
Refining the mechanism of CO2 and H2 activation over gold-ceria catalysts by IR modulation excitation spectroscopy†
Jakob
Weyel
and
Christian
Hess
 *
*
Eduard-Zintl-Institute of Inorganic and Physical Chemistry, Technical University of Darmstadt, Peter-Grünberg-Str. 8, 64287 Darmstadt, Germany. E-mail: christian.hess@tu-darmstadt.de
First published on 31st January 2024
Abstract
The activation and utilization of the greenhouse gas CO2 is of great interest for the energy transition as a fossil-free carbon source for mitigating climate change. CO2 hydrogenation via the reverse water–gas shift reaction (RWGSR) converts CO2 to CO, a crucial component of syngas, enabling further transformation by means of the Fischer–Tropsch process. In this study, we unravel the detailed mechanism of the RWGSR on low-loaded Au/CeO2 catalysts using IR modulation excitation spectroscopy (MES), by periodically modulating the concentration of the reactants, followed by phase-sensitive detection (PSD). Applying such a MES-PSD approach to Au/CeO2 catalysts during RWGSR gives direct spectroscopic evidence for the active role of gold hydride, bidentate carbonate and hydroxyl species in the reaction mechanism, while disproving the participation of other species such as formate. Our results highlight the potential of modulation excitation spectroscopy combined with phase-sensitive detection to provide new mechanistic insight into catalytic reactions not accessible by steady-state techniques, including a profound understanding of the sequence of reaction steps.
Introduction
The reverse water–gas shift reaction (RWGSR) over Au/CeO2 catalysts is of great interest for the energy transition,1–5 enabling the conversion of CO2 to CO, the key component of syngas besides H2. Syngas can subsequently be used as a platform chemical to produce chemical energy storage materials like methanol or even alkanes via the Fischer–Tropsch mechanism.2,3,6 The RWGSR can therefore make use of emitted CO2 and mitigate its effect on global warming by simultaneously providing a new source for syngas production.Our previous studies on RWGSR have demonstrated that low-loaded Au/CeO2 catalysts operate mainly according to an associative mechanism,7 in contrast to comparable Cu/CeO2 where a redox mechanism dominates.8 Nevertheless, the RWGSR mechanism over Au/CeO2 catalysts, in particular the sequence of steps is not fully understood, leading to a demand for development and application of spectroscopic techniques, which can enhance our mechanistic understanding.
Modulation excitation spectroscopy (MES) in combination with the mathematical tool of phase-sensitive detection (PSD)9,10 and applied to diffuse reflectance infrared Fourier transform spectroscopy (ME-DRIFTS) can differentiate between surface species that appear to be actively participating and those that are merely spectators. ME-DRIFTS has previously been used to gain detailed insight into the mechanistic behavior of heterogeneously catalyzed reactions, such as (preferential) CO oxidation,11–14 CO2 hydrogenation,15–18 SCR19–21 or propane ODH22 among others.23–25 In a previous study, we applied the ME-DRIFTS technique to the CO oxidation over low-loaded Au/CeO2 catalysts, allowing us to elucidate new details on the CO oxidation mechanism,11 not accessible by steady-state spectroscopy. In the context of the RWGSR we expect ME-DRIFTS to provide new insight into the adsorption, desorption and transformation of surface species such as carbonates, formates and hydroxides, which have previously been proposed to play a crucial role in the RWGSR.5,7,26,27
In this contribution, we address the mechanism of the RWGSR over low-loaded Au/CeO2 catalysts by using IR modulation excitation spectroscopy. By studying the catalysts’ behavior during different modulation approaches and by applying phase-sensitive detection, we develop a comprehensive and temporally-resolved picture of the surface processes relevant to the catalysis, which has not been available previously by using steady-state approaches. While illustrated here for the CO2 activation over gold–ceria, our study highlights the more general potential of the MES-PSD approach to refine the mechanistic understanding of heterogeneous catalysts.
Experimental section
Catalyst preparation and characterization.
Polycrystalline ceria sheets were prepared by thermal decomposition of Ce(NO3)3·6H2O (Alfa Aesar, 99.5%) at 600 °C, as described in detail previously.7,28,29 To load the ceria sheets with gold electrolyte deposition is used. After dispersing the ceria samples in deionized water at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :150, the pH is adjusted to 9 using a 0.1 M NaOH solution (98%, Grüssing GmbH). Then, a 10−3 M solution of HAuCl4·3H2O (99.9%, Carl Roth), adjusted to pH 9, is added to the ceria dispersion to attain a loading of 0.5 wt% Au. The reaction mixture is heated to 65 °C for two hours, cooled, and then placed in an ultrasonic bath for 30 min. Lastly, the residue is centrifuged, washed four times with deionized water, and then dried in a drying oven at 85 °C for about 24 h. Details on the characterization of the gold–ceria samples can be found elsewhere.7
:150, the pH is adjusted to 9 using a 0.1 M NaOH solution (98%, Grüssing GmbH). Then, a 10−3 M solution of HAuCl4·3H2O (99.9%, Carl Roth), adjusted to pH 9, is added to the ceria dispersion to attain a loading of 0.5 wt% Au. The reaction mixture is heated to 65 °C for two hours, cooled, and then placed in an ultrasonic bath for 30 min. Lastly, the residue is centrifuged, washed four times with deionized water, and then dried in a drying oven at 85 °C for about 24 h. Details on the characterization of the gold–ceria samples can be found elsewhere.7
Modulation excitation (ME) DRIFTS
The basic experimental setup for conventional DRIFTS and ME-DRIFTS was described in detail in our previous studies.7,11,25,30 The setup used in this study (see Fig. 1) consisted of a Bruker Vertex 70 spectrometer with a liquid nitrogen cooled mercury cadmium telluride (MCT) detector and a commercial reaction cell (Praying Mantis™ High Temperature Reaction Chamber, Harrick Scientific Products). Typically, 25–35 mg of material was placed in a stainless-steel sample holder (∅: 8 mm; depth: 0.5 mm). In ME-DRIFTS experiments, during each period, the concentration of either CO2 was switched between 0 and 2 mol%, while the concentration of H2 was kept constant at 4 mol%, or vice versa, i.e., the concentration of H2 was switched between 0 and 4 mol% at a constant flow of 2 mol% CO2. Ar was used as inert carrier gas and the total flow rate was set to 100 mL min−1. The samples were treated for 15 min at the reaction temperature of 250 °C in a reductive 4 mol% H2/Ar environment (for CO2 modulation) or a more oxidative 2 mol% CO2/Ar environment (for H2 modulation). Background spectra were measured at the reaction temperature after one modulation period as a pretreatment. The modulation experiments consisted of 20 periodic concentration changes of 123 s length each. Each interferogram yielded a spectrum, so that approximately every 1.5 s a spectrum was acquired, which was used as the experimental uncertainty for the derived time values, unless stated otherwise.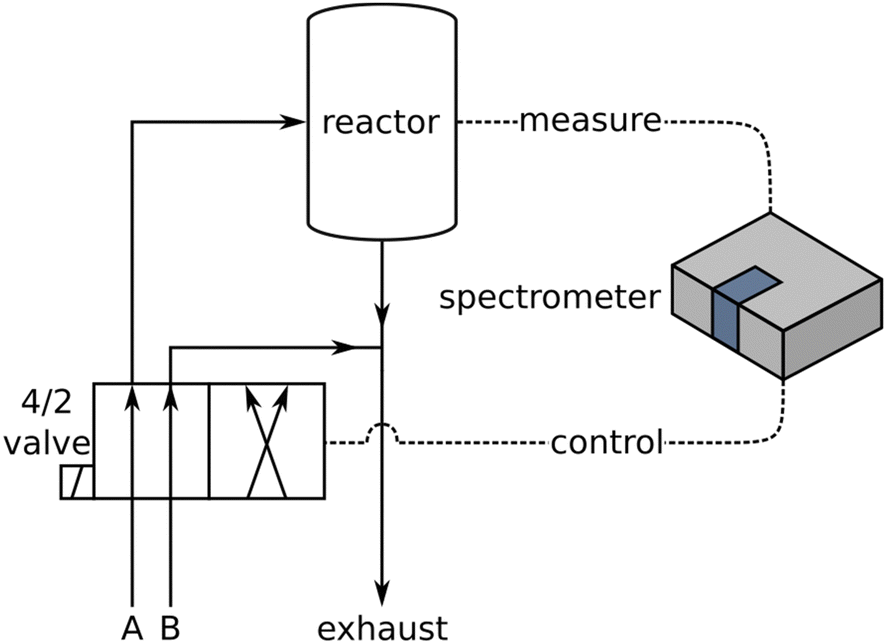 | ||
| Fig. 1 Scheme of the experimental ME-DRIFTS setup. For details see text. | ||
Based on previous steady-state reactivity experiments at 250 °C the Au/CeO2 sheets with a loading of 0.31 wt% gold, which were primarily used in this work, exhibit a CO2 conversion of 5.4% at 100% selectivity towards CO, while the bare ceria sheets are also active showing a CO2 conversion of 1.6% and 100% CO selectivity under identical operating conditions.7
Phase angles (φ in °) extracted via PSD are translated into the discussed time values (tφ in s) via the following formula
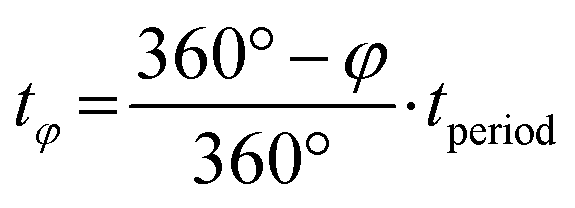
Results and discussion
CO2 modulation experiments
Fig. 2 shows a comparison of time-resolved DRIFT spectra (top) recorded during RWGSR over Au/CeO2 at 250 °C and the resulting PSD spectra (bottom). In these CO2 modulation experiments, the CO2 concentration was switched between 0 and 2 mol%, while the H2 concentration was kept constant at 4 mol%. In general, the use of PSD reduces the presence of noise in the spectra (see also Fig. S1, ESI†), making it possible to investigate the occurrence of small signals, such as CO adsorbate-related signals.11 However, the set of PSD spectra at the bottom of Fig. 2 reveals that, apart from the two gaseous species CO2 (reactant) and CO (product), there are no features within the region of 2000 to 2200 cm−1, which is typical for CO adsorbates, that are useful in clarifying reaction mechanisms by means of tracking adsorption sites and oxidation states as has been reported earlier.11,31–34 On the other hand, further signals in the PSD spectra are located in the carbonate region below 1800 cm−1, in the C–H stretching region at around 2800 cm−1, and in the hydroxyl region above 3500 cm−1. When the view is enlarged, a faint signal at 1953 cm−1 also becomes visible (see Fig. S1, ESI†), which, as DFT calculations have shown,7 is assigned to a Au–H2 species, whose asymmetry towards higher wavenumbers indicates the presence of H2 adsorbed to larger gold clusters (Aun–H2) as well.7,35Table 1 provides a summary of all detected signals together with their relative time values. The presence of the other product, H2O, was not observed in the CO2 modulation spectra, but will be discussed in the context of complementary H2 modulation experiments (see Fig. 3).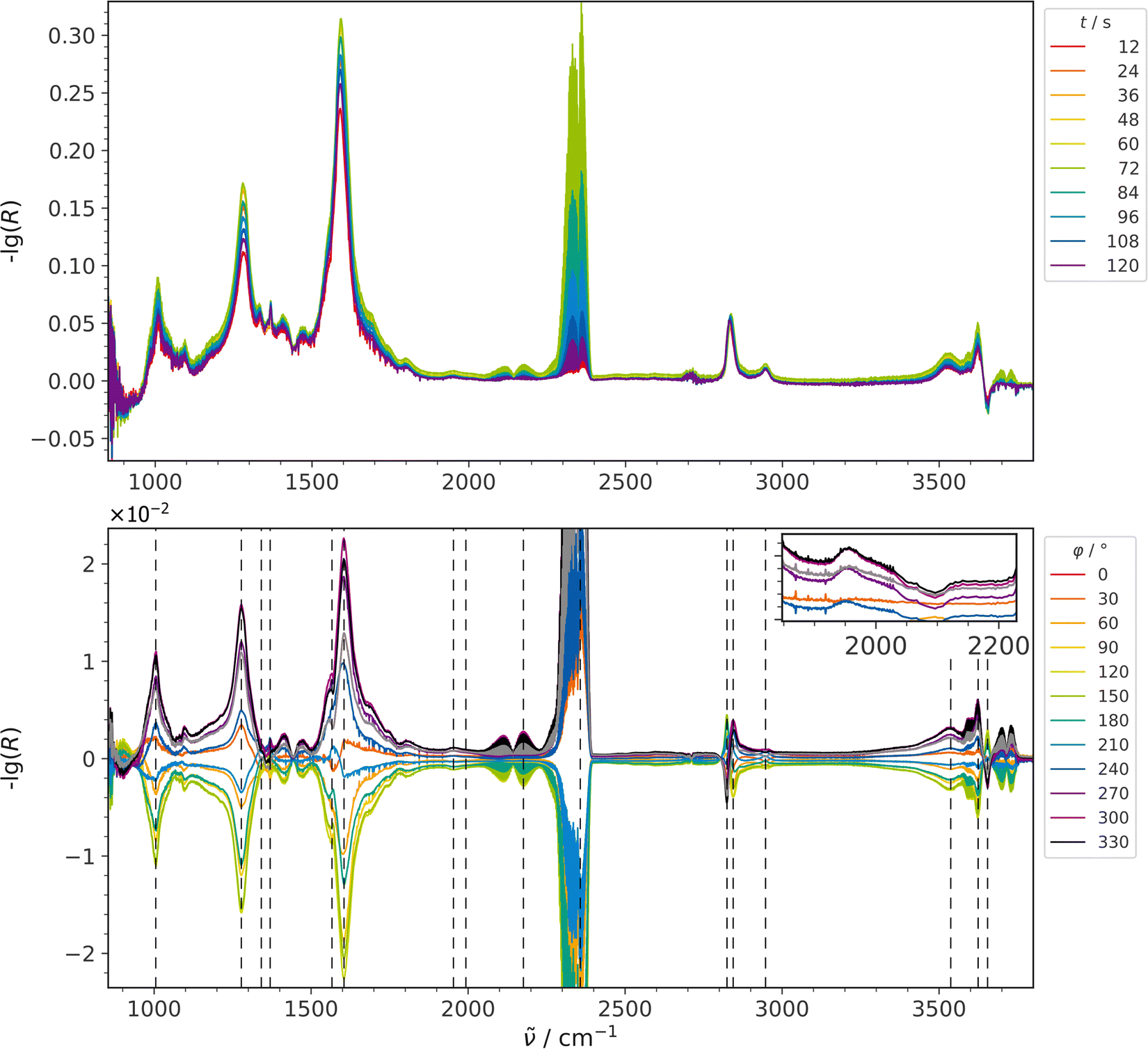 | ||
| Fig. 2 ME-DRIFT spectra during RWGSR over Au/CeO2 for CO2 modulation conditions, that is, switching the CO2 concentration between 0 and 2 mol%, while keeping the H2 concentration constant at 4 mol%. Top: Selected spectra from the time-resolved data set before applying PSD. Bottom: PSD spectra in an enlarged view cutting the intensity of the gas-phase CO2 signal. The inset provides an enlarged view of the Au–H2 signal. Signal positions from Table 1 are indicated by dashed lines. | ||
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) /cm−1 /cm−1 |
t φ /s | Assignment |
|---|---|---|
| 2824 | (−) 12 | Formate |
| 2010 | 14 | Aun–H2 |
| 1953 | 15 | Au–H2 |
| 2363 | 16 | CO2 (g) |
| 1007 | 17 | Bidentate carbonate |
| 1278 | 17 | Bidentate carbonate |
| 3530 | 17 | OH, vicinity to Au |
| 3613 | 18 | OH(III) |
| 1604 | 19 | Bidentate carbonate |
| 2173 | 19 | CO (g) |
| 3654 | (−) 20 | OH (II*-B) |
| 1570 | 23 | Formate |
| 2847 | 24 | Formate |
| 1332 | 26 | Formate |
| 2946 | 27 | Formate |
| 1369 | 41 | Formate |
 | ||
| Fig. 3 ME-DRIFT spectra during RWGSR over Au/CeO2 for CO2 modulation conditions, that is, switching the H2 concentration between 0 and 4 mol%, while keeping the CO2 concentration constant at 2 mol%. Top: Selected spectra from the time-resolved data set before applying PSD. Bottom: PSD spectra. The inset provides an enlarged view of the Au–H2 signal. Signal positions from Table 2 are indicated by dashed lines. | ||
Table 1 lists the detected features according to their time values. There is a number of signals occurring prior to product formation (CO). The signal of the reactant CO2 appears at 16 s together with signals assigned to molecularly adsorbed hydrogen on gold clusters and single gold atoms,7 considering the experimental uncertainty of each signal of 1.5 s. They are followed by two carbonate signals at 17 s, signals of OH in the vicinity to Au at 17 s,7 a triple (III) bridged OH group at 18 s,36 and yet another carbonate signal at 19 s, before the appearance of the CO signal at 20 s. The signal of the OH vibration observed at 20 s, which can be attributed to OH located on a more reduced ceria surface (II*-B),36 should also be taken into account regarding the mechanism because its error range of 1.5 s overlaps with that of CO. Its presence would be consistent with a more reduced ceria surface (before CO2 is switched on) and its possible transformation into an OH II-B species on a more oxidized surface (after CO2 is switched on) as has been discussed in the literature but the adjacent OH (II-B) signal, expected at about 3630 to 3640 cm−1, is not visible in this data set.7,36 Based on the literature, the carbonate signals located at 1278 (17 s), 1007 (17 s) and 1604 cm−1 (19 s) can be attributed to a bidentate carbonate forming on ceria.7,37–39 The difference in time values falls within the experimental uncertainty of each signal of 1.5 s, suggesting coincidence and thus the presence of different vibrations of the same molecule.
Previously, besides the signal at 1953 cm−1, another hydrogen-related signal was observed around 2134 cm−1,7,35 and, also with the aid of DFT calculations,7 attributed to dissociatively adsorbed hydrogen. Furthermore the 2134 cm−1 signal is not clearly detected for the Au/CeO2 sheets investigated here. Also, in non-modulated experiments (See Fig. S1, ESI†) there is almost no difference observed in the region around 2134 cm−1 when comparing Au/CeO2 samples with different loadings and bare CeO2. However, there is a direct correlation between gold loading and the intensity of the signal assigned to gold hydrides at approximately 1953 cm−1 visible in that particular data set. This observation is in stark contrast to samples with different nanoparticle shape, for example, Au/CeO2 polyhedra.7 While the assignment of the Au hydrides was confirmed by DFT calculations,7 further experimental confirmation by D2 exchange experiments is not possible, as the bands shift into the carbonate/formate region and the Au–H2 region is covered by C–D vibrations of formates (not shown).
The signal at 2824 cm−1, which originates from the C–H stretching vibration of a formate,5,7,37,38,40 appears before that of the modulated reactant CO2. Previously, this signal has been reported to reversibly shift its position upon changes in the gas environment, which may result in a distorted appearance of the signal during modulation experiments.7 Such a behavior of the time resolved C–H signals can also be seen in the data shown here (see Fig. 2), leading to the twisted behavior of the modulation signals mentioned. Thus, the early appearance of the 2824 cm−1 signal at (−) 12 s, which is 4 s before the reactant CO2 is introduced, can be attributed to a potential overlap of two opposing signals. As noted in one of our previous studies,11 the overlap of neighboring signals leads to a blurring of the adjacent time values extracted through PSD. Consequently, these time values may be prone to significant errors, requiring caution in their interpretation or even exclusion.
Following product (CO) formation, several signals persist, which are primarily due to formate vibrations with time values between 23 s and 41 s. The most isolated formate signal at 2946 cm−1 (27 s) has no neighbor signals and is therefore considered to be least prone to errors according to the discussion above. Moreover, signals located at 1570 cm−1 (23 s), 2847 cm−1 (24 s), 1332 cm−1 (26 s) and possibly also at 1369 cm−1 (41 s), which were discussed in previous studies,5,7,37,38,40 may be linked to the same species but due to the temporal deviations observed this cannot be stated without doubt. The relevant information of the time of formate appearance in the reaction mechanism can still be retrieved as will be discussed later.
H2 modulation experiments
To obtain a more detailed picture of the reaction mechanism, we reversed the modulation procedure by periodically pulsing H2 while keeping the CO2 flow constant. The DRIFT spectra collected under these conditions are depicted in Fig. 3. All major signals that were visible for CO2 modulation were also detected for H2 modulation and are summarized in Table 2, together with their corresponding time values.|
/cm−1 |
t φ /s | Assignment |
|---|---|---|
| 1291 | 3 | Carbonate |
| 1609 | (−) 9 | Bidentate carbonate |
| 2363 | (−) 17 | CO2 (g) |
| 1940 | 19 | Au–H2 |
| 2015 | 19 | Aun–H2 |
| 3655 | 20 | OH (II*-B) |
| 3517 | 20 | OH, vicinity to Au |
| 3745 | 22 | H 2 O (g) |
| 3275 | 22 | H 2 O (ads) |
| 2173 | 23 | CO (g) |
| 3614 | 24 | OH(III) |
| 2946 | 25 | Formate |
| 2829 | 26 | Formate |
| 1270 | (−) 29 | Bidentate carbonate |
| 1583 | 36 | Formate |
| 2850 | 36 | Formate |
| 1368 | 37 | Formate |
| 1337 | 39 | Formate |
Comparing time- and phase-resolved spectra in Fig. 3 reveals a strong decrease in the intensity of some of the carbonate-related signals within 850–1800 cm−1 after application of PSD. Besides, there is a broad feature due to weakly adsorbed water, extending from about 1700 cm−1 to 3500 cm−1, which is prominent in the time- and phase-resolved spectra. Such broad features adversely affect the sensitivity of MES-PSD since any signal in the range covered by this broad responsive feature will also appear in the PSD spectra due to its non-selective nature in amplifying each spectral position if there is any response to the modulation frequency. This applies even to signals (caused by spectator species), which are superimposed on the broad absorption feature. The underlying dynamics of the broad active contribution cannot be separated from additional signals appearing above it by conventional PSD, so the latter are “transported” into the PSD spectra via the broad active feature.11 However, in contrast to the situation caused by inseparable neighboring signals, broad periodic background effects can be handled by background subtraction prior to PSD, as described previously.24
In the present case, such a procedure does not provide additional mechanistic insights as compared to the data shown in Fig. 2. All signals within the range of the broad water feature can be eliminated as potential active species based on the CO2 modulation experiments (see discussion below), or have been confirmed to be active in previous unmodulated transient experiments,7 in which Au–H2 adsorbates occurred under both reductive and oxidative conditions. For these species, CO2 modulation experiments provided clear evidence of their active participation in the RWGSR (see Table 1).
For H2 modulation, the broad band at around 2010 cm−1 originating from Aun–H2 species, which was observed as a shoulder of the Au–H2 signal during CO2 modulation, is present as a rather confined signal at 2015 cm−1 (see Fig. S1, ESI†), which is separated from the symmetrical signal of Au–H2 at 1940 cm−1, indicating the participation of less Aun–H2 species under these conditions.
While the time values for the features that overlap with the broad water signal between about 1700 and 3500 cm−1 seem to be in agreement with the findings from Fig. 2, the absolute numbers need to be treated with care, as they may be subject to errors due to their overlap with the broad feature.
The remaining signals, which are located primarily in the carbonate and hydroxyl region, exhibit an intriguing behavior. It should be mentioned that under these modulation conditions the second product, water, has become observable. However, water is not only present in its adsorbed form, centered around 3275 cm−1 and characterized by a time delay of 22 s, but also as a gas-phase species (evidenced by the 3745 cm−1 feature at 22 s). The CO gas-phase signal located at 2173 cm−1 appears at 23 s, so the formation of both product molecules seems to occur at about the same time.
The signals at 1337 cm−1 (39 s), 1368 cm−1 (36 s), and 1583 cm−1 (36 s) can be attributed to formates.5,7,37,38,40 This behavior reveals that formates are formed only after the reaction in a hydrogen-rich environment. The signals from C–H stretch vibrations of formate are located at 2829, 2850, and 2946 cm−1 and are characterized by time values of 26, 36, and 25 s, respectively. While these time values are higher than those of the products, they should be considered with reservation, due to the overlap of the C–H-related features with the broad water band.
Fig. 3 shows additional signals at 1270 and 1609 cm−1, which decrease when H2 is switched on and can be attributed to bidentate carbonate.7,37–39 Clearly assigning these signals to the same species is not as straightforward as in case of the CO2 modulation experiments as time values diverge although matching signal positions are observed, as discussed in more detail in the following. The signal located at 1270 cm−1 answers to the external trigger at 29 s, while the one at 1609 cm−1 answers after only 9 s, which is even earlier than the response of the signal of the reactant CO2 is visible (17 s), with the latter signal also decreasing when H2 is present and the reaction takes place. This significant difference in time values may be caused by the (partial) overlap with neighboring signals (at 1290 and 1583 cm−1) of similar height. As a consequence, the error of the time values may significantly increase, as discussed above.
At first glance, it is surprising that CO2 does not appear until 17 s, while signals at 1291 and 1609 cm−1 appear at 3 and 9 s, respectively. However, caution must be exercised when calculating time values for all three cases due to signal overlap. For instance, the CO2 signal is located amid a broad background feature that may render the calculated time value ambiguous. The other two signals at 1270 and 1583 cm−1 overlap with intense features that behave in an opposite manner, suggesting a possible mixing of their time values.
In the OH region, three signals are observed, located at 3655 cm−1 (20 s), originating from double bridged hydroxyl species on a reduced surface (II*-B), at 3517 cm−1 (20 s), assigned to OH in close vicinity to the Au site, and at 3614 cm−1 (24 s), assigned to triple bridged OH(III) (see Table 2). All three signals respond at about the same time as the products. As expected, in contrast to the CO2 modulation experiments, the OH (II*-B) signal exhibits an increase during the more reducing conditions of the reaction phase of H2 modulation, following a phase with only CO2 being present (see above). The corresponding OH II-B species is not detected as has already been the case for the CO2 modulation. The behavior of the double bridged hydroxyl species (II-*B) may therefore serve as an indicator of the reducing properties of the gas environment acting on the catalyst surface relative to its preceding gas atmosphere.
Discussion of the reaction mechanism
Combining the MES-PSD results from the CO2 and H2 modulation experiments allows to gain important new mechanistic insight into the RWGSR over gold–ceria catalysts, highlighting the potential of MES-PSD to develop and/or refine reaction mechanisms.Typically discussed intermediates in the context of the RWGSR are CO, formates, and carbonates. Due to its complete absence in the experiments, adsorbed CO can be ruled out as active species in the reaction over gold–ceria catalysts. In the case of formates, the situation is more demanding as formates are observed under both modulation conditions. However, using a temporal analysis based on PSD spectra, the active involvement of formates can be excluded, as their signals appear exclusively after product formation. In contrast, CO2 modulation experiments emphasize the importance of bidentate carbonates for the reaction mechanism, while during H2 modulation a broad background signal from water is detected, which hampers the determination of reliable time values for this species. Our MES-PSD results confirm the direct involvement of gold hydrides in the reaction, based on the CO2 modulation experiments. Similar to carbonate detection, in the H2 modulation experiments, intense background absorption prevents a meaningful temporal evaluation of hydride signals. The results from the CO2 modulation experiments presented here are supported by our preliminary studies using non-modulated transient DRIFTS (see Fig. S1, ESI†), highlighting the crucial role of gold hydrides in the associative RWGSR pathway.
The potential participation of double and triple bridged hydroxyl groups as well as hydroxyl groups located in close vicinity to gold sites in the reaction could be clarified by the combination of CO2 and H2 modulation experiments. Based on our observations we propose that OH in close vicinity to gold, due to its signal increase prior to product formation in both modulation experiments, plays an important role in the reaction. But also the triple bridged OH(III) species, which also show an increase in both modulation experiments, may be actively participating in the catalytic cycle, even though in case of H2 modulation, its signals appears slightly later than product (CO) formation (see Table 2) but within the error range of 1.5 s. The difference in signal position, that is, 3530 cm−1 in case of the CO2 and 3517 cm−1 in case of the H2 modulation, may be assigned to differences caused by the degree of reduction of the surface during the respective modulation experiment.36 The double bridged OH species (II*-B) is proposed to give signals in the MES-PSD not because of its direct participation in product formation but rather due to its response to changes in the gas environment.36
The H2 modulation experiments can detect both products, CO and water. According to Table 2, gaseous and weakly adsorbed water appear at about the same time (22 s) as CO (23 s), which lies within the error range of each signal. We can therefore conclude that the formation of both products is nearly synchronous.
Fig. 4 summarizes the proposed reaction mechanism for the associative RWGSR over Au/CeO2 catalysts based on the MES-PSD data: first, H2 is molecularly adsorbed to single gold atoms and clusters on the surface. Then, CO2 from the gas phase adsorbs into an oxygen vacancy on the surface in proximity to the Au–H2 species, forming a stable bidentate carbonate by inclusion of a neighboring surface oxygen atom. Next, transfer of a hydrogen atom to the bidentate carbonate causes its decomposition into CO and a surface hydroxyl, with gas-phase CO leaving the reaction site. The catalytic cycle is completed by the formation of the second product, H2O. This may occur via a fast spillover of the hydrogen atom, which is still attached to the gold, onto the newly formed OH group in its proximity. Water can then desorb into the gas phase, leaving the surface in its initial state.
 | ||
| Fig. 4 Proposed mechanism for the RWGSR over Au/CeO2 based on the presented ME-DRIFTS results. | ||
Conclusion
The results of this study demonstrate the potential of ME-DRIFTS to provide valuable insight into the reaction mechanism of the reverse water–gas shift (RWGSR). In particular, ME-DRIFTS allows a reduction of the number of signals present in the IR spectra, while preserving the signals of species that are actively responding to the external periodic stimulus (concentration modulation), which gives a strong indication of their active participation in the investigated reaction. Moreover, the capability of MES-PSD to extract further temporal information allows to develop a detailed mechanistic picture including the sequence of reaction steps or, as in the present case, to prove and refine pre-existing concepts of reaction mechanisms from the literature by direct spectroscopic evidence on actively participating surface species.Applying the MES-PSD approach to the RWGSR over gold–ceria catalysts confirms the suggested literature concept of an adsorbate-mediated associative reaction mechanism.7 Our findings provide spectroscopic evidence for the active participation of gold hydride, bidentate carbonate and specific hydroxyl species in the RWGSR. The potential involvement of formate, which has also been suggested in the literature on supported Au and Pt catalysts,1,7,26,41 can be excluded for the gold–ceria system as it apparently forms after the reaction.
Ultimately, transient spectroscopic techniques such as ME-DRIFTS significantly improve our comprehension of heterogeneously catalyzed reactions and thus facilitate knowledge-based design of promising catalytic materials such as CeO2-supported noble metal catalysts, which may become important in future processes needed to meet the increasing demands of alternative carbon-sources posed by the energy transition, such as the utilization of CO2 in hydrogenation reactions.1–3,6,42
Code availability
The software used for calculation of PSD spectra is available at https://github.com/Ja-We/Phase_Sensitive_Detection_for_Spectroscopy.Author contributions
Jakob Weyel: investigation, methodology, validation, formal analysis, writing – original draft, visualization. Christian Hess: conceptualization, writing – review & editing, supervision, project administration, funding acquisition.Conflicts of interest
The authors do not have conflicts of interest to declare.Acknowledgements
We thank Karl Kopp for technical support. Jakob Weyel gratefully acknowledges a scholarship from the Fonds der Chemischen Industrie im Verband der Chemischen Industrie e.V.References
- X. Chen, Y. Chen, C. Song, P. Ji, N. Wang, W. Wang and L. Cui, Recent Advances in Supported Metal Catalysts and Oxide Catalysts for the Reverse Water–Gas Shift Reaction, Front. Chem., 2020, 8, 709 CrossRef CAS PubMed.
- A. Jess, P. Kaiser, C. Kern, R. Unde and C. von Olshausen, Considerations concerning the Energy Demand and Energy Mix for Global Welfare and Stable Ecosystems, Chem. Ing. Technol., 2011, 83, 1777–1791 CrossRef CAS.
- P. Kaiser, R. B. Unde, C. Kern and A. Jess, Production of Liquid Hydrocarbons with CO2 as Carbon Source based on Reverse Water–Gas Shift and Fischer–Tropsch Synthesis, Chem. Ing. Technol., 2013, 85, 489–499 CrossRef CAS.
- A. Rezvani, A. M. Abdel-Mageed, T. Ishida, T. Murayama, M. Parlinska-Wojtan and R. J. Behm, CO2 Reduction to Methanol on Au/CeO2 Catalysts: Mechanistic Insights from Activation/Deactivation and SSITKA Measurements, ACS Catal., 2020, 10, 3580–3594 CrossRef CAS.
- D. Vovchok, C. Zhang, S. Hwang, L. Jiao, F. Zhang, Z. Liu, S. D. Senanayake and J. A. Rodriguez, Deciphering Dynamic Structural and Mechanistic Complexity in Cu/CeO2/ZSM-5 Catalysts for the Reverse Water–Gas Shift Reaction, ACS Catal., 2020, 10, 10216–10228 CrossRef CAS.
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, The teraton challenge. A review of fixation and transformation of carbon dioxide, Energy Environ. Sci., 2010, 3, 43–81 RSC.
- M. Ziemba, J. Weyel and C. Hess, Elucidating the mechanism of the reverse water–gas shift reaction over Au/CeO2 catalysts using operando and transient spectroscopies, Appl. Catal., B, 2022, 301, 120825 CrossRef CAS.
- M. Ziemba and C. Hess, Unravelling the mechanism of CO2 activation over low-loaded Cu/CeO2(111) catalysts using operando and transient spectroscopies, Catal. Sci. Technol., 2023, 13, 2922–2926 RSC.
- D. Baurecht and U. P. Fringeli, Quantitative modulated excitation Fourier transform infrared spectroscopy, Rev. Sci. Instrum., 2001, 72, 3782–3792 CrossRef CAS.
- P. Müller and I. Hermans, Applications of Modulation Excitation Spectroscopy in Heterogeneous Catalysis, Ind. Eng. Chem. Res., 2017, 56, 1123–1136 CrossRef.
- J. Weyel, M. Ziemba and C. Hess, Elucidating Active CO–Au Species on Au/CeO2(111): A Combined Modulation Excitation DRIFTS and Density Functional Theory Study, Top. Catal., 2022, 65, 779–787 CrossRef CAS.
- A. Aguirre, R. Zanella, C. Barrios, S. Hernández, A. Bonivardi and S. E. Collins, Gold Stabilized with Iridium on Ceria–Niobia Catalyst: Activity and Stability for CO Oxidation, Top. Catal., 2019, 62, 977–988 CrossRef CAS.
- E. del Río, S. E. Collins, A. Aguirre, X. Chen, J. J. Delgado, J. J. Calvino and S. Bernal, Reversible deactivation of a Au/Ce0.62Zr0.38O2 catalyst in CO oxidation: A systematic study of CO2-triggered carbonate inhibition, J. Catal., 2014, 316, 210–218 CrossRef.
- R. Kydd, D. Ferri, P. Hug, J. Scott, W. Y. Teoh and R. Amal, Temperature-induced evolution of reaction sites and mechanisms during preferential oxidation of CO, J. Catal., 2011, 277, 64–71 CrossRef CAS.
- A. Gau, J. Hack, N. Maeda and D. M. Meier, Operando Spectroscopic Monitoring of Active Species in CO2 Hydrogenation at Elevated Pressure and Temperature: Steady-State versus Transient Analysis, Energy Fuels, 2021, 35, 15243–15246 CrossRef CAS.
- L. P. L. Gonçalves, J. Mielby, O. S. G. P. Soares, J. P. S. Sousa, D. Y. Petrovykh, O. I. Lebedev, M. F. R. Pereira, S. Kegnæs and Y. V. Kolen’ko, In situ investigation of the CO2 methanation on carbon/ceria-supported Ni catalysts using modulation-excitation DRIFTS, Appl. Catal., B, 2022, 312, 121376 CrossRef.
- F. Hemmingsson, A. Schaefer, M. Skoglundh and P.-A. Carlsson, CO2 Methanation over Rh/CeO2 Studied with Infrared Modulation Excitation Spectroscopy and Phase Sensitive Detection, Catalysts, 2020, 10, 601 CrossRef CAS.
- I. C. ten Have, J. J. G. Kromwijk, M. Monai, D. Ferri, E. B. Sterk, F. Meirer and B. M. Weckhuysen, Uncovering the reaction mechanism behind CoO as active phase for CO2 hydrogenation, Nat. Commun., 2022, 13, 324 CrossRef PubMed.
- A. G. Greenaway, A. Marberger, A. Thetford, I. Lezcano-González, M. Agote-Arán, M. Nachtegaal, D. Ferri, O. Kröcher, C. R. A. Catlow and A. M. Beale, Detection of key transient Cu intermediates in SSZ-13 during NH3-SCR deNOx by modulation excitation IR spectroscopy, Chem. Sci., 2020, 11, 447–455 RSC.
- R. J. G. Nuguid, D. Ferri, A. Marberger, M. Nachtegaal and O. Kröcher, Modulated Excitation Raman Spectroscopy of V2O5/TiO2: Mechanistic Insights into the Selective Catalytic Reduction of NO with NH3, ACS Catal., 2019, 9, 6814–6820 CrossRef CAS.
- R. J. G. Nuguid, D. Ferri and O. Kröcher, Design of a Reactor Cell for Modulated Excitation Raman and Diffuse Reflectance Studies of Selective Catalytic Reduction Catalysts, Emiss. Control Sci. Technol., 2019, 5, 307–316 CrossRef.
- L. Schumacher, J. Weyel and C. Hess, Unraveling the Active Vanadium Sites and Adsorbate Dynamics in VOx/CeO2 Oxidation Catalysts Using Transient IR Spectroscopy, J. Am. Chem. Soc., 2022, 144, 14874–14887 CrossRef CAS PubMed.
- P. D. Srinivasan, B. S. Patil, H. Zhu and J. J. Bravo-Suárez, Application of modulation excitation-phase sensitive detection-DRIFTS for in situ/operando characterization of heterogeneous catalysts, React. Chem. Eng., 2019, 4, 862–883 RSC.
- M. Pfeiffer and C. Hess, Application of Transient Infrared Spectroscopy To Investigate the Role of Gold in Ethanol Gas Sensing over Au/SnO2, J. Phys. Chem. C, 2022, 126, 3980–3992 CrossRef CAS.
- M. Ziemba, J. Weyel and C. Hess, Approaching C1 Reaction Mechanisms Using Combined Operando and Transient Analysis: A Case Study on Cu/CeO2 Catalysts during the LT-Water–Gas Shift Reaction, ACS Catal., 2022, 12, 9503–9514 CrossRef CAS.
- L. F. Bobadilla, J. L. Santos, S. Ivanova, J. A. Odriozola and A. Urakawa, Unravelling the Role of Oxygen Vacancies in the Mechanism of the Reverse Water–Gas Shift Reaction by Operando DRIFTS and Ultraviolet–Visible Spectroscopy, ACS Catal., 2018, 8, 7455–7467 CrossRef CAS.
- X. Lu, W. Wang, S. Wei, C. Guo, Y. Shao, M. Zhang, Z. Deng, H. Zhu and W. Guo, Initial reduction of CO2 on perfect and O-defective CeO2 (111) surfaces: towards CO or COOH?, RSC Adv., 2015, 5, 97528–97535 RSC.
- A. Filtschew, K. Hofmann and C. Hess, Ceria and Its Defect Structure: New Insights from a Combined Spectroscopic Approach, J. Phys. Chem. C, 2016, 120, 6694–6703 CrossRef CAS.
- C. Schilling and C. Hess, Elucidating the Role of Support Oxygen in the Water–Gas Shift Reaction over Ceria-Supported Gold Catalysts Using Operando Spectroscopy, ACS Catal., 2019, 9, 1159–1171 CrossRef CAS.
- C. Schilling and C. Hess, CO Oxidation on Ceria Supported Gold Catalysts Studied by Combined Operando Raman/UV–Vis and IR Spectroscopy, Top. Catal., 2017, 60, 131–140 CrossRef CAS.
- P. G. Lustemberg, C. Yang, Y. Wang, C. Wöll and M. V. Ganduglia-Pirovano, Vibrational frequencies of CO bound to all three low-index cerium oxide surfaces: A consistent theoretical description of vacancy-induced changes using density functional theory, J. Chem. Phys., 2023, 159, 1–14 CrossRef PubMed.
- L. Caulfield, E. Sauter, H. Idriss, Y. Wang and C. Wöll, Bridging the Pressure and Materials Gap in Heterogeneous Catalysis: A Combined UHV, In Situ, and Operando Study Using Infrared Spectroscopy, J. Phys. Chem. C, 2023, 127, 14023–14029 CrossRef CAS.
- C. Yang, M. Capdevila-Cortada, C. Dong, Y. Zhou, J. Wang, X. Yu, A. Nefedov, S. Heißler, N. López, W. Shen, C. Wöll and Y. Wang, Surface Refaceting Mechanism on Cubic Ceria, J. Phys. Chem. Lett., 2020, 11, 7925–7931 CrossRef CAS PubMed.
- C. Schilling, M. Ziemba, C. Hess and M. V. Ganduglia-Pirovano, Identification of single-atom active sites in CO oxidation over oxide-supported Au catalysts, J. Catal., 2020, 383, 264–272 CrossRef CAS.
- R. Juárez and S. F. Parker, P. Concepción, A. Corma and H. García, Heterolytic and heterotopic dissociation of hydrogen on ceria-supported gold nanoparticles. Combined inelastic neutron scattering and FT-IR spectroscopic study on the nature and reactivity of surface hydrogen species, Chem. Sci., 2010, 1, 731 RSC.
- A. Badri, C. Binet and J. C. Lavalley, An FTIR study of surface ceria hydroxy groups during a redox process with H2, J. Chem. Soc., Faraday Trans., 1996, 92, 4669–4673 RSC.
- G. N. Vayssilov, M. Mihaylov, P. S. Petkov, K. I. Hadjiivanov and K. M. Neyman, Reassignment of the Vibrational Spectra of Carbonates, Formates, and Related Surface Species on Ceria: A Combined Density Functional and Infrared Spectroscopy Investigation, J. Phys. Chem. C, 2011, 115, 23435–23454 CrossRef CAS.
- F. Romero-Sarria, L. M. Martínez T, M. A. Centeno and J. A. Odriozola, Surface Dynamics of Au/CeO2 Catalysts during CO Oxidation, J. Phys. Chem. C, 2007, 111, 14469–14475 CrossRef CAS.
- Y. Denkwitz, A. Karpenko, V. Plzak, R. Leppelt, B. Schumacher and R. Behm, Influence of CO2 and H2 on the low-temperature water–gas shift reaction on Au/CeO2 catalysts in idealized and realistic reformate, J. Catal., 2007, 246, 74–90 CrossRef CAS.
- W. O. Gordon, Y. Xu, D. R. Mullins and S. H. Overbury, Temperature evolution of structure and bonding of formic acid and formate on fully oxidized and highly reduced CeO2(111), Phys. Chem. Chem. Phys., 2009, 11, 11171–11183 RSC.
- X. Chen, X. Su, B. Liang, X. Yang, X. Ren, H. Duan, Y. Huang and T. Zhang, Identification of relevant active sites and a mechanism study for reverse water gas shift reaction over Pt/CeO2 catalysts, J. Energy Chem., 2016, 25, 1051–1057 CrossRef.
- L. C. Wang, D. Widmann and R. J. Behm, Reactive removal of surface oxygen by H2, CO and CO/H2 on a Au/CeO2 catalyst and its relevance to the preferential CO oxidation (PROX) and reverse water gas shift (RWGS) reaction, Catal. Sci. Technol., 2015, 5, 925–941 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp05102a |
| This journal is © the Owner Societies 2024 |