
Homo- and heterobimetallic transition metal cluster derived from [Cp*Fe(η5-E5)] (E = P, As) – unprecedented structural motifs of the resulting polypnictogen ligands†
Sabrina B.
Dinauer
a,
Robert
Szlosek
a,
Martin
Piesch
a,
Gábor
Balázs
a,
Stephan
Reichl
a,
Lukas
Prock
a,
Christoph
Riesinger
a,
Marc D.
Walter
 b and
Manfred
Scheer
*a
b and
Manfred
Scheer
*a
aInstitute of Inorganic Chemistry, University of Regensburg, Universitätsstr. 31, 93053 Regensburg, Germany. E-mail: Manfred.Scheer@chemie.uni-regensburg.de
bInstitute of Inorganic and Analytical Chemistry, Technical University of Braunschweig, 38106 Braunschweig, Germany. E-mail: mwalter@tu-bs.de
First published on 27th May 2024
Abstract
A general synthetic procedure to neutral homo- and heterobimetallic cage compounds exhibiting various structural motifs of the polypnictogen ligands starting from [Cp*Fe(η5-E5)] (E = P (1), As (2); Cp* = C5Me5) is reported. The impact of the implemented transition metal precursors {Cp′′′M} (M = Cr, Mn, Fe, Ni; Cp′′′ = 1,2,4-tBu3C5H2) emphasises the variability of the isolated complexes exhibiting a broad variety of structural motifs of the pnictogen ligands. Spectroscopic, crystallographic, and theoretical investigations provide insight into the structure of the partially unprecedented polypnictogen ligands.
Introduction
Transition metal polypnictides (En ligand complexes) have been extensively studied for almost 50 years.1,2 During this time, a plethora of neutral compounds featuring polyphosphides coordinated to various transition metals have emerged.3–5 Most of these compounds were generated by thermolysis or photolysis reactions of a transition metal precursor with E4 (E = P, As).6 Nevertheless, complexes with the heavier congener arsenic are less frequently encountered. Additionally, thermolytic conditions often yield complex mixtures of mono- and bimetallic complexes.4,6 For example, in the thermolysis reaction of [Cp*Fe(CO)2]2 (Cp* = C5Me5) and yellow arsenic, the complexes [Cp*Fe(η5-As5)] and [{Cp*Fe}3(μ,η4:4:4-As6)] were isolated.7,8 Even more challenging is the controlled implementation of a second or third transition metal, different to the first one, into a complex featuring an En ligand. For example, upon reaction with a suitable transition metal fragment precursor, the cyclo-E5 moiety in [Cp*Fe(η5-E5)] (E = P (1), As (2)) undergoes either simple coordination, rearrangement, or fragmentation. Nevertheless, in 1998, Kudinov et al. demonstrated that the coordination of cationic fragments can be achieved while the central pentaphospholyl ligand is conserved as exemplified by the synthesis of e.g. [(Cp*Fe)(μ,η5:5-P5)(MLn)][BF4] (I, {MLn} = {Cp*Fe}, {CpRu}, {Cp*Ru}, Fig. 1).9 Our group extended this methodology to transition metal fragments of group 6 to 10 to yield cationic triple-decker complexes (I, {MLn} = {MCp*}, M = Cr, Mn, Fe, Co, Ni; Fig. 1).10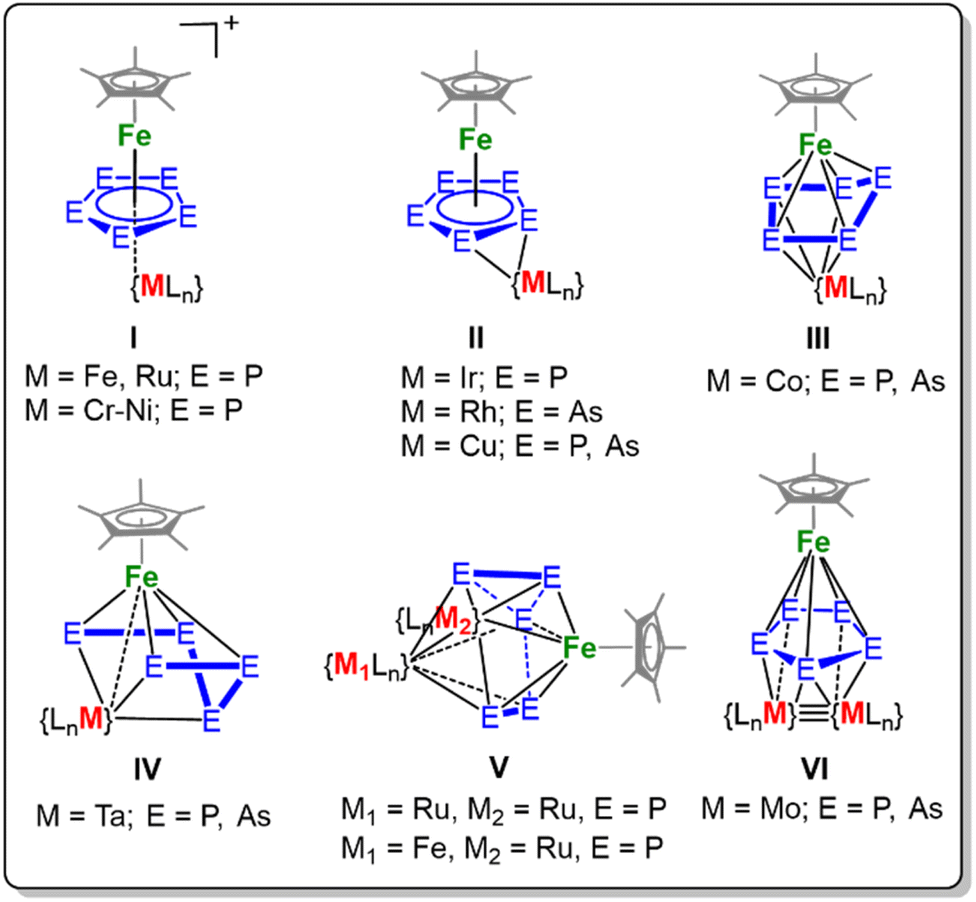 | ||
| Fig. 1 Selected examples reported for heterometallic polypnictogen complexes containing E5 ligands derived from 1 and 2, respectively. (I: {MLn} = {CpFe}, {CpRu}, {Cp*Ru}, {Cp′′′Cr}, {Cp′′′Mn}, {Cp′′′Fe}, {Cp′′′Co}, {Cp′′′Ni}; II: {MLn} = {Cp*Ir(Co)}, {Cp*Rh(CO)}, {nacnacCu}; III: {MLn} = {Cp′′′Co}; IV: {MLn} = {Cp′′Ta}; V: {MLn} = {Cp′′Ru}, {Cp*Fe}; VI: {MLn}2 = Mo2[μ,κ2-Me2Si(N-2,6-iPr2C6H3)2]2; Cp′′ = 1,3-tBu2C5H3, Cp′′′ = 1,2,4-tBu3C5H2, Cp* = C5Me5, nacnac = {N(C6H3Me-2,6)CMe}2CH. | ||
Furthermore, complexes of the type II are known in which the {MLn} moiety coordinates in an η2 fashion side-on to the cyclo-E5 ring (Fig. 1). Hence, under rather mild conditions, group 9 ({Cp*Ir(CO)}, {Cp*Rh(CO)})11 and group 11 ([({N(C6H3Me2-2,6)CH}2CH)Cu])12 metal fragments can be introduced. Moreover, the coordination of the 14VE fragment {Cp′′′Co} induces a distortion of the E5 moiety to adopt an envelope-type conformation within the heterobimetallic triple-decker complexes [(Cp*Fe)(Cp′′′Co)(μ,η5:η4-E5)] (E = P,13 As;14 Cp′′′ = 1,2,4-tBu3C5H2) (III, Fig. 1). The rupture of one E–E bond in the envelope-type structural motif III results in a catena-E5 chain, which was realised within complexes of type IV ({MLn} = {Cp′′Ta}, Cp′′ = 1,3-tBu2C5H3; Fig. 1).15 Moreover, the fragmentation into partially separated P2 and P3 units within the coordination sphere of group 8 metals such as Fe and Ru was realised by Scherer et al. (V, {MLn} = {Cp′′Ru} or {Cp′′Fe}; Fig. 1). These trimetallic species V were synthesised under thermolytic conditions and are only known for the lighter homologue phosphorus.16 An alternative synthetic approach was realised independently by the groups of Roesky and Tsai. Under reductive conditions, they achieved the formation of di or trimetallic complexes of 1 or 2 and f-block17,18 or d-block metals,19 respectively. For example, the highly reducing quadruple bond within the dimolybdenum complex Mo2[μ,κ2-Me2Si(N-2,6-iPr2C6H3)2]2 reacts with 1 or 2 to the trimetallic species VI revealing a novel structural motif (Fig. 1). In VI the cyclo-E5 ligand remains almost planar, coordinating towards the Fe fragment and the Mo2 fragment in a η5:2:2 fashion. Complexes I-VI were isolated by various synthetic procedures and transition metal (TM) precursors, and the question arises as to how a more systematic synthetic protocol and utilisation of isostructural TM precursors from group 6 to 10 affects product formation. Herein, we report a general procedure to di- or trinuclear multimetallic cage complexes starting from 1 and 2, respectively.
Results and discussion
Reduction of 1 and 2 and subsequent reactivity
As demonstrated previously, [Cp*Fe(η5-P5)] (1) can readily be reduced to the dianionic species [Cp*Fe(η4-P5)]2− (ref. 20) that can subsequently be reacted with suitable electrophiles.13,21 Following this pathway, for example the neutral triple-decker complex [(Cp*Fe)(Cp′′′Co)(μ,η5:4-P5)] was accessible.13 The heavier congener [Cp*Fe(η5-As5)] (2) is prone to undergo reduction, but in a rather unselective manner, giving rise to mixtures of various reaction products of anionic species containing As4, As10, As14 and As18 units.22 Nevertheless, when the reduction of 2 is performed in