
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Appropriation of group II metals: synthesis and characterisation of the first alkaline earth metal supported transition metal carbonite complexes†
Siad
Wolff
 ,
Annabelle
Ponsonby
,
André
Dallmann
,
Christian
Herwig
,
Fabian
Beckmann
,
Beatrice
Cula
and
Christian
Limberg
*
,
Annabelle
Ponsonby
,
André
Dallmann
,
Christian
Herwig
,
Fabian
Beckmann
,
Beatrice
Cula
and
Christian
Limberg
*
Institut für Chemie, Humboldt-Universität zu Berlin, Brook-Taylor-Straße 2, 12489 Berlin, Germany. E-mail: Christian.limberg@chemie.hu-berlin.de
First published on 7th May 2024
Abstract
Nickel carbonite complexes supported by alkaline earth metals have been accessed via salt-metathesis of the corresponding alkali metal precursors. The new complexes undergo Schlenk-like exchange reactions in solution which have been investigated by NMR spectroscopy. Also their reactivity towards epoxides and carbon monoxide was studied.
Due to its high abundancy CO2 represents a readily available feedstock for the chemical industry.1 Hence, current research efforts strongly focus on the development of often metal-based catalysts to transform the molecule into reduced C1 building blocks.2 However, issues like poor selectivity, low turnover numbers or high overpotentials prevent large scale applications. To overcome these problems, it is necessary to understand how chemical and structural modification can affect the performance of a catalyst. Therefore, chemists have designed molecular systems for the activation of CO2, which gave access to stabilised metal CO2 adducts and allowed to elucidate the relation between structure and reactivity of these key intermediates.3 Due to its ambiphilic nature CO2 is highly eligible for bifunctional activation. Hence, systems which combine a reducing metallic Lewis base with a Lewis acidic site, leading to a push–pull interaction, have proved very efficient in facilitating the transforming of the CO2 molecule into a carbonite dianion (CO22−).4 Transition metal-based systems often involve alkali metals (AM) as Lewis acidic support being introduced during the synthesis.5 In our recent reports we have described the synthesis of nickel carbonite complexes [LtBuNi(CO2)AM] (AM = Li, Na, K), IAM (Fig. 1), supported by a β-diketiminate ligand, which represent intermediates in a Ni-mediated CO2 reduction process leading to CO.6 It was found that even a change from Na to K had significant effects on the reactivity of the system, so that we were interested to extend the palette to earth alkaline metal ions as counterions. Surprisingly, alkaline earth (AE) metals have not received as much attention in the construction of metal CO2 adducts7 despite their high natural abundance and strong oxophilic character. Although, Jones et al. reported the first reductive disproportion of CO2 at reduced Mg(I) sites already 10 years ago,8 no structurally characterised metal carbonite featuring an interaction with AE metal cations has been described to date. Herein we report the first representatives, accessed by exchange of the AM counter ions in [LtBuNi(CO2)AM] complexes against group II metal ions (Fig. 1).
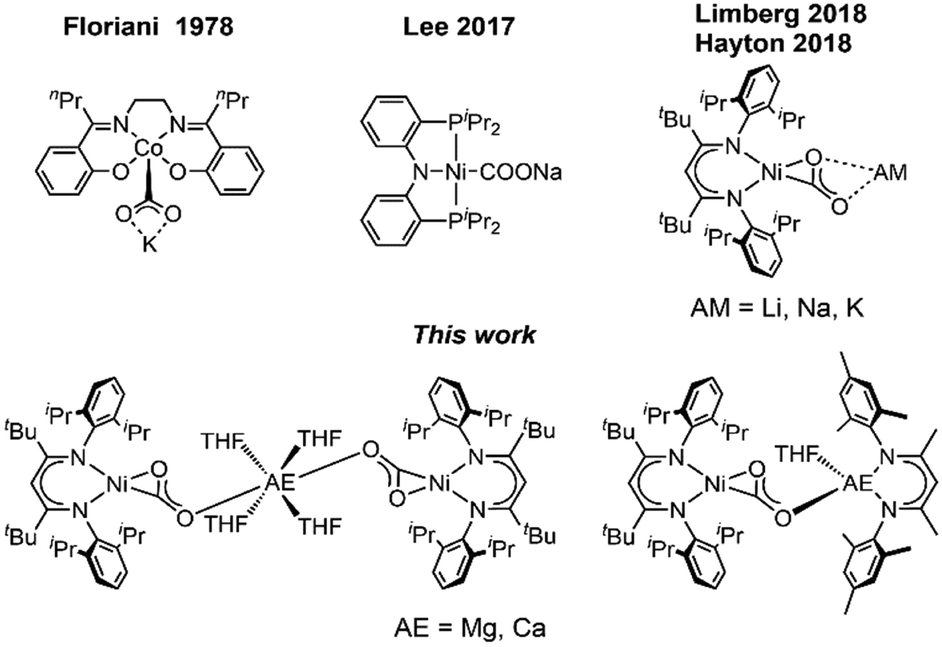 | ||
| Fig. 1 Selected examples of Alkali metal stabilised CO2 complexes. | ||
First attempts to create such compounds via a simple salt-metathesis approach starting from the previously described Li complex [LtBuNi(CO2)Li], ILi, and a respective AE iodide salt (AE = Mg, Ca) failed; no reaction could be observed at all. Presumably, the tendency of the Li+ ion to develop covalent bonding contributions together with its highly oxophilic character prevented any exchange reactions. Therefore, INa was used instead as the precursor compound leading to the formation of two new complexes [Mg(THF)4(LtBuNiCO2)2], 1, and [Ca(THF)4(LtBuNiCO2)2], 2 (see Scheme 1).
