens the P–Hads bond, ultimately leading to efficient H2 production.
Ir single atom incorporation into and Zr doping of cobalt phosphide could modify the electronic structure, improve the conductivity, optimize the adsorption energy with intermediates, and enhance the performance for the HER and OER. Ngo et al.61 observed that Ir@Zr–CoP exhibited higher activity and lower charge transfer resistance (Rct: 1.97 Ω) than those of Zr–CoP for the OER in 1 M KOH, while it exhibited a cell voltage of 1.88 V at a huge current density of 1000 mA cm−2 for a large, scalable, anion exchange membrane water electrolyzer with high stability. It was obtained by hydrothermal treatment of carbon cloth followed by wet impregnation and phosphidation (Fig. 4(a)).
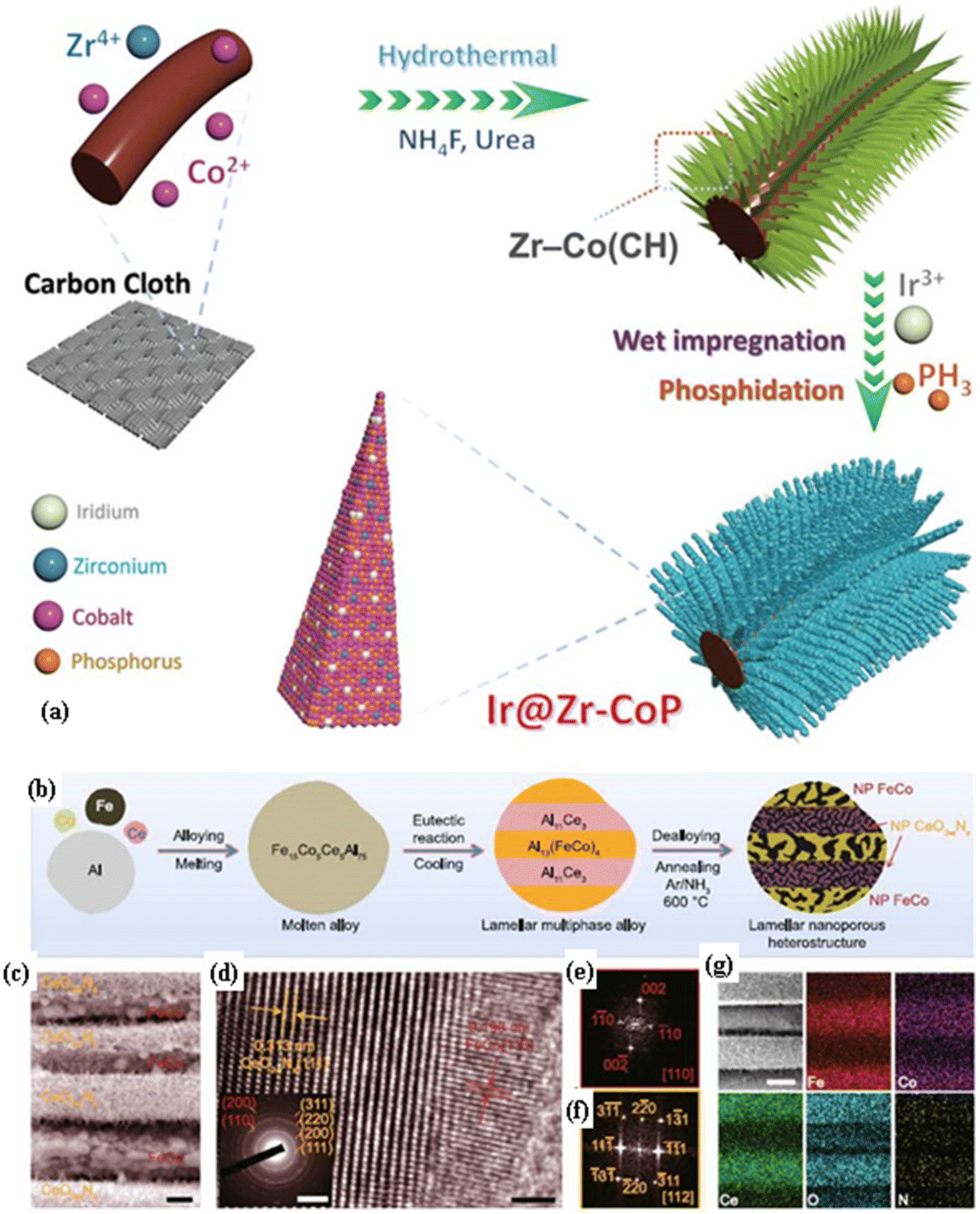 | ||
| Fig. 4 (a) Schematic diagram illustrating the synthesis process for Ir@Zr–CoP. (b) Schematic diagram with microstructures depicting the synthesis process for the lamellar nanoporous FeCo/CeO2−xNx composite electrode. (c) SEM image of the FeCo/CeO2−xNx composite (scale bar: 200 nm), where the hierarchical nanoporous FeCo alloy and nanoporous CeO2−xNx lamellas alternate and are periodically arranged. (d) HRTEM image of FeCo/CeO2−xNx depicts the heterostructured interface with a scale bar of 1 nm, while the inset shows the corresponding SAED patterns with a scale bar of 5 nm−1. FFT patterns of (e) FeCo alloy and (f) CeO2−xNx obtained from the selected areas in the FeCo/CeO2−xNx heterostructure in (d). (g) SEM image with the corresponding EDS elemental mapping images of FeCo/CeO2−xNx for N, O, Ce, Fe, and Co elements with a scale bar of 200 nm (reproduced with permission from ref. 21. Copyright 2023, Springer Nature). | ||
Furthermore, creating nanoporous bimetallic alloy/OOH and metal oxynitride heterolamellas enhances the OER performance by exposing numerous active sites, improving conductivity, optimizing the adsorption energy with intermediates, and facilitating gas evolution. Zeng et al.21 synthesized FeCo/CeO2−xNx through a method involving alloying and thermal nitridation. FeCo/CeO2−xNx exhibits a significantly higher activity (η of 360 mV at a huge current density of 3940 mA cm−2) and lower charge transfer resistance (Rct: 1.3 Ω; RP: 1.6 Ω) than those of FeCo/Ce–O for OER in 1 M KOH, while it delivers ∼1900 mA cm−2 for about 1000 h, suggesting its robust stability.
The insulating hexagonal boron nitride with an optimal thickness (one-atom-thick) can be used as a co-catalyst to enhance the activity for the OER. Lu et al.62 reported that hexagonal BN-NiFeOxHy exhibited a significantly higher activity (η of 230 mV at 10 mA cm−2; η of 490 mV at a huge current density of 2000 mA cm−2) for the OER in 1 M KOH compared to NiFeOxHy, and it afforded a low charge transfer resistance for the OER. The high activity of hexagonal BN-NiFeOxHy is attributed to the covering of NiFeOxHy with one-atom-thick hexagonal boron nitride as a co-catalyst, where the oxygen-containing intermediates can adsorb insulating boron nitride, and localized electrons can enhance the deprotonation processes at the electrode. However, the catalytic activity of multilayer hexagonal boron nitride is significantly reduced due to its insulating properties.
2.4. Carbon/alloy/MOF/organic based nano-electrocatalysts
Fabricating pyrrolic-N-doped graphitized porous carbon loaded with Ru nanoparticles could enhance the conductivity and provide an optimal adsorption energy with intermediates, and boost the performance for the HER. Shin et al.63 observed that Ru–N-GPC (pyrrolic-N-doped graphitized porous carbon loaded with Ru nanoparticles) exhibited a much higher activity (η of −9.6 mV at −10 mA cm−2) and lower charge transfer resistance than those of Ru–N-rGO (rGO: reduced graphene oxide) for the HER in 1 M KOH; it was obtained by pyrolyzing g-C3N4 in the presence of Mg metal. The improvement in the Tafel step can be attributed to the strengthened coupling between the Ru nanoparticles and the pyrrolic-N moieties present in the support, which promote an optimal adsorption energy with intermediates on the Ru nanoparticles.Cu doping can amplify the Kirkendall effect, and the fabrication of double-shell hollow Cu-doped CoP nanoparticles anchored to carbon arrays can optimize the adsorption energy with intermediates and improve the conductivity, thereby enhancing the HER and OER performance. Wang et al.44 found that DH–CuCo–P@NC/CC outperformed H–CuCo–P@NC/CC in terms of the HER activity and charge transfer resistance (Rct: 2.4 Ω) in 1 M KOH; it also showed notable activity for water electrolysis. This improvement is attributed to the structure of DH–CuCo–P@NC/CC, featuring double-shell hollow Cu-doped CoP nanoparticles supported on carbon arrays, which intensify the Kirkendall effect and optimize the adsorption energy and conductivity for better HER and OER outcomes. Furthermore, integrating high-polarity F, CoFe alloy, and N-doped carbon nanotubes enhances the conductivity and adsorption with intermediates, boosting the OER performance. Kuang et al.64 demonstrated that CoFe@NCNTs–F exhibited superior activity and lower charge transfer resistance (Rct: 31 Ω) compared to CoFe@NCNTs in 1 M KOH. The enhanced properties are due to the CoFe alloy/fluoride composition interconnected by bamboo-like N-doped carbon nanotubes, which improve the conductivity and adsorption, thus elevating the OER efficiency. Constructing Mn-doped bimetallic hydroxide nanostructures intertwined with carbon nanotubes modifies the electronic structure and optimizes the adsorption energy, leading to increased conductivity and improved OER performance. Wu et al.65 observed that 1.5Mn–NiCo HNS/CNT showed higher activity and lower charge transfer resistance than both NiCo HNS/CNT and 1.5Mn–NiCo HNS in 1 M KOH. The composition of Mn-doped NiCo hydroxide nanosheets intertwined with carbon nanotubes enhances the electronic structure and adsorption energy, significantly improving the OER efficiency. The fabrication of nanostructured transition bimetallic alloy/OOH heterostructures can offer complex synergistic effects, rapid mass and charge transfer, and abundant exposed active sites, leading to superior performance for the HER and OER. Lv et al.45 observed that cNiFe/aNiFeOOH@NiMo (a: amorphous; c: crystalline) exhibited higher activity and lower charge transfer resistance (for the HER, Rct: 0.946 Ω; for the OER, Rct: 0.962 Ω; Table 2) than those of cNiFe/aNiFeOOH and NiMo for the HER and OER in 1 M KOH. It was prepared by a two-step electrodeposition process. cNiFe/aNiFeOOH@NiMo is composed of ultrafine NiFe nanocrystals with numerous edges and exceptional electronic conductivity, amorphous NiFeOOH enriched with defects and nanosized pores, and NiMo alloy as a firm grasp for nanostructured cNiFe/aNiFeOOH, which could provide complex synergistic effects, fast mass and charge transfer, and expose/provide abundant active sites, leading to enhanced performance for the HER and OER.
| OER electrocatalysts | η at 10 mA cm−2 (mV) | R ct or Rct1 | R P or Rct2 | Stability | Ref. |
|---|---|---|---|---|---|
| OER: oxygen evolution reaction; η: overpotential; Rct or Rct1 or Rct2: charge transfer resistance; RP: pore resistance; NA: not applicable. | |||||
| cNiFe/aNiFeOOH@NiMo | 133.2 at 20 mA cm−2 | 0.962 Ω | NA | 100 h (100 mA cm−2) | 45 |
| Ru–Ni3N/NiO | 181 | NA | NA | 1000 h (500 mA cm−2) | 40 |
| FeNiS | 191 | 17.1 Ω cm−2 | NA | 550 h (500 mA cm−2) | 60 |
| Fe@Co/Se2 | 200 | 15.04 Ω | NA | 20 h (10 mA cm−2) | 14 |
| Ce/NiV LDH | 209 | 0.6 Ω | NA | 20 h (50 mA cm−2) | 58 |
| Fe4N/Nb4N5 | 224 | 0.206 Ω | NA | NA | 39 |
| Hexagonal BN-NiFeOxHy | 230 | NA | NA | Current density varies less than 10% over 150 h (2000 mA cm−2) | 62 |
| CoFe@NCNTs–F | 231 | 31 Ω | NA | 20 h (10 mA cm−2) | 64 |
| FeCoCrCuOx | 255 | NA | NA | 165 h (500 mA cm−2) | 41 |
| Ni(OH)2Fe(III) | 269 at 100 mA cm−2 | NA | NA | 600 h (1000 mA cm−2) | 16 |
| Ir@Zr–CoP | 292 | 1.97 Ω | NA | 50 h (100 mA cm−2) | 61 |
| FeCo/CeO2−xNx | 297 at 1000 mA cm−2 | 3.3 Ω | 1.6 Ω | 1000 h (1900 mA cm−2) | 21 |
| Cu–RuO2activated | 316 at 100 mA cm−2 | NA | NA | NA | 4 |
| (Cr1/5Mn1/5Fe1/5Co1/5Ni1/5)3O4 | 360 | NA | NA | 3 h (40 mA cm−2) | 57 |
| Co–Ni3S2 1 M NaOH | 440 | 33.12 Ω or Ω cm−2 | NA | 24 h (50 mA cm−2) | 59 |
Cr doping of FeMOF with Ru nanoclusters could manipulate the d orbitals of the Ru sites and enhance the conductivity, which could enhance the performance for the HER and OER. Zhao et al.66 observed that Ru@Cr-FeMOF (MOF: metal–organic frameworks) exhibited higher activity and lower charge transfer resistance than those of Ru@FeMOF for the HER and OER in 1 M KOH, while it exhibited a cell voltage of 1.72 V at a huge current density of 1000 mA cm−2 for overall water electrolysis, suggesting its excellent activity. Ru@Cr-FeMOF is composed of Ru nanoclusters anchored onto Cr-doped Fe-metal–organic frameworks. This unique structure enhances electron transfer and enables the manipulation of the highest occupied d orbitals and d-band centers of the Ru sites, thereby boosting the catalytic activity for both the HER and OER.
An organic coating on Ni foam can significantly improve the conductivity and optimize the adsorption energy with intermediates, thereby boosting the hydrogen evolution reaction (HER) activity. Das et al.67 found that Ni foam coated with a dual-layer of N,N-dimethylaminopropylamine (DMAPA) and 2-(dimethylamine)ethyl acrylate (DMAEA) outperformed the HER performance of bare Ni foam in 1 M KOH, exhibiting a superior current density of 1270 mA cm−2 (at −0.9 V), an overpotential value of −250 mV (at 100 mA cm−2) and reduced charge transfer resistance. This enhancement is attributed to the conductivity and adsorption properties of the DMAPA and DMAEA coating, which provides a non-adhesive superaerophobicity with a nucleated bubble adhesion force of 4.6 ± 0.3 μN, which is favorable for the HER.
3. Water electrolysis in an acidic environment
3.1. Oxide based nano-electrocatalysts
The fabrication of strained Ir incorporated into MnO2 could modify the electronic structure, facilitate the conductivity, and provide an optimal adsorption energy with intermediates, which could enhance the performance for the OER. Zhao et al.1 successfully prepared strained-Mn0.98Ir0.02O2 through a hydrothermal method and observed that it exhibited higher activity (η of 105 mV at 10 mA cm−2 (Fig. 5(a)); mass activity: 300 mV at 5681 A g−1) and lower charge transfer resistance (Fig. 5(c)) than those of unstrained-Mn0.98Ir0.02O2 for the OER in 0.5 M H2SO4, while it exhibited high stability for 168 h. The introduction of Ir into MnO2 induces strain, resulting in an elongated Mn–O bond length and a shift of the d-band centre closer to the Fermi level. This modified electronic structure facilitates the transfer of holes from the O 2p orbitals to the catalyst surface, accelerating charge transfer and enhancing the catalytic activity for the OER. | ||
| Fig. 5 (a) LSV curves (inset plot depicts LSV curves normalized by ECSA), (b) corresponding Tafel slopes, and (c) Nyquist plots of sl-Mn0.98Ir0.02O2 and its counterparts in 0.5 M H2SO4 (reproduced with permission from ref. 1. Copyright 2023, Wiley-VCH GmbH). (d) Schematic diagram depicting the synthesis process for Fe2−xRuxNb2O6/carbon cloth. Chemical structures of (e) Fe2Nb2O6, (f) Fe2−xRuxNb2O6, and (g) Fe2−xRuxNb2O6/carbon cloth depicting the site occupancies in columbite (reproduced with permission from ref. 26. Copyright 2023, Wiley-VCH GmbH). | ||
Similarly, doping Fe2Nb2O6 with Ru, as detailed by Bacirhonde et al.,26 alters its electronic structure to enhance both the hydrogen evolution reaction (HER) and OER performances. The product Fe2−xRuxNb2O6 (FRNO) demonstrates superior activity (with an overpotential of −30 mV at −10 mA cm−2 for the HER and 200 mV at 10 mA cm−2 for the OER) compared to its undoped counterparts in 0.5 M H2SO4, alongside lower charge transfer resistances, as shown in Tables 3 and 4. This enhancement is credited to a low-temperature hydrogel crosslinking and hydrothermal encapsulation process, which not only boosts intrinsic conductivity but also optimizes the adsorption energy and facilitates water dissociation on its surface through effective charge transfer. DFT calculations and operando X-ray adsorption studies reveal that Fe2−xRuxNb2O6, with its high intrinsic conductivity, enhances the adsorption and dissociation of H2O on its surface. This is achieved by modulating the charge distribution, which facilitates charge transfer to the coordinated surface oxygen. This can stabilize the adsorption of the *OOH intermediate on Ru and Fe sites, and further enhances the activity for the OER.
| HER electrocatalysts | η at −10 mA cm−2 (mV) | R ct | Stability | Ref. |
|---|---|---|---|---|
| HER: hydrogen evolution reaction; η: overpotential; Rct: charge transfer resistance; NA: not applicable; DETA = diethylenetriamine; HCDs: hollow carbon dodecahedrons. | ||||
| Pd2RuOx | −12 | NA | 20 h (10 mA cm−2) | 74 |
| RuP2–C@RuP2–C | −17 | 9.10 Ω | 20 h (10 mA cm−2) | 77 |
| Ru/Co–N–C | −17 | NA | 20 h (100 mA cm−2) | 78 |
| PtCo@PtSn | −21 | NA | 100 h (80 mA cm−2) | 79 |
| Ru-30 | −24 | NA | 10 h (10 mA cm−2) | 76 |
| IrIn2/C | −25 | NA | NA | 80 |
| IrCo@CNT | −26 | 3.78 Ω | 90 h (10 mA cm−2) | 81 |
| Mo8O26–NbNxOy/NG | −27 | 3.79 Ω | 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s 000 s |
73 |
| Fe2−xRuxNb2O6 | −30 | 2.72 Ω | 80 h (10 mA cm−2) | 26 |
| Co@CNTs|Ru | −32 | 51.46 Ω | NA | 82 |
| Ni2P–Ni12P5@Ni3S2 | −46 | NA | 24 h (10 mA cm−2) | 83 |
| Ru–g-CN | −54.5 | NA | 120 h (10 mA cm−2) | 84 |
| Ru@MoO(S)3 | −63 | NA | 24 h | 69 |
| Mn2P–MnP/PNC | −63 | NA | 96 h | 85 |
| NCAG/Ru-3 | −65 | NA | 30 h (10 mA cm−2) | 86 |
| La–RuO2 | −71 | NA | 50 h (50 mA cm−2) | 70 |
| Pb–CoSe2–DETA | −74 | 4.8 Ω | 20 h (10 mA cm−2) | 87 |
| Ni2P/Ni5P4 | −78 | NA | 100 h (50 mA cm−2) | 3 |
| N-IrP2 | −80 mV at −100 mA cm−2 | NA | 10 h (60 mA cm−2) | 28 |
| WC–W2C–HCDs | −96 | 20.1 Ω | 150 h (10 mA cm−2) | 88 |
| Co2P/CoP@Co@NCNT | −136 | 10.3 Ω | 12 h (10 mA cm−2) | 51 |
| Holey-VO2 (R) | −184 | NA | NA | 89 |
| V–Ti4N3Tx | −330 | 70 Ω | NA | 90 |
| OER electrocatalysts | η at 10 mA cm−2 (mV) | R ct | Stability | Ref. |
|---|---|---|---|---|
| OER: oxygen evolution reaction; η: overpotential; Rct: charge transfer resistance; NA: not applicable. | ||||
| Strained-Mn0.98Ir0.02O2 | 105 | NA | 48 h (10 mA cm−2) | 1 |
| N-IrP2 | 160 | NA | 50 h (100 mA cm−2) | 28 |
| Ru-30 | 181 | NA | 10 h (10 mA cm−2) | 76 |
| GB-Ta0.1Tm0.1Ir0.8O2−δ | 198 | NA | 500 h (10 mA cm−2) | 91 |
| Fe2−xRuxNb2O6 | 200 | 4 Ω | 24 h (200 mA cm−2) | 26 |
| La–RuO2 | 208 | NA | 50 h (10 mA cm−2) | 70 |
| Ru@MoO(S)3 | 226 | NA | 24 h | 69 |
| Ru/Co–N–C | 232 | NA | 20 h (10 mA cm−2) | 78 |
| IrCo@CNT | 241 | NA | 90 h (10 mA cm−2) | 81 |
| IrIn2/C | 254 | NA | 55 h (10 mA cm−2) | 80 |
| CeO2–Co2NiP0.03Ox | 262 | 7.7 Ω | 30 h (5 mA cm−2) | 75 |
| Holey-VO2 (R) | 282 | NA | NA | 89 |
Developing acid stable OER electrocatalysts for high performance water splitting is of great significance but still remains challenging. To this end, a Ru/MnO2 electrocatalyst where patches of Ru atom arrays were supported on α-MnO2 was prepared by Lin et al.68 The catalyst follows an oxygen evolution mechanism involving only *O and *OH species as intermediates, which enables direct O–O radical coupling for O2 evolution. This results in high activity (161 mV at 10 mA cm−2) and outstanding stability, with minimal degradation after 200 hours of operation, making Ru/MnO2 one of the best-performing acid-stable OER catalysts. Operando vibrational and mass spectroscopy measurements validate the reaction intermediates and gaseous products, confirming the OER pathway. First-principles calculations revealed a cooperative catalysis mechanism with a reduced energy barrier. Additionally, time-dependent elemental analysis demonstrated an in situ dynamic cation exchange reaction during the OER, which triggered the reconstruction of Ru atoms into a highly durable ordered array.
Besides, research by Chen et al.69 indicates that Ru@MoO(S)3 exhibits higher activity and lower charge transfer resistance than those of MoO(S)3 in acidic medium for the HER and OER. In this structure, Ru nanoparticles are harmoniously encapsulated within a sulfur-modulated molybdenum oxide (MoO(S)3) shell. This all-in-one integration enables Ru to maintain its intrinsic catalytic activity and rapid mass transfer capabilities while preventing electrochemical oxidation, thereby ensuring recyclability. Notably, Ru@MoO(S)3 facilitates water splitting in 0.5 M H2SO4 and 1 M KOH at a current density of 10 mA cm−2, requiring cell voltages of only 1.522 V and 1.526 V, respectively, with nearly 100% faradaic efficiency. Moreover, Ru@MoO(S)3 also demonstrates satisfactory stability during a 24 h continuous water splitting test. In another study, Wu et al.70 synthesized La-doped RuO2 (La–RuO2) nanocrystals with oxygen vacancies. La–RuO2 exhibits a remarkable electrocatalytic performance, with ultra-low overpotentials of 208 mV for the OER and 71 mV for the HER at a current density of 10 mA cm−2, and demonstrates robust long-term stability in 0.5 M H2SO4. Furthermore, the La–RuO2 nanocrystals require only 1.53 V to drive overall water splitting, maintaining exceptional stability. DFT calculations reveal that the synergistic effects of La doping and oxygen vacancy engineering tune the d-band center of Ru active sites and optimize the Gibbs free energy of intermediate states, enhancing the catalytic activity of RuO2.
