
Coinage metal-ethylene complexes of sterically demanding 1,10-phenanthroline ligands†
Deepika V.
Karade
 ,
Vo Quang Huy
Phan
and
H. V. Rasika
Dias
*
,
Vo Quang Huy
Phan
and
H. V. Rasika
Dias
*
Department of Chemistry and Biochemistry, The University of Texas at Arlington, Box 19065, Arlington, Texas 76019-0065, USA. E-mail: dias@uta.edu
First published on 19th April 2024
Abstract
Phenanthroline-based ligands with bulky aryl groups flanking the metal binding pocket enabled the synthesis and detailed investigation of ethylene complexes of copper(I), silver(I), and gold(I), including structural data of [{2,9-bis(2,4,6-triisopropylphenyl)-1,10-phenanthroline}M(C2H4)][SbF6] (M = Cu, Ag, Au), Additionally, a related copper(I)-ethylene complex with a highly fluorinated ligand is also reported. Gold(I) affects the ethylene moiety significantly as evident from the notable upfield coordination shifts of ethylene carbon signals in the NMR and lengthening of the ethylene C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond length. Silver(I) forms the weakest bond with ethylene in this series of isoleptic, group 11 metal-ethylene complexes. Preliminary catalytic investigations underscore the potential of copper complexes, particularly those with weakly coordinating supporting ligands, as effective catalysts for C(sp3)–H functionalization through trifluoromethyl carbene insertion.
C bond length. Silver(I) forms the weakest bond with ethylene in this series of isoleptic, group 11 metal-ethylene complexes. Preliminary catalytic investigations underscore the potential of copper complexes, particularly those with weakly coordinating supporting ligands, as effective catalysts for C(sp3)–H functionalization through trifluoromethyl carbene insertion.
Introduction
1,10-Phenanthrolines are very popular supporting ligands for d-block chemistry.1–3 They have a planar, rigid ligand backbone with two inward pointing electron donor sites perfectly oriented for metal ion chelation, affording entropic advantage over other more flexible bidentate nitrogen-based ligands.3,4 1,10-Phenanthroline (phen) decorated with several different backbone substituents are known, and they as well as the parent phen have been utilized in a variety of applications including the development of luminescent materials,1,5–10 mechanochromic indicators,11 homogeneous catalysts,8,12–18 molecular chemo-sensors for anions and metal cations,19,20 and DNA intercalating and antibacterial and anticancer agents.21–25We have been utilizing nitrogen-based chelators such as bis(pyrazolyl)borates,26–36 bis(pyrazolyl)methanes,30,34,37 and bis(pyridyl)borates38,39 in coinage metal (Cu, Ag, Au) chemistry, in particular, to develop homogeneous catalysts,27,29,40 achieve olefin-paraffin separations,35,36 and to stabilize molecules with CO, ethylene and acetylene, and larger alkenes and alkynes on these metal ions26,28–30,36,37,40–42 for bonding investigations and to serve as models for reaction intermediates in catalytic processes involving these metal-gas combinations. In this work, we report the use of two sterically demanding phen ligands, 2,9-bis(2,4,6-triisopropylphenyl)-1,10-phenanthroline (L1) and 2,9-bis(2,4,6-trifluoromethylphenyl)-1,10-phenanthroline (L2) in a systematic study of coinage metal ethylene chemistry (Fig. 1). Backbone rigidity and flanking bulky aryl groups on these ligands L1 and L2 are particularly attractive, as they could provide ideal coordination pockets for stabilizing coinage metal-ethylene moieties for detailed studies.
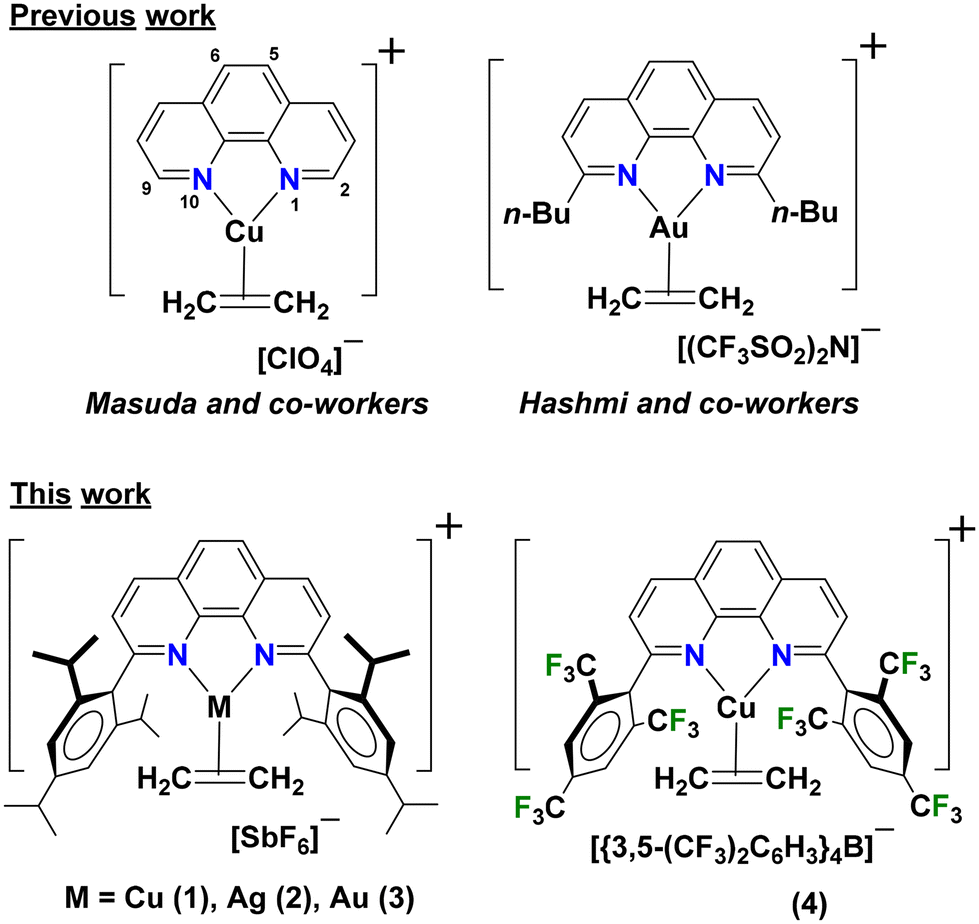 | ||
| Fig. 1 Structurally characterized ethylene adducts of coinage metals supported by 1,10-phenanthroline ligands in the literature and reported in this work, [L1M(C2H4)][SbF6] (M = Cu (1), Ag (2), Au (3)) and [L2Cu(C2H4)][BArF] (4). | ||
Note that the isolation of such species is often plagued by the lability and the facile loss of bound ethylene.43 Notably, despite the wide use of phen with coinage metals, there are only two examples of phenanthroline supported cationic coinage metal-ethylene complexes in the literature with X-ray structural data to our knowledge. Masuda et al. reported the first copper ethylene complex supported by the parent 1,10-phenanthroline, [(phen)Cu(C2H4)][ClO4] in 1987 to understand the effect of phenanthroline on the bonding between copper(I) and ethylene (Fig. 1).44 In 2019, Hashmi and coworkers reported 2,9-bis(n-butyl)-1,10-phenanthroline supported Au(I)-ethylene complex for performing Au/Ag bimetallic catalysis to pursue C–H activation of cyclopropenes for direct alkynylation.18 Quite surprisingly, structurally authenticated coinage metal complexes of even the larger olefins are quite rare, and only four such species45,46 in the Cambridge Structural Database.47 Herein we report syntheses, structures, properties of coinage metal ethylene complexes 1–4 supported by bulky phen ligands L1 and L2, and the C–H bond functionalization via carbene insertion mediated by the copper(I) complexes 1 and 4.
Results and discussion
The 1,10-phenanthroline ligands L1 and L2 were synthesized using literature procedures.16,48 The coinage metal ethylene complexes supported by L1, [L1M(C2H4)][SbF6] (M = Cu (1), Ag (2), Au (3)) were prepared successfully by first generating the tris(ethylene) copper(I), silver(I), and gold(I) hexafluoroantimonate complex in situ,49,50 followed by addition of L1 in dichloromethane. Compounds 1–3 have been isolated as crystalline solids in 75–96% yield. They do not lose ethylene under reduced pressure at room temperature. The [L2Cu(C2H4)][BArF] (4) was prepared by treating L2CuI with Na[BArF] under ethylene (∼1 atm) for 3 h at room temperature. The product [L2Cu(C2H4)][BArF] ([BArF] = [{3,5-(CF3)2C6H3}4B]) was isolated and dried using an ethylene stream. In contrast to [L1M(C2H4)][SbF6], the loss of bound ethylene was observed in [L2Cu(C2H4)][BArF] if placed under reduced pressure.Compounds 1–3 were characterized by several analytical techniques including 1H and 13C NMR spectroscopy. The key NMR spectroscopic features of olefinic ligand bound to the coinage metal ions are summarized in Table S1.† In comparison to the free ethylene, the 1H and 13C{1H} NMR spectra of copper(I) complex 1 and the analogous silver(I) complex 2 in CDCl3 show coordination induced upfield shifts of 2.17 ppm and 36.89 ppm and 1.90 ppm and 21.18 ppm for the ethylene protons and carbons, respectively, from the corresponding chemical shift of the free ethylene. The gold(I) complex 3 displayed a noticeably large upfield shift of the ethylene proton and carbon signals (3.04 ppm (1H) and 62.02 ppm (13C) relative to the corresponding chemical shift of the free ethylene). The relative magnitude of the upfield shifts in ethylene carbons due to coordination reflects the σ-acceptor and π-donor abilities of the coinage metal atom and the extent of M-ethylene π-back bonding that is believed to exist in these molecules. Although the trend of coordination shifts of the metal bound ethylene protons in 1H NMR spectra are also in agreement, those peak positions are likely affected by the ring current effects of the flanking aryl groups, preventing a direct analysis. For example, ethylene protons of the flanking aryl group free [(phen)Au(C2H4)][N(SO2CF3)2] in 1H NMR spectrum has been observed at δ 3.97 ppm,18 while the corresponding resonance in [L1Au(C2H4)][SbF6] (3) that has flanking aryl groups appears at a significantly upfield region, at δ 2.34 ppm, pointing to extra chemical shielding in the latter. The 13C chemical shift of the ethylene carbons, however, is very similar in the two adducts, at δ 61.91 and 61.08 ppm, respectively. Better donor solvents like acetone, benzene, and tetrahydrofuran do not result in the removal of the attached ethylene moiety in 1–3, as evident from their NMR data in these solvents.
