 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Developing deprotectase biocatalysts for synthesis†
Lisa Kennedy,
Mariyah Sajjad,
Michael A. Herrera,
Peter Szieber,
Natasza Rybacka,
Yinan Zhao,
Craig Steven,
Zainab Alghamdi ,
Ivan Zlatkov,
Julie Hagen,
Chloe Lauder,
Natalie Rudolfova,
Magdalena Abramiuk,
Karolina Bolimowska,
Daniel Joynt,
Angelica Lucero,
Gustavo Perez Ortiz,
Annamaria Lilienkampf,
Alison N. Hulme and
Dominic J. Campopiano*
,
Ivan Zlatkov,
Julie Hagen,
Chloe Lauder,
Natalie Rudolfova,
Magdalena Abramiuk,
Karolina Bolimowska,
Daniel Joynt,
Angelica Lucero,
Gustavo Perez Ortiz,
Annamaria Lilienkampf,
Alison N. Hulme and
Dominic J. Campopiano*
School of Chemistry, University of Edinburgh, David Brewster Road, Edinburgh, EH9 3FJ, UK. E-mail: Dominic.Campopiano@ed.ac.uk
First published on 1st March 2024
Abstract
Organic synthesis often requires multiple steps where a functional group (FG) is concealed from reaction by a protecting group (PG). Common PGs include N-carbobenzyloxy (Cbz or Z) of amines and tert-butyloxycarbonyl (OtBu) of acids. An essential step is the removal of the PG, but this often requires excess reagents, extensive time and can have low % yield. An overarching goal of biocatalysis is to use “green” or “enzymatic” methods to catalyse chemical transformations. One under-utilised approach is the use of “deprotectase” biocatalysts to selectively remove PGs from various organic substrates. The advantage of this methodology is the exquisite selectivity of the biocatalyst to only act on its target, leaving other FGs and PGs untouched. A number of deprotectase biocatalysts have been reported but they are not commonly used in mainstream synthetic routes. This study describes the construction of a cascade to deprotect doubly-protected amino acids. The well known Bacillus BS2 esterase was used to remove the OtBu PG from various amino acid substrates. The more obscure Sphingomonas Cbz-ase (amidohydrolase) was screened with a range of N-Cbz-modified amino acid substrates. We then combined both the BS2 and Cbz-ase together for a 1 pot, 2 step deprotection of the model substrate CBz-L-Phe OtBu to produce the free L-Phe. We also provide some insight into the residues involved in substrate recognition and catalysis using docked ligands in the crystal structure of BS2. Similarly, a structural model of the Cbz-ase identifies a potential di-metal binding site and reveals conserved active site residues. This new biocatalytic cascade should be further explored for its application in chemical synthesis.
Introduction
The introduction and subsequent removal of protecting groups (PGs) are often important and widely executed steps in synthetic routes towards complex organic molecules such as peptides, nucleotides, oligosaccharides and natural products. Suitable protecting groups are necessary to prevent the formation of undesired bonds and side reactions. Certainly, a diverse range of protecting groups have been developed for this purpose and chemical methods for both their protection and deprotection are well-established.1–3 Regularly, the issue arises where a particular functional group requires selective protection/deprotection in the presence of similarly reactive groups. In such instances, the mildest reaction conditions and highest selectivity are desirable. The application of biocatalysts in protecting group chemistry offers an excellent alternative to chemical methods, with mild reaction conditions and high selectivity as notable advantages. Therefore, it is surprising that biocatalysts are not more commonly used for this purpose.Protection of the α-amino group of amino acids in peptide chemistry is important to prevent polymerization of the amino acid once activated.1 The first modern protecting group, N-carbobenzyloxy (Cbz or Z) group is often used to protect amino groups during organic synthesis.1 The deprotection step for this group frequently involves catalytic hydrogenation (Fig. 1). Issues with the selectivity of this method can arise when other reactive functional groups exist in the protected substrate such as carbon–carbon double bonds, thiols or sulfides, which can interfere.4 It should also be noted that the toxicity of palladium becomes an issue in the synthesis of peptide drugs. Some examples of enzymatic hydrolysis of Cbz-protected substrates have been reported previously through the use of soil microorganisms,5 an immobilized penicillin acylase,6 and an amidohydrolase from Sphingomonas paucimobilis (Fig. 1).4 The latter was reported by Patel et al. in which the biocatalyst was isolated from the cell extracts of S. paucimobilis, and shown to successfully deprotect a number of Cbz-protected amino acids with enantioselectivity for the (L)-enantiomer.4 Kinetic resolutions of the D/L-Cbz-substrates resulted in the isolation of both the deprotected L-amino acid and the Cbz-D-amino acid in high ee (>99%).7 In subsequent years, it was also reported that a complimentary D-specific Cbz hydrolase existed from Burkholderia phenazinium.8 Unfortunately, no further characterisation of this potentially useful biocatalyst has been reported. While these studies have highlighted the potential for high activity and selectivity in the deprotection of Cbz-substrates, it is surprising that this class of ‘deprotectases’ have not received more attention in both academic and industrial settings.
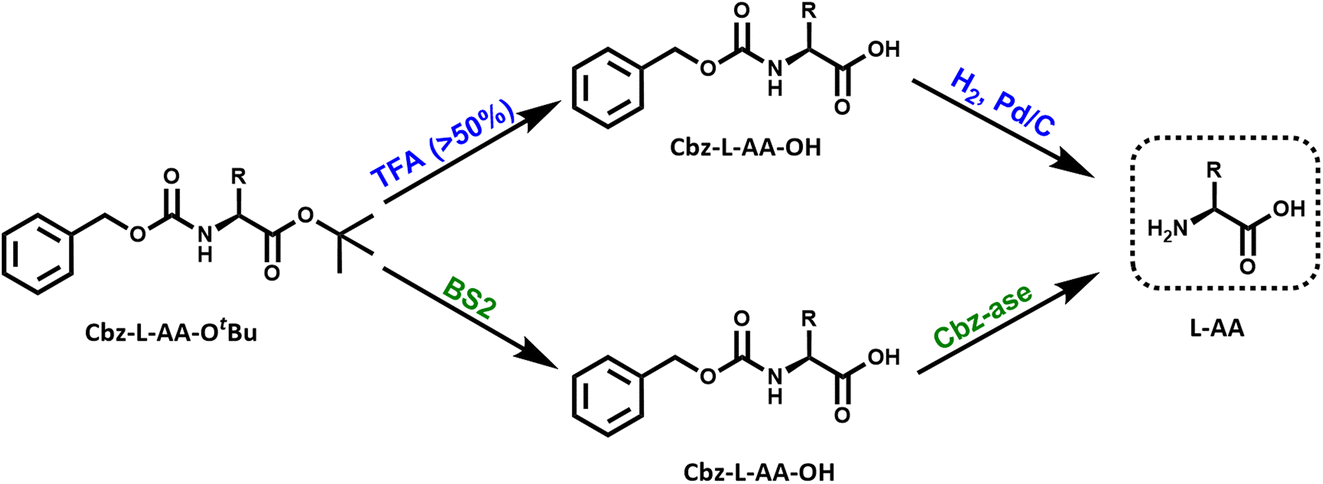 | ||
| Fig. 1 Reaction schemes for the chemical and biocatalytic deprotection of a doubly protected amino acid (Cbz-L-AA-OtBu). Top route (blue): treatment with trifluoroacetic acid (% TFA) removes the OtBu ester protecting group from the acid that yields the Cbz-protected amino acid. The Cbz group is removed by catalytic hydrogenation over palladium. This results in the free L-amino acid. Bottom route (green): the same sequential reactions are catalysed by the combination of BS2 esterase and Cbz-ase biocatalysts. | ||
Many examples of enzymatic deprotection exist, the literature in this area particularly focuses on carbohydrate protection/deprotection.9 However, BS2, an esterase from Bacillus subtilis has been shown to deprotect a number of carboxyl protecting groups; the tert-butyl group,10 benzyl and methyl groups,11 and allyl and chloroethyl groups.12 The carboxyl protecting groups are also important moieties in peptide chemistry for the protection of the C-terminal carboxylic acid or the side chain carboxylic acid of glutamate or aspartate residues.1 tert-Butyl (tBu) protection is often used in both solid and solution-phase peptide synthesis. However, deprotection of this group requires harsh conditions of acid or base,13 high temperatures and organic solvents or the use of expensive transition metal complexes.14 Acidic conditions (trifluoroacetic acid, TFA) are most commonly employed (Fig. 1), however this may pose a challenge if there are acid-sensitive functional groups present within the protected compound. The use of an enzyme, such as BS2, for the deprotection allows the reaction to occur under mild conditions with high selectivity.
One of the major advantages of biocatalysis is the ability to combine reactions in a one-pot cascade due to the compatibility of biocatalysts with mild conditions and their inherent selectivity.15 A cascade is defined as “the combination of at least two chemical steps in a single reaction vessel without the isolation of any intermediate(s)”.16 This study aims to further explore two of the existing deprotectases and their combined application in a deprotection cascade to prepare a full deprotected product (Fig. 1). As proof of concept we describe the cloning and characterisation of recombinant BS2 and Cbz-ase and their use in the two-step deprotection of Cbz-L-Phe-OtBu to L-Phe.
Results and discussion
Enzymatic deprotection of the N-carbobenzyloxy (Cbz or Z) group
A synthetic gene encoding the Cbz-ase was ordered from Genscript based on the published sequence.7 The original paper reported cloning the gene from Sphingomonas paucimobilis SC 16113. However, we could not find an accession code for this, but did find a related amidohydrolase from a Sphingobium yanoikuyae strain YC-JY1 (GenBank: QHD65840). The synthetic gene (Fig. S1A†) was cloned in a pET28a plasmid containing an N-terminal 6xhistidine tag to express a recombinant Cbz-ase (447aa, 48 kDa). This plasmid was used to transform E. coli BL21 (DE3) cells and the Cbz-ase was overexpressed by addition of IPTG (final conc. 0.2 mM) at O.D. (600 nm) = 0.6. The expression was carried out for 20 h at 16 °C. The cells were harvested by centrifugation and the Cbz-ase was purified by immobilised metal affinity chromatography (IMAC). The enzyme was well expressed and we isolated ∼10 mg litre−1 of culture. The recombinant Cbz-ase was analysed by ESI-MS and a molecular weight of 47![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 930 Da was in good agreement with the predicted mass minus the N-terminal methionine residue (47927 Da, Fig. S4†). The enzyme was judged to be pure enough for activity assay after IMAC (Fig. S2†) for use in the deprotection of Cbz-L-phenylalanine (Z-L-Phe) and detection of the L-Phe product through a coupled colorimetric assay.17 This method uses the FAD-dependent L-amino acid oxidase (L-AAO) and the hydrogen peroxide formed is easily monitored using horseradish peroxidase (HRP) and 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid (ABTS), measuring absorbance at 420 nm (Fig. S3†). The purified Cbz-ase was active in this coupled assay (Fig. S5B†) using Z-L-Phe, and the biocatalyst was then tested in the deprotection of various Cbz-protected amino acids by RP-HPLC to detect the formation of the free amino acid (L-Phe, L-Tyr, L-Trp and Gly-L-Phe, Fig. S19–S27,† and Table 1). The hydrolysis of the Cbz group results in the direct formation of both the free amino acid and benzyl alcohol. This direct production of benzyl alcohol allowed the monitoring of deprotection of non-UV active substrates via HPLC (e.g. L-Ala, L-Lys, L-Pro, L-Glu, Fig. S19–S27† and Table 1).
930 Da was in good agreement with the predicted mass minus the N-terminal methionine residue (47927 Da, Fig. S4†). The enzyme was judged to be pure enough for activity assay after IMAC (Fig. S2†) for use in the deprotection of Cbz-L-phenylalanine (Z-L-Phe) and detection of the L-Phe product through a coupled colorimetric assay.17 This method uses the FAD-dependent L-amino acid oxidase (L-AAO) and the hydrogen peroxide formed is easily monitored using horseradish peroxidase (HRP) and 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid (ABTS), measuring absorbance at 420 nm (Fig. S3†). The purified Cbz-ase was active in this coupled assay (Fig. S5B†) using Z-L-Phe, and the biocatalyst was then tested in the deprotection of various Cbz-protected amino acids by RP-HPLC to detect the formation of the free amino acid (L-Phe, L-Tyr, L-Trp and Gly-L-Phe, Fig. S19–S27,† and Table 1). The hydrolysis of the Cbz group results in the direct formation of both the free amino acid and benzyl alcohol. This direct production of benzyl alcohol allowed the monitoring of deprotection of non-UV active substrates via HPLC (e.g. L-Ala, L-Lys, L-Pro, L-Glu, Fig. S19–S27† and Table 1).
| Substrate | Conversiona (%) |
|---|---|
| a The Z-amino acid substrates (5 mM) and Cbz-ase (0.5 mg mL−1) were incubated at 37 °C, 250 rpm for 24 hours. Conversion was determined using HPLC analysis through the quantification of benzyl alcohol. | |
| Z-L-Phe | 100 |
| Z-L-Tyr | 100 |
| Z-L-Ala | 99 |
| Z-L-Lys | 83 |
| Z-L-Gly-Phe | 65 |
| Z-L-Trp | 7 |
| Boc-L-Lys(Z)-OH | 3 |
| Z-L-Glu(OtBu)-OH | 1 |
| Z-L-Pro | 0 |
The substrate activity we have measured is in good agreement with that reported in the original study by Patel et al. (L-Phe, L-Tyr, L-Leu, L-Pro, L-Lys).7 They also reported little to no activity with the D-enantiomers of these amino acid substrates although this analysis was carried out with E. coli cell extracts expressing the Cbz-ase rather than purified enzyme. HPLC analysis revealed a time-dependent formation of L-Phe (9.6 min) with the depletion of the Cbz-L-Phe substrate (20.1 min, Fig. 2B) and the appearance of the benzyl alcohol by product (14.2 min, Fig. 2B and C). The reaction was complete after 90 min (Fig. 2B and C). The formation of this by-product in the enzymatic hydrolysis differs from the by-product produced in the metal-catalysed hydrogenation that is commonly employed for the deprotection of Cbz groups. The production of benzyl alcohol in enzymatic Cbz-deprotection was also noted by Morozova et al. in 2023 through their study of Cbz deprotection by penicillin acylases.18
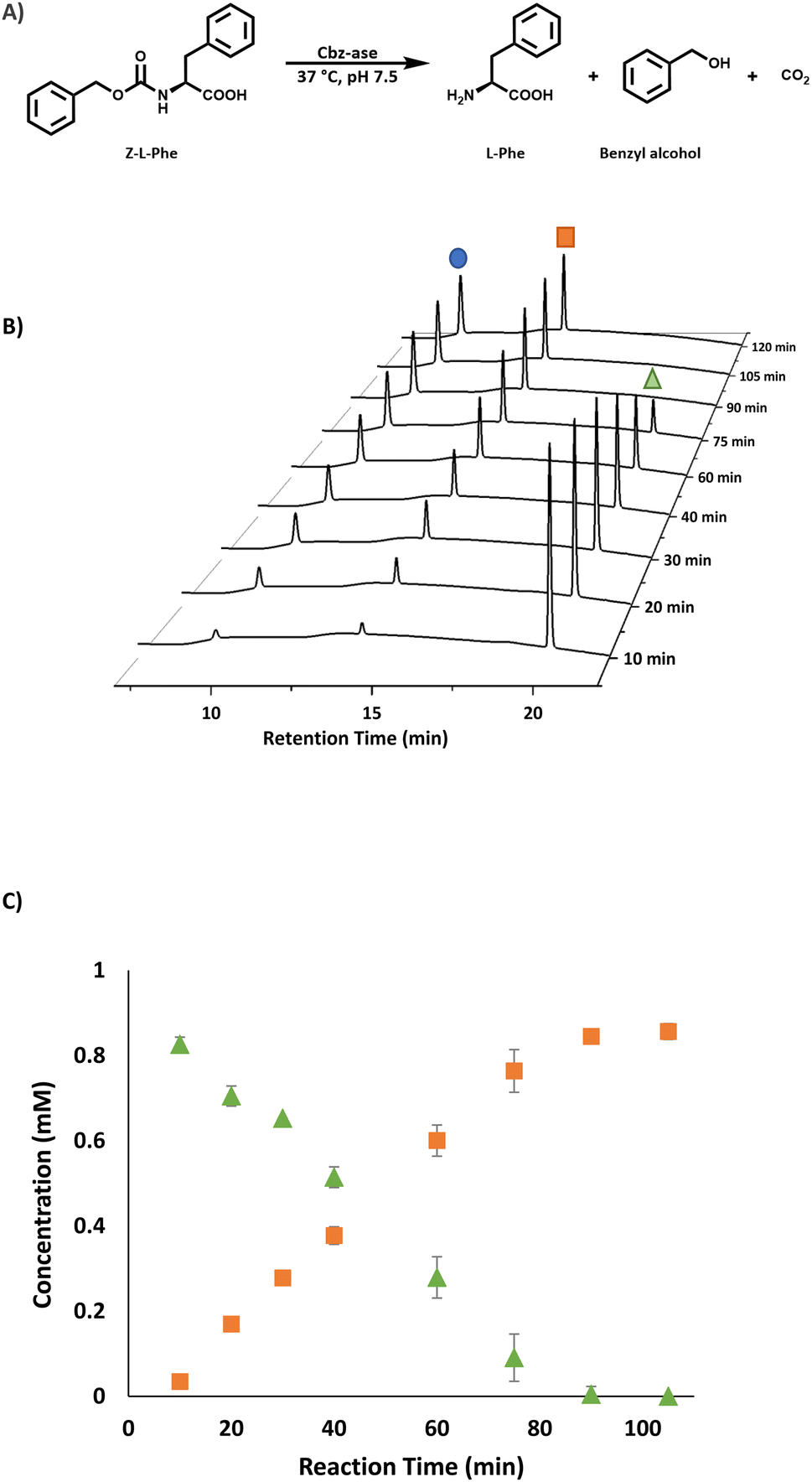 | ||
| Fig. 2 Analysis of the Cbz-ase catalysed reaction. (A) Reaction scheme for the Cbz-ase catalysed deprotection of Z-L-Phe. (B) Reaction monitoring using HPLC analysis showing the formation of L-Phe (9.6 min, blue circle) and benzyl alcohol (14.2 min, orange square) and the disappearance of the Z-L-Phe substrate (20.1 min, green triangle). (C) Plot of concentration (mM) vs. reaction time (min) displaying the direct formation of by-product benzyl alcohol (orange square) in relation to the depletion of Z-L-Phe (green triangle). Reaction conditions: Z-L-Phe (10 mM), Cbz-ase (0.075 mg mL−1) in sodium phosphate buffer (50 mM, pH 7.5), 1 mL reaction volume, 37 °C, 250 rpm. Samples (100 μL) taken at specified time intervals, quenched with TFA (2 μL, 10% in water) and diluted to 1 mL with water. | ||
Enzymatic deprotection of the tert-butyl ester group
We chose the well characterised esterase from Bacillus subtilis (BS2) as the biocatalyst for the tert-butyl (tBu) deprotection of the carboxyl group.10 The synthetic gene from Genscript was cloned in a pET28a plasmid containing a C-terminal histidine tag and TEV cleavage site. The sequence (Fig. S1B†) of this clone was based on the Bacillus subtillis gene sequence and the PDB code (WP_326228118 and 1QE3). This plasmid was used to transform E. coli BL21 (DE3) cells and the protein was overexpressed by the addition of IPTG (final conc. 0.2 mM) at O.D. (600 nm) = 0.6. The expression was carried out for 20 h at 16 °C. The cells were harvested by centrifugation and lysed by sonication. The BS2 was purified by immobilised metal affinity chromatography (IMAC). The enzyme was expressed very well and we isolated ∼45 mg litre−1 of culture. The recombinant BS2 (∼56 kDa) was analysed by ESI-MS analysis and a molecular weight of 55540 Da (Fig. S7†) was in good agreement with the predicted mass minus the N-terminal methionine residue (55537 Da). The activity of the purified, recombinant BS2 esterase was verified using the p-nitrophenolate assay, by monitoring absorbance at 405 nm resulting from the hydrolysis of p-nitrophenyl-acetate substrate (Fig. S6†). As reported by Bornscheuer, Kokotos et al., BS2 is capable of hydrolysing a wide range of N-protected tert-butyl esters.10–12 In accordance with the substrate chosen for the Cbz-ase and for ease of detection by HPLC, L-Phe-OtBu was tested as a substrate for BS2. It was shown that BS2 hydrolyses the carboxyl protecting group efficiently with full conversion to form the unprotected L-Phe (9.6 min) from L-Phe-OtBu (16.6 min) in 24 h (Fig. 3).
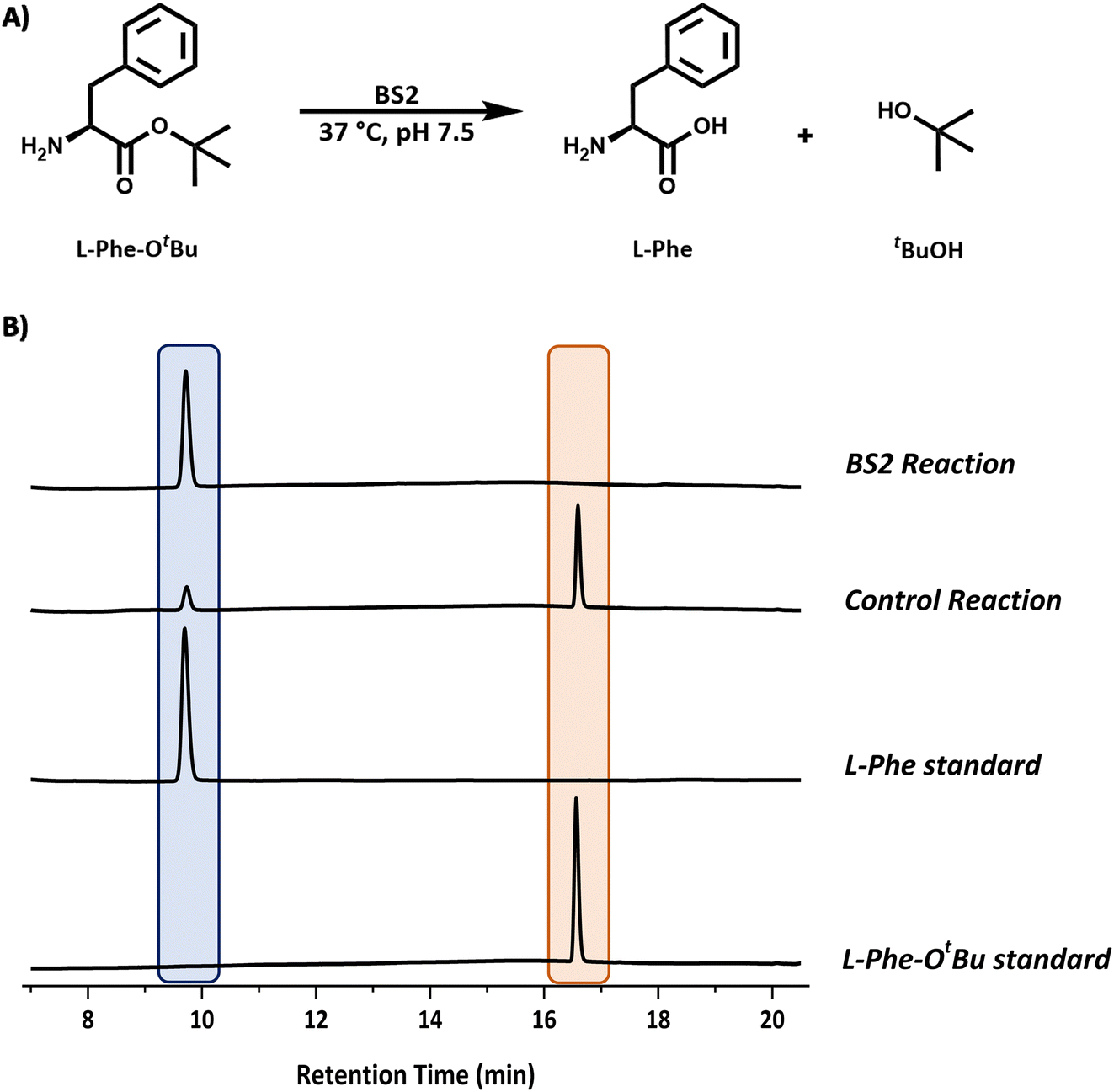 | ||
| Fig. 3 Analysis of the BS2 catalysed reaction. (A) Reaction scheme for the BS2-catalysed deprotection of L-Phe-OtBu. (B) Reaction monitoring by HPLC analysis showing the formation of L-Phe (9.6 min, blue) from the L-Phe-OtBu substrate (16.6 min, peach). Reaction conditions: L-Phe-OtBu (10 mM), BS2 (0.9 mg mL−1) in sodium phosphate buffer (50 mM, pH 7.5), 1 mL reaction volume, 37 °C, 250 rpm. Samples (200 μL) taken and quenched with acetonitrile (200 μL) and diluted to 1 mL with water. | ||
Biocatalytic cascade for the deprotection of a doubly protected amino acid
In light of the successful deprotection of the singly protected amino acids, it was proposed that a novel one-pot deprotection cascade could be employed for doubly protected substrates. The strategy was to use recombinant BS2 to firstly hydrolyse the tert-butyl group, then the recombinant Cbz-ase removes the Cbz group. As a proof of concept, the doubly protected phenylalanine (Z-L-Phe-OtBu) was chosen since both biocatalysts had been shown to fully deprotect the corresponding singly-protected L-Phe as described above. The Z-L-Phe-OtBu substrate used in the cascade reaction was synthesised in 59% yield using standard methods. The solubility of Z-L-Phe-OtBu in the biocatalysis reaction buffer was a challenge, and to prevent precipitation the reaction was performed with low substrate concentration (2 mM), high enzyme loading (2.5 mg BS2, 0.3 mg Cbz-ase) and 10% AcCN to dissolve the substrates. The cascade reaction was carried out in a sequential manner; firstly using BS2 to deprotect the tert-butyl group and generate Z-L-Phe over a 24 h period. Despite the low substrate concentration, the disappearance of the Z-L-Phe-OtBu was observed. This was accompanied by the appearance of the Z-L-Phe product at 20 min (Fig. 4A). The first step to deprotect Z-L-Phe-OtBu with BS2 appeared to not go to completion as some of the doubly-protected Z-L-Phe-OtBu substrate was still observed and Z-L-Phe was formed with 26% conversion. The low conversion in this step can somewhat be attributed to the insolubility of Z-L-Phe-OtBu, as precipitation could be observed in the reaction. It is also worth noting that the benzyl alcohol by-product was detected suggesting that, under these conditions, BS2 displays some activity in hydrolysing the Cbz group. Then, addition of the Cbz-ase resulted in the deprotection of the Cbz group (over 6 h) from the Z-L-Phe “intermediate” to produce the unprotected L-Phe (at 9.6 min, Fig. 4B) in 89% conversion from the Z-L-Phe intermediate, with 23% overall conversion. The benzyl alcohol by-product was also observed (14.2 min, Fig. 4B). Taken together, this analysis provides supporting evidence that the combination of BS2 with Cbz-ase has produced L-Phe from a doubly-protected amino acid.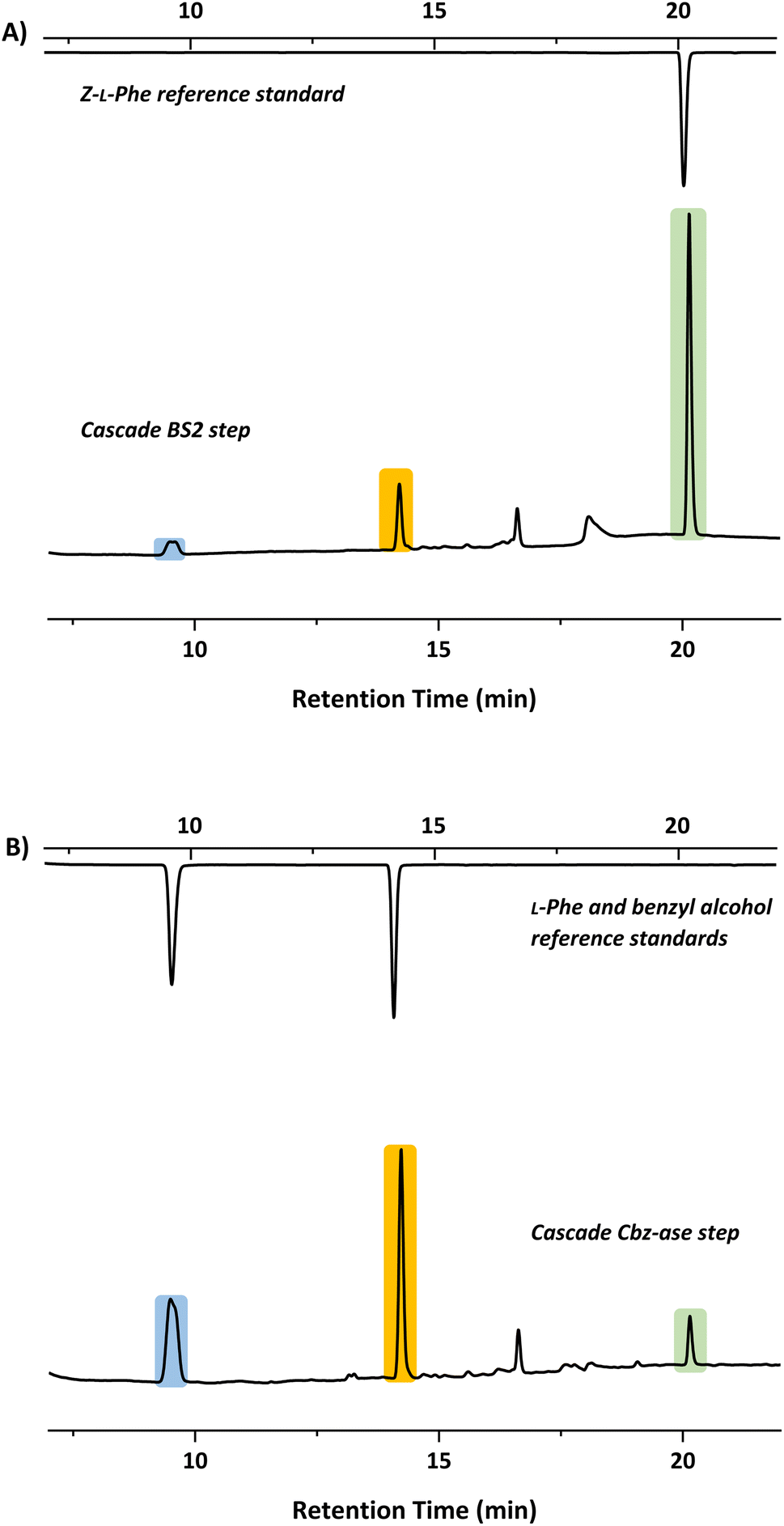 | ||
| Fig. 4 HPLC chromatograms showing product formation after each step of the sequential biocatalytic deprotection cascade. (A) BS2 step in which Z-L-Phe-OtBu is deprotected to form Z-L-Phe (green, 20.1 min). (B) Cbz-ase step in which the Z-Phe formed in step A is converted to L-Phe (blue, 9.6 min) and benzyl alcohol (orange, 14.2 min). | ||
Structural analysis of the BS2 and Cbz-ase biocatalysts
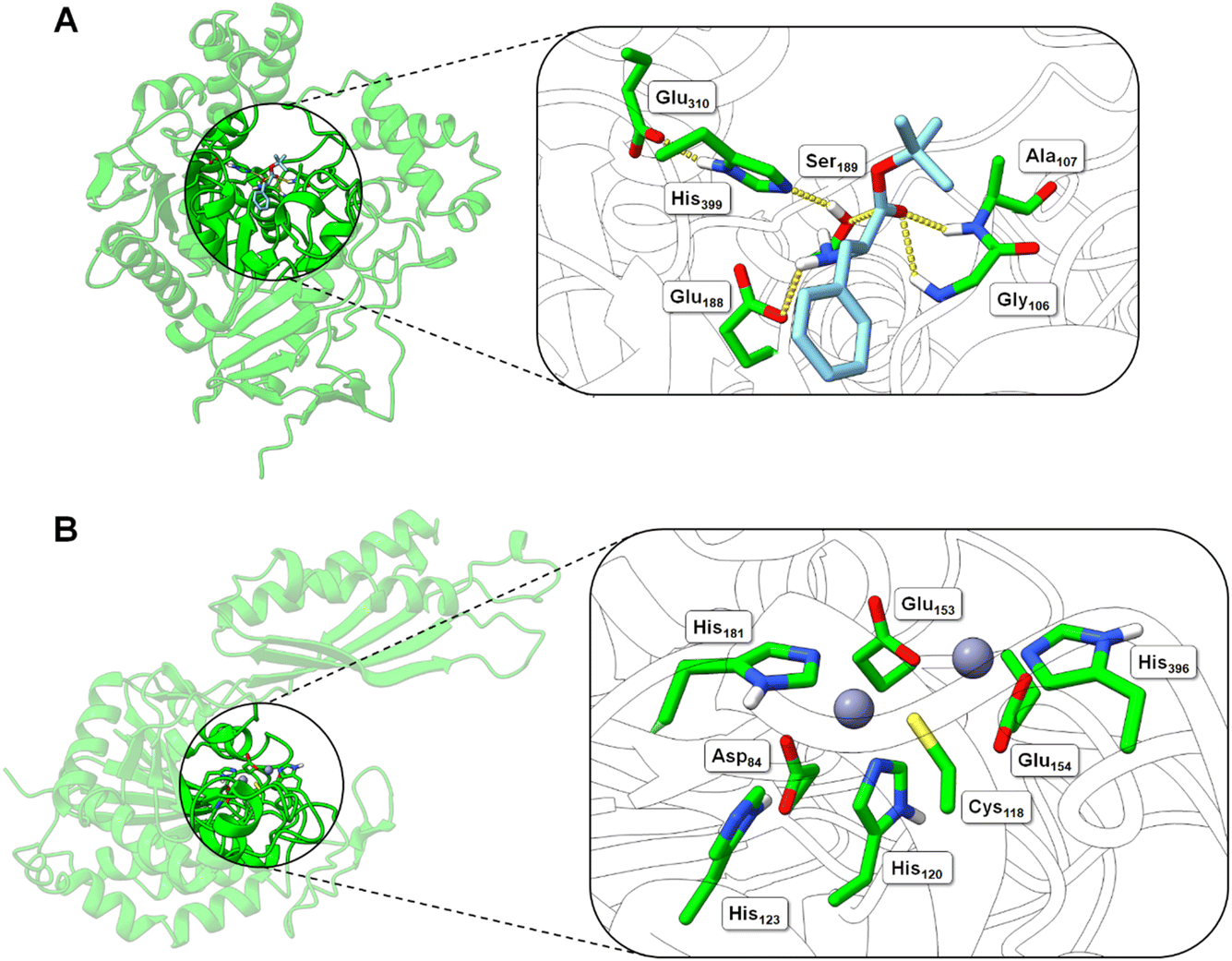 | ||
| Fig. 5 Structural models of the BS2 and Cbz-ase biocatalysts. (A) Model of the BS2 based on the X-ray crystal structure (PDB: 1QE3). The inset shows the putative active with the catalytic triad (Ser189, Glu310, His399) with the key Ser189 nucleophile highlighted. The docked L-Phe-OtBu ligand is shown to H-bond with the side chains of the potential oxyanion hole (Gly106 and Ala107) and the amine of the substrate engages in an ionic interaction with Glu188. The Phe side chain binds in a hydrophobic pocket. (B) Model of the Cbz-ase structure with two Zn2+ ions docked in putative metal-binding pockets. Putative binding residues are shown. A highly conserved Cys118 is highlighted and Zn2+ ions are shown as grey-blue spheres. | ||
In the absence of a crystal structure, we instead built a structural model of S. paucimobilis Cbz-ase using the powerful structural prediction tool ColabFold.21,22 Cbz-ase is predicted to structurally resemble members of the M20 peptidase family of enzymes (pLDDT = 96.2, pTM = 0.91),23 including N-α-acylglutamine aminoacylase from Corynebacterium sp. (6SLF),24 and amidohydrolases from Staphylococcus aureus (4EWT),25 Bacillus subtilis (1YSJ) and Arabidopsis thaliana (1XMB).26 Further homologues were identified in several Sphingomonas and Bacillus species, with more distant homologues identified in other bacterial phyla by phylogenetic analysis (Fig. S8B†).27 The predicted Cbz-ase architecture comprises a 124aa ferredoxin-like “satellite” domain (N201–N324), which protrudes mid-sequence from the larger α/β/α sandwich fold that defines the catalytic domain (Fig. S10†). It is understood that oligomerisation of M20 peptidases occurs via this satellite domain; therefore, by excising and docking this satellite domain against itself, we predict that Cbz-ase assumes a homotetrameric organisation (pTM = 0.96) that closely resembles structural homologues PDB: 4EWT and 6SLF (Fig. S11 and S12†). This is supported by size exclusion chromatography data (Fig. S32†) which is consistent with a homotetrameric structure.
Within the catalytic domain, two metal-binding centres were inferred by structural homology, within which Zn2+ could be comfortably docked using the MIB prediction and docking server (Fig. 5B).28 The Zn2+ ions remain tightly bound in the predicted Cbz-ase structure during a 10 ns molecular dynamics simulation (MDS, Fig. S13†).29–31 Of note, the Zn2+ ions are coordinated by a strictly-conserved bi-dentate cysteine (Cys118) which is also featured in structural homologues of PDB – 4EWT and 6SLF – and sequence homologues identified by evolutionary conservation analysis.32 Furthermore, we predict that several highly-conserved acidic (Asp84, Glu153, Glu154) and histidine residues (His120/123/181/396) likely contribute to the coordination spheres of the Zn2+ ions within the Cbz-ase active site (Fig. S31†). The role(s) of the Zn2+ ions in the catalytic mechanism has not been explored for this particular enzyme and further studies should determine if one or both are involved in the hydrolysis of the Cbz group. Our model predicts which residues should be targeted for site directed mutagenesis to probe their role in substrate binding and catalysis.
Conclusion
The expanded use of biocatalysts in various roles in synthetic chemistry requires well characterised enzymes, available from academic labs, as well as commercial suppliers. For example lipases are used routinely since they are produced on a large scale in highly active forms. Biocatalysts have the potential to be used for removal of commonly used organic protecting groups but their advantages over chemical methods must be emphasised. One such advantage is to be able to couple biocatalysts in a cascade to sequentially remove orthogonal protecting groups. Another is the ability to engineer/evolve a biocatalyst for bespoke synthetic application. Here we describe the combination of a well known BS2 esterase with a more obscure Cbz-ase. Our methods illustrate the ease of application and versatility. In future studies, immobilisation of theses deprotecting enzymes could be carried out to allow for reactions to take place in different solvents and accommodate the solubility of complex substrates. Cascade reactions using natural and engineered biocatalysts are already delivering complex organic molecules at scale.33 Moreover, biocatalyst databases such as RetroBioCat are being expanded to construct novel biocatalytic routes to important drug molecules and commodity chemicals.34,35Experimental
General
All chemicals and solvents were purchased from Sigma Aldrich or Fisher Scientific and used as received, without further purification. Competent cells were purchased from New England Biolabs. Plasmids were designed and ordered from Genscript.High performance liquid chromatography (HPLC)
HPLC analysis was carried out using a Shimadzu instrument fitted with an autosampler (SIL-20A HT), pump (LC-20AD), UV/visible detector (SPD-20A), system controller (CBM-20Alite) and a column oven (CTO-40C). The method for HPLC analysis was as follows; Luna 5 μm C-18 column (250 × 4.6 mm), 30 °C, water (0.1% TFA)/acetonitrile (0.1% TFA) in a gradient at 1 mL min−1; 0–3 min 10% acetonitrile (ACN), 3–7 min 20% ACN, 18 min 75% ACN, 21 min 75% ACN, 25 min 10% ACN.General method for biocatalyst expression
Commercial E. coli BL21 (DE3) competent cells (10 μL) were transformed via heat shock transformation with plasmid DNA (2 μL) and selection was carried out on an LB agar plate containing kanamycin (30 μg mL−1). The plate was incubated overnight at 37 °C. One colony was used to inoculate a seed culture LB media (250 mL) containing kanamycin (30 μg mL−1) and incubated overnight at 37 °C, 200 rpm. The seed culture inoculated LB media (1 L) containing kanamycin (30 μg mL−1) to an OD600 of 0.1. The cells were grown at 37 °C, 200 rpm until an OD600 of 0.6–1.0 was achieved. Protein expression was induced using iso-propyl-β-D-1-thiogalactopyranoside (IPTG) (final concentration 0.2 mM), and ZnSO4 (final concentration 0.25 mM) was added in the case of Cbz-ase. The temperature lowered to 16 °C, 180 rpm overnight. The cells were harvested by centrifugation (Thermo Scientific Multicentrifuge X3R, 3500g, 20 min, 4 °C, 4 × 1000 rotor). The cell pellets were resuspended in phosphate buffer, centrifuged (Thermo Scientific Multicentrifuge X3R, 4000g, 45 min, 4 °C, 8 × 50 rotor) and cell pellets were stored at −20 °C.General method for biocatalyst purification
The cell pellet was defrosted on ice and resuspended in binding buffer (sodium phosphate (pH 7.4, 50 mM), NaCl (300 mM), imidazole (20 mM)). Cell lysis was carried out by sonication (30 s on 30 s off, 15 cycles) and the cell debris was collected by centrifugation (Thermo Scientific Multicentrifuge X3R, 9000g, 50 min, 4 °C, 8 × 50 rotor). The cell lysate was filtered using Millex HA filters (0.45 μm) and loaded onto a pre-equilibrated His Trap nickel affinity column (5 mL) using an ÄKTA explorer (Cytiva Lifesciences, UK) monitoring at 280 nm. The column was washed with 20 column volumes of binding buffer (sodium phosphate (pH 7.4, 50 mM), NaCl (300 mM), imidazole (20 mM)). Elution buffer (sodium phosphate (pH 7.4, 50 mM), NaCl (300 mM), imidazole (300 mM)) were applied with a gradient of 0 to 100% over 10 min, then held at 100% for 10 min, 5 mL min−1. Analysis of fractions with high UV/Vis absorbance by 12% SDS-PAGE was carried out. Collected fractions were concentrated and dialysed overnight in dialysis buffer (sodium phosphate (pH 7.4, 50 mM), NaCl (300 mM), 10% glycerol).Synthesis of Z-Phe-OtBu
The method for synthesis was devised from Strazzolini et al.36 tert-Butanol (270 mg, 3.67 mmol, 2.2 eq.), cbz-L-phenylalanine (500 mg, 1.67 mmol, 1 eq.) and 4-dimethylaminopyridine (DMAP, 40 mg, 0.33 mmol, 0.2 eq.) were added to a round-bottom flask. Anhydrous dichloromethane (DCM, 4 mL) was added and the solution was stirred to dissolve the solid reagents at 0 °C. N,N′-Dicyclohexylcarbodiimide (DCC, 1 g, 4.85 mmol, 2.9 eq.) was added to a beaker and dissolved in anhydrous DCM (3 mL). The DCC solution was cooled in an ice bath before being slowly transferred to the reaction flask. The mixture was stirred in the ice bath for 1 hour, then stirring was continued at room temperature overnight. The mixture was filtered and concentrated under reduced pressure giving a colourless oil. The oil was dissolved in diethyl ether (30 mL) and transferred to a separator funnel. The solution was washed with (i) HCl (0.5 M, 3 × 10 mL), (ii) NaHCO3 (5% aq., 3 × 10 mL) and (iii) NaCl (30% aq.). The organic layer was dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography (hexane/ethyl acetate 9:1). Z-Phe-OtBu was isolated as a white solid (330 mg, 59% yield).
Deprotection cascade
A 20 mM stock solution of Z-L-Phe-OtBu was prepared in acetonitrile. A solution of purified BS2 (900 μL, 2.8 mg mL−1) in sodium phosphate buffer (50 mM, pH 7.5) was prepared. To this, Z-L-Phe-OtBu (100 μL, 10 mM) was added in 4 × 25 μL volumes over 4 hours at 37 °C. This was done to prevent precipitation of Z-L-Phe-OtBu. A control reaction was also prepared in the same way but without the addition of BS2. The reaction was shaken at 500 rpm, 37 °C for 24 hours. A sample of this reaction was taken (150 μL) and analysed by HPLC. Cbz-ase (500 μL, 1 mg mL−1) was added to the reaction and left to react at 500 rpm, 37 °C for 24 hours. The reaction was analysed by reverse-phase HPLC.Author contributions
LK and MS carried out biocatalyst purification, assay, chemical synthesis, HPLC analysis and wrote the manuscript. MAH carried out modelling studies and wrote the manuscript. GPO and PS carried out modelling and phylogenetic analysis. NRy, YZ, CS, ZA, IZ, JH, CL, NRu, MA, KB, DJ, ALu carried out the biocatalyst purification, assay, chemical synthesis, HPLC analysis. AL and ANH designed the experiments and analysed data. DJC initiated the project, designed the experiments, analysed data and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was carried out as part of a 4th year undergraduate research project course in the area of biocatalysis (Chem4P, 2022/23 and 2023/24). LK thanks the School of Chemistry and Syngenta UK for PhD funding. MS thanks the School of Chemistry and the Derek Stewart Trust for PhD funding. PS and DJC thank the Industrial Biotechnology Innovation Centre (IBIOIC) for funding. DJC thanks the BBSRC for funding (grant code: BB/V001620/1).References
- A. Isidro-Llobet, M. Álvarez and F. Albericio, Chem. Rev., 2009, 109, 2455–2504 CrossRef CAS PubMed.
- M. Schelhaas and H. Waldmann, Angew. Chem., Int. Ed., 1996, 35, 2056–2083 CrossRef CAS.
- S. Vidal, Protecting Groups: Strategies and Applications in Carbohydrate Chemistry, John Wiley & Sons, 2019 Search PubMed.
- R. N. Patel, V. Nanduri, D. Brzozowski, C. McNamee and A. Banerjee, Adv. Synth. Catal., 2003, 345, 830–834 CrossRef CAS.
- M. Maurs, F. Acher and R. Azerad, J. Mol. Catal., 2012, 84, 22–26 CrossRef CAS.
- G. Alvaro, J. A. Feliu, G. Caminal, J. López-Santín and P. Clapés, Biocatal. Biotransform., 2000, 18, 253–258 CrossRef CAS.
- V. B. Nanduri, S. Goldberg, R. Johnston and R. N. Patel, Enzyme Microb. Technol., 2004, 34, 304–312 CrossRef CAS.
- L. N. Chu, V. B. Nanduri, R. N. Patel and A. Goswami, J. Mol. Catal., 2013, 85–86, 56–60 CrossRef CAS.
- D. Kadereit and H. Waldmann, Chem. Rev., 2001, 101, 3367–3396 CrossRef CAS PubMed.
- M. Schmidt, E. Barbayianni, I. Fotakopoulou, M. Höhne, V. Constantinou-Kokotou, U. T. Bornscheuer and G. Kokotos, J. Org. Chem., 2005, 70, 3737–3740 CrossRef CAS PubMed.
- E. Barbayianni, I. Fotakopoulou, M. Schmidt, V. Constantinou-Kokotou, U. T. Bornscheuer and G. Kokotos, J. Org. Chem., 2005, 70, 8730–8733 CrossRef CAS PubMed.
- I. Fotakopoulou, E. Barbayianni, V. Constantinou-Kokotou, U. T. Bornscheuer and G. Kokotos, J. Org. Chem., 2007, 72, 782–786 CrossRef CAS PubMed.
- P. G. M. Mutts and T. W. Greene, in Greene's Protective Groups in Organic Synthesis, 2006, ch. 5, pp. 533–646 Search PubMed.
- S. Hanada, A. Yuasa, H. Kuroiwa, Y. Motoyama and H. Nagashima, Eur. J. Org Chem., 2010, 2010, 1021–1025 CrossRef.
- E. L. Bell, W. Finnigan, S. P. France, A. P. Green, M. A. Hayes, L. J. Hepworth, S. L. Lovelock, H. Niikura, S. Osuna, E. Romero, K. S. Ryan, N. J. Turner and S. L. Flitsch, Nat. Rev. Methods Primers, 2021, 1, 46 CrossRef CAS.
- J. H. Schrittwieser, S. Velikogne, M. Hall and W. Kroutil, Chem. Rev., 2018, 118, 270–348 CrossRef CAS PubMed.
- G. Sánchez-Carrón, T. Fleming, K. E. Holt-Tiffin and D. J. Campopiano, Anal. Chem., 2015, 87, 3923–3928 CrossRef PubMed.
- I. A. Morozova, D. F. Guranda, N. V. Panin and V. K. Švedas, Acta Naturae, 2023, 15, 69–73 CrossRef CAS PubMed.
- B. Spiller, A. Gershenson, F. H. Arnold and R. C. Stevens, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12305–12310 CrossRef CAS PubMed.
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61, 3891–3898 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- M. Mirdita, K. Schütze, Y. Moriwaki, L. Heo, S. Ovchinnikov and M. Steinegger, Nat. Methods, 2022, 19, 679–682 CrossRef CAS PubMed.
- C. A. Orengo, A. D. Michie, S. Jones, D. T. Jones, M. B. Swindells and J. M. Thornton, Structure, 1997, 5, 1093–1108 CrossRef CAS PubMed.
- A. Natsch and R. Emter, Philos. Trans. R. Soc., B, 2020, 375, 20190269 CrossRef CAS PubMed.
- T. S. Girish, B. Vivek, M. Colaco, S. Misquith and B. Gopal, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2013, 69, 103–108 CrossRef CAS PubMed.
- E. Bitto, C. A. Bingman, L. Bittova, N. L. Houston, R. S. Boston, B. G. Fox and G. N. Phillips, Proteins: Struct., Funct., Bioinf., 2009, 74, 61–71 CrossRef CAS PubMed.
- C. M. Seibert and F. M. Raushel, Biochemistry, 2005, 44, 6383–6391 CrossRef CAS PubMed.
- Y.-F. Lin, C.-W. Cheng, C.-S. Shih, J.-K. Hwang, C.-S. Yu and C.-H. Lu, J. Chem. Inf. Model., 2016, 56, 2287–2291 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- J. Huang and A. D. Mackerell, J. Comput. Chem., 2013, 34, 2135–2145 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- H. Ashkenazy, S. Abadi, E. Martz, O. Chay, I. Mayrose, T. Pupko and N. Ben-Tal, Nucleic Acids Res., 2016, 44, W344–W350 CrossRef CAS PubMed.
- A. Fryszkowska and P. N. Devine, Curr. Opin. Chem. Biol., 2020, 55, 151–160 CrossRef CAS PubMed.
- W. Finnigan, L. J. Hepworth, S. L. Flitsch and N. J. Turner, Nat. Catal., 2021, 4, 98–104 CrossRef CAS PubMed.
- W. Finnigan, M. Lubberink, L. J. Hepworth, J. Citoler, A. P. Mattey, G. J. Ford, J. Sangster, S. C. Cosgrove, B. Z. da Costa, R. S. Heath, T. W. Thorpe, Y. Yu, S. L. Flitsch and N. J. Turner, ACS Catal., 2023, 13, 11771–11780 CrossRef CAS PubMed.
- P. Strazzolini, M. Scuccato and A. G. Giumanini, Tetrahedron, 2000, 56, 3625–3633 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4fd00016a |
| This journal is © The Royal Society of Chemistry 2024 |