In a recent study, Song et al.71 utilized a lattice-matching strategy to construct a RuO2–CeO2 electrocatalyst for the OER. The interfacial Ru–O–Ce bridge structure facilitates electron transfer between Ru and Ce, creating lattice stress that distorts the local structure of RuO2. This RuO2–CeO2 catalyst demonstrated exceptional stability with negligible decay after 1000 hours of the OER in 0.5 M H2SO4, and high activity with an overpotential of only 180 mV at 10 mA cm−2. In situ attenuated total reflectance surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS), in situ differential electrochemical mass spectrometry (DEMS), and density functional theory (DFT) calculations were employed to reveal that the interface and noninterface RuO2 sites enabled an oxide path mechanism (OPM) and an enhanced adsorbate evolution mechanism (AEM-plus), respectively, during the OER. This dual-pathway approach, guided by lattice matching, enhances electrocatalyst design for the OER in acidic media.
It is claimed that current reliance on iridium-based OER catalysts is unsustainable due to their high cost and limited availability. Recently, Wu et al.72 fabricated a nickel-stabilized ruthenium dioxide (Ni–RuO2) catalyst as a promising alternative, demonstrating significant improvements in both activity and durability for the acidic OER in PEM–WE applications. The Ni–RuO2 catalyst showed exceptional stability, maintaining its performance for over 1000 hours at a water-splitting current of 200 mA cm−2. In contrast, pristine RuO2 degraded rapidly under similar conditions. The incorporation of nickel into the RuO2 lattice significantly stabilized both surface Ru and subsurface oxygen, extending the catalyst's durability by more than an order of magnitude. The improved stability was confirmed by density functional theory (DFT) studies and operando differential electrochemical mass spectroscopy (DEMS), which highlighted the critical role of Ni dopants in enhancing the lattice stability and supporting the adsorbate-evolving mechanism (AEM) on Ni–RuO2. It is revealed by DFT calculations that the Ni dopant prefers to replace the coordination bridge Ru site of the RuO2(110) surface. Moreover, the limiting potential for the OER following an AEM is 1.87 eV lower than that of the lattice oxygen mechanism (LOM).
Moreover, Yang et al.73 observed that Mo8O26–NbNxOy/NG (G: graphene) afforded an η of −27 mV at −10 mA cm−2 for the HER in 0.5 M H2SO4, suggesting both high activity and low charge transfer resistance (3.79 Ω). It is composed of heterogeneous Mo8O26–NbNxOy, which is supported on N-doped graphene. It is prepared by hydrothermal treatment followed by a nitridation process. In Mo8O26–NbNxOy/NG, the O-exposed Mo8O26 clusters are covalently bound to NbNxOy nanodomains, leading to a unique interface configuration with an optimal electronic structure, and the totally exposed multiple active sites provide superior HER activity in acidic medium, which is almost the same as that of a commercial Pt/C catalyst at −75 mA cm−2. DFT calculations reveal that facilitated initial H2O adsorption and near-optimal hydrogen adsorption energy are achieved due to the Mo8O26–NbNxOy interface with a modified electronic structure. The terminal O atoms of the Mo8O26 clusters interact with the Nb atoms to enhance the initial H2O adsorption, and then decrease the H2O dissociation energy, facilitating the entire HER kinetics. In addition, Do et al.74 observed that Pd2RuOx afforded an η of −12 mV at −10 mA cm−2 for the HER in 0.5 M H2SO4, showing satisfactory activity and durability (negligible decay after 10000 cycles of CV at −140 mA cm−2), as well as a low charge transfer resistance. The Pd2RuOx material is comprised of atomically thin Pd–PdO nanodomains that are encapsulated within amorphous Ru metallene oxide (RuO2). This multi-component heterostructure, along with the atomically thin nanosheet morphology and coexistence of defective crystalline and amorphous phases, collectively regulate the electronic structure, provide an optimal adsorption energy for the intermediates at the active sites, expose/provide abundant active sites, and significantly enhance the HER performance.
Additionally, Liu et al.75 discovered that the precious-metal-free CeO2–Co2NiP0.03Ox electrocatalyst delivered an exceptional OER performance, achieving an overpotential of 262 mV at 10 mA cm−2 in 0.5 M H2SO4, along with notable stability and a low charge transfer resistance of 7.7 Ω. This innovation, leveraging a CeO2 nanorod core with a Co–Ni–P oxide shell, benefits from the addition of trace P atoms and heterojunction interfaces that effectively adjust Co valence states to +2.51. This modification enhances the transformation of surface Co2+–O sites into more reactive Co3+–O sites, improving water molecule deprotonation and OER kinetics. The enhanced durability of CeO2–Co2NiP0.03Ox is further reinforced by an increased vacancy formation energy at the Co site, reducing its dissolution in acidic environments.
Similarly, the study by Yan et al.76 highlights Ru-30's outstanding OER and HER performance in 0.5 M H2SO4, achieving overpotentials of 181 mV and −24 mV at 10 mA cm−2, respectively. This performance is coupled with substantial stability over 10 h and sustained durability, demonstrating minimal degradation after extensive cyclic voltammetry (500 cycles for the OER and 5000 cycles for the HER). For overall water splitting, Ru-30 achieves a potential of 1.468 V at 10 mA cm−2, maintaining its effectiveness over 16 h. Ru-30, a partially oxidized 3D porous ruthenium aerogel, boasts a distinct Ru/RuO2 interface and a unique porous architecture. This unique combination facilitates rapid mass and electron transfer channels, offering an abundance of electrochemically active sites. Furthermore, it modifies the electronic structure and optimizes the adsorption energy with water splitting intermediates. Consequently, Ru-30 significantly enhances the performance for the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER) in acidic environments.
Najafi et al.89 reported that holey-VO2 (R) achieved an overpotential of 282 mV at 10 mA cm−2 for the OER in 0.5 M H2SO4, highlighting its exceptional activity, along with displaying reasonable stability over 150 h. For the HER, this material reaches an overpotential of −184 mV at −10 mA cm−2, underscoring its high activity and maintaining robust stability for 120 h. Moreover, it demonstrates a high mass activity of 972.3 A g−1 at −0.300 V versus RHE and features a low charge transfer resistance, showcasing its efficiency at electrochemical water splitting. As-prepared VO2 (R) is composed of rutile VO2 nanosheets, while it is a 2D metallic non-layered material and stable at room temperature. It is prepared under a reducing Ar–H2 atmosphere through topochemically transforming liquid-phase exfoliated VSe2, whereas the 2D holey-VO2 (R) nanosheets with a porous morphology were obtained through pretreating the VSe2 nanosheets using O2 plasma. Metallic holey-VO2 (R) with a high specific surface area and defective sites exhibits accelerated electron/mass transfer, enhanced conductivity, and results in good performance for the HER and OER. Moreover, Hao et al.91 observed that torsion-strained GB-Ta0.1Tm0.1Ir0.8O2−δ (GB: abundant grain boundaries) afforded an η of 198 mV at 10 mA cm−2 for the OER in 0.5 M H2SO4, showing a laudable activity, robust stability, and low charge transfer resistance. X-ray absorption spectroscopy, microstructural analyses, and DFT calculations reveal that abundant trigeminal grain boundaries are formed due to the doping induced ligand effect and torsion-strained Ir–O bonds, which could modulate the adsorption energy of the intermediates thereby improving the activity for the OER. A proton exchange membrane electrolyser delivers a remarkably high current density of 1.5 A cm−2 for 500 h at a low cell voltage of 1.766 V using GB-Ta0.1Tm0.1Ir0.8O2−δ as a catalyst for the OER at a low mass loading of 0.2 mg cm−2 with an estimated cost of US$1 per kg of hydrogen, which is much lower than the target of US$2 per kg per H2 (fixed by the US Department of Energy).
3.2. Phosphide/sulfide/selenide/nitride based nano-electrocatalysts
The nanoscale interface-engineered transition metal phosphide heterostructure can enhance the conductivity, modify the electronic structure, and expose/afford abundant active sites for the HER. Lyu et al.3 observed that the Ni2P/Ni5P4 heterostructure nanosheets exhibited a higher activity (η of −78 mV at −10 mA cm−2; Fig. 6(a)) and lower charge transfer resistance (Fig. 6(b)) than those of Ni2P and Ni5P4 for the HER in 0.5 M H2SO4. It was obtained by a solvothermal method and phosphorization treatment. The interface-engineered Ni2P/Ni5P4 heterostructure with a porous nanosheet morphology can enhance the conductivity, and expose/afford abundant active sites for the HER. DOS simulations and UPS characterization reveal that a built-in electric field is observed at the heterostructure interface region, where electrons have been transferred to Ni5P4 from Ni2P. Theoretical calculations reveal that the lowest water dissociation energy barrier and optimal H* adsorption free energy are observed for the active sites at the interface domain of Ni2P/Ni5P4 when compared to that of single-phase Ni5P4 and Ni2P, which could optimize the electronic structure and redistribute the electrons, leading to an enhancement of the activity of Ni2P/Ni5P4 for the HER.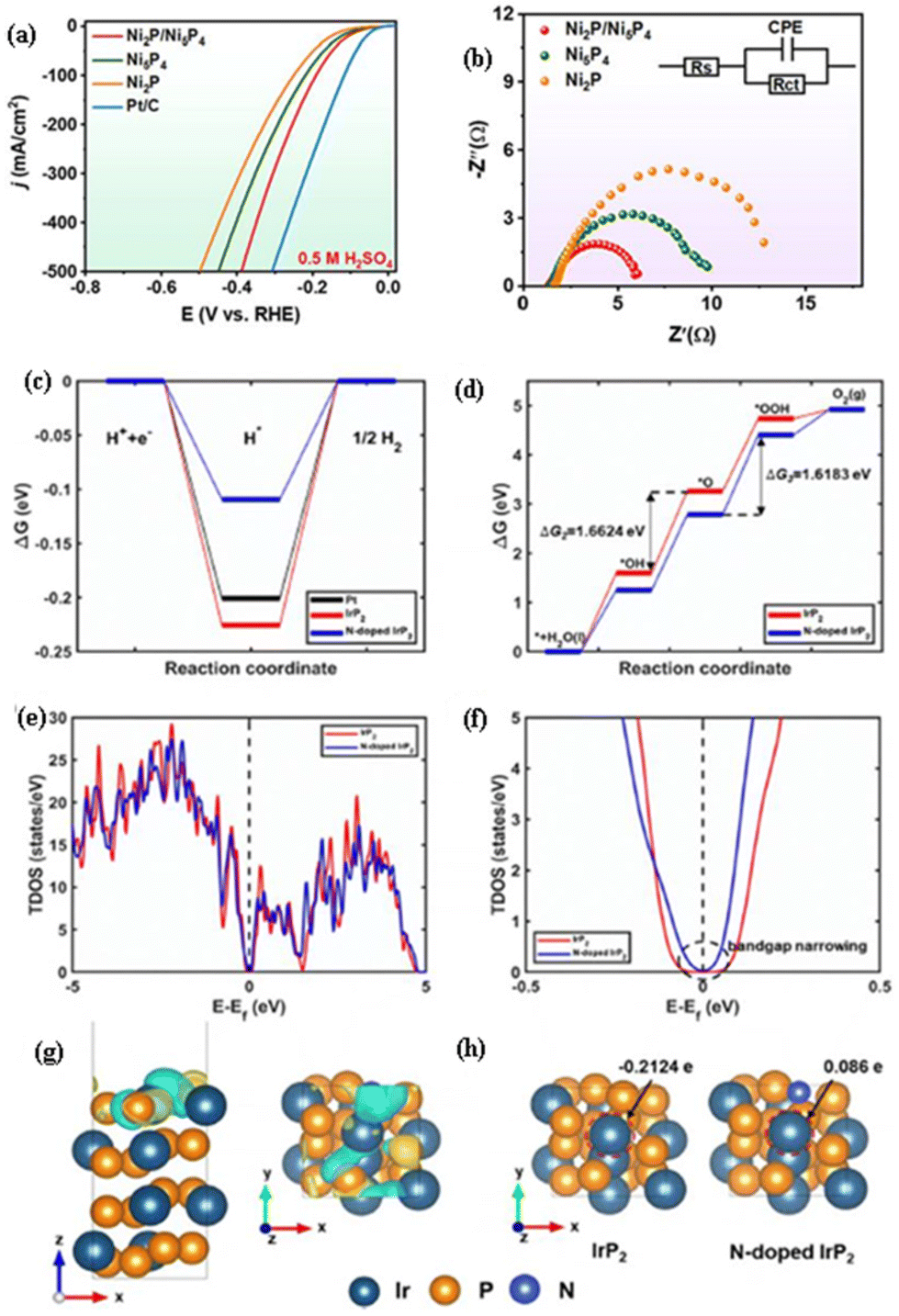 | ||
| Fig. 6 (a) LSV curve and (b) Nyquist plot for the HER of Ni2P/Ni5P4 in 0.5 M H2SO4 in comparison with those of Ni2P and Ni5P4 (reproduced with permission from ref. 3. Copyright 2023, Elsevier B.V.). The Gibbs free energy difference (ΔG) for (c) the HER process on N-doped IrP2 in comparison with those of Pt and IrP2, and (d) the OER process on N-doped IrP2 in comparison with that of IrP2. (e) The TDOS of N-doped IrP2 in comparison with that of IrP2; (f) enlarged view of the TDOS of N-doped IrP2 in comparison with that of IrP2 near the Fermi level, where the bandgap of N-doped IrP2 is narrowed when compared to that of IrP2, while the bandgap had been calculated from TDOS having a tolerance of 0.1 states per eV. (g) The difference in the charge densities between IrP2 and the N-doped IrP2 structure, where cyan and yellow colors indicate electron density depletion and enrichment with an iso-value of 0.01 e Bohr−3. (h) Bader charge analysis of IrP2 and N-doped IrP2, where the charge value of the Ir atom increases from −0.2124 e in IrP2 to 0.086 e in N-doped IrP2 as indicated by the red dashed circle (reproduced with permission from ref. 28. Copyright 2022, Elsevier B.V.). | ||
The introduction of N dopants into IrP2 effectively modifies the charge distribution on the IrP2 surface and adjusts the band (d and p) centers of P and Ir. This modulation narrows the bandgap, enhances conductivity, and modifies the electronic structure. Additionally, it provides an optimal adsorption energy for intermediates, ultimately leading to improved performance for the HER and OER. Roy et al.28 observed that N-IrP2 exhibited higher activity and lower charge transfer resistance than those of IrP2 for the HER and OER in 0.5 M H2SO4, while it exhibited a low cell voltage of 1.41 V at 10 mA cm−2 (Table 5) for overall water electrolysis, suggesting its markedly higher activity in an acidic environment. As shown in Fig. 6(c), the Gibbs free energy differences (ΔG) for the HER processes of all the catalysts were almost zero, while N-doped IrP2 exhibited a much lower ΔG (much closer to zero) than those of Pt and IrP2, suggesting high activity for N-doped IrP2, which was corroborated by the experimental results. Fig. 6(d) depicts the ΔG values of N-doped IrP2 in comparison with IrP2. The OER process is composed of four elementary reaction steps, where the rate determining step for the overall OER process is determined by the largest step value of ΔG among the four steps. In IrP2, the H dissociation step (OH → O), with a ΔG value of 1.6624 eV, serves as the rate-determining step, while in N-doped IrP2, it shifts to OH adsorption by the O atom (O → OOH) with a slightly lower ΔG of 1.6183 eV. Thus, N doping induces a shift towards a different rate-limiting process with a reduced ΔG value, which aligns well with the experimental results. To investigate the bandgap, the total density of states (TDOS) of N-doped IrP2 in comparison with IrP2 is shown in Fig. 6(e), with an enlarged view shown in Fig. 6(f). The N-doped IrP2 catalyst exhibits a narrower bandgap (0.1381 eV) when compared to that of IrP2 (0.2194 eV), suggesting enhanced electron transfer, leading to an enhancement in the activity of N-doped IrP2 for the HER and OER. Fig. 6(g) depicts the difference in charge densities between IrP2 and the N-doped IrP2 structure. DFT calculations revealed that the N dopant in IrP2 could modify the charge distribution of the IrP2 surface and the band (d and p) centers of P and Ir, leading to high activity for water electrolysis. The Bader charge analysis presented in Fig. 6(h) reveals a significant increase in the positive charge of Ir atoms on the surface of N-doped IrP2, from −0.2124 e to 0.086 e. This positive charge augmentation endows the Ir atoms with enhanced activity for the chemisorption of OER intermediates. Consequently, N doping of IrP2 leads to a reduction in ΔG and subsequent enhancement of the OER activity.
| Bifunctional electrocatalysts for HER and OER | Overall water splitting (V at 10 mA cm−2) | Stability | Ref. |
|---|---|---|---|
| N-IrP2 | 1.41 | 50 h (100 mA cm−2) | 28 |
| Ru-30 | 1.468 | 10 h (10 mA cm−2) | 76 |
| Ru/Co–N–C | 1.49 | 20 h (10 mA cm−2) | 78 |
| IrCo@CNT | 1.5 | 90 h (10 mA cm−2) | 81 |
| IrIn2/C | 1.51 | 55 h (10 mA cm−2) | 80 |
| Ru@MoO(S)3 | 1.522 | 24 h | 69 |
| La–RuO2 | 1.53 | 50 h (10 mA cm−2) | 70 |
| Fe2−xRuxNb2O6 | 1.55 | 24 h (200 mA cm−2) | 26 |
Besides, the work of Wu et al.85 proved that Mn2P–MnP/PNC exhibited a higher activity than those of MnP/PNC and Mn2P/PNC for the HER in 0.5 M H2SO4 and 1 M KOH, while it exhibited a lower charge transfer resistance than those of MnP/PNC and Mn2P/PNC for the HER in 1 M KOH. Moreover, Luo et al.77 observed that RuP2–C@RuP2–C exhibited a higher activity than that of RuP2 for the HER in 0.5 M H2SO4, while it exhibited a low charge transfer resistance for the HER in 0.5 M H2SO4. In contrast, Lu et al.51 observed that Co2P/CoP@Co@NCNT exhibited a higher activity than those of Co2P@Co@NCNT and CoP@Co@NCNT for the HER in 0.5 M H2SO4 and 1 M KOH, whereas Co2P@Co@NCNT (Rct: 8.9 Ω) exhibited a lower charge transfer resistance than those of Co2P/CoP@Co@NCNT (Rct: 10.3 Ω) and CoP@Co@NCNT (Rct: 12.4 Ω) for the HER in 1 M KOH. The decrease in the relative metallic cobalt content and increase of the P content in Co2P/CoP@Co@NCNT can be attributed to the decrease in the electrical conductivity of Co2P/CoP@Co@NCNT compared to that of Co2P@Co@NCNT.
Moreover, Yang et al.83 found that Ni2P–Ni12P5@Ni3S2 demonstrated a remarkable HER activity with an overpotential of −46 mV at −10 mA cm−2 in 0.5 M H2SO4, alongside notable stability over 24 h and minimal charge transfer resistance. This heterostructure, featuring Ni2P–Ni12P5 nanorod arrays on Ni3S2 film, is crafted through simultaneous corrosion, sulfidation, and phosphidation. In/ex situ analyses and DFT studies reveal strong interfacial coupling between Ni12P5 and Ni2P, with the Ni2P–Ni12P5 nanorods’ stability being significantly bolstered by the Ni3S2 film, thereby optimizing water adsorption/dissociation energies and hydrogen adsorption, enhancing electron/proton transfer, and ultimately, boosting the HER efficiency.
Similarly, Wu et al.87 highlighted Pb–CoSe2–DETA's exceptional HER performance, showing an overpotential of −74 mV at −10 mA cm−2 in 0.5 M H2SO4, excellent stability for 20 h, and a low charge transfer resistance of 4.8 Ω. This material, a Pb single-atom-doped CoSe2–DETA nanobelt, is closest to the peak of the HER activity in the “volcano plot,” indicating an optimal Co–N to Co–Se bond ratio. The unique Co atomic configuration, induced by Pb doping, optimizes the adsorption energy with intermediates, significantly enhancing the HER performance. Yu et al.84 reported that Ru–g-CN exhibited remarkable HER activity in 0.5 M H2SO4, achieving an overpotential of −54.5 mV at −10 mA cm−2. It demonstrates a high TOF (12.9 s−1 at 100 mV), mass activity (24.55 A mg−1 at η of 100 mV), apparent current density, and stability (lasting 120 h). Furthermore, it displays a low charge transfer resistance and a high photocatalytic H2 evolution rate of 489.7 mmol H2 g−1 h−1. Ru–g-CN is composed of Ru single atoms anchored on mesoporous graphitic carbon nitride. Theoretical calculations reveal that anchoring Ru single atoms on g-CN can tune the electronic structure, decrease the bandgap, facilitate electrical conductivity, enhance electron transfer, reduce the Gibbs free energy, and thereby enhance the HER performance.
Djire et al.90 noted V–Ti4N3Tx's superior HER activity in 0.5 M H2SO4, with an overpotential of −330 mV at −10 mA cm−2 and a low charge transfer resistance of 70 Ω. V–Ti4N3Tx is prepared from pristine exfoliated Ti4N3Tx MXene with V salt through a solution-based method, while the mixed transition metal nitride MXene with rich metal loading optimizes the electrochemistry, increases the conductivity, and enhances the HER performance. The electrochemical activity of individual MXene flakes was investigated by scanning electrochemical microscopy (SECM), which revealed that the basal plane of MXene exhibited much higher HER activity than the flake edge. This may be ascribed to the high exposure of metallic sites for proton adsorption.
3.3. Carbon/alloy based electrocatalysts
The engineering of the electronic structure of single-atom Ru sites through Co–N4 sites can provide an optimal adsorption energy with intermediates, enhance the conductivity, and promote the performance for the HER and OER. Rong et al.78 reported that Ru/Co–N–C exhibited higher activity, reduced charge transfer resistance, and improved stability compared to that of Ru–N–C. DFT calculations reveal that the introduction of Co–N4 sites into Ru/Co–N–C fine-tunes the electronic structure of Ru by enhancing its electron density and strengthening the Ru–O covalency. This modulation results in an optimal adsorption energy for HER and OER intermediates on Ru sites, thereby boosting the performance for both reactions. Additionally, the incorporation of Co–N4 sites prompts electron redistribution around Ru–N4, thereby enhancing the corrosion resistance of Ru/Co–N–C during acidic electrolysis.The fabrication reparation of IrIn2 intermetallics having a low Ir content with an unconventional face-centered orthorhombic phase could modify the electronic structure, enhance the conductivity, and provide optimal adsorption of intermediates, which could enhance the performance for the HER and OER. He et al.80 observed that IrIn2/C exhibited higher activity and lower charge transfer resistance than those of Ir/C for the HER and OER in 0.5 M H2SO4, and it also showed high activity and stability for overall water electrolysis in 0.5 M H2SO4. IrIn2 intermetallics having a low Ir content of 33% with an unconventional face-centered orthorhombic phase were obtained by a high-temperature annealing reduction method. Theoretical calculations reveal that an upward shift in the d-band center of IrIn2 is observed when compared to that of metallic Ir, leading to an enhancement in the adsorption of intermediates. Moreover, the ordered Ir–In bonds in IrIn2 intermetallic compounds could enhance charge transfer and enable resistance to dissolution and atom migration during electrolysis.
Moreover, Chen et al.82 observed that Co@CNTs|Ru afforded an η of −32 mV at −10 mA cm−2 for the HER in 0.5 M H2SO4, demonstrating excellent activity and stability as well as a low charge transfer resistance (51.46 Ω). Co@CNTs|Ru is composed of Co nanoparticles, which are in situ confined inside bamboo-like carbon nanotubes, where an ultralow Ru loading (∼2.6 μg per electrode area ∼ cm−2) is homogeneously deposited on their exterior walls. DFT calculations and atomic-scale structural analysis reveal that outer loaded Ru and inner confined Co can induce charge redistribution and synergistic electron coupling, which can provide an optimal adsorption energy of H intermediates (ΔGH*) and enhance electron/mass transfer, finally improving the HER performance.
In addition, Wang et al.81 observed that IrCo@CNT afforded an η of 241 mV at 10 mA cm−2 for OER in 0.5 M H2SO4, suggesting high activity and stability over 90 h. IrCo@CNT demonstrates an overpotential of −26 mV at −10 mA cm−2 for the hydrogen evolution reaction (HER) in 0.5 M H2SO4, highlighting exceptional activity and maintaining high stability over 90 h with a low charge transfer resistance of 3.78 Ω. In the context of overall water splitting in the same acidic medium, IrCo@CNT achieves a potential of 1.5 V at 10 mA cm−2, utilizing an ultra-low Ir loading of 0.027 mg cm−2, which underlines its remarkable efficiency. Additionally, its performance remains consistent without any decay throughout the 90-hour test period, confirming its durable stability. It was obtained on carbon cloth through a MOF-templated and dicyandiamide-assisted pyrolysis strategy. IrCo@CNT is composed of IrCo nanoparticles, which are encapsulated by high-density carbon nanotubes (CNTs). The superior performance of IrCo@CNT can be attributed to the synergistic interaction between Ir and Co, coupled with the protective role played by CNTs. Notably, the IrCo nanoparticles within IrCo@CNT remain intact after the HER, whereas they transform into Co-doped IrO2 nanoparticles following the OER. Theoretical calculations reveal that the energy barriers for the HER and OER can be lowered due to the optimal electronic structure obtained through Co doping of Ir and IrO2, respectively.
Sun et al.88 reported that WC–W2C–HCDs (HCDs: hollow carbon dodecahedrons) exhibited a remarkable HER activity in 0.5 M H2SO4, achieving an overpotential of −96 mV at −10 mA cm−2. Additionally, they demonstrate high stability over 150 h and a low charge transfer resistance of 20.1 Ω. WC–W2C–HCDs are composed of dual-phase WC–W2C nanocrystals, which are encapsulated in hollow carbon dodecahedrons. The heterointerfaces between WC and W2C facilitate strong electronic coupling, optimizing the adsorption energy for reaction intermediates. Concurrently, the hollow carbon dodecahedrons enhance the catalytic activity and stability for the HER by exposing abundant active sites and enhancing the electrical conductivity.
He et al.86 discovered that NCAG/Ru-3 delivered an overpotential of −65 mV at −10 mA cm−2 for the hydrogen evolution reaction (HER) in 0.5 M H2SO4, highlighting its outstanding activity and stability, alongside a low charge transfer resistance. NCAG/Ru-3, a composite of carbon aerogels doped with atomically isolated Ru nanoclusters and RuNx moieties, was synthesized through the pyrolysis of biomass hydrogels. DFT calculations suggest that the high activity originates from the RuNx sites located at zigzag edges and nanowrinkles. Similarly, Chen et al.79 reported that PtCo@PtSn achieved an overpotential of −21 mV at −10 mA cm−2 for the HER in 0.5 M H2SO4, demonstrating exceptional activity and remarkable durability, with negligible decay observed after 50000 cycles of cyclic voltammetry (CV), and maintaining a low charge transfer resistance. The superior performance of PtCo@PtSn is credited to the presence of a Pt-based bimetallic alloy and heterojunction, with DFT calculations indicating that interface engineering optimizes the surface electronic structure of Pt. This optimization enhances the water dissociation capability and reduces the bond strength of Pt–H, achieving an optimal H* Gibbs free energy (ΔGH*), thereby boosting HER efficiency.
4. Electrolysis in seawater
4.1. Oxide/hydroxide/(oxy)hydroxide/layered double hydroxide based nano-electrocatalysts
Liang et al.25 demonstrated highly stable seawater electrolysis lasting 100 h, employing a MnOx cathode and a PtRuTiOx (PRT) anode in a single-compartment cell without the need for a membrane. As schematically depicted in Fig. 7(a), the use of a selective chlorine oxidation reaction (ClOR) anode and a HER-selective cathode effectively eliminates the chlorine reduction reaction (ClRR) and the oxygen evolution reaction (OER). Consequently, pure H2 gas was produced, and the chlorine generated remained in the aqueous phase. The PtRuTiOx catalyst exhibits superior performance in saline water, boasting a high faradaic efficiency (FE) of approximately 100% for the chlorine oxidation reaction (ClOR) (Fig. 7(d)), as well as reduced charge transfer resistance (Fig. 7(b and e)). Its high activity (Fig. 7(c)) compared to other catalysts indicates a selectivity towards the ClOR over the oxygen evolution reaction (OER). The as-annealed PRT catalyst exhibits high stability for the ClOR for 500 h at a higher current density of 800 mA cm−2 (Fig. 7(f)). The MnOx catalyst demonstrates a remarkable performance for the hydrogen evolution reaction (HER). In saline water with chlorine species (HClO/ClO−), it exhibits a high faradaic efficiency (FE) of approximately 100% (Fig. 7(g)) and reduced charge transfer resistance (Fig. 7(g)), outperforming other catalysts. Its high activity further underscores its selectivity towards the HER over the chlorine reduction reaction (ClRR), making it an ideal candidate for efficient seawater electrolysis.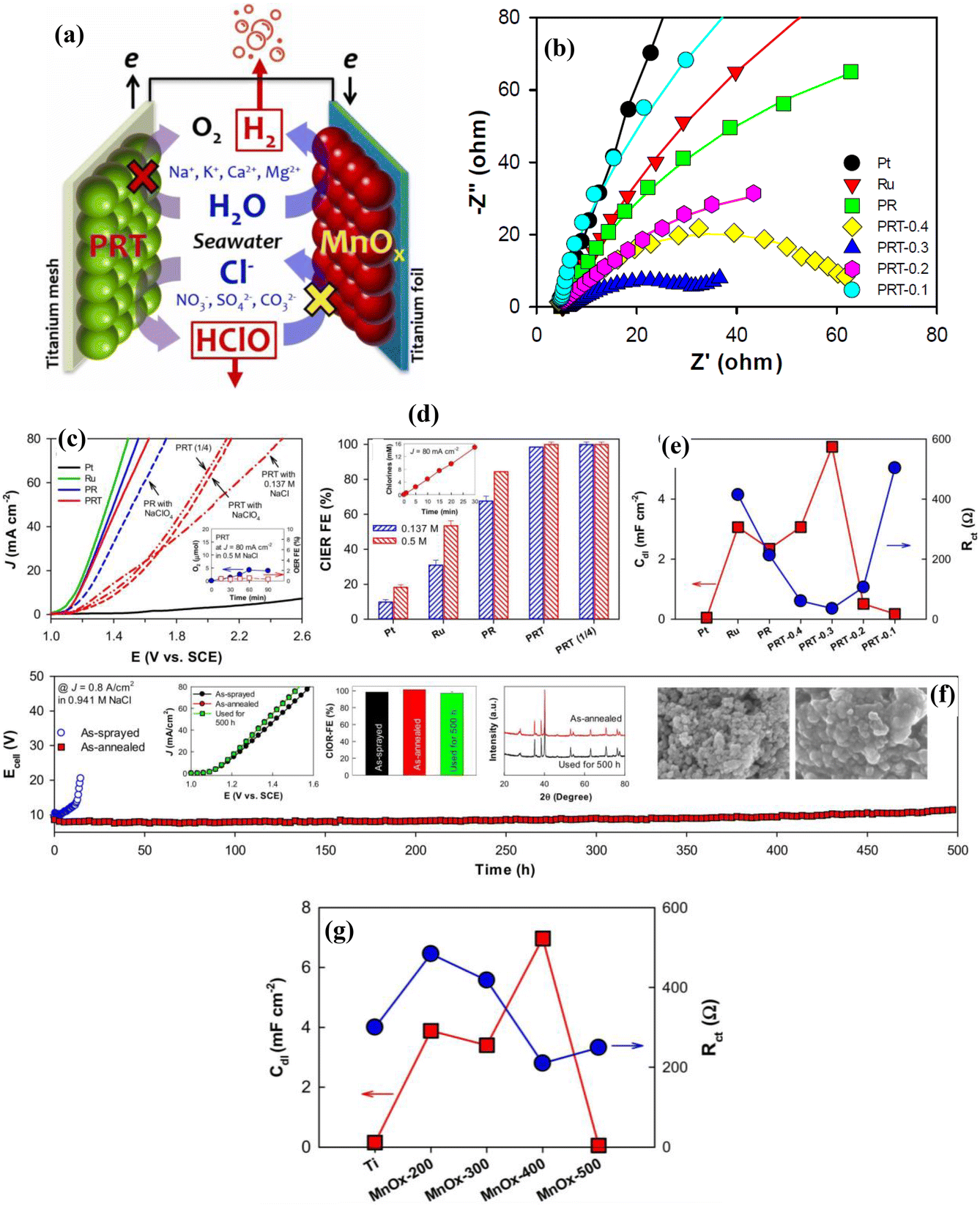 | ||
| Fig. 7 (a) Schematic diagram illustrating seawater electrolysis using the MnOx cathode and PtRuTiOx (PRT) anode without using a membrane in a single compartment cell, where the ClOR selective anode and HER selective cathode could eliminate the ClRR, while pure H2 gas could be formed, where the chlorine generated could be in the aqueous form. (b) Nyquist plot of Pt0.06Ru0.24Ti0.7Oy in comparison with those of its counterparts for ClOR in simulated seawater electrolyte. (c) LSV curves for ClOR of PRT in comparison with those of its counterparts in 0.5 M NaCl (pH 6), 0.137 M NaCl and 0.5 M NaClO4 (pH 6), where PRT (1/4) indicates a mass loading of 0.83 mg cm−2, while the inset depicts the ClOR and faradaic efficiency (FE) of PRT at 80 mA cm−2 in 0.5 M NaCl. (d) FE of PRT in comparison with those of its counterparts for ClOR at 80 mA cm−2, where the inset depicts the production of free chlorine over time at 80 mA cm−2. (e) Rct and Cdl values obtained from the Nyquist plot of Pt0.06Ru0.24Ti0.7Oy in comparison with those of its counterparts in simulated seawater electrolyte. (f) Long-term stability test for ClOR of as-annealed PRT in comparison with that of as-sprayed PRT at 80 mA cm−2 in 0.941 M NaCl, where the inset depicts the LSV curves and FE for ClOER of the as-sprayed and as-annealed materials and PRT used for 500 h at 80 mA cm−2 in 0.5 M NaCl, while the inset further shows the XRD patterns of the as-annealed material and PRT used for 500 h, and the inset further depicts the SEM images of the as-annealed material (left) and PRT used for 500 h (right). (g) Rct and Cdl values obtained from the Nyquist plot of MnOx-400 in comparison with those of its counterparts for the HER in simulated seawater electrolyte (reproduced with permission from ref. 25. Copyright 2022, Elsevier B.V.). | ||
The design of an electrocatalyst with a dual interface can reinforce the built-in electric field, enhance the conductivity, and provide an optimal adsorption energy for intermediates, which could enhance chlorine-free seawater oxidation. Zhang et al.92 observed that the (Ni, Fe)OOH@NixP‖NixP electrolyzer exhibited higher activity (1.71 V at 100 mA cm−2 and 1.81 V at 500 mA cm−2) and stability, and a lower charge transfer resistance than those of (Ni, Fe)OOH‖NixP and NixP‖NixP for overall seawater splitting in alkaline natural seawater. Moreover, almost no hypochlorite was detected for a long time in the seawater electrolyte at a cell voltage of ≥1.72 V, where this operating voltage was larger than that of the theoretical ClOR potential. (Ni, Fe)OOH@NixP is comprised of a dual-interface between (Ni, Fe)OOH and Ni12P5/Ni2P. This interface enhances the intrinsic driving force, facilitating interfacial electron transport and generating a robust built-in electric field with asymmetric charge distribution at the (Ni, Fe)OOH/Ni12P5 boundary. This configuration boosts the kinetics for the oxygen evolution reaction (OER) while mitigating the adsorption capacity of chloride ions (Cl−), thus optimizing the catalyst's performance in electrolysis.
Moreover, Ren et al.93 reported that Cu–MoO2 achieved an overpotential of −330.2 mV at −10 mA cm−2 for the hydrogen evolution reaction (HER) in artificial seawater (neutral pH), indicating exceptional activity. Furthermore, it demonstrates significant durability, with minimal decay observed after 1000 cycles of cyclic voltammetry (CV), and maintains a low charge transfer resistance. It was in situ obtained from polyoxometallate-based metal–organic frameworks (POMOFs). Cu–MoO2 is a hybrid material with a Schottky heterojunction, which could modulate the electronic structure, provide an optimal adsorption energy with intermediates, and enhance the performance for the HER.
In addition, Liu et al.94 observed that Fe–Mo–Co–O afforded an η of 365 mV at 50 mA cm−2 for the OER in 1 M KOH + seawater, suggesting its very high activity, while it exhibited low charge transfer resistance. It affords an η of −250 mV at −50 mA cm−2 for the HER in 1 M KOH + seawater, suggesting its very high activity. For overall water splitting in 1 M KOH + seawater, Fe–Mo–Co–O//Fe–Mo–Co–O affords a potential of 1.59 V at 10 mA cm−2, suggesting its very high activity, while it affords high stability for 180 h. Fe–Mo–Co–O is composed of Fe-doped Co2Mo3O8/MoO3/Co3O4 nanosheets, where the enhanced performance is attributed to the combination of highly active Fe dopant and corrosion-resistant Mo–O species.
Jin et al.95 highlighted that CoCr0.7Rh1.3O4 reached an overpotential of 273 mV at 10 mA cm−2 for the OER in 1 M KOH + 0.5 M NaCl, indicating outstanding activity and stability for 12 h, along with minimal charge transfer resistance. It is composed of nanofibers of single-phase trimetallic spinel CoCr0.7Rh1.3O4. DFT calculations reveal that the synergistic effects of Co, Cr, and Rh can decrease the energy barriers for water dissociation, which could enhance the performance for the OER.
Wen et al.96 revealed that Fe1/Mn_NiO and Pt1/Mn_NiO exhibited overpotentials of 290 mV for the OER and −129 mV for the HER, respectively, at 100 mA cm−2 in 0.5 M KOH + seawater, both maintaining stability for 30 h. The introduction of Mn into low polarized NiO nanosheets can alter the surface polarization, which can regulate the electronic metal–support interactions between the NiO support and anchored Pt or Fe single atoms, while the resulting change in coordination structure and charge redistribution in Fe1/Mn_NiO and Pt1/Mn_NiO can decrease the Gibbs free energy for the rate-determining steps in the OER and HER, respectively, which can enhance the performance.
In addition, Hemmati et al.97 observed that NRAHM–NiO afforded an η of 70 mV at 10 mA cm−2 for the OER in 1 M KOH + 0.5 M NaCl, suggesting its very high activity, while it exhibited a low charge transfer resistance (Rct: 3.4 Ω; Rp: 0.5611 Ω). NRAHM–NiO affords an η of −51 mV at −10 mA cm−2 for the HER in 1 M KOH + 0.5 M NaCl, suggesting its very high activity, while it exhibits a low charge transfer resistance (Rct: 3.015 Ω; Rp: 0.4327 Ω). For overall water splitting in 1 M KOH + 0.5 M NaCl, NRAHM–NiO//NRAHM–NiO affords a potential of 1.66 V at 100 mA cm−2, suggesting its very high activity, while it affords high stability for 168 h; the integrated photolysis system powered by a single solar cell exhibits a high solar-to-hydrogen (STH) efficiency of 9.9%. It exhibits high selectivity (99% faradaic efficiency) for the OER in seawater without generating any chlorine species. NRAHM–NiO is composed of hydrophilic urchin-like nanorod array-based hierarchical NiO microspheres, which can facilitate gas evolution, enhance the charge transfer ability, afford high intrinsic electroactive sites, and improve the corrosion resistance, and thus could enhance the performance for the HER and OER.
Furthermore, Zhuo et al.98 reported the synthesis of Ni–Fe hydroxides through a cost-effective, ultrafast method. The catalyst demonstrates low OER overpotentials of 240 mV and 254 mV at 10 mA cm−2 in 1 M KOH and alkaline seawater, respectively, while maintaining excellent electrochemical stability. In situ Raman spectroscopy and other physical characterization studies reveal that the incorporation of Fe and the transformation of Ni(Fe)(OH)2 into Ni(Fe)OOH are key to the enhanced OER performance. Additionally, the Ni–Fe hydroxide catalyst can be synthesized in just 1 min and scaled up easily. Even at a size of 2000 cm2, the catalyst remains electrochemically uniform.
A study by Du et al.99 showed that Co,Cl–Cu(OH)2 demonstrated an overpotential of 310 mV at 100 mA cm−2 for the OER and Ru,Cl–Cu(OH)2 achieved −37 mV at −10 mA cm−2 for the HER in 1 M KOH + seawater, highlighting exceptional activity for both reactions. Moreover, both catalysts show high stability and low charge transfer resistance, indicating their efficiency at electrolytic water splitting. For overall water splitting in 1 M KOH + seawater, Co,Cl–Cu(OH)2OER//Ru,Cl–Cu(OH)2HER affords a potential of 1.55 V at 10 mA cm−2, suggesting its very high activity, while it affords high stability for 100 h. It was obtained by in situ corrosive engineering and cation/anion exchange at ambient temperature. Co,Cl–Cu(OH)2 is composed of Cu-based hollow nanotubes, where the rough surfaces are coupled with a chloridion-enriched environment. The enriched chloride surface can inhibit the attack of chlorine species, while the hollow nanotubes with rough surfaces can expose abundant active sites and facilitate gas evolution, which could enhance the performance for the OER.
Sun et al.100 identified that Pt2/Ni(OH)2, derived from a MOF, catalyzed the HER with an overpotential of −19 mV at −10 mA cm−2 in 1 M KOH + seawater, showcasing high activity, low charge transfer resistance, and a solar-to-hydrogen efficiency of 13.5% when powered by perovskite solar cells. A low cell voltage of 1.46 V at 10 mA cm−2 is achieved from the anion exchange membrane (AEM) seawater electrolyzer with a Pt2/Ni(OH)2 cathode and Ni foam anode, while it exhibits low energy consumption to generate 1 m3 of H2 at 200 mA cm−2 (3.8 kW h). Pt2/Ni(OH)2 is composed of Ni(OH)2 nanosheets, which are decorated with Pt nanoparticles. It is derived from a MOF. It exhibits SMSIs (strong metal–support interactions) of Pt–O–Ni bonds at the interface, a high superhydrophilic surface area, and the synergistic effect of Pt–O vacancy (OV) sites, which could enhance the performance for the HER. Furthermore, DFT calculations reveal that SMSIs at the interface and OV can alter the electronic structure and localized charge density of Ni(OH)2. The synergistic effect of Pt–OV sites can afford optimal adsorption and desorption of H*, while adsorption of H2O with high efficiency can be achieved by OV, which can regulate the spillover of hydrogen to Pt from Ni(OH)2, and this could enhance the performance for the HER.
Pan et al.101 found that s-NiFeCrMo–OH achieved an overpotential of 242 mV for the OER at 100 mA cm−2 in 1 M KOH + 0.5 M NaCl, with exceptional stability and a low charge transfer resistance. A low cell voltage of 1.82 V at a huge current density of 500 mA cm−2 over 1000 h at ambient temperature was achieved in an overall alkaline seawater electrolyzer using s-NiFeCrMo–OH as the anode, while the floating solar seawater splitting device exhibited a high STH efficiency of 20.61 ± 0.77%. s-NiFeCrMo–OH is a 2D quaternary metal hydroxide nanosheet, which is composed of Ni, Fe, Cr, and Mo elements; partial Mo element leaching morphologically transforms the catalyst by in situ electrochemical activation. The existence of a metal with high valence states and abundant O vacancies is attributed to the enhanced catalytic performance and corrosion resistance.
Moreover, Zhou et al.102 observed that Ru/NiFeOOH afforded an η of 330 mV at a huge current density of 500 mA cm−2 for the OER in 1 M KOH + seawater, suggesting its very high activity, and it exhibited high stability for >95 h with a low charge transfer resistance. Ru/NiFeOOH is prepared on NiFe foam through a one-step bimetallic substrate induction strategy to construct supported metal catalysts (SMCs) integrally as a binder-free electrode; this synthesis strategy (bimetallic substrate induction) can also be applied using seawater rather than pure water. The NiFe foam substrate is a source of Ni and Fe metals, acts as a reducing agent for the in situ growth of NiFeOOH nanosheets with Ru nanoparticles, and affords strong interactions between the NiFe substrate and SMCs. Experimental studies and DFT calculations reveal that in situ formed metal–support interactions can provide optimal electronic states and increase the chemical stability.
In addition, Zhang et al.109 demonstrated that the Ni based anode suffered more corrosion by Br− in seawater than that of Cl− in seawater due to the faster corrosion kinetics in bromine. Simulated and experimental studies reveal that extensive etching of Br− leads to the formation of shallow-wide pits, while localized corrosion of Cl− leads to the generation of narrow-deep pits, which can be ascribed to the lower reaction energy of Br− and the fast diffusion kinetics of Cl− in the passivation layer. Furthermore, NiFe LDH exhibits rapid performance degradation due to the extensive spalling of the catalyst layer induced by Br−, suggesting the importance of designing anti-Br− corrosion anodes along with anti-Cl− corrosion anodes for seawater electrolysis.
Enkhtuvshin et al.106 observed that c-NiFe/a-NiFe LDH (crystalline and amorphous layered double hydroxide) demonstrated an overpotential of 300 mV at 100 mA cm−2 for the oxygen evolution reaction (OER) in 1 M KOH + 0.5 M NaCl, showcasing exceptional activity and maintaining its stability over 100 h. Similarly, for the hydrogen evolution reaction (HER) under the same conditions, it achieves an overpotential of −200 mV at −100 mA cm−2, again proving its high activity and enduring stability, coupled with a low charge transfer resistance of approximately 5 Ω. In applications involving overall water splitting in 1 M KOH + seawater, the c-NiFe/a-NiFe LDH system requires about 1.69 V at 10 mA cm−2 to operate, maintaining its performance and stability for 100 h. This LDH structure, comprising a Ni3Fe alloy phase within a 2D network topped by a ∼2 nm ultrathin, low-crystalline NiFeOOH phase, facilitates enhanced catalytic activity for both cathodic and anodic reactions, alongside robust corrosion resistance. Notably, c-NiFe/a-NiFe LDH can be efficiently scaled up to 25 cm2, demonstrating its potential for hydrogen production from saline water using a low-intensity, lab-made photovoltaic cell, highlighting its scalability and efficiency in practical applications.
4.2. Phosphide/sulfide/carbon/alloy based nano-electrocatalysts
Creating phosphorus vacancies in cobalt phosphide embedded in N-doped carbon can modify the electronic structure, provide an optimal adsorption energy with intermediates, enhance the conductivity, and enable the rapid reconstruction of active species for the OER, which could enhance the performance for the HER and OER. Wang et al.15 reported that CoxPv@NC demonstrated superior activity and lower charge transfer resistance compared to CoxP@NC, CoxP, and CoxPv for both the HER and OER in seawater containing 1 M KOH. Specifically, it achieves an impressive HER performance with an η of −260 mV at a high current density of −1000 mA cm−2 (Table 6) and an OER η of 386 mV at 1000 mA cm−2 (Table 7). Furthermore, CoxPv@NC exhibits remarkable activity and stability for overall seawater electrolysis, achieving a voltage of 1.88 V at 1000 mA cm−2 (Table 8; Fig. 8(j)) and maintaining its stability for 100 h (Fig. 8(k)). It was obtained on Ni foam by the following steps (Fig. 8(a)): first, the MOF (Co(OH)F@ZIF-67) was prepared by hydrothermal treatment followed by immersion; then, CoxP@NC was obtained by phosphidation; finally, CoxPv@NC was obtained by NaBH4 solution treatment. CoxPv@NC possesses a nanowire morphology, where tiny lamella structures are observed on the surface (SEM image; Fig. 8(b)). The ultrathin nanosheets further wrapped the outer layer of nanowires (TEM image; Fig. 8(c)). The surface of CoxP is wrapped in a NC shell with ∼0.5 nm thickness (HRTEM image; Fig. 8(d)). The lattice fringes of Co2P and CoP inside the carbon layers reveal d-spacings of 0.142 nm and 0.125 nm, which correspond to the (330) and (303) lattice planes of Co2P and CoP, respectively (HRTEM image; Fig. 8(e)), while abundant vacancies and lattice distortions are observed. The marked red circles show the anionic P vacancies (STEM-BF image; Fig. 8(f)), while the corresponding intensity profile depicts the anionic P vacancies (Fig. 8(g)). CoxPv@NC is polycrystalline, where the diffraction rings correspond to the facets of CoP and Co2P (SAED pattern; Fig. 8(h)), while it contains Co, P, N, and C, which are uniformly distributed (Fig. 8(i)), 40.4 wt% CoxPv. DFT calculations and in situ Raman spectra reveal that the enhancement of the catalytic performance is attributed to the quick reconstruction of active species for the OER, and P vacancies provide an optimal adsorption energy with intermediates for the HER. | ||
| Fig. 8 (a) Schematic diagram illustrating the synthesis process for CoxPv@NC; (b) SEM image and (c) TEM image of CoxPv@NC; (d and e) HRTEM images and (f) STEM-BF image of CoxPv@NC; (g) intensity profile obtained from the corresponding areas in part (f); (h) SAED pattern and (i) EDS mapping of CoxPv@NC for Co, P, N and C; (j) LSV curves for CoxPv@NC//CoxPv@NC in comparison with those of the RuO2‖Pt/C couple in 1 M KOH with seawater; (k) chronopotentiometry curve of CoxPv@NC//CoxPv@NC in 6 M KOH with seawater at 60 °C (reproduced with permission from ref. 15. Copyright 2023, Elsevier Ltd). | ||
| HER electrocatalysts | η (mV) | j (mA cm−2) | R ct | R p | Electrolyte | Stability | Ref. |
|---|---|---|---|---|---|---|---|
| Ref.: reference; Rct: charge transfer resistance; Rp: polarization resistance; HER: hydrogen evolution reaction; η: overpotential; NA: not applicable; j (mA cm−2): current density (mA cm−2); NRAHM–NiO: nanorod array based hierarchical NiO microspheres; c: crystalline; a: amorphous. | |||||||
| Pt2/Ni(OH)2 | −19 | −10 | NA | NA | 1 M KOH + seawater | 200 h | 100 |
| Ru,Cl–Cu(OH)2 | −37 | −10 | NA | NA | 1 M KOH + seawater | 12 h (500 mA cm−2) | 99 |
| NRAHM–NiO | −51 | −10 | 3.015 Ω | 0.4327 Ω | 1 M KOH + 0.5 M NaCl | 100 h (500 mA cm−2) | 97 |
| Ni@CNTs–MoxC/Ni2P | −65.9 | −10 | NA | NA | 1 M KOH + seawater | 50 h (50 mA cm−2) | 103 |
| Pt1/Mn_NiO | −129 | −100 | NA | NA | 0.5 M KOH + seawater | 30 h (100 mA cm−2) | 96 |
| Co–N,P-HCS | −164 | −10 | NA | NA | 1 M KOH + seawater | 100 h | 50 |
| Cu2S@NiS@Ni–NiMo | −177 | −500 | 2.16 Ω | NA | 1 M NaOH + 0.5 M NaCl | 2000 h (500 mA cm−2) | 104 |
| Cr–CoXP | −194 | −100 | NA | NA | 1 M KOH + seawater | 140 h | 105 |
| c-NiFe/a-NiFe LDH | −200 | −100 | ≈5 Ω | NA | 1 M KOH + 0.5 M NaCl | 100 h (500 mA cm−2) | 106 |
| CoFe–Ni2P | −241 | −100 | NA | NA | 1 M KOH + seawater | 100 h (500 mA cm−2) | 107 |
| Fe–Mo–Co–O | −250 | −50 | NA | NA | 1 M KOH + seawater | 72 h (10 mA cm−2) | 94 |
| CoxPv@NC | −260 | −1000 | NA | NA | 1 M KOH + seawater | 100 h (800 mA cm−2) | 15 |
| Cu–MoO2 | −330.2 | −10 | NA | NA | Artificial seawater (neutral pH) | 10 h | 93 |
| Mn–Ni2P–Fe2P | −470 | −1000 | NA | NA | 1 M KOH + seawater | 100 h (500 mA cm−2) | 108 |
| OER electrocatalysts | η (mV) | j (mA cm−2) | R ct | R p | Electrolyte | Stability | Ref. |
|---|---|---|---|---|---|---|---|
| Ref.: reference; Rct: charge transfer resistance; Rp: polarization resistance; OER: oxygen evolution reaction; η: overpotential; NA: not applicable; j (mA cm−2): current density (mA cm−2); NRAHM–NiO: nanorod array based hierarchical NiO microspheres; c: crystalline; a: amorphous. | |||||||
| NRAHM–NiO | 70 | 10 | 3.4 Ω | 0.5611 Ω | 1 M KOH + 0.5 M NaCl | 100 h (500 mA cm−2) | 97 |
| CoFe–Ni2P | 224 | 10 | NA | NA | 1 M KOH + seawater | 130 h (500 mA cm−2) | 107 |
| Ni@CNTs–MoxC/Ni2P | 230 | 10 | NA | NA | 1 M KOH + seawater | 50 h (50 mA cm−2) | 103 |
| s-NiFeCrMo–OH | 242 | 100 | NA | NA | 1 M KOH + 0.5 M NaCl | 400 h (500 mA cm−2) | 101 |
| Ni(Fe)(OH)2 | 257 | 10 | NA | NA | 1 M KOH + seawater | 100 h (250 mA cm−2) | 98 |
| CoFePBA–Co2P | 257 | 10 | 0.57 Ω | NA | 1 M NaOH + 0.5 M NaCl | 100 h (2000 mA cm−2) | 110 |
| CoCr0.7Rh1.3O4 | 273 | 10 | NA | NA | 1 M KOH + 0.5 M NaCl | 12 h (20 mA cm−2) | 95 |
| Fe1/Mn_NiO | 290 | 100 | NA | NA | 0.5 M KOH + seawater | 30 h (100 mA cm−2) | 96 |
| RuMoNi | 291 | 100 | NA | NA | 1 M KOH + seawater | 3000 h (500 mA cm−2) | 111 |
| c-NiFe/a-NiFe LDH | 300 | 100 | NA | NA | 1 M KOH + 0.5 M NaCl | 100 h (500 mA cm−2) | 106 |
| Co,Cl–Cu(OH)2 | 310 | 100 | NA | NA | 1 M KOH + seawater | 12 h (500 mA cm−2) | 99 |
| (Ni, Fe)OOH@NixP | 318 | 500 | 0.85 Ω | NA | 1 M KOH + seawater | 100 h (500 mA cm−2) | 92 |
| Co–N,P-HCS | 320 | 10 | NA | NA | 1 M KOH + seawater | 100 h | 50 |
| Ru/NiFeOOH | 330 | 500 | NA | NA | 1 M KOH + seawater | 400 h (100 mA cm−2) | 102 |
| Cr–CoXP | 334 | 100 | NA | NA | 1 M KOH + seawater | 140 h | 105 |
| Mn–Ni2P–Fe2P | 358 | 1000 | NA | NA | 1 M KOH + seawater | 100 h (500 mA cm−2) | 108 |
| Fe–Mo–Co–O | 365 | 50 | NA | NA | 1 M KOH + seawater | 72 h (10 mA cm−2) | 94 |
| CoxPv@NC | 386 | 1000 | NA | NA | 1 M KOH + seawater | 100 h (800 mA cm−2) | 15 |
| Co–(NiFe)N@NiSx | 425 | 100 | 0.14 Ω | NA | Artificial seawater (neutral pH) | 50 h (100 mA cm−2) | 112 |
| NiS2pSx | 460 | 10 | 33.7 Ω | NA | 0.5 M NaCl (neutral pH) | 90 h | 113 |
| Bifunctional electrocatalysts for HER and OER | Overall water splitting (V) | j (mA cm−2) | Electrolyte | Stability | Ref. |
|---|---|---|---|---|---|
| nb: non-bifunctional electrocatalyst; Ref.: reference; j (mA cm−2): current density (mA cm−2); NRAHM–NiO: nanorod array based hierarchical NiO microspheres. | |||||
| nbFe1/Mn_NiOOER//Pt1/Mn_NiOHER | 1.44 | 10 | 0.5 M KOH + seawater | 30 h (100 mA cm−2) | 96 |
| Cr–CoXP | 1.54 | 10 | 1 M KOH + seawater | 160 h | 105 |
| nbCo,Cl–Cu(OH)2OER//Ru,Cl–Cu(OH)2HER | 1.55 | 10 | 1 M KOH + seawater | 100 h (200 mA cm−2) | 99 |
| Ni@CNTs–MoxC/Ni2P | 1.56 | 10 | 1 M KOH + seawater | 50 h (50 mA cm−2) | 103 |
| Fe–Mo–Co–O | 1.59 | 10 | 1 M KOH + seawater | 72 h (10 mA cm−2) | 94 |
| Mn–Ni2P–Fe2P | 1.64 | 10 | 1 M KOH + seawater | 120 h (500 mA cm−2) | 108 |
| NRAHM–NiO | 1.66 | 100 | 1 M KOH + 0.5 M NaCl | 168 h (240 mA cm−2) | 97 |
| CoFe–Ni2P | 1.738 | 100 | 1 M KOH + seawater | 350 h (1000 mA cm−2) | 107 |
| nb(Ni, Fe)OOH@NixPOER//NixPHER | 1.81 | 500 | 1 M KOH + seawater | 50 h (500 mA cm−2) | 92 |
| CoxPv@NC | 1.884 | 1000 | 1 M KOH + seawater | 100 h (200 mA cm−2) | 15 |
| Co–N,P-HCS | ≈2.12 | 200 | 1 M KOH + seawater | 1000 h (10 mA cm−2) | 50 |
| Ni–B–P | 2.76 | 500 | 1 M KOH + seawater | 8 h (500 mA cm−2) | 114 |
The fabrication of asymmetric Co–N3P1 implanted into carbon can provide symmetry-breaking electronic structures, enhance the conductivity, enable the affinity of strong oxygen-containing intermediates with weak Cl− adsorption and moderate H adsorption, and that could enhance the performance for HER and OER. Wang et al.50 observed that Co–N,P-HCS (HCS: hollow carbon spheres) exhibited higher activity and lower charge transfer resistance than those of HCS, Co–HCS, and Co–N-HCS for the HER and OER in seawater with 1 M KOH, while it exhibited high stability for 1000 h. Co–N,P-HCS is composed of an asymmetric Co–N3P1 structure, which is implanted on hollow carbon spheres. The DFT calculations disclose that the asymmetric Co–N3P1 sites with symmetry-breaking electronic structures can be formed due to P-doping into Co–N–C, enabling the affinity of strong oxygen-containing intermediates with weak Cl− adsorption and moderate H adsorption.
Bahuguna et al.113 reported that NiS2pSx achieved an overpotential of 460 mV at 10 mA cm−2 for the oxygen evolution reaction (OER) in 0.5 M NaCl (neutral pH), highlighting its exceptional activity. It also maintains high stability over 90 h and presents a low charge transfer resistance of 33.7 Ω. 3D NiS2pSx is a binder-free, catenated sulphur type Ni polysulfide-based chlorine-phobic electrocatalyst. NiS2pSx produces pure oxygen without the formation of any chlorine in 0.5 M NaCl. This high selectivity towards oxygen evolution is attributed to the limited accessibility of the electrostatically and sterically hindered Ni metal centers to the larger Cl− ions. Furthermore, the generation of catenated sulphur type polysulphide species (pSn2−/S2− = 2.1) due to the rapid electrochemical evolution of the NiS2pSx surface can further facilitate the intrinsic chlorine-phobicity of the electrocatalyst, which can lead to pure oxygen evolution at current densities up to 300 mA cm−2.
Additionally, Badreldin et al.112 reported that the Co–(NiFe)N@NiSx catalyst exhibited a remarkable performance for the OER in artificial seawater at neutral pH. It achieves an η of 425 mV at 100 mA cm−2, indicating its exceptionally high activity. Furthermore, it maintains a current retention of 84.3% to deliver approximately 200 mA cm−2 after 168 h, highlighting its remarkable stability. The catalyst also exhibits a low charge transfer resistance (0.14 Ω) and a high faradaic efficiency of 87.4% at 1.75 V (vs. RHE), further confirming its outstanding performance for oxygen evolution reactions in seawater. Co–(NiFe)N@NiSx is composed of Co–(NiFe) oxide/nitride with NiSx as an interlayer. The formation of stable anionic [O–N]δ− and cationic [Co–(NiFe)]δ+ surface species coupled with the NiSx interlayer can facilitate the dissociation of H2O, which could improve the activity and kinetics for the OER.
Xu et al.104 demonstrated that Cu2S@NiS@Ni–NiMo achieved an impressive −177 mV overpotential for the HER at −500 mA cm−2 in 1 M NaOH + 0.5 M NaCl, showcasing excellent activity. It maintains a stable performance without decay for 2500 h at the same current density and exhibits a low charge transfer resistance of 2.16 Ω, underscoring its exceptional durability and conductive efficiency. The synthesis route to Cu2S@NiS@Ni–NiMo on Cu foam can be scalable to over 100 cm2. The enhanced performance for the HER of Cu2S@NiS@Ni–NiMo is attributed to the following factors: (1) the unique boundary interface can enhance the high intrinsic activity, (2) the superaerophobic nanoarray architecture can facilitate the evolution of gas and improve mass transport, (3) the polyanion-rich passivating layers can improve the corrosion resistance.
Huang et al.107 reported that CoFe–Ni2P achieved an overpotential of 224 mV at 10 mA cm−2 for the OER in 1 M KOH + seawater, indicating exceptional activity. Remarkably, it maintains its stability, with no decay observed over 500 h at a current density of 500 mA cm−2, highlighting its superior stability and minimal charge transfer resistance. For the HER, CoFe–Ni2P reaches an overpotential of −241 mV at −100 mA cm−2, further confirming its high efficiency. In the context of overall water splitting in the same medium, CoFe–Ni2P achieves a potential of 1.738 V at 100 mA cm−2, showcasing its outstanding activity. Moreover, it demonstrates enduring stability over 100 h at a current density of 500 mA cm−2, underscoring its durability and efficient performance. CoFe–Ni2P is composed of nickel phosphide, which is modified by Co and Fe, where the conductivity of Ni2P can be enhanced by Fe atoms to facilitate electron transfer, while the self-reconstruction process can be enhanced by Co atoms to form CoFe–NiOOH species in situ on the surface of the electrode. Furthermore, the selective adsorption of OH− ions, which significantly hinders the adsorption of Cl− ions, can be achieved by the in situ-generated CoFe–NiOOH species, and this could enhance the performance for seawater splitting even at a huge current density.
Besides, Liu et al.110 found that CoFePBA–Co2P demonstrated an overpotential of 257 mV at 10 mA cm−2 for the OER in 1 M NaOH + 0.5 M NaCl, indicating exceptional activity. Remarkably, this catalyst maintains its performance, generating a current density of 1000 mA cm−2 over 1000 h in 20 wt% NaOH + seawater and sustaining a current density of 2000 mA cm−2 for 100 h in 20 wt% NaOH + saturated NaCl, highlighting its outstanding stability. Additionally, it shows a low charge transfer resistance of 0.57 Ω, further confirming its efficient electrochemical behavior. CoFePBA–Co2P is composed of cobalt ferricyanide/cobalt phosphide with a cap/pin structure. Molecular dynamics simulations and experimental investigations reveal that PO43− and Fe(CN)63− formed by the electrode can effectively hinder Cl− through dense coverage and electrostatic repulsion, leading to about a 5-fold decrease in Cl− adsorption, thereby enhancing the corrosion resistance.
Notably, Song et al.105 discovered that Cr–CoXP delivered an overpotential of 334 mV at 100 mA cm−2 for the OER and −194 mV for the HER at −100 mA cm−2 in 1 M KOH + seawater, indicating outstanding activity for both processes. This catalyst maintains its performance with remarkable stability over 140 h for both the OER and HER, alongside demonstrating a low charge transfer resistance. In the context of overall water splitting in the same medium, Cr–CoXP achieves a potential of 1.54 V at 10 mA cm−2, maintaining exceptional activity and extending its stability to 160 h, highlighting its efficiency and durability under challenging conditions. Cr–CoXP is composed of CoxP, which is doped with vein-like Cr. DFT calculations and experimental studies reveal that the doping of CoxP with Cr can regulate the electronic structure of CoxP and effectively obviate the work function of CoxP, which can facilitate electron transfer between absorbates and the catalyst surface, and decrease the energy barriers for water dissociation and the rate-determining steps for the OER and HER. Furthermore, the doping of CoxP with Cr can also prevent Co sites with robust valence states to retain their high performance during the OER process, which can enhance the performance for seawater electrolysis.
Luo et al.108 demonstrated that the Mn–Ni2P–Fe2P catalyst, synthesized via a hydrothermal–phosphorization method, exhibited an outstanding electrocatalytic performance for both oxygen and hydrogen evolution reactions (OER and HER) in 1 M KOH + seawater. Specifically, it achieves an overpotential of 358 mV at a current density of 1000 mA cm−2 for the OER and −470 mV at the same density for the HER, highlighting its exceptional activity. Remarkably, this catalyst shows high stability, maintaining around 500 mA cm−2 for 200 h in the OER and for over 100 h in the HER, with minimal decay observed. Furthermore, in overall water splitting, Mn–Ni2P–Fe2P achieves a potential of 1.64 V at 10 mA cm−2, sustaining nearly 500 mA cm−2 for 120 h, indicating its durability and low charge transfer resistance. This combination of high activity and stability underlines the effectiveness of the Mn–Ni2P–Fe2P system for sustainable water electrolysis in seawater. The enhanced performance of Mn–Ni2P–Fe2P for seawater electrolysis is attributed to the following factors: (1) Mn doping and Ni2P/Fe2P heterointerfaces can provide optimal electronic structures that can provide an optimal adsorption energy for intermediates to enhance the intrinsic HER and OER activities. (2) Mn doping and heterointerfaces can generate abundant active sites, while the 3D nanoflower structure can expose abundant active sites. (3) Nickel–iron phosphides can provide high corrosion resistance.
In addition, Ahasan Habib et al.114 observed that Ni–B–P//Ni–B–P afforded a potential of 2.76 V at a huge current density of 500 mA cm−2, suggesting substantial activity and stability for overall water splitting in 1 M KOH + seawater. It was obtained through a hydrothermal approach. Ni–B–P is composed of nickel–boride–phosphide microspheres, which can modify the electronic structure, enhance the conductivity, afford/expose abundant active sites, and enhance the corrosion resistance, which could enhance the performance for seawater electrolysis.
Wang et al.103 found that Ni@CNTs–MoxC/Ni2P delivered an overpotential of 230 mV at 10 mA cm−2 for the OER and −65.9 mV for the HER at −10 mA cm−2 in 1 M KOH + seawater, indicating its outstanding activity for both reactions. This catalyst also maintains its performance with high stability over 50 h and demonstrates a low charge transfer resistance. In terms of overall water splitting in the same medium, Ni@CNTs–MoxC/Ni2P achieves a potential of 1.56 V at 10 mA cm−2, maintaining its excellent activity and stability for 50 h, showcasing its efficiency and durability in electrolysis applications. Ni@CNTs–MoxC/Ni2P is composed of hierarchically structured molybdenum carbide/nickel phosphide micro-pillar heterostructures, which are bridged with nickel nanoparticles encapsulated in carbon nanotubes that can enhance the conductivity, provide abundant multiple electroactive sites and an optimal electronic structure to elevate the density of states near the Fermi level, afford an optimal adsorption free energy, and enhance the performance for seawater electrolysis.
Moreover, Kang et al.111 reported that RuMoNi achieved an overpotential of 291 mV at 100 mA cm−2 for the OER in 1 M KOH + seawater, highlighting its exceptional activity. Impressively, it maintains strong stability over 3000 h at 500 mA cm−2 and demonstrates a low charge transfer resistance, underscoring its efficiency and endurance under electrolysis conditions. RuMoNi exhibits the in situ formation of molybdate ions on its surface, which can repel chloride ions, and enhance the corrosion resistance and performance for seawater electrolysis. An anion exchange membrane electrolyzer with the RuMoNi catalyst delivers a huge current density of 1000 mA cm−2 at 1.72 V with an energy conversion efficiency of 77.9%, exhibiting a GGE of $0.85, which is far lower than the GGE of $2.0 ($2.0 GGE: 2026 technical target of the United Stated Department of Energy; GGE: price per gallon of gasoline equivalent for H2 generation).
5. Photocatalytic water splitting
5.1. Nano-photocatalysts with a heterojunction/cocatalyst loading/defect engineering/homojunction
Spatial separation of suitable cocatalysts on a Z-scheme heterostructure can enhance the surface reaction rate and make charge separation and transfer of the photogenerated electrons and holes efficient, which can enhance photocatalytic H2 evolution. Moon et al.7 prepared a Z-scheme Pt/g-C3N4/TiO2/IrOx (PCTI) hollow sphere photocatalyst through electrostatic interactions. PCTI with efficient light absorption and interfacial charge separation abilities shows a much higher H2 evolution rate (8.15 mmol h−1 g−1; Table 9) and a considerably higher apparent quantum yield (24.3% at 330 nm) with 0.5 wt% Pt cocatalyst on g-C3N4 and 1.2 wt% IrOx cocatalyst on TiO2. Moreover, to further facilitate the reaction kinetics and surface charge separation, Pt and IrOx, as two cocatalysts, have been spatially divided along the Z-scheme charge-transfer pathway. The photoelectrochemical measurements demonstrate the role of Pt and IrOx cocatalysts on g-C3N4/TiO2. The Pt and IrOx cocatalysts loaded on g-C3N4/TiO2 (PCTI) exhibit significantly higher transient photocurrent density when compared to that of other cocatalysts loaded on g-C3N4/TiO2 as shown in Fig. 9(a), where the chopped photocurrent–time response of the photocatalysts was recorded at 0.0 V versus Ag/AgCl in 0.5 M Na2SO4 (sequential illumination: 20 s on/off time interval). Enhanced surface charge separation of PCTI is observed, where the photogenerated electrons and photoexcited holes can be trapped at Pt on the external surface of CT and IrOx on the internal surface of CT, respectively, which could enhance the performance for the water redox reaction. Moreover, the Pt and IrOx cocatalysts loaded on g-C3N4/TiO2 (PCTI) exhibit lower charge transfer resistance (Rct = 1.52 kΩ) when compared to that of other cocatalysts loaded on g-C3N4/TiO2 as shown in Fig. 9(b), for Nyquist plots obtained at 0.6 V versus Ag/AgCl in 0.5 M Na2SO4 under light irradiation. This suggests a faster charge transfer process in PCTI. These results further reveal efficient charge separation and transfer for spatially separated cocatalysts. The spatial charge separation in PCTI under irradiation was further demonstrated by photo-KPFM analysis (KPFM: Kelvin probe force microscopy). Fig. 9(c) depicts the KPFM topography mapping of PCTI, while the corresponding surface potential (SP) mapping of PCTI in darkness and under light irradiation is depicted in Fig. 9(d) and (e), respectively. Moreover, Fig. 9(f) depicts ΔSP (difference in SP) of PCTI in comparison with TI and CTI, which is calculated based on structural evolution to analyze the photoresponse and photogenerated electron–hole pairs of the photocatalyst. In general, if light strikes a semiconductor, the photoexcited electrons can accumulate at the material's surface, leading to the negative shift in SP, whereas a positive shift in SP can be observed if the electron density decreases. The ΔSP of 40 mV (highly increased SP) for TI after light illumination denotes a decrease in the electron density at the surface, suggesting the transfer of electrons to the internal IrOx particles from the external TiO2 shell due to the large work function of IrOx. Nevertheless, the ΔSP of 6 mV (slightly increased SP) for CTI is much lower than that of the ΔSP of 40 mV (highly increased SP) for CTI after light illumination due to the formation of a heterojunction of TI with g-C3N4, where the photoexcited electrons can be accumulated on the outermost g-C3N4 layer and the inward migration of electrons can be decreased because of the Z-scheme charge transfer. In contrast, the ΔSP of −33 mV (highly decreased SP) for PCTI after light illumination denotes that photoexcited electrons can migrate to g-C3N4 and the external PCTI surface, where the migrated electrons can be particularly trapped by the Pt cocatalyst. The efficiency of the H2 evolution rate can be primarily determined by the surface reaction rate and the efficiency of photogenerated carrier separation and migration. Thus, the significantly higher H2 evolution rate (8.15 mmol h−1 g−1) of PCTI can be attributed to the following factors, as shown in Fig. 9(g) and (h): (1) enhanced charge separation and migration efficiency at the bulk phase and surface because of the synergy of the Z-scheme CT heterostructure; (2) separately loaded Pt and IrOx cocatalysts. A built-in electric field can be generated adjacent to the PCTI heterojunction because of the mismatch in Fermi energy levels in the case of Z-scheme charge transfer within PCTI, leading to the accumulation of electrons and holes at the interfaces, which can cause band bending. The efficient separation and transportation of photogenerated electrons and holes can occur due to interfacial band bending and the internal electric field, which can retain a strong reduction and oxidation ability, where the electrons are in the conduction band of g-C3N4 and holes are in the valence band of TiO2. Moreover, the separately loaded Pt and IrOx cocatalysts on the surfaces of the hollow spheres can enhance the surface reaction kinetics, where the cocatalysts can decrease surface recombination and reduce the activation energy for oxidation and reduction reactions in water splitting. If Pt and IrOx cocatalysts are loaded in reverse, then this could reverse the reactions to retard water splitting. Hence, the loading of Pt on g-C3N4 and IrOx on the TiO2 surface could be essential to the enhancement of the photocatalytic H2 evolution activity. | ||
| Fig. 9 (a) Chopped photocurrent–time response of PCTI in comparison with those of other photocatalysts recorded at 0.0 V versus Ag/AgCl in 0.5 M Na2SO4, where sequential illumination of 20 s on/off time intervals were used. (b) Nyquist plot of PCTI in comparison with other photocatalysts obtained at 0.6 V versus Ag/AgCl in 0.5 M Na2SO4 under light irradiation (inset depicts the corresponding equivalent circuit). (c) Kelvin probe force microscopy (KPFM) topography mapping of PCTI, (d) corresponding surface potential (SP) mapping of PCTI in darkness, and (e) corresponding SP mapping of PCTI under light irradiation. (f) ΔSP of PCTI in comparison with TI and CTI, which was calculated based on evolution of the structure. (g) Schematic diagram illustrating the physical structure of PCTI. (h) Schematic diagram illustrating the charge-transfer process within PCTI under light irradiation (reproduced with permission from ref. 7. Copyright 2022, Wiley-VCH GmbH). | ||
| Photocatalysts with/without cocatalysts | Light source | Reaction medium | H2 evolution rate (mmol h−1 g−1) | AQY | Ref. |
|---|---|---|---|---|---|
| Ref.: reference; NA: not applicable; AQY: apparent quantum yield; TSP: twin S-scheme photocatalyst comprising of 2D graphitic carbon nitride nanosheets, which is inserted between anatase TiO2 nanoparticles and H-doped rutile TiO2 nanorods, HAp: hydroxyapatite; CIZS/MoS2/CDs: Cu–In–Zn–S quantum dots/MoS2/carbon dots; QDs: quantum dots; CDs/CCN: carbon dots/C-doping g-C3N4; 21.3a%@420 nm for Au@MoS2; Au@MoS2–ZnOb: piezo-photocatalyst (H2 evolution was obtained under light and ultrasonic waves); 32.87c: photothermal-assisted photocatalytic H2 evolution rate of 32.87 mmol h−1 g−1; 19.654d: photothermal-assisted photocatalytic H2 evolution rate of 19.654 mmol h−1 g−1. | |||||
| N–NaTaO3@Ta3N5/Rh@Cr2O3 | 300 W Xe lamp | Pure water | 25.12 | 9.32%@350 nm | 11 |
| Ru–In SA/TiO2 | 300 W Xe lamp (λ ≥ 300 nm) | Pure water | ∼4.8362 | NA | 8 |
| PCN–Ni–CoP | 300 W Xe lamp | Pure water | 0.143 | 2.7%@380 nm | 116 |
| Ni@NiSx–CdS | 300 W Xe lamp (λ > 420 nm) | Na2S + Na2SO3 | 78.7 | 36.74%@420 nm | 117 |
| NiSeS/ZnSe | Visible light (λ > 400 nm) | Ascorbic acid | 73.54 | 57.68@400 nm | 118 |
| TSP | 300 W Xe lamp (200 nm < λ < 1000 nm) | Methanol | 62.37 | 45.9%@365 nm | 115 |
| Pt–CdSx–T | 300 W Xe lamp (λ ≥ 420 nm) | Lactic acid | 42.7 | 43.6%@470 nm | 119 |
| RuNi/g-C3N4 | 300 W Xe lamp (>400 nm) | Triethanolamine | 35.1 | 1.27%@380 nm | 120 |
| ZrC@ZnIn2S4 | 300 W Xe lamp (>800 nm) | Triethanolamine | 32.87c | NA | 121 |
| NiCo2S4/ZnIn2S4 | 300 W Xe lamp (λ > 420 nm) | Ascorbic acid | 19.654d | 56.7%@400 nm | 17 |
| 15% CoS2/CdS-L | 5 W LED | Lactic acid | 19.22 | 22.6%@420 nm | 55 |
| Au@MoS2–ZnOb | AM 1.5 G filter | Na2SO3 | 13.09 | 21.3a%@420 nm | 122 |
| Ru@Nb2O5/Nb2C | 300 W Xe lamp | Glycerol | 10.11 | 41.25%@313 nm | 123 |
| Zn–Ni5P4/CdS | 300 W Xe lamp | Lactic acid | 8.969 | NA | 124 |
| CDs/CCN | 300 W Xe lamp | Triethanolamine | 8.682 | 1.41%@420 nm | 125 |
| Pt/g-C3N4/TiO2/IrOx | 300 W Xe lamp | Methanol | 8.153 | 24.3%@330 nm | 7 |
| Protonated NH2-MIL-125 | 300 W Xe lamp | Acetonitrile + triethanolamine | 6.7628 | NA | 126 |
| CN/CNQDs | 300 W Xe lamp | Triethanolamine | 4.0877 | 6.74%@420 nm | 127 |
| CIZS/MoS2/CDs | LED light (λ ≥ 420 nm) | L-Ascorbic acid | 3.706 | 34.23%@450 nm | 128 |
| TiO2–Pt/FeHAp | 300 W Xe lamp | Triethanolamine | 3.026 | 4.72%@370 nm | 129 |
| PtO@Ti3C2/TiO2 | 300 W Xe lamp | Methanol | 2.54 | 4.2%@365 nm | 130 |
| Amorphous-NiB/TiO2 | 4 LED lamps (3 W; 365 nm) | Lactic acid | 2.334 | 16.30% | 131 |
| Bi20TiO32/Bi4Ti3O12 | 250 W Xe lamp (λ > 400 nm) | Methanol | 1.89 | NA | 132 |
| BOC–CNF@CNT | 300 W Xe lamp | Triethanolamine | 0.02542 | NA | 133 |
Using only pure water without any sacrificial agent, Lu et al.11 observed that the N–NaTaO3@Ta3N5/Rh@Cr2O3 photocatalyst demonstrated a remarkably high photocatalytic H2 evolution rate of 25.12 mmol h−1 g−1 when used for overall photocatalytic water splitting, achieving a high apparent quantum yield of 9.32% at 350 nm. Furthermore, this photocatalyst exhibits a low charge transfer resistance. The ultrathin Ta3N5 shell (thickness: ≈5 nm) can enhance the lifetimes of the photogenerated charges, while the core–shell heterojunction with type-I band alignment can enable smooth charge flow from N–NaTaO3 to Ta3N5, especially with the aid of the shared communal Ta atoms at the interface, and the efficiency can be further enhanced by the in situ deposition of a core–shell structured Rh@Cr2O3 cocatalyst.
Moreover, Ruan et al.115 synthesized a twin S-scheme (step scheme) photocatalyst (TSP) comprised of 2D graphitic carbon nitride nanosheets, which were inserted between anatase TiO2 nanoparticles and H-doped rutile TiO2 nanorods. The TSP photocatalyst shows an exceptionally high H2 evolution rate of 62.37 mmol h−1 g−1 and an impressive quantum yield of 45.9% at 365 nm, while maintaining a low charge transfer resistance. The enhanced photocatalytic performance of TSP is ascribed to optimal charge separation and transfer due to the unique twin S-scheme structure. Time-resolved characterization studies reveal that rapid electron trapping and suppressed recombination of photogenerated carriers contribute to TSP enhanced carrier separation and transfer efficiency. Moreover, the S-scheme pathway of charge transfer enables electrons and holes to remain in the CB of C3N4 and the VB of TiO2, respectively, ensuring a robust redox ability.
In addition, Wang et al.55 observed that the 15% CoS2/CdS-L photocatalyst exhibited a much higher photocatalytic H2 evolution rate (19.22 mmol h−1 g−1) with a much higher apparent quantum yield of 22.6% at 420 nm, while it exhibited a low charge transfer resistance. Theoretical and experimental studies reveal that the fully exposed (002) surface of CdS-L (leaf-like CdS) possesses superior activity for H2 evolution, while the electron flow between the CdS-L and CoS2 S-scheme heterojunction can be efficiently enhanced by the utilization of the interface heterojunction strategy, which can tune the potential barrier difference. Thus, the design of a photocatalyst with more exposed crystal planes on the basis of fabricating S-scheme heterojunctions can afford abundant active sites leading to enhanced reactivity.
Das et al.132 discovered that the Bi20TiO32/Bi4Ti3O12 photocatalyst achieved a remarkable photocatalytic hydrogen evolution rate of 1.89 mmol h−1 g−1, accompanied by a low charge transfer resistance. This enhanced performance is attributed to a synergistic type-I bridged coupled Z-scheme electron migration process, facilitating prolonged excited state lifetimes, increased interfacial contact, rapid electron channeling, and improved optical absorption. Similarly, Shu et al.129 found that the TiO2–Pt/FeHAp (hydroxyapatite) photocatalyst reached a high hydrogen evolution rate of 3.026 mmol h−1 g−1, with an impressive apparent quantum yield of 4.72% at 370 nm, while also displaying a low charge transfer resistance. This indicates its efficiency for photocatalytic hydrogen production. TiO2–Pt/FeHAp is obtained by the following steps: first, TiO2–Pt was prepared by photoreduction of 1 wt% Pt clusters onto sub-15 nm TiO2 nanoparticles using 20 vol% methanol. Finally, TiO2–Pt/FeHAp was obtained by an electrostatic assembly method. Thus, a built in electric field is formed in the TiO2–Pt/FeHAp heterojunction, which drives the photogenerated electrons to flow from FeHAp on TiO2 nanoparticles to the Pt cocatalyst via a type-II path. The improved photocatalytic performance of TiO2–Pt/FeHAp is ascribed to enhanced interfacial charge migration and separation due to the heterostructure, a large specific surface area stemming from sub-15 nm TiO2, and facile H2 production kinetics associated with the Pt cocatalyst.
Moreover, Chen et al.128 used Cu–In–Zn–S quantum dots (CIZS QDs), MoS2 and carbon dots (CDs) to construct the CIZS/MoS2/CDs photocatalyst, which showed an impressive photocatalytic H2 evolution rate (3.706 mmol h−1 g−1) with a high apparent quantum yield of 34.23% at 450 nm and a low charge transfer resistance. The transient photovoltage measurements reveal that MoS2 has a negligible effect on the charge extraction rate, whereas the CDs significantly elevate the attenuation constant of the charge recombination process from 0.178 ms to 0.260 ms because of its electron sink effect. In addition, various studies along with an electrocatalytic investigation reveal that MoS2 plays a vital role in the hydrogen evolution reaction, whereas CIZS facilitates a light harvesting process and CDs enhance the charge sink effect.
Xu et al.123 demonstrated that the Ru@Nb2O5/Nb2C photocatalyst achieved a remarkable hydrogen evolution rate of 10.11 mmol h−1 g−1, coupled with an impressive apparent quantum yield of 41.25% at 313 nm, and maintained a low charge transfer resistance, highlighting its superior photocatalytic efficiency. The Ru@Nb2O5/Nb2C catalyst was synthesized through a one-pot hydrothermal method, during which the Nb2CTx MXene's reducibility was crucial to the oxidation process, actively producing H2 from water splitting. This process reduces Ru3+ to Ru nanoparticles in situ, which are then deposited on Nb2O5 nanowires formed through the partial hydrothermal oxidation of Nb2CTx, illustrating the seamless integration of catalytic materials for enhanced photocatalytic hydrogen production. The valence state of the noble metal Ru/Pt obtained through this one-pot hydrothermal synthesis method was closer to the metallic state when compared to that of a traditional photodeposition method, and this decreased the charge transfer resistance by 82.5%.
Han et al.120 demonstrated the RuNi/g-C3N4 photocatalyst's outstanding performance, delivering a hydrogen evolution rate of 35.1 mmol h−1 g−1 and an apparent quantum yield of 1.27% at 380 nm. This catalyst also features a low charge transfer resistance, highlighting its efficacy for photocatalytic hydrogen generation. RuNi/g-C3N4 with about 2.3% Ru content exhibits a higher photocatalytic H2 evolution rate (>twice) than that of the Pt/g-C3N4 photocatalyst with about 2.3% Pt content under simulated sunlight irradiation. It is composed of a 2D structured bimetallic RuNi alloy as a co-catalyst of g-C3N4 nanosheets. The integration of RuNi with g-C3N4 effectively reduces the transfer barrier for photogenerated electrons and provides ample reaction sites for photocatalytic H2 generation. Additionally, the synergistic interaction between the Ni and Ru metals in the RuNi alloy enhances its performance as a co-catalyst, outperforming Pt in this role.
Besides, Yang et al.130 found that the PtO@Ti3C2/TiO2 photocatalyst achieved a notable hydrogen evolution rate of 2.54 mmol h−1 g−1 and an apparent quantum yield of 4.2% at 365 nm, showcasing its effectiveness at photocatalytic hydrogen production alongside featuring a low charge transfer resistance. The in situ grown TiO2 nanosheets on Ti3C2 MXene can enhance charge separation, while the deposited PtO nanodots with a higher oxidation state can decrease the hydrogen back oxidation reaction, and lead to improved photocatalytic H2 generation. In the PtO@Ti3C2/TiO2 photocatalyst, photogenerated electrons and holes on TiO2 are transferred to PtO and Ti3C2, respectively. Simultaneously, photogenerated holes from PtO migrate to Ti3C2, while electrons remain on PtO, facilitating efficient charge separation. Then, the holes on Ti3C2 can be consumed by reacting with the sacrificial agent methanol, while the electrons on PtO can react with protons to form H2, where the hydrogen back oxidation reaction can be suppressed.
Moreover, Zhang et al.117 reported that the Ni@NiSx–CdS photocatalyst achieved an impressive hydrogen evolution rate of 78.7 mmol h−1 g−1 and an apparent quantum yield of 36.74% at 420 nm, demonstrating efficient charge separation with a low charge transfer resistance. The crystalline CdS nanorod was prepared by a solvothermal method, while the amorphous Ni@NiSx co-catalyst was prepared and assembled onto CdS through a partial reduction strategy. The Ni@NiSx–CdS photocatalyst exhibits a unique combination of amorphous and crystalline phases, resulting from the encapsulation of Ni clusters within an amorphous NiSx layer. Its gradient work function variation enhances the charge extraction efficiency, effectively suppressing carrier recombination. Furthermore, it regulates the S p-band center away from the Fermi level, optimizing the Gibbs free energy for atomic H2 adsorption.
In addition, research by Ma et al.127 shows that the CN/CNQD (quantum dot) photocatalyst delivers a notable hydrogen evolution rate of 4.0877 mmol h−1 g−1 and achieves an apparent quantum yield of 6.74% at 420 nm, showcasing effective charge separation, as indicated by its low charge transfer resistance. The CN/CNQD composite is constructed from a planar carbon nitride (CN) structure, which is seamlessly integrated with carbon nitride quantum dots (CNQDs). This unique composite is achieved through a novel phototriggered self-assembly strategy, where covalent bonds are forged at the atomic junctions between CN and CNQDs. This approach differs from conventional interfacial modification methods that rely solely on hydrogen bonding or van der Waals forces. Consequently, the CN/CNQD hybrid boasts superior interfacial interactions, enabling efficient carrier migration between its components. Additionally, the presence of a continuous π-conjugated structure within CN/CNQDs provides an ideal milieu for carrier transport. Furthermore, an inherent in-plane electric field acts as a driving force for the directional transfer of electrons and holes, thereby enhancing the separation and transportation of photogenerated carriers.
Besides, Long et al.131 observed that the amorphous-NiB/TiO2 photocatalyst exhibited a much higher photocatalytic H2 evolution rate (2.334 mmol h−1 g−1) with a much higher apparent quantum yield of 16.30%, while it exhibited a low charge transfer resistance. Amorphous-NiB/TiO2 is composed of amorphous NiB nanodots (0.5 to 1 nm), which are anchored onto the TiO2 surface through a novel light-induced route. Theoretical and experimental investigations reveal that the amorphization of NiB can induce reverse electron transfer from B to Ni, which contributes to an optimal electronic structure on Ni active sites and facilitates the hydrogen desorption process on the Ni active sites, leading to enhanced efficiency for H2 generation.
Moreover, Zeng et al.118 observed an exceptionally high photocatalytic H2 evolution rate of 73.54 mmol h−1 g−1 for the NiSeS/ZnSe photocatalyst, coupled with a noteworthy apparent quantum yield of 57.68% at 400 nm, while maintaining a low charge transfer resistance. The NiSeS cocatalyst with dual Ni–Se vacancies is integrated with ZnSe to obtain NiSeS/ZnSe heterojunctions, where a local polarization electric field can be induced by the dual Ni–Se vacancies, greatly enhancing surface charge transfer and effectively reducing the reaction barrier within the photocatalytic process.
Tang et al.119 precisely anchored Pt sub-nanoclusters (SNCs) onto the tandem CdS homojunction (CdSx-T) through an in situ photoreduction route. The CdSx-T photocatalyst, enriched with coordination-unsaturated S atoms, serves as an ideal anchor for Pt single nanoclusters (SNCs), promoting the formation of interfacial Pt–S bonds. These bonds enhance the directional transfer of photocarriers at the metal–semiconductor interface. Additionally, the CdSx-T homojunction, characterized by a tandem potential barrier, promotes the efficient separation of photocarriers through an internal electrostatic field. The size of Pt SNCs plays a pivotal role in the hydrogen ion (H*) adsorption capacity and electron transfer kinetics at the metal–semiconductor interface. Consequently, the Pt–CdSx-T photocatalyst demonstrates an impressive photocatalytic hydrogen evolution rate of 42.7 mmol h−1 g−1, along with a remarkable apparent quantum yield of 43.6% at 470 nm. Furthermore, it exhibits a low charge transfer resistance, highlighting its superior photocatalytic performance.
The sluggish kinetics of photocatalytic H2 evolution can be enhanced by efficient replenishment through the photothermal effect, where hot electrons can produce a photothermal effect by means of phonon–phonon scattering. Nevertheless, the hot electrons have short lifetimes due to fast relaxation kinetics, leading to a low energy utilization efficiency. The fabrication of hollow-structured ZnIn2S4 can enhance the efficiency of interfacial electron transfer at the NiCo2S4/ZnIn2S4 photocatalyst junction, extending the hot electron lifetimes for efficient solar energy utilization and significantly improving the hydrogen evolution rate. Guo et al.17 observed that the NiCo2S4/ZnIn2S4 hollow nanocage (NCS/ZIS-HNC) photocatalyst demonstrated a remarkable hydrogen evolution rate of 19.654 mmol g−1 h−1 and an impressive apparent quantum efficiency of 56.7% at 400 nm, when exposed to visible and near-infrared (NIR) light. Hollow-structured ZnIn2S4 with surface S vacancies obtained by a soft template method can control the electron accumulation/consumption ability of interfacial atoms to enhance the interfacial interaction with NiCo2S4. Hollow-structured ZnIn2S4 enhances the number of photogenerated electrons through multiple light scattering, facilitating their efficient transfer at the interface with NiCo2S4. This transfer accumulates photogenerated electrons on the surface of NiCo2S4, which are further excited to hot electrons by the localized surface plasmon resonance (LSPR) effect. This process continuously generates hot electrons by intrinsic excitation, prolonging their lifetimes for efficient solar energy utilization. Fig. 10(h) depicts the photocatalytic hydrogen evolution rates of NiCo2S4/ZnIn2S4 in comparison with its counterparts in the presence and absence of cooling water at an indoor temperature of 25 °C. NiCo2S4/ZnIn2S4 in the absence of cooling water exhibits a H2 evolution rate of 19.654 mmol g−1 h−1, which is 5.98 times higher than that in the presence of cooling water (3.2838 mmol g−1 h−1). This further demonstrates that the photothermal effect plays a pivotal role in the substantial enhancement of the H2 evolution rate for the NiCo2S4/ZnIn2S4 photocatalyst. Moreover, compared to ZIS-HNCs, lower charge transfer resistance and higher transient photocurrent density were obtained for NCS/ZIS-HNCs, suggesting effective migration of photogenerated electrons at the interface. The high resolution Zn 2p XPS spectrum obtained for NCS/ZIS-HNCs (Fig. 10(a)) shows higher binding energies compared to those of ZIS-HNCs, indicating the existence of strong interfacial interactions between ZIS-HNCs and NCS. ZIS-HNCs were derived from ZIF-8-YS, and therefore ZIF-8-YS was characterized. Fig. 10(b) depicts the ESR spectrum of ZIF-8-YS in comparison with that of ZIF-8. A noticeable ESR response at a g-value of 2.004 is observed for ZIF-8-YS, which can be ascribed to the generation of abundant defects during the hollowing process. In this process, the organic frameworks coordinated with Zn2+ can be destroyed by free H+ ions released from tannic acid to generate abundant defects, leading to a disordered crystal structure for ZIF-8-YS. Moreover, even after releasing H+ ions, tannic acid possesses numerous electrons, enabling it to form a new coordination bond with Zn2+. This, in turn, partially mitigates the subsequent sulphuration reaction of thioacetamide. When compared to ZIS, ZIS-NCs, and NCS, both NCS/ZIS-HNCs and ZIS-HNCs exhibit a pronounced ESR response at a g-value of 2.002 (Fig. 10(c)), which is attributed to sulfur vacancies resulting from unpaired electrons with low spin states in the S 3p orbital, suggesting the formation of sulfur vacancies around Zn atoms during the preparation of hollow-structured ZIS. In addition, DFT calculations were conducted to investigate the role of sulfur vacancies in enhancing strong interfacial interactions. Fig. 10(d) depicts the ZIS crystal structure with the S vacancy formation energy at different sites, which reveals that the S vacancies around Zn atoms have a lower formation energy of 4.47 eV, compared to that around In atoms (4.74 eV). Fig. 10(e) compares the electron density differences of NCS/VS-ZIS and NCS/ZIS. Both NCS/VS-ZIS and NCS/ZIS afford electron consumption on ZIS (cyan region) and electron accumulation on NCS (yellow region). Notably, NCS/VS-ZIS with S vacancies shows higher interfacial electron accumulation/consumption as shown in Fig. 10(f), suggesting a stronger interfacial interaction caused by S vacancies. Moreover, the Bader charge of interfacial S atoms was calculated to understand this phenomenon. As shown in Fig. 10(g), a decreasing trend is observed for the Bader charges of all interfacial S atoms (labeled as SZ1–SZ6) at the ZIS part, whereas an increasing trend is observed for all the S atoms (labeled as SN1–SN6) at the NCS part. This suggests that the interfacial S atoms with S vacancies can improve the electron accumulation/consumption ability and enhance efficient transfer of more photogenerated carriers. Subsequently, the transferred electrons are excited to hot electrons through the LSPR effect to constantly enable the intrinsic excitation of NCS, promoting the photothermal effect and ultimately enhancing photocatalytic H2 evolution.
 | ||
| Fig. 10 (a) High resolution XPS spectra of Zn 2p obtained from NCS/ZIS-HNCs in comparison with its counterparts; (b) ESR spectrum of ZIF-8-YS in comparison with that of ZIF-8; (c) ESR spectrum of NCS/ZIS-HNCs in comparison with those of its counterparts; (d) ZIS crystal structure depicting the S vacancy formation energy at different sites; (e) electron density difference of NCS/VS-ZIS (right) in comparison with that of NCS/ZIS (left); (f) planar-averaged electron density difference Δρ(z) of NCS/VS-ZIS in comparison with that of NCS/ZIS; (g) Bader charge of interfacial atoms of NCS/VS-ZIS in comparison with that of NCS/ZIS; (h) photocatalytic hydrogen evolution rates of NCS/ZIS-HNCs in comparison with those of its counterparts in the presence and absence of cooling water (reproduced with permission from ref. 17. Copyright 2023, The Royal Society of Chemistry). | ||
Shi et al.121 prepared ZrC@ZnIn2S4 through a facile water bath method. The formation of a Schottky junction in the ZrC@ZnIn2S4 photocatalyst can kinetically accelerate photogenerated carrier transfer and separation, enhancing photocatalytic water splitting for H2 formation. Moreover, the photothermal effect induced by the LSPR effect (LSPR: local surface plasmon resonance) of metallic ZrC particles can thermodynamically decrease the activation energy of the reaction and enable the forward reaction in the ZrC@ZnIn2S4 heterostructure system. Finally, a desirable H2 evolution rate (32.87 mmol h−1 g−1) with a much higher photothermal conversion efficiency of 43.54% using NIR irradiation (λ > 800 nm) was obtained. Alternatively, Guo et al.122 observed that the Au@MoS2–ZnO piezo-photocatalyst exhibited a much higher piezo-enhanced photocatalytic H2 evolution rate (13.09 mmol h−1 g−1) under light and ultrasonic waves, but it exhibited a low charge transfer resistance. It was obtained by a hydrothermal method. DFT calculations and photoelectric characterization reveal that the ultrasonic field's spatial compression and cavitation effect can enhance the dynamics of mass transfer. Specifically, the cavitation effect enhances the chemical bond breakage–reformation process, thereby accelerating the surface redox reaction, and the ultrasonic field can tune the built-in electric field, thereby facilitating transfer of the photoinduced carriers.
5.2. Nano-photocatalyst with elemental doping/metal free/carbon based materials/MOFs
A metal-free photocatalyst can be prepared by integrating melon with highly reduced graphene oxide, which could enhance the visible light utilization, narrow the bandgap, enable charge redistribution at the interface, and improve the performance for photoelectrochemical hydrogen evolution. Ashraf et al.54 demonstrated that photoelectrochemical hydrogen evolution could be enhanced by bandgap engineering of melon with highly reduced graphene oxide (HRG), where melon is the uncondensed form of polymeric carbon nitrides. The 1% HRG@melon nanocomposite exhibits a higher photocurrent density (71 μA cm−2) than that of melon (24 μA cm−2) under alkaline conditions, while the photocurrent density is further increased to 630 μA cm−2 with the addition of triethanolamine as a hole scavenger, and it exhibits a low charge transfer resistance. The addition of HRG can reduce the bandgap of melon from 2.6 eV to 2.4 eV, enhance visible light utilization and charge redistribution at the interface between melon and HRG, and finally lead to improved performance for photoelectrochemical hydrogen evolution.The decoration of TiO2 with Ru/In dual-single atoms can produce oxygen vacancies and elevate the Ti3+ concentration, accelerating charge separation and ultimately promoting photocatalytic H2 evolution. Peng et al.8 observed that Ru–In SA/TiO2 (TiO2 doped with Ru/In single atoms) could alter the separation of photogenerated carriers during photocatalytic H2 evolution using pure water. Ru–In SA/TiO2 exhibits a much higher photocatalytic H2 evolution rate of 174.1 μmol h−1 and a lower charge transfer resistance than those of In SA/TiO2, Ru SA/TiO2, and TiO2. The decoration of Ru/In dual-single atoms on TiO2 increases the oxygen vacancies and elevates Ti3+, leading to accelerated charge separation. Femtosecond transient absorption spectroscopy reveals that the distribution separation of photogenerated electrons and holes of TiO2 are accelerated by doping with Ru and In single atoms, respectively.
Besides, a carbon doped and carbon dot loaded graphitic carbon nitride (CDs/CCN) photocatalyst was obtained by Yuan et al. using a direct thermal polymerization route.125 CDs/CCN shows a boosted photocatalytic H2 evolution rate (8.682 mmol h−1 g−1) with a high apparent quantum yield of 1.41% at 420 nm. Transient absorption spectroscopy investigations reveal that C-doping of g-C3N4 induced shallow trap states that could confine photoinduced electrons to alleviate direct recombination and deep trapping of photogenerated carriers. The integration of CDs with CCN can create a fast hole transfer path and provide additional reaction sites for the oxidation of triethanolamine (serving as a sacrificial agent) via extracted holes, giving rise to efficient spatial separation of electron–hole pairs.
Moreover, Luo et al.124 found that doping Ni5P4 with Zn could lower its capacitance and enhance the release of photoexcited electrons. Besides, Zn-doped Ni5P4 exhibits a low overpotential and high specific surface area, which can provide abundant active sites and facilitate the kinetics for H2 production. The constructed Zn–Ni5P4/CdS photocatalyst demonstrates an exceptionally high rate of photocatalytic H2 evolution (8.969 mmol h−1 g−1), which is 2.4 times higher than that of Ni5P4/CdS (3.668 mmol h−1 g−1). Specifically, Zn doping decreases the capacitance of Ni5P4 from 680 to 540 mF g−1 while enhancing electron transport, which in turn mitigates the negative effects of capacitance and promotes the release of photoexcited electrons.
Zhang et al.116 precisely anchored Ni-doped cobalt phosphide nanoparticles (∼4 nm) on phosphatized carbon nitride nanosheets through an absorption–phosphidation strategy to obtain the PCN–Ni–CoP photocatalyst. It achieves a remarkably high photocatalytic H2 evolution rate of 0.143 mmol h−1 g−1 and impressive apparent quantum yield of 2.7% at 380 nm through overall water splitting, and maintains a low charge transfer resistance. PCN–Ni–CoP is composed of Ni-doped cobalt phosphide nanoparticles (∼4 nm), which are precisely anchored on phosphatized carbon nitride nanosheets through an absorption–phosphidation strategy. In the PCN–Ni–CoP photocatalyst, the CoP nanoparticles with atomic-scale doped Ni element can effectively facilitate the surface catalytic reaction kinetics and act as the active center for the redox reaction to generate H2. The PCN nanosheets with higher electrical conductivity and a lower band gap energy can efficiently enhance photogenerated charge separation and transfer. Moreover, the Co–N coordination effect between Ni–CoP and PCN can also contribute to the acceleration of interfacial electron transfer.
In addition, Zhou et al.133 utilized BiOCl (BOC) semiconductor and cellulose nanofibers@carbon nanotubes (CNF@CNT) to construct a 2D lamellar membrane, BOC–CNF@CNT, which showed a satisfactory photocatalytic H2 evolution rate (0.02542 mmol h−1 g−1) and a low charge transfer resistance. The CNTs effectively function as transfer channels for photogenerated carriers, optimizing the separation of electrons and holes while minimizing interfacial charge transfer resistance and mitigating carrier recombination. Consequently, this enhances the photocatalytic H2 production performance of BOC–CNF@CNT.
Moreover, Luo et al.126 synthesized the NH2-MIL-125 photocatalyst using HCl vapor as a protonating reagent through natural volatilization of hydrochloric acid. The –NH3+Cl− group (a moiety with charge and ample hydrogen) is introduced into NH2-MIL-125, which leads to efficient charge carrier separation and a high photocatalytic H2 evolution rate (6.7628 mmol h−1 g−1). Characterization studies including PL, FT-IR, and in situ DRIFTS demonstrate that the protonation effect not only enhances the electron migration rates but also effectively accelerates the separation of photogenerated electron–hole pairs during the photocatalytic reaction.
6. Photocatalytic HX splitting
6.1. Lead free perovskite based photocatalysts
Photocatalytic H2 production using hydrohalic acid (HX) is a promising strategy, where value-added chemicals (such as X2/X3−) can also be generated along with clean, renewable H2 fuel. However, developing environmentally friendly lead-free photocatalysts with strong acid resistance and a visible light response is a challenging task. The in situ formation of a bismuth-based perovskite heterojunction has recently been proved as an effective way to attain better photocatalytic HI splitting performance. Tang et al.5 successfully fabricated a lead-free bismuth-based hybrid perovskite by utilizing a solvent engineering technique, which enabled the in situ growth of heterojunctions at the interface of MA3Bi2I9 (methylammonium bismuth iodide) and DMA3BiI6 (tri(dimethylammonium)hexa-iodobismuthate). The well-matched type-II heterostructure of BBP-5 gives rise to a much higher H2 evolution rate of 198.2 μmol h−1 g−1 (Table 10; Fig. 11(b)) and apparent quantum efficiency, compared to that of its counterparts, without using noble metal co-catalysts under the illumination of 100 mW cm−2 visible light (λ ≥ 420 nm) in an aqueous hydrogen iodide solution. Besides, there are no side products from the photocatalytic HI splitting (Fig. 11(c)). Moreover, MA3Bi2I9/DMA3BiI6 (BBP-5) can maintain a high photocatalytic H2 evolution activity for 100 h (Fig. 11(d)). Photoelectrochemical characterization studies illustrate that BBP-5 possesses a lower charge transfer resistance (Rct: 1.15 kΩ cm2) and photoluminescence (PL) peak intensity, compared to that of its counterparts (other Bi-based perovskites), suggesting suppression of photogenerated charge recombination and a facilitated charge diffusion rate at the heterojunction interface. BBP-5 exhibits higher PL lifetimes (short-lived lifetime, τ1: 14.63 ns; long-lived lifetime, τ2: 115.67 ns; average lifetimes, τ: 37.98 ns) compared to that of its counterparts, suggesting an extended exciton diffusion lifetime because of the reduction of nonradiative recombination in the MA3Bi2I9/DMA3BiI6 heterojunctions. This reduction can facilitate the H2 evolution activity due to the efficient charge separation of BBP-5 (Fig. 11(a)).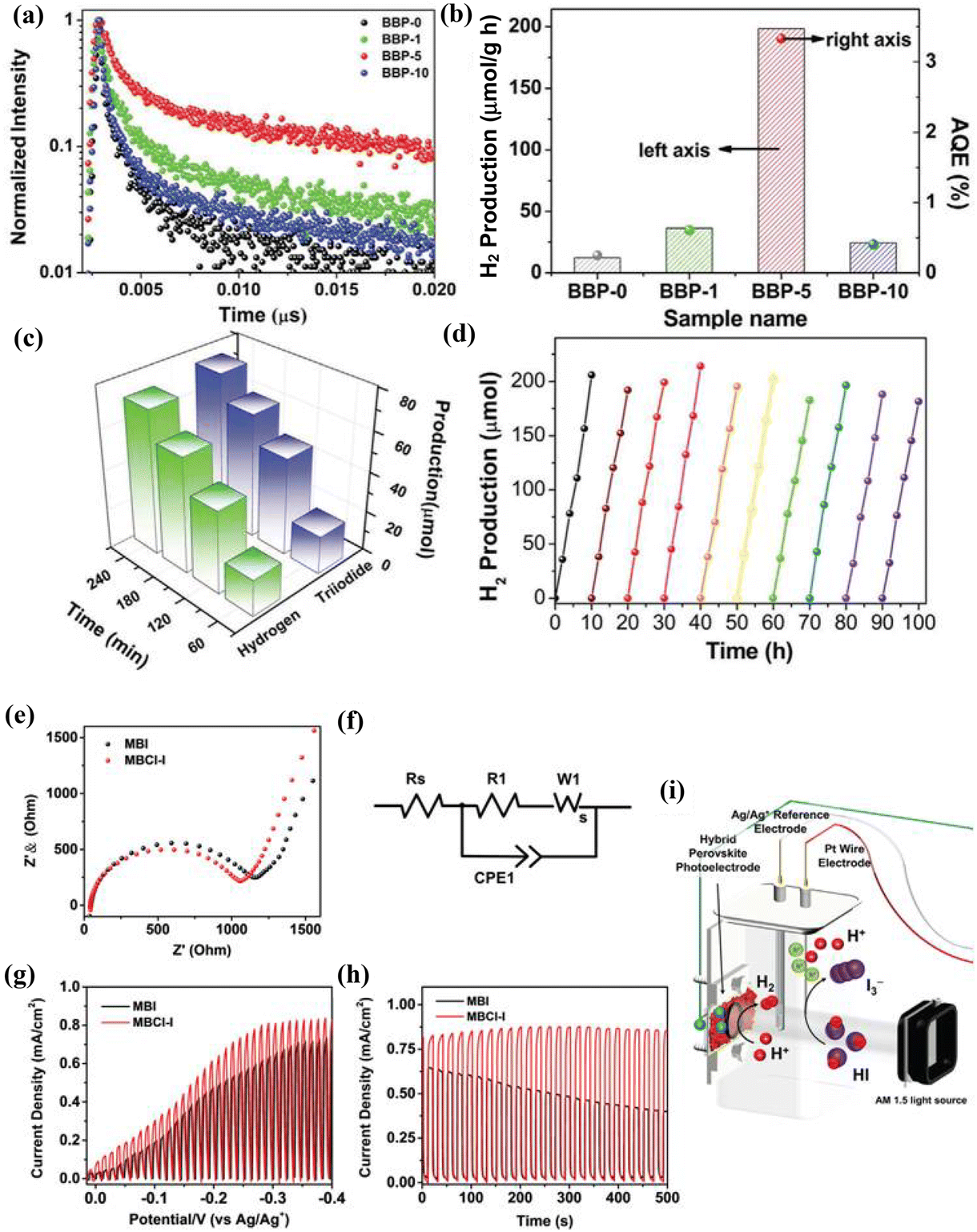 | ||
| Fig. 11 (a) Time-resolved photoluminescence (TRPL) spectrum of BBP-5 in comparison with those of its counterparts obtained at 650 nm; (b) photocatalytic H2 production along with the apparent quantum efficiency (AQE) of BBP-5 in comparison with those of its counterparts in HI saturated solution; (c) comparison of the amount of H2 (green) and I3− (purple) generated over time using the BBP-5 photocatalyst; (d) stable photocatalytic H2 evolution over 100 h using the BBP-5 photocatalyst in HI saturated solution, where H3PO2 was added to the HI solution (reproduced with permission from ref. 5. Copyright 2020, Wiley-VCH GmbH). (e) Nyquist plot of MBCl-I in comparison with that of MBI recorded at 0.14 V versus Ag/Ag+ electrode in CH2Cl2 solution containing 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) under visible-light illumination, where (f) depicts the corresponding equivalent electrical circuit; (g) chopped photocurrent–voltage (J–V) curve of MBCl-I in comparison with that of MBI measured under visible-light irradiation; (h) chopped photocurrent response of MBCl-I in comparison with that of MBI recorded at −0.3 V vs. Ag/Ag+ under visible-light irradiation; (i) schematic diagram illustrating the working principle of the photoelectrochemical HI splitting cell using MBCl-I|FTO (or MBI|FTO) as a photoelectrode (reproduced with permission from ref. 9. Copyright 2022, Wiley-VCH GmbH). | ||
| Pb-free/Pb-based perovskite photocatalysts with/without cocatalysts | Light source | Reaction solution | H2 evolution rate (mmol h−1 g−1) | AQY | Ref. |
|---|---|---|---|---|---|
| Ref.: reference; NA: not applicable; AQY: apparent quantum yield; SA: single atom; aMAPb(I1−xBrx)3: MAPb(I1−xBrx)3 with x = 0.10; BP: black phosphorus; rGO: reduced graphene oxide. | |||||
| MAPbI3/MoS2 | 280 W Xe lamp (λ ≥ 420 nm) | HI and H3PO2 | 29.389 | 22.1%@500 nm | 134 |
| MoSe2/MAPbBr3−xIx | 300 W Xe lamp (λ > 420 nm) | HBr, HI and H3PO2 | 22.935 | 37.67%@530 nm | 10 |
| PtSA/FAPbBr3−xIx | 300 W Xe lamp (AM 1.5G) | HBr, HI and H3PO2 | 6.826 | 33.4%@530 nm | 32 |
| MAPbI3/BP | 300 W Xe lamp (λ ≥ 420 nm) | HI and H3PO2 | 3.742 | 23.2%@420 nm | 135 |
| Ni3C/MAPbI3 | 300 W Xe lamp (λ ≥ 420 nm) | HI and H3PO2 | 2.362 | 16.6%@420 nm | 136 |
| MAPbI3/CoP | 150 W Xe lamp (λ ≥ 420 nm) | HI and H3PO2 | 2.0875 | NA | 137 |
| MAPbI3/MoS2 | 9 LED lamp (10 W; 380 nm ≤ λ ≤ 780 nm) | HI and H3PO2 | ∼2.061 | NA | 138 |
| aMAPb(I1−xBrx)3 | 300 W Xe lamp (λ ≥ 420 nm) | HI, HBr and H3PO2 | 1.471 | NA | 2 |
| CH3NH3PbI3/rGO | 300 W Xe lamp (λ > 420 nm) | HI and H3PO2 | 0.9389 | 1.4%@450 nm | 139 |
| MA3Bi2Cl9−xIx/Pt | 300 W Xe lamp (λ ≥ 420 nm) | HCl, HI and H3PO2 | ∼0.682 | 1.25%@435 nm | 140 |
| PtSA/Cs2SnI6 | 300 W Xe lamp (λ ≥ 420 nm) | HI and H3PO2 | 0.43 | NA | 18 |
| Cs2AgBiBr6/N–C | 300 W Xe lamp (λ ≥ 420 nm) | HBr and H3PO2 | 0.38 | 0.59%@420 nm | 141 |
| MA3Bi2I9/DMA3BiI6 | 300 W Xe lamp (λ ≥ 420 nm) | HI and H3PO2 | 0.1982 | >3% | 5 |
| MoS2/Cs2AgBiBr6 | 300 W Xe lamp (λ ≥ 420 nm) | HBr and H3PO2 | 0.0875 | 0.20%@450 nm | 142 |
The fabrication of a mixed-halide perovskite with a bandgap funnel structure is another viable strategy to improve the photocatalytic H2 evolution performance. Tang et al.140 prepared an environmentally friendly lead-free MA3Bi2Cl9−xIx photocatalyst with a bandgap funnel structure. Because of the gradient neutral formation energy within the mixed halide perovskite, the content of iodide element diminishes from the surface to the interior across the MA3Bi2Cl9−xIx perovskite, resulting in the formation of a graded bandgap funnel structure, which can facilitate photoinduced charge transfer from the interior to the surface. Consequently, an enhanced photocatalytic H2 evolution rate of ≈341 ± 61.7 μmol h−1 with a Pt co-catalyst was achieved under the illumination of visible light, which was higher when compared to that of MA3Bi2Cl9. The content of iodide element diminishes from the surface to the interior of the MA3Bi2Cl9−xIx perovskite because of the gradient neutral formation energy within the mixed halide perovskite. Thus, a graded bandgap funnel structure has been formed due to the aligned energy levels of the iodide/chloride-mixed perovskite (MA3Bi2Cl9−xIx), which facilitate photoinduced charge transfer to the surface from the interior, leading to an enhanced photocatalytic redox reaction.
Besides, understanding the dynamic interfacial interactions of bandgap funneling in Bi-based mixed-halide perovskites in the photoelectrochemical system are crucial. Hence, Tang et al.9 employed photophysical and photoelectrochemical characterization studies to reveal that MA3Bi2Cl9−yIy with a bandgap funnel structure exhibited a higher photocurrent density (Fig. 11(g and h)) and lower charge transfer resistance (Fig. 11(e)) compared to those of MA3Bi2I9. The photophysical and (photo-)electrochemical phenomena of solid–solid and solid–liquid interfaces for MA3Bi2Cl9−yIy and MA3Bi2I9 are confirmed using electrochemical voltammetric and temperature dependent transient photoluminescence techniques. The stronger electronic coupling of MA3Bi2Cl9−yIy, stemming from the facilitated overlap of electronic wavefunctions, results in a higher diffusion coefficient and electron-transfer rate, ultimately enhancing photoelectrochemical H2 production.
