 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the biocatalytic synthesis of silicone polymers
Yuqing Lu ab and
Lu Shin Wong*ab
ab and
Lu Shin Wong*ab
aManchester Institute of Biotechnology, University of Manchester, 131 Princess Street, Manchester M1 7DN, UK. E-mail: l.s.wong@manchester.ac.uk
bDepartment of Chemistry, University of Manchester, Oxford Road, Manchester M13 9PL, UK
First published on 13th February 2024
Abstract
Polysiloxanes, with poly(dimethyl)siloxane (PDMS) being the most common example, are widely used in various industrial and consumer applications due to the physicochemical properties imparted by their Si–O–Si backbone structure. The conventional synthesis of PDMS involves the hydrolysis of dichlorodimethylsilane, which raises environmental concerns due to the usage of chlorinated compounds. Herein, a biocatalytic approach for PDMS synthesis is demonstrated using silicatein-α (Silα), an enzyme from marine sponges that is known to catalyse the hydrolysis and condensation of Si–O bonds. Using dialkoxysilane precursors, it was found that Silα catalyses the formation of PDMS in non-aqueous media, yielding polymers with higher molecular weights (approximately 1000–2000 Da). However, on prolonged exposure, the gradual degradation of the polymers was also observed. Overall these observations indicate that Silα catalyses the formation polysiloxanes, demonstrating the potential of biocatalysis for more sustainable polysiloxane production.
Introduction
Polysiloxane ‘silicone’ polymers, characterised by the Si–O–Si backbone motif (Fig. 1), have found widespread use in a range of industrial and consumer applications including as lubricants, antifoaming agents, pharmaceuticals, and personal care products.1–3 An estimated 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 122000 metric tons of silicone products were produced in 2013 and this demand continues to escalate with growing applications in the transport, construction, energy and electronics sectors.2 Of these silicones, polydimethylsiloxane (PDMS) is the most widely used and forms the bulk of most products. However, other silicones such as polymethylphenylsiloxane are also employed in various specialist technical applications such as separation media and biomedical devices.4,5
122000 metric tons of silicone products were produced in 2013 and this demand continues to escalate with growing applications in the transport, construction, energy and electronics sectors.2 Of these silicones, polydimethylsiloxane (PDMS) is the most widely used and forms the bulk of most products. However, other silicones such as polymethylphenylsiloxane are also employed in various specialist technical applications such as separation media and biomedical devices.4,5
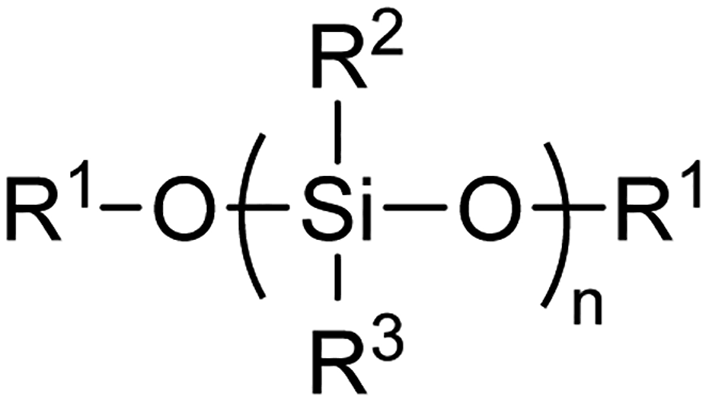 | ||
| Fig. 1 General structure of silicone or siloxane polymer backbone. In methoxy-terminated PDMS, R1 = R2 = R3 = Me. | ||
However, the existing route of PDMS production suffers from many drawbacks. Currently, PDMS is industrially produced via the controlled hydrolysis of dichlorodimethylsilane, a process which is not only environmentally undesirable but also energy-intensive.6,7 The use of chlorinated compounds in this process is problematic due to their high toxicity and necessitates specific engineering measures for their containment and processing. Additionally, the siloxane and chlorinated by-products are also hazardous,8 and unless properly managed, can contribute to further secondary pollution. Thus, alternative routes to the production of PDMS that address these issues would be desirable. In this regard, biocatalytic methods for the formation and cleavage of Si–O bonds may offer more sustainable approaches to the synthetic manipulation of siloxanes.
Very few enzymes are known to be involved in the metabolism of silicon-containing compounds,9,10 and of these the silicateins from marine hexactinellid sponges are the most well studied.11,12 These silicateins catalyse the condensation of soluble silicates to inorganic silica, which the sponges deposit in their skeletons.13 Crucially, recent work with silicatein-α (Silα), the most common isoform of this protein, has demonstrated that it is able to catalyse the condensation of a variety of organosilyl ethers (i.e. compounds bearing organic substituents around the silicon atom) from the corresponding silanol and alcohol.14,15 These findings therefore demonstrate a means to form Si–O bonds while circumventing the need for chlorosilanes, and allude to the possibility of employing Silα to catalyse the formation of polysiloxanes.
This study therefore aimed to investigate the biocatalytic synthesis of polysiloxane polymers (including PDMS) using dialkoxysilanes as precursor monomers and recombinant Silα as a model biocatalyst.
Results and discussion
Polysiloxane polymerisation
To investigate the silicatein-catalysed polymerisation reactions, the dialkoxysilane precursors dimethyldimethoxysilane 1, dimethoxymethylphenylsilane 2, and bis(4-methoxyphenoxy)dimethylsilane 3 were chosen as the substrates (Fig. 2). The dimethoxy 1 and 2 were chosen as they were structurally simple and readily available precursors for PDMS and polymethylphenylsiloxane, respectively. Precursor 3 was chosen as the (4-methoxy)phenoxy moieties have previously been shown to be accepted as a substrate by Silα.15 As the biocatalyst, the previously reported Silα fusion protein was used,14,15 bearing an N-terminal trigger factor protein and a C-terminal Strep II-tag (referred to henceforth as TF-Silα-Strep).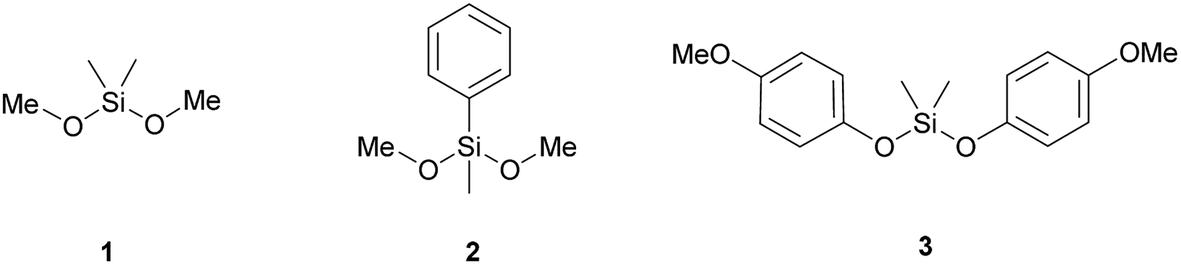 | ||
| Fig. 2 Structures of the precursor molecules used as enzyme substrates in this study. | ||
Initially, the enzymatic synthesis of PDMS was investigated with the dialkoxysilane 1. The reactions were carried out using TF-Silα-Strep in the form of a lyophilised solid (in a matrix of potassium salts and 18-crown-6), as this preparation has previously been shown to be effective for the catalysis of silyl ether condensations at 75 °C in anhydrous non-polar solvents.14 The reaction was conducted using toluene as the reaction medium and the reaction products analysed by MALDI-MS after 48 h to ascertain the presence of any polymeric products.
In the control reaction where the enzyme was omitted, a single population distribution of polymers was observed, with a weight-averaged molecular weight (![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n) of 703 Da and individual peaks separated by an m/z value of 74 Da corresponding to a single Si(Me)2O repeating unit (Fig. 3A and Table 1). Trace amounts of these polymers were already observable in the substrate sample prior to the reaction (Fig. 3A), so their presence was attributed to a small amount of polymerisation resulting from non-specific acid–base catalysis arising from the presence of the additives used to lyophilise the enzyme (i.e. in the negative control, the additives were still present, only the enzyme was omitted). However, in the reaction containing the enzyme, two population distributions of polymers were found (labelled as ‘i’ and ‘ii’ in Fig. 3A). The lower molecular weight population is analogous to that which was observed in the control reactions noted above, but a second population with a higher n of 1471 Da was found, which was attributed to enzyme-specific catalysis. Furthermore, the new polymers also exhibited a distribution that somewhat favoured longer polymer chains, as evidenced by the higher weight average and Z-average molecular weights (w and z, respectively) and the larger polydispersity (Table 1).
n) of 703 Da and individual peaks separated by an m/z value of 74 Da corresponding to a single Si(Me)2O repeating unit (Fig. 3A and Table 1). Trace amounts of these polymers were already observable in the substrate sample prior to the reaction (Fig. 3A), so their presence was attributed to a small amount of polymerisation resulting from non-specific acid–base catalysis arising from the presence of the additives used to lyophilise the enzyme (i.e. in the negative control, the additives were still present, only the enzyme was omitted). However, in the reaction containing the enzyme, two population distributions of polymers were found (labelled as ‘i’ and ‘ii’ in Fig. 3A). The lower molecular weight population is analogous to that which was observed in the control reactions noted above, but a second population with a higher n of 1471 Da was found, which was attributed to enzyme-specific catalysis. Furthermore, the new polymers also exhibited a distribution that somewhat favoured longer polymer chains, as evidenced by the higher weight average and Z-average molecular weights (w and z, respectively) and the larger polydispersity (Table 1).
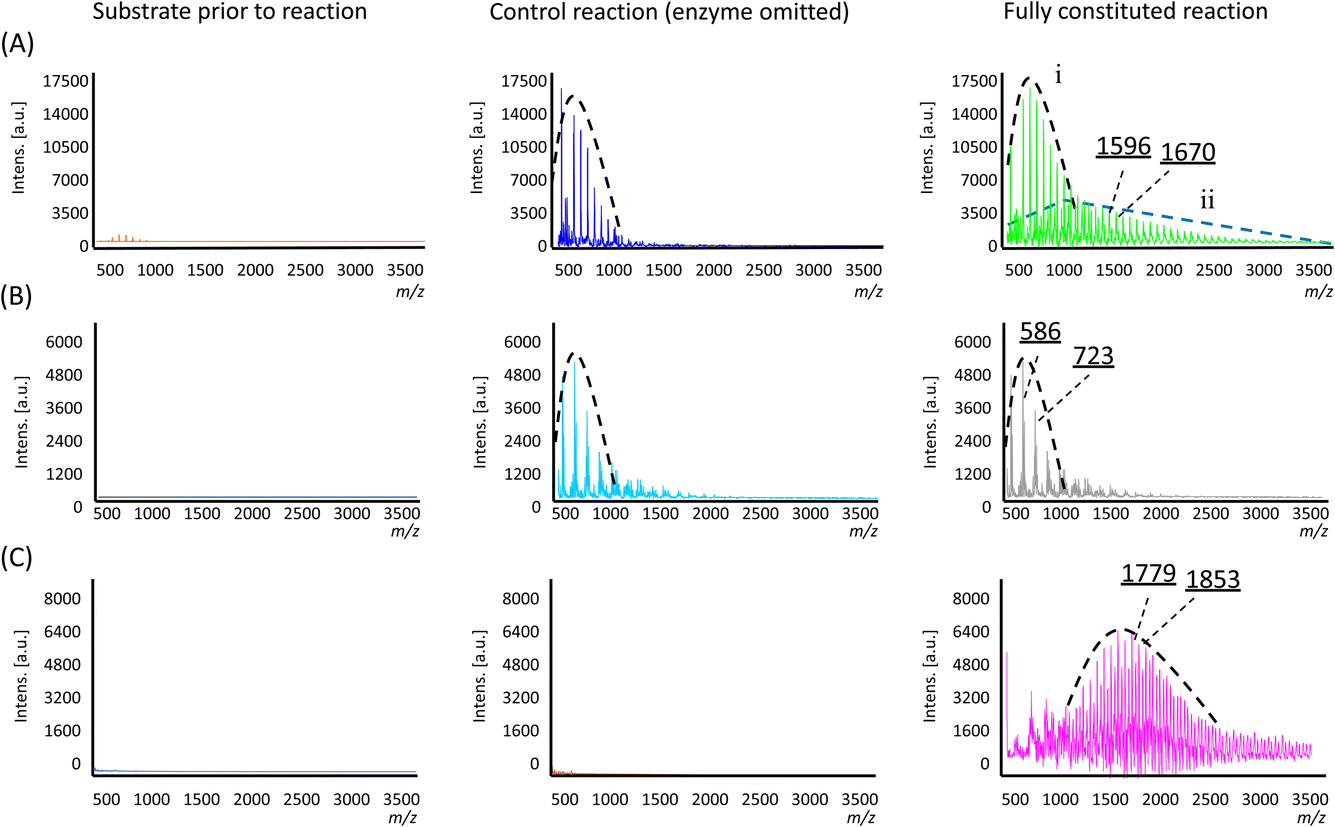 | ||
| Fig. 3 MALDI mass spectra of the products of the polymerisation reactions for substrate 1 (A), 2 (B), 3 (C) with population distributions indicated. Illustrative m/z values are also shown, the difference of which corresponds to a Si(Me)2O or SiMePhO repeating unit. | ||
| Substrate | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| Reaction | Negative control | Fully constituteda | Negative control | Fully constituted | Negative controlb | Fully constituted |
| a “i” and “ii” refer to the two population distributions of polymers observed in Fig. 3A.b n.d. = not determined, no polymer detected. | ||||||
| n |
703 | (i) 782 | 652 | 680 | n.d. | 1798 |
| (ii) 1471 | ||||||
| w |
721 | (i) 814 | 697 | 732 | n.d. | 1822 |
| (ii) 1682 | ||||||
| z |
740 | (i) 846 | 743 | 785 | n.d. | 1845 |
| (ii) 1906 | ||||||
| Polydispersity | 1.02 | (i) 1.04 | 1.07 | 1.08 | n.d. | 1.01 |
| (ii) 1.14 | ||||||
In examining the interplay between substituents in the substrate structure and enzyme catalysis, reactions were carried out with substrate 2 and analysed in a similar manner. Here, both the fully constituted reaction and the negative control experiment (enzyme omitted) gave essentially the same result. In both cases, polymers with individual mass differences of 137 Da corresponding to SiMePhO repeating units were indeed produced (Fig. 3B) but only at relatively short lengths (n 680 Da, Table 1). Thus, in contrast with substrate 1, the addition of TF-Silα-Strep did not give rise to any additional biocatalysis. This result suggests that 2 is not accepted by the enzyme, likely due to its larger steric bulk.
For substrate 3, under identical conditions the fully constituted reaction produced a polymer with an n of 1798 Da (Table 1). No polymer formation was observed in the negative control or the original monomer sample (Fig. 3C), indicating that the substrate was generally more stable under these operating conditions, likely due to the increased steric hindrance around the scissile Si–O bonds.
In principle, the synthesis of PDMS requires at least two steps, hydrolysis of at least one alkoxide to give the corresponding silanol followed by condensation of the silanols to construct the Si–O–Si siloxane chain (Scheme 1). If so, at least a catalytic amount of water is required for polymerisation. The results with 3 further implies that the enzyme is catalysing its hydrolysis to the silanol, since in the absence of the enzyme no polymerisation is observed.
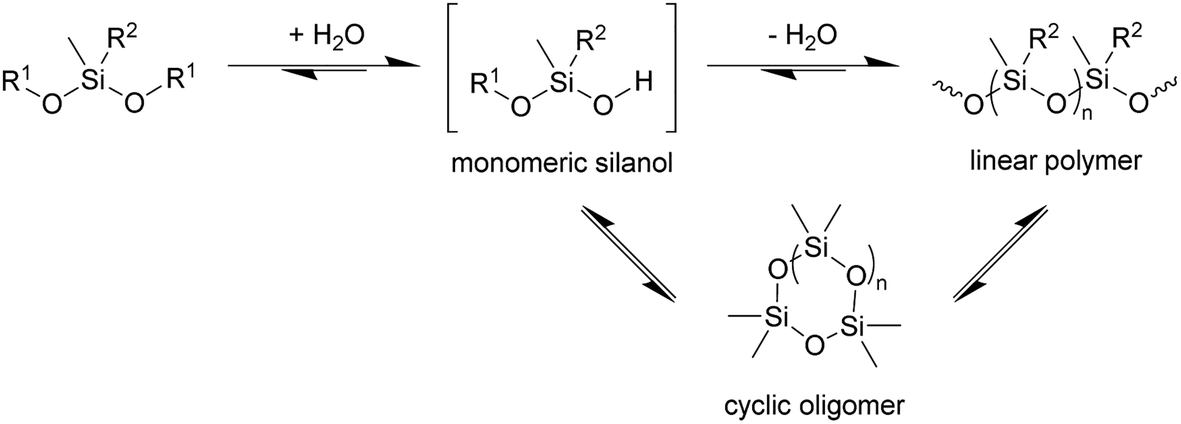 | ||
| Scheme 1 Condensation of siloxanes. Reaction conditions: TF-Silα-Strep (cat.), toluene, 70 °C. R1 = Me or (4-methoxy)phenyl; R2 = Me or Ph; n = 1–3. | ||
Silα-catalysed polymerisation of PDMS has been reported previously16 using substrate 1 under aqueous conditions. However, only short polymers were produced (estimated n 600–700 Da). This result is unsurprising since the aqueous conditions would favour the rapid hydrolysis of the substrate, but would also equally favour the hydrolysis of the polymer. In contrast, the present experiments gave polymers that were approximately double in size (Table 1). This result is consistent with the use of anhydrous solvent, whereby the only source of water would be the residual amounts that were present in the lyophilised enzyme preparation,17 and hence the equilibrium position would favour the formation of polymeric materials.
Cyclic oligosiloxane formation
In addition to the linear polymers, the formation of cyclic oligomers as intermediates may also occur since their formation is thermodynamically favourable18 (Scheme 1). To investigate their presence under these reaction conditions, the reaction mixture where 1 was used as the model substrate was analysed by GC-MS. Here, any new chromatographic peaks and their corresponding mass spectra were compared to authentic standards of the cyclic trimers, tetramers and pentamers (the so-called “D3”, “D4” and “D5”, respectively). It was found that all three compounds were detected after 48 h (Fig. 4), with the relative amounts of D3 being highest followed by D4 and D5 (ratio of each oligomer 18:8:1 by chromatogram peak area). In comparison, in the control reaction, the same oligomers were also detected, but in much lower quantities, with D3, D4 and D5 being present in approximately 4, 3 and 3-fold less (by peak area), respectively, compared to the fully constituted reaction.
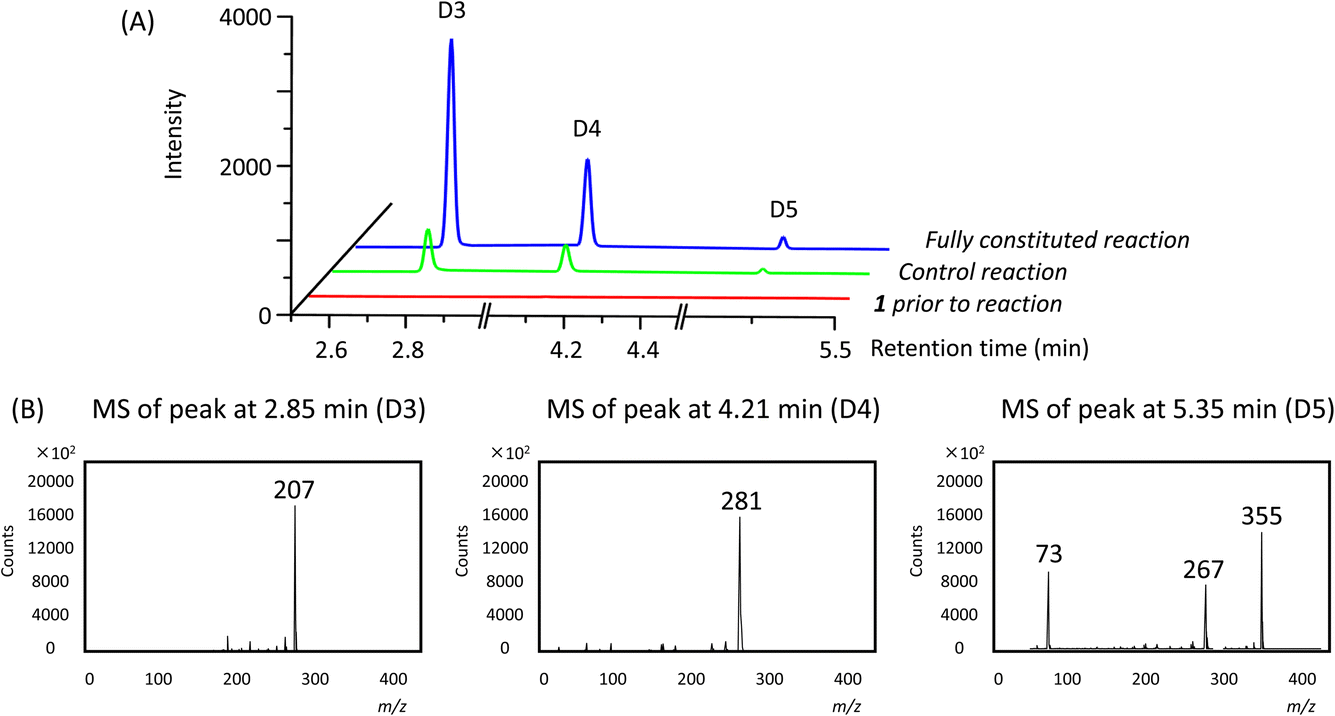 | ||
| Fig. 4 Gas chromatograms (A) and mass spectra (B) of the reaction mixtures for substrate 1. The fragmentation patterns of D3, D4 and D5 are consistent with the electron ionisation fragmentation patterns observed for authentic standards. | ||
The presence of these cyclic compounds alongside the expected linear polymers suggests two possible scenarios: their formation is an intermediate step during the generation of linear polymers; or that they are formed as a result of the gradual degradation of the linear polymers by a thermally activated backbiting reaction.18,19 Thus, in order to investigate whether the cyclic oligomers are derived from the thermal degradation of the linear polymer, the reaction was carried out for an extended duration (to 120 h) and the product profile monitored at 24 h intervals after the formation of the second distribution of polymers (i.e. after 48 h from the start of the original experiment). It was found that the molecular weight values of the linear polymers in the enzymatic reaction decreased over time, while the net conversion of the cyclic polymers gradually increased (Fig. 5). These results show that the linear polymers degrade over longer timeframes and that most of the cyclic siloxanes are likely by-products of the backbiting reaction. The changes in n of the linear polymers appear to have stabilised after 96 h and this size may therefore represent the thermodynamically defined size under these reaction conditions.
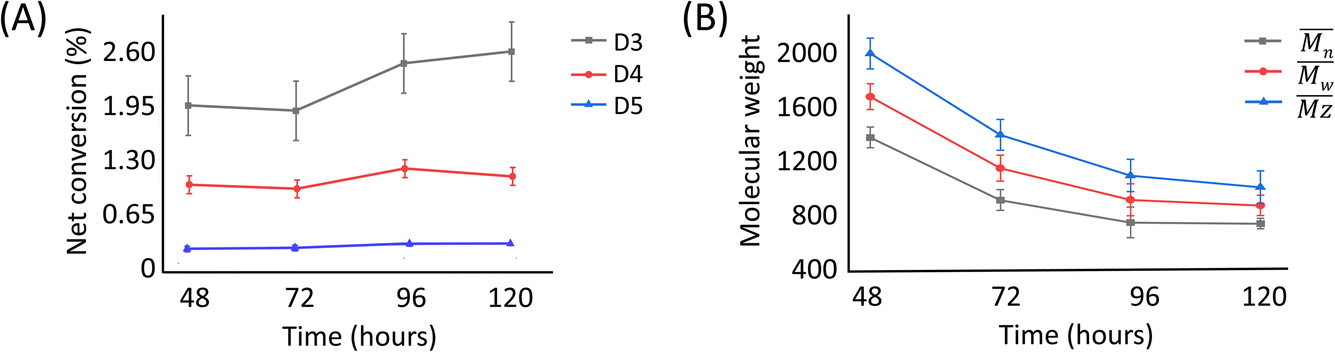 | ||
| Fig. 5 Plots of (A) net conversion of the cyclic oligomers against time and (B) the molecular weight of the linear polymer. Net conversion = conversion from enzyme-catalysed reaction minus conversion from negative control reaction. | ||
Experimental
Materials and methods
All reagents, authentic samples and solvents were purchased from either Sigma Aldrich or Fisher Scientific in their highest available purity. The commercially sourced 1 and 2 were found to be essentially free of polymeric materials by MALDI-MS and GC-MS (Fig. 3 and 4) and used without further purification. Substrate 3 was synthesised in-house according to the procedure below. The TF-Silα-Strep was heterologously produced in E. coli and the lyophilised enzyme prepared as previously described.15 The condensation reactions were carried out in crimp-sealable 8 mm vials (Chromacol C4008-741) that were heated and shaken on an Eppendorf Thermomixer 5350. GC-MS analyses were carried out on an Agilent 7890B GC system with 5977A mass detector according to previously reported methods.15 MALDI-MS analyses were conducted with a Bruker Biotyper Sirius and the mass spectra were calibrated using the ACTH peptide fragment 18–39 and oxidised insulin B chain.Bis(4-methoxyphenoxy)dimethylsilane, 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) 20
20
4-Methoxyphenol (496.6 mg, 4 mmol) and Et3N (0.56 mL, 4 mmol) were dissolved in anhydrous toluene (10 mL). Me2SiCl2 (0.24 mL, 2 mmol) was added to the reaction mixture and stirred at room temperature. Reaction progress was monitored by TLC (hexane/EtOAc, 10:1) and was observed to be complete after 16 h. To the reaction mixture was then added 1 N aqueous NaOH (in three 10 mL portions). The aqueous phase was removed, and the organic phase washed twice with water (10 mL each) and once with brine (10 mL). The organic phase was dried with Na2SO4, filtered and evaporated under reduced pressure. The residue was purified by silica gel chromatography to yield the desired product as white crystals (318 mg, 53%); Rf 0.46 (hexane/EtOAc, 10:1); νmax (solid)/cm−1 2930 (C–H), 1281 (Si–O), 1251 (C–O), 824 (Si–CH3); δH (400 MHz, CDCl3) 6.88 (d, J = 8.3 Hz, 4H), 6.79 (d, J = 8.3 Hz, 4H), 3.76 (s, 6H), 0.33 (s, 6H); δC (100 MHz, CDCl3) 152.69 (CO), 148.02 (C), 120.51 (CH), 114.76 (CH), 55.75 (OCH3), −2.28 (SiCH3). m/z (EI+) 304 (M˙+, 100%). Data are consistent with ref. 20.
Enzymatic polymerisation reactions
100 μL of a stock solution of the desired substrate (0.7 M in anhydrous toluene) was added into each vial containing the 500 μg lyophilized enzyme and the vial crimp sealed. The reaction mixtures were then heated at 75 °C while shaking at 650 rpm. For the control reaction where the enzyme was omitted, the lyophilised enzyme was substituted with only the lyophilised additives (potassium salts and 18-crown-6).15 All reactions were performed in triplicate.After the desired reaction time, the vials were removed from heating, 100 μL of pentane was added and the mixture centrifuged (17000g, 10 min) to separate the solid matter. 10 μL of the supernatant was removed and mixed with 10 μL of matrix solution (10 mg of 2,5-dihydroxybenzoic acid in 1 mL of 50% v/v acetonitrile, 50% H2O and 0.1% TFA). 2 μL of this mixture was applied to the MALDI-MS plate and dried for analysis. The remaining 190 μL of the supernatant was transferred to a clean vial and subjected to GC-MS analysis. For quantification of the cyclic oligomers, the GC-MS was first calibrated using the authentic samples of the products.
Conclusions
In summary, a preliminary investigation into the biocatalytic synthesis of polysiloxanes was carried out, using TF-Silα-Strep as a model biocatalyst. It was found that the enzyme was able to catalyse the formation of PDMS in an organic solvent, at a level above that of non-specific polymerisation. This biocatalytic polymerisation was apparently only successful when the substituents around the Si atom were small, as no polymer was formed with the methylphenylsilane under analogous conditions. However, the size of the alkoxy leaving groups appeared to be less important, with even the relatively bulky (4-methoxy)phenoxy group giving polymeric products. Indeed, the use of monomer 3 bearing these groups may be preferable as they exhibited no background (non-enzymatic) hydrolysis.A more detailed analysis of the reaction products over an extended time course identified that cyclic oligomers were also produced. The molecular weight of the linear polymers decreased with time as the formation of cyclic polymers gradually increased, indicating that cyclic polymers may result from the backbiting mechanism of chain polymerization, or that both products may also be capable of reversible reactions at elevated temperatures. However, further mechanistic studies are still needed to elucidate the relative contributions of the individual reaction steps.
This biocatalytic polymerization mechanism presents an opportunity to develop more sustainable approaches for on-demand production of polymeric PDMS materials, and the observed backbiting reaction underscores the enzyme's potential to degrade PDMS materials.
Author contributions
Conceptualisation, Y. L and L. S. W.; methodology, Y. L. and L. S. W.; validation, Y. L.; formal analysis, Y. L and L. S. W.; investigation, Y. L. and L. S. W.; data curation, Y. L. and L. S. W.; writing—original draft preparation, Y. L.; writing—review and editing, Y. L. and L. S. W.; visualisation, Y. L.; supervision, L. S. W.; project administration, L. S. W.; all authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analysis, or interpretation of data; in the writing of the manuscript or in the decision to publish the results.Acknowledgements
This research was supported by the UK Engineering and Physical Sciences Research Council under grant EP/S013539/1.References
- M. Andriot, S. H. Chao, A. Colas, S. Cray, F. d. Buyl, J. V. DeGroot, A. Dupont, T. Easton, J. L. Garaud, E. Gerlach, F. Gubbels, M. Jungk, S. Leadley, J. P. Lecomte, B. Lenoble, R. Meeks, A. Mountney, G. Shearer, S. Stassen, C. Stevens, X. Thomas and A. T. Wolf, in Inorganic Polymers, ed. R. De Jaeger and M. Gleria, Nova Science Publishers, Hauppauge, 2007, pp. 61–161, https://isbnsearch.org/isbn/1600216560 Search PubMed.
- C. Corden, D. Tyrer, H. Menadue, J. Calero, J. Dade and R. Leferink, The Socioeconomic Impact of the Silicones Industry in Europe, Global Silicones Council, Washington DC, 2015, https://www.silicones.eu/wp-content/uploads/2018/12/ces_socio-economic_study_eu_final.pdf Search PubMed.
- C. Rücker and K. Kümmerer, Chem. Rev., 2015, 115, 466–524, DOI:10.1021/cr500319v.
- A. C. M. Kuo, in Polymer Data Handbook, ed. J. E. Mark, Oxford University Press, Oxford, 2nd edn, 2009, pp. 838–845, DOI:10.1093/oso/9780195181012.003.0145.
- C. E. Brunchi, A. Filimon, M. Cazacu and S. Ioan, High Perform. Polym., 2009, 21, 31–47, DOI:10.1177/0954008308088737.
- H.-H. Moretto, M. Schulze and G. Wagner, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley, 2012, vol. 32, pp. 675–712, DOI:10.1002/14356007.a24_057.
- Y. Ikeda, W. Huang and A. Oku, Green Chem., 2003, 5, 508–511, 10.1039/b304483c.
- J. M. McKim, P. C. Wilga, W. J. Breslin, K. P. Plotzke, R. H. Gallavan and R. G. Meeks, Toxicol. Sci., 2001, 63, 37–46, DOI:10.1093/toxsci/63.1.37.
- K. Shimizu, T. Amano, M. R. Bari, J. C. Weaver, J. Arima and N. Mori, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11449–11454, DOI:10.1073/pnas.1506968112.
- M. B. Frampton and P. M. Zelisko, Chem.–Asian J., 2017, 12, 1153–1167, DOI:10.1002/asia.201700214.
- J. N. Cha, K. Shimizu, Y. Zhou, S. C. Christiansen, B. F. Chmelka, G. D. Stucky and D. E. Morse, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 361–365, DOI:10.1073/pnas.96.2.361.
- W. E. G. Müller, U. Schloßmacher, X. Wang, A. Boreiko, D. Brandt, S. E. Wolf, W. Tremel and H. C. Schröder, FEBS J., 2008, 275, 362–370, DOI:10.1111/j.1742-4658.2007.06206.x.
- R. L. Brutchey and D. E. Morse, Chem. Rev., 2008, 108, 4915–4934, DOI:10.1021/cr078256b.
- S. Y. T. Dakhili, S. A. Caslin, A. S. Faponle, P. Quayle, S. P. De Visser and L. S. Wong, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E5285–E5291, DOI:10.1073/pnas.1613320114.
- E. I. Sparkes, C. S. Egedeuzu, B. Lias, R. Sung, S. A. Caslin, S. Y. Tabatabaei Dakhili, P. G. Taylor, P. Quayle and L. S. Wong, Catalysts, 2021, 11, 879, DOI:10.3390/catal11080879.
- S. E. Wolf, U. Schlossmacher, A. Pietuch, B. Mathiasch, H.-C. Schröder, W. E. G. Müller and W. Tremel, Dalton Trans., 2010, 39, 9245–9249, 10.1039/B921640E.
- C. Ó'Fágáin, in Protein Purification Protocols, ed. P. Cutler, Humana Press, Totowa, 2nd edn, 2004, vol. 244, pp. 309–321, DOI:10.1385/1-59259-655-X:309.
- N. D. Vu, A. Boulègue-Mondière, N. Durand, J. Raynaud and V. Monteil, Green Chem., 2023, 25, 3869–3877, 10.1039/d3gc00293d.
- J. P. Lewicki and R. S. Maxwell, in Concise Encyclopedia of High Performance Silicones, ed. A. Tiwari and M. D. Soucek, Scrivener Publishing, Beverly, 2014, pp. 191–210, DOI:10.1002/9781118938478.ch13.
- M. Shi, K. Yamamoto, Y. Okamoto and S. Takamuku, Phosphorus, Sulfur Silicon Relat. Elem., 1991, 60, 1–14, DOI:10.1080/10426509108233919.
| This journal is © The Royal Society of Chemistry 2024 |