 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Concluding remarks: biocatalysis
Uwe T. Bornscheuer *
*
Department of Biotechnology & Enzyme Catalysis, Institute of Biochemistry, University of Greifswald, Felix-Hausdorff-Str. 4, 17489 Greifswald, Germany. E-mail: uwe.bornscheuer@uni-greifswald.de
First published on 10th June 2024
Abstract
Biocatalysis is a rapidly evolving field with increasing impact in organic synthesis, chemical manufacturing and medicine. The Faraday Discussion reflected the current state of biocatalysis, covering the design of de novo enzymatic activities, but especially methods for the improvement of enzymes targeting a broad range of applications (i.e., hydroxylations by P450 monooxygenases, enzymatic deprotection of organic compounds under mild conditions, synthesis of chiral intermediates, plastic degradation, silicone polymer synthesis, and peptide synthesis). Central themes have been how to improve an enzyme using methods of rational design and directed evolution, informed by computer modelling and machine learning, and the incorporation of new catalytic functionalities to create hybrid and artificial enzymes.
It is a great pleasure and honour for me to summarise the Faraday Discussion on biocatalysis, which took place May 22–24 2024 at the Royal Society of Chemistry at Burlington House in London, UK. This meeting was organized by Adrian Mulholland (University of Bristol, UK) and Nicholas Turner (Manchester Institute of Biotechnology, UK) serving as Chairs of the Scientific Committee. As I had never attended a Faraday Discussion meeting before, I was initially surprised that for all presentations given, the authors had to submit a manuscript beforehand, distributed to all participants in advance, and that they were allocated only 5 min presentation time for their specific topic, followed by a 25 min discussion with the audience (and some online participants). This worked out perfectly and I was very pleased to have seen very intensive, stimulating and constructive discussions throughout the entire conference. In addition, almost 60 posters were presented and approximately 20 poster presenters got the opportunity to present their work orally in 2 min before the actual poster sessions took place. I was also very pleased that the (fully booked) meeting was attended by a large proportion of undergraduate and PhD students, as well as many young postdocs, who all actively participated in the discussions and for sure learned a lot about the field of biocatalysis during these very intensive days.
Biocatalysis is a rapidly evolving field with increasing impact in organic synthesis, chemical manufacturing and medicine as documented in recent reviews.1–3 Enzyme engineering, the application of rational design and directed evolution supported by computational tools, has substantially boosted the development of the field in the past decade, as summarized in my own review articles4–6 and those by others.3,7 Notably, the Nobel prize in Chemistry was awarded in 2018 to Frances H. Arnold for the “directed evolution of enzymes”.8
The programme started with an outstanding introductory lecture given by Donald Hilvert (ETH Zürich, Switzerland) about “De novo design of enzymes” (https://doi.org/10.1039/D4FD00139G). Hilvert demonstrated in his contribution that enzymes can now be designed from scratch, with astonishing catalytic activity. This was shown for several examples, including a Zn-dependent hydrolase,9 where the initial design had very low performance, which was then boosted by several orders of magnitudes using methods of directed evolution, resulting in a final variant with a kcat/Km of 106 M−1 s−1. Hilvert also presented a de novo designed Diels–Alderase10 and a novel metallo aldolase. Notably, all these new activities could be designed into naïve proteins without any prior catalytic function. He emphasised the importance of machine learning and deep mutational scanning. He also provided a roadmap11 for better enzyme design including design–build–test–learn cycles. His Spiers memorial lecture was thus an excellent starting point for the subsequent session on “Enzyme evolution, engineering and design: mechanism and dynamics”. The first three speakers all reported on P450-monooxygenases, but with different intentions. Joelle Pelletier (University of Montreal, Canada) targeted the synthesis of indigo (Scheme 1) using the P450-enzyme BM3. First, 42 positions were subjected to site-saturation mutagenesis and about 1000 single-mutation variants were identified by screening – colourful indigo-producing – E. coli strains. Promising mutations were then combined and more active variants of P450-BM3 were found, which also converted anisole and naphthalene (Scheme 1) (https://doi.org/10.1039/D4FD00017J). Zhiqi Cong (Qingdao Institute of Bioenergy and Bioprocess Technology, Qingdao, China) focused on improving the (usually undesired) peroxygenase activity of a P450 enzyme by targeting redox-sensitive residues (Scheme 1). In addition, to understand how hydroxylation or peroxygenase activity can be controlled, his P450 variants may also represent a useful alternative to known peroxygenase enzymes, such as horseradish peroxygenase or unspecific peroxygenases (https://doi.org/10.1039/D4FD00008K).12,13 Finally, Jeremy Harvey (KU Leuven, Belgium) explored the selectivity of cytochrome P450 enzymes to enable the synthesis of novel anticancer agents (https://doi.org/10.1039/D4FD00004H). These three lectures exemplified the broad range of reactions catalyzed by P450-monooxygenases and at the same time the participants learned not only about different motivations for the presented research, but especially about various concepts on how to plan enzyme-engineering campaigns and which range of experimental and computational tools can be used to create the desired improved biocatalysts.
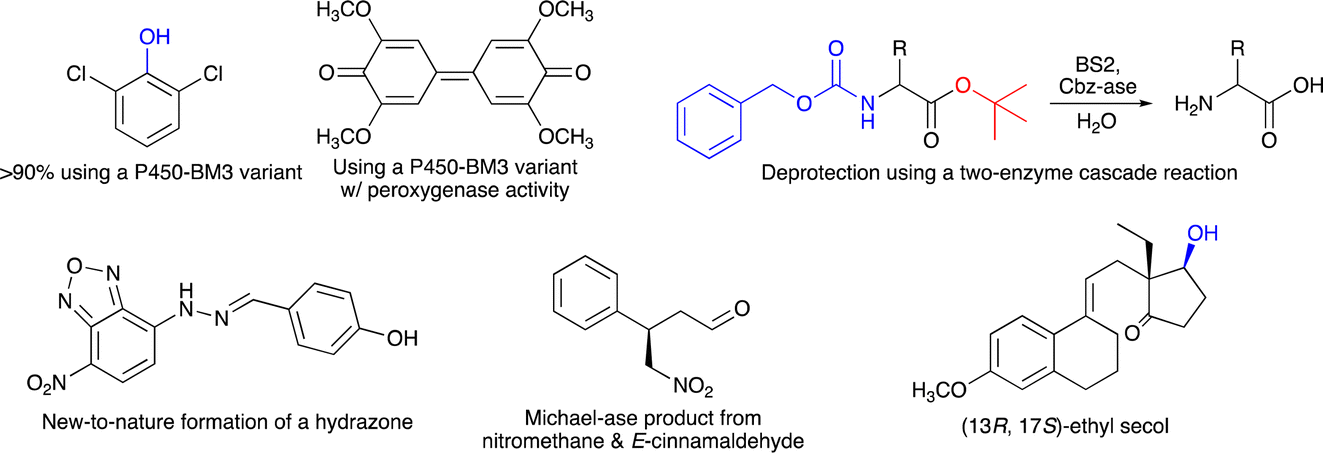 | ||
| Scheme 1 Selected examples of (chiral) products made using biocatalysis, as shown during the Faraday Discussion meeting. | ||
In his impressive talk, Florian Hollfelder (University of Cambridge, UK) combined two methods, which at a first glance are commonly applied separately in directed evolution campaigns: ultrahigh-throughput screening (to identify desired hits out of several millions of variants of a given enzyme or to find novel activities in metagenome libraries) and machine learning (used to predict a handful of mutations for the enzyme of interest). Exemplified for an imine reductase (IRED), Hollfelder showed that his setup enables rapid screening of IRED variants followed by deep mutational scanning, which then provides the information required to create sequence–function maps for fitness predictions. This calculation lead to position-dependent mutability and combinability scores of mutations and indeed, they could identify an IRED with >23-fold improvement in catalytic rate (https://doi.org/10.1039/D4FD00065J). Moreover, this whole enzyme-engineering campaign could be performed within a few weeks. The final talk in Session 1 was given by Mikael Widersten (Uppsala University, Sweden) and he added another concept for directed evolution to the array of methods presented before: phage display. Commonly, phage display is used to identify novel antibodies with desired affinity for a given target, such as an antigen. Catalytically active enzymes are very difficult to identify using phage-display methods, but Widersten showed that indeed it is possible to find an alkylhalide halogenase using this technique, as the first half-reaction leads to a suicide link in the screening of the displayed variants for which he could later confirm experimentally that indeed this mutant is active on the desired substrate (https://doi.org/10.1039/D4FD00001C).
In the discussions related to Session 1, the key points reflected the current challenges we have to deal with when we wish to identify and especially improve enzymes using rational design, directed evolution and/or combinations of both concepts. General questions are, for instance: how to predict the best (smart) mutant library, i.e., can we foresee which mutations (and especially which combinations of mutations) lead to improved enzyme performance, are neutral or are even detrimental? It is especially challenging to discover or predict epistasis effects. A recent publication addresses this point nicely for a β-lactamase.14,15 The roadmap to “fully programmable protein catalysis”11 presented by Hilvert was without doubt very helpful, and stimulated the discussion. The audience also intensively discussed whether machine learning/artificial intelligence may solve these problems in the near future, but this requires a lot of high-quality experimental data, which is difficult (or even almost impossible) to retrieve from scientific publications. Notably, the Pelletier group had just reported their extensive P450-BM3 database, which allows interactive analysis of >1000 mutations collected from several hundred publications, with data collected for a broad range of target compounds.16
As recently pointed out in our review published in Science,4 the reliable calculation of 3D protein structures made a huge step forward with the creation of AlphaFold, but a tool that is still missing is the prediction of enzyme properties from a given protein structure (or even sequence). Scientists active in biocatalysis aim to apply enzymes and therefore knowledge about properties like substrate scope, stereopreference, optical purity of product (% ee), solvent tolerance, pH and temperature optima, and even kcat/Km-predictions would save an enormous amount of experimental time to have the desired synthetically and practically useful enzyme at hand.
Further aspects included the question: what defines an “evolvable” protein? It was discussed that in principle every protein/enzyme can be evolved, but this is rather a matter of the starting point (a lousy catalyst can for sure be improved) and the final target (a highly active enzyme is more difficult to improve, and the theoretically highest activity is set by the diffusion limit). I pointed out that the key natural enzyme in photosynthesis required to fix carbon dioxide, RuBisCo (ribulose-1,5-diphosphate-carboxylase-oxygenase), is a very slow enzyme and despite billions of years of natural evolution it still cannot distinguish properly between CO2 and O2. Obviously, there was no evolutionary pressure and nature overcame the need for RuBisCo improvement simply by making more of the enzyme, making it the most abundant protein on the planet. Another practically not yet solved problem is that recombinant expression of a target enzyme is not always easy (e.g., causing formation of inclusion bodies in E. coli), which becomes even more important when the functional expression of mutant libraries fails, so that improved enzyme variants escape the assay used for screening in microtiter plates or ultrahigh-throughput assays. Clearly, we need better prediction tools for protein solubility and adapted expression systems ensuring that desired enzymes and mutants can always be experimentally identified.
The central theme of Session 2 was “Biocatalytic pathways, cascades, cells and systems”. Pimchai Chaiyen (VISTEC, Rayong, Thailand) presented how they could enhance the production of essential cofactors required for in vivo biocatalysis, as cofactor imbalance often represents an issue in metabolically engineered cells. Indeed, just the overexpression of a xylose reductase enhanced the performance of recombinantly expressed enzymes significantly (4-fold) as shown for a carboxylic acid reductase (CAR), a luciferase and the production of alkanes, as now ATP and NADPH-cofactors could be more efficiently recycled (https://doi.org/10.1039/D4FD00013G).17 In the discussion round, it was questioned whether this concept has a negative impact on E. coli cells, as the initial reduction should cause some penalty to make the aldehyde. It was also asked how much (NADPH/ATP) cofactor is not used in the cell. It was also recommended to include flux analysis in further studies.
Organic chemists regularly have to use protective groups in multistep synthesis and thus also need to ensure that they can be removed selectively and under mild reaction conditions. An alternative for chemical deprotection is the use of enzymes, and Dominic Campopiano (University of Edinburgh, UK) informed the audience about his two-step cascade reaction, where he successfully used the esterase BS2 to hydrolyze a t-butyl group and a second enzyme (Cbz-ase) to remove the Cbz-group in the same molecule (Scheme 1) (https://doi.org/10.1039/D4FD00016A). This lecture raised considerable interest amongst the audience and it was discussed why biocatalytic deprotection is not used more frequently and also for other protecting groups. It was recommended that we should also consider establishing protecting groups designed for enzymes (for both introduction and removal) rather than focusing on currently used protecting groups, as they were designed by chemists and for chemists in the past. Clare Megarity (University of Manchester, UK) presented her electrochemical research on the photosynthetic enzyme ferredoxin NADP+ reductase (FNR), originating from the green alga Chlamydomonas reinhardtii, for which she could invert cofactor preference to enable recycling of NADPH by coupling a FNR variant to downstream NAD(H)-dependent enzymes, co-entrapped in the porous electrode of this so-called e-leaf (https://doi.org/10.1039/D4FD00020J). This electrochemical concept represents an interesting alternative to well-established NAD(P)H recycling systems required for in vitro biocatalysis. Neil Marsh (University of Michigan, USA) studies aromatic decarboxylations catalyzed by prenylated-flavin-dependent enzymes. These UbiD-like enzymes have attracted considerable interest as they (de)carboxylate a broad range of aromatic substrates. He used sequence similarity network analyses to discover novel enzymes and to identify potential substrates based on detecting enzyme-catalysed solvent deuterium exchange into potential substrates, as exemplified for ferulic acid decarboxylase as a model system (https://doi.org/10.1039/D4FD00006D). This presentation provided very interesting insights on how novel enzyme sequences and their catalytic function may be explored.
The sustainable production of hydrogen by means of biocatalysis represents a highly important research area, but the in principle very active [FeFe] hydrogenases required for this are highly oxygen-sensitive, hindering their practical application. Francesca Valetti (University of Torino, Italy) addressed this problem by studying a new class of oxygen-resistant [FeFe] hydrogenases, which opens up new opportunities, not only for hydrogen production, but also for inclusion of these enzymes in CO2 conversion and NAD(P)H recycling. She demonstrated how protein engineering (via point mutations and the creation of chimeric enzymes) can lead to improved variants and provided further information about the mechanism and oxygen-resistance of these interesting enzymes (https://doi.org/10.1039/D4FD00010B).
Session 3 was fully devoted to the creation and study of artificial, biomimetic and hybrid enzymes. Gerard Roelfes (University of Groningen, The Netherlands) started this session and showed how computation-guided engineering of distal mutations enabled the creation of an artificial enzyme, starting from the lactococcal multidrug resistance regulator (LmrR) protein as a scaffold. First, the genetically encoded non-canonical amino acid p-aminophenylalanine (pAF), serving as a catalytic residue, was introduced via stop-codon suppression. Then the incorporation of only a few further mutations – in silico predicted using Zymspot – with >11 Å distance from the active site led to a substantial increase in the formation of a hydrazone from 4-hydroxybenzaldehyde and 4-hydrazino-7-nitro-2,1,3-benzoxadiazole (Scheme 1) (https://doi.org/10.1039/D4FD00069B). Ivana Drienovská (VU Amsterdam, The Netherlands) also used LmrR-pAF as a scaffold (and another protein, TOYE) to design an enzyme catalyzing a Michael-addition using nitromethane and E-cinnamaldehyde as substrates. Notably, both designs were active and stereoselective and even showed opposite stereopreference (Scheme 1) (https://doi.org/10.1039/D4FD00057A). In these two lectures, the incorporation of a non-canonical amino acid (ncAA) was crucial to enable new-to-nature chemistry. Another relevant and well-studied ncAA is Nδ-methylhistidine (MeHis), and Anthony Green (University of Manchester, UK) showed in his lecture how MeHis-containing enzymes can be produced on a larger scale (and not only by small-scale in vitro transcription/translation or E. coli cultures). For this, they developed a highly efficient aminoacyl tRNA synthetase, which operates even at MeHis concentrations of only ∼0.1 mM (https://doi.org/10.1039/D4FD00019F). This achievement makes a scalable and economical production of proteins bearing ncAAs much more feasible. It also fills an important research gap, as with only cheap biosynthesis, newly designed artificial enzymes can indeed reach the application level in biocatalysis.
The next three lectures in this session dealt with computational studies to design and understand enzymes better. Sílvia Osuna (University of Girona, Spain) presented the “shortest path map” method to estimate conformational dynamics landscapes in enzyme catalysis and exemplified this for a beta subunit of a Trp synthase. She showed that her concept can more efficiently capture the effect of distal mutations (https://doi.org/10.1039/D3FD00156C). Computational studies were also used by Vicent Moliner (University Jaume I, Castellón, Spain) to study the mechanism of a “polyurethane esterase” from Pseudomonas chlororaphis, which can hydrolyse ester bonds in mixed polyester-polyurethanes, such as Impranil as studied here. He showed that the enzyme PueA has a typical carboxylesterase catalytic triad composed of Ser–His–Asp (https://doi.org/10.1039/D4FD00022F). As pointed out in the discussion of this lecture, it must be emphasised that PueA is not acting on the carbamate bond present in polyurethanes, for which only recently have true urethanases belonging to an amidase signature family been described.18 Finally, Lynn Kamerlin (GeorgiaTech, Atlanta, USA) presented two computational tools (key interactions finder, KIF, and key interaction networks, KIN), which she had recently developed (https://doi.org/10.1039/D4FD00018H). KIF allows the relation of non-covalent interactions in structural ensembles and KIN can be used to analyse evolutionary groups in the context of protein structures and in interaction networks. She exemplified the usefulness of these tools in the engineering of a β-lactamase, as this helped to prioritize residues and networks. This concept was well-received by the audience also because she could show that generalist enzymes can be turned into specialist enzymes or vice versa.
The last session focussed on “Biocatalysis for industry, medicine and the circular economy”. Two presentations dealt with enzymatic degradation of polyethylene terephthalate (PET). As microplastic particles (MPs) represent an important environmental issue and it was recently reported that MPs have even been found in human blood, Per-Olof Syrén (KTH Stockholm, Sweden) investigated whether an esterase (IsPETase mutant S2382A) can hydrolyse PET in human serum. This mutant was created to act on trans-configured PET and thus it should also act on crystalline PET polymer fibers. Syrén showed that indeed hydrolysis took place and using HEK cells, he clarified that the viability of the cells is unaffected by the presence of the PETase (https://doi.org/10.1039/D4FD00014E). In the discussion, it was pointed out that MPs are composed of various plastics and not only PET. Hence, for their degradation, different enzymes would be needed. Another point was the potential immunogenicity of the enzyme and potential harmful effects of the degradation products, such as terephthalic acid and ethylene glycol. For recycling of PET from waste materials such as drinking bottles, the company Carbios reported in 2020 about the engineering of a highly active and thermostable enzyme, a variant of a leaf-branch compost cutinase, LCC. This quadruple mutant (LCC-ICCG)19 could already be used for large-scale recycling of PET on the multi-ton scale; the current state of enzymatic polymer degradation and recycling has been recently summarized.20 Bruce Lichtenstein (University of Portsmouth, UK) wished to further improve PET hydrolysis and investigated the effect of fusion partners (using the SpyCatcher:SpyTag complex) for three PETases (IsPETase, TfCut1 and LCC-ICCG). Although a positive effect on thermal stability was found, unfortunately none of the constructs showed higher catalytic activity on PET films (https://doi.org/10.1039/D4FD00067F). As an alternative to polymer hydrolysis, Lu Shin Wong (University of Manchester, UK) presented his research on the biocatalytic synthesis of silicone polymers. He used the enzyme silicatein-α from marine sponges as a starting enzyme and demonstrated that it indeed can make (or break) Si–O bonds, using dialkoxysilanes as starting materials (https://doi.org/10.1039/D4FD00003J). Thus, he demonstrated the potential of biocatalysis for polysiloxane synthesis.
A completely different biocatalytic application was presented by Louis Luk (Cardiff University, UK). He reported on a peptide asparaginyl ligase (PAL) that is useful to make modified peptides and proteins. A challenge in its application is the difficult expression of active PAL. To overcome this problem, Luk used a lumazine-synthetase-based compartmentalization, which allows encapsulation of PAL in a “container” in E. coli. He showed a proof-of-concept for these artificial organelles, but it turned out that the PAL activity was significantly lower (https://doi.org/10.1039/D4FD00002A).
The enzymatic synthesis of active pharmaceutical intermediates (API) has a long tradition in biocatalysis. Here, Daniel Dourado (Almac Sciences, UK) presented the identification, design and application of a carbonyl reductase to make (13R,17S)-ethyl secol (Scheme 1), the key intermediate in the synthesis of etonogestrel and levonorgestrel, two modern contraceptive APIs. Starting from their in-house panel of reductases, they then improved one enzyme to meet the process requirements, which finally enabled complete conversion of the corresponding ketone in this asymmetric reduction at 90 g L−1 substrate loading (https://doi.org/10.1039/D4FD00011K). A completely different application of biocatalysis was shown by Stefan Lutz (Codexis, Redwood City, USA). He exemplified how the engineering of a T7 RNA polymerase allows efficient co-transcriptional capping with reduced dsRNA byproducts in the synthesis of mRNA molecules (https://doi.org/10.1039/D4FD00023D). Thus, not only enzymatic DNA synthesis is currently explored as a new emerging application field, but also RNA synthesis can be advanced using suitable enzymes combined with the many tools available for enzyme engineering.
The Faraday Discussion ended with my concluding remarks. Several aspects of my presentation are already mentioned in the summary of the four sessions as covered above. Further points included the question of how to capture CO2 efficiently using biocatalysis and how we can recycle/upcycle/degrade (micro)plastics better. It was also discussed whether we need more political steps to push the knowledge about biocatalysis as modern, safe, sustainable, and environmentally friendly method following also the 12 Green Chemistry principles. We also felt that basic knowledge about biocatalysis, its tools and applications must find much more prominent ways into teaching courses for undergraduate (and PhD) students (especially in organic chemistry) to make them aware of this highly advanced field, so that they learn that not only classical organic synthesis routes exist, especially for making chiral products.
Conclusions
The Faraday Discussion on biocatalysis was an outstanding and highly successful event, which should be obvious from reading these concluding remarks, as well as the many original publications from the speakers in this issue. Other aspects, not or less covered during this meeting, include for instance (chemo-)enzymatic cascades, retrosynthesis, and the integration of biocatalysis into synthetic biology/metabolic engineering efforts. Eventually, these could be topics for a future Faraday Discussion event, which I happily will attend.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
UTB wrote this review article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
I am very grateful to Adrian Mulholland and Nicholas Turner for critical reading of this article.References
- D. Yi, T. Bayer, C. P. S. Badenhorst, S. Wu, M. Doerr, M. Höhne and U. T. Bornscheuer, Chem. Soc. Rev., 2021, 50, 8003–8049 RSC.
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef CAS PubMed.
- E. L. Bell, W. Finnigan, S. P. France, A. P. Green, M. A. Hayes, L. J. Hepworth, S. L. Lovelock, H. Niikura, S. Osuna, E. Romero, K. S. Ryan, N. J. Turner and S. L. Flitsch, Nat. Rev. Methods Primers, 2021, 1, 46 CrossRef CAS.
- R. Buller, S. Lutz, R. J. Kazlauskas, R. Snajdrova, J. C. Moore and U. T. Bornscheuer, Science, 2023, 382, eadh8615 CrossRef CAS PubMed.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS PubMed.
- U. T. Bornscheuer, Philos. Trans. R. Soc., A, 2018, 376, 20170063 CrossRef PubMed.
- M. T. Reetz, Angew. Chem., Int. Ed., 2011, 50, 138–174 CrossRef CAS PubMed.
- F. H. Arnold, Angew. Chem., Int. Ed., 2019, 58, 14420–14426 CrossRef CAS PubMed.
- S. Studer, D. A. Hansen, Z. L. Pianowski, P. R. E. Mittl, A. Debon, S. L. Guffy, B. S. Der, B. Kuhlman and D. Hilvert, Science, 2018, 362, 1285–1288 CrossRef CAS PubMed.
- S. Basler, S. Studer, Y. Zou, T. Mori, Y. Ota, A. Camus, H. A. Bunzel, R. C. Helgeson, K. N. Houk, G. Jiménez-Osés and D. Hilvert, Nat. Chem., 2021, 13, 231–235 CrossRef CAS PubMed.
- S. L. Lovelock, R. Crawshaw, S. Basler, C. Levy, D. Baker, D. Hilvert and A. P. Green, Nature, 2022, 606, 49–58 CrossRef CAS PubMed.
- Y. Jiang, C. Wang, N. Ma, J. Chen, C. Liu, F. Wang, J. Xu and Z. Cong, Catal. Sci. Technol., 2020, 10, 1219–1223 RSC.
- N. Ma, W. Fang, C. Liu, X. Qin, X. Wang, L. Jin, B. Wang and Z. Cong, ACS Catal., 2021, 11, 8449–8455 CrossRef CAS.
- C. Fröhlich, H. A. Bunzel, K. Buda, A. J. Mulholland, M. W. van der Kamp, P. J. Johnsen, H.-K. S. Leiros and N. Tokuriki, Nat. Catal., 2024, 7, 499–509 CrossRef PubMed.
- G. Dotta and A. J. Vila, Nat. Catal., 2024, 7, 467–468 CrossRef CAS.
- D. J. Fansher, J. N. Besna, A. Fendri and J. N. Pelletier, ACS Catal., 2024, 14, 5560–5592 CrossRef CAS PubMed.
- J. Jaroensuk, C. Sutthaphirom, J. Phonbuppha, W. Chinantuya, C. Kesornpun, N. Akeratchatapan, N. Kittipanukul, K. Phatinuwat, S. Atichartpongkul, M. Fuangthong, T. Pongtharangkul, F. Hollmann and P. Chaiyen, J. Biol. Chem., 2024, 300, 105598 CrossRef CAS PubMed.
- Y. Branson, S. Söltl, C. Buchmann, R. Wei, L. Schaffert, C. P. S. Badenhorst, L. Reisky, G. Jäger and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2023, 62, e202216220 CrossRef CAS PubMed.
- V. Tournier, C. M. Topham, A. Gilles, B. David, C. Folgoas, E. Moya-Leclair, E. Kamionka, M. L. Desrousseaux, H. Texier, S. Gavalda, M. Cot, E. Guémard, M. Dalibey, J. Nomme, G. Cioci, S. Barbe, M. Chateau, I. André, S. Duquesne and A. Marty, Nature, 2020, 580, 216–219 CrossRef CAS PubMed.
- V. Tournier, S. Duquesne, F. Guillamot, H. Cramail, D. Taton, A. Marty and I. André, Chem. Rev., 2023, 123, 5612–5701 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |