
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxygen sensitivity of [FeFe]-hydrogenase: a comparative study of active site mimics inside vs. outside the enzyme†
Shanika
Yadav
 a,
Rieke
Haas
b,
Esma Birsen
Boydas
c,
Michael
Roemelt
c,
Thomas
Happe
b,
Ulf-Peter
Apfel
*ad and
Sven T.
Stripp
*e
a,
Rieke
Haas
b,
Esma Birsen
Boydas
c,
Michael
Roemelt
c,
Thomas
Happe
b,
Ulf-Peter
Apfel
*ad and
Sven T.
Stripp
*e
aInorganic Chemistry I, Ruhr-Universität Bochum, Universitätsstraße 150, 44801 Bochum, Germany. E-mail: ulf.apfel@rub.de
bFaculty of Biology & Biotechnology, Ruhr-Universität Bochum, Universitätsstrasse 150, 44801 Bochum, Germany
cInstitute of Chemistry, Humboldt-Universität zu Berlin, Brook-Taylor Str.2, 12489, Berlin, Germany
dDepartment of Electrosynthesi, Fraunhofer UMSICHT, Osterfelder Str. 3, 46047 Oberhausen, Germany
eBiophysical Chemistry, Technical University Berlin, Strasse des 17. Juni 124, 10623 Berlin, Germany. E-mail: s.stripp@tu-berlin.de
First published on 26th June 2024
Abstract
[FeFe]-hydrogenase is nature's most efficient proton reducing and H2-oxidizing enzyme. However, biotechnological applications are hampered by the O2 sensitivity of this metalloenzyme, and the mechanism of aerobic deactivation is not well understood. Here, we explore the oxygen sensitivity of four mimics of the organometallic active site cofactor of [FeFe]-hydrogenase, [Fe2(adt)(CO)6−x(CN)x]x− and [Fe2(pdt)(CO)6−x(CN)x]x− (x = 1, 2) as well as the corresponding cofactor variants of the enzyme by means of infrared, Mössbauer, and NMR spectroscopy. Additionally, we describe a straightforward synthetic recipe for the active site precursor complex Fe2(adt)(CO)6. Our data indicate that the aminodithiolate (adt) complex, which is the synthetic precursor of the natural active site cofactor, is most oxygen sensitive. This observation highlights the significance of proton transfer in aerobic deactivation, and supported by DFT calculations facilitates an identification of the responsible reactive oxygen species (ROS). Moreover, we show that the ligand environment of the iron ions critically influences the reactivity with O2 and ROS like superoxide and H2O2 as the oxygen sensitivity increases with the exchange of ligands from CO to CN−. The trends in aerobic deactivation observed for the model complexes are in line with the respective enzyme variants. Based on experimental and computational data, a model for the initial reaction of [FeFe]-hydrogenase with O2 is developed. Our study underscores the relevance of model systems in understanding biocatalysis and validates their potential as important tools for elucidating the chemistry of oxygen-induced deactivation of [FeFe]-hydrogenase.
Introduction
Renewable energy sources such as solar, wind, and hydropower offer sustainable alternatives to fossil fuels but suffer from limitations in storage and transportation, impelling researchers to look for other sources of energy. Molecular hydrogen (H2) is a promising clean energy storage and transport molecule;1–4 however, cost-efficient and sustainable generation of H2 is yet to be achieved. Biotechnological H2 generation via the enzyme hydrogenase5 is one potential pathway to produce “green fuel”. Although [NiFe]- and [FeFe]-hydrogenases are amongst the most effective H2-forming enzymes (TOF of up to ∼21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s−1),6–8 biocatalytic H2 production is at a very early technology readiness level.
000 s−1),6–8 biocatalytic H2 production is at a very early technology readiness level.
The efficiency of [FeFe]-hydrogenases is attributed to their organometallic cofactor. The so-called H-cluster is an assembly of two distinct iron–sulfur sites connected via a cysteine ligand: a [4Fe–4S] cluster, which participates in electron transfer during catalysis and a unique diiron site, where the proton reduction reaction occurs (Fig. 1). The H-cluster carries an “azadithiolate” ligand (adt) and two μCO-bridged iron ions, each of which is coordinated additionally to a terminal CN− and CO ligand. Fig. 1 illustrates how the cyanide ligands may interact with residues in the active site.9 The adt ligand functions as an internal hydrogen-bonding donor and proton relay site, coupling proton transfer across the protein fold to an open coordination site at the distal iron ion (Fed) of the diiron site.10 The proton transfer pathway comprises cysteine, glutamic acid, serine, and arginine residues (Fig. 1). Despite their catalytic efficiency, large-scale applications are limited due to the O2 sensitivity of [FeFe]-hydrogenase. Investigating the mechanism of aerobic deactivation is crucial for understanding and addressing these challenges and will inspire strategies of enhancing enzyme stability and functionality in the presence of O2 expanding the applicability of [FeFe]-hydrogenases.
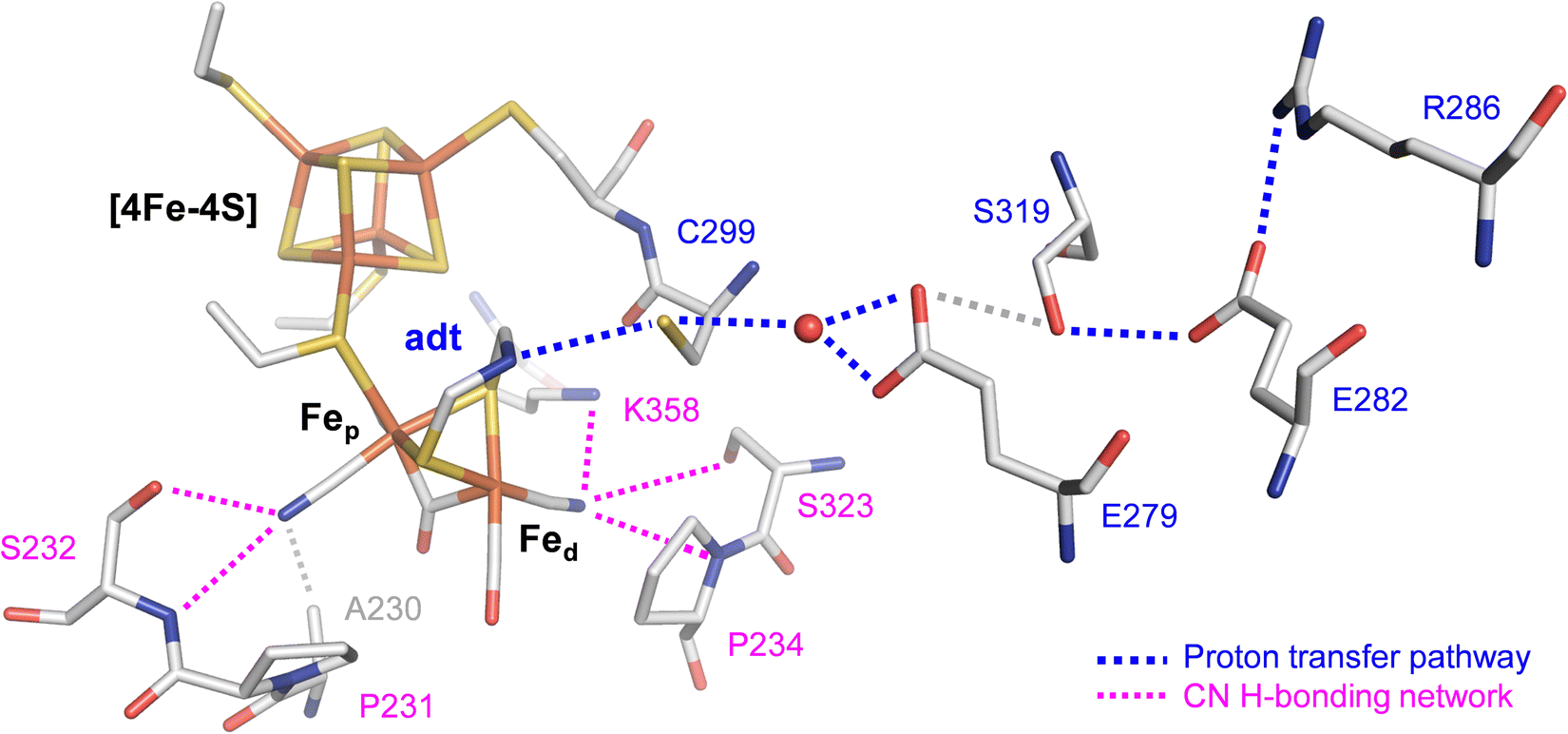 | ||
| Fig. 1 Stick representation of the active site and the proton transfer pathway (PTP) of [FeFe]-hydrogenase CpI (PDB ID 4XDC).11 The H-cluster consists of a [4Fe–4S] cluster connected to a diiron site with an open site at Fed where O2, H2, or CO can bind. Via the azadithiolate ligand (adt), the H-cluster exchanges protons with the PTP (blue traces) that connects active site and bulk solvent. The cyanide ligands of the H-cluster are hydrogen-bonded to residues in the 2nd coordination sphere (magenta traces). | ||
To understand the reaction of [FeFe]-hydrogenase with O2, numerous theoretical and spectroscopic studies have shown diffusion of O2 through hydrophobic gas channels.12–14 We and others have suggested that O2 undergoes reductive activation at the H-cluster and is converted to reactive oxygen species (ROS), which subsequently damage the diiron site, the [4Fe–4S] cluster, or both.15–18 In addition, CO inhibition experiments revealed that CO shields the H-cluster against aerobic deactivation suggesting that Fed is the initial binding site for both O2 and H2 (Fig. 1).17–19 In similar fashion, the [FeFe]-hydrogenase CbA5H is protected from aerobic deactivation by interconversion from the Hox state to the sulfur-inhibited Hintact state in the presence of O2.20–22 Another mechanism applies to the sensory [FeFe]-hydrogenase TamHydS that forms a stable intermediate due to insufficient proton transfer as a sub-step in the reaction with O2.23,24 Oxygen tolerance (i.e., hydrogen turnover in the presence of O2) and a catalytic reduction of O2 to water like in certain [NiFe]-hydrogenases25–29 has not been observed with [FeFe]-hydrogenase.
With respect to synthetic mimics of the H-cluster, past research was focused on studying the reaction of carbonyl-type diiron complexes with O2 or ROS. However, there has been a lack of investigation into the oxygen reactivity of [Fe2(xdt)(CO)4(CN)2]2− type complexes. This may be due to the demanding synthesis of such compounds. For example, the synthetic route towards model complex [Fe2(adt)(CO)4(CN)2]2− involves condensation of Fe2(SH)2(CO)6 with premixed solution of (NH4)2CO3 and paraformaldehyde to afford the diiron azadithiolate precursor Fe2(adt)(CO)6, which renders the desired mimic after CO/CN− ligand exchange ready to be inserted in hydrogenase apoenzyme (“artificial maturation”).30–33 Despite the widespread utilization of this protocol, the process suffers from several challenges. For example, the yields of Fe2(adt)(CO)6 have been low and inconsistent. Additionally, the use of Fe2S2(CO)6, which is acquired through a challenging synthetic pathway, further undermines the appeal of this method.31,34,35 Addressing these obstacles, we herein describe a straightforward and scalable synthetic scheme for the precursor complex Fe2(adt)(CO)6 (Fig. S1, ESI†).
Previously, several diiron site mimics have been investigated towards their proton reduction mechanism, while only a few reports highlight the reactivity with O2.7,36–39 For example, Darensbourg et al. reported on the site-specificity of the oxygenation of Fe2(pdt)(CO)6−x(L)x (pdt = S2(CH2)3, L = CO, PMe3, PPh3; x = 1 or 2) with m-chloroperoxybenzoic acid. Although density functional theory (DFT) calculations suggested formation of a Fe–O–Fe bond species, experimental studies revealed a dithiolate-centred oxidation, resulting exclusively in S-oxygenated reaction products.39 Similarly, Weigand et al. reported on the chemical sulfur oxidation of Fe2(sdt)(CO)6 type models with varying equivalents of dimethyldioxirane (sdt = S2(CH2)2S).38,40 Furthermore, Dey et al. reported hexacarbonyl diiron mimics with a 4-bromoaniline dithiolate ligand that electrocatalytically convert O2 at reducing potentials.36 Here, formation of H2O2 was observed and the bridgehead amine was suggested to be of key importance to protect these H-cluster mimics from further aerobic deactivation. Along these lines, Berggren and Hammerström et al. conducted oxidative studies on the hexacarbonyl diiron mimics Fe2(adt)(CO)6 and Fe2(pdt)(CO)6 with O2 and ROS. Here, the interaction of Fe2(adt)(CO)6 with O2 in presence of chemical reductants lead to short-lived species with oxygenated thiol groups.37
As the individual steps of cluster degradation remained unidentified, we investigate the oxygen sensitivity of biomimetic complexes [Fe2(adt)(CO)6−x(CN)x]x− and [Fe2(pdt)(CO)6−x(CN)x]x− with x = 2 (ADTCN[1] and PDTCN[2]) and x = 1 (ADTmono-CN[3] and PDTmono-CN[4]) in this work to receive a deeper understanding of the mechanism of aerobic deactivation in [FeFe]-hydrogenase (Fig. 2). We emphasize that these four active site models are direct mimics of the H-cluster, and ADTCN[1] and PDTCN[2] are commonly utilized for artificial maturation of [FeFe]-hydrogenase.30,31
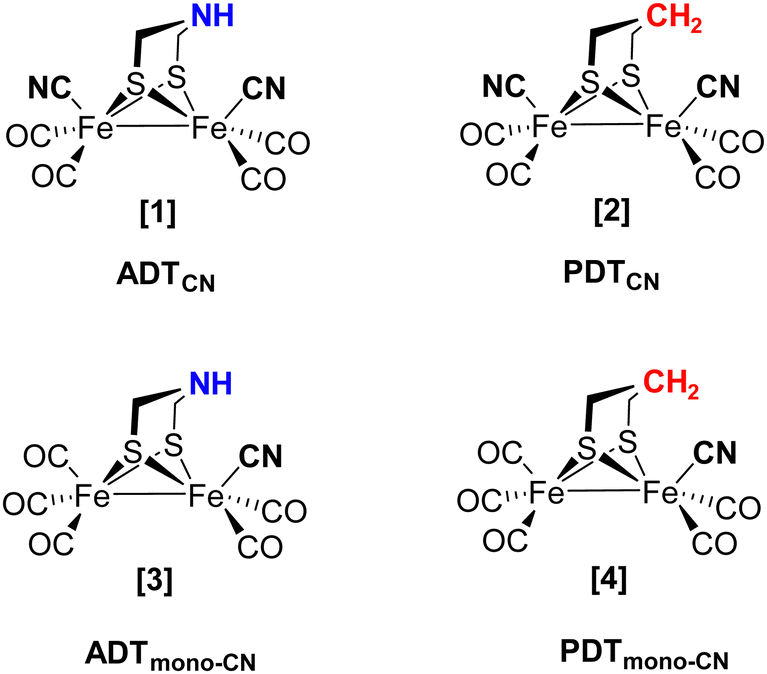 | ||
| Fig. 2 Schematic drawings of the active site models investigated in this study. Complex [1] closely resembles the H-cluster while the natural adt ligand (headgroup NH, blue) is replaced by a pdt ligand in [2] (headgroup CH2, red). | ||
Among the various oxygen species, superoxide (O2−) is the most likely ROS affecting iron–sulfur clusters, and it is a much stronger one-electron oxidant than O2 (+0.9 V and −0.1 V vs. RHE, respectively).41 Additionally, formation of H2O2 is not uncommon in natural systems and considered a harmful substrate for [FeFe]-hydrogenase in particular.42–44 Following these considerations, we report the interactions of complexes [1–4] with O2, H2O2, and superoxide by means of FTIR and Mössbauer spectroscopy, both in the presence and absence of protons. In the next step, model complexes [1–4] were probed as part of the H-cluster within the [FeFe]-hydrogenase of Chlamydomonas reinhardtii, CrHydA1. Four cofactor variants CrHydA1adt, CrHydA1pdt, CrHydA1mono-adt, and CrHydA1mono-pdt were spectroscopically investigated towards O2-induced deactivation. Additionally, CrHydA1adt and CrHydA1pdt variants were treated with H2O2. We find that proton transfer and hydrogen-bonding with the adt ligand critically influence the reactivity with O2 but barely affect H2O2-mediated oxidation. Our results facilitate understanding the initial steps of O2 deactivation in the biological system and show that the model complexes directly reflect the reaction of [FeFe]-hydrogenase with O2 and ROS.
Materials and methods
Synthesis of Fe2(adtCbz)(CO)6
Under inert conditions, Fe3(CO)12 (100 mg, 0.20 mmol) was dissolved in 20 ml THF. Then, adtCbz (58.4 mg, 0.24 mmol) was added dropwise via a syringe to the solution. The reaction was refluxed for 2 h, whereby the color of the reaction mixture changed from dark blue to reddish-brown. Afterwards the reaction mixture was allowed to cool to room temperature and the volatiles were removed in vacuo. Column chromatography using SiO2 (DCM:PE 3:1) was conducted. The red fraction was collected, dried over MgSO4, filtered, and concentrated. After removal of the solvent in vacuo the dark red product was obtained with a yield of 80% (83 mg). Crystals for X-ray diffraction analysis (Fig. S1, ESI†) were grown upon slow evaporation of saturated solution of Fe2(adtCbz)(CO)6 in pentane at 4 °C. The FTIR spectrum is shown in Fig. S2A (ESI†).
Synthesis of Fe2(adt)(CO)6 from Fe2(adtCbz)(CO)6
Under inert conditions, Fe2(adtCbz)(CO)6 (100 mg, 0.19 mmol) was dissolved in 5 ml DCM. Then, degassed Me2S (0.37 ml, 5.15 mmol) and BF3·Et2O (0.48 ml, 48%) were added and the mixture was stirred for 1.5 h at RT until the red solution brightened up a little. Hereafter, additional Me2S (0.30 ml, 4.20 mmol) was added, and the mixture was allowed to stir for another 2 h at RT. Next, 3.5 ml degassed H2O was added followed by addition of 3.5 ml 10% NH4OH. After shortly stirring the mixture, the compound was extracted with 3 × 5 ml DCM. The organic phase was dried over MgSO4 and filtered. After removal of the solvent in vacuo the crude product was obtained as reported before.45 Following column chromatography (SiO2) DCM:PE 1:1, the red fraction was collected, dried over MgSO4 (which was filtered off) and the organic phase was dried in vacuo yielding Fe2(adt)(CO)6 as a red powder with a yield of 60% (44 mg). The FTIR spectrum is shown in Fig. S2B (ESI†).
Synthesis of ADTCN[1] [Fe2(adt)(CO)4(CN)2][Et4N]2
Fe2(adt)(CO)6 (0.1 g, 0.26 mmol) was dissolved in CH3CN (2 ml) and cooled to 0 °C. A cold solution of [Et4N][CN] (0.085 g, 0.55 mmol) in CH3CN (1 ml) was introduced via a gas-tight syringe to the Fe2(adt)(CO)6 solution. The reaction mixture instantaneously turned dark red and released CO gas as previously reported.32 The reaction was allowed to warm to room temperature and further allowed stirring for 3 h. The solvent was removed in vacuo to give a dark red residue. The complex was transferred to the glovebox and was washed with 6 ml of diethylether (Et2O), followed by washing with Et2O:MeCN (20:1). The complex was dried, resulting in the dark red solid (0.113 g, 68%). A similar procedure was followed for the synthesis of PDTCN[2] [Fe2(pdt)(CO)4(CN)][Et4N]2 from Fe2(pdt)(CO)6. See Fig. S3 (ESI†) for a schematic depiction of the synthetic route.
Synthesis of ADTmono-CN[3] [Fe2(adt)(CO)5(CN)][Et4N]
A mixture of Fe2(adt)(CO)6 (0.1 g, 0.26 mmol) and Me3NO (0.0195 g, 0.26 mmol) was dissolved in CH3CN (2 ml) and cooled to −40 °C. A cold solution of [Et4N][CN] (0.028 g, 0.26 mmol) in CH3CN (1 ml) was introduced via a gas-tight syringe to the Fe2(adt)(CO)6 + Me3NO solution. The reaction mixture turned dark red and released CO gas as previously reported.46 The reaction mixture was allowed to warm to room temperature and further stirred for 3 h. The solvent was removed in vacuo to give a dark blackish red, sticky residue. Inside a glove box, the complex was washed with 6 ml of Et2O, followed by washing with Et2O:MeCN (20:1). The complex was dried resulting in the dark blackish-red solid with a yield of 62% (0.085 g). Similar procedure was followed for the synthesis of PDTmono-CN[4] [Fe2(pdt)(CO)5(CN)][Et4N] from Fe2(pdt)(CO)6.
See ESI† for details on further Material and methods.
Results and discussion
Reaction of the complexes with O2
Under an inert argon atmosphere, complex ADTCN[1] and PDTCN[2] reveal nearly identical FTIR spectra between 1800–2150 cm−1 (Fig. 3). The five observed bands have been assigned to the coupled CN− and CO stretching vibrations and suggest reduced FeI–FeI complexes.47 The response toward O2 was studied by purging pure oxygen gas through the individual solutions in MeCN. A mean shift of Δ = 96 cm−1 and Δ = 92 cm−1 towards higher wavenumbers was observed (Table 1) but while the CO and CN− bands drastically lost intensity for ADTCN[1] the overall band intensity of PDTCN[2] was less affected.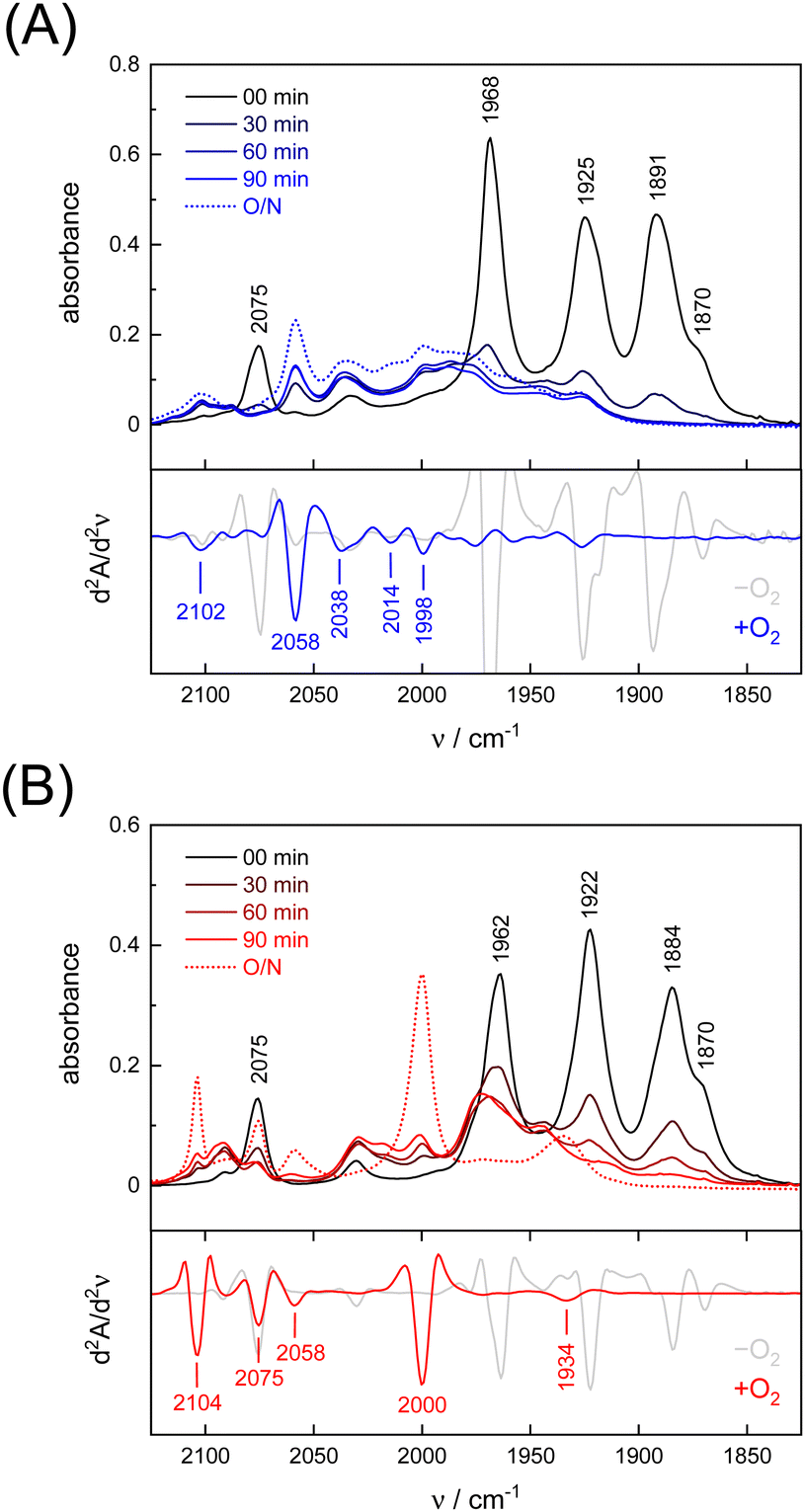 | ||
| Fig. 3 (A) FTIR spectra of ADTCN[1] and (B) PDTCN[2] in MeCN upon O2 purging for 30 seconds, recorded every 30 minutes and after overnight incubation (O/N = 23 h). Below each absorbance spectrum, the second derivative spectrum at 0 min (−O2) and after 23 h (+O2) is plotted to identify the bands of the O2-oxidised complexes. | ||
| Model | [1] | [1] | Δ | [2] | [2] | Δ |
|---|---|---|---|---|---|---|
| −O2 | +O2 | −O2 | +O2 | |||
| νCO | 1870 | 1998 | 128 | 1870 | 1934 | 64 |
| νCO | 1891 | 2014 | 123 | 1884 | 2000 | 116 |
| νCO | 1925 | 2038 | 113 | 1922 | 2058 | 136 |
| νCO | 1968 | 2058 | 90 | 1962 | 2075 | 113 |
| νCN− | 2075 | 2102 | 27 | 2075 | 2104 | 29 |
| Mean | 1946 | 2045 | 96 | 1943 | 2024 | 92 |
The spectrum of O2-treated complex [1] after 90 minutes indicates a number of broad features spanning from 1930–2100 cm−1 along with better defined bands at 1998, 2014, 2038, and 2058 cm−1 that we assign to the CO ligands. The second derivative FTIR spectrum in Fig. 3(A) suggests a high-frequency band at 2102 cm−1 assigned to the CN− ligands. For complex [2], sharper and more defined CO signals emerge at 1934, 2000, 2058, and 2075 cm−1, with one CN− band at 2104 cm−1 (second derivative FTIR spectrum in Fig. 3(B)), notably only after overnight incubation in the presence of O2. The reaction with O2 does not lead to complete loss of CO and CN− ligands, which is evident from the FTIR spectra measured after up to 23 hours (Fig. S4, ESI†). Interestingly, complex PDTCN[2] seems to be particularly O2-stable. For both complexes, the mean difference in CO/CN− frequency is small under anaerobic conditions (−O2, Δ = 3 cm−1) but the difference increases under aerobic conditions (+O2, Δ = 21 cm−1) reflecting the ten times stronger up-shift for complex ADTCN[1] (Table 1).
Interestingly, we observed that both the nature of the dithiolate head group and the composition of diatomic ligands affect the susceptibility for O2 damage. Fig. 4(A) shows the intensity of the CO band at 1891 cm−1 and 1884 cm−1 of the reduced complex [1] and [2] as a function of time, which allows comparing the decay velocity and illustrates the faster degradation of ADTCN[1]. For ADTmono-CN[3] a comparatively slow decrease in band intensity upon exposure to O2 is observed; however, no oxidized product accumulates. Besides a small loss in intensity, the FTIR spectrum of PDTmono-CN[4] remains largely unaltered under O2 (Fig. S5, ESI†). These observations are in line with previous reports, which state that complex [1] and [2] are easier to oxidize than their mono-cyanide derivates.48
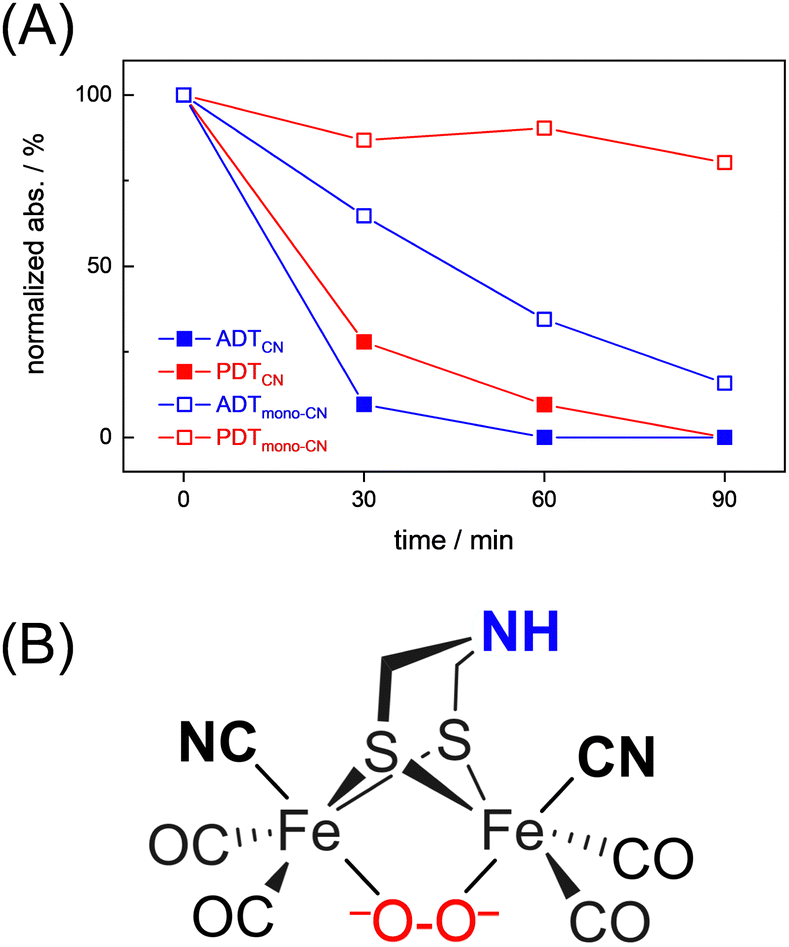 | ||
| Fig. 4 (A) Comparison of the O2-induced decay of complexes [1–4]. At t0, the band intensity at 1891 cm−1 (ADTCN[1]), 1884 cm−1 (PDTCN[1]), and 1975 cm−1 (ADTmono-CN[3] and PDTmono-CN[4]) was normalized to 1 and plotted against time (spectral traces in Fig. 3 and Fig. S5, ESI†). The graph illustrates how the adt-type complexes decay faster than the pdt-type complexes, and how di-cyanide complexes decay faster than mono-cyanide complexes. (B) Most probable structure of a di-ferrous peroxide dianion intermediate of ADTCN according to DFT calculations. | ||
In variance to earlier observations,37 no oxygenation of the thiol group were observed, at least within the timeframe of our experiments (Fig. S4, ESI†). The low-frequency end of the IR spectrum below 1200 cm−1 is sensitive for S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibrations.49 Instead, we presume that O2 reacts at the metal sites. While direct spectroscopic evidence of O2 binding at the metal centers is lacking, the large CO/CN− band shifts towards higher wavenumbers for complex ADTCN[1] and PDTCN[2] indicates oxidation of the diiron site.
O vibrations.49 Instead, we presume that O2 reacts at the metal sites. While direct spectroscopic evidence of O2 binding at the metal centers is lacking, the large CO/CN− band shifts towards higher wavenumbers for complex ADTCN[1] and PDTCN[2] indicates oxidation of the diiron site.
To shed light on the nature of the observed products, a comprehensive density functional theory (DFT) investigation has been conducted. Initial states of the complexes [1] and [2] with two antiferromagnetically coupled local spins leading to a spin singlet was modelled by means of broken-symmetry DFT. The oxygenation reaction has been explored through three primary routes, as illustrated in Fig. S19 and S20 (ESI†). These routes encompass both terminal and bridging coordination modes, while the latter of which can occur in either “end-on” or “side-on” fashion. For all different oxygen attack modes, complex [1] exhibited lower activation barriers, approximately 2–3 kcal mol−1 lower than that of complex [2], confirming the reactive nature of [1]. When one electron is transferred from the diiron cluster to O2, the resulting FeII–FeI superoxo complex emerges as a reactive intermediate for both complexes across all conceivable binding modes. According to the modeled reaction mechanism, it was determined that the terminal coordination of oxygen is unlikely. Instead, the thermodynamically favored product contains a peroxide dianion that bridges the two FeII centers as depicted in Fig. 4(B).
In addition to the aforementioned considerations of the reaction energetics, the calculated CO-stretch frequencies were compared to the experimentally observed IR bands. As depicted in the correlation analysis shown in Fig. S23 (ESI†), formation of a bridging hydride is the most probable form of the protonated compounds, i.e., [1,2] + H+. Results for the oxygenated products, on the other hand, are not as clear and straightforward. The oxygenation at one of the sulfur atoms did not induce a significant difference in the CO/CN-vibration regime compared to the initial XDTCN–O2 system. This result was anticipated since the coordination environment of the iron centers remains unchanged in this binding mode. Conversely, terminal coordination of the O2 moiety consistently led to a highly asymmetric shift in CO-vibrations across all cases, which contradicts the experimentally observed differences, as shown in Tables S3 and S4 (ESI†). Considering the reaction profiles alongside the IR data, it can be inferred that terminal binding of oxygen is highly improbable. The bridging coordination of the peroxide dianion to FeII–FeII is distinguished by a consistent, progressively positive shift (relative to the initial states) averaging around 90 cm−1. This observed shift, consistent across both experimental data and simulations, aligns closely with the experimental findings. Further details involving the computational methodology, structural isomers (apical-basal) of [1–2], spin isodensity plots depicting the possible intramolecular magnetic exchange interactions, reaction profiles, and an analysis of IR bands can be found in ESI.†
Complexes [1] and [2] were found to be EPR inactive, both in the absence and presence of O2 (data not shown). This hints at a two-electron oxidation process, converting a diamagnetic starting state [FeI–FeI] into a diamagnetic product state [FeII–FeII]. The strong IR up-shift of 92–96 cm−1 (Table 1) and the DFT evaluation (Fig. S22, ESI†) agrees with this explanation. In the next step, we analyzed the reaction with O2 using Mössbauer spectroscopy to demonstrate metal oxidation directly. The MeCN solutions of complex [1] and [2] were treated with O2 for 30 seconds. Then, the samples were frozen in liquid nitrogen and measured. Mössbauer spectra (based on the natural abundance of 57Fe) for complex [1] and [2] after O2 exposure result in two quadrupole doublets (Fig. 5, bottom traces) that significantly differ from the respective starting materials (Fig. 5, top traces and Table 2).
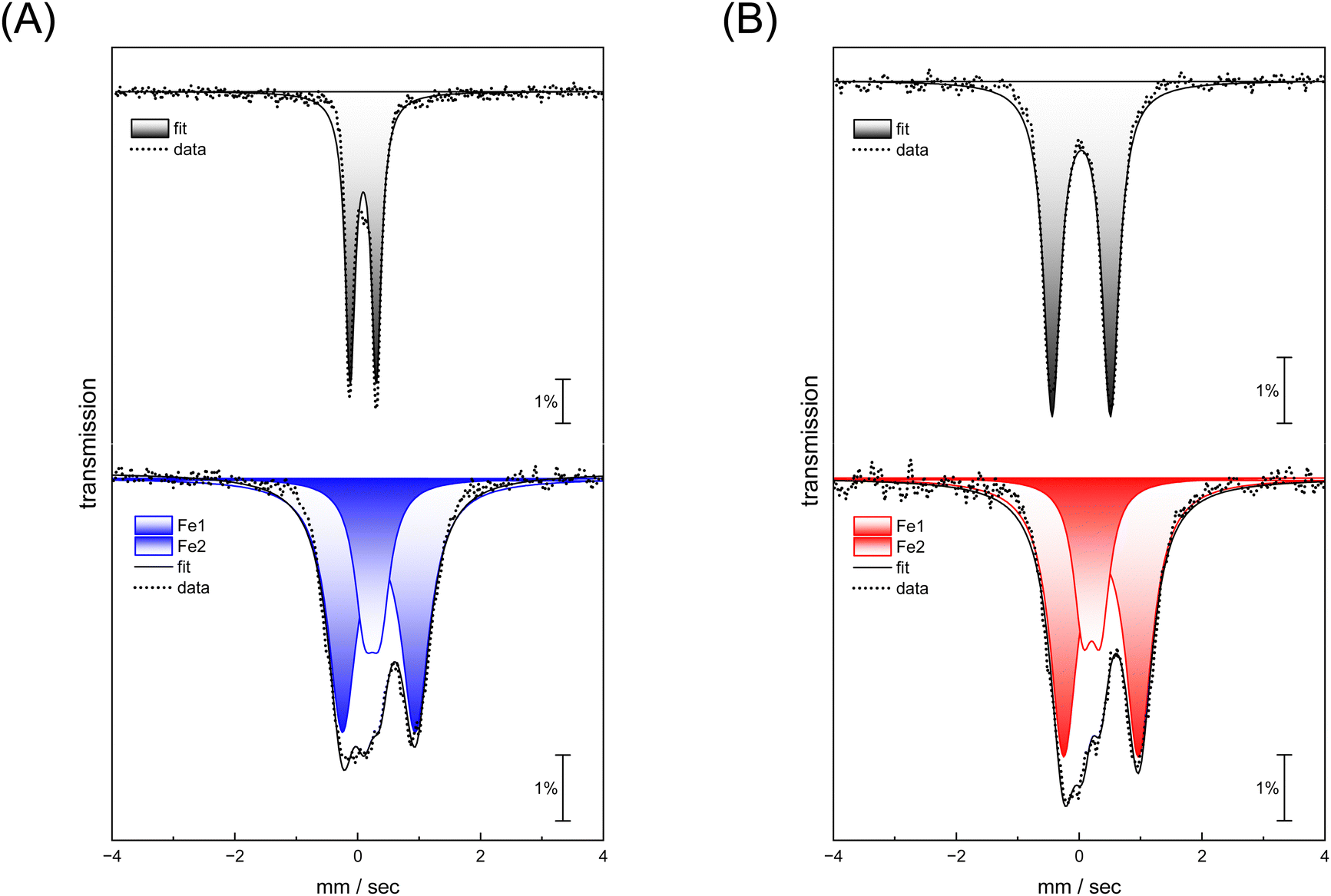 | ||
| Fig. 5 (A) Mössbauer spectra of complex ADTCN[1] and (B) PDTCN[2] under anaerobic conditions (top traces) and recorded after purging the complexes with O2 for 30 seconds each (lower traces, blue ADTCN[1] and red ADTCN[2]). | ||
| Model | [1] | [1] | [2] | [2] | ||
|---|---|---|---|---|---|---|
| –O2 | +O2 | –O2 | +O2 | |||
| Iron | Fe1 | Fe1 | Fe2 | Fe1 | Fe1 | Fe2 |
| δ | 0.037 | 0.238 | 0.345 | 0.035 | 0.204 | 0.358 |
| ΔEq | 1.109 | 0.253 | 1.188 | 0.951 | 0.288 | 1.219 |
For both complexes, an increase of isomer shift is observed indicating oxidation from FeI to FeII. However, while one site reveals a lower quadrupole splitting and isomer shift (Fe1 in Table 2), the second site shows higher values for both parameters (Fe2 in Table 2) suggesting that the metal centers in both complex [1] and [2] have a similar oxidation state. This agrees with a formal redox state [FeII–FeII]. These measurements demonstrate that iron participates in the reaction with O2, and that the coordination environment is clearly altered.
The high reactivity of complex [1] and [2] with O2 made an isolation of the degradation products challenging. The obtained FTIR and Mössbauer spectra as well as DFT calculations now support the oxidation of the metal centers with subsequent loss of ancillary ligands under aerobic conditions, in particular for the adt-type complexes [1] and [3]. Such observations have been previously reported for the [FeFe]-hydrogenase CrHydA1, and the formation of superoxide at the H-cluster has been concluded.17 In a cysteine-to-alanine variant impaired for proton transfer, the corresponding superoxide-binding state (Hox–O2) was trapped and suggested to be unreactive provided no further protonation occurs.50
Reaction of the complexes with O2 in presence of protons.
As a proton-reducing enzyme, [FeFe]-hydrogenase cycles through various catalytic intermediates while shuttling protons. Moreover, the O2 sensitivity significantly depends on the availability of protons at the H-cluster.16,51 Therefore, the active site mimics were tested towards O2 sensitivity in the presence of protons. Acids with varying strength were employed. Firstly, studies were carried out using acetic acid (pKa = 23 in MeCN). The presence of one equivalent of acetic acid had no influence on the CO/CN− band positions, which means that no protonation is observed, and both ADTCN[1] and PDTCN[2] react with O2 in a similar manner as in the absence of protons (Fig. S6, ESI†).Subsequently, the stronger acid p-toluene sulfonic acid (TsOH, pKa = 8.3 in MeCN) was used. The FTIR spectra of complex [1] and [2] are dramatically influenced in the presence of TsOH. After addition of one equivalent of acid, a mean IR up-shift of 98 cm−1 (νCO) and 32 cm−1 (νCN) was recorded (Fig. 6(A), (B) and Table 3). This indicates protonation and/or oxidation of the complexes.52,53 In variance to the reactions with O2, note that both band intensity and band pattern are conserved in the absence and presence of protons. To further identify complex [1] and [2] in the presence of TsOH, 1H NMR spectroscopy was performed. Fig. 6(C) shows high field 1H signals at −19.15 ppm and −19.75 ppm for complex [1] and complex [2], respectively, directly indicating formation of a bridging hydride ligand (μH) that differs from the terminal hydride at the H-cluster.52,54 Both complexes show additional signals at −15.65 and −16.25 ppm indicating protonation of the CN− ligands.55 We note that the strong IR up-shift ligands may result from both protonation of the ligands and oxidation of the complexes due to hydride formation. In [FeFe]-hydrogenase, the CO ligands shift about ten times stronger than the CN− ligands, e.g., comparing the structurally conservative Hox and Hhyd states.56–58 The CO/CN− shift ratio of only three observed here may reflect the stronger up-shift of the CN− ligands upon protonation.
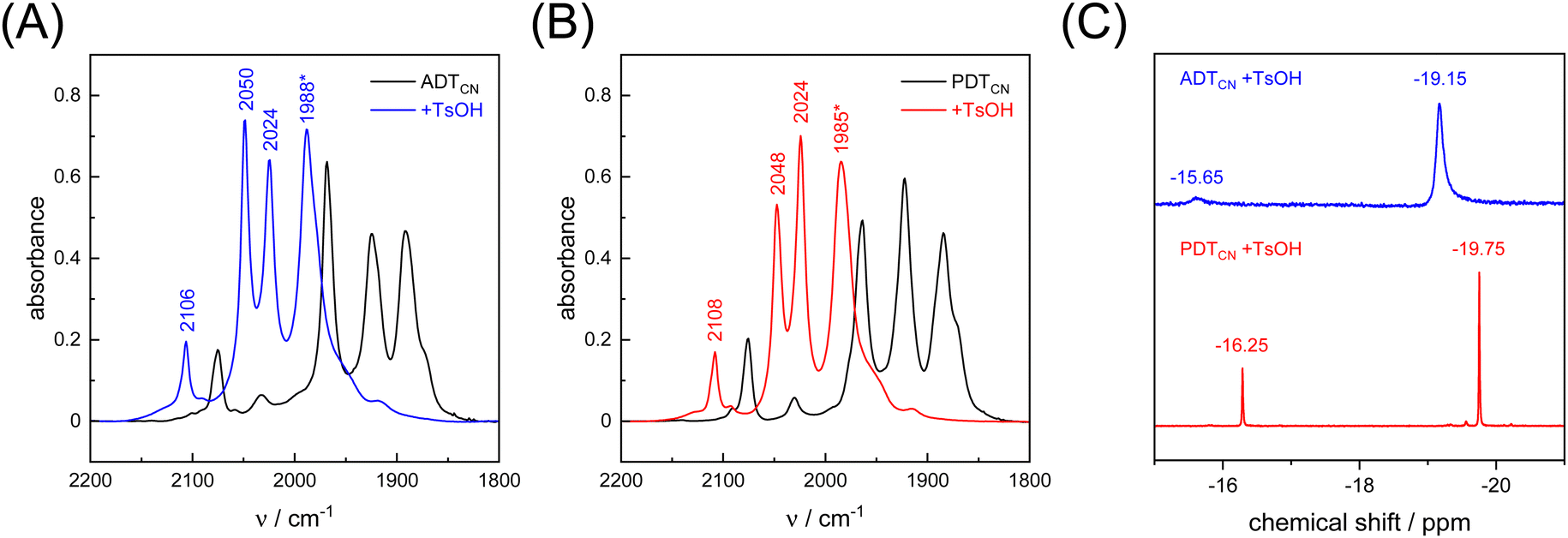 | ||
| Fig. 6 FTIR spectra of (A) ADTCN[1] and (B) PDTCN[2] measured in the absence and presence of 1 equivalent of TsOH (black and blue/red traces). Asterisk: the bands at 1988 and 1985 cm−1 show a shoulder at 1977 and 1978 cm−1, respectively. (C) 1H NMR (CD3CN, 400 MHz) spectra of ADTCN[1] and PDTCN[2] obtained upon reaction with 1 equivalent of TsOH. Signals at −19.15 and −19.75 stem from a μH ligand. Signals at −15.65 and −16.25 indicate protonation of the CN− ligands. | ||
| Model | [1] | [1] | Δ | [2] | [2] | Δ |
|---|---|---|---|---|---|---|
| −H+ | +H+ | −H+ | +H+ | |||
| νCO | 1870 | 1977 | 107 | 1870 | 1978 | 108 |
| νCO | 1891 | 1988 | 97 | 1884 | 1985 | 101 |
| νCO | 1925 | 2024 | 99 | 1922 | 2024 | 102 |
| νCO | 1968 | 2050 | 82 | 1962 | 2048 | 86 |
| νCN− | 2075 | 2106 | 31 | 2075 | 2108 | 33 |
The catalytically relevant H-cluster state Hhyd carries a terminal hydride at Fed.56–58 In variance, the bridging hydride geometry of complex [1] and [2] is comparable to H-cluster states Hred and Hsred where the diiron site formally adopts the FeII–FeII state and has been suggested to bind a μH ligand instead of a μCO ligand.59–61 In these intermediates, [FeFe]-hydrogenase shows a prolonged resistance to O2 and CO that we have explained by the presence of a fifth terminal ligand (CO or CN−) occupying the open coordination site at Fed.61,62 This model is not generally accepted, though. Under cryogenic conditions, ligand rotation is prohibited so that Hred and Hsred retain the μCO geometry and can be photoisomerized into Hhyd-like states.63 It is conceivable that line broadening of the μCO band shows a stronger dependence on temperature than the terminal ligands, making the band “disappear” at room temperature.64 This would explain the rather conservative, temperature-independent IR profile of the terminal ligands in Hred and Hsred.59 However, the upshift of μCO in Hred and Hsred relative to Hox is yet to explain,63–65 and the presence of a third CO band between 1950–1960 cm−1 argues against a conservative μCO geometry.60 Therefore, we suggest that complex [1] and [2] can serve as models for Hred and Hsred. After formation of the μH geometry, the model complexes display no further reactivity with O2 and can be kept under aerobic conditions without further changes. Notably, these protonated states were also found to be resistant toward other oxidizing reagents such as H2O2 (Fig. S7, ESI†). Our observations highlight the similarities between the natural system and its respective mimic.53,66
Reaction of the complexes with hydrogen peroxide
Previous work on aerobic H-cluster deactivation suggested superoxide as an intermediate that requires further protonation. In aqueous solution, ˙O2− is known to disproportionate into hydrogen peroxide (H2O2) and O2 at physiologically relevant rates.67 Therefore, H2O2 can be anticipated to be a major ROS involved in the decomposition of the H-cluster. First, we monitored the reactivity of complexes [1–4] with 30% H2O2 by IR spectroscopy. Fig. S8 (ESI†) show the differences in reactivity of complexes [1] and [2] with 2–10 equivalents of H2O2. Sub-stochiometric amounts of H2O2 were found to result in very long reaction times (hours) and not considered. Again, ADTCN[1] is more susceptible toward oxidation as it reacts with four equivalents of H2O2 while it takes at least six equivalents of H2O2 to convert PDTCN[2] into an oxidized species. A quantitative titration was not attempted. The Mössbauer spectra observed for both complexes upon H2O2 treatment (Fig. S9, ESI†) are comparable to the ones observed after reaction with O2 (Table S1, ESI†). Based on previous reports and our own data, di-ferrous complexes can be concluded.68,69 Additionally, ADTmono-CN[3] degrades in presence of eight equivalents of H2O2, whereas no significant shifts in the IR spectra of PDTmono-CN[4] were observed (data not shown).For both complex [1] and [2], there are several possibilities of H2O2 to interact with the metal centers as well as with the adt or pdt ligand. Based on calculations by Reiher et al., the reaction of the bridging sulfur atoms with H2O2 is most likely43 but within the timeframe of our experiments, no oxygenation of the thiol groups were observed (Fig. S8, ESI†). Considering the large shifts in the FTIR spectra, a direct interaction of H2O2 with the iron sites seems plausible. Although the reaction with H2O2 should be independent of additional proton sources, we speculate that the diminished reactivity of the pdt-type complex [2] and [4] reflects the absence of the amine bridgehead that may stabilize an intermediate apical oxygen ligand.
Reaction of the enzyme with O2 and H2O2
To probe the reactivity of complexes [1–4] as part of the H-cluster within [FeFe]-hydrogenase, we conducted aerobic deactivation experiments on CrHydA1 matured with mimics [1–4] producing four cofactor variants namely CrHydA1adt, CrHydA1pdt, CrHydA1mono-adt, and CrHydA1mono-pdt. In preparation of the FTIR experiment, the enzymes were kept under an inert N2 atmosphere to promote accumulation of the Hox state, which was achieved for CrHydA1adt and CrHydA1pdt (Fig. S10, ESI†). However, for CrHydA1mono-adt and CrHydA1mono-pdt a mixture of unknown states was obtained (Fig. S11, ESI†) presumably due to different isomers of the model complexes [3] and [4] that were used to mature the apo-enzyme.47,70 The CN− ligands are important anchor points for the diiron site (Fig. 1). After oxidation of the enzymes, 20% O2 v/v (or 5% H2O2 m/v) was introduced and changes of the H-cluster were monitored by in situ ATR FTIR spectroscopy.In line with the results observed for the active site mimics the enzyme matured with ADTCN[1] (CrHydA1adt, resembling wild-type enzyme) displayed faster degradation compared to CrHydA1pdt (Fig. 7(A)). FTIR difference spectra revealed a complete disintegration of the H-cluster in case of CrHydA1adt while a damaged cluster suggesting formation of a monometallic species with two CO and one CN− ligands with bands at 1930, 1985, and 2095 cm−1 was observed for CrHydA1pdt (Fig. 8(A) and Fig. S10, ESI†). The ‘survival’ of an H-cluster fragment in CrHydA1pdt is reminiscent of sensory [FeFe]-hydrogenases TamHydS, where similar traces were observed after contact with O2.23 Just like CrHydA1pdt this hydrogenase shows a discontinued proton transfer pathway.24,71 In case of CrHydA1mono-adt, the O2 treatment revealed a significantly slower oxidation (Fig. S11, ESI†) much in agreement with an earlier report on CrHydA1mono-adt.70 Notably, the H-cluster in CrHydA1mono-pdt was resistant to O2-induced oxidation as inferred from the largely unchanged FTIR spectra in Fig. S11 (ESI†).
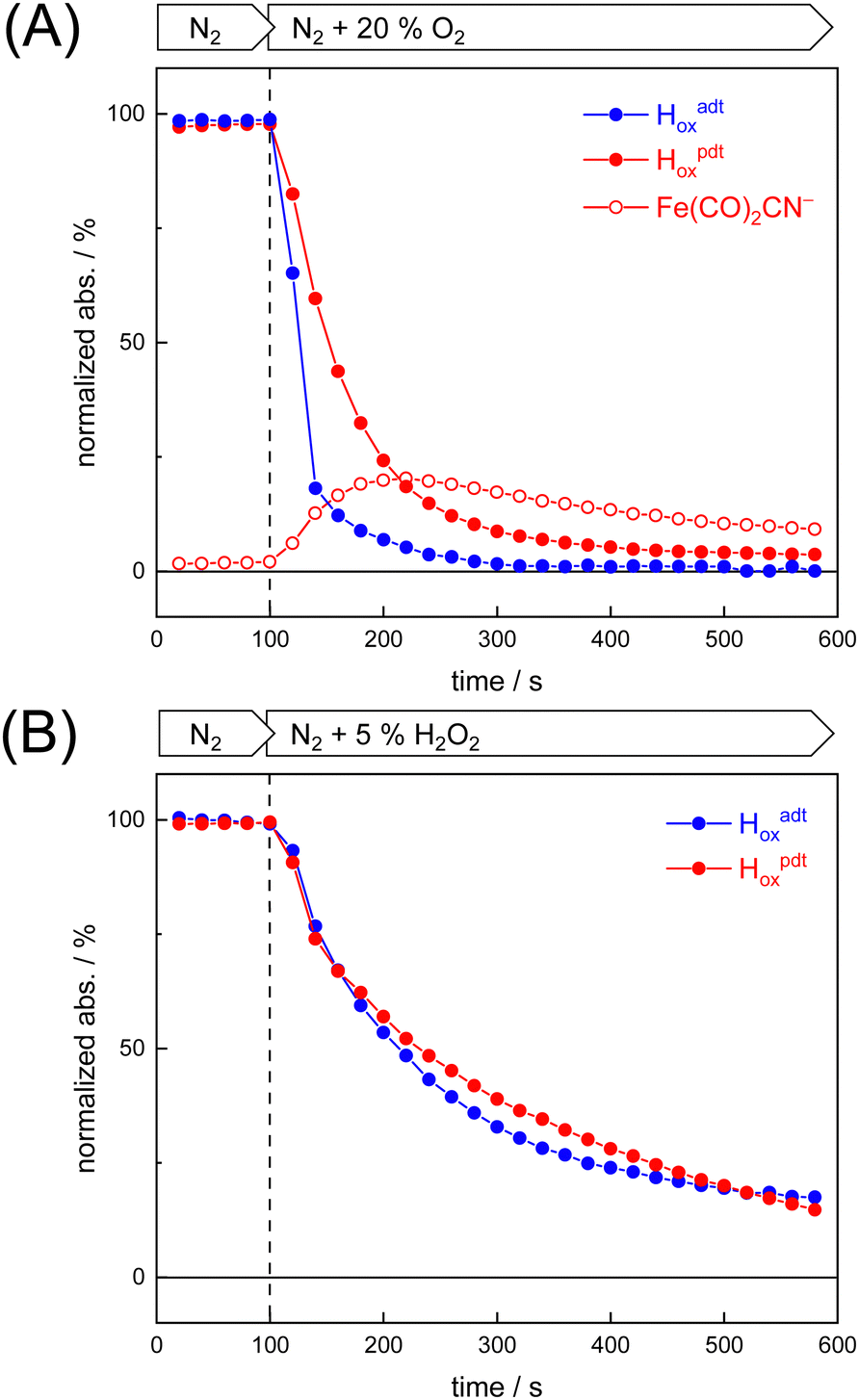 | ||
| Fig. 7 Kinetics of oxidative damage to the H-cluster. The reaction of [FeFe]-hydrogenase CrHydA1adt and CrHydA1pdt with O2 and H2O2 was monitored by in situ ATR FTIR spectroscopy. The normalized integral of all five H-cluster bands is plotted against time. (A) CrHydA1adt (Hox, blue traces) was quickly deactivated in the presence of 20% O2 v/v while cofactor variant CrHydA1pdt (Hox, red traces) was deactivated notably slower and formed a Fe(CO)2CN− intermediate (open red symbols). (B) No meaningful differences between CrHydA1adt and CrHydA1pdt were observed with 5% H2O2 m/v. | ||
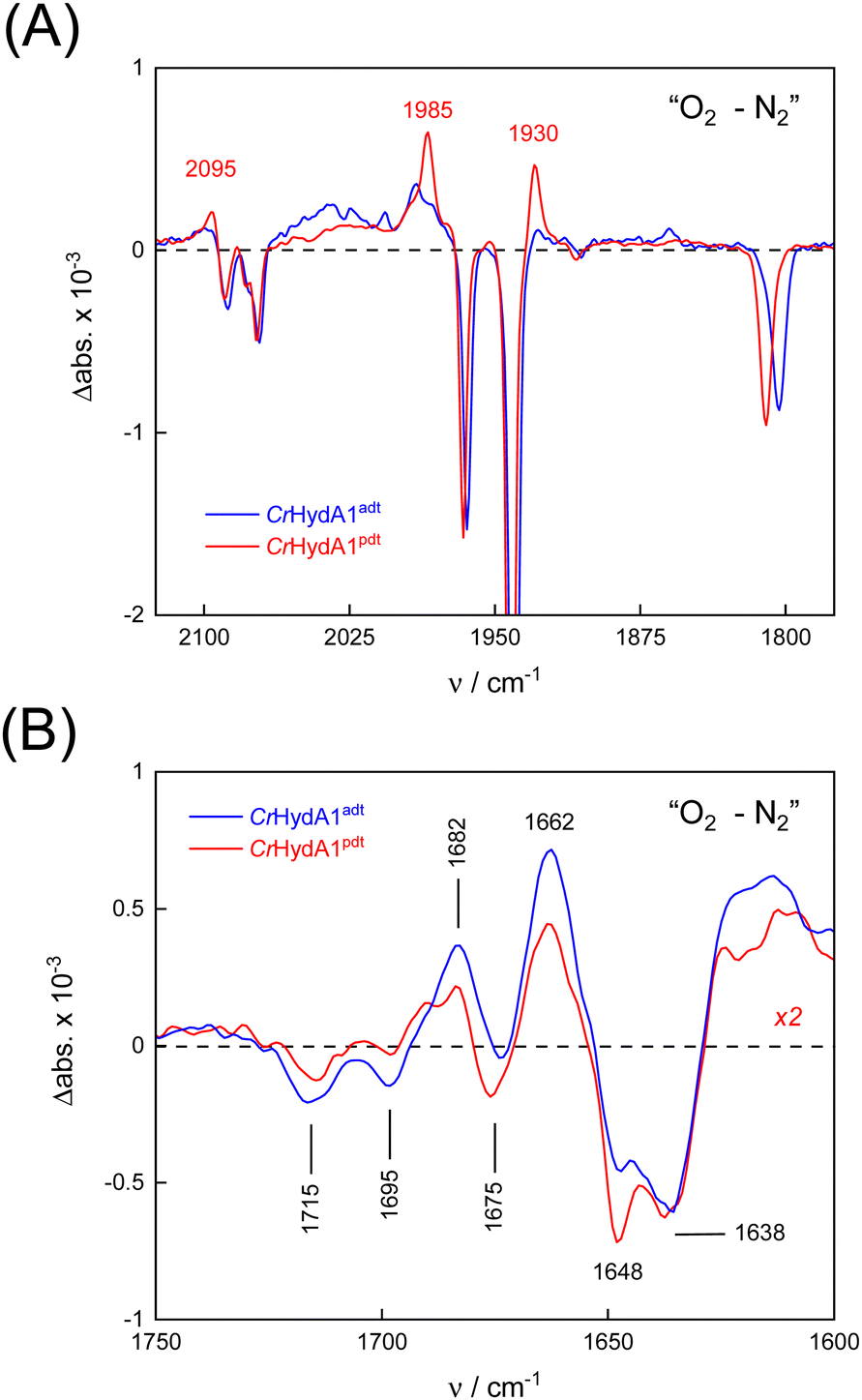 | ||
| Fig. 8 FTIR difference spectra reveal details of the reaction with O2. (A) In the CO/CN− frequency regime of the H-cluster, O2–N2 spectra show the decrease of the Hox state (negative bands). Note the accumulation of a Fe(CO)2CN− intermediate exclusive to CrHydA1pdt (positive bands, red traces). (B) At lower frequencies, bands have been assigned to glutamic acid residues E141 and E144 (1715 and 1695 cm−1) as well as arginine R148 (1682 cm−1). Other feature may be related to protein structural changes. Note that the traces for CrHydA1adt and CrHydA1pdt are very similar. | ||
At lower frequencies, protonation and hydrogen-bonding changes shape the FTIR difference spectra. Fig. 8(B) highlights the deprotonation of glutamic acid residues E141 and E144 (negative bands at 1715 cm−1 and 1695 cm−1) and the protonation of R148 (positive band at 1682 cm−1) upon contact with O2. These residues have been assigned in CrHydA1 previously and correspond to E279, E282, and R286 in Fig. 1.72,73 The reaction is potentially accompanied by protein structural changes (1675–1638 cm−1). Surprisingly, both CrHydA1adt and CrHydA1pdt show very similar FTIR signatures, indicating proton transfer toward the H-cluster in the O2 deactivation reaction. When scaled to the absorbance of the (negative) Hox bands, the amino acid signals in CrHydA1pdt are half as intense as in CrHydA1adt. This results from hindered proton transfer in the pdt variant and can explain the observed differences deactivation kinetics (see Fig. S12, ESI† for the time-resolved evolution of FTIR difference spectra.).
Summing up, we found that the different CrHydA1 cofactor variants follow the same O2 degradation trend as observed with the model complexes. However, introduction of 5% H2O2 did not lead to a faster H-cluster degradation in CrHydA1adt as compared to CrHydA1pdt (Fig. 7(B)). Unlike model complexes [1–4] that show pronounced differences in H2O2 sensitivity amongst each other (Fig. S8, ESI†) the enzyme variants interact with H2O2 irrespective of the bridgehead ligand. Both enzymes reacted equally slow, arguably due to the smaller H2O2 concentration in the aerosol, and no traces of Fe(CO)2CN− were observed.
Comparison between model complexes and enzyme
The differences in the rate of oxygen-induced degradation of ADTCN[1] and PDTCN[2] can be explained as follows. In both cases, O2 initially interacts with the metal centers forming a metal–oxygen transient state. Based on the DFT calculations (Fig. S22 and Table S4, ESI†) and our data, a bridging peroxide dianion ligand (μ(O−)2) is most likely (Fig. S19 and S20, ESI†). This geometry is not optimal for proton transfer via the adt ligand and subsequent H2O2 formation. We speculate that this is the reason why even the most O2-sensitive complex [1] is stable under air for several minutes. However, H2O2 formation is not suppressed completely, and our NMR data suggest that the CN− ligands may assist in proton transfer toward μO2− (Fig. 6(C)). The later is true for complex [2] as well, which is why the increased O2 stability of PDTCN can only be explained by the lack of an internal hydrogen-boning donor.42,74 The dithiolate ligand has limited effect on the electronic structure, as evidenced by the similar IR and Mössbauer spectra. Therefore, significant structural differences between complex [1] and [2] are excluded explaining the diverging kinetics. The mono-cyanide complexes [3] and [4] generally show higher Lewis acidity than the di-cyanide complexes [1] and [2].75 According to the model explained above, a lower pKa of the ADT ligand can explain the resistance to aerobic deactivation in the mono-cyanide complexes. Similar arguments have been used to explain the necessity of CN− ligands at the H-cluster.76–80 Remarkably, our data on the diiron complexes suggest that the interaction with O2 is not limited to the presence of an open binding site but rather is controlled by the ligand environment. The obtained results show that the bridgehead amine is not a mere spectator in oxygen-induced degradation but plays a crucial role in disintegration process.A model consisting of O2 reduction and protonation is applicable to [FeFe]-hydrogenase as well (Scheme 1). First, O2 binds to the H-cluster in the Hox state and is reduced to a superoxide ligand in the Hox–O2 state as characterized earlier.50 The diiron site is oxidized from [FeII–FeI] to [FeII–FeII] in the process. In variance to the μ(O−)2-binding complexes, the oxygen ligand is found in apical position in the enzyme; here, hydrogen-bonding with the adt ligand may promote the susceptibility to O2 binding, reduction, and protonation thus slowing down the reaction velocity of CrHydA1pdt. Similar second coordination sphere effects have been observed for the formation and stabilization of the apical H− ligand in the hydride state Hhyd,50,57,62 which cannot be accumulated in the pdt variant. In the second step, protonation and disproportionation of superoxide to H2O2 will demand an additional superoxide radical (˙O2−). We speculate that this may stem from a second population of enzyme in the Hox–O2 state; such ligand transfer is common in [FeFe]-hydrogenase, e.g., in the formation of the Hox–CO state (“cannibalization”).81 Oxidation of ˙O2− in the disproportionation reaction produces O2, which may re-bind the intact H-cluster. For CrHydA1pdt and its parent complex PDTCN[2], the absence of a hydrogen-bonding donor impedes further ROS formation, hence a slower response towards O2 is observed (Fig. 4 and 7). This observation in agreement with electrochemical O2 reduction experiments by Dey et al., where the formation of a catalytic peroxide species critically depends on hydrogen-bonding with an adt-type ligand.36
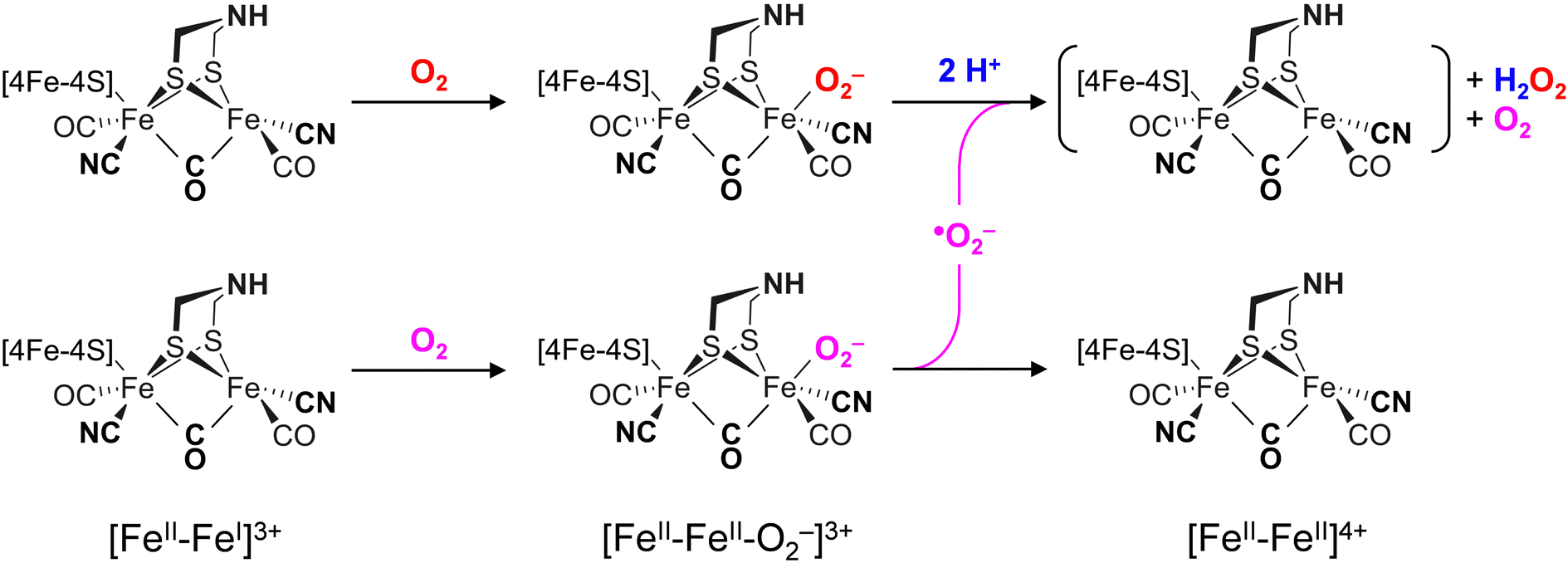 | ||
| Scheme 1 Binding and reduction of O2 at the H-cluster in the [FeII–FeI] state (Hox) produces the [FeII–FeII–O2−] state (Hox–O2) stabilized by hydrogen bonding between ligands adt and O2− (dashed lines). Protonation and disproportionation of superoxide leads to H2O2 formation and regeneration of O2. We presume that the [FeII–FeII] state is unstable and will continue decaying. | ||
Reacting CrHydA1adt and CrHydA1pdt with H2O2 directly (Fig. 7(B)) allows bypassing the need for the explicit protonation step necessary in O2-induced deactivation. We emphasize that these results agree with superoxide as an intermediate species in O2-induced deactivation. Scheme 1 depicts how H2O2 formation may produce an unstable H-cluster species in the +4 state, not protected by a ligand in apical position at Fed. Eventually, H2O2 will continue to disintegrate the H-cluster and other metal centers in the enzyme.15–18
Conclusions
We have probed active site mimics of [FeFe]-hydrogenase and the respective enzyme variants toward O2 deactivation and their reactivity with various reactive oxygen species (ROS). Our results show that in the case of the model complexes, the primary deleterious interaction occurs at the iron center(s). Concomitant O2 binding and oxidation of the diiron site is required. Thereafter, a rapid degenerative pathway follows, which is influenced by the nature of the bridgehead ligand. The ADTCN complex [1], featuring an amine bridgehead, exhibits higher response towards O2, while the PDTCN complex [2], with a methylene bridgehead, shows reduced reactivity. Furthermore, the reactivity of mono- and di-cyanide substituted complexes correlates with increased O2 and H2O2 susceptibility, presumably due to a decrease in acidity proportional to the number of cyanide ligands.75In [FeFe]-hydrogenase fine tuning via the amine bridgehead and the diatomic ligands of the H-cluster is necessary for the supreme catalytic activity. This includes proton transfer, the formation of a frustrated Lewis pair between adt and Fed, hydrogen bonding between CN− and protein (Fig. 1), and other “outer” coordination sphere effects.9 However, this unique arrangement comes at the expense of O2 sensitivity where electron transfer precedes proton transfer resulting in the sub-stochiometric production of harmful H2O2 (Scheme 1). While [FeFe]-hydrogenases treat O2 essentially like a proton, [NiFe]-hydrogenases are reversibly inhibited by O282 or have evolved to fully reduce O2 to two equivalents of H2O, e.g., in Knallgas bacteria.25–29 Some [FeFe]-hydrogenases adopt inhibited states in the presence of O220–24 but from a physiological perspective, [FeFe]-hydrogenase may fail at O2 tolerance due to the lack of evolutionary pressure.83 Our study now establishes reactivity patterns of [FeFe]-hydrogenase mimics, offering insights into the initial steps of oxygen deactivation in the enzyme. We propose a model in which O2 binds to the H-cluster, is reduced to an apical superoxide ligand and reacts to H2O2 upon proton transfer and disproportionation. Such species then continue to deactivate the [FeFe]-hydrogenase.15–18
Remarkably, the active site mimics react with protons resulting in a bridging hydride state (FeII–μH–FeII), which is unresponsive towards O2 and H2O2. This observation agrees with the enhanced O2-stability of certain H-cluster intermediates62 where a μH ligand is formed at the reduced diiron site and an apical CO ligand slows down aerobic deactivation.59–61 The identification of an O2-stable FeII–μH–FeII state may inspire the design of new O2 tolerant catalysts.
Abbreviations
| adt | Azadithiolate |
| pdt | Propanedithiolate |
| ROS | Reactive oxygen species |
| FTIR | Fourier-transform infrared |
| NMR | Nuclear magnetic resonance |
| EPR | Electron paramagnetic resonance |
| DFT | Density functional theory |
Author contributions
SY synthesized complexes, performed spectroscopy, and wrote the manuscript; RH produced and activated the enzyme; EBB performed DFT calculations; MR analyzed DFT calculations; TH reviewed the manuscript; UPA analyzed data and wrote the manuscript; STS performed spectroscopy, analyzed data, and wrote the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Melanie Heghmanns and Müge Kasanmascheff for assistance with the EPR measurements of the samples. STS acknowledges Joachim Heberle for providing access to his laboratory and spectrometers at Freie Universität Berlin. We thank Moritz Senger for fruitful discussions. SY gratefully acknowledges the German Academic Exchange Service (DAAD) for a scholarship. This work was supported by the Deutsche Forschungsgemeinschaft (DFG, RO 5688/1, AP242/12-1, and STR 1554/5-1). This work was likewise supported by the Fraunhofer Internal Programs under Grant No. Attract 097-602175 and the DFG under Germany's Excellence Strategy – EXC-2033 – Projektnummer 390677874 “RESOLV”.References
- International Energy Agency, Global Hydrogen Review 2021, OECD, 2021.
- B.-F. M. for E. A. and C. Action, “The National Hydrogen Strategy,” can be found under https://www.bmwk.de/Redaktion/EN/Publikationen/Energie/the-national-hydrogen-strategy.html, n.d.
- D. Siegmund, S. Metz, V. Peinecke, T. E. Warner, C. Cremers, A. Grevé, T. Smolinka, D. Segets and U.-P. Apfel, JACS Au, 2021, 1, 527–535 CrossRef CAS PubMed.
- M. Yue, H. Lambert, E. Pahon, R. Roche, S. Jemei and D. Hissel, Renewable Sustainable Energy Rev., 2021, 146, 111180 CrossRef.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- C. Madden, M. D. Vaughn, I. Díez-Pérez, K. A. Brown, P. W. King, D. Gust, A. L. Moore and T. A. Moore, J. Am. Chem. Soc., 2012, 134, 1577–1582 CrossRef CAS PubMed.
- J. T. Kleinhaus, F. Wittkamp, S. Yadav, D. Siegmund and U.-P. Apfel, Chem. Soc. Rev., 2021, 50, 1668–1784 RSC.
- J. A. Birrell, P. Rodríguez-Maciá, E. J. Reijerse, M. A. Martini and W. Lubitz, Coord. Chem. Rev., 2021, 449, 214191 CrossRef CAS.
- S. T. Stripp, B. R. Duffus, V. Fourmond, C. Léger, S. Leimkühler, S. Hirota, Y. Hu, A. Jasniewski, H. Ogata and M. W. Ribbe, Chem. Rev., 2022, 122, 11900–11973 CrossRef CAS PubMed.
- H. Tai, S. Hirota and S. T. Stripp, Acc. Chem. Res., 2021, 54, 232–241 CrossRef CAS PubMed.
- J. Esselborn, N. Muraki, K. Klein, V. Engelbrecht, N. Metzler-Nolte, U.-P. Apfel, E. Hofmann, G. Kurisu and T. Happe, Chem. Sci., 2016, 7, 959–968 RSC.
- A. Kubas, C. Orain, D. De Sancho, L. Saujet, M. Sensi, C. Gauquelin, I. Meynial-Salles, P. Soucaille, H. Bottin, C. Baffert, V. Fourmond, R. B. Best, J. Blumberger and C. Léger, Nat. Chem., 2017, 9, 88–95 CrossRef CAS PubMed.
- M. Mohammadi and H. Vashisth, J. Phys. Chem. B, 2017, 121, 10007–10017 CrossRef CAS PubMed.
- J. Cohen, K. Kim, P. King, M. Seibert and K. Schulten, Structure, 2005, 13, 1321–1329 CrossRef CAS PubMed.
- M. T. Stiebritz and M. Reiher, Inorg. Chem., 2009, 48, 7127–7140 CrossRef CAS PubMed.
- J. Esselborn, L. Kertess, U.-P. Apfel, E. Hofmann and T. Happe, J. Am. Chem. Soc., 2019, 141, 17721–17728 CrossRef CAS PubMed.
- S. T. Stripp, G. Goldet, C. Brandmayr, O. Sanganas, K. A. Vincent, M. Haumann, F. A. Armstrong and T. Happe, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(41), 17331–17336 CrossRef CAS PubMed.
- K. D. Swanson, M. W. Ratzloff, D. W. Mulder, J. H. Artz, S. Ghose, A. Hoffman, S. White, O. A. Zadvornyy, J. B. Broderick, B. Bothner, P. W. King and J. W. Peters, J. Am. Chem. Soc., 2015, 137, 1809–1816 CrossRef CAS PubMed.
- G. Goldet, C. Brandmayr, S. T. Stripp, T. Happe, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2009, 131, 14979–14989 CrossRef CAS PubMed.
- S. Morra, M. Arizzi, F. Valetti and G. Gilardi, Biochemistry, 2016, 55, 5897–5900 CrossRef CAS PubMed.
- M. Winkler, J. Duan, A. Rutz, C. Felbek, L. Scholtysek, O. Lampret, J. Jaenecke, U.-P. Apfel, G. Gilardi, F. Valetti, V. Fourmond, E. Hofmann, C. Léger and T. Happe, Nat. Commun., 2021, 12, 756 CrossRef CAS PubMed.
- H. Land, M. Senger, G. Berggren and S. T. Stripp, ACS Catal., 2020, 10, 7069–7086 CrossRef CAS.
- H. Land, A. Sekretareva, P. Huang, H. J. Redman, B. Németh, N. Polidori, L. S. Mészáros, M. Senger, S. T. Stripp and G. Berggren, Chem. Sci., 2020, 11, 12789–12801 RSC.
- P. R. Cabotaje, K. Walter, A. Zamader, P. Huang, F. Ho, H. Land, M. Senger and G. Berggren, ACS Catal., 2023, 13, 10435–10446 CrossRef CAS PubMed.
- J. A. Cracknell, A. F. Wait, O. Lenz, B. Friedrich and F. A. Armstrong, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20681–20686 CrossRef PubMed.
- J. Fritsch, P. Scheerer, S. Frielingsdorf, S. Kroschinsky, B. Friedrich, O. Lenz and C. M. T. Spahn, Nature, 2011, 479, 249–252 CrossRef CAS PubMed.
- S. Frielingsdorf, J. Fritsch, A. Schmidt, M. Hammer, J. Löwenstein, E. Siebert, V. Pelmenschikov, T. Jaenicke, J. Kalms, Y. Rippers, F. Lendzian, I. Zebger, C. Teutloff, M. Kaupp, R. Bittl, P. Hildebrandt, B. Friedrich, O. Lenz and P. Scheerer, Nat. Chem. Biol., 2014, 10, 378–385 CrossRef CAS PubMed.
- P. Wulff, C. C. Day, F. Sargent and F. A. Armstrong, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6606–6611 CrossRef CAS PubMed.
- L. Lauterbach and O. Lenz, J. Am. Chem. Soc., 2013, 135, 17897–17905 CrossRef CAS PubMed.
- H. Li and T. B. Rauchfuss, J. Am. Chem. Soc., 2002, 124, 726–727 CrossRef CAS PubMed.
- J. L. Stanley, T. B. Rauchfuss and S. R. Wilson, Organometallics, 2007, 26, 1907–1911 CAS.
- A. L. Cloirec, S. C. Davies, D. J. Evans, D. L. Hughes, C. J. Pickett, S. P. Best and S. Borg, Chem. Commun., 1999, 2285–2286 RSC.
- E. J. Lyon, I. P. Georgakaki, J. H. Reibenspies and M. Y. Darensbourg, Angew. Chem., Int. Ed., 1999, 38, 3178–3180 CrossRef CAS PubMed.
- L. Mond and C. Langer, J. Chem. Soc., Trans., 1891, 59, 1090–1093 RSC.
- W. Hieber and J. Gruber, Z. Anorg. Allg. Chem., 1958, 296, 91–103 CrossRef CAS.
- S. Dey, A. Rana, D. Crouthers, B. Mondal, P. K. Das, M. Y. Darensbourg and A. Dey, J. Am. Chem. Soc., 2014, 136, 8847–8850 CrossRef CAS PubMed.
- V. C.-C. Wang, C. Esmieu, H. J. Redman, G. Berggren and L. Hammarström, Dalton Trans., 2020, 49, 858–865 RSC.
- J. Windhager, R. A. Seidel, U.-P. Apfel, H. Görls, G. Linti and W. Weigand, Chem. Biodiversity, 2008, 5, 2023–2041 CrossRef CAS PubMed.
- T. Liu, B. Li, M. L. Singleton, M. B. Hall and M. Y. Darensbourg, J. Am. Chem. Soc., 2009, 131, 8296–8307 CrossRef CAS PubMed.
- M. Y. Darensbourg and W. Weigand, Eur. J. Inorg. Chem., 2011, 994–1004 CrossRef CAS.
- J. A. Imlay, Mol. Microbiol., 2006, 59, 1073–1082 CrossRef PubMed.
- A. R. Finkelmann, M. T. Stiebritz and M. Reiher, Inorg. Chem., 2014, 53, 11890–11902 CrossRef CAS PubMed.
- M. K. Bruska, M. T. Stiebritz and M. Reiher, J. Am. Chem. Soc., 2011, 133, 20588–20603 CrossRef CAS PubMed.
- M. Schieber and N. S. Chandel, Curr. Biol., 2014, 24, R453–R462 CrossRef CAS PubMed.
- L. Kertess, F. Wittkamp, C. Sommer, J. Esselborn, O. Rüdiger, E. J. Reijerse, E. Hofmann, W. Lubitz, M. Winkler, T. Happe and U.-P. Apfel, Dalton Trans., 2017, 46, 16947–16958 RSC.
- C. Esmieu and G. Berggren, Dalton Trans., 2016, 45, 19242–19248 RSC.
- J. F. Siebel, A. Adamska-Venkatesh, K. Weber, S. Rumpel, E. Reijerse and W. Lubitz, Biochemistry, 2015, 54, 1474–1483 CrossRef CAS PubMed.
- F. Gloaguen, J. D. Lawrence, M. Schmidt, S. R. Wilson and T. B. Rauchfuss, J. Am. Chem. Soc., 2001, 123, 12518–12527 CrossRef CAS PubMed.
- S. Detoni and D. Hadzi, Spectrochim. Acta, 1956, 11, 601–608 CrossRef.
- S. Mebs, R. Kositzki, J. Duan, L. Kertess, M. Senger, F. Wittkamp, U.-P. Apfel, T. Happe, S. T. Stripp, M. Winkler and M. Haumann, Biochim. Biophys. Acta, Bioenerg., 2018, 1859, 28–41 CrossRef CAS PubMed.
- J. Noth, R. Kositzki, K. Klein, M. Winkler, M. Haumann and T. Happe, Sci. Rep., 2015, 5, 13978 CrossRef CAS PubMed.
- X. Zhao, I. P. Georgakaki, M. L. Miller, J. C. Yarbrough and M. Y. Darensbourg, J. Am. Chem. Soc., 2001, 123, 9710–9711 CrossRef CAS PubMed.
- F. Gloaguen, J. D. Lawrence, T. B. Rauchfuss, M. Bénard and M.-M. Rohmer, Inorg. Chem., 2002, 41, 6573–6582 CrossRef CAS PubMed.
- S. Rumpel, E. Ravera, C. Sommer, E. Reijerse, C. Farès, C. Luchinat and W. Lubitz, J. Am. Chem. Soc., 2018, 140, 131–134 CrossRef CAS PubMed.
- A. Jablonskytė, J. A. Wright and C. J. Pickett, Dalton Trans., 2010, 39, 3026 RSC.
- D. W. Mulder, Y. Guo, M. W. Ratzloff and P. W. King, J. Am. Chem. Soc., 2017, 139, 83–86 CrossRef CAS PubMed.
- M. Winkler, M. Senger, J. Duan, J. Esselborn, F. Wittkamp, E. Hofmann, U.-P. Apfel, S. T. Stripp and T. Happe, Nat. Commun., 2017, 8, 16115 CrossRef CAS PubMed.
- E. J. Reijerse, C. C. Pham, V. Pelmenschikov, R. Gilbert-Wilson, A. Adamska-Venkatesh, J. F. Siebel, L. B. Gee, Y. Yoda, K. Tamasaku, W. Lubitz, T. B. Rauchfuss and S. P. Cramer, J. Am. Chem. Soc., 2017, 139, 4306–4309 CrossRef CAS PubMed.
- S. T. Stripp, S. Mebs and M. Haumann, Inorg. Chem., 2020, 59, 16474–16488 CrossRef CAS PubMed.
- M. Haumann and S. T. Stripp, Acc. Chem. Res., 2018, 51, 1755–1763 CrossRef CAS PubMed.
- S. Mebs, M. Senger, J. Duan, F. Wittkamp, U.-P. Apfel, T. Happe, M. Winkler, S. T. Stripp and M. Haumann, J. Am. Chem. Soc., 2017, 139, 12157–12160 CrossRef CAS PubMed.
- J. Duan, S. Mebs, K. Laun, F. Wittkamp, J. Heberle, T. Happe, E. Hofmann, U.-P. Apfel, M. Winkler, M. Senger, M. Haumann and S. T. Stripp, ACS Catal., 2019, 9, 9140–9149 CrossRef CAS.
- C. Lorent, S. Katz, J. Duan, C. J. Kulka, G. Caserta, C. Teutloff, S. Yadav, U.-P. Apfel, M. Winkler, T. Happe, M. Horch and I. Zebger, J. Am. Chem. Soc., 2020, 142, 5493–5497 CrossRef CAS PubMed.
- J. A. Birrell, V. Pelmenschikov, N. Mishra, H. Wang, Y. Yoda, K. Tamasaku, T. B. Rauchfuss, S. P. Cramer, W. Lubitz and S. DeBeer, J. Am. Chem. Soc., 2020, 142, 222–232 CrossRef CAS PubMed.
- M. W. Ratzloff, J. H. Artz, D. W. Mulder, R. T. Collins, T. E. Furtak and P. W. King, J. Am. Chem. Soc., 2018, 140, 7623–7628 CrossRef CAS PubMed.
- F. Gloaguen, J. D. Lawrence and T. B. Rauchfuss, J. Am. Chem. Soc., 2001, 123, 9476–9477 CrossRef CAS PubMed.
- Y. Sheng, I. A. Abreu, D. E. Cabelli, M. J. Maroney, A.-F. Miller, M. Teixeira and J. S. Valentine, Chem. Rev., 2014, 114, 3854–3918 CrossRef CAS PubMed.
- F. Roncaroli, E. Bill, B. Friedrich, O. Lenz, W. Lubitz and M.-E. Pandelia, Chem. Sci., 2015, 6, 4495–4507 RSC.
- A. S. Pereira, P. Tavares, I. Moura, J. J. G. Moura and B. H. Huynh, J. Am. Chem. Soc., 2001, 123, 2771–2782 CrossRef CAS PubMed.
- M. Lorenzi, J. Gellett, A. Zamader, M. Senger, Z. Duan, P. Rodríguez-Maciá and G. Berggren, Chem. Sci., 2022, 13, 11058–11064 RSC.
- A. Fasano, H. Land, V. Fourmond, G. Berggren and C. Léger, J. Am. Chem. Soc., 2021, 143, 20320–20325 CrossRef CAS PubMed.
- M. Senger, V. Eichmann, K. Laun, J. Duan, F. Wittkamp, G. Knör, U.-P. Apfel, T. Happe, M. Winkler, J. Heberle and S. T. Stripp, J. Am. Chem. Soc., 2019, 141, 17394–17403 CrossRef CAS PubMed.
- J. Duan, M. Senger, J. Esselborn, V. Engelbrecht, F. Wittkamp, U.-P. Apfel, E. Hofmann, S. T. Stripp, T. Happe and M. Winkler, Nat. Commun., 2018, 9, 4726 CrossRef PubMed.
- A. Kubas, D. De Sancho, R. B. Best and J. Blumberger, Angew. Chem., Int. Ed., 2014, 53, 4081–4084 CrossRef CAS PubMed.
- A. Nayek, S. Dey, S. Patra, A. Rana, P. N. Serrano, S. J. George, S. P. Cramer, S. G. Dey and A. Dey, Chem. Sci., 2024, 15, 2167–2180 RSC.
- F. Wang, M. Wang, X. Liu, K. Jin, W. Dong and L. Sun, Dalton Trans., 2007, 3812–3819 RSC.
- F. Wang, M. Wang, X. Liu, K. Jin, W. Dong, G. Li, B. Åkermark and L. Sun, Chem. Commun., 2005, 3221–3223 RSC.
- G. Eilers, L. Schwartz, M. Stein, G. Zampella, L. de Gioia, S. Ott and R. Lomoth, Chem. – Eur. J., 2007, 13, 7075–7084 CrossRef CAS PubMed.
- Ö. F. Erdem, M. Stein, S. Kaur-Ghumaan, E. J. Reijerse, S. Ott and W. Lubitz, Chem. – Eur. J., 2013, 19, 14566–14572 CrossRef PubMed.
- T. B. Rauchfuss, Acc. Chem. Res., 2015, 48, 2107–2116 CrossRef CAS PubMed.
- W. Roseboom, A. L. De Lacey, V. M. Fernandez, E. C. Hatchikian and S. P. J. Albracht, J. Biol. Inorg. Chem., 2006, 11, 102–118 CrossRef CAS PubMed.
- K. A. Vincent, A. Parkin, O. Lenz, S. P. J. Albracht, J. C. Fontecilla-Camps, R. Cammack, B. Friedrich and F. A. Armstrong, J. Am. Chem. Soc., 2005, 127, 18179–18189 CrossRef CAS PubMed.
- S. Morra, Front. Microbiol., 2022, 13 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp06048a |
| This journal is © the Owner Societies 2024 |