 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-throughput selection of (new) enzymes: phage display-mediated isolation of alkyl halide hydrolases from a library of active-site mutated epoxide hydrolases†
Marija Blazic‡
 ,
Candice Gautier‡,
Thomas Norberg and
Mikael Widersten*
,
Candice Gautier‡,
Thomas Norberg and
Mikael Widersten*
Department of Chemistry – BMC, Uppsala University, Box 576, SE-751 23 Uppsala, Sweden. E-mail: mikael.widersten@kemi.uu.se
First published on 3rd June 2024
Abstract
Epoxide hydrolase StEH1, from potato, is similar in overall structural fold and catalytic mechanism to haloalkane dehalogenase DhlA from Xanthobacter autotrophicus. StEH1 displays low (promiscuous) hydrolytic activity with (2-chloro)- and (2-bromo)ethanebenzene producing 2-phenylethanol. To investigate possibilities to amplify these very low dehalogenase activities, StEH1 was subjected to targeted randomized mutagenesis at five active-site amino acid residues and the resulting protein library was challenged for reactivity towards a bait chloride substrate. Enzymes catalyzing the first half-reaction of a hydrolytic cycle were isolated following monovalent phage display of the mutated proteins. Several StEH1 derived enzymes were identified with enhanced dehalogenase activities.
Introduction
Functional development of proteins may be explained by a model of gene duplication resulting in relaxed constraints on conservation of the original function encoded by the duplicate gene copy.1 In microbial communities emergence of new functional traits can be fast, where an externally applied selection pressure will accelerate ‘evolution’ of proteins with required functions. An example is the development of resistance to the herbicide Dalapon, 2,2-dichloropropionic acid,2 later referred to the action of an α-haloacid dehalogenase (c.f. ref. 3).Divergent evolution within a protein superfamily can be driven by enhancement of low, non-physiologically relevant (promiscuous) activities that are amplified as a result of mutagenesis events,4 where an already existing catalytic machinery may be exploited for an alternative chemical transformation or substrate scope.5
The enzyme superfamily of α/β-hydrolases6 maintains a wide range of peptidases, esterases, lactonases, epoxide hydrolases and dehalogenases. Enzymes within the family display conserved structural features as well as catalytic. The shared core catalytic machinery consists of a nucleophilic group (hydroxyl, thiolate or carboxylate), a general base (His) and a charge-relay residue (carboxylate). The catalytic triads are highly similar in arrangement to corresponding groups found in structurally unrelated enzymes such as subtilisin or the pancreatic serine hydrolases. The respective substrate and reaction scopes depend on the chemical nature of the nucleophilic group and additional auxiliary groups.
We have focused on two enzyme representatives, haloalkane dehalogenase DhlA from the soil bacterium Xanthobacter autotrophicus11 and an epoxide hydrolase from potato, StEH1.12 The enzymes are approximately 20% identical at the linear amino acid sequence level but share a conserved tertiary fold of the α/β-hydrolase domains (Fig. 1A) and the respective catalytic triads (Fig. 1B). Both enzymes act upon alkyl substrates breaking either carbon–halogen (Cl or Br) or carbon–oxygen bonds in the respective first half-reactions of catalysis. The catalytic cycles begin with nucleophilic attack by Asp carboxylates on an electron deficient carbon, generating a covalent ester intermediate (Fig. 2). General-base dependent hydrolysis of the respective alkylenzyme intermediate(s) completes the turnover.13
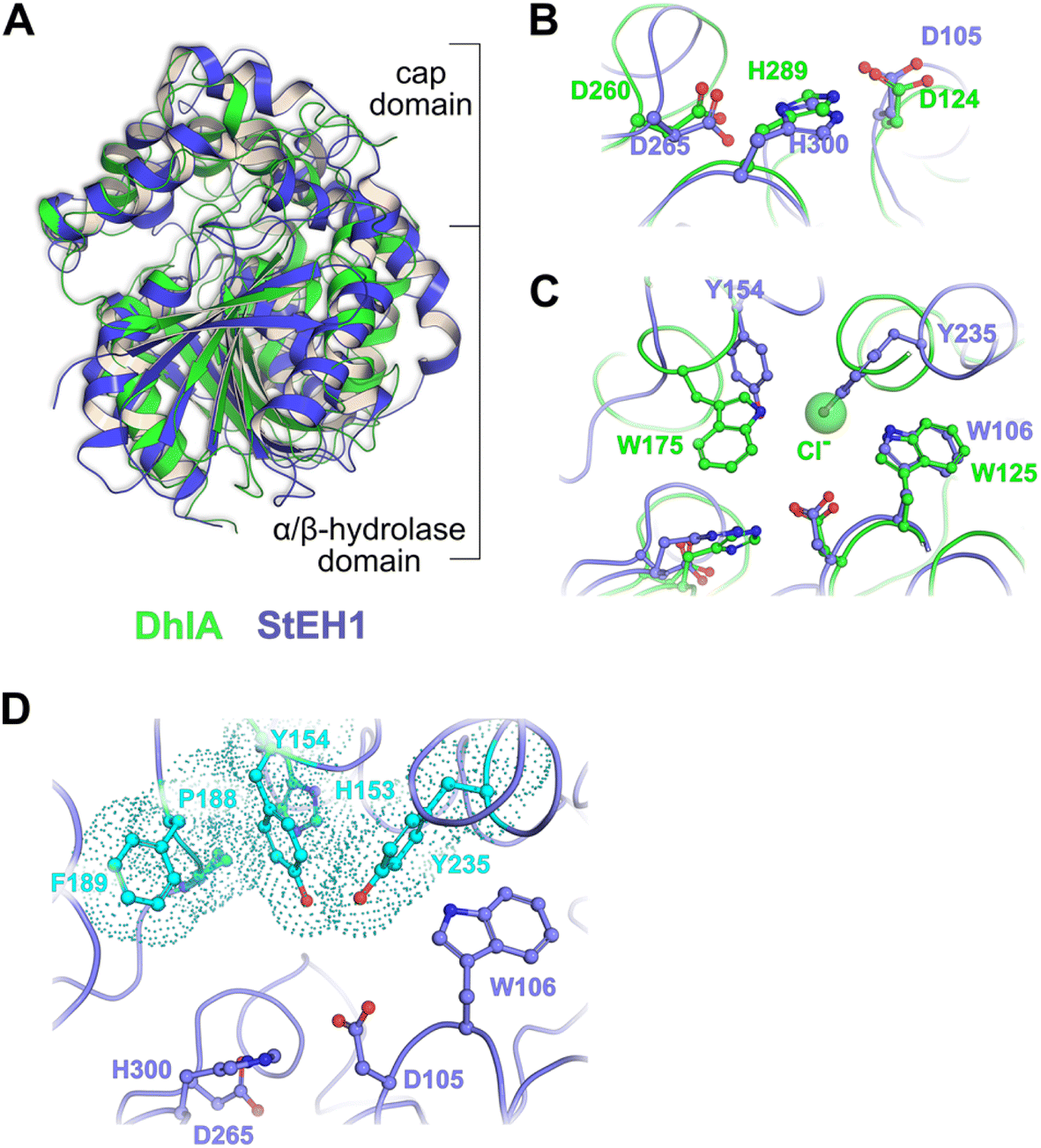 | ||
| Fig. 1 (A) Superposition of the crystal structures of DhlA from X. autotrophicus (green, PDB entry: 2dhe7) and StEH1 from S. tuberosum (slate blue, PDB entry: 2cjp8). The RMSD was 2.93 Å3 from superposition of 1250 atoms. (B) Amino acid residues forming the respective catalytic triads in DhlA (green carbons) and StEH1 (slate blue carbons). (C) The chloride binding site of DhlA consists of the indole NH groups of W125 and W175 (green carbons).9 The position of chloride ion in the crystal structure (pdb entry: 2dhe) superimposes on the phenol oxygen of Y235 of StEH1. Tyrosines 154 and 235 in StEH1 function as catalytic acids in the catalytic epoxide hydrolysis.10 (D) Amino acid residues in the active site of StEH1 (cyan colored carbons) that were subjected to random mutagenesis in the construction of the library of StEH1 variants selected by phage display. Image created in Pymol 2.5.5. | ||
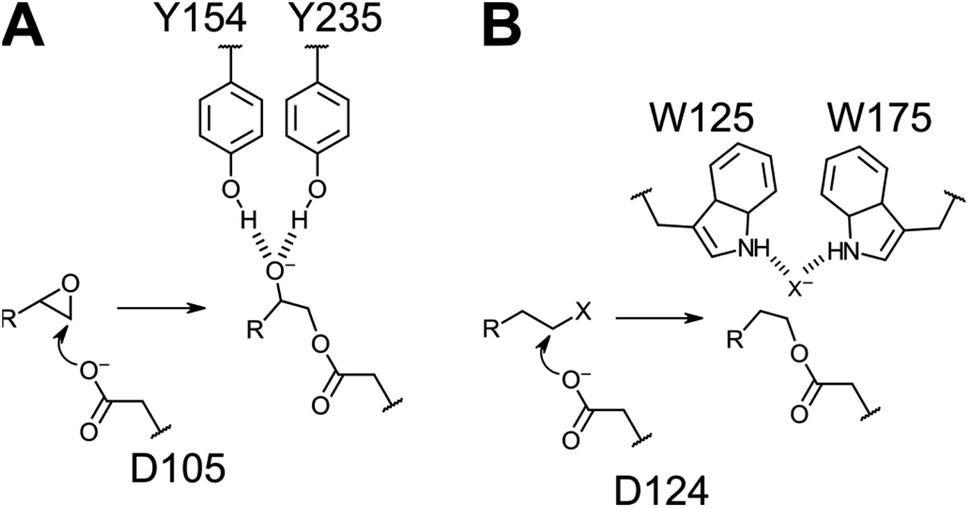 | ||
| Fig. 2 Similarities in reaction mechanisms in the first half-reaction of epoxide (A) and alkyl halide (B) hydrolysis catalyzed by StEH1 and DhlA, respectively. Auxiliary groups that are differing between the enzymes are Tyr acids in the epoxide hydrolase and a halide binding site, formed by two Trp indoles, in the dehalogenase. | ||
Although the sequence similarities between these enzymes is modest, the structural and mechanistic similarities invites questions regarding possible ancestral origin. Furthermore, StEH1 exhibits low but detectable promiscuous hydrolytic activity with model alkyl halide substrate 9 (Chart 1) (∼40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold lower hydrolysis rate as compared to the activity with a structurally similar epoxide). These combined observations led us to test the possibility to amplify the alkyl halide hydrolysis activity of StEH1 via directed evolution driven by targeted random mutagenesis of active-site amino acid residues. Inspired by the active-site architecture of DhlA five residues were chosen for mutagenesis, resulting in a theoretical number of 3.2 × 106 individual StEH1 derived structures, a number too large for direct screening methods. Thus, a high-throughput method was developed that relied on monovalent phage display of the protein library during selection.
000-fold lower hydrolysis rate as compared to the activity with a structurally similar epoxide). These combined observations led us to test the possibility to amplify the alkyl halide hydrolysis activity of StEH1 via directed evolution driven by targeted random mutagenesis of active-site amino acid residues. Inspired by the active-site architecture of DhlA five residues were chosen for mutagenesis, resulting in a theoretical number of 3.2 × 106 individual StEH1 derived structures, a number too large for direct screening methods. Thus, a high-throughput method was developed that relied on monovalent phage display of the protein library during selection.
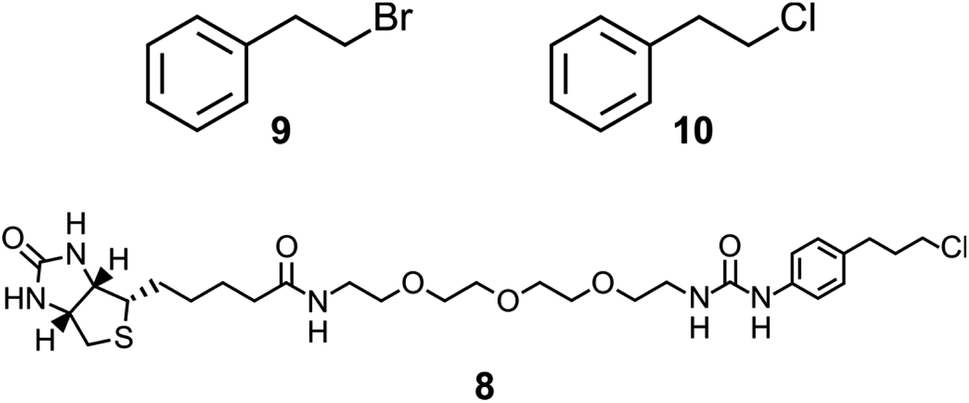 | ||
| Chart 1 Compounds 9 and 10 are model substrates used for assaying halide hydrolysis. Compound 8 is the bait ligand applied in the phage display selection for StEH1 variants capable of catalyzing breaking of a carbon–chlorine bond. | ||
Results and discussion
Macromolecular display methodologies, providing facile screening of relatively large protein libraries (106–109), cannot easily be applied to enzyme engineering. As the selection criteria are usually based on ligand–receptor interactions, these methods are poorly suited for searching enzyme libraries; catalysis is rarely critically dependent on interactions between enzyme and ligands in their ground states. However, if the chemical mechanism of interest involves the formation of a catalytically relevant intermediate species that may be trapped and isolated, direct selection for catalysis may be possible.Phage display afforded selection for catalytically competent variant proteins
In the case of the epoxide hydrolysis and dehalogenation reactions studied here, both mechanisms involve the formation of an intermediate alkylenzyme. This is an obligatory step for the continuation towards the hydrolysis product. Thus, if the aim is to identify and isolate modified epoxide hydrolase variant(s) able to break the carbon–halogen bond, a selection based on the ability to perform the first half-reaction of the catalytic cycle, the nucleophilic attack by the carboxylate nucleophile, breaking the carbon–halogen bond and releasing the halide would be sufficient. The alkylenzyme, however, is short-lived with half-lives on the milliseconds time scale. An obvious approach to prolong the alkylenzyme half-life would be to slow down the rate of its decay (hydrolysis and/or reverse reaction back to the Michaelis complex). Previous studies on StEH1 have demonstrated that mutating basic groups in the active site, the main general base H300 (H300N13b) or an auxiliary basic group, E35 to Gln,14 slows down the hydrolysis rate significantly. A double mutant carrying both substitutions exhibits, in a tested epoxide hydrolysis reaction, very slow reverse formation of the epoxide from the alkylenzyme, and undetectable hydrolysis.15 The alkylation rate is also slowed down approximately three orders of magnitude by these mutations but remains at a level where alkylation is feasible within practical experimental limitations of a phage display afforded selection.The strategy for selection was thus to construct an StEH1 cDNA library on an E35Q/H300 mutant background, cloned into a phagemid vector that would allow for monovalent phage display of produced variant proteins.16 Five residues lining the active site cavity of StEH1 (Fig. 1D), including tyrosines Y154 and Y235, known to be involved in the mechanism of epoxide hydrolysis, were randomly mutated into any of the 20 proteinaceous amino acids using a non-biased codon set. Phage display was performed for five cycles where the StEH1 variants were challenged for substituting the D105 carboxylate for the chlorine in the bait ligand, compound 8 in Chart 1. Bound phage-StEH1 particles attached to the bait ligand were trapped onto streptavidin coated paramagnetic beads as outlined in Fig. 3. Phage clones that had passed through selection rounds 3–5 were randomly picked and their amino acid sequences deduced from the respective cDNA sequences (Table S2†).
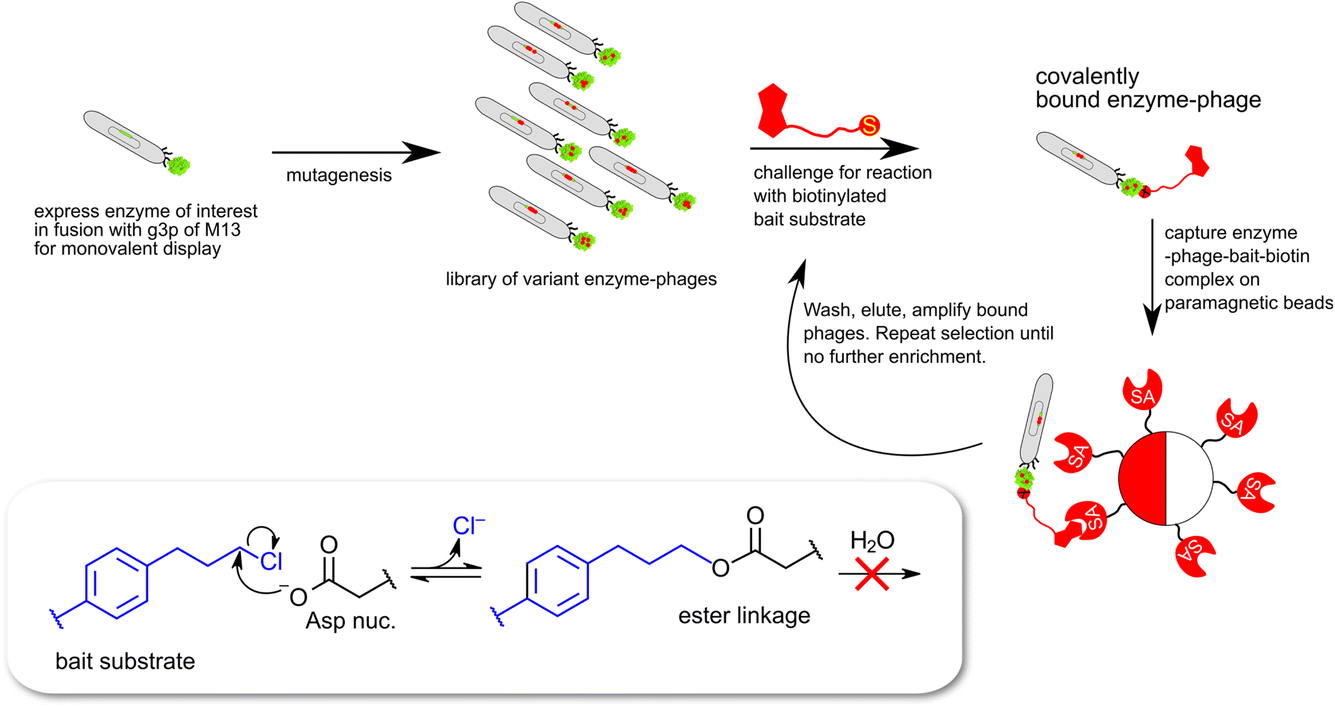 | ||
| Fig. 3 Schematic description of monovalent phage-display expression of epoxide hydrolase derived proteins. The selection traps enzymes capable of catalyzing a nucleophilic attack leading to cleavage of the carbon–chlorine bond in the bait substrate (8 in Chart 1) and the formation of a covalent ester linkage between enzyme and biotinylated bait. | ||
Dehalogenase activities of selected proteins
A selected number of clones were cured of the E35Q/H300N mutations and subcloned into an expression vector for production of soluble proteins, freed of phage (Table S2†). The proteins were affinity purified and tested for their ability to catalyze the hydrolysis of compounds 9 and 10.Several of the isolates displayed hydrolase activity with chloro compound 10 with the Y235I mutant displaying highest relative activity (Fig. 4). The same variant also displayed highest relative activity also with the bromo substrate 9. Thus, the variant that was among the highest enriched during the phage display selection also displayed the highest hydrolytic activity. It should be noted that the assay conditions (overnight incubation of the reaction mixtures with relatively high concentrations of enzyme) did not provide pseudo-first order reaction conditions, but was applied for screening purposes of (any) hydrolysis activity. Hence, the obtained values require validation by more stringent steady-state kinetics. The results, however, strongly indicate that the selection criteria during phage display expression indeed trapped functional dehalogenases from the large pool of proteins in the library. The phage display selection was based on the formation of an alkylenzyme involving the carboxylate of D105. This design originated from the presumption that the mechanism of epoxide hydrolysis would be retained also in the hydrolysis of the bait chlorine substrate. The observation that a double D105A/Y235 mutant is inactive with either 9 or 10 supports that the chemical mechanism of hydrolysis is indeed retained and includes an alkylenzyme intermediate also in this engineered variant.
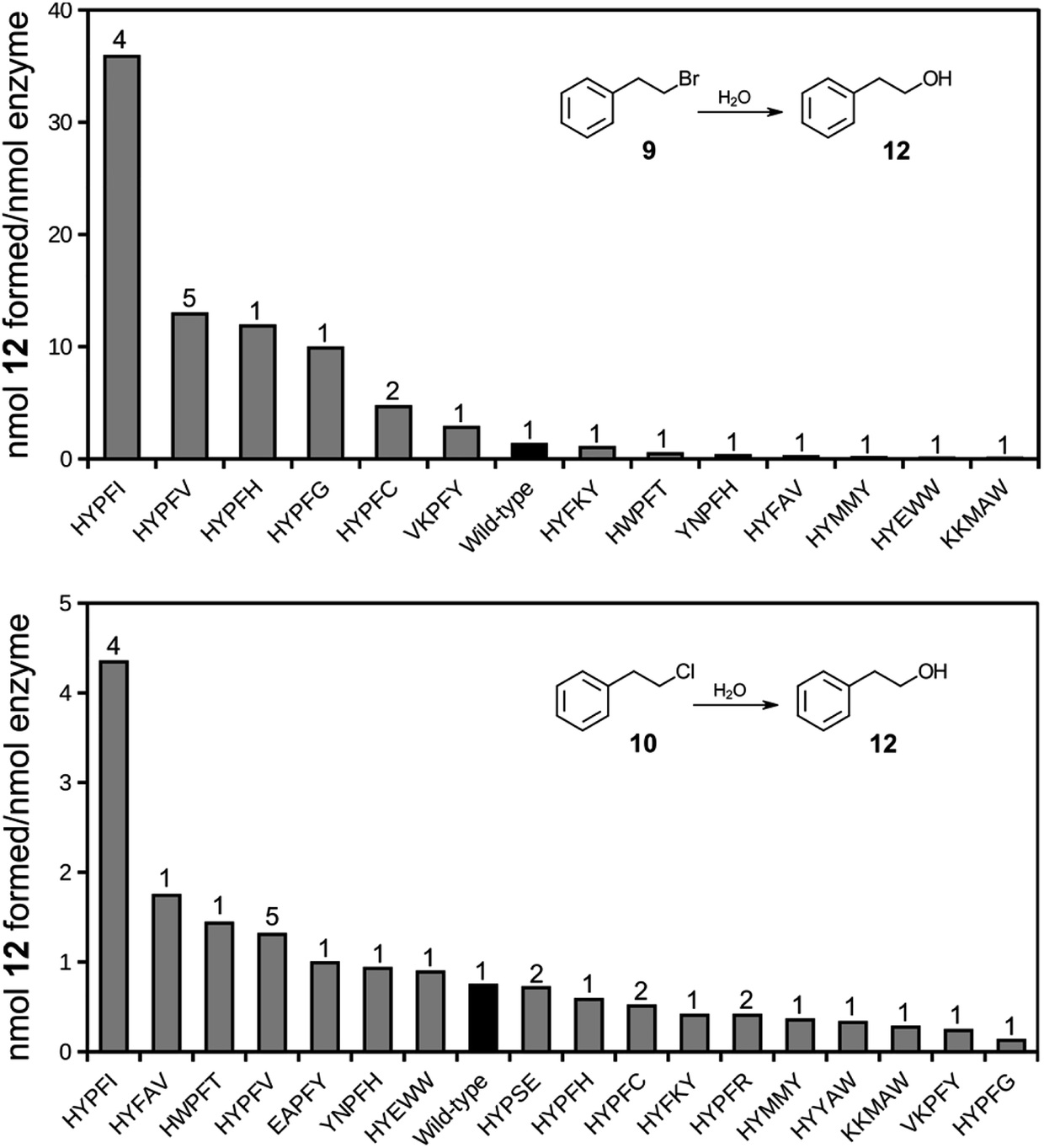 | ||
| Fig. 4 Bar charts of formation of hydrolysis product 12 following incubation of halide substrates 9 (top) or 10 (bottom) in the presence of StEH1 variants isolated following phage display selection, restoration of catalytic base functions and purification. Black bars indicate wild type StEH1 and numbers on top of bars represent number of identical clones isolated during phage display selections. See Table S2† for detailed sequence information. | ||
Experimental
Construction of StEH-QN-mutant cDNA library for monovalent phage display selection
The cDNA library was assembled step-wise from PCR-amplified gene fragments, with the StEH1-QN cDNA15 as template, as follows: (1) fragment 1 encoding the 5′ XhoI site and the N-terminus of the protein was amplified with primers pUC-Forward, annealing upstream of the Tac promoter in the vector, and the HYX1 reverse primer (Table S1†). Fragment A, introducing NDT, VMA, ATG and TGG randomization at H153 and Y154 was amplified with primers forward, HY(N1A, V1B, A1C, T1D), and reverse, PF1. Fragment B, introducing NDT, VMA, ATG and TGG randomization at P188 and F189 was amplified with primers forward, PF(N1A, V1B, A1C, T1D), and reverse, TYR1. Fragment C, encoding the C-terminus of the protein and the 3′ SpeI site and introducing NDT, VMA, ATG, TGG randomization at Y235 was amplified with primers forward, TYR(N1A, V1B, A1C and T1D), and reverse, pUC-Reverse. pUC-Reverse anneal to vector sequence downstream of the SpeI cloning site.(2) The cDNA library was assembled by subsequent overlap PCR and PCR amplifications of fragments 1 + A, with primers pUC_EH-Forward and ReverseBamHI, yielding fragment 1A. Another overlap was done with fragments B and C with ForwardBamHI, introducing a silent mutation to obtain a BamHI site at position R184 (cgc-cgg) and reverse pUC-EH, yielding fragment BC. Final overlap was done with fragment 1A and fragment BC using forward PCEH and reverse BCEH primers.
(3) The mutated cDNA pool was subsequently digested with XhoI/SpeI and subcloned into the same sites of pComb3ΔNX.16b Randomization of targeted codons was confirmed by DNA sequencing of a mixed cDNA pool sample.
Synthesis of bait substrate
The overall synthesis schemes are shown in Fig. S1 and S2.† Concentrations were performed at reduced pressure (bath temperature <40 °C). NMR spectra were recorded with Agilent or Varian Innova NMR spectrometers operating at 400, 500 or 600 MHz (proton) at 298 K, using residual protium solvent signals (D2O: δH = 4.793, CHCl3: δH = 7.250), DSS (sodium trimethylsilylpropane sulfonate, δC = 0.00 in deuterium oxide) or actual solvent signals (middle CDCl3 triplet signal δC = 77.0) as chemical shift reference unless otherwise stated. Coupling constants (J) are given in Hz.Analytical LC/MS was performed on an Agilent 1100/Waters Micromass ZQ instrument using a 3.0 × 50 mm C-18 column. Mobile phase and gradient: acetonitrile/water (5–95%) to acetonitrile/water (95–5%), always with 0.1% formic acid added. The MS was operated in both positive and negative ion mode, and UV data was collected between 210 and 625 nm. TLC was performed on Silica Gel F254 plates (Merck, Darmstadt, Germany) (Fig. S3†). The plates were eluted with the indicated solvent mixtures and the spots were visualized with UV light and/or by dipping/charring with 0.5% ninhydrin in 100:3 ethanol/acetic acid. Preparative silica gel open-column chromatography was on Matrix silica gel 60 (35–70 μm) using the indicated amounts and eluants. All reagents were, unless otherwise stated, purchased from Merck-Sigma-Aldrich (Darmstadt, Germany) and used as purchased without further purification.
:1. Re-crystallization of 4.2 g crude product from chloroform (40 ml) gave pure (1H-NMR, Fig. S4†) compound 2 (1.84 g, 35%). Another pure batch of 2 (0.26 g, 5%) precipitated from the mother liquor in the refrigerator over a few days. The 1H-NMR was again satisfactory. Thus, the total yield of the para-isomer of 2 was 2.1 g = 40%. Theoretical yield: 5.2 g.17:1 ratio.:1, Fig. S3G†) showed the presence of a major new spot (Rf 0.6, ninhydrin- and UV positive) and only traces of the (Rf 0.4) starting material spot. The mixture was diluted with dichloromethane (3 ml) and washed with 2 M aq sulfuric acid. The organic phase was passed through a Pasteur pipette stoppered with glass wool and containing a cm of dry magnesium sulfate powder, the filtrate was evaporated, concentrated, and passed through a silica gel (10 g) column, packed in toluene and eluted first with toluene, then with toluene–ethyl acetate 9:1. The appropriate fractions were pooled and concentrated, the solid residue of pure 6 (30 mg, 90%) was co-evaporated once with chloroform and once with deuterochloroform, then checked with 1H- and 13C-NMR (Fig. S7†), which showed the expected signals, plus some aromatic impurities. The (somewhat reactive) material was deemed pure enough for direct use in the next step.:3:3:2, 20:3:3:2 and 6:3:3:2, Fig. S3H†) indicated formation of a new, UV-absorbing spot and reduced intensity of the biotin-PEG3-amine amine-spot. Additional 6 (16 mg, 0.05 mmol) dissolved in DMF (400 μl) was added. After 2 h and some gentle warming, the reaction mixture TLC indicated a complete reaction (no amine left, Fig S3H†). The mixture was neutralized with acetic acid (0.25 mmol = 15 μl), concentrated to a small (<100 μl, 5 torr, 30 °C) volume, diluted with EtOAc–MeOH–HOAc–H2O, 80:3:3:2 (0.5 ml) and applied to a column of silica gel (4.0 g, packed in EtOAc–MeOH–HOAc–H2O, 80:3:3:2). Elution with first the packing solvent (20 ml), then EtOAc–MeOH–HOAc–H2O, 40:3:3:2 (15 ml), and finally 20:3:3:2 (20 ml). The tubes containing the product (as determined by TLC, Fig. S3H†) were pooled. Evaporation, co-evaporation twice with chloroform and drying (2 torr for several hours) left syrupy 8 (38 mg = 88%). The 1H and 13C-NMR’s showed the expected signals (Fig. S8†). Analysis by ES-MS showed a major UV-absorbing peak which gave a strong m/z = 614.4/616.5 positive ion (M+H)+.Phage display selection for dehalogenase activity
The pC3ΔNX-StEH1-QN library was transformed by electroporation into E. coli XL1-Blue cells (Agilent Tech.). After a recovery period of 1 hour in 2TY medium (16 g l−1 bacto tryptone, 10 g l−1 bacto yeast extract, 5 g l−1 NaCl) and under shaking at 200 rpm and 37 °C, the number of transformants were titered by plating aliquots on LB-agar plates (10 g l−1 bacto tryptone, 5 g l−1 bacto yeast extract, 10 g l−1 NaCl) fortified with 100 μg ml−1 carbenicillin. The culture was expanded by addition of 100 ml 2TY and ampicillin and tetracycline were added to the liquid culture to final concentrations of 50 and 35 μg ml−1, respectively, and incubation was continued for 1 additional hour. Then, helper phages M13KO7, ∼1011 pfu, were added. After two hours incubation, kanamycin was added to 70 μg ml−1 and incubation was continued overnight to allow for phage particle proliferation. Harvesting and titration of phagemid containing particles (‘IN’ phages) was performed as described previously.19 Approximately 1010 cfu phagemid particles were entered into the selection, as follows: phage particles dissolved in 50 mM sodium phosphate, pH 7.5, 0.5% (w/v) bovine serum albumin (BSA), were incubated with 10 μM of biotinylated bait substrate 8 (Chart 1) for 24 hours at room temperature. Following incubation, phages were precipitated as per previous protocol16b and resuspended in reaction buffer. At this point, 0.1 ml of a suspension of streptavidin coated paramagnetic beads (MagnaSphere®, Promega # Z5481), pre-incubated in 50 mM sodium phosphate, pH 7.5, and 0.5% (w/v) BSA, were added and incubation was continued with periodical mixing by gently tapping the tube for 30 minutes. The beads were trapped in a magnetic stand and were washed by addition of 5 × 700 μl of 50 mM sodium phosphate, pH 7.5, with 0.5% (w/v) BSA. Phages still captured on the beads after washing were allowed to infect log phase E. coli XL1-Blue cells, and proliferated overnight as described above. An aliquot of selected phages was used for titration of ‘OUT’ phages. The selection was repeated for three additional rounds (four in total) following the same protocol with the difference that the initial incubation time with bait substrate was shortened from 24 to 3 hours, from the second selection round and onward.Clones from selection rounds 1 to 3 were randomly picked (from the OUT-phage titration plates) and the StEH1 variant cDNAs were sequenced (Table S2†). Fragments containing the mutated regions were PCR amplified from selected cDNAs in the presence of oligonucleotide primers PauI_fp and Rp_PUC, (Table S1†). The fragments were digested with NcoI and PauI, purified and ligated into a modified version of pGTacStEH1-5H,13b digested with the same enzymes. The used derivative of pGTacStEH1-5H contained silent mutations that had been introduced at codons H31 and L109 by a modified version of QuikChange® (Agilent Technol.) using oligonucleotide primers NcoI_fp and PauI_fp (Table S1†). The introduced mutations removed an upstream NcoI site and introduced a unique PauI site (Fig. S9†).
Site-directed mutagenesis of HYPFI D105A
A D105A mutation was introduced using the cDNA encoding the StEH1 Y235I mutant as a template with oligodeoxynucleotides fp_D105A and rp_D105A primers (Table S1†), by a modified version of QuikChange. Successful substitution was proven by DNA sequencing of plasmids isolated from transformed cells.Protein expression and purification
Mutant cDNAs in plasmid pGTac-StEH1-5H were transformed into XL1-Blue cells and plated on LB with 100 μg ml−1 ampicillin. Transformants were inoculated into 5 ml 2TY (with 100 μg ml−1 ampicillin) and grown overnight. Overnight cultures were further expanded to 500 ml 2TY/100 μg ml−1 ampicillin the following day. At OD600 ∼0.3, protein expression was induced by addition of 1 mM isopropyl β-thiogalactoside. The protein was expressed overnight at 30 °C when cells were harvested by centrifugation.For purification, cells were resuspended in lysis buffer (10 mM sodium-phosphate, pH 7.0, 500 mM NaCl, 20 mM imidazole 10 μg ml−1 DNase I and a cocktail of protease inhibitors (Thermo Scientific)) and cells were lysed by ultrasonication in a Vibra Cell Sonifier, (power setting 70%, for 3 × 30 s pulses interrupted by 30 s of cooling on ice), after which cell debris was pelleted by centrifugation. Lysate was filtered using 0.45 μm filter and incubated with Ni(II) charged Chelating Sepharose® (Cytiva) equilibrated with binding buffer (lysis buffer minus DNase I and protease inhibitors) under rotation at 6 °C for one hour. Sepharose beads were washed with 5 column volumes of binding buffer, followed by washing with an additional 5 column volumes of wash buffer (binding buffer with 100 mM imidazole). Retained protein was eluted with elution buffer (binding buffer with 300 mM imidazole). Protein containing fractions were pooled, concentrated to a volume of 2.5 ml by ultrafiltration (PES, cut-off 10 kDa, Sartorius) and desalted through a PD-10 column (Cytiva) pre-equilibrated in 0.1 M sodium phosphate, pH 7.4.
Detection of halide hydrolysis activity
Enzyme (1–20 μM) was mixed with 2 mM of either 9 or 10 (Chart 1) in 0.1 M sodium-phosphate, pH 7.5, and incubated for 22 h at 30 °C. Reactions were stopped by addition of methanol, containing 2 mM benzyl alcohol as internal standard, to 50% (v/v). Mixtures were vortexed for 20 seconds and insoluble debris was pelleted by centrifugation at 17000g for 30 minutes. Cleared reaction mixtures were subsequently separated by reversed phase HPLC (Ascentis C-18, 25 × 0.46 cm, 5 μm bead size) on a Shimadzu Providence system equipped with an LC-20AD pump. The solvent system consisted of methanol and 50 mM sodium phosphate, pH 3.0. Samples (20 μl) were injected with a SIL-20A autosampler and eluted with a gradient of 40–90% methanol over 50 min at a flow rate of 0.5 ml min−1. Eluted components were detected at 212 nm using an SPD-M20A diode array detector. The retention times for the internal standard, benzyl alcohol and the product phenylethyl alcohol (12), were 17 and 24 minutes, respectively. Amount of product formation was determined from a standard curve of phenylethyl alcohol run under identical conditions (Fig. S10†).
Conclusions
We show that a promiscuous dehalogenase activity can be amplified dramatically by surprisingly few substitutions within the active site of an epoxide hydrolase. The isolation of the described first-generation enzyme variants has been possible due to a high-throughput selection system afforded by monovalent phage display and tailored to identify catalytically competent enzyme variants. More detailed kinetic analyses, together with structural studies, remain to better understand the structure–activity relationships revealed by the isolated proteins in catalysis. The variant enzyme exhibiting the highest degree of improvement in halide hydrolysis activity (Y235I) will be employed as the parent structure for repeated library construction and phage display selection along the lines described in this work.Author contributions
MB and CG conducted the main part of the experimental work of protein selection, isolation and characterization. TN designed and synthesized the ligands. MW contributed to the characterization of isolated proteins and was responsible for overall planning. All authors contributed to writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by Byggmästare Olle Engkvists Stiftelse (#218-0061). The authors thank Belén Hervás Povo, Hanna Osterholz and Hannes Pålsson for assisting in protein productions.Notes and references
- (a) R. L. Watts and D. C. Watts, Nature, 1968, 217, 1125 CrossRef CAS PubMed; (b) S. D. Copley, J. Biol. Chem., 2012, 287, 3 CrossRef CAS PubMed.
- E. Senior, A. T. Bull and J. H. Slater, Nature, 1976, 263, 476 CrossRef CAS PubMed.
- See J. W. Schmidberger, J. A. Wilce, A. J. Weightman, J. C. Whisstock and C. J. Wilce, J. Mol. Biol., 2008, 378, 284 CrossRef CAS PubMed , for an example.
- (a) O. Khersonsky, C. Roodveldt and D. S. Tawfik, Curr. Opin. Chem. Biol., 2006, 10, 498 CrossRef CAS PubMed; (b) S. D. Copley, Trends Biochem. Sci., 2015, 40, 72 CrossRef CAS PubMed.
- G. J. Bartlett, N. Borkakoti and J. M. Thornton, J. Mol. Biol., 2003, 331, 829 CrossRef CAS PubMed.
- D. L. Ollis, E. Cheah, M. Cygler, B. Dijkstra, F. Frolow, S. M. Franken, M. Harel, S. J. Remington, I. Silman, J. Schrag, J. L. Sussman, K. H. G. Verschueren and A. Goldman, Protein Eng., 1992, 5, 197 CrossRef CAS PubMed.
- K. H. G. Verschueren, F. Seljee, H. J. Rozeboom, K. H. Kalk and B. W. Dijkstra, Nature, 1993, 363, 693 CrossRef CAS PubMed.
- S. L. Mowbray, L. T. Elfström, K. M. Ahlgren, E. C. Andersson and M. Widersten, Protein Sci., 2006, 15, 1628 CrossRef CAS PubMed.
- C. Kennes, F. Pries, G. H. Krooshof, E. Bokma, J. Kingma and D. B. Janssen, Eur. J. Biochem., 1995, 228, 403 CrossRef CAS PubMed.
- L. T. Elfström and M. Widersten, Biochemistry, 2006, 45, 205 CrossRef PubMed.
- D. B. Janssen, F. Pries, J. van der Ploeg, B. Kazemier, P. Terpstra and B. Witholt, J. Bacteriol., 1989, 171, 6791 CrossRef CAS PubMed.
- A. Stapleton, J. K. Beetham, F. Pinot, J. E. Garbarino, B. D. Hammock and W. R. Belknap, Plant J., 1994, 6, 251 CrossRef CAS PubMed.
- (a) F. Pries, J. Kingma, G. H. Krooshof, C. M. Jeronimus-Stratingh, A. P. Bruins and D. B. Janssen, J. Biol. Chem., 1995, 270, 10405 CrossRef CAS PubMed; (b) L. T. Elfström and M. Widersten, Biochem. J., 2005, 390, 633 CrossRef PubMed.
- A. Thomaeus, J. Carlsson, J. Åqvist and M. Widersten, Biochemistry, 2007, 46, 2466 CrossRef CAS PubMed.
- B. A. Amrein, P. Bauer, F. Duarte, Å. J. Carlsson, A. Baworyta, S. L. Mowbray, M. Widersten and S. C. L. Kamerlin, ACS Catal., 2015, 5, 5702 CrossRef CAS PubMed.
- (a) C. F. Barbas III, A. S. Kang, R. A. Lerner and S. J. Benkovic, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 7978 CrossRef PubMed; (b) M. Widersten and B. Mannervik, J. Mol. Biol., 1995, 250, 115 CrossRef CAS PubMed.
- O. Pinto, J. Srdinha, P. D. Vaz, F. Piedade, M. J. Calhorda, R. Abramovitch, N. Nazareth, M. Pinto, M. S. J. Nascimento and A. P. Rauter, J. Med. Chem., 2011, 54, 3175 CrossRef CAS PubMed.
- P. G. Baraldi, B. Cacciari, R. Romagnoli, G. Spalluto, A. Monopoli, E. Ongini, K. Varani and P. A. Borea, J. Med. Chem., 2002, 45, 115 CrossRef CAS PubMed.
- M. T. I. Nilsson and M. Widersten, Biochemistry, 2004, 43, 12038 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4fd00001c |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2024 |