situ, the generated anionic intermediates might be reacted further by using transition metal complexes. Note that the follow-up reactivity of the anionic arsenic species has remained unexplored. In order to stabilise novel polypnictogen moieties, TM halide dimers of the type [Cp′′′M(L)(μ-X)]2 (M = Cr, Mn, Fe, Ni; L = thf (for M = Mn), X = Cl, Br, I)23–26 were selected. The sterically crowded Cp′′′ ligand is particularly suited for stabilising various TM complexes featuring pnictogen ligands.Development of a general synthetic procedure
In a general procedure, [Cp*Fe(η5-E5)] (E = P, As) were reduced by KC8 and subsequently reacted with [Cp′′′M(μ-X)]2 in salt metathesis reactions in DME or THF to yield the neutral complexes 1-Cr to 2-Ni (Scheme 1). To identify the best reaction conditions, all reactions were performed initially with 1.0 eq. 1 or 2, 2.5 eq. KC8 and 1.0 eq. [Cp′′′M(μ-X)]2 and then optimised (see ESI†). Notably, the in situ reduction of 2 and the subsequent reaction with [Cp′′′Mn(thf)(μ-I)]2 were also investigated. However, no single crystals were isolated in this reaction and the spectroscopic data are not reliable enough to unequivocally characterise any product. Even so, the formation of [(Cp′′′)2Mn]27 and HCp′′′ can be observed (see ESI†). For the other reactions, after column chromatographic workup under inert conditions (except for 1-Mn), the highly air- and moisture-sensitive compounds 1-Cr to 2-Ni can be isolated in crystalline yields of up to 44%. All products are extremely soluble in both non-polar (e.g. n-hexane) and polar solvents (acetonitrile) and cannot be precipitated from concentrated solutions (with the exception of 2-Fe). Interestingly to note that once crystallised all compounds are hard to re-dissolve, most likely due to effective packing and lattice energy effects in the solid state.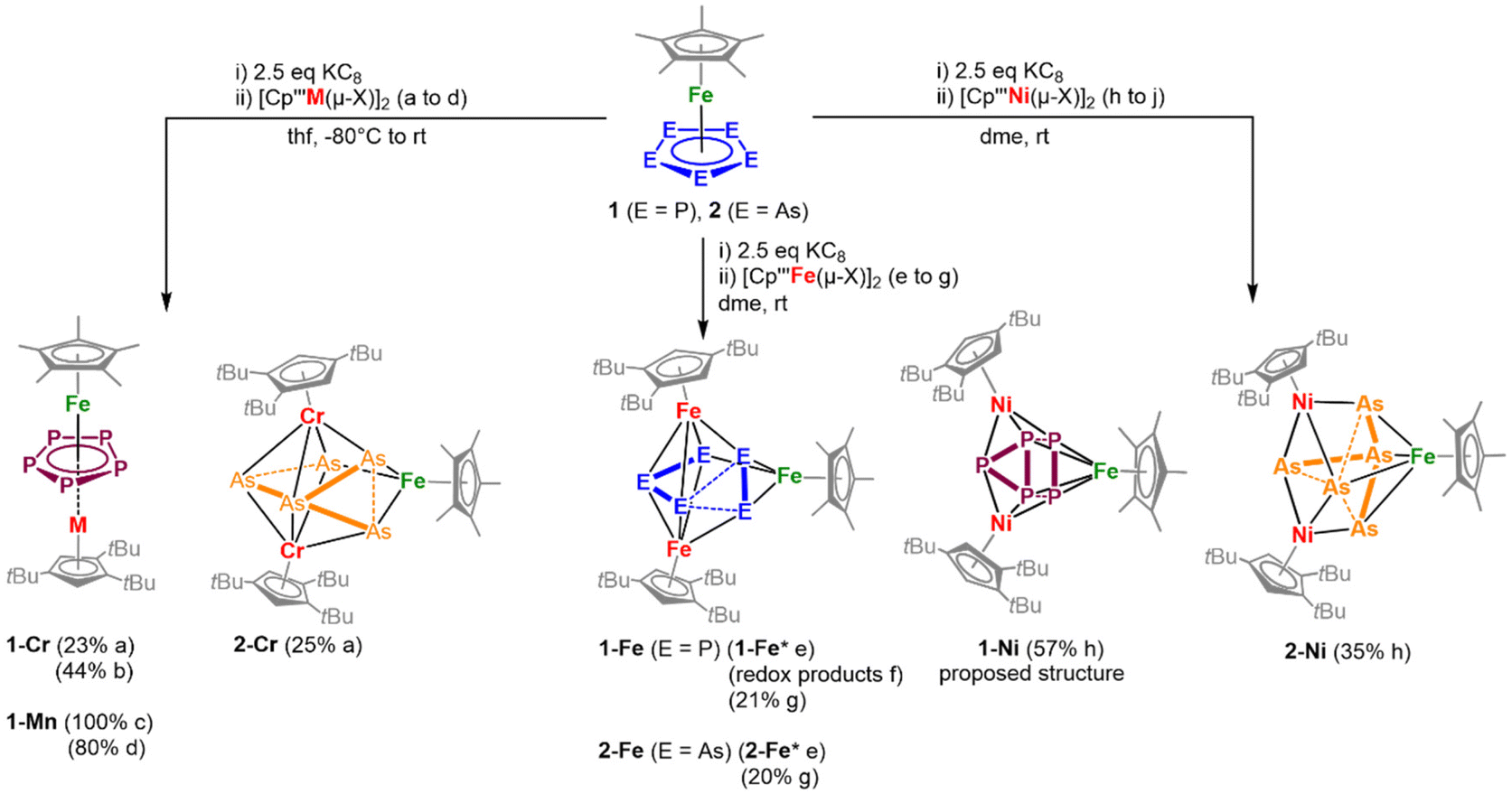 | ||
| Scheme 1 Synthesis of the triple-decker (1-Cr, 1-Mn) and cluster-type complexes (2-Cr, 1-Fe, 2-Fe, 1-Ni, 2-Ni) starting from [Cp*Fe(η5-E5)] (E = P (1), As (2)). Yields after workup are given in parentheses; 1-Ni was isolated as powder and the proposed structure was determined by coordination to silver (3-Ag, vide infra). Reaction conditions and yields: (a) 1.0 eq. [Cp′′′Cr(μ-Cl)]2; (b) 0.5 eq. [Cp′′′Cr(μ-Cl)]2; (c) 1.0 eq. [Cp′′′Mn(THF)(μ-I)]2 (quantitative conversion according to 31P NMR spectroscopy, isolation in an analytically pure manner as a few crystals); (d) 0.5 eq. [Cp′′′Mn(THF)(μ-I)]2 (1-Mn and formation of redox products); (e) 1.0 eq. [Cp′′′Fe(μ-Br)]2, 3 h; (f) 0.5 eq. [Cp′′′Fe(μ-Br)]2, 16 h; (g) 0.8 eq. 1.0 eq. [Cp′′′Fe(μ-Br)]2, 3 h; (h) 1.0 eq. [Cp′′′Ni(μ-Br)]2, 30 min (1-Ni), 3 h (2-Ni); (j) 0.5 eq. [Cp′′′Ni(μ-Br)]2 (no characterisable products); (* = minor impurities of [Cp′′′Fe(μ-Br)]2 are still present). For further information see ESI.† | ||
Despite several attempts, no suitable single crystals for 1-Ni could be received. To gain insight into the composition of the latter compound, 1-Ni was coordinated to Ag+ and the adduct 3-Ag was isolated (vide infra).
Work-up for 1-Mn
Column chromatography was not possible for 1-Mn, which was attributed to the air and moisture sensitivity of 1-Mn. Complex 1-Mn was, however, synthesised selectively as monitored by 31P NMR spectroscopy (δ(31P) = −38.4 ppm), but a second (minor) – not yet identified – species is also visible in the 31P NMR spectrum (δ(31P) = −34.3 ppm).‡ Due to a very similar solubility of 1-Mn and the by-product, their separation by crystallisation is extremely difficult. However, an analytically pure sample of 1-Mn was obtained by cooling a concentrated solution in n-pentane. But only few crystals of 1-Mn could be isolated by this method and a considerable amount of 1-Mn was lost.Alternative synthetic pathway
The phosphorus-containing complexes 1-Cr, 1-Ni, and 1-Mn were also obtained starting from isolated [K(dme)2]2[Cp*Fe(η4-P5)]20 and the respective TM halide dimer (see Fig. S22–S24, see ESI†), which shows that the in situ generation method is also selective. According to 31P NMR spectroscopy in these reactions always 1 is observed besides the formation of the respective product. Furthermore, this method does not work for the synthesis of 1-Fe, since only 1 can be detected.Changes in stoichiometry
Due to the found ratio for the isolated triple-decker complexes 1-Mn and 1-Cr, the use of 0.5 eq. of the TM halide dimer instead of 1.0 eq. was investigated. When 0.5 eq. of the Mn dimer is used, the ratio of the by-products of 1-Mn is increased. 1-Mn is formed, but additionally the formation of 1 along with the redox-product [Cp*Fe(η4-P5)]2− is observed (see ESI, Fig. S5–S7†). Using 0.5 eq. [Cp′′′M(μ-X)]2 yields 1-Cr in a selective manner, and in a crystalline yield of 44% (referred to the Cr dimer) that is slightly higher than in the initial synthetic approach (one equivalent of the dimer). Furthermore, decreasing the amount of the TM halide dimer, the chromatographic workup can be avoided (see ESI†).Furthermore, the utilisation of 1.0 eq. [Cp′′′Fe(μ-Br)]2 in the reaction with in situ generated 1.0 eq. [Cp*Fe(η4-P5)]2− provided slightly contaminated 1-Fe and 2-Fe by residual [Cp′′′Fe(μ-Br)]2, even after column chromatographic workup (see ESI, see Fig. S28†). Varying the amount of [Cp′′′Fe(μ-Br)]2 and measuring X-band EPR and 31P NMR spectra of the reaction mixture support the requirement of 0.8 eq. [Cp′′′Fe(μ-Br)]2 for a clean formation of 1-Fe and 2-Fe.
Decreasing the amount of [Cp′′′Fe(μ-Br)]2 to 0.5 eq. in the reaction with 1.0 eq. [Cp*Fe(η4-P5)]2− did not give 1-Fe but redox products (see ESI, Fig. S12†). Interestingly, in the reaction of 1.0 eq. [Cp*Fe(η4-P5)]2− with 0.5 eq. [Cp′′′Ni(μ-Br)]2, another product is formed exhibiting a broad resonance in the 31P NMR spectrum (δ/ppm = 35.8) and no signals for 1-Ni are observed. So far, a structural elucidation of this product and further spectroscopic insight were not possible, regardless of numerous attempts.
Structural characterisation in the solid state
Structurally, all obtained complexes reveal a neutral framework of the pnictogen ligand coordinating to one {Cp*Fe} and one or two {Cp′′′M} (M = Cr–Ni) fragments, respectively. In most of the products, the former cyclo-E5 unit is rearranged or fragmented and a broad variety of structural motifs of the pnictogen ligand are observed.Structural elucidation in the solid state of the triple-decker complexes
Both 1-Cr and 1-Mn display neutral triple-decker complexes of the type [(Cp*Fe)(Cp′′′M)(μ,η5:5-P5)]. The pentaphospholyl ligand remains intact in accordance with its isostructural cationic complexes (Fig. 2).10 The P–P bond lengths in both complexes are in the range of 2.1782(6) to 2.1936(6) Å (1-Cr) and 2.1442(8) to 2.1557(7) Å (1-Mn) and thus slightly shorter than in their corresponding cationic complexes.10 One P atom of each compound is slightly bent out of the P5 plane by 4.3° (1-Mn) or 4.8° (1-Cr), respectively. Note that 1-Mn unveils, to the best of our knowledge, the first neutral heterobimetallic manganese polypnictogen complex. The calculated structure for the singlet state of 1-Mn coincides with the structure determined by X-ray crystallography.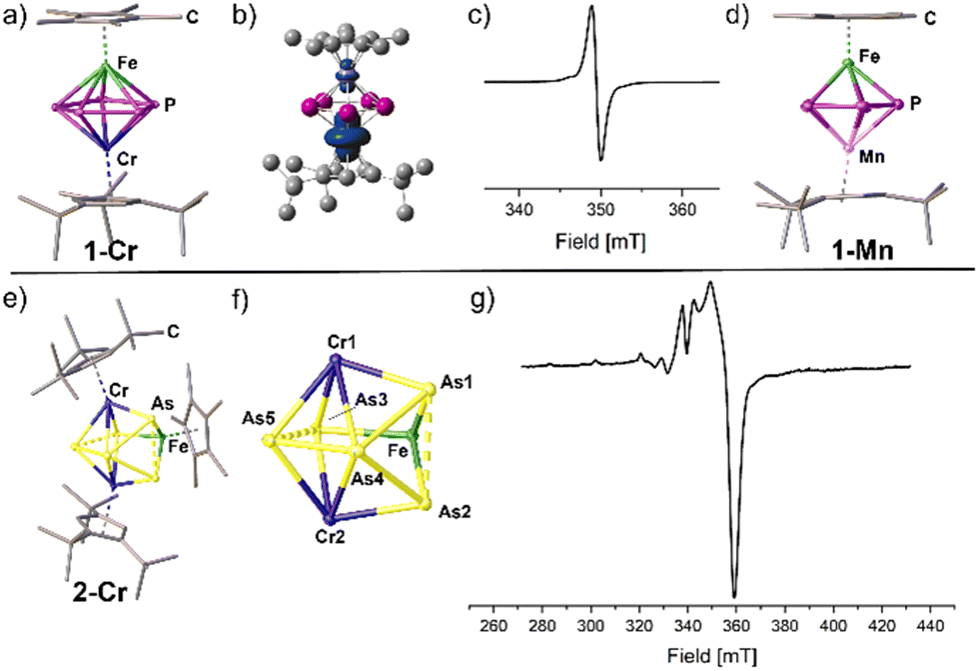 | ||
| Fig. 2 (a) Solid-state structure of the triple-decker complex 1-Cr; (b) calculated spin density of 1-Cr (at BP86/Def2-TZVP); (c) experimental X-band EPR spectrum of 1-Cr in thf solution at 298 K (giso = 1.918); (d) solid-state structure of the triple-decker complex 1-Mn; (e) solid-state structure of the cluster 2-Cr; (f) FeCr2As5 structural core motif of 2-Cr in the solid state; (g) experimental X-band EPR spectrum of 2-Cr in thf solution at 77 K. H atoms are omitted for clarity, ellipsoids are depicted at the 50% probability level. | ||
Structural elucidation in the solid state of the FeM2E5 complexes
Especially, the arsenic-containing complexes are of great interest to expand the field of heterobimetallic arsenic cage compounds. For 2-Cr, 1-Fe, 2-Fe and 2-Ni, geometries resembling distorted cubane frameworks of the type FeM′2E5 are observed (Fig. 3). In each cubane, two distorted tetragons built up by the pnictogen atoms and the transition metal atoms are connected. In 2-Cr, the distorted cubane is constructed by two parallelogram-like faces of As3–Fe–As2–Cr2 and Cr1–As1–As4–As5 which are staggered. Focusing on the pnictogen moiety, it is best described as an iso-tetraarsane unit§ coordinating to three {CpRM} fragments along with an As1 ligand in μ3 coordination mode (Fig. 2). Moreover, the isostructural homometallic complexes 1-Fe and 2-Fe expand the series of complexes containing catena-E3⋯E2 ligands, in which 2-Fe exhibits a rare example for the heavier arsenic homologue (Fig. 3). In both complexes, the E2 moiety is perpendicular to the E3 unit. However, the fragmentation does not lead to completely independent E3⋯E2 units as the E⋯E distances and WBIs for both compounds suggest weak interactions (P1–P3 2.3991(6) Å, P2–P3 2.3962(5) Å, WBI 0.56–0.57; As1–As3 2.6081(3) Å, As2–As3 2.6097(4) Å, WBI 0.56 for both), which are shorter than the sum of the van der Waals radii (ΣvdW(P⋯P) = 3.80 Å; ΣvdW(As⋯As) = 3.76 Å).28 Within the E3 and the E2 units, respectively, the E–E bond lengths are in the range of 2.1689(3)–2.2143(6) Å (WBI 0.87–0.97) for the P–P bonds in 1-Fe and 2.3833(6)–2.4596(4) Å (WBI 0.82–0.93) for the As–As bonds in 2-Fe, exhibiting single bond character.29 The homotrimetallic cage complexes can be described as well as distorted cubanes consisting of two staggered tetragons (Fe3–E2–Fe1–E5 and E1–E3–E4–Fe2). Surprisingly, in 1-Ni, a P2 unit is extruded from the former P5 ring and a cyclo-P3 ring is formed representing a rare structural motif of the polyphosphorus ligand, which was structurally characterised as a coordination compound of Ag+ in 3-Ag, and supported by 31P NMR spectroscopy (Fig. 4, vide infra). The Asn moiety in 2-Ni resembles the moiety in 2-Cr consisting of an iso-tetraarsane part and of one As atom in a μ3 coordination mode between the three metal fragments (Fig. 3). However, in 2-Ni, the core motif can be described as a cubane with two congruent tetragon faces (Ni1–As1–Ni2–As3 and As4–Fe–As2–As5). The As1 atom is slightly disordered over two positions with occupancies of 0.91 and 0.09. The part with the higher occupancy will be discussed further down. The As1 atom coordinates to the three metal fragments with Ni–As distances of 2.381(3) Å (Ni1–As1), 2.413(3) Å (Ni2–As1) and 2.459(4) Å. The As–As bond lengths in the As4 unit are in the range of elongated single bonds both for 2-Cr (2.4762(3)–2.5550(3) Å, WBI 0.63–0.75) and 2-Ni (2.4500(3)–2.5576(3) Å, WBI 0.78–0.80). The polyarsenic structural motif in 2-Ni bears resemblance to the arsenic units in the homo-trimetallic complexes of the type [(CpRNi)3(μ3-As)(As4)] (CpR = Cp*,30 Cp′′′31). These complexes exhibit a μ3 bridging As atom and an iso-tetraarsane entity containing shorter As–As bond lengths of 2.4464(8) to 2.4567(8) Å (CpR = Cp′′′)31 and 2.436 Å to 2.444 Å (CpR = Cp*).30 Complexes 2-Ni and 2-Cr emphasise the impact of lower E–E bond energy for the heavier pnictogens and their tendency to rearrange and form cluster-type structures.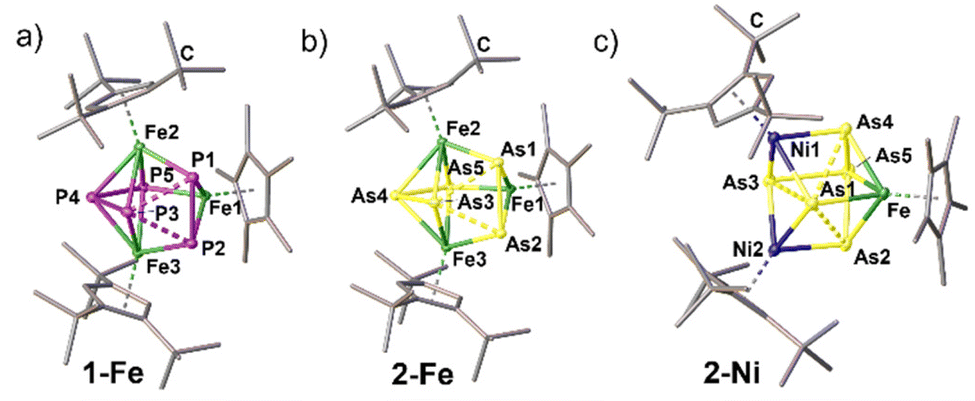 | ||
| Fig. 3 Solid-state structures for 1-Fe (a), 2-Fe (b) and 2-Ni (c); H atoms are omitted for clarity, ellipsoids are depicted at the 50% probability level. | ||
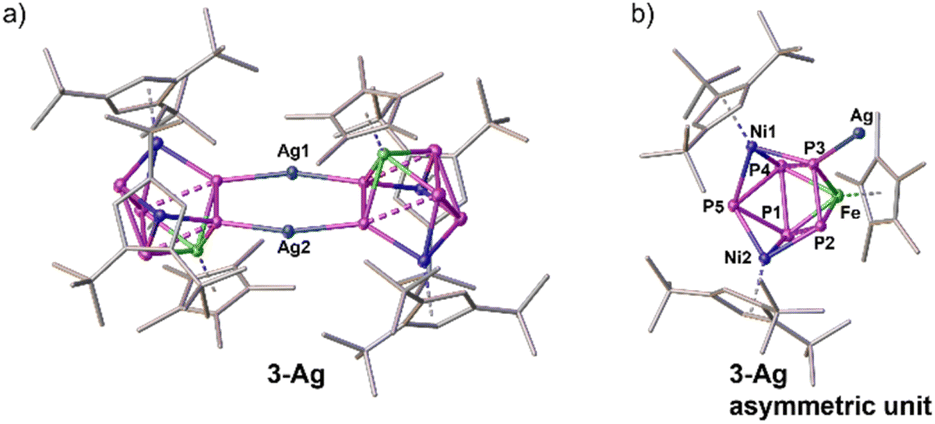 | ||
| Fig. 4 Solid-state structure of the cationic coordination complex 3-Ag. Anion and H atoms are omitted for clarity, ellipsoids are depicted at the 50% probability level. (a) Side view of the dimeric cation; (b) top view of the asymmetric unit in 3-Ag. | ||
Structural elucidation in solution of 1-Cr and 2-Cr
Spectroscopic investigations in solution indicate the paramagnetic character of the chromium complexes 1-Cr and 2-Cr. Broad and shifted signals in the corresponding 1H NMR spectra (see Fig. S1 and S2, ESI†) along with the effective magnetic moments obtained in solution by the Evans method (1-Cr: μeff = 1.21; 2-Cr: μeff = 1.36; corresponding to roughly one unpaired electron each) suggest doublet ground states. This is further confirmed by X-band EPR spectra recorded in solution (1-Cr and 2-Cr) and in the solid state (1-Cr), which show an isotropic signal (1-Cr, g = 1.918) or a rhombic signal (2-Cr, Fig. 2) at room temperature in thf solution, respectively. For 1-Cr upon cooling of the thf solution to 77 K and in the solid state an anisotropy of the g-tensors can be observed (see Fig. S26 and S27, ESI†). Notably, for 2-Cr, the hyperfine coupling in the EPR spectrum in thf solution remains unresolved even upon cooling to 77 K.Structural elucidation in solution of 1-Mn, 1-Fe and 2-Fe
For the diamagnetic complex 1-Mn, a sharp singlet at δ/ppm = −38.9 in the 31P NMR spectrum can be assigned to the cyclo-P5 moiety. The homotrimetallic group 8 complexes 1-Fe and 2-Fe show diamagnetic behaviour in solution as well. 1-Fe exhibits an A2BMX spin system in the 31P NMR spectrum (δ/ppm = 385.4, 376.5, −66.6 and −231.8) with an integral ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:1. The chemical shifts and P–P coupling constants of 1-Fe are in line with the data reported by Scherer et al. for similar Ru3P5 complexes.16 In the 1H NMR spectrum of 1-Fe, well-resolved resonances for the Cp* and Cp′′′ ligands are observed, whereas the signals for 2-Fe in the 1H NMR spectrum are broadened.
:1:1:1. The chemical shifts and P–P coupling constants of 1-Fe are in line with the data reported by Scherer et al. for similar Ru3P5 complexes.16 In the 1H NMR spectrum of 1-Fe, well-resolved resonances for the Cp* and Cp′′′ ligands are observed, whereas the signals for 2-Fe in the 1H NMR spectrum are broadened.
Structural elucidation in solution of 1-Ni and 2-Ni
In the 31P NMR spectrum of 1-Ni, resonances at δ/ppm = 390.0 (t), 280.7 (m) and 152.8 (m) with an integral ratio of 1:2:2 are observed at 253 K. The broad multiplet at 152.8 ppm is assigned to the P2 dumbbell, whereas a splitting into a triplet and a not completely resolved multiplet with a P–P coupling constant of JP–P = 158 Hz is observed for the cyclo-P3 unit. Furthermore, a smaller coupling constant of JP–P = 23 Hz is detected between the multiplets indicating the presence of a high order spin system in solution. However, the multiplets observed cannot be resolved by further cooling. Instead, cooling of the solution to 193 K leads to a broadening of the signals. At higher temperatures, the 31P NMR spectrum of 1-Ni reveals a temperature dependent broadening, which could arise from a dynamic rearrangement (see Fig. S16, ESI†). These assumptions are in accordance with the single crystal X-ray structure of the corresponding coordination complex 3-Ag (vide infra). The 1H NMR spectrum of 2-Ni reveals sharp singlets for the Cp* and Cp′′′ ligands indicating its diamagnetic character.
Coordination of 1-Ni towards Ag+: characterisation in solution and solid state
As mentioned above, it was unfortunately not possible to obtain single crystals for complex 1-Ni from concentrated solutions in toluene, n-hexane or n-pentane or by layering a concentrated solution of 1-Ni in CH2Cl2 with CH3CN at various temperatures. Thus, 1-Ni was reacted with one equivalent of the silver salt of the weakly coordinating anion [FAl] ([FAl] = [FAl{O(1-C6F5)C6F10}3]−) in o-DFB at ambient temperature. After work-up and layering with n-hexane, single crystals of the dimeric coordination compound [{(Cp*Fe)(Cp′′′Ni)2(μ3,η2:1:1-P2)(μ3,η2:2:2-P3)}Ag]2[FAl]2 (3-Ag) (cf.Fig. 4) were isolated in 51% yield. The X-ray structure of complex 3-Ag shows two {FeNi2P5} units connected via two Ag atoms. The Ag1–Ag2 distance of 2.9711(7) Å is significantly below the sum of the van der Waals radii of two Ag ions (ΣvdW(Ag⋯Ag) = 3.44 Å) demonstrating the possible existence of argentophilic interactions.32 Within the {FeNi2P5} units, a partial separation of the pnictogen ligand into a cyclo-P3 and a P2 fragment is perceived, with one P atom of the P2 dumbbell coordinating to Ag. The P–P distances within the cyclo-P3 unit are in the range of 2.2967(8) to 2.3362(10) Å and thus represent slightly elongated single bonds with WBIs of 0.64 to 0.80.29 The P2–P3 distance within the P2 unit amounts to 2.1335(7) Å, which is in between a P–P single and a double bond.29,33 However, the corresponding WBI of 1.09 confirms the presence of some double bond character. The distances of P1–P2 (2.6545(7) Å, WBI 0.42) and P3–P4 (2.5436(7) Å, WBI 0.26) indicate weak interactions between both pnictogen units. This P3/P2 fragmentation with weak interactions between them bears resemblance to the literature-known complexes [{Cp′′Ru}2{Cp*Fe}P5] and [{Cp*Fe}2{Cp′′Ru}P5] reported by Scherer et al., in which the distances between the phosphorus units are in the range of 2.4524(12) to 2.504(4) Å.16 In the corresponding 31P NMR spectrum of 3-Ag, slightly down-field shifted signals for the cyclo-P3 unit (δ/ppm = 430.6, 248.8) are observed compared to the ones in 1-Ni, whereas the signal for the P2 unit (δ/ppm = 88.2) is very broad.¶ Due to the very similar chemical shift in the 31P NMR spectra of 3-Ag and 1-Ni, it is assumed that, upon coordination towards Ag, the structural change within the P3 unit is neglectable. The optimised geometry of 1-Ni calculated at the BP86-D3(BJ)/def2-TZVP level of theory based on the X-ray coordinates of 3-Ag indicates that separated P3 and P2 units are present. The calculated P–P distances within the cyclo-P3 unit are in the range of 2.3502 to 2.3904 Å and amount to 2.1649 Å within the P2 unit. Relying on this spectroscopic information and the structure of 3-Ag in the solid state, the structure of the P5 unit in 1-Ni can be proposed as depicted in Scheme 1.Conclusions
In summary, a general synthetic approach for polypnictogen cage complexes starting from cyclo-E5 ligands was developed. In situ reduction of 1 and 2, respectively, and subsequent reaction with transition metal halide dimers give rise to a broad range of differently structured complexes 1-Cr to 2-Ni. The structural motifs of these complexes and cage compounds, respectively, differ in most of the cases for the phosphorus and arsenic derivatives. Due to the lower E–E bond energy, the arsenic containing complexes show more fragmentation compared to the phosphorus derivatives. Interestingly to note, that the structural motif of the isolated cage compound or the triple-decker complex cannot be predicted including the number of {Cp′′′M} fragments attached. Within the isolated products, a simple coordination (1-Cr, 1-Mn) of one {Cp′′′M} fragment or a complete rearrangement of the former E5 unit (1-Fe, 2-Fe, 2-Cr, 2-Ni) are realised. In this regard, 1-Mn displays a rare example of neutral heterobimetallic manganese complexes containing a Pn ligand. Furthermore, new As4–As1 (2-Cr) or E3–E2 (1-Fe, 2-Fe) ligands in the coordination environment of homotrimetallic or heterotrimetallic frameworks were realised expanding the library of heterometallic arsenic cage compounds. Surprisingly, for the homotrimetallic cage compounds 1-Fe and 2-Fe, isostructural motifs were obtained. Both complexes might serve as interesting starting materials for further studies like the oxidation of the iron atoms. For group 10, a rare example of a cyclo-P3–P2 separation was observed exhibiting weak interactions still present. To confirm the molecular structure of 1-Ni, its Ag+ complex 3-Ag was synthesised and structurally characterised. The complexes 2-Ni and 2-Cr feature an As4–As1 ligand, consisting of an iso-tetraarsane unit and the μ3-bridging As1 atom. The Ni fragments are coordinated similarly by three arsenic atoms each and the Fe fragment by four arsenic atoms. Multinuclear NMR spectroscopy along with EPR spectroscopy and mass spectrometry verify the features of the isolated complexes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft within the project Sche 384/40-1. C.R. is greatful to the Studienstiftung des Deutschen Volkes for a PhD fellowship as it is R.S. to the Fonds der Chemischen Industrie.References
- O. J. Scherer, Angew. Chem., Int. Ed. Engl., 1985, 24, 924 CrossRef
.
- O. J. Scherer, Acc. Chem. Res., 1999, 32, 751 CrossRef CAS
- B. M. Cossairt, N. A. Piro and C. C. Cummins, Chem. Rev., 2010, 110, 4164 CrossRef CAS
- M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. Rev., 2010, 110, 4178 CrossRef CAS PubMed
- C. M. Hoidn, D. J. Scott and R. Wolf, Chem. – Eur. J., 2021, 27, 1886 CrossRef CAS PubMed
- M. Seidl, G. Balázs and M. Scheer, Chem. Rev., 2019, 119, 8406 CrossRef CAS PubMed
- O. J. Scherer, C. Blath and G. Wolmershäuser, J. Organomet. Chem., 1990, 387, C21–C24 CrossRef CAS
- C. Riesinger, L. Dütsch and M. Scheer, Z. Anorg. Allg. Chem., 2022, 648, e202200102 CrossRef CAS
- A. R. Kudinov, D. A. Loginov, P. V. Petrovskii and M. I. Rybinskaya, Russ. Chem. Bull., 1998, 47, 1583 CrossRef CAS
- C. Riesinger, D. Röhner, I. Krossing and M. Scheer, Chem. Commun., 2023, 59, 4495 RSC
- M. Detzel, G. Friedrich, O. J. Scherer and G. Wolmershäuser, Angew. Chem., Int. Ed. Engl., 1995, 34, 1321 CrossRef CAS
- M. Haimerl, M. Piesch, G. Balázs, P. Mastrorilli, W. Kremer and M. Scheer, Inorg. Chem., 2021, 60, 5840 CrossRef CAS PubMed
- M. Piesch, F. Dielmann, S. Reichl and M. Scheer, Chem. – Eur. J., 2020, 26, 1518 CrossRef CAS PubMed
- M. Piesch and M. Scheer, Organometallics, 2020, 39, 4247 CrossRef CAS
- K. Mast, J. Meiers, O. J. Scherer and G. Wolmershäuser, Z. Anorg. Allg. Chem., 1999, 625, 70 CrossRef CAS
- B. Koch, O. J. Scherer and G. Wolmershäuser, Z. Anorg. Allg. Chem., 2000, 626, 1797 CrossRef CAS
- S. Schäfer, S. Kaufmann, E. S. Rösch and P. W. Roesky, Chem. Soc. Rev., 2023, 52, 4006 RSC
- N. Arleth, M. T. Gamer, R. Köppe, S. N. Konchenko, M. Fleischmann, M. Scheer and P. W. Roesky, Angew. Chem., Int. Ed., 2016, 55, 1557 CrossRef CAS PubMed
- M.-W. Lee, G. Balázs, S. Reichl, L. Zimmermann, M. Scheer and Y.-C. Tsai, Chem. Commun., 2023, 59, 11192 RSC
- M. V. Butovskiy, G. Balázs, M. Bodensteiner, E. V. Peresypkina, A. V. Virovets, J. Sutter and M. Scheer, Angew. Chem., Int. Ed., 2013, 52, 2972 CrossRef CAS PubMed
- S. Reichl, E. Mädl, F. Riedlberger, M. Piesch, G. Balázs, M. Seidl and M. Scheer, Nat. Commun., 2021, 12, 5774 CrossRef CAS PubMed
- M. Schmidt, D. Konieczny, E. V. Peresypkina, A. V. Virovets, G. Balázs, M. Bodensteiner, F. Riedlberger, H. Krauss and M. Scheer, Angew. Chem., Int. Ed., 2017, 56, 7307 CrossRef CAS PubMed
- M. W. Wallasch, F. Rudolphi, G. Wollmershäuser and H. Sitzmann, Z. Naturforsch., B: J. Chem. Sci., 2009, 64, 11 CrossRef CAS
- M. Kreye, C. G. Daniliuc, M. Freytag, P. G. Jones and M. D. Walter, Dalton Trans., 2014, 43, 9052 RSC
- M. Schär, D. Saurenz, F. Zimmer, I. Schädlich, G. Wolmershäuser, S. Demeshko, F. Meyer, H. Sitzmann, O. M. Heigl and F. H. Köhler, Organometallics, 2013, 32, 6298 CrossRef
- M. Maekawa, M. Römelt, C. G. Daniliuc, P. G. Jones, P. S. White, F. Neese and M. D. Walter, Chem. Sci., 2012, 3, 2972 RSC
- M. D. Walter, C. D. Sofield, C. H. Booth and R. A. Andersen, Organometallics, 2009, 28, 2005 CrossRef CAS
- S. Alvarez, Dalton Trans., 2013, 42, 8617 RSC
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 186 CrossRef
- O. J. Scherer, J. Braun and G. Wolmershäuser, Chem. Ber., 1990, 123, 471 CrossRef CAS
- V. Heinl, M. Schmidt, M. Eckhardt, M. Eberl, A. E. Seitz, G. Balázs, M. Seidl and M. Scheer, Chem. – Eur. J., 2021, 27, 11649 CrossRef CAS
- H. Schmidbaur and A. Schier, Angew. Chem., Int. Ed., 2015, 54, 746 CrossRef CAS PubMed
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 12770 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2347421–2347427. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01160k |
| ‡ The yield in percentage cannot be determined, as the number of P atoms within the by-product is unknown. |
| § The As1–As2 distance within 2-Cr amounts to 2.5546(5) Å. The value of the corresponding Wiberg bond index of 0.39 indicates solely the presence of an interaction. Thus, this interaction is described as a dashed line within Scheme 1 and Fig. 2. |
| ¶ Due to the very similar chemical shift of both species, a check for their dynamic behaviour in solution was performed via variable temperature NMR spectroscopy, which confirmed the presence of two different species, but no dynamic behaviour could be detected (see ESI†). |
| This journal is © The Royal Society of Chemistry 2024 |