Zhou et al.18 integrated Pt single atoms into Cs2SnI6 to improve the photocatalytic activity. The PtSA/Cs2SnI6 catalyst prepared exhibits enhanced tolerance towards HI aqueous solutions and achieves outstanding photocatalytic H2 production, which is about 176.5 times higher when compared to that of Pt nanoparticles supported on the Cs2SnI6 perovskite. As a result, PtSA/Cs2SnI6 exhibits a significantly higher photocatalytic activity for H2 production (turnover frequency of 70.6 per h per Pt), which is about 176.5 times higher when compared to that of Pt nanoparticles supported on the Cs2SnI6 perovskite, while it exhibits a significantly higher cycling durability. The PL technique was used to determine the role of Pt single atoms on the kinetics of charge carrier transfer and recombination on Cs2SnI6. Fig. 12(a) depicts the steady-state photoluminescence spectrum of the PtSA/Cs2SnI6 photocatalyst in comparison with Cs2SnI6 and PtNP/Cs2SnI6 (NP: nanoparticles). Characterization studies including PL, TRPL, photocurrent, and EIS were conducted to investigate the intriguing properties of PtSA/Cs2SnI6. The results indicated that PtSA/Cs2SnI6 exhibited a lower PL intensity, faster average decay lifetime, higher photocurrent density, and reduced electrochemical impedance, demonstrating that the evenly distributed atomic Pt species could significantly promote the separation and transfer of charge carriers. DFT calculations illustrated maps of the charge density differences before and after the photoexcitation of PtSA/Cs2SnI6 (Fig. 12(f)) in comparison with PtNP/Cs2SnI6 (Fig. 12(e)). The added photogenerated electrons tend to be distributed over the entire Pt nanoparticles in PtNP/Cs2SnI6 (Fig. 12(e)), which can decrease the electron density per Pt atom in Pt nanoparticles, whereas the electrons in PtSA/Cs2SnI6 are only located between the Pt single atoms and three neighboring I atoms (Fig. 12(f)), suggesting the existence of high electron density on the Pt–I3 sites, which can be ascribed to a strong metal–support interaction (SMSI) effect, leading to higher HER activity on PtSA/Cs2SnI6. The SMSI effect, which is conducive to photogenerated charge transfer between the photocatalyst and cocatalyst, was also supported by the calculated PDOS of PtSA/Cs2SnI6 in comparison with PtNP/Cs2SnI6 (Fig. 12(g)), where most 5d states of PtSA are observed below the Fermi level, suggesting an electron-rich state in PtSA. The integrated PDOS areas of uncaptured Pt 5d states above the Fermi level in PtSA are calculated to be 0.71, which is lower than that of PtNP (1.19), further suggesting an electron-rich state in PtSA. This implies that the PtSA species has a superior ability to capture electrons from Cs2SnI6, resulting in higher H2 production of PtSA/Cs2SnI6. PtSA exhibits a significantly lower energy barrier (0.11 eV) than that of PtNP (0.92 eV; Fig. 12(h)), and the enhanced electronic properties of PtSA on Cs2SnI6 lead to elevated photocatalytic H2 production on PtSA/Cs2SnI6. Thus, the electronic property and unique coordination structure of Pt–I3 sites as well as strong metal–support interactions collectively reduce the reaction energy barrier and contribute to an excellent photocatalytic H2 evolution performance.
 | ||
| Fig. 12 (a) Steady-state photoluminescence spectrum, (b) time-resolved transient photoluminescence decay, (c) photocurrent response spectrum, and (d) Nyquist plot of the PtSA/Cs2SnI6 photocatalyst in comparison with those of Cs2SnI6 and PtNP/Cs2SnI6; the maps depict the charge density differences before and after photoexcitation of (f) PtSA/Cs2SnI6 and (e) PtNP/Cs2SnI6, where the isosurface of charge density is 0.001 e Å−3, while the insets depict the top view, and the additional electron distribution is indicated by the yellow region; the model adds an excess electron to describe the photogenerated electron; (g) the PDOS (5d states) of PtSA/Cs2SnI6 in comparison with that of PtNP/Cs2SnI6, where the dashed line indicates the Fermi level; (h) the calculated energy profile for hydrogen generation on PtSA/Cs2SnI6 in comparison with that of PtNP/Cs2SnI6 (reproduced with permission from ref. 18. Copyright 2021, Springer Nature). | ||
Moreover, Chen et al.56 prepared lead-free Cs3Bi0.6Sb1.4I9 perovskites with a high photocatalytic H2 evolution rate and a low charge transfer resistance. Cs3Bi2xSb2–2xI9 doped with Sb can effectively diminish the contribution of Bi3+ to the conduction band, which can suppress the impact of Bi vacancies on the band structure. Therefore, Cs3Bi2xSb2–2xI9 exhibits an optimal bandgap and higher optical absorption when compared to those of Cs3Bi2I9 and Cs3Sb2I9, leading to enhanced performance for photocatalytic H2 evolution. Besides, Zhang et al.142 observed that the MoS2/Cs2AgBiBr6 photocatalyst exhibited a high photocatalytic H2 evolution rate through HX splitting, while it exhibited a low charge transfer resistance. The enhanced performance of MoS2/Cs2AgBiBr6 can be attributed to the kinetics-facilitated heterostructure comprised of stable MoS2 and Cs2AgBiBr6. In addition, Jiang et al.141 found that the addition of N–C with a high surface area could inhibit the aggregation of Cs2AgBiBr6 nanoparticles and afford further pathways for the migration of photoinduced carriers. Doping with nitrogen can enhance photoinduced electron transfer from Cs2AgBiBr6 to N–C, leading to spatially separated electrons and holes with a prolonged electron life time, and boost the photocatalytic performance.
6.2. Lead based perovskites as photocatalysts
The introduction of Br into MAPbI3 can enhance charge separation and transfer, which can enhance photocatalytic H2 evolution. Zhao et al.2 reported that MAPb(I1−xBrx)3 (x = 0.10) exhibited the highest H2 evolution rate of 1471 μmol h−1 g−1 under visible light illumination (λ ≥ 420 nm) in HI/HBr aqueous solutions without using Pt cocatalyst; this was about 40 times larger than that of pure MAPbI3. And it also maintains high stability for photocatalytic H2 production for 252 h (more than 60 runs) without changes to the perovskite structure. MAPb(I1−xBrx)3 exhibits an obvious current increase under visible light irradiation, as indicated by the photocurrent responses and decreased semicircle in the Nyquist plot, suggesting superior separation and migration of charge carriers. The enhancement in the photocatalytic H2 evolution of MAPb(I1−xBrx)3 (x = 0.10) is ascribed to the Br-incorporated perovskite with a lower Pb–H energy and lead at the defect site being exposed to amine cations.Liu et al.10 innovatively integrated photocatalytic HI splitting with electrocatalysis, creating an efficient closed-loop system for pure water splitting. This system is bifurcated into two parts: the first one involves the generation of H2 using the MoSe2/MAPbBr3−xIx photocatalyst via photocatalytic HI splitting. Subsequently, the I3− ions obtained from this photocatalytic process are subjected to electrolysis for electrocatalytic I3− reduction, resulting in the production of O2. This integrated approach is schematically depicted in Fig. 13(c), highlighting the coherence and efficiency of the pure water splitting process. The 3.0 wt% MoSe2/MAPbBr3−xIx catalyst demonstrates an outstanding photocatalytic hydrogen production rate of 22.935 mmol h−1 g−1 under visible light illumination, coupled with reduced charge transfer resistance, indicating its superior charge separation and transfer capabilities. The enhanced photocatalytic H2 production activity is attributed to efficient charge separation, abundant active sites for H2 generation, and a small energy barrier for HI splitting. Hence, the electrolyzer can operate efficiently at a low cell voltage of 1.22 V and a current density of 10 mA cm−2, facilitating the electrocatalytic reduction of I3− and the production of O2. This voltage is notably lower than the theoretical requirement of 1.23 V for electrocatalytic pure water splitting. Simultaneously, this approach significantly reduces the dependence on H3PO2 as a sacrificial agent in the photocatalytic HI splitting system.
 | ||
| Fig. 13 (a) Chopped photocurrent responses and (b) Nyquist plots of MAPb(I1−xBrx)3 photocatalysts in saturated HI/HBr solution, where x = 0, 0.05, 0.10, 0.15, and 0.20 (reproduced with permission from ref. 2. Copyright 2019, Elsevier B.V.). (c) Schematic diagram depicting the mechanism of pure water splitting using tandem photocatalytic H2 generation and electrocatalytic O2 generation with I3−/I− as a redox shuttle mediator (reproduced with permission from ref. 10. Copyright 2023, Wiley-VCH GmbH). (d) The difference in the charge densities of Pt/FAPbBr3−xIx, where the inset depicts its local structure, while the isosurface value is set at 0.001 e Å−3 (positive charge: yellow color; negative charge: cyan color); (e) projected density of states (PDOS) of the photocatalyst (Pt/FAPbBr3−xIx); (f) calculated free energy of H* adsorption for Pt/FAPbBr3−xIx in comparison with those of Pt and FAPbBr3−xIx, where the inset depicts the corresponding schematic mechanism; (g) schematic diagram illustrating the photocatalytic process for Pt/FAPbBr3−xIx (reproduced with permission from ref. 32. Copyright 2022, The Royal Society of Chemistry). | ||
The integration of single Pt atoms into FAPbBr3−xIx can enhance the charge density around the Pt atom, afford optimal charge separation and transfer, and decrease the energy barrier for H2 production, leading to enhanced photocatalytic H2 production. Wu et al.32 observed that PtSA/FAPbBr3−xIx (SA: single-atoms; FA = CH(NH2)2) exhibited a much higher photocatalytic H2 evolution rate of 682.6 mmol h−1 under simulated sunlight irradiation with a high STH of 4.50%. Using a self-adsorption and photoreduction method, Pt single atoms are immobilized individually and coordinated with surface halide anions of FAPbBr3−xIx through replacing formamidinium groups. Higher photocurrent responses and the smaller semicircle in the Nyquist plot demonstrate efficient charge separation and transfer in PtSA/FAPbBr3−xIx. Time-/space-resolved photoelectron formation and migration characteristics were investigated with an advanced PL microscope at the single-particle level. This study reveals that photogenerated electrons from the perovskite can be effectively captured and trapped by individual Pt atoms, and then be preserved for the reduction of protons in the photocatalysis process. The difference in the charge densities of Pt/FAPbBr3−xIx shown in Fig. 13(d) reveals that the loading of single Pt atoms can induce increased charge density. The projected density of states (PDOS) (Fig. 13(e)) shows that orbital hybridization can occur between the coordinated Br/I atoms and the Pt introduced due to the loading of single Pt atoms, which can facilitate charge transfer to the Pt site from the perovskite, leading to enhancement of the proton reduction process. Fig. 13(f) depicts the calculated free energy of H* adsorption (ΔGH*) for PtSA/FAPbBr3−xIx in comparison with those of Pt and FAPbBr3−xIx. PtSA/FAPbBr3−xIx exhibits the lowest adsorption energy of 0.04 eV (almost close to zero), when compared to those of FAPbBr3−xIx (1.18 eV) and bare Pt (−0.09 eV), suggesting the promising proton reduction ability of PtSA/FAPbBr3−xIx.
Li et al.135 constructed the MAPbI3/BP (BP: black phosphorus) type I heterojunction, where BP could function as an electron promoter to catch electrons from MAPbI3 and facilitate charge carrier separation, leading to an impressive photocatalytic H2 evolution rate (3.742 mmol h−1 g−1) and an apparent quantum yield of 23.2% at 420 nm. Besides, Zhao et al.136 observed that the Ni3C/MAPbI3 photocatalyst exhibited a high photocatalytic H2 evolution rate (2.362 mmol h−1 g−1) through HX splitting with a high apparent quantum yield of 16.6%@420 nm, while it exhibited a low charge transfer resistance. The enhanced performance of Ni3C/MAPbI3 is ascribed to the enhanced capabilities of charge carrier transfer and separation along with the enormous reactive centers on the surface of MAPbI3 due to the decoration of Ni3C. In addition, Cai et al.137 observed that the MAPbI3/CoP photocatalyst exhibited a high photocatalytic H2 evolution rate (2.0875 mmol h−1 g−1) through HX splitting, while it exhibited a low charge transfer resistance. The CoP nanoparticle co-catalyst can effectively extract photogenerated electrons from MAPbI3 to facilitate photoinduced charge separation, and enhance the interfacial catalytic reaction. Moreover, Wang et al.138 observed that the MAPbI3/MoS2 photocatalyst exhibited a high photocatalytic H2 evolution rate (∼2.061 mmol h−1 g−1) through HX splitting, while it exhibited a low charge transfer resistance. The integration of the MoS2 co-catalyst with MAPbI3 can efficiently transfer photogenerated electrons from MAPbI3 to MoS2, causing efficient separation of photogenerated carriers, while photogenerated electrons on MoS2 can migrate to the surface active sites, leading to an enhancement in the reduction of protons to H2. Besides, Guan et al.134 observed that the MAPbI3/MoS2 photocatalyst exhibited a significantly higher photocatalytic H2 evolution rate (29.389 mmol h−1 g−1) through HX splitting with a considerably higher solar HI splitting efficiency of 7.35% and much higher apparent quantum yield of 22.1%@500 nm, while it exhibited a low charge transfer resistance. The integration of MoS2 nanoflowers, with abundant active sites, an optimal band gap and unsaturated species, with MAPbI3 can efficiently facilitate charge separation and this can enhance the reduction process for H2 production. Wu et al.139 announced that the CH3NH3PbI3/rGO photocatalyst exhibited a high photocatalytic H2 evolution rate of 0.9389 mmol h−1 g−1 through HX splitting, demonstrating remarkable stability over 200 h. The integration of rGO with CH3NH3PbI3 enables the efficient transfer of photogenerated electrons from MAPbI3 to rGO sites, significantly enhancing the reduction process for protons on rGO's surface. This results in improved H2 production rates, further highlighting the photocatalyst's performance and stability.
7. Summary and perspectives
Boosting the production of clean hydrogen fuel has been regarded as one of the most promising strategies to address the increasing energy crisis and environmental pollution. Electrocatalysis and photocatalysis are highly anticipated to achieve efficient H2 production by using catalysts with desirable properties. With the aim of prompting further research in this field, this review comprehensively articulated the recent advances in various nanostructured electrocatalysts and photocatalysts. In detail, electrocatalysts including oxide/hydroxide/layered double hydroxide, sulfide/selenide, phosphide/nitride, and carbon/alloy/MOF/organic based materials applied in different solution environments (e.g. acidic, alkaline, and seawater electrolyte) were discussed. And modification strategies aimed at an enhanced HER and OER performance have been evaluated. Subsequently, photocatalysts explored for water and HX splitting are also reviewed by elaborating modification approaches like heterojunction/cocatalyst loading/defect engineering/homojunction/elemental doping. More importantly, the enhancement mechanisms of H2 evolution were investigated from the catalyst's design to characterization. Accordingly, we propose the following issues that may be conducive to a deeper understanding of complex mechanisms and better development of potential nano-electro- or -photocatalysts.(1) Adopting appropriate design and modification strategies for electrocatalysts according to specific reaction conditions is important. By tailoring the physicochemical properties of electrocatalysts, such as their composition, morphology, and surface structure, researchers can significantly improve the catalysts’ activity, stability, and durability under operating conditions.
(2) The photocatalytic H2 evolution process involves initial light absorption, charge carrier generation, and subsequent conversion of H2O/H+ into H2. Therefore, fabricating photocatalysts with efficient exciton separation and highly active surface catalytic sites is essential. Additional investigation of the complex relationship between the light exciton process and hydrogen evolution process on photocatalysts is also necessary. Understanding their interactions at a fundamental level will enable the development of photocatalysts with tailored properties, such as optimized band structures, enhanced charge carrier mobility, and increased surface reactivity, all of which are essential for improving the efficiency and scalability of H2 production from solar energy.
(3) It is encouraged to gain deeper insights into the underlying reaction mechanisms through the employment of characterization studies and theoretical calculations, to design and synthesize desired electrocatalysts and photocatalysts. With the aid of powerful characterization methods, detailed insights into the morphology, crystallinity, composition, surface states, and interfacial charge carrier dynamics of catalysts are readily obtained. Combined with predictive insights into the energetics, kinetics, and mechanisms of catalytic reactions that are offered by DFT, we can foster a feedback loop where experimental data inform theoretical models, which in turn predict new materials and modifications that can be tested experimentally.
Author contributions
M.-I. J. and H.-Y. H. conceptualized the manuscript. M.-I. J. and H. T. wrote the original draft. M.-I. J., H. T., S. P. S., W. N., J.-J. K., C.-W. H., K.-C. C., F.-F. L., B. H., J. C. C. and H.-Y. H. contributed to the revision of the manuscript. H.-Y. H. supervised the writing and revision of the manuscript and provided the resources.Data availability
Data availability does not apply to this article because no new data are generated or analyzed in this study.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors acknowledge financial support from the Research Grants Council of Hong Kong (grant nos. CityU 21203518 and F-CityU106/18), the Innovation and Technology Commission (grant no. MHP/104/21), the Shenzhen Science Technology and Innovation Commission (grant nos. JCYJ20210324125612035, R-IND12303, and R-IND12304), the City University of Hong Kong (grant nos. 7005289, 7005580, 7005720, 9667213, 9667229, 9680331 and 9678291), and the National Natural Science Foundation of China (Grant no. 21974131, 22071070, and 51901119).References
- W. Zhao, F. Xu, L. Liu, M. Liu and B. Weng, Adv. Mater., 2023, 2308060, DOI:10.1002/adma.202308060.
- Z. Zhao, J. Wu, Y.-Z. Zheng, N. Li, X. Li, Z. Ye, S. Lu, X. Tao and C. Chen, Appl. Catal., B, 2019, 253, 41–48 CrossRef CAS.
- C. Lyu, C. Cao, J. Cheng, Y. Yang, K. Wu, J. Wu, W.-M. Lau, P. Qian, N. Wang and J. Zheng, Chem. Eng. J., 2023, 464, 142538 CrossRef CAS.
- W. Xi, L. Jin, A. Mahmood, W. Zhang, Y. Li, H. Li, P. An, J. Zhang, T. Ma, S. Liu and J. Yan, Adv. Energy Mater., 2023, 2302668, DOI:10.1002/aenm.202302668.
- Y. Tang, C. H. Mak, R. Liu, Z. Wang, L. Ji, H. Song, C. Tan, F. Barrière and H.-Y. Hsu, Adv. Funct. Mater., 2020, 30, 2006919 CrossRef CAS.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- H. S. Moon, K.-C. Hsiao, M.-C. Wu, Y. Yun, Y.-J. Hsu and K. Yong, Adv. Mater., 2023, 35, 2200172 CrossRef CAS PubMed.
- H. Peng, T. Yang, H. Lin, Y. Xu, Z. Wang, Q. Zhang, S. Liu, H. Geng, L. Gu, C. Wang, X. Fan, W. Chen and X. Huang, Adv. Energy Mater., 2022, 12, 2201688 CrossRef CAS.
- Y. Tang, C. H. Mak, J. Zhang, G. Jia, K.-C. Cheng, H. Song, M. Yuan, S. Zhao, J.-J. Kai, J. C. Colmenares and H.-Y. Hsu, Adv. Mater., 2023, 35, 2207835 CrossRef CAS.
- X. Liu, Q. Zhang, S. Zhao, Z. Wang, Y. Liu, Z. Zheng, H. Cheng, Y. Dai, B. Huang and P. Wang, Adv. Mater., 2023, 2208915, DOI:10.1002/adma.202208915.
- K. Lu, F. Xue, F. Liu, M. Li, W. Fu, H. Peng, C. Zhang, J. Huang, Z. Gao, H. Huang and M. Liu, Adv. Energy Mater., 2023, 13, 2301158 CrossRef CAS.
- M. I. Jamesh, J. Power Sources, 2016, 333, 213–236 CrossRef CAS.
- M.-I. Jamesh and X. Sun, J. Power Sources, 2018, 400, 31–68 CrossRef CAS.
- S. Ibraheem, G. Yasin, A. Kumar, M. A. Mushtaq, S. Ibrahim, R. Iqbal, M. Tabish, S. Ali and A. Saad, Appl. Catal., B, 2022, 304, 120987 CrossRef CAS.
- X. Wang, X. Liu, S. Wu, K. Liu, X. Meng, B. Li, J. Lai, L. Wang and S. Feng, Nano Energy, 2023, 109, 108292 CrossRef CAS.
- Z. Huang, A. Reda Woldu, X. Peng, P. K. Chu, Q.-X. Tong and L. Hu, Chem. Eng. J., 2023, 477, 147155 CrossRef CAS.
- Y. Guo, J. Sun, Y. Tang, X. Jia, Y. Nie, Z. Geng, C. Wang, J. Zhang, X. Tan, D. Zhong, J. Ye and T. Yu, Energy Environ. Sci., 2023, 16, 3462–3473 RSC.
- P. Zhou, H. Chen, Y. Chao, Q. Zhang, W. Zhang, F. Lv, L. Gu, Q. Zhao, N. Wang, J. Wang and S. Guo, Nat. Commun., 2021, 12, 4412 CrossRef CAS.
- H.-Y. Wang, M.-L. Sun, J.-T. Ren and Z.-Y. Yuan, Adv. Energy Mater., 2023, 13, 2203568 CrossRef CAS.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- S.-P. Zeng, H. Shi, T.-Y. Dai, Y. Liu, Z. Wen, G.-F. Han, T.-H. Wang, W. Zhang, X.-Y. Lang, W.-T. Zheng and Q. Jiang, Nat. Commun., 2023, 14, 1811 CrossRef CAS.
- J. Guo, Y. Zheng, Z. Hu, C. Zheng, J. Mao, K. Du, M. Jaroniec, S.-Z. Qiao and T. Ling, Nat. Energy, 2023, 8, 264–272 CAS.
- W. Tong, M. Forster, F. Dionigi, S. Dresp, R. Sadeghi Erami, P. Strasser, A. J. Cowan and P. Farràs, Nat. Energy, 2020, 5, 367–377 CrossRef CAS.
- M.-I. Jamesh and X. Sun, J. Energy Chem., 2019, 34, 111–160 CrossRef.
- N.-N. Liang, D. S. Han and H. Park, Appl. Catal., B, 2023, 324, 122275 CrossRef CAS.
- P. M. Bacirhonde, A. Y. Mohamed, B. Han, D.-Y. Cho, S. Devendra, J.-W. Choi, C.-R. Lim, E. O. Afranie, K.-H. Baik, K. Kang, S. Lee, E.-S. Jeong, N. Komalla, N. Y. Dzade, C. H. Park and C. S. Kim, Adv. Energy Mater., 2023, 13, 2300174 CrossRef CAS.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS.
- S. B. Roy, S. Moon, K. Hee kim, A. Patil, M. A. Rehman, S. Yoo, Y. Seo, J. H. Park, K. Kang and S. C. Jun, Appl. Catal., B, 2022, 319, 121906 CrossRef CAS.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, Science, 2016, 353, 1011–1014 CrossRef CAS PubMed.
- M. I. Jamesh and M. Harb, J. Energy Chem., 2021, 56, 299–342 CrossRef CAS.
- J. Luo, J.-H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N.-G. Park, S. D. Tilley, H. J. Fan and M. Grätzel, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed.
- Y. Wu, Q. Wu, Q. Zhang, Z. Lou, K. Liu, Y. Ma, Z. Wang, Z. Zheng, H. Cheng, Y. Liu, Y. Dai, B. Huang and P. Wang, Energy Environ. Sci., 2022, 15, 1271–1281 RSC.
- M.-I. Jamesh, Y. Kuang and X. Sun, ChemCatChem, 2019, 11, 1550–1575 CrossRef CAS.
- M. Rashid, M. K. Al Mesfer, H. Naseem and M. Danish, Int. J. Eng. Adv. Technol., 2015, 4, 2249–8958 Search PubMed.
- S. Stiber, N. Sata, T. Morawietz, S. A. Ansar, T. Jahnke, J. K. Lee, A. Bazylak, A. Fallisch, A. S. Gago and K. A. Friedrich, Energy Environ. Sci., 2022, 15, 109–122 RSC.
- M.-I. Jamesh and M. Harb, Mater. Sci. Energy Technol., 2020, 3, 780–807 Search PubMed.
- S. Dresp, F. Dionigi, M. Klingenhof, T. Merzdorf, H. Schmies, J. Drnec, A. Poulain and P. Strasser, ACS Catal., 2021, 11, 6800–6809 CrossRef CAS.
- F. Dionigi, T. Reier, Z. Pawolek, M. Gliech and P. Strasser, ChemSusChem, 2016, 9, 962–972 CrossRef CAS PubMed.
- S. Seenivasan and J. Seo, Chem. Eng. J., 2023, 454, 140558 CrossRef CAS.