Compared to L1, the highly fluorinated L2 is a weaker donor and should make the metal sites supported by this ligand relatively electron poor and more electrophilic. The copper(I)-ethylene complex 4, indeed shows relatively smaller upfield shifts of the ethylene 1H signal due to metal ion coordination, suggesting a somewhat lower level of metal-ethylene back bonding relatively to the related complex 1. A broad ethylene peak of 4 in 1H NMR suggests the presence of rather labile ethylene group in solution, consistent with the facile ethylene loss from solids under reduced pressure. Our attempt to synthesize [L2Cu(C2H4)][SbF6] via an analogous route used for [L1M(C2H4)][SbF6] yielded a product with very poor solubility in weakly-coordinating solvents such as dichloromethane, chloroform, and hexanes. This product is soluble in polar solvents like acetone-d6 and DMSO-d6, but they displace the bound ethylene as evident from the 1H NMR data. To address the solubility issue, counter-anion screening was conducted. Notably, the product 4 involving the larger, [{3,5-(CF3)2C6H3}4B]− ([BArF]−) counter anion afforded improved solubility in halogenated solvents. Additionally, X-ray quality crystals were successfully obtained at −20 °C from dichloromethane, facilitating the confirmation of the structure. These crystals were used to obtain the 1H NMR as well, but the solubility of [L2Cu(C2H4)][BArF] in CDCl3 is still poor to observe 13C signals. Solutions of these olefin complexes in dichloromethane are air-stable for several hours.
The X-ray crystal structures of [L1M(C2H4)][SbF6] (M = Cu (1), Ag (2), Au (3)) and [L2Cu(C2H4)][BArF] (4) are illustrated in Fig. 2. Compounds 1–3 represent a rare, complete series of closely related, isoleptic coinage metal-ethylene complexes with structural data.37,51 They are three-coordinate metal complexes with two flanking aryl groups protecting the metal-ethylene core, and feature discrete cationic and anionic moieties. The ethylene coordinates to metal in a familiar η2-fashion. Table 1 summarizes selected structural parameters. The sum of angles about the metal center in 1–3 is 360°, indicating the trigonal planer geometry at the metal site. The Cu–N < Au–N < Ag–N bond length trend of 1–3 follows the covalent radii of the M,52 as the covalent radius of silver(I) is larger than those of gold(I) and copper(I). The M–C bond lengths of 1–3 also follow the same trend. The ethylene CC bond is longest for [L1Au(C2H4)]+, followed by [L1Cu(C2H4)]+ and [L1Ag(C2H4)]+ with bond lengths of 1.400 (av.), 1.364, and 1.326 Å, respectively. The metal bound ethylene 13C shifts and CC distances suggest that the gold interacts strongest with ethylene, followed by copper while silver having the weakest interaction with ethylene, consistent with previous observations involving bis(pyridyl)borate systems.53
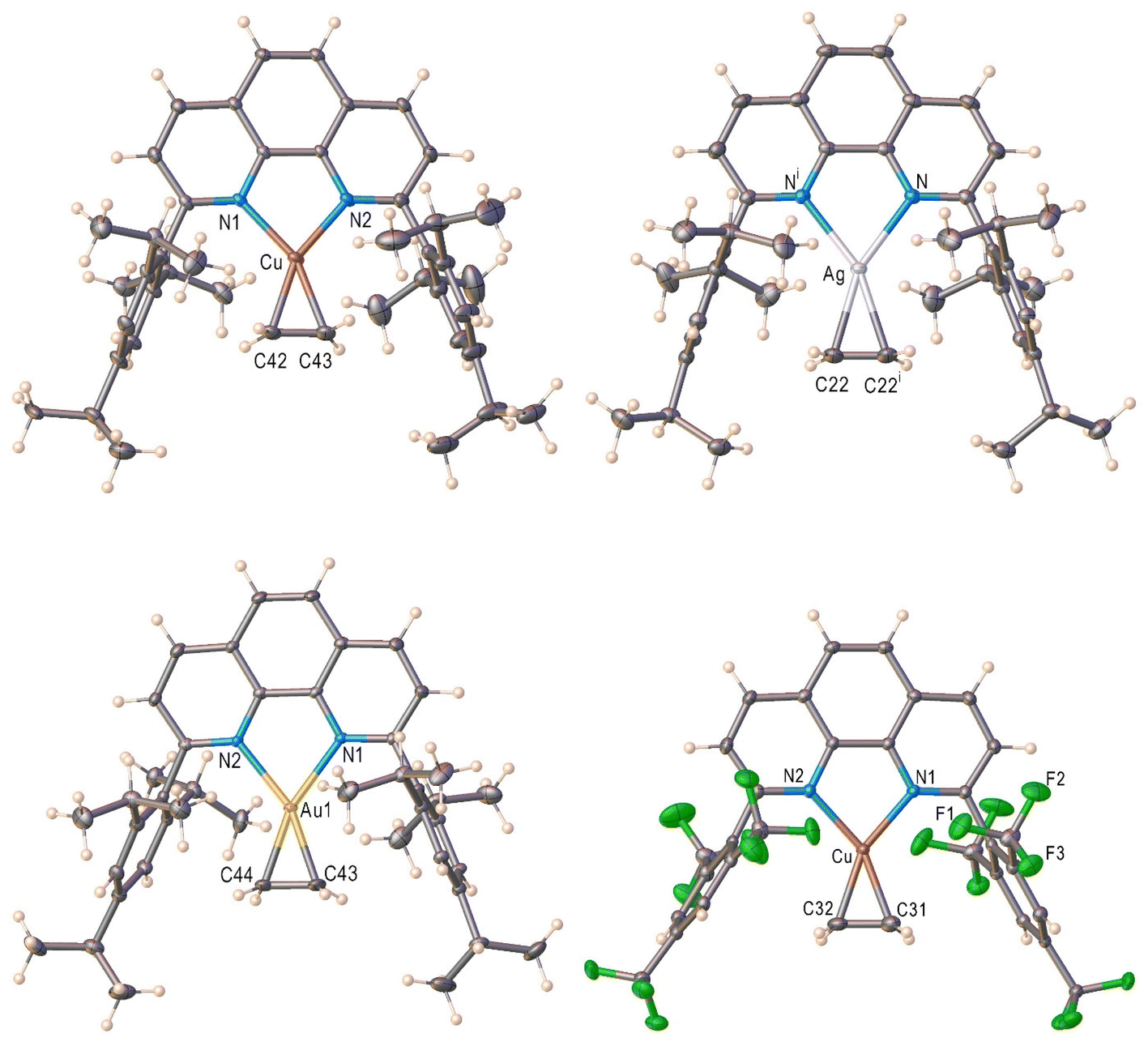 | ||
| Fig. 2 Molecular structures of the cationic moieties of [L1Cu(C2H4)]+ (top-left), [L1Ag(C2H4)]+ (top-right), [L1Au(C2H4)]+ (bottom-left), and [L2Cu(C2H4)]+ (bottom-right). The counter ions have been omitted for clarity. | ||
| Complex | [L1Cu(C2H4)]+ | [L1Ag(C2H4)]+ | [L1Au(C2H4)]+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
[L2Cu(C2H4)]+ |
|---|---|---|---|---|
| a There are two chemically identical molecules of [L1Au(C2H4)][SbF6] in the asymmetric unit. Metrical parameters of the second molecules are given in italics. b Sum of the angles at the metal ion. | ||||
| CC |
1.364(6) | 1.326(4) | 1.394(5) | 1.339(3) |
| 1.405(5) | ||||
| M–N | 2.011(2) | 2.2843(15) | 2.208(3) | 2.0059(14) |
| 2.013(2) | 2.2842(15) | 2.214(3) | 2.0179(14) | |
| 2.206(3) | ||||
| 2.216(3) | ||||
| M–C | 2.027(3) | 2.283(2) | 2.099(4) | 2.024(2) |
| 2.024(3) | 2.283(2) | 2.112(3) | 2.0011(19) | |
| 2.099(4) | ||||
| 2.112(3) | ||||
| ∠NMN | 83.90(10) | 73.42(8) | 75.31(10) | 83.59(6) |
| 75.15(11) | ||||
| ∠CMC | 39.34(16) | 33.77(11) | 38.99(15) | 38.86(9) |
| 38.66(15) | ||||
| ∑ at Mb | 360.1 | 360 | 360.1 | 360.6 |
| 360.0 | ||||
We have also managed to crystallize and characterize [L2Cu(C2H4)][BArF] (4) that has a highly fluorinated phenanthroline supporting ligand, using single crystal X-ray crystallography (Fig. 2). Basic structural features are similar between [L1Cu(C2H4)][SbF6] and [L2Cu(C2H4)][BArF]. The [L2Cu(C2H4)][BArF] is also a three-coordinate, trigonal planar metal complex. Interestingly, the effects of aryl group fluorination are not reflected in Cu–C or Cu–N distances (Table 1).
The functionalization of C(sp3)–H bonds to obtain fluorinated molecules via the insertion of fluorinated carbene moieties is quite attractive, yet rare and of significant current interest.54 We set out to probe this chemistry utilizing 2,2,2-trifluorodiazoethane as the carbene source and the copper(I) catalysts 1 and 4, following the recent important contributions of Daugulis involving “sandwich” diimine copper adducts in related chemistry.55 The adamantane was chosen as the hydrocarbon substrate.
As summarized in Scheme 1, cationic copper ethylene complexes 1 and 4 (supported by L1 and L2, respectively) are active catalysts affording a mixture of products resulting from the carbene insertion into secondary and tertiary C–H bonds. Reactions were performed at room temperature using 1 mol% of the catalyst, CF3CHN2 (1.0 equiv., solution in CH2Cl2) and adamantane (5.0 equiv.) and the product yields were determined by 19F NMR with an internal standard. The combined product yields of the two isomers were 39 and 84% for 1 and 4 catalyzed chemistry, with the selectivity of 90:10 (3°/2°) and 77:23 (3°/2°), respectively. These reactions produce bis(trifluoromethyl)ethylene isomers as the byproduct. For comparison, diimine supported copper-ethylene catalyst has produced analogous carbene insertion products with 79% total yield, and selectivity of 86:14 (3°/2°).55 Mechanistic work by Daugulis et al. suggests that the ethylene exchange with CF3CHN2 on copper is the first step and the presence of free ethylene hinders the carbene insertion. Thus, in order to see if we could improve the carbene insertion product yields of the reaction catalyzed by 1, the ethylene-free [L1Cu][SbF6] was also prepared and tested as a catalyst. However, it essentially gave the same carbene insertion product yields as observed with 1. This suggests that the high electrophilicity at the copper center, as in [L2Cu][BArF] is important to drive the reaction between the likely copper-carbene intermediate {[L2Cu]CHCF3}+ and adamantane effectively affording the desired carbene insertion products before it reacts competitively with another molecule of 2,2,2-trifluorodiazoethane54 producing undesired bis(trifluoromethyl)ethylene byproducts. Interestingly, silver(I) and gold(I) complexes 2 and 3 did not produce carbene insertion products to adamantane under these conditions. A black precipitate and mostly unreacted diazo reagent were observed in these reaction mixtures.
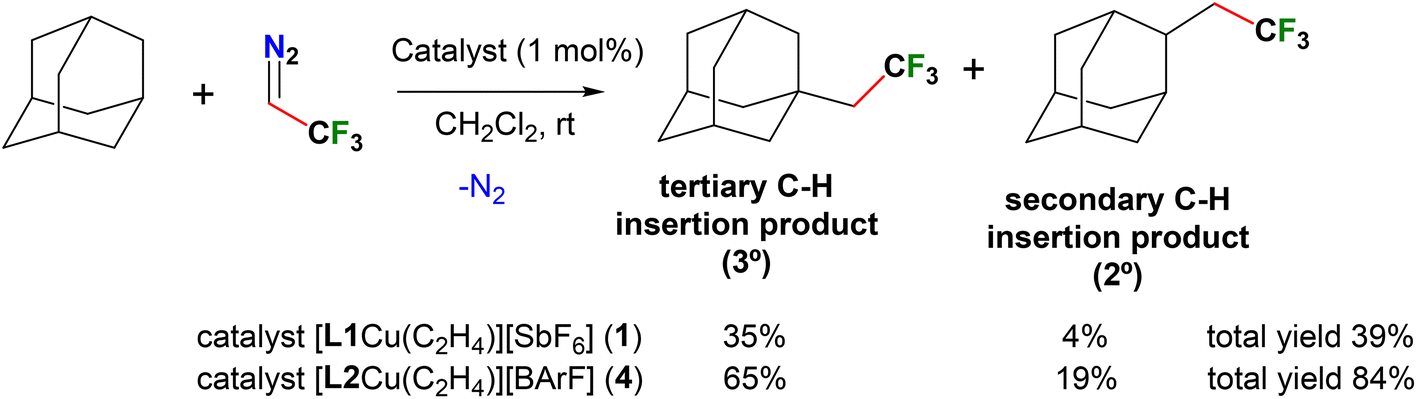 | ||
| Scheme 1 Functionalization of C(sp3)–H bonds via carbene insertion mediated by 1 and 4. | ||
Complexes 2–4 display luminescence in solution and solid state which hindered our ability to obtain Raman spectroscopic data. This observation prompted us to collect photophysical data on these compounds along with those of the ethylene-free [L1M][SbF6] complexes (M = Cu, Ag) (Fig. S13 and Table S7†). In general, upon excitation of dichloromethane solution of these compounds at 360 nm, they exhibited emissions in the visible region, with a maximum wavelength ranging between 428 and 502 nm. The complex 1 displayed very weak photoluminescence with λmax = 502 nm, while the 2 and 3 complexes revealed their emission bands with λmax = 490 and 487 nm, respectively. In contrast to the feeble luminescence observed in 1, compound 4 revealed a more intense emissions with λmax at 428 nm, displaying a notable impact due to ligand fluorination. These emissions are likely a result of a ligand-to-metal charge transfer (LMCT) process.8,56 To explore the impact of ethylene on photoluminescence properties of 1 and 2, we also collected the luminescence spectra of [L1M][SbF6] (M = Cu, Ag). Notably, both [L1Cu][SbF6] and [L1Ag][SbF6] complexes exhibited much higher photoluminescence intensities with λmax at 497 and 495 nm in dichloromethane solution (Fig. S13b and c†) relative to those of 1 and 2. Thus, the ethylene binding to Cu(I) and Ag(I) in these complexes leads to significant quenching of the photoluminescence.
Overall, the phenanthroline ligand systems L1 and L2 enabled a detailed investigation of coinage metal ethylene complexes including the solid state structural data on a rare isoleptic series [L1M(C2H4)]+ (M = Cu, Ag, Au). This includes the first well-authenticated silver(I)-ethylene adduct supported by a phen. We were also able to stabilize a copper-ethylene complex [L2Cu(C2H4)]+ supported by an encumbered fluorinated phen ligand. Structural and NMR spectroscopic data of 1–3 show that gold(I) has a significant effect on the ethylene moiety while the silver(I) forms the weakest bond. Preliminary catalytic investigations of these complexes suggest that the copper complexes, especially [L2Cu(C2H4)]+ with a weakly coordinating supporting ligand and more electrophilic copper site, are effective catalysts for C(sp3)–H functionalization via trifluoromethyl carbene insertion. We are currently pursuing further studies involving coinage metals and the bulky phen ligands L1 and L2.
Experimental section
General information
All preparations and manipulations were carried out under an atmosphere of purified nitrogen using standard Schlenk techniques or in a vacuum atmosphere single-station dry box equipped with a −25 °C refrigerator. Dichloromethane and hexane were dried by passing HPLC-grade solvent through a solvent purification system (SPS, Innovative Technologies Inc.) and stored in Straus flasks. Commercially available solvents were purified and dried by standard methods. Glassware was oven dried overnight at 150 °C. NMR spectra were acquired at 25 °C (unless noted) on a JEOL Eclipse 500 spectrometer (1H, 500 MHz; 13C, 126 MHz; 19F, 471 MHz), and a JEOL Eclipse 400 spectrometer (1H, 400 MHz; 13C, 100 MHz; 19F, 376 MHz), and all the spectral data were processed on MNova. 19F NMR values were referenced to external CFCl3. 1H and 13C NMR spectra were referenced internally to solvent signals (CDCl3, 7.26 ppm for 1H NMR, 77.16 ppm for 13C NMR). 1H, 13C, and 19F NMR chemical shifts are reported in ppm, and coupling constants (J) are reported in hertz (Hz). NMR solvents were purchased from Cambridge Isotopes Laboratories and used as received. Ethylene gas was purchased from Matheson. Raman data were collected on a Thermo Scientific DXR3 Raman microscope with laser source of 633 nm. Fluorescence spectroscopy was recorded in RF-5301 spectrofluorometer with a 150 W xenon lamp source. Panorama software was used to collect the fluorescence data. All the recorded fluorescence data was plotted on OriginPro 8.5 software. All emission spectra were recorded in freshly collected dichloromethane from SPS, followed by further drying with freeze pump thaw and sparging with nitrogen. High-resolution (HR) mass spectra were recorded at Shimadzu Center Laboratory for Biological Mass Spectrometry at UTA. Heating was accomplished by either a heating mantle or a silicone oil bath. The 2,9-bis(2,4,6-triisopropylphenyl)-1,10-phenanthroline (L1),48 2,9-bis(2,4,6-trifluoromethylphenyl)-1,10-phenanthroline (L2),16 copper(I), silver(I), and gold(I) tris(ethylene) hexafluoroantimonate(V) complexes,49,50 and trifluorodiazoethane57 (solution in CH2Cl2) were synthesized following literature procedures. All other reagents were obtained from commercial sources and used as received. Silver and gold complexes were prepared in reaction vessels protected from light using aluminum foil.Caution: Diazocompounds are toxic and shock sensitive. Therefore, extreme caution must be taken when handling and working with diazocompounds, and large-scale reactions must be avoided when possible. All used equipment (glassware and syringes) was washed with acetic acid in toluene (v/v 1/2) inside the hood to quench any residual diazoalkane prior to discarding. In the following experiments and during the synthesis of the starting materials, no incidents occurred.
Excess NaBArF (103.68 mg, 0.117 mmol) and L2CuI (99.00 mg, 0.106 mmol) were loaded into a 50 mL Schlenk flask. Under an ethylene atmosphere, 8 mL of ethylene saturated dichloromethane was added. Immediate color change from red solution to colorless solution was observed. The colorless solution was allowed to stir at room temperature for 3 hours. This mixture was cannula filtered through a Celite-packed frit to remove NaI. The colorless solution was dried under a stream of ethylene to obtain 4. X-ray quality crystals were obtained by layering dichloromethane solution of the abovementioned complex with hexane and cooling to −20 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 8.70 (s, 2H), 8.39 (s, 4H), 8.02 (s, 4H), 7.68 (s, 8H), 7.45 (s, 4H), 3.56 (s, 4H, C2H4). 19F NMR (471 MHz, CDCl3) δ (ppm) −57.08 (s), −62.05 (s), −62.85 (s). HR-MS [ESI, positive ion mode ESI-TOF]: m/z for [C32H14N2CuF18]+ 831.0165 (predicted), 802.9867 (found, matches ethylene dissociated [C30H10N2CuF18]+ 802.9852).
Catalytic studies
To a mixture of adamantane (232 mg, 1.70 mmol, 5.00 equiv.) and a catalyst (1 mol%, 0.01 equiv.) in a 50 mL Schlenk flask, was added anhydrous dichloromethane (12.5 mL) under nitrogen atmosphere. To the resulting solution trifluorodiazoethane (1.36 mL of 0.25 M solution in dichloromethane,0.34 mmol, 1.00 equiv.) was added with an aid of a syringe pump over the period of 30 min. After the addition was complete, stirring was continued for an additional 3 hours. Yields of the 3° and 2° insertion products were determined using 19F NMR data55 (by comparing to previous reports) and 1,3,5-tris(trifluoromethyl)benzene was used as an internal standard. 19F NMR (273 MHz, CDCl3) δ (ppm) −58.52 (t, J = 12.4 Hz, 1-(2,2,2-trifluoroethyl)adamantane), −63.88 (t, J = 11.7 Hz, 2-(2,2,2-trifluoroethyl)adamantane).X-ray structure determinations
A suitable crystal covered with a layer of hydrocarbon/Paratone-N oil was selected and mounted on a Cryo-loop, and immediately placed in the low temperature nitrogen stream. The X-ray intensity data of [L1Cu(C2H4)][SbF6] and [L1Ag(C2H4)][SbF6] were measured at 100 K, on a SMART APEX II CCD area detector system equipped with an Oxford Cryosystems 700 series cooler, a graphite monochromator, and a Mo Kα fine-focus sealed tube (λ = 0.71073 Å). The X-ray intensity data of [L1Au(C2H4)][SbF6] and [L2Cu(C2H4)][{3,5-(CF3)2C6H3}4B] were measured at 100 K on a Bruker D8 Quest equipped with a PHOTON II 7 CPAD detector and an Oxford Cryosystems 700 series cooler, a Triumph monochromator, and a Mo Kα fine-focus sealed tube (λ = 0.71073 Å). Data were processed using the Bruker Apex program suite. Absorption corrections were applied by using SADABS.58 Initial atomic positions were located by SHELXT,59 and the structures of the compounds were refined by the least-squares method using SHELXL60 within Olex2 GUI.61 All the non-hydrogen atoms were refined anisotropically. Hydrogen atoms, except those on ethylene moiety of [L1Cu(C2H4)][SbF6], were included at calculated positions and refined using appropriate riding models. Ethylene hydrogen atoms of [L1Cu(C2H4)][SbF6] were located in the difference map and were incorporated into the final refinement. The [L1Ag(C2H4)][SbF6] sits on a 2-fold rotation axis. The unit cell of [L1Ag(C2H4)][SbF6] contained a badly disordered, partially occupied hexane molecule, that could not be modeled satisfactorily. It was, therefore, removed from the electron density map using Olex2 mask routine. The [L1Au(C2H4)][SbF6] crystallizes in the Pna21 space group with two molecules in the asymmetric unit. It shows racemic twinning which was resolved satisfactorily. X-ray structural figures were generated using Olex2.61 The CCDC 2334609–2334612† files contain the supplementary crystallographic data. These data files have been deposited at the Cambridge Crystallographic Data Centre (CCDC), 12 Union Road, Cambridge, CB2 1EZ, UK. Additional details are provided in ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the support from National Science Foundation under grant number (CHE-1954456, HVRD) and American Floral Endowment (HVRD).References
- G. Accorsi, A. Listorti, K. Yoosaf and N. Armaroli, Chem. Soc. Rev., 2009, 38, 1690–1700 RSC.
- P. G. Sammes and G. Yahioglu, Chem. Soc. Rev., 1994, 23, 327–334 RSC.
- A. Bencini and V. Lippolis, Coord. Chem. Rev., 2010, 254, 2096–2180 CrossRef CAS.
- A. T. Bui and F. N. Castellano, in Comprehensive Coordination Chemistry III, ed. E. C. Constable, G. Parkin and L. Que Jr, Elsevier, Oxford, 2021, pp. 78–89, DOI:10.1016/B978-0-08-102688-5.00068-4.
- D. Felder, J.-F. Nierengarten, F. Barigelletti, B. Ventura and N. Armaroli, J. Am. Chem. Soc., 2001, 123, 6291–6299 CrossRef CAS PubMed.
- M. W. Blaskie and D. R. McMillin, Inorg. Chem., 1980, 19, 3519–3522 CrossRef CAS.
- N. Armaroli, Chem. Soc. Rev., 2001, 30, 113–124 RSC.
- J. Beaudelot, S. Oger, S. Peruško, T.-A. Phan, T. Teunens, C. Moucheron and G. Evano, Chem. Rev., 2022, 122, 16365–16609 CrossRef CAS PubMed.
- D. V. Scaltrito, D. W. Thompson, J. A. O'Callaghan and G. J. Meyer, Coord. Chem. Rev., 2000, 208, 243–266 CrossRef CAS.
- C.-W. Chan, W.-T. Wong and C.-M. Che, Inorg. Chem., 1994, 33, 1266–1272 CrossRef CAS.
- J. L. Barilone, J. Tůma, S. Brochard, K. Babková and M. Krupička, ACS Omega, 2022, 7, 6510–6517 CrossRef CAS PubMed.
- Y. Zhang, R. P. Hsung, M. R. Tracey, K. C. M. Kurtz and E. L. Vera, Org. Lett., 2004, 6, 1151–1154 CrossRef CAS PubMed.
- Z. Li, D. e. A. Capretto, R. Rahaman and C. He, Angew. Chem., Int. Ed., 2007, 46, 5184–5186 CrossRef CAS PubMed.
- J. W. Rigoli, C. D. Weatherly, J. M. Alderson, B. T. Vo and J. M. Schomaker, J. Am. Chem. Soc., 2013, 135, 17238–17241 CrossRef CAS PubMed.
- F. Wu, J. Xie and Z. Zhu, Appl. Organomet. Chem., 2020, 34, e5926 CrossRef CAS.
- C. W. Hamilton, D. S. Laitar and J. P. Sadighi, Chem. Commun., 2004, 1628–1629 RSC.
- J. Wang, Y. Hu, Q. Zhou, L. Hu, W. Fu and Y. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 44466–44473 CrossRef CAS PubMed.
- Y. Yang, P. Antoni, M. Zimmer, K. Sekine, F. F. Mulks, L. Hu, L. Zhang, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2019, 58, 5129–5133 CrossRef CAS PubMed.
- P. Alreja and N. Kaur, RSC Adv., 2016, 6, 23169–23217 RSC.
- C. S. Smith, C. W. Branham, B. J. Marquardt and K. R. Mann, J. Am. Chem. Soc., 2010, 132, 14079–14085 CrossRef CAS PubMed.
- D. S. Sigman, Acc. Chem. Res., 1986, 19, 180–186 CrossRef CAS.
- S. Masuri, P. Vanhara, M. G. Cabiddu, L. Moran, J. Havel, E. Cadoni and T. Pivetta, Molecules, 2022, 27, 49 CrossRef CAS PubMed.
- J. M. Veal and R. L. Rill, Biochemistry, 1991, 30, 1132–1140 CrossRef CAS PubMed.
- M. C. Aragoni, E. Podda, V. Caria, S. A. Carta, M. F. Cherchi, V. Lippolis, S. Murgia, G. Orru, G. Pippia, A. Scano, A. M. Z. Slawin, J. D. Woollins, A. Pintus and M. Arca, Inorg. Chem., 2023, 62, 2924–2933 CrossRef CAS PubMed.
- M. A. Cinellu, L. Maiore, M. Manassero, A. Casini, M. Arca, H.-H. Fiebig, G. Kelter, E. Michelucci, G. Pieraccini, C. Gabbiani and L. Messori, ACS Med. Chem. Lett., 2010, 1, 336–339 CrossRef CAS PubMed.
- H. V. R. Dias, S. A. Richey, H. V. K. Diyabalanage and J. Thankamani, J. Organomet. Chem., 2005, 690, 1913–1922 CrossRef CAS.
- T. T. Ponduru, Z. Sun, T. R. Cundari and H. V. R. Dias, ChemCatChem, 2019, 11, 4966–4973 CrossRef CAS.
- A. Noonikara-Poyil, S. G. Ridlen and H. V. R. Dias, Inorg. Chem., 2020, 59, 17860–17865 CrossRef CAS PubMed.
- A. Noonikara-Poyil, A. Munoz-Castro, A. Boretskyi, P. K. Mykhailiuk and H. V. R. Dias, Chem. Sci., 2021, 12, 14618–14623 RSC.
- A. S. Asundi, A. Noonikara-Poyil, V. Q. H. Phan, H. V. R. Dias and R. Sarangi, Inorg. Chem., 2023, 62, 19298–19311 CrossRef CAS PubMed.
- M. A. Omary, M. A. Rawashdeh-Omary, H. V. K. Diyabalanage and H. V. R. Dias, Inorg. Chem., 2003, 42, 8612–8614 CrossRef CAS PubMed.
- H. V. R. Dias and H.-L. Lu, Inorg. Chem., 2000, 39, 2246–2248 CrossRef CAS PubMed.
- H. V. R. Dias and J. D. Gorden, Inorg. Chem., 1996, 35, 318–324 CrossRef CAS PubMed.
- A. Noonikara-Poyil, S. G. Ridlen, I. Fernández and H. V. R. Dias, Chem. Sci., 2022, 13, 7190–7203 RSC.
- A. Noonikara-Poyil, H. Cui, A. A. Yakovenko, P. W. Stephens, R.-B. Lin, B. Wang, B. Chen and H. V. R. Dias, Angew. Chem., Int. Ed., 2021, 60, 27184–27188 CrossRef CAS PubMed.
- A. Noonikara-Poyil, H. Cui, B. Wang, Y. Shi, B. Chen and H. V. R. Dias, Small, 2023, 19, 2206984 CrossRef CAS PubMed.
- J. Mehara, B. T. Watson, A. Noonikara-Poyil, A. O. Zacharias, J. Roithova and H. V. R. Dias, Chem. – Eur. J., 2022, 28, e202103984 CrossRef CAS PubMed.
- B. T. Watson, M. Vanga, A. Noonikara-Poyil, A. Muñoz-Castro and H. V. R. Dias, Inorg. Chem., 2023, 62, 1636–1648 CrossRef CAS PubMed.
- M. Vanga, A. Noonikara-Poyil, J. Wu and H. V. R. Dias, Organometallics, 2022, 41, 1249–1260 CrossRef CAS.
- A. Noonikara-Poyil, A. Munoz-Castro and H. V. R. Dias, Molecules, 2022, 27, 16 CrossRef CAS PubMed.
- S. G. Ridlen, N. V. Kulkarni and H. V. R. Dias, Inorg. Chem., 2017, 56, 7237–7246 CrossRef CAS PubMed.
- A. H. Elashkar, D. Parasar, A. Munoz-Castro, C. M. Doherty, M. G. Cowan and H. V. R. Dias, ChemPlusChem, 2021, 86, 349 CrossRef CAS.
- H. V. R. Dias and J. Wu, Eur. J. Inorg. Chem., 2008, 2008, 509–522 CrossRef.
- H. Masuda, N. Yamamoto, T. Taga, K. Machida, S. Kitagawa and M. Munakata, J. Organomet. Chem., 1987, 322, 121–129 CrossRef CAS.
- J. Zhang, R.-G. Xiong, J.-L. Zuo, C.-M. Che and X.-Z. You, J. Chem. Soc., Dalton Trans., 2000, 2898–2900 RSC.
- M. A. Cinellu, M. Arca, F. Ortu, S. Stoccoro, A. Zucca, A. Pintus and L. Maiore, Eur. J. Inorg. Chem., 2019, 2019, 4784–4795 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- L. Kohler, R. G. Hadt, D. Hayes, L. X. Chen and K. L. Mulfort, Dalton Trans., 2017, 46, 13088–13100 RSC.
- M. Fianchini, C. F. Campana, B. Chilukuri, T. R. Cundari, V. Petricek and H. V. R. Dias, Organometallics, 2013, 32, 3034–3041 CrossRef CAS.
- H. V. R. Dias, M. Fianchini, T. R. Cundari and C. F. Campana, Angew. Chem., Int. Ed., 2008, 47, 556–559 CrossRef CAS PubMed.
- K. Klimovica, K. Kirschbaum and O. Daugulis, Organometallics, 2016, 35, 2938–2943 CrossRef CAS PubMed.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- M. Vanga, A. Muñoz-Castro and H. V. R. Dias, Dalton Trans., 2022, 51, 1308–1312 RSC.
- T. V. Le, G. G. Ramachandru and O. Daugulis, Chem. – Eur. J., 2024, e202303190 CrossRef CAS PubMed.
- K. Klimovica, J. X. Heidlas, I. Romero, T. V. Le and O. Daugulis, Angew. Chem., Int. Ed., 2022, 61, e202200334 CrossRef CAS PubMed.
- O. Green, B. A. Gandhi and J. N. Burstyn, Inorg. Chem., 2009, 48, 5704–5714 CrossRef CAS PubMed.
- S. Hyde, J. Veliks, B. Liégault, D. Grassi, M. Taillefer and V. Gouverneur, Angew. Chem., Int. Ed., 2016, 55, 3785–3789 CrossRef CAS PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2334609–2334612. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00822g |
| This journal is © The Royal Society of Chemistry 2024 |