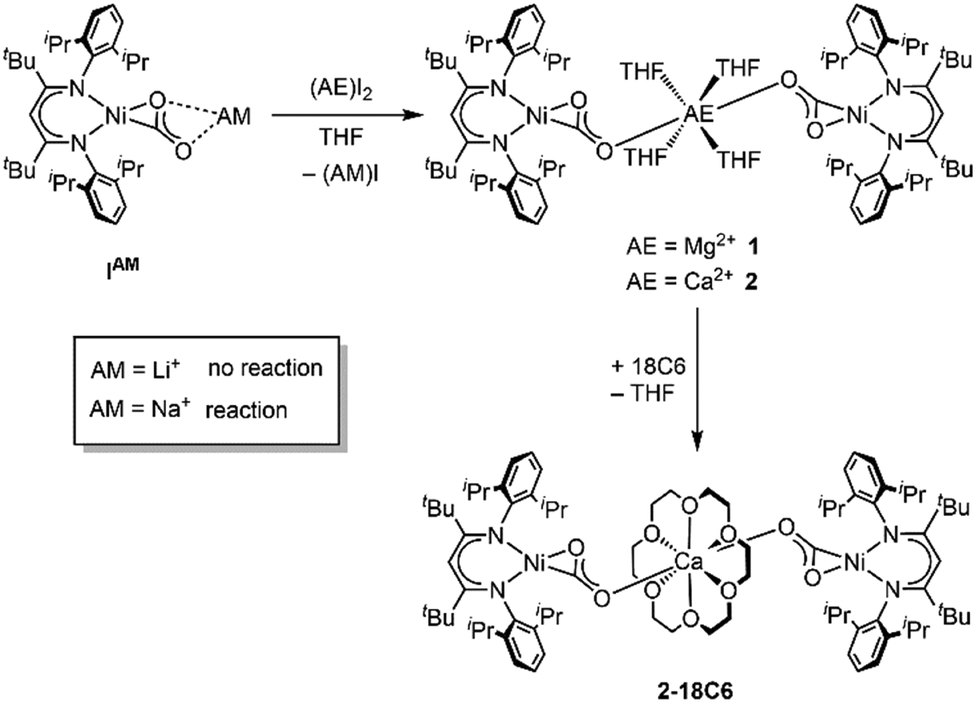 | ||
| Scheme 1 Synthetic routes to the complexes 1, 2 and 2-18C6. | ||
X-Ray diffraction analyses of single crystals grown of 1 and 2 revealed that in both cases two [LtBuNiCO2]− fragments are bridged by a central AE2+ ion, coordinated in a η1-κO fashion by the distal O atom of the carbonite ligand (for 1 see Fig. 2, left structure). The AE2+ ion is further coordinated by four equatorially arranged THF molecules leading to an octahedral geometry. In case of 2 the disorder around the NiII–CO22− moieties together with the high symmetry within the unit cell prevented complete refinement of the molecular structure and all attempts to grow single crystals of higher quality failed.
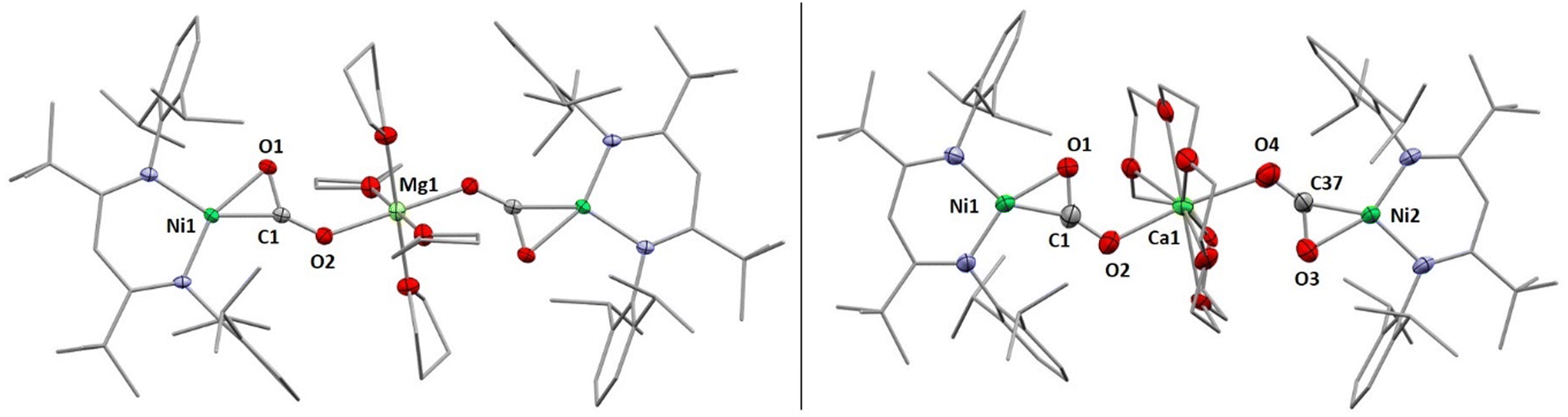 | ||
| Fig. 2 Molecular structures of 1 (left) and 2-18C6 (right) as determined by X-ray diffraction analysis. H atoms and co-crystallised solvent molecules omitted for clarity. 1 exhibits Ci symmetry where the inversion centre is located at the Mg-position. Selected bond lengths (Å) and angles (°): (for 1) Ni1–C1 1.822(3), Ni1–O1 1.932(2), C1–O1 1.275(3), C1–O2 1.239(3), O1–C1–O2 130.5(3), O2–Mg1 2.039; (for 2-18C6) Ni1–C1 1.779(7), Ni2–C37 1.820(7), Ni1–O1 1.909(5), Ni2–O3 1.925(6), C1–O1 1.243(8), C37–O3 1.270(9), C1–O2 1.243(8), C37–O4 1.201(8), O1–C1–O2 127.8(6), O3–C37–O4 132.0(6), O2–Ca1 2.191(7), O4–Ca1 2.384(7). | ||
Hence, replacement of the THF ligands around the Ca2+ ion by a chelating crown ether was pursued through the reaction of 2 with 18-crown-6. Indeed this afforded [Ca(18C6)(LtBuNiCO2)2], 2-18C6, in quantitative yields and an X-ray diffraction analysis of single crystals led to a far better data set yielding a structure similar to the one determined for 1 (see Fig. 2, right structure).
Although an excess of the respective AE salts had been used for the cation exchange reaction starting from INa, both derivatives 1 and 2 feature a Ni/AE ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. However, monitoring the reaction of INa with CaI2 by NMR spectroscopy it was observed that two species are present in solution (see ESI,† Section S3). The same product mixture was also generated when reacting isolated 2 at room temperature with an excess of CaI2 indicating that 2 is participating in a kind of Schlenk equilibrium9 (see Scheme 2). Indeed the minor product was identified as 2 while for the main product a composition of [CaI(THF)4(LtBuNiCO2)], 2′, is suggested (see below). By contrast, reaction of 1 with an excess of MgI2 was only observed at temperatures above 50 °C, leading to the formation of [MgI(THF)4(LtBuNiCO2)], 1′, in around 40% yield; cooling gave back 1 as the single product. However, the reverse reaction did not proceed continuously throughout the cooling process. The ratio between 1 and 1′ remained unchanged until 0 °C, while at −20 °C MgI2 precipitated from solution, accompanied by the disappearance of all 1H NMR signals of 1′. Apparently, the conversion of the complexes 1 or 2 to their respective asymmetric counterparts 1′ or 2′ and vice versa does not proceed within a dynamic equilibrium but is rather determined by the amount of AE iodide salt, which is dissolved in solution.
:1. However, monitoring the reaction of INa with CaI2 by NMR spectroscopy it was observed that two species are present in solution (see ESI,† Section S3). The same product mixture was also generated when reacting isolated 2 at room temperature with an excess of CaI2 indicating that 2 is participating in a kind of Schlenk equilibrium9 (see Scheme 2). Indeed the minor product was identified as 2 while for the main product a composition of [CaI(THF)4(LtBuNiCO2)], 2′, is suggested (see below). By contrast, reaction of 1 with an excess of MgI2 was only observed at temperatures above 50 °C, leading to the formation of [MgI(THF)4(LtBuNiCO2)], 1′, in around 40% yield; cooling gave back 1 as the single product. However, the reverse reaction did not proceed continuously throughout the cooling process. The ratio between 1 and 1′ remained unchanged until 0 °C, while at −20 °C MgI2 precipitated from solution, accompanied by the disappearance of all 1H NMR signals of 1′. Apparently, the conversion of the complexes 1 or 2 to their respective asymmetric counterparts 1′ or 2′ and vice versa does not proceed within a dynamic equilibrium but is rather determined by the amount of AE iodide salt, which is dissolved in solution.
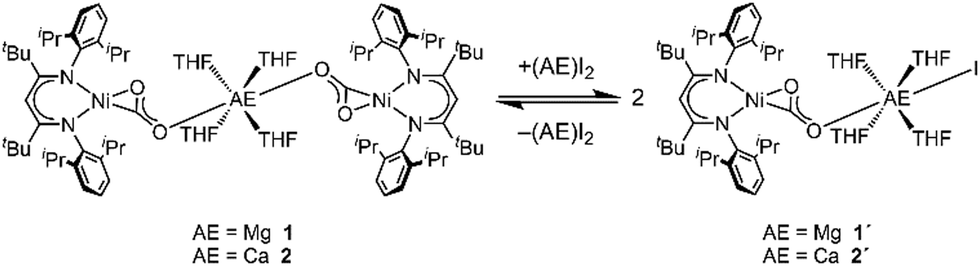 | ||
| Scheme 2 Schlenk equilibrium of 1 or 2 when treated with (AE)I2. | ||
To support the assignment of the second species in the solutions discussed above at r.t. to 2′ a comprehensive DOSY NMR study was performed (see ESI†). It was found that – as should be expected – 2′ exhibits a smaller hydrodynamic radius than its parent compound 2, supporting the formulation of a mixed carbonite-iodide compound.
All attempts to isolate 1′ or 2′ failed, as (AE)I2 consistently precipitated when cooling down the solutions or reducing its volumes, leading to the isolation of 1 or 2 as the sole products. Therefore, DFT calculations were carried out to model the suggested structures of the heteroleptic complexes. Furthermore, reaction energies for the Schlenk equilibria were calculated and found to be close to zero (see ESI† Section S5).
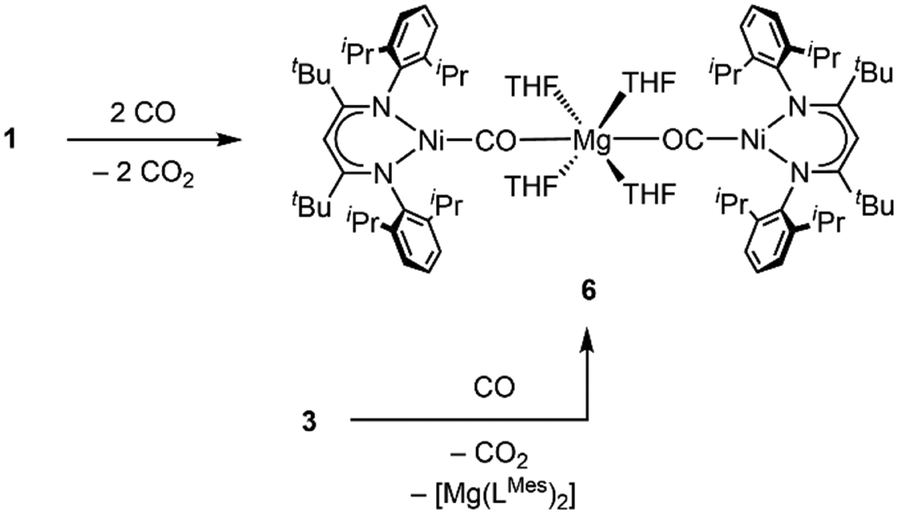
Having found that counterions of the carbonites can be exchanged by reaction with AE iodide salts we were interested whether this approach can also be adapted to AE complexes supported by β-diketiminate ligands to yield Ni/AE 1:1 complexes. Indeed, reaction of INa with [LMesMg(Et2O)I] led to the selective formation of the heterodinuclear complex [LtBuNiCO2Mg(THF)LMes], 3 (Scheme 3). Single crystal X-ray diffraction analysis revealed the expected tetrahedral coordination sphere around the Mg2+ ion composed of LMes, a η1-κO coordinated carbonite ligand and a THF molecule (see Fig. 3).
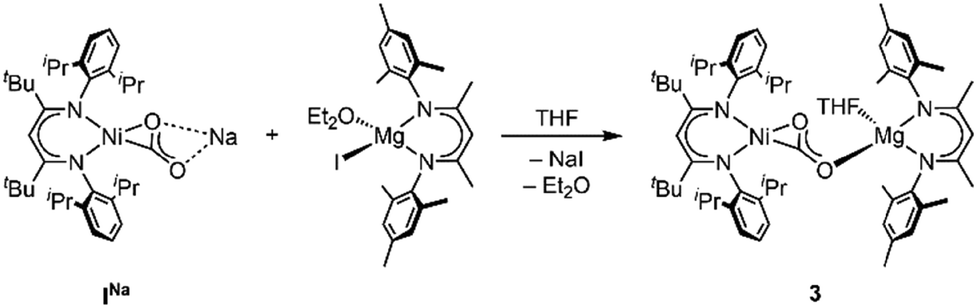 | ||
| Scheme 3 Synthesis of the heterobimetallic complex 3. | ||
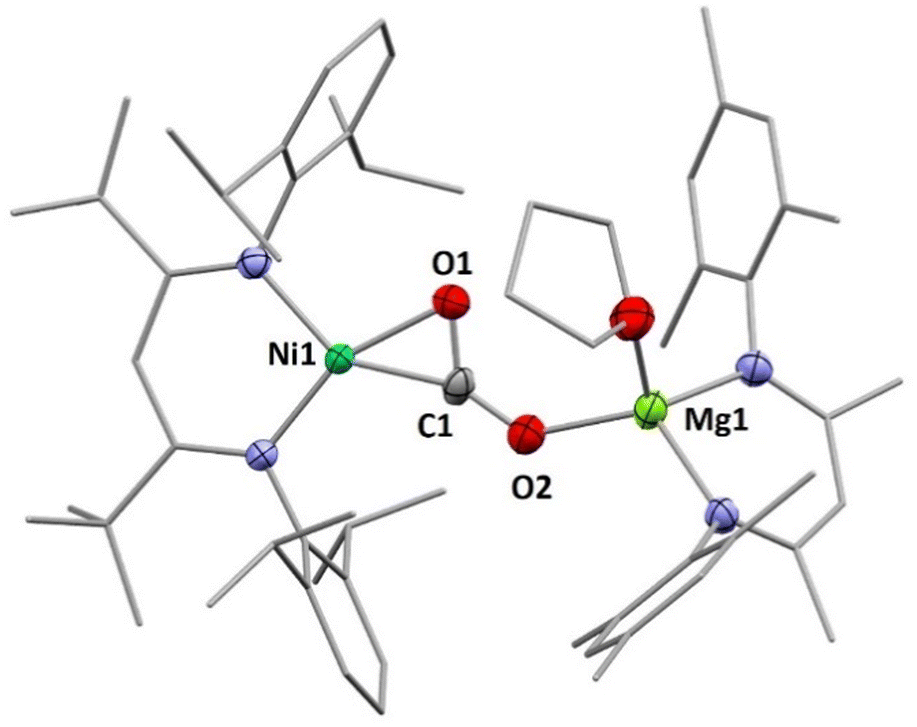 | ||
| Fig. 3 Molecular structure of 3. H atoms and co-crystallised solvent molecules omitted for clarity. Selected bond lengths (Å) and angles (°): Ni1–C1 1.839(2), Ni1–O1 1.925(2), C1–O1 1.269(4), C1–O2 1.236(3), O1–C1–O2 129.3(3), O2–Mg1 1.974(2). | ||
Synthesis of the corresponding Ca derivative [LtBuNiCO2Ca(THF)LDipp], 4, was also targeted, but, as Ca exhibits a stronger tendency to undergo ligand exchange,9a,c,10 treatment of INa with [LDippCa(THF)I] afforded a complicated mixture: performing the reaction in THF 2 precipitated, while NMR spectroscopic analysis revealed the presence of 2′ and [Ca(LDipp)2] in solution as well as one further species. Analysis via DOSY NMR measurements confirmed that the new complex exhibits a very similar hydrodynamic radius as compared to 3 suggesting an assignment of this complex to 4 (Scheme 4). Similar as in the case of 2′ all attempts to isolate 4 failed as all manipulations of the reaction mixture repeatedly triggered ligand exchange reactions. DFT structure optimisation of the hypothetical complex 4 led to a structure similar to the one of 3 (see ESI† Section S5).
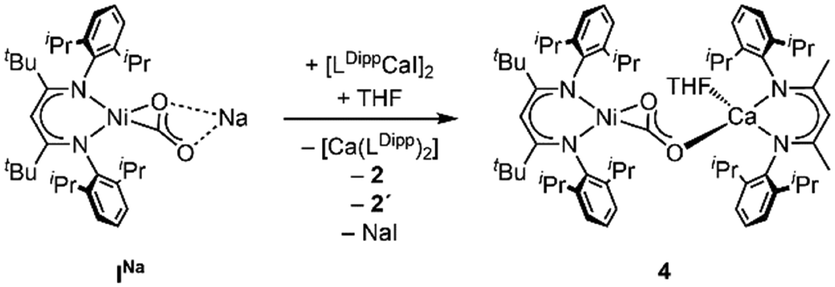 | ||
| Scheme 4 In situ generation of complex 4. | ||
Having synthesised a series of AE metal supported carbonite complexes the dependence of the degree of CO2 activation on the counter ions was analysed, which is reflected by the 13C NMR shift (δ13CCO2) and the C–O stretching frequencies (νCO2)6c (data is summarised in ESI,† Table S1). Generally, the spectroscopic data suggest that the AE stabilised derivatives exhibit a stronger CO2 activation compared to the AM derivatives. This was also supported by a stronger tendency of the AE complexes to decompose under C–O bond cleavage reactions. While the AM derivates are stable under inert conditions it was observed that the AE derivates steadily decompose at room temperature yielding [LtBuNiCO] among other products. The strongest activation of the CO22− ligand was observed within 3. The low coordination number and the Ni:Mg ratio of 1:1 favour a strong interaction of the oxophilic metal with the carbonite ligand leading to strong push–pull activation within the Ni–CO2–Mg core.
In the context of CO2 utilisation AE metals are often employed as catalysts for copolymerisation reactions with epoxides.11 Hence, the reactivity of complexes 1 and 2 towards propylene oxide was tested. In both cases an immediate conversion occurred leading to new diamagnetic compounds, for which NMR spectra suggested symmetric square planar ligand arrangement around the Ni site. The solid-state structure of the product derived from 2 as determined by single crystal X-ray diffraction revealed that the carbonite ligand had been transformed into carbonate yielding [AE(THF)4(LtBuNiCO3)2], 5AE (see Scheme 5 and ESI†). Following the reaction by 1H NMR spectroscopy confirmed the formation of propylene as the side product, showing that the propylene oxide has acted as an O atom transfer reagent. Apparently, the carbonite ligand is sufficiently reducing to favour a reaction of the complexes via deoxygenation of the epoxide over a ring-opening at the AE ions.
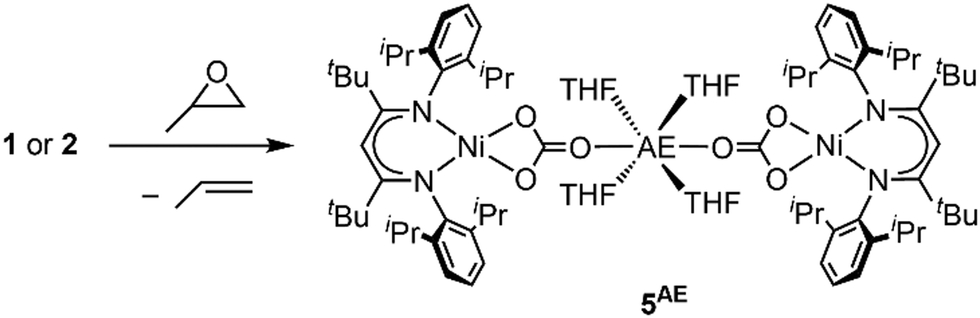 | ||
| Scheme 5 Oxidation of 1 or 2 with propylene oxide. | ||
Next, we investigated whether the exchange of the AM counterions by AE metal ions can alter the reactivity of the carbonite towards substrates that had already been studied as reaction partners. As previously described, exposure of IAM to an atmosphere of CO triggers the formation of [LtBuNiCO] regardless of the AM counterion present in the complex.6a While 2 is reacting in the same way as the AM derivates, the reaction of 1 led to a different result. After exposure to CO a crystalline precipitate formed within a few minutes, which was analysed by single crystal X-ray diffraction. The solid-state structure (Fig. 4) revealed the formation of [Mg(THF)4(LtBuNiCO)2], 6, where a Mg2+ cation bridges two [LtBuNi0CO]− units via the carbonyl O atoms, that is, through a very rare isocarbonyl coordination.12 The CO contact thus triggers a redox event in which two electrons of the original carbonite ligand are transferred to the Ni(II) centre with concomitant release of CO2 (the released CO2 was detected during NMR spectroscopic monitoring of the reaction, see ESI†), while CO coordinates instead. Interestingly, exposure of 3 to CO led to the precipitation of the same product, meaning that a ligand scrambling had occurred (see Scheme 6). Apparently, the low solubility and precipitation of 6 represents a driving force towards this product.
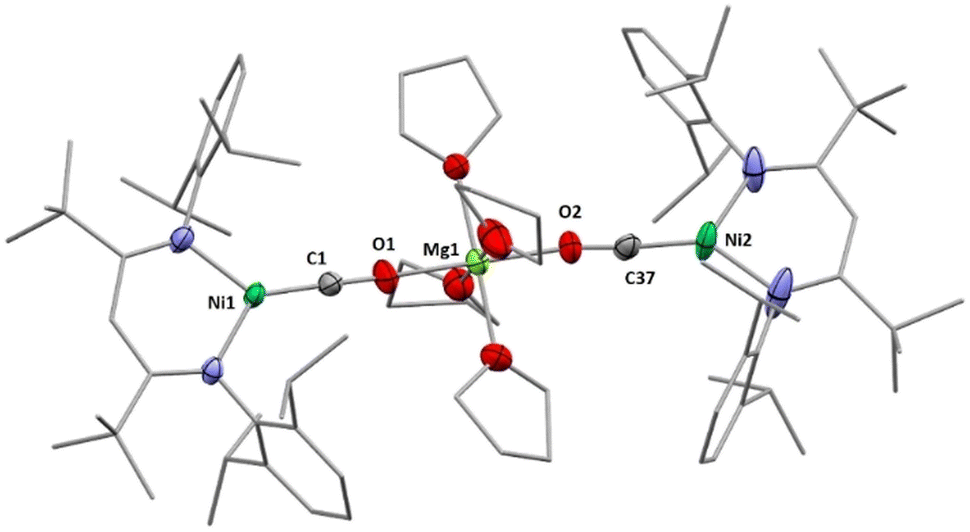 | ||
| Fig. 4 Molecular structures of 6. H atoms and co-crystallised solvent molecules omitted for clarity. Selected bond lengths (Å) and angles (°): Ni1–C1 1.659(2), Ni2–C37 1.658(2), C1–O1 1.197(3), C37–O2 1.199(3), O1–Mg1 2.015(2), O2–Mg1 2.003(2), O1–C1–Ni1 173.6(2), Ni2–C37–O2 178.8(2). | ||
 | ||
| Scheme 6 CO2/CO ligand displacement of Mg-supported Ni carbonite complexes. | ||
In summary, we report the isolation of the first structurally characterised AE metal supported transition metal carbonite complexes. It was shown that the complexes undergo Schlenk-type exchange reactions in solution which have been especially pronounced in case of the Ca derivatives. Reactivity studies with epoxides indicated that the carbonite ligand can act as a reductant, preventing typical ring-opening reactions. Furthermore, for AE = Mg reactivity towards CO leads to a Ni0 complex with rare isocarbonyl bridges. Future work will aim at expanding the palette of Lewis acidic support towards redox active transition metals.
Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2008/1 – 390540038.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) R. Schlögl, Angew. Chem., Int. Ed., 2022, 61, e202007397 CrossRef PubMed; (b) J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed.
- (a) H. Q. Liang, T. Beweries, R. Francke and M. Beller, Angew. Chem., Int. Ed., 2022, 61, e202200723 CrossRef CAS PubMed; (b) S.-T. Bai, G. De Smet, Y. Liao, R. Sun, C. Zhou, M. Beller, B. U. W. Maes and B. F. Sels, Chem. Soc. Rev., 2021, 50, 4259–4298 RSC; (c) N. W. Kinzel, C. Werle and W. Leitner, Angew. Chem., Int. Ed., 2021, 60, 11628–11686 CrossRef CAS PubMed.
- (a) P. M. Jurd, H. L. Li, M. Bhadbhade and L. D. Field, Organometallics, 2020, 39, 2011–2018 CrossRef CAS; (b) T. T. Adamson, S. P. Kelley and W. H. Bernskoetter, Organometallics, 2020, 39, 3562–3571 CrossRef CAS; (c) I. Castro-Rodriguez, H. Nakai, L. N. Zakharov, A. L. Rheingold and K. Meyer, Science, 2004, 305, 1757–1759 CrossRef CAS PubMed.
- (a) M. Perez-Jimenez, H. Corona, F. de la Cruz-Martinez and J. Campos, Chem. – Eur. J., 2023, 29, e202301428 CrossRef CAS PubMed; (b) H. Corona, M. Perez-Jimenez, F. de la Cruz-Martinez, I. Fernandez and J. Campos, Angew. Chem., Int. Ed., 2022, 61, e202207581 CrossRef CAS PubMed; (c) J. A. Buss, D. G. VanderVelde and T. Agapie, J. Am. Chem. Soc., 2018, 140, 10121–10125 CrossRef CAS PubMed; (d) Y.-E. Kim, J. Kim and Y. Lee, Chem. Commun., 2014, 50, 11458–11461 RSC; (e) A. Paparo and J. Okuda, J. Organomet. Chem., 2018, 869, 270–274 CrossRef CAS.
- (a) G. Fachinetti, C. Floriani and P. F. Zanazzi, J. Am. Chem. Soc., 1978, 100, 7405–7407 CrossRef CAS; (b) N. J. Hartmann, G. Wu and T. W. Hayton, Chem. Sci., 2018, 9, 6580–6588 RSC; (c) C. Yoo and Y. Lee, Chem. Sci., 2017, 8, 600–605 RSC; (d) C. Yoo and Y. Lee, Inorg. Chem. Front., 2016, 3, 849–855 RSC.
- (a) P. Zimmermann, D. Ar, M. Rößler, P. Holze, B. Cula, C. Herwig and C. Limberg, Angew. Chem., Int. Ed., 2021, 60, 2312–2321 CrossRef CAS PubMed; (b) P. Zimmermann, S. Hoof, B. Braun-Cula, C. Herwig and C. Limberg, Angew. Chem., Int. Ed., 2018, 57, 7230–7233 CrossRef CAS PubMed; (c) S. Wolff, V. Pelmenschikov, R. Müller, M. Ertegi, B. Cula, M. Kaupp and C. Limberg, Chem. – Eur. J., 2024, 30, e202303112 CrossRef CAS PubMed.
- (a) C.-C. Chang, M.-C. Liao, T.-H. Chang, S.-M. Peng and G.-H. Lee, Angew. Chem., Int. Ed., 2005, 44, 7418–7420 CrossRef CAS PubMed; (b) D. Hong, T. Kawanishi, Y. Tsukakoshi, H. Kotani, T. Ishizuka and T. Kojima, J. Am. Chem. Soc., 2019, 141, 20309–20317 CrossRef CAS PubMed.
- R. Lalrempuia, A. Stasch and C. Jones, Chem. Sci., 2013, 4, 4383–4388 RSC.
- (a) S. P. Sarish, S. Nembenna, S. Nagendran and H. W. Roesky, Acc. Chem. Res., 2011, 44, 157–170 CrossRef CAS PubMed; (b) C. Helling and C. Jones, Chem. – Eur. J., 2023, 29, e202302222 CrossRef CAS PubMed; (c) P. Rinke, H. Görls and R. Kretschmer, Inorg. Chem., 2021, 60, 5310–5321 CrossRef CAS PubMed.
- (a) S. Harder, Chem. Rev., 2010, 110, 3852–3876 CrossRef CAS PubMed; (b) C. Ruspic and S. Harder, Inorg. Chem., 2007, 46, 10426–10433 CrossRef CAS PubMed.
- (a) A. C. Deacy, A. F. R. Kilpatrick, A. Regoutz and C. K. Williams, Nat. Chem., 2020, 12, 372–380 CrossRef CAS PubMed; (b) W. Gruszka and J. A. Garden, Nat. Commun., 2021, 12, 3252 CrossRef CAS PubMed; (c) J. Liang, S. Ye, S. Wang, M. Xiao and Y. Meng, Polym. J., 2020, 53, 3–27 CrossRef; (d) X. Liang, F. Tan and Y. Zhu, Front. Chem., 2021, 9 DOI:10.3389/fchem.2021.647245.
- (a) E. S. Akturk, G. P. A. Yap and K. H. Theopold, Chem. Commun., 2015, 51, 15402–15405 RSC; (b) M. Fritz, S. Demeshko, C. Würtele, M. Finger and S. Schneider, Eur. J. Inorg. Chem., 2023, e202300011 CrossRef CAS; (c) Z. Zhang, Q.-s Li, Y. Xie, R. B. King and H. F. Schaefer, III, J. Phys. Chem. A, 2010, 114, 4672–4679 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2346585–2346589. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc01682c |
| This journal is © The Royal Society of Chemistry 2024 |