- R. Liu, M. Sun, X. Liu, Z. Lv, X. Yu, J. Wang, Y. Liu, L. Li, X. Feng, W. Yang, B. Huang and B. Wang, Angew. Chem., Int. Ed., 2023, 62, e202312644 CrossRef CAS PubMed.
- C. Liu, J. Feng, P. Zhou, D. Liu, L. Qiao, D. Liu, Y. Cao, S.-C. Su, H. Liu and H. Pan, Chem. Eng. J., 2023, 476, 146710 CrossRef CAS.
- H. Fan, J. Jia, D. Wang, J. Fan, J. Wu, J. Zhao and X. Cui, Chem. Eng. J., 2023, 455, 140908 CrossRef CAS.
- H.-Y. Wang, L. Wang, J.-T. Ren, W.-W. Tian, M.-L. Sun and Z.-Y. Yuan, Nano-Micro Lett., 2023, 15, 155 CrossRef CAS PubMed.
- X. Wang, H. Huang, J. Qian, Y. Li and K. Shen, Appl. Catal., B, 2023, 325, 122295 CrossRef CAS.
- Q. Lv, B. Yao, W. Zhang, L. She, W. Ren, L. Hou, Y. Fautrelle, X. Lu, X. Yu and X. Li, Chem. Eng. J., 2022, 446, 137420 CrossRef CAS.
- P. P. Dhakal, U. N. Pan, M. R. Kandel, R. B. Ghising, T. H. Nguyen, V. A. Dinh, N. H. Kim and J. H. Lee, Chem. Eng. J., 2023, 473, 145321 CrossRef CAS.
- H. Xiang, W. Chen, T. Li, J. Huang, G. Chen, T. Gong and K. Ken Ostrikov, Chem. Eng. J., 2022, 446, 137419 CrossRef CAS.
- M. Mekete Meshesha, D. Chanda, S. Gwon Jang and B. Lyong Yang, Chem. Eng. J., 2023, 474, 145708 CrossRef CAS.
- J. Feng, C. H. Mak, L. Yu, B. Han, H. H. Shen, S. P. Santoso, M. Yuan, F. F. Li, H. Song, J. C. Colmenares and H. Y. Hsu, Small Methods, 2024, 8, e2300429 CrossRef PubMed.
- X. Wang, X. Zhou, C. Li, H. Yao, C. Zhang, J. Zhou, R. Xu, L. Chu, H. Wang, M. Gu, H. Jiang and M. Huang, Adv. Mater., 2022, 34, 2204021 CrossRef CAS.
- Z. Lu, Y. Cao, J. Xie, J. Hu, K. Wang and D. Jia, Chem. Eng. J., 2022, 430, 132877 CrossRef CAS.
- M. I. Jamesh, J. Power Sources, 2020, 448, 227375 CrossRef.
- M.-I. Jamesh, A. Akila, D. Sudha, K. Gnana Priya, V. Sivaprakash and A. Revathi, Sustainability, 2022, 14, 16359 CrossRef CAS.
- M. Ashraf, R. Ali, I. Khan, N. Ullah, M. S. Ahmad, T. Kida, S. Wooh, W. Tremel, U. Schwingenschlögl and M. N. Tahir, Adv. Mater., 2022, 2301342, DOI:10.1002/adma.202301342.
- X. Wang and Z. Jin, Appl. Catal., B, 2024, 342, 123373 CrossRef CAS.
- G. Chen, P. Wang, Y. Wu, Q. Zhang, Q. Wu, Z. Wang, Z. Zheng, Y. Liu, Y. Dai and B. Huang, Adv. Mater., 2020, 32, 2001344 CrossRef CAS PubMed.
- C. Triolo, K. Moulaee, A. Ponti, G. Pagot, V. Di Noto, N. Pinna, G. Neri and S. Santangelo, Adv. Funct. Mater., 2023, 2306375, DOI:10.1002/adfm.202306375.
- K. Zeng, M. Chao, M. Tian, J. Yan, M. H. Rummeli, P. Strasser and R. Yang, Adv. Funct. Mater., 2023, 2308533, DOI:10.1002/adfm.202308533.
- Y. Li, Y. Duan, K. Zhang and W. Yu, Chem. Eng. J., 2022, 433, 134472 CrossRef CAS.
- S. He, K. Wang, B. Li, H. Du, Z. Du, T. Wang, S. Li, W. Ai and W. Huang, Adv. Mater., 2023, 2307017, DOI:10.1002/adma.202307017.
- Q. P. Ngo, T. T. Nguyen, Q. T. T. Le, J. H. Lee and N. H. Kim, Adv. Energy Mater., 2023, 2301841, DOI:10.1002/aenm.202301841.
- Y. Lu, B. Li, N. Xu, Z. Zhou, Y. Xiao, Y. Jiang, T. Li, S. Hu, Y. Gong and Y. Cao, Nat. Commun., 2023, 14, 6965 CrossRef CAS PubMed.
- C.-H. Shin, T. H. Yu, H.-Y. Lee, B.-J. Lee, S. Kwon, W. A. Goddard and J.-S. Yu, Appl. Catal., B, 2023, 334, 122829 CrossRef CAS.
- Y. Kuang, R. He, X. Gu, F. Yang, X. Tian and L. Feng, Chem. Eng. J., 2023, 456, 141055 CrossRef CAS.
- K. Wu, C. Cao, K. Li, C. Lyu, J. Cheng, H. Li, P. Hu, J. Wu, W.-M. Lau, X. Zhu, P. Qian and J. Zheng, Chem. Eng. J., 2023, 452, 139527 CrossRef CAS.
- C. Zhao, J. Wang, Y. Gao, J. Zhang, C. Huang, Q. Shi, S. Mu, Q. Xiao, S. Huo, Z. Xia, J. Zhang, X. Lu and Y. Zhao, Adv. Funct. Mater., 2023, 2307917, DOI:10.1002/adfm.202307917.
- J. Das, S. Mandal, A. Borbora, S. Rani, M. Tenjimbayashi and U. Manna, Adv. Funct. Mater., 2023, 2311648, DOI:10.1002/adfm.202311648.
- C. Lin, J.-L. Li, X. Li, S. Yang, W. Luo, Y. Zhang, S.-H. Kim, D.-H. Kim, S. S. Shinde, Y.-F. Li, Z.-P. Liu, Z. Jiang and J.-H. Lee, Nat. Catal., 2021, 4, 1012–1023 CrossRef CAS.
- D. Chen, R. Yu, D. Wu, H. Zhao, P. Wang, J. Zhu, P. Ji, Z. Pu, L. Chen, J. Yu and S. Mu, Nano Energy, 2022, 100, 107445 CrossRef CAS.
- Y. Wu, R. Yao, Q. Zhao, J. Li and G. Liu, Chem. Eng. J., 2022, 439, 135699 CrossRef CAS.
- H. Song, X. Yong, G. I. N. Waterhouse, J. Yu, H. Wang, J. Cai, Z. Tang, B. Yang, J. Chang and S. Lu, ACS Catal., 2024, 14, 3298–3307 CrossRef CAS.
- Z.-Y. Wu, F.-Y. Chen, B. Li, S.-W. Yu, Y. Z. Finfrock, D. M. Meira, Q.-Q. Yan, P. Zhu, M.-X. Chen, T.-W. Song, Z. Yin, H.-W. Liang, S. Zhang, G. Wang and H. Wang, Nat. Mater., 2023, 22, 100–108 CrossRef CAS PubMed.
- Y. Yang, L. Liu, S. Chen, W. Yan, H. Zhou, X.-M. Zhang and X. Fan, Angew. Chem., Int. Ed., 2023, 62, e202306896 CrossRef CAS PubMed.
- V.-H. Do, P. Prabhu, V. Jose, T. Yoshida, Y. Zhou, H. Miwa, T. Kaneko, T. Uruga, Y. Iwasawa and J.-M. Lee, Adv. Mater., 2023, 35, 2208860 CrossRef CAS PubMed.
- J. Liu, T. Wang, X. Liu, H. Shi, S. Li, L. Xie, Z. Cai, J. Han, Y. Huang, G. Wang and Q. Li, ACS Catal., 2023, 13, 5194–5204 CrossRef CAS.
- S. Yan, W. Liao, M. Zhong, W. Li, C. Wang, N. Pinna, W. Chen and X. Lu, Appl. Catal., B, 2022, 307, 121199 CrossRef CAS.
- J. Luo, J. Wang, Y. Guo, J. Zhu, H. Jin, Z. Zhang, D. Zhang, Y. Niu, S. Hou, J. Du, D. He, Y. Xiong, L. Chen, S. Mu and Y. Huang, Appl. Catal., B, 2022, 305, 121043 CrossRef CAS.
- C. Rong, X. Shen, Y. Wang, L. Thomsen, T. Zhao, Y. Li, X. Lu, R. Amal and C. Zhao, Adv. Mater., 2022, 34, 2110103 CrossRef CAS PubMed.
- J. Chen, G. Qian, H. Zhang, S. Feng, Y. Mo, L. Luo and S. Yin, Adv. Funct. Mater., 2022, 32, 2107597 CrossRef CAS.
- C. He, C. Ma, J. Xia, H. Zhang, S. Han, Y. Tian, A.-L. Wang, X. Meng, W. Cao and Q. Lu, Adv. Funct. Mater., 2023, 2311683, DOI:10.1002/adfm.202311683.
- X. Wang, Z. Qin, J. Qian, L. Chen and K. Shen, ACS Catal., 2023, 13, 10672–10682 CrossRef CAS.
- J. Chen, Y. Ha, R. Wang, Y. Liu, H. Xu, B. Shang, R. Wu and H. Pan, Nano-Micro Lett., 2022, 14, 186 CrossRef CAS PubMed.
- H. Yang, P. Guo, R. Wang, Z. Chen, H. Xu, H. Pan, D. Sun, F. Fang and R. Wu, Adv. Mater., 2022, 34, 2107548 CrossRef CAS PubMed.
- Z. Yu, Y. Li, A. Torres-Pinto, A. P. LaGrow, V. M. Diaconescu, L. Simonelli, M. J. Sampaio, O. Bondarchuk, I. Amorim, A. Araujo, A. M. T. Silva, C. G. Silva, J. L. Faria and L. Liu, Appl. Catal., B, 2022, 310, 121318 CrossRef CAS.
- W. Wu, Y. Huang, X. Wang, P. K. Shen and J. Zhu, Chem. Eng. J., 2023, 469, 143879 CrossRef CAS.
- T. He, Y. Song, Y. Chen, X. Song, B. Lu, Q. Liu, H. Liu, Y. Zhang, X. Ouyang and S. Chen, Chem. Eng. J., 2022, 442, 136337 CrossRef CAS.
- R. Wu, J. Xu, C.-L. Zhao, X.-Z. Su, X.-L. Zhang, Y.-R. Zheng, F.-Y. Yang, X.-S. Zheng, J.-F. Zhu, J. Luo, W.-X. Li, M.-R. Gao and S.-H. Yu, Nat. Commun., 2023, 14, 2306 CrossRef CAS PubMed.
- S.-C. Sun, F.-X. Ma, H. Jiang, M.-X. Chen, P. Xu, L. Zhen, B. Song and C.-Y. Xu, Chem. Eng. J., 2023, 462, 142132 CrossRef CAS.
- L. Najafi, R. Oropesa-Nuñez, S. Bellani, B. Martín-García, L. Pasquale, M. Serri, F. Drago, J. Luxa, Z. Sofer, D. Sedmidubský, R. Brescia, S. Lauciello, M. I. Zappia, D. V. Shinde, L. Manna and F. Bonaccorso, ACS Nano, 2022, 16, 351–367 CrossRef CAS PubMed.
- A. Djire, X. Wang, C. Xiao, O. C. Nwamba, M. V. Mirkin and N. R. Neale, Adv. Funct. Mater., 2020, 30, 2001136 CrossRef CAS.
- S. Hao, H. Sheng, M. Liu, J. Huang, G. Zheng, F. Zhang, X. Liu, Z. Su, J. Hu, Y. Qian, L. Zhou, Y. He, B. Song, L. Lei, X. Zhang and S. Jin, Nat. Nanotechnol., 2021, 16, 1371–1377 CrossRef CAS PubMed.
- S. Zhang, Y. Wang, X. Wei, L. Chu, W. Tian, H. Wang and M. Huang, Appl. Catal., B, 2023, 336, 122926 CrossRef CAS.
- Z. Ren, S. Wang, J. Yu, F. Mao, K. Wang and H. Wu, Chem. Eng. J., 2023, 470, 144107 CrossRef CAS.
- W. Liu, W. Que, R. Yin, J. Dai, D. Zheng, J. Feng, X. Xu, F. Wu, W. Shi, X. Liu and X. Cao, Appl. Catal., B, 2023, 328, 122488 CrossRef CAS.
- D. Jin, H. Woo, S. Prabhakaran, Y. Lee, M. H. Kim, D. H. Kim and C. Lee, Adv. Funct. Mater., 2023, 33, 2301559 CrossRef CAS.
- N. Wen, D. Zhang, X. Zhao, X. Jiao, Y. Xia and D. Chen, ACS Catal., 2023, 13, 7868–7878 CrossRef CAS.
- K. Hemmati, A. Kumar, A. R. Jadhav, O. Moradlou, A. Z. Moshfegh and H. Lee, ACS Catal., 2023, 13, 5516–5528 CrossRef CAS.
- Y. Zhuo, D. Liu, L. Qiao, S. Chen, J. Lu, W. F. Ip, H. Pan and Z. Wang, Adv. Energy Mater., 2023, 13, 2301921 CrossRef CAS.
- Y. Du, Q. Li, M. Liu, D. Liu, W. Xiao, Z. Xiao, Z. Li, Y. Yamauchi, S. M. Osman, Z. Wu and L. Wang, Chem. Eng. J., 2023, 475, 146057 CrossRef CAS.
- J. Sun, Z. Zhang and X. Meng, Appl. Catal., B, 2023, 331, 122703 CrossRef CAS.
- S. Pan, R. Li, J. Wang, Q. Zhang, M. Wang, B. Shi, P. Wang, Y. Zhao and X. Zhang, ACS Nano, 2023, 17, 4539–4550 CrossRef CAS PubMed.
- L. Zhou, Z.-H. Huang, F. Kang and R. Lv, Chem. Eng. J., 2023, 458, 141457 CrossRef CAS.
- J. Wang, D. T. Tran, K. Chang, S. Prabhakaran, J. Zhao, D. H. Kim, N. H. Kim and J. H. Lee, Nano Energy, 2023, 111, 108440 CrossRef CAS.
- W. Xu, T. Ma, H. Chen, D. Pan, Z. Wang, S. Zhang, P. Zhang, S. Bao, Q. Yang, L. Zhou, Z. Tian, S. Dai and Z. Lu, Adv. Funct. Mater., 2023, 33, 2302263 CrossRef CAS.
- Y. Song, M. Sun, S. Zhang, X. Zhang, P. Yi, J. Liu, B. Huang, M. Huang and L. Zhang, Adv. Funct. Mater., 2023, 33, 2214081 CrossRef CAS.
- E. Enkhtuvshin, S. Yeo, H. Choi, K. M. Kim, B.-S. An, S. Biswas, Y. Lee, A. K. Nayak, J. U. Jang, K.-H. Na, W.-Y. Choi, G. Ali, K. H. Chae, M. Akbar, K. Y. Chung, K. Yoo, Y.-C. Chung, T. H. Shin, H. Kim, C.-Y. Chung and H. Han, Adv. Funct. Mater., 2023, 33, 2214069 CrossRef CAS.
- C. Huang, Q. Zhou, L. Yu, D. Duan, T. Cao, S. Qiu, Z. Wang, J. Guo, Y. Xie, L. Li and Y. Yu, Adv. Energy Mater., 2023, 13, 2301475 CrossRef CAS.
- Y. Luo, P. Wang, G. Zhang, S. Wu, Z. Chen, H. Ranganathan, S. Sun and Z. Shi, Chem. Eng. J., 2023, 454, 140061 CrossRef CAS.
- S. Zhang, Y. Wang, S. Li, Z. Wang, H. Chen, L. Yi, X. Chen, Q. Yang, W. Xu, A. Wang and Z. Lu, Nat. Commun., 2023, 14, 4822 CrossRef CAS PubMed.
- W. Liu, J. Yu, M. G. Sendeku, T. Li, W. Gao, G. Yang, Y. Kuang and X. Sun, Angew. Chem., Int. Ed., 2023, 62, e202309882 CrossRef CAS PubMed.
- X. Kang, F. Yang, Z. Zhang, H. Liu, S. Ge, S. Hu, S. Li, Y. Luo, Q. Yu, Z. Liu, Q. Wang, W. Ren, C. Sun, H.-M. Cheng and B. Liu, Nat. Commun., 2023, 14, 3607 CrossRef CAS PubMed.
- A. Badreldin, J. Abed, N. Hassan, A. El-Ghenymy, W. Suwaileh, Y. Wubulikasimu, Z. K. Ghouri, K. Youssef, D. Kumar, K. Elsaid, E. H. Sargent and A. Abdel-Wahab, Appl. Catal., B, 2023, 330, 122599 CrossRef CAS.
- G. Bahuguna, B. Filanovsky and F. Patolsky, Nano Energy, 2023, 111, 108439 CrossRef CAS.
- M. Ahasan Habib, R. Mandavkar, S. Lin, S. Burse, T. Khalid, M. Hasan Joni, J.-H. Jeong and J. Lee, Chem. Eng. J., 2023, 462, 142177 CrossRef CAS.
- X. Ruan, C. Huang, H. Cheng, Z. Zhang, Y. Cui, Z. Li, T. Xie, K. Ba, H. Zhang, L. Zhang, X. Zhao, J. Leng, S. Jin, W. Zhang, W. Zheng, S. K. Ravi, Z. Jiang, X. Cui and J. Yu, Adv. Mater., 2023, 35, 2209141 CrossRef CAS PubMed.
- L. Zhang, W. Dong, Y. Zhang, X. Song and H. Jiang, Chem. Eng. J., 2023, 472, 144898 CrossRef CAS.
- X. Zhang, F. Wu, G. Li, L. Wang, J. Huang, A. Meng and Z. Li, Appl. Catal., B, 2024, 342, 123398 CrossRef CAS.
- R. Zeng, C. Cheng, F. Xing, Y. Zou, K. Ding and C. Huang, Appl. Catal., B, 2022, 316, 121680 CrossRef CAS.
- Y. Tang, X. Jia, Y. Guo, Z. Geng, C. Wang, L. Liu, J. Zhang, W. Guo, X. Tan, T. Yu and J. Ye, Adv. Energy Mater., 2023, 13, 2203827 CrossRef CAS.
- X. Han, T. Si, Q. Liu, F. Zhu, R. Li, X. Chen, J. Liu, H. Sun, J. Zhao, H. Ling, Q. Zhang and H. Wang, Chem. Eng. J., 2021, 426, 130824 CrossRef CAS.
- W. Shi, Z. Chen, J. Lu, X. Sun, Z. Wang, Y. Yan, F. Guo, L. Chen and G. Wang, Chem. Eng. J., 2023, 474, 145690 CrossRef CAS.
- S. Guo, J. Su, H. Luo, X. Duan, Q. Shen, J. Xue, B. Wei and X.-M. Zhang, ACS Catal., 2023, 13, 296–307 CrossRef CAS.
- W. Xu, X. Li, C. Peng, G. Yang, Y. Cao, H. Wang, F. Peng and H. Yu, Appl. Catal., B, 2022, 303, 120910 CrossRef CAS.
- L. Luo, J. Tian, W. Hu, P. Han, W. Wang and B. Ma, Appl. Catal., B, 2023, 321, 122008 CrossRef CAS.
- H. Yuan, H. Sun, Y. Shi, J. Wang, A. Bian, Y. Hu, F. Guo, W. Shi, X. Du and Z. Kang, Chem. Eng. J., 2023, 472, 144654 CrossRef CAS.
- S. Luo, C. Zhang, X. Liu, Y. Li, L. Tang, M. Fu, S. Wang, J. Wu, M. Xu, X. Wang and Y. He, Chem. Eng. J., 2022, 432, 134244 CrossRef CAS.
- X. Ma and H. Cheng, Appl. Catal., B, 2023, 339, 123101 CrossRef CAS.
- Q. Chen, Y. Liu, X. Gu, D. Li, D. Zhang, D. Zhang, H. Huang, B. Mao, Z. Kang and W. Shi, Appl. Catal., B, 2022, 301, 120755 CrossRef CAS.
- K. Shu, C. Chuaicham and K. Sasaki, Chem. Eng. J., 2023, 477, 146994 CrossRef CAS.
- J.-X. Yang, W.-B. Yu, C.-F. Li, W.-D. Dong, L.-Q. Jiang, N. Zhou, Z.-P. Zhuang, J. Liu, Z.-Y. Hu, H. Zhao, Y. Li, L. Chen, J. Hu and B.-L. Su, Chem. Eng. J., 2021, 420, 129695 CrossRef CAS.
- H. Long, D. Gao, P. Wang, X. Wang, F. Chen and H. Yu, Appl. Catal., B, 2024, 340, 123270 CrossRef CAS.
- K. Das, R. Bariki, D. Majhi, A. Mishra, K. K. Das, R. Dhiman and B. G. Mishra, Appl. Catal., B, 2022, 303, 120902 CrossRef CAS.
- W. Zhou, H. Huang, Y. Wu, J. Wang, Y. Yamauchi, J. Kim, S. M. Osman, X. Xu, L. Wang, C. Wang and Z. Yuan, Chem. Eng. J., 2023, 471, 144395 CrossRef CAS.
- W. Guan, Y. Li, Q. Zhong, H. Liu, J. Chen, H. Hu, K. Lv, J. Gong, Y. Xu, Z. Kang, M. Cao and Q. Zhang, Nano Lett., 2021, 21, 597–604 CrossRef CAS PubMed.
- R. Li, X. Li, J. Wu, X. Lv, Y.-Z. Zheng, Z. Zhao, X. Ding, X. Tao and J.-F. Chen, Appl. Catal., B, 2019, 259, 118075 CrossRef CAS.
- Z. Zhao, J. Wu, Y.-Z. Zheng, N. Li, X. Li and X. Tao, ACS Catal., 2019, 9, 8144–8152 CrossRef CAS.
- C. Cai, Y. Teng, J.-H. Wu, J.-Y. Li, H.-Y. Chen, J.-H. Chen and D.-B. Kuang, Adv. Funct. Mater., 2020, 30, 2001478 CrossRef CAS.
- F. Wang, X. Liu, Z. Zhang and S. Min, Chem. Commun., 2020, 56, 3281–3284 RSC.
- Y. Wu, P. Wang, X. Zhu, Q. Zhang, Z. Wang, Y. Liu, G. Zou, Y. Dai, M.-H. Whangbo and B. Huang, Adv. Mater., 2018, 30, 1704342 CrossRef PubMed.
- Y. Tang, C. H. Mak, C. Wang, Y. Fu, F.-F. Li, G. Jia, C.-W. Hsieh, H.-H. Shen, J. C. Colmenares, H. Song, M. Yuan, Y. Chen and H.-Y. Hsu, Small Methods, 2022, 6, 2200326 CrossRef CAS PubMed.
- Y. Jiang, K. Li, X. Wu, M. Zhu, H. Zhang, K. Zhang, Y. Wang, K. P. Loh, Y. Shi and Q.-H. Xu, ACS Appl. Mater. Interfaces, 2021, 13, 10037–10046 CrossRef CAS PubMed.
- Y. Zhang, Z. Sun, Z. Wang, Y. Zang and X. Tao, Int. J. Hydrogen Energy, 2022, 47, 8829–8840 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |