
Spin crossover of a Fe(II) mononuclear complex induced by intermolecular factors involving chloride and solvent ordering†
Kenneth
Zhang‡
a,
Matthew J.
Wallis‡
a,
Alexander R.
Craze
b,
Shinya
Hayami
 c,
Hyunsung
Min
ad,
Daniel J.
Fanna
d,
Mohan M.
Bhadbhade
e,
Ruoming
Tian
e,
Christopher E.
Marjo
e,
Leonard F.
Lindoy
f and
Feng
Li
*a
c,
Hyunsung
Min
ad,
Daniel J.
Fanna
d,
Mohan M.
Bhadbhade
e,
Ruoming
Tian
e,
Christopher E.
Marjo
e,
Leonard F.
Lindoy
f and
Feng
Li
*a
aSchool of Science, Western Sydney University, Locked Bag 1797, Penrith, NSW, Australia. E-mail: Feng.Li@westernsydney.edu.au
bDepartment of Chemistry, University of Oxford, Oxford OX1 3TA, UK
cDepartment of Chemistry, Graduate School of Science and Technology, Kumamoto University, 2-39-1 Kurokami, Chuo-ku, Japan
dAdvanced Materials Characterisation Facility, Western Sydney University, Locked Bag 1797, Penrith, NSW, Australia
eMark Wainwright Analytical Centre, University of New South Wales, Kensington, NSW, Australia
fSchool of Chemistry, The University of Sydney, NSW 2006, Australia
First published on 3rd May 2024
Abstract
Two new salts of a mononuclear tripodal Fe(II) complex were prepared, using ClO4− and Cl−. The ClO4− sample (1) remained HS at low temperatures, similar to the previously reported BF4− analogue. Crystallising with the Cl− anion (2) led to a markedly different crystal packing arrangement, and engendered SCO activity. This has been correlated to the lower crystal packing density in 2 and the coordination complex conformational differences arising due to the packing motifs of 1 and 2. Further, solvent ordering effects have been proposed to facilitate spin transition behaviour in 2.
Introduction
Materials that exhibit spin crossover (SCO) continue to receive extensive multidisciplinary attention1–5 and have demonstrated potential utility in applications such as sensing, displays and data storage.2,5–9 Transition metals with electron configurations of d4–d7, under an appropriately balanced ligand field, may undergo a transition between the low-spin (LS) and the high-spin (HS) states, often as a result of perturbations in temperature, pressure or light.1,6,7,10 Six-coordinate complexes of iron(II) with nitrogen donor ligands (Fe(II)N6) characteristically experience a large modification of the magnetic moment and major structural changes upon switching between the diamagnetic LS 1A1 (S = 0) and the paramagnetic HS 5T2 (S = 2) states.4,11 As a result, complexes with an Fe(II)N6 coordination sphere have commonly been studied, often producing materials with cooperative spin-transitions (ST) and interesting structural chemistry.4,11Contrasting with SCO behaviour in the solution state, the SCO transition characteristics are usually drastically influenced in the solid state by the supramolecular environment. Thus, by ‘tuning’ factors such as the counter-ion identity12–18 and/or the presence of lattice solvent molecules,16,19,20 potentially important properties such as the abruptness and completeness of a spin transition (ST), the transition temperature (T1/2) and the number of SCO steps can be modified.13,15,17–23 Further to this is the effect of co-crystallising other components on the material's crystal structure,13,15,17,24 as varied crystal packing motifs will often impose different steric constraints on the SCO complexes, which need to be afforded enough steric freedom if a spin transition is to occur. Previous studies have shown that the nature of a SCO material can be altered by the size and electronic characteristics of counter-ions present, while particular studies have demonstrated a change of counter-ion can completely change the crystal packing arrangement, thus drastically altering the associated magnetic behaviour.12,15,22–26 Further, the degree of solvation often plays a crucial role in determining the supramolecular environment, and thus also can be used to modify the magnetic behaviour of SCO materials.
The loss of solvent within a crystal lattice affects its constituent intermolecular interactions and may have drastic effects on the SCO behaviour of compounds, for example, due to the shrinkage of the lattice volume27,28 and in some cases a change of phase.23,29,30 The loss of solvation may influence the cooperativity of the crystal system in various ways, with reports of magnetic switching behaviour being either enhanced or quenched. Enhancements of magnetism from desolvation may occur from enhanced elastic cooperativity resulting in a denser crystal lattice,23,27,29,31 or by lowering of the steric hindrance between SCO complexes and solvent, enabling intramolecular adaptations typical of ST.4 On the other hand, quenching of magnetism may be caused by the formation of anti-cooperative interactions,19,30,32 or by an increase in steric hindrance in the crystal lattice between adjacent complexes and/or counter-ions.28,33–35 The effect of desolvation on a SCO system is heavily dependent on the contacts formed between the solvent and other units in the crystal and, in particular, the position of the solvent in the lattice relative to the SCO complexes.
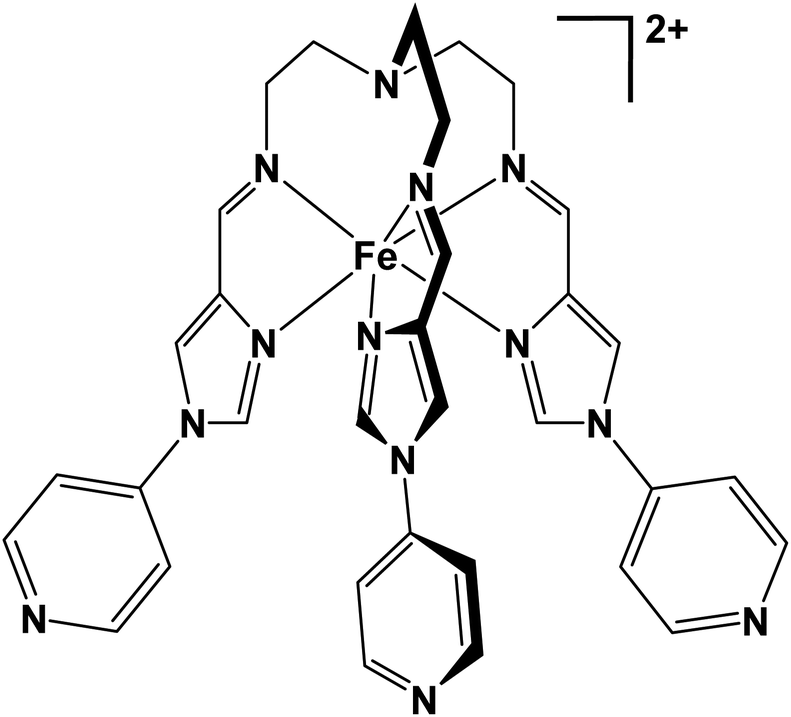
Tripodal metalloligands incorporating Fe(II) and the triethylamine linker moiety have been widely investigated for their SCO capabilities. Often, these complexes bear coordinating domains such as imidazole-imine18,36–38 or thiazole-imine.39–41 We have recently reported the application of a similar semi-rigid tripodal metalloligand [FeL]2+ (see below) for the self-assembly of heteronuclear tetradecanuclear cubic cages.42,43 When [FeL]2+ was combined with a secondary metal ion, cubic cages were formed, whereby eight Fe(II) ions occupy the corners of the cube, and six secondary metal ions are arranged on the cubic faces. The [Fe8Pd6L8]28+ cubic cage synthesised from [FeL](BF4)2 did not exhibit SCO, (and neither did the [FeL](BF4)2 metalloligand). When the identity of the face-occupying Pd(II) was replaced with Ni(II), forming [Fe8Ni6L8(CH3CN)12]28+, the product was observed to undergo SCO.
In this work, in order to gain further insight into the emergence of SCO in this system, we focus on the metalloligand material [FeL]X2 (where X = ClO4− or Cl−), and how SCO can be elicited in this building block, rather than the more complex cage architecture. The material [FeL](ClO4)2 (1) portrayed similar magnetic and structural behaviour to the [FeL](BF4)2 metalloligand species described above, not exhibiting SCO. Several key similarities in regard to important intermolecular parameters and the crystallographic packing motifs were observed between these two HS species. On the other hand, [FeL]Cl2 (2) exhibited a spin transition that was solvent dependant. The transition was two-step in the solvated and desolvated samples. Additionally, the residual HS fraction at low temperatures was higher in the desolvated sample. Magnetostructural correlations are presented, to give possible explanations as to how the [FeL]X2 material becomes SCO active when the identity of the counter-ion X is changed to Cl−. These materials were characterised by CHN, SEM-EDS, HR ESI-MS, TGA-DSC, PXRD, magnetometry and variable temperature single crystal X-ray diffraction (VT-SCXRD).
Results and discussion
Materials and methods
All reactions were conducted under an inert atmosphere. Ligand (L) was synthesised as previously described.42 All other reagents were used as received.Caution! Perchlorate salts are highly explosive and should be handled with care and in small amounts.
Physical measurements
Simultaneous thermal analysis (STA) experiments were conducted on a Netzsch STA-449C Jupiter instrument. STA measurements were acquired using argon for both the protective and purge gases at a flow rate of 25 mL min−1. The sample was weighed into a pierced aluminium crucible and measured between 303–500 K at a heating rate of 10 K min−1.
Powder X-ray diffraction measurements were conducted on a Bruker D8 ADVANCE X-ray diffractometer fitted with a Cu source (Cu Kα1 at 1.54 Å). The tube accelerating voltage and current were set to 40 kV and 40 mA, respectively. A LYNXEYE XE-T position sensitive detector (PSD) with energy discriminator settings optimised to reject Fe fluorescence was employed. All scans were collected at room temperature in Bragg–Brentano geometry. The scan for 1 was conducted with a fixed divergent slit opening of 0.16°, a 5–90° 2θ scan range with a step size of 0.005° and a dwell time of 0.5 seconds per step. The scan for air dried sample of 2 (2·6.5H2O) was conducted with motorised slits on primary optics set to fixed illumination mode with sample beam illumination set to 10 mm. Samples were scanned at room temperature across a 2.5–50°2θ scan range with a step size of 0.02° and a dwell time of 0.25 seconds per step. These scan parameters were optimised for this sample to achieve better intensity and lower amount of sample degradation. Details of sample preparation and data processing are available in the ESI (S1†).
Magnetic susceptibility measurements were performed using a Quantum Design SQUID magnetometer calibrated against a standard palladium sample under an applied field of 0.5 T. Air dried sample (2·6.5H2O) was used for measurement. Magnetic susceptibility measurements were conducted between 5 and 400 K at scan rates of 1 and 4 K min−1 in heating and cooling modes. Desolvation of the air-dried sample (2·6.5H2O) was achieved by holding the samples at 400 K for 60 minutes in situ, after which additional measurements were performed in heating and cooling modes at 1 and 4 K min−1. Light-induced excited spin state trapping (LIESST) studies were performed with red (800 nm) and green (532 nm) laser irradiation on the air dried sample. The sample was irradiated with one excitation wavelength at 5 K until the magnetic signal saturation and then heated at 1 K min−1.
Single crystal data (SCXRD) for all complexes were collected at the Australian Synchrotron from the MX1 beamline using silicon double crystal monochromated radiation (λ = 0.71073 Å). Variable temperature SCXRD experiments were conducted on crystals (2-CH3OH) from 100 K to 300 K at 50 K intervals. All data sets consisted of two collections consisting of a sweep through θ of 360° but differing by the setting of κ either 0° or 180°. XDS44 software at the Australian Synchrotron was utilised for merging, data integration, processing, scaling and the merging of raw datasets. Absorption corrections were applied using SADABS.45 The structures were solved and refined using a suite of ShelX programs46,47via the Olex248 interface. Octahedral distortion parameters were calculated using OctaDist.49 The crystallographic data in CIF format has been deposited at the Cambridge Crystallographic Data Centre with CCDC numbers 2338622–2338627.†
Synthesis and characterisation
The synthesis of L has been reported previously by our group.42 The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 reaction of L with FeX2 (X = ClO4− and Cl−) in CH3OH resulted in the formation of [FeL](ClO4)2 (1) and [FeL]Cl2 (2) respectively. For 1, the reaction was left for 1 hour, during which time orange crystalline precipitate formed. The precipitate was isolated using gravity filtration, air-dried, and collected. The reaction of 2 for 1 hour resulted in a reddish orange solution. This was subjected to vapour diffusion of diethyl ether which yielded reddish orange crystals of 2 (2-CH3OH), which were air dried for bulk analysis. Both samples were utilised for the characterisation described herein. HR ESI-MS, scanning SEM-EDS and SCXRD (see below) confirmed the formation of 1 and 2. HR ESI-MS showed peaks that fit well with the simulated m/z peaks of 1 and 2 (S3) while SEM-EDS portrayed that the product is homogenous and identified the expected elements for the metal complexes (S3). Powder X-ray diffraction (PXRD) patterns of 1 correspond well to the simulated single-crystal powder pattern confirming that the air-dried sample is pure and representative through the single crystal structures (Fig. S1†). CHN data indicates the air-dried sample (2·6.5H2O) occurs with 6.5 H2O molecules per formula unit, differing from the findings of SCXRD data in 2-CH3OH. STA analysis (S4) was in accord with the CHN elemental analysis. PXRD indicates that the crystal packing of air-dried 2·6.5H2O is significantly different to the simulated powder pattern of the single crystal (2-CH3OH) at 300 K, indicating that a phase change may have occurred upon air drying this sample (Fig. S2†).
:1 reaction of L with FeX2 (X = ClO4− and Cl−) in CH3OH resulted in the formation of [FeL](ClO4)2 (1) and [FeL]Cl2 (2) respectively. For 1, the reaction was left for 1 hour, during which time orange crystalline precipitate formed. The precipitate was isolated using gravity filtration, air-dried, and collected. The reaction of 2 for 1 hour resulted in a reddish orange solution. This was subjected to vapour diffusion of diethyl ether which yielded reddish orange crystals of 2 (2-CH3OH), which were air dried for bulk analysis. Both samples were utilised for the characterisation described herein. HR ESI-MS, scanning SEM-EDS and SCXRD (see below) confirmed the formation of 1 and 2. HR ESI-MS showed peaks that fit well with the simulated m/z peaks of 1 and 2 (S3) while SEM-EDS portrayed that the product is homogenous and identified the expected elements for the metal complexes (S3). Powder X-ray diffraction (PXRD) patterns of 1 correspond well to the simulated single-crystal powder pattern confirming that the air-dried sample is pure and representative through the single crystal structures (Fig. S1†). CHN data indicates the air-dried sample (2·6.5H2O) occurs with 6.5 H2O molecules per formula unit, differing from the findings of SCXRD data in 2-CH3OH. STA analysis (S4) was in accord with the CHN elemental analysis. PXRD indicates that the crystal packing of air-dried 2·6.5H2O is significantly different to the simulated powder pattern of the single crystal (2-CH3OH) at 300 K, indicating that a phase change may have occurred upon air drying this sample (Fig. S2†).
Magnetic susceptibility
Magnetic susceptibility studies were conducted on air-dried samples of 2·6.5H2O in the range 5–400 K with heating and cooling cycles of 1 and 4 K min−1. Increasing the scan rate from 1 K min−1 to 4 K min−1 resulted in a higher residual magnetic moment at lower temperatures for both the solvated and desolvated samples. Heating and cooling cycles revealed full reversibility of the spin transition in all cases (Fig. S7†).At a scan rate of 4 K min−1, the air-dried sample (2·6.5H2O) exhibits an incomplete two-step ST (Fig. 1) that occurs between 362 to 90 K. The first step occurs with a TSCO value of 292 K (2.61 cm3 K mol−1) (Fig. S8†). Between 250 and 190 K the profile adopts a shallow gradient between the two steps. The second spin transition event occurs with a TSCO value at 133 K (1.88 cm3 K mol−1). At temperatures under 100 K, the curve flattens out with an approximate residual χMT of 1.57 cm3 K mol−1, indicating a residual HS fraction of ∼53%. Desolvation (desolv. 2) further reduced the completeness of the transition. Polynomial fitting suggests two-step dynamics are maintained, with TSCO temperatures of 272 K (2.68 cm3 K mol−1) and 150 K (2.39 cm3 K mol−1) (Fig. S9†) at a scan rate of 4 K min−1. The higher residual magnetic susceptibility of ∼2.34 cm3 K mol−1 at low temperatures, corresponds to a residual HS fraction of ∼80% in desolv. 2. Additionally, LIESST studies were performed, indicating that 2·6.5H2O experiences photomagnetic behaviour in response to irradiation with red (800 nm) and green (532 nm) light (Fig. S10†).
 | ||
| Fig. 1 Magnetic susceptibility plot for air-dried (2·6.5H2O) and desolvated (desolv. 2) samples at a heating rate of 4 K min−1. | ||
Single crystal X-ray diffraction
1 packs in the monoclinic P21/c space group with one metal complex, two perchlorate anions and one methanol per asymmetric unit, with no additional voids found. All non-hydrogen atoms were refined anisotropically and hydrogen atoms were fixed using a riding model.All structures obtained for single crystal (2-CH3OH) were solved in the monoclinic C2/c space group. At 100 K, one metal complex and two chloride anions were found, as well as several solvent molecules. Two well-ordered methanols and one disordered methanol were found, as well as two water molecules in occupancies of 1 and 0.5. A solvent void occurring at a special position was masked, attributed to a water molecule with an occupancy of 0.8. This solvent composition was used to assign residual electron density in higher temperature structures, where thermal motion prohibited modelling of several solvent molecules (S6). In all structures, the metal complex and chloride anions were refined anisotropically and hydrogen atoms were fixed using a riding model. The restraints DFIX, DANG, FLAT and RIGU were used to model disordered pyridyl rings where appropriate. Solvent atoms were modelled anisotropically with hydrogen atoms fixed where possible, though in many cases were left isotropic and/or without hydrogen atoms fixed. DFIX and RIGU were applied to model disordered methanols.
Magnetostructural correlations
Single crystal X-ray diffraction (SCXRD) experiments confirmed the formation and structure of both the above compounds. 1 crystallises in the monoclinic P21/c space group with two ClO4− anions and one CH3OH per formula unit. The ClO4− anions are each observed to participate in three intermolecular O⋯π contacts and one O⋯HC contact with the triethylamine backbone and pyridine terminal groups of four adjacent [FeL]2+ cations (Table S3†). A distorted octahedral coordination sphere is formed around the central Fe(II) ion by three imidazolylimine groups with both Δ and Λ isomers found in the crystal lattice. The average Fe–N bond length of 2.21 Å and angular distortion parameters, Σ and Θ with values of 104.0° and 280.3° respectively (Table 1), are consistent with HS Fe(II) metal centres. Further, the distance between the apical nitrogen (N1) and Fe(II) metal centre is 2.947 Å and the average C–N1–C angle is 114.1° correlates to literature values of HS triethylamine-linked Fe(II) complexes.39 A series of key structural similarities between 1 and the previously reported HS metalloligand [FeL](BF4)2, aligns well with 1 remaining fully HS at 100 K. In both lattices, the crystal packing may be characterised by one-dimensional chains of tripodal complex ion units, arranged along the a-axis (Fig. 2). This arrangement is stabilised by intermolecular contacts between metal complex enantiomers. One Npyridine⋯HCimidazole contact forms between the distal pyridine N of the ligand B arm and the imidazole CH proximal to the coordination sphere on the ligand C arm of an adjacent unit (D⋯A distance 3.394 Å). Further, the pyridyl group of arm B is involved in an intermolecular π⋯π contact across an inversion centre. Another weaker dipole–dipole contact is observed between the distal pyridyl N of ligand C and the imidazole moiety of ligand arm B.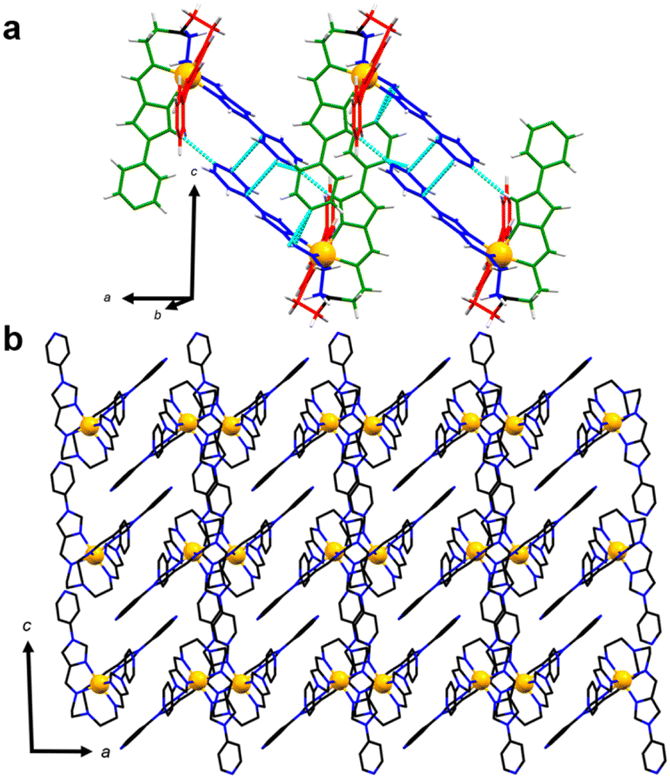 | ||
| Fig. 2 (a) Molecular structure of 1-CH3OH with intermolecular contacts illustrating N⋯HC and π⋯π interactions between enantiomeric pairs. Solvent and counter-ions are omitted for clarity. The colour codes for each ligand arm are red (A), blue (B) and green (C). The apical nitrogen is coloured black and hydrogens white. (b) Extended view of the packing motif along the b-axis highlighting the π⋯π interactions and the packing arrangement of dimers. | ||
| Sample | [FeL](BF4)2a | 1 | 2·3CH3OH, 2.3H2O (2-CH3OH) | ||||
|---|---|---|---|---|---|---|---|
| Temperature (K) | 100 | 100 | 100 | 150 | 200 | 250 | 300 |
| a [FeL](BF4)2 has been previously reported by our group, and is included here for structural comparison against the compounds studied in this work. | |||||||
| Average Fe⋯N bond length (Å) | 2.20 | 2.2054(15) | 1.982(22) | 1.983(20) | 1.994(17) | 2.056(19) | 2.152(38) |
| ζ (Å) | 0.16 | 0.155 | 0.031 | 0.027 | 0.027 | 0.043 | 0.025 |
| Σ (°) | 105.0 | 104.0 | 53.6 | 53.5 | 54.6 | 64.1 | 77.4 |
| Θ (°) | 280.4 | 280.3 | 174.3 | 174.4 | 179.0 | 208.4 | 246.4 |
| Volume (Å3) | 3806.1(12) | 3880.2(12) | 8289(5) | 8313(5) | 8427(5) | 8600(5) | 8598(5) |
| Packing coefficient (%) | 54.57 | 53.57 | 49.56 | 49.41 | 48.66 | 47.58 | 47.71 |
Previous investigations of the [Fe8Ni6L8(CH3CN)12]28+ coordination cage concluded that in order for the Fe(II) metalloligand to undergo SCO, the pyridyl termini must be able to spread apart from one another (see Fig. 3 for structural overlap). In the two structures that do not undergo SCO, 1 and its BF4− analogue, the packing arrangement is such that intermolecular contacts favour a more compact arrangement of the distal pyridyl units (specifically by N⋯HC and π⋯π contacts between B and C). The transition to LS may have a restrictive conformational barrier, or may be anticooperative with transitions in neighbouring units. Overall, the effect of these arrangements is stabilisation of the HS state in the P21/c lattices of 1 and [FeL](BF4)2, the ClO4− and BF4− analogues.
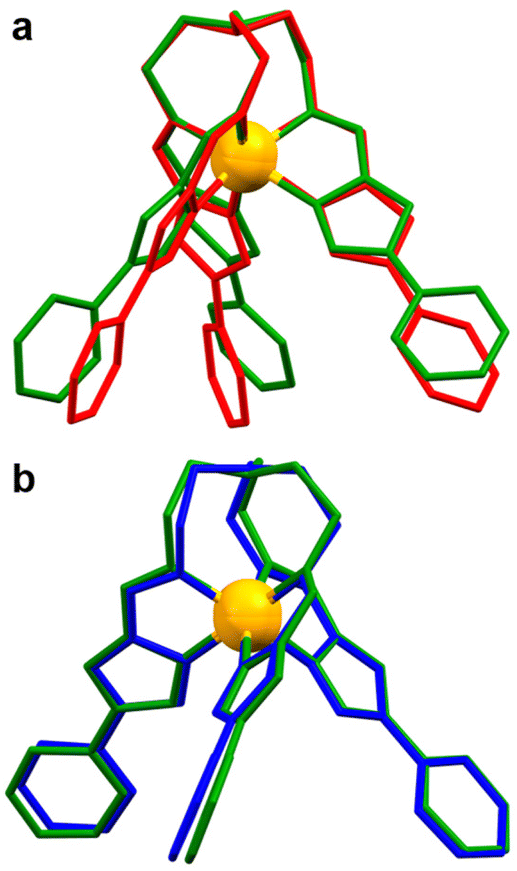 | ||
| Fig. 3 (a) An overlay of the crystal structure of 1 (red) with 2-CH3OH (green) at 300 K, (b) is an overlay of 2-CH3OH at 100 K (blue) and 300 K (green). Both overlays are presented with a side view of the [FeL]2+ cation. | ||
In order to probe how the structure of 2 changed as a result of SCO, VT-SCXRD between 100–300 K was conducted on the crystals (2-CH3OH). Similar to 1, a distorted octahedral coordination sphere around the Fe(II) centre is formed by three imidazolylimine groups and both optical isomers are present within the crystal lattice. The compound is observed to be in the monoclinic C2/c space group with one complete Fe(II) metal complex, two chloride anions, three methanol molecules and three water positions, adding up to 2.3 waters per formula unit. A solvent mask was applied in all structures (S6). Interestingly, the distance between Fe(II) and the apical nitrogen atom (N1) shortens from 3.489 Å to 3.222 Å across the temperature series 100–300 K. A tightening of the average C–N1–C from 120° to 117° is also observed. The changing of these parameters is caused by the transition towards HS. The lone pair of the apical nitrogen is directed towards the t2g orbitals of the metal centre, which bear a lower occupancy in the HS state.39
To note, fewer direct interactions between adjacent [FeL]2+ cations were observed, in comparison to the tight N⋯HC and π⋯π interactions observed in 1. The crystal lattice packs as 1D chains through Cl⋯HC hydrogen bonding along the b-axis in an undulating sequence of ΔΛ complexes. Hydrogen bonding between the terminal pyridines of ligand arms B and C (N4B⋯C1C) forms voids, which are continuous along the c-axis (Fig. 4) and contain all solvent molecules. Along the c-axis, Cl− ions hydrogen bond with solvent ions in the pore. Further, hydrogen bonding between tripodal complexes and Cl− anions links CH groups of imidazole and pyridyl moieties of adjacent complexes (Fig. 4d). The Cl− ion designated Cl1 links the imidazole CH proximal to the coordination sphere of arm A with the imidazole CH groups distal to the coordination sphere on arms B and C. Meanwhile, Cl2 links the imidazole CH distal to the coordination sphere of arm A to the imidazole proximal to the coordination sphere of arm C.
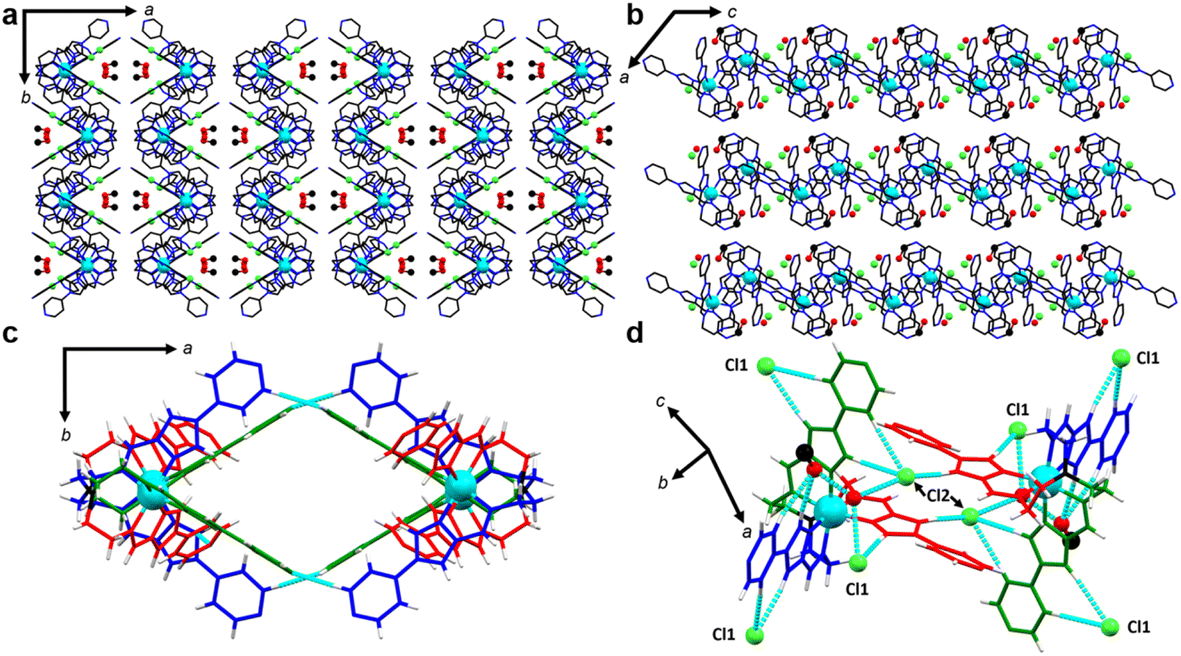 | ||
| Fig. 4 (a) The crystal packing motif of 2-CH3OH viewed along the c-axis illustrating the pore structure. (b) Crystal packing observed along the b-axis showing the void space, as well as the resolved counter-ion and solvent positions at 300 K. Colour code for (a) and (b): Fe (cyan), C (black), N (blue), O (red), Cl (green). Hydrogen atoms omitted for clarity. Anions and solvent atoms shown as balls with Cl (green), C (black) and O (red). (c) A closeup view on the formation of the major void space and the intermolecular interactions between adjacent metal complexes via ligand arms B and C. (d) A depiction of the hydrogen bonding between Cl− and [FeL]2+ cations. Colour code for c and d: Fe (cyan), ligand arm A (red), arm B (blue), arm C (green), H (white). Anions and solvent atoms shown as balls with Cl (green), C (black) and O (red). | ||
Beginning at 100 K, the average Fe–N bond lengths (1.982 Å) and octahedral distortion parameters Σ and Θ49 of 53.6° and 174.3°, respectively, are consistent with the Fe(II) metal centre being in the LS state. The octahedral distortion values remain consistent up to 200 K, implying that the ST occurs primarily between 200 and 300 K, with the most significant change in octahedral distortions occurring between 250–300 K. At 300 K, the metalloligand demonstrated larger octahedral distortion parameter values (Fe–N = 2.15 Å, Σ = 77.4° and Θ = 246.4°, Table 1). Comparison of the HS values observed in 1 to the 300 K values of the single crystal (2-CH3OH), illustrates that the latter material has not reached a fully HS state at 300 K. This is in accord with the magnetic susceptibility data of the air dried sample (2·6.5H2O), at which point χMT = 2.70 cm3 K mol−1. This indicates a mixed spin fraction is present in single crystals of 2-CH3OH at 300 K. Based on the average coordinate bond lengths at this temperature of 2.152 Å, with respect to those observed in the fully HS (2.205 Å) and fully LS structures (1.982 Å), we estimate that at 300 K a HS fraction of ∼76% persists in the crystal, which is in agreement with the HS fraction in magnetic susceptibility measurements of the air-dried material at 300 K. This means that the higher temperature step of the HS > LS transition cannot be structurally characterised with the available crystallographic data. Below we show our findings which we attribute to the magnetostructural dynamics of the lower temperature step of spin transition in 2 (Fig. 1).
Of note in the manifestation of SCO in this material is the lower packing density of complex ions in the crystal lattice of 2-CH3OH, with respect to those found in the BF4− and ClO4− analogues (Table 1). This was gauged by measuring the metal complex crystal packing coefficient, as calculated in Olex248 using the ‘molinfo’ command. This provides a measure of the percentage of the unit cell volume which is occupied by coordination complexes. The lower crystal packing coefficient in 2-CH3OH correlates to metalloligand complexes occupying less of the total lattice volume, suggesting that the metal complexes have more space to undergo the conformational changes which must occur upon spin transition (as seen in comparing HS and LS structures). Interestingly, the evolution of the crystal lattice parameters in 2-CH3OH was non linear, with the volumes of the 250 K (8600 Å3) and 300 K (8598 Å3) lattices being within experimental error of one another. This occurs primarily due to growth of the ac plane at 250 K, with the a- and c-axis lengths at 300 K of 32.281(7) Å and 20.748(4) Å growing to 32.450(7) Å and 20.800(4) Å.
Inductive effects from intermolecular interactions between complexes and counter-ions may contribute to the ligand field of the Fe(II) centres impacting the overall magnetic profile. However, analysis of contacts between Cl− anions and metalloligands across the experimental temperature range has revealed that contact distances remain relatively stable (Tables S4–8†) with maximum differences between all temperatures ranging from 0.01–0.13 Å. The largest difference in hydrogen bonding distance of 0.13 Å (donor⋯acceptor) was ascribed to the interaction between a pyridyl CH (C10B) and Cl1. In fact, only three of these hydrogen bond distances in total exhibited changes greater than 0.1 Å, attributed in each case to pyridyl CH's. This suggests these contacts change primarily as a result of pyridyl groups twisting with respect to their respective imidazole counterparts, as opposed to a large-scale supramolecular reconfiguration.
As in the air-dried material (2·6.5H2O), solvent appears to play a key role in the magnetic behaviour of single crystals (2-CH3OH). In the 300 K structure, a high degree of thermal disorder prohibited the modelling of all but two solvent molecules – one methanol which is hydrogen bonded to the imidazole CH proximal to the coordination sphere of arm B, and a water molecule which is likely involved in hydrogen bonding interactions with that methanol and both Cl− anions. Both of these solvents are modelled with ¾ occupancy at 300 K, affording Ueq values approximately 2–2.5 times the Ueq values of main residue atoms. At lower temperatures, the occupancy of these two solvent positions increases to 1. Two additional methanol molecules are able to be modelled with partial occupancies at 250 K, which reach full occupancy in the 200 K structure. These latter methanol molecules occupy a pocket in the pore structure which contacts the triethylamine linker components of four Fe(II) complexes (Fig. 5b). Concomitant with the ordering of solvent molecules, the spacing of metal complexes aligned approximately with the a-axis direction increases (Fig. 5c). This effect pushes complexes outward from the solvent pocket, causing the increase in the a-axis as the temperature is lowered to 250 K. As temperature is lowered further, this spacing steadily decreases with reduced thermal motion. Meanwhile, the contact between the enantiomeric pair interacting via ligand arms B and C (running along the a-axis), remains largely unchanged (Tables S4–8†).
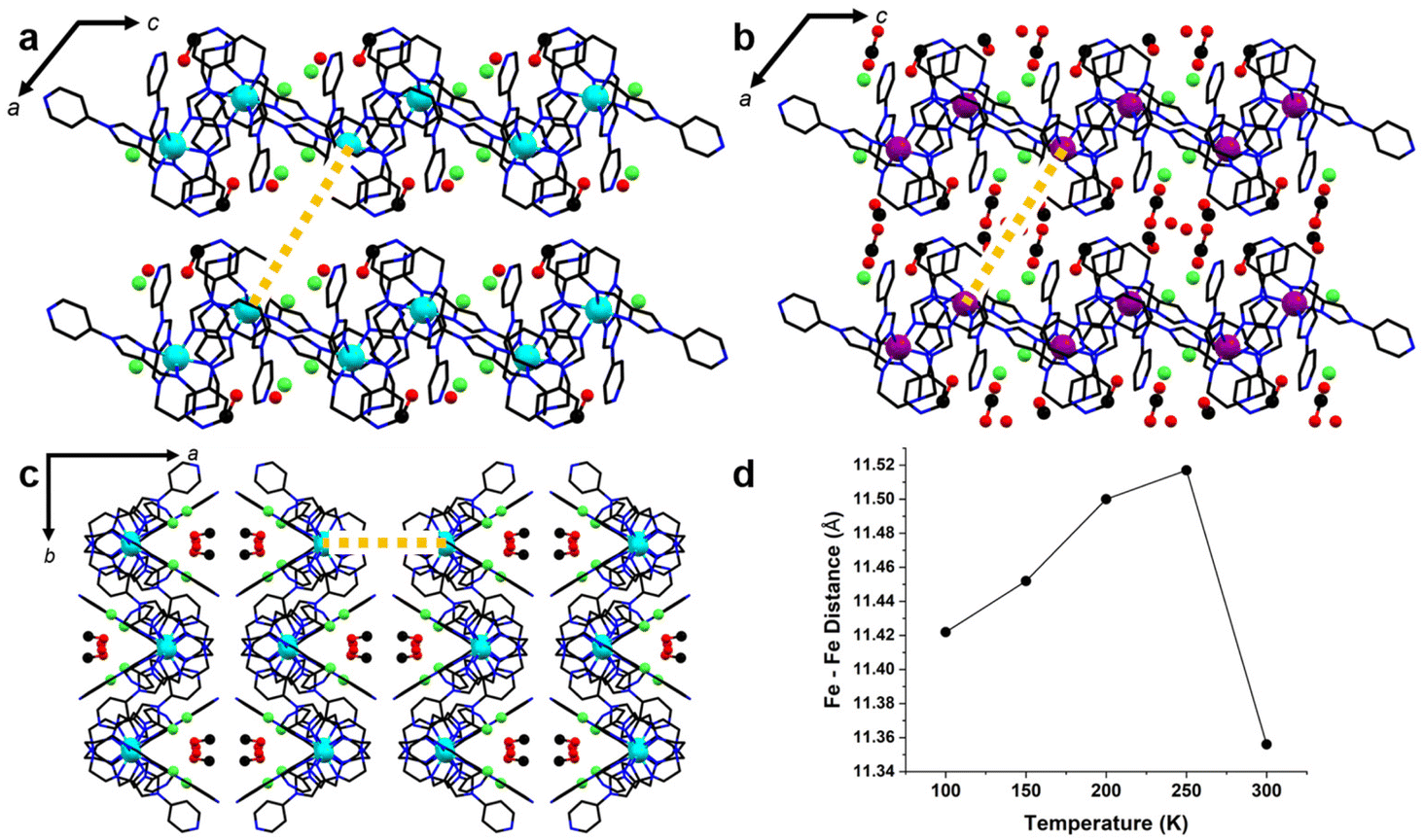 | ||
| Fig. 5 Effect of solvent ordering on the intermetallic Fe⋯Fe distance (gold dashed line) through the solvent pocket and along the a-axis. (a) 300 K structure (viewed along the b-axis) in which many solvent molecules in the pore could not be modelled, (b) 100 K structure (viewed along the b-axis), in which several methanol molecules along the Fe⋯Fe distance were modelled, (c) The same feature viewed along the c-axis, (d) plot of the intermetallic distance observed between between the two Fe(II) metal centres portrayed by (a)–(c). | ||
The effect of solvent ordering on the lattice positions of the SCO complexes affords more steric freedom to the terminal pyridyl groups on ligand arm A, providing more space to reconform as necessary for a transition to LS. This was quantified by calculating the mutual angles between the secondary bonding vectors of the terminal pyridyl units, defined as the interval between N and opposite C (S9). By measuring the angles between these intervals, we are able to quantify the degree of spread between the three different ligand arms, as we have done in our previous work.42,50
The mutual bond angles between the ligand arms in the HS trapped structure 1 have a much lower average value (55.8°) than the mixed spin structure of the single crystal (2-CH3OH) at 300 K (74.6°). As was found in our previous study36 of the coordination cage [Fe8Ni6L8(CH3CN)12]28+, a contributing factor to SCO behaviour in the [FeL]2+ metalloligand scaffold is the spread of the terminal pyridyl groups. The necessary distortions of the coordination sphere upon ST are more easily incorporated into the transitioning complexes with sufficient angular spread and steric freedom.
Regarding the SCO dynamics in the crystal (2-CH3OH), at 300 K the mutual bond angle (Fig. 6) between arms B and C is measured to be 74.2°, with slightly lower values at lower temperatures and a minimum value of 73.6° at 150 K. This implies that the terminal pyridyl groups of B and C slightly approach colinearity at lower temperatures. Meanwhile, the mutual bond angle values between A and B, as well as that between A and C both give their respective minimum values at 300 K, and both rise slightly to maximum values at 200 K, plateauing at lower temperatures (Fig. 6). Ultimately, these results indicate that the distortion of the octahedral coordination sphere on ST brings about a reconformation of ligand arms, and due to the contact strength between arms B and C, the relatively unhindered ligand arm A is more easily able to accommodate the change in coordination sphere size.
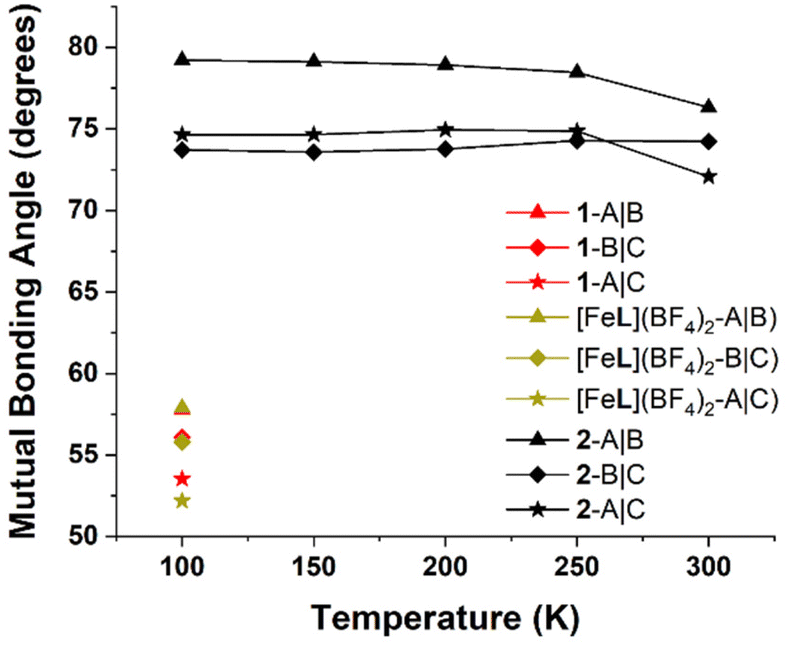 | ||
| Fig. 6 Mutual bonding angles of the ligand arms in 2-CH3OH from 100 to 300 K. The notation I|J refers to the angle between the secondary bonding vectors of the Ith and Jth ligand arms. For full details and calculated values, see section S9.† | ||
In the air-dried material 2·6.5H2O, it is likely that solvent ordering/interactions may be a contributing factor to the second spin transition event. The degree of solvation would certainly affect the pore structure in this material, and whether the crystallographic location of solvent pockets remains equivalent between the crystal (2-CH3OH) and the air-dried sample (2·6.5H2O) is ambiguous at this stage, as PXRD results demonstrate that a phase change occurs upon air drying (Fig. S2†). In the fully desolvated material (desolv. 2), the loss of solvent molecules may result in the pore structure collapsing, forcing the material to adopt a higher crystal packing coefficient and thus a higher degree of steric crowding. This may also force molecules in the lattice to adopt intermolecular contacts which are anticooperative with a spin transition to LS, as was noted in the structures of the BF4− and ClO4− analogues.
Conclusion
In an attempt to define structure–function correlations regarding the nature of SCO in a tripodal metalloligand used in the metal-templated self-assembly of heteronuclear coordination cages, two new salts of this previously investigated mononuclear Fe(II) complex have been synthesised and characterised. The ClO4− salt (1) crystallised in the same space group as the previously reported BF4− salt, remaining HS at low temperatures. This was found to be a result of tight interactions between adjacent pyridyl arms, driven by N⋯HC and π⋯π interactions, to provide a more tightly packed crystal lattice. On the other hand, the compound formed from crystallisation with Cl− as the counter-ion (2) resulted in a different crystal packing arrangement that enabled SCO. This was proposed to be due to more steric freedom being afforded to the coordination complexes in this case. Furthermore, solvent dependence was observed in the spin transition of 2. Single crystals (2-CH3OH) achieved full transition to LS while two-step behaviour was observed in the air-dried solvated material (2·6.5H2O) and fully desolvated 2 (desolv. 2), reaching completions of ∼47% and ∼20%, respectively. Future work will involve detailed structural characterisation of the air-dried material and potentially TIESST studies.Author contributions
K. Z. and H. M. performed the synthetic work and majority of the characterisation. K. Z. and M. J. W. solved and refined crystallographic data and M. J. W. performed structural analysis. K. Z., H. M., R. T., M. M. B. and C. E. M collected the SCXRD data. PXRD data was collected and refined by K. Z., H. S. M. and D. J. F. Magnetic susceptibility and CHN data was collected by S. H. The manuscript was drafted by K. Z., M. J. W. and A. R. C. All authors edited the manuscript. L. F. L and F. L. conceptualised the project and F. L. directed the work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research conduced was supported by Western Sydney University (WSU). The authors acknowledge the Advanced Materials Characterisation Facility (AMCF) located at WSU, Mark Wainwright Analytical Centre at UNSW and Kumamoto University where magnetic susceptibility and CHN was performed. The crystallographic experiments performed on the MX1 beamline at the Australian Synchrotron, Clayton, Victoria, Australia. We thank the Australian Synchrotron for travel support and their staff for assistance on the beamline.References
- P. Gütlich, A. Hauser and H. Spiering, Angew. Chem., Int. Ed. Engl., 1994, 33, 2024–2054 CrossRef.
- A. Bousseksou, G. Molnár, L. Salmon and W. Nicolazzi, Chem. Soc. Rev., 2011, 40, 3313–3335 RSC.
- K. S. Murray, The Development of Spin-Crossover Research, ed. M. A. Halcrow, Wiley-Blackwell, United Kingdom, 2013, vol. 1, pp. 1–54 Search PubMed.
- M. A. Halcrow, Chem. Soc. Rev., 2011, 40, 4119–4142 RSC.
- K. S. Kumar and M. Ruben, Coord. Chem. Rev., 2017, 346, 176–205 CrossRef.
- J. A. Real, A. B. Gaspar and M. C. Muñoz, Dalton Trans., 2005, 2062–2079 RSC.
- P. Gütlich and H. A. Goodwin, Spin Crossover in Transition Metal Compounds, Springer, 2004 Search PubMed.
- Z. Ni and M. P. Shores, J. Am. Chem. Soc., 2009, 131, 32–33 CrossRef CAS PubMed.
- C. Faulmann, K. Jacob, S. Dorbes, S. Lampert, I. Malfant, M. Doublet, L. Valade and J. A. Real, Inorg. Chem., 2007, 46, 8548–8559 CrossRef CAS PubMed.
- P. Gütlich, Y. Garcia and H. A. Goodwin, Chem. Soc. Rev., 2000, 29, 419–427 RSC.
- E. König, Structural Changes Accompanying Continuous and Discontinuous Spin-State Transitions, ed. S. J. Lippard, John Wiley & Sons, Inc., 1987, pp. 527–622 Search PubMed.
- Y. Luo, G. Wen, L. Gu, M. Wang and B. Sun, Polyhedron, 2017, 121, 101–106 CrossRef CAS.
- C. M. Klug, A. M. McDaniel, S. R. Fiedler, K. A. Schulte, B. S. Newell and M. P. Shores, Dalton Trans., 2012, 41, 12577–12585 RSC.
- C. Wu, J. Jung, P. K. Gantzel, P. Gϋtlich and D. N. Hendrickson, Inorg. Chem., 1997, 36, 5339–5347 CrossRef CAS.
- A. R. Craze, M. M. Bhadbhade, Y. Komatsumaru, C. E. Marjo, S. Hayami and F. Li, Inorg. Chem., 2020, 59, 1274–1283 CrossRef CAS PubMed.
- B. A. Leita, S. M. Neville, G. J. Halder, B. Moubaraki, C. J. Kepert, J. Létard and K. S. Murray, Inorg. Chem., 2007, 46, 8784–8795 CrossRef CAS PubMed.
- H. S. Scott, T. M. Ross, S. R. Batten, I. A. Gass, B. Moubaraki, S. M. Neville and K. S. Murray, Aust. J. Chem., 2012, 65, 874–882 CrossRef CAS.
- M. Yamada, H. Hagiwara, H. Torigoe, N. Matsumoto, M. Kojima, F. Dahan, J. Tuchagues, N. Re and S. Iijima, Chem. – Eur. J., 2006, 12, 4536–4549 CrossRef CAS PubMed.
- W. Zhang, F. Zhao, T. Liu, M. Yuan, Z. Wang and S. Gao, Inorg. Chem., 2007, 46, 2541–2555 CrossRef CAS PubMed.
- Z. Ni and M. P. Shores, Inorg. Chem., 2010, 49, 10727–10735 CrossRef CAS PubMed.
- K. S. Kumar, Y. Bayeh, T. Gebretsadik, F. Elemo, M. Gebrezgiabher, M. Thomas and M. Ruben, Dalton Trans., 2019, 48, 15321–15337 RSC.
- W. Phonsri, D. S. Macedo, C. G. Davies, G. N. L. Jameson, B. Moubaraki and K. S. Murray, Dalton Trans., 2017, 46, 7020–7029 RSC.
- U. Habarakada, T. Boonprab, P. Harding, K. S. Murray, W. Phonsri, S. M. Neville, M. Ahmed and D. J. Harding, Cryst. Growth Des., 2022, 22, 4895–4905 CrossRef CAS.
- Z. Yu, S. Zhao, Y. Wang, P. Xu, C. Qin, Y. Li, X. Zhou and S. Wang, Dalton Trans., 2021, 50, 15210–15223 RSC.
- J. A. Kitchen, N. G. White, G. N. L. Jameson, J. L. Tallon and S. Brooker, Inorg. Chem., 2011, 50, 4586–4597 CrossRef CAS PubMed.
- S. Athira, D. J. Mondal, S. Shome, B. Dey and S. Konar, Dalton Trans., 2022, 51, 16706–16713 RSC.
- L. J. K. Cook, R. Kulmaczewski, O. Cespedes and M. A. Halcrow, Chem. – Eur. J., 2015, 22, 1789–1799 CrossRef PubMed.
- X. Zhao, Y. Deng, J. Huang, M. Liu and Y. Zhang, Inorg. Chem. Front., 2024, 11, 808–816 RSC.
- R. Díaz-Torres, T. Boonprab, S. Gómez-Coca, E. Ruiz, G. Chastanet, P. Harding and D. J. Harding, Inorg. Chem. Front., 2022, 9, 5317–5326 RSC.
- H. Wang, C. Sinito, A. Kaiba, J. S. Costa, C. Desplanches, P. Dagault, P. Guionneau, J. Létard, P. Negrier and D. Mondieig, Chem. – Eur. J., 2014, 4927–4933 CAS.
- R. J. Archer, H. S. Scott, M. I. J. Polson, C. Mathonière, M. Rouzières, R. Clérac and P. E. Kruger, Chem. – Asian J., 2019, 14, 2225–2229 CrossRef CAS PubMed.
- W. Li, X. Li, K. Robeyns, M. Wolff, J. Kfoury, J. Oláh, R. Herchel, S. Demeshko, F. Meyer and Y. Garcia, Dalton Trans., 2024, 53, 1449–1459 RSC.
- F. Yang, W. Wu, Y. Wang, X. Chen, B. Liang, H. Mi, G. Zhang, X. Chen and Y. Shi, Cryst. Growth Des., 2021, 21, 6671–6683 CrossRef CAS.
- I. Šalitroš, O. Fuhr and M. Ruben, Materials, 2016, 9, 585 CrossRef PubMed.
- A. R. Craze, M. M. Bhadbhade, C. J. Kepert, L. F. Lindoy, C. E. Marjo and F. Li, Crystals, 2018, 8, 376 CrossRef.
- M. Seredyuk, M. C. Muñoz, V. Ksenofontov, P. Gütlich, Y. Galyametdinov and J. A. Real, Inorg. Chem., 2014, 53, 8442–8454 CrossRef CAS PubMed.
- M. Yamada, E. Fukumoto, M. Ooidemizu, N. Bréfuel, N. Matsumoto, S. Iijima, M. Kojima, N. Re, F. Dahan and J. Tuchagues, Inorg. Chem., 2005, 20, 6967–6974 CrossRef PubMed.
- G. Brewer, C. Luckett, L. May, A. M. Beatty and W. R. Scheidt, Inorg. Chim. Acta, 2004, 357, 2390–2396 CrossRef CAS.
- G. Brewer, Magnetochemistry, 2020, 6, 28 CrossRef CAS.
- R. Kulmaczewski, O. Cespedes and M. A. Halcrow, Inorg. Chem., 2017, 56, 3144–3148 CrossRef CAS PubMed.
- N. Struch, N. Wagner, G. Schnakenburg, R. Weisbarth, S. Klos, J. Beck and A. Lützen, Dalton Trans., 2016, 45, 14023–14029 RSC.
- H. Min, A. R. Craze, T. Taira, M. J. Wallis, M. M. Bhadbhade, R. Tian, D. J. Fanna, R. Wuhrer, S. Hayami, J. K. Clegg, C. E. Marjo, L. F. Lindoy and F. Li, Chemistry, 2022, 4, 535–547 CrossRef CAS.
- H. Min, A. R. Craze, M. J. Wallis, R. Tokunaga, T. Taira, Y. Hirai, M. M. Bhadbhade, D. J. Fanna, C. E. Marjo, S. Hayami, L. F. Lindoy and F. Li, Chem. – Eur. J., 2023, 29, e202203742 CrossRef CAS PubMed.
- W. Kabsch, J. Appl. Crystallogr., 1993, 26, 795–800 CrossRef CAS.
- SADABS, version 2014/5, Bruker AXS Inc., Madison, WI, USA, 2001 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- R. Ketkaew, Y. Tantirungrotechai, P. Harding, G. Chastanet, P. Guionneau, M. Marchivie and D. J. Harding, Dalton Trans., 2021, 50, 1086–1096 RSC.
- M. J. Wallis, H. Min, L. F. Lindoy and F. Li, Molecules, 2023, 28, 1404 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: HR ESI-MS, SEM-EDS, PXRD, STA, solvent masking details, intermolecular interaction tables, calculation of parameters and crystallographic data. CCDC 2338622–2338627. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00706a |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |