
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hierarchical peroxiredoxin assembly through orthogonal pH-response and electrostatic interactions†
Eduardo
Anaya-Plaza
 *a,
Zulal
Özdemir
b,
Zdenek
Wimmer
b and
Mauri A.
Kostiainen
*a
*a,
Zulal
Özdemir
b,
Zdenek
Wimmer
b and
Mauri A.
Kostiainen
*a
aDepartment of Bioproducts and Biosystems, School of Chemical Engineering, Aalto University, Kemistintie 1, Finland. E-mail: eduardo.anaya@aalto.fi; mauri.kostiainen@aalto.fi
bDepartment of Chemistry of Natural Compounds, University of Chemistry and Technology in Prague, Technická 5, 16628 Prague 6, Czech Republic
First published on 14th November 2023
Abstract
Morpheeins are proteins that adapt their morphology and function to the environment. Therefore, their use in nanotechnology opens up the bottom-up preparation of anisotropic metamaterials, based on the sequential use of different stimuli. A prominent member of this family of proteins is peroxiredoxins (Prx), with dual peroxidase and chaperone function, depending on the pH of the media. At high pH, they show a toroidal morphology that turns into tubular stacks upon acidification. While the toroidal conformers have been explored as building blocks to yield 1D and 2D structures, the obtention of higher ordered materials remain unexplored. In this research, the morpheein behaviour of Prx is exploited to yield columnar aggregates, that are subsequently self-assembled into 3D anisotropic bundles. This is achieved by electrostatic recognition between the negatively charged protein rim and a positively charged porphyrin acting as molecular glue. The subsequent and orthogonal input lead to the alignment of the monodimensional stacks side-by-side, leading to the precise assembly of this anisotropic materials.
Introduction
Nature has developed, by means of selection and evolution, highly specialised biomolecules to address a plethora of functions and processes. Among them, proteins arise as a versatile and multifunctional family, performing structural, recognition, transport, and catalytic tasks. Interestingly, a selected group of proteins has evolved to fulfil more than one task.1 For instance, morpheeins are proteins that, upon external stimuli (temperature, pH, ionic strength, or oxidating/reducing agents), undergoes conformational changes on the secondary and ternary structure. These environmentally triggered changes have a direct impact on the quaternary structure, thus dictating the protein morphology and function. A prominent example of morpheeins are peroxiredoxins (Prx).2,3 These ubiquitous proteins have a primary peroxidase activity based on cysteine chemistry.4,5 In reducing conditions, the Prx homodimer forms a (do)decamer named low molecular weight (LMW) ring. Upon certain in vivo or in vitro stimuli, co-facial dimers, trimers, and higher stacks are formed (also known as high molecular weight (HMW) stacks). These are found to act as chaperones, ensuring the correct folding of other proteins. The exploitation of the dual and orthogonal functionality (i.e. structural–functional) in bioinspired materials is an exciting avenue, taking advantage of the ready-optimized building blocks from nature.Highly-ordered protein assemblies have received growing interest as bottom-up strategy to yield biodegradable and biocompatible materials.6–9 Their well-defined structure, presenting precisely positioned building blocks and regular pore sizes enables technologies such as metamaterial preparation,10–16 catalytic systems,17,18 templated (porous)materials,19,20 biomedical applications,21 and waste-water treatment.22 The potential Prx have not been missed in this approach.23 The monomeric ring, as well as other toroidal proteins such as stable protein one (SP1), GroEl, or tobacco mosaic virus (TMV) toroids has been applied as building block for discrete aggregates,24,25 1D,26–34 or 2D assemblies.35–37 However, preparation of well-defined 3D structures, while explored for other toroids,38 remains a challenge for peroxiredoxins.
Among the different building blocks employed in protein self-assembly, and electrostatic self-assembly in particular, porphyrinoids are highly versatile.39 They have been shown to direct the assembly of other proteins such as protein cages40 or virus-like particles.41 The main representative of the family, porphyrins (Por) are broadly applied in fields ranging from biomedical to catalysis.42–44 Their chemical structure, based on a tetrapyrrolic ring, can host a range of metals in their inner cavity. Their optical properties can be finely tuned, showing a wide absorption spectrum ranging from the ultraviolet to the near-infrared region.45–48 Their excitation leads to emitting properties (e.g. fluorescence) or intersystem crossing that further lead to optoelectronics or singlet oxygen generation.
In this manuscript, we take advantage of the morpheein behaviour of Prx to prepare 1D assemblies and subsequently assemble them through orthogonal electrostatic interactions. This is achieved by employing a positively charged Por to mediate the hierarchical self-assembly of Prx into highly ordered and anisotropic 3D bundles.
Materials and methods
Methods
The 1H NMR and the 13C NMR spectra were recorded on a Bruker AVANCE 600 MHz spectrometer at 600.13 MHz and 150.90 MHz, respectively, in deuterochloroform or deuterium oxide. Tetramethylsilane was used as an internal reference δ = 0.0. The 1H NMR data is presented in the following order: chemical shift (δ) expressed in ppm, multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; and bs, broad singlet), coupling constants in Hertz (Hz), and number of protons. Samples were prepared by dissolving the solids directly into the deuterated solvents in concentrations of ca. 1 mM.Infrared spectra (IR) were measured using a Nicolet iS5 FT-IR spectrometer with the Attenuated Total Reflection (ATR) method. Peak intensity is noted as strong (s), medium (m), and weak (w). Samples were measured as neat solids through the aforementioned ATR method.
Mass spectra (MS) were measured with a Waters ZMD mass spectrometer in ESI mode (from solution in acetonitrile and catalytic amounts of formic acid) or with an Autoflex Speed MALDI-TOF/TOF mass spectrometer (Bruker Daltonics), of 2,5-dihydroxybenzoic acid as matrix, acetonitrile/water/trifluoro-acetic acid (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:0.001). External calibration was carried out using peptide calibration standard II (Bruker Daltonics). The mass spectra were manually collected by Flex Control software (version 3.4, Bruker Daltonics) in reflector positive mode at the range of 700–4500 Da. Individual spectra acquired on at least ten different position of sample crystals were summarized to one resulting spectrum for each sample. The resulting spectrum was then processed in mMass software 5.5.0.49
:1:0.001). External calibration was carried out using peptide calibration standard II (Bruker Daltonics). The mass spectra were manually collected by Flex Control software (version 3.4, Bruker Daltonics) in reflector positive mode at the range of 700–4500 Da. Individual spectra acquired on at least ten different position of sample crystals were summarized to one resulting spectrum for each sample. The resulting spectrum was then processed in mMass software 5.5.0.49
Solutions of 0.1 mg mL−1 of Prx were prepared diluting from a stock solution in MilliQ water into buffer (20 mM HEPES, 2 mM of TCEP) at the selected pH (from pH 4 to 8), and small aliquots of 1 were added. Dilution correction was not performed, given the small overall dilution factor (typically less than 5%). Following, the final sample was titrated with 0.01–0.5 M NaCl to disassemble the complex. Beam position, attenuators, and experiment acquisition time was fixed through the experiment.
The molar concentration Prx has been determined by either using the calculated extinction coefficient of (ε280nm = 11375) or with the molecular weight of 23300 Da.
Electrostatic potential surface calculation. The electrostatic potential of Prx at different pH has been calculated with the help of the APBS web service.50
Transmission electron microscopy (TEM) images were taken using a FEI Tecnai 12 Bio-Twin instrument operated at an acceleration voltage of 120 kV. Further, the images were processed using Gatan Digital Micrograph software. The TEM samples were prepared on Formvar carbon coated copper grids (400 mesh, Electron Microscopy Sciences) and plasma cleaned for 20 seconds using a Gatan Solarus. Samples were stained with uranyl formate 2%.
The samples were prepared by either depositing a Prx solution (0.1 mg mL−1) in the selected buffer, or a mixture of Prx/1 (0.1:0.2 mg mL−1) and incubated overnight at +4 °C. 3 μL of the sample solution was applied on the carbon-coated side of the grid for 45 seconds. After the incubation, the excess solution was drained from the edge using filter paper. First, the grid was immersed into a 5 μL droplet of uranyl formate solution, and excess liquid was directly drained off from the edge using filter paper, followed by a subsequent immersion into a 20 μL droplet of uranyl formate solution. The excess amount of stain was removed after 45 seconds from the edge of the grid using filter paper. After this process, the grid was dried at room temperature for at least 30 minutes before imaging.
Materials
Human peroxiredoxin III (Prx) was purchased from ABCAM (UK). All chemicals were purchased from commercial sources in analytical grade, and the solvents were purified by general methods before use: 5,10,15,20-tetrakis(4-hydroxyphenyl)-21H,23H-porphyrin (P) ((75%), Aldrich Chemistry (CZ)), propargyl chloride (Merck/Sigma Aldrich (CZ)), potassium carbonate (Penta (CZ)), TBTA (tris((1-benzyl-4-triazolyl)methyl)amine) (Bioconjugate Chemistry (USA)), sodium ascorbate 98% (Merck/Sigma Aldrich (CZ)), CuSO4·5H2O 98% (Merck/Sigma Aldrich (CZ)), 1 M HCl (g) in EtOAc (Acros Organics (BE)), Zn(OAc)2 (Merck/Sigma Aldrich (CZ)). Silica gel 60 (0.063–0.200 mm) from Merck (CZ) was used for column chromatography. Thin layer chromatography (TLC) was carried out on silica gel plates (Merck 60F254; CZ), and the visualization was performed by ultraviolet (UV) detection and by spraying with a methanolic solution of phosphomolybdic acid (5%) followed by heating.Synthesis of tetracationic Por 1
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), 2923 (w), 2359 (w), 2342 (w), 2112 (m) ν(CC), 1605 (s), 1603 (s), 1498 (s), 1471 (s), 1381 (m) δs (CH3), 1349 (m) δs (CH3), 1288 (m) ν(C–O),1213 (s), 1173 (s) ν(C–O), 1109 (m), 1024 (s), 980 (m), 964 (s), 925 (m), 843 (m), 800 (s), 786 (s), 734 (s), 660 (m) γ (CH) (CH), 539 (s), 484 (m). MS (m/z): 831.0 [M + H]+ (ESI+, coin voltage 30 V). 1H NMR (600.13 MHz, CDCl3): δ [ppm] 8.80 (bs, 1H, 3-CH), 8.07–8.10 (m, 1H, 6-CH), 7.29–7.32 (m, 1H, 7-CH), 4.92 (d, 2H, J = 2.5 Hz, 9-CH2), 2.63 (t, 1H, J = 2.5 Hz, 11-CH), 2.83 (bs, 1H, NH). 13C NMR (150.92 MHz, CDCl3): δ [ppm] 129.4 (d, C-2), a (C-3), 119.56 (s, C-4), 135.47 (s, C-5), 135.56 (d, C-6), 113.11 (d, C-7), 157.43 (s, C-8), 56.15 (t, C-9), 78.66 (s, C-10), 75.86 (d, C-11).
CH), 2919 (w), 2359 (m), 2341 (m), 2123 (w) ν(CC), 1600 (m), 1603 (s), 1488 (s), 1375 (m) δs(CH3), 1337 (m) δs (CH3), 1280 (m) ν(C–O), 1204 (s), 1173(s) ν(C–O), 1108 (m), 1069 (m), 1026 (m), 993 (s), 928 (m), 848 (m), 796 (s), 715 (m), 668 (m) γ (CH) (CH), 634 (s), 532 (m), 424 (m). MS (m/z): 892.0 [M + H]+ (ESI+, coin voltage 25 V). 1H NMR (600.13 MHz, CDCl3): δ [ppm] 8.93 (s, 1H, 3-CH), 8.02–8.13 (m, 1H, 6-CH), 7.29–7.34 (m, 1H, 7-CH), 4.94 (d, 2H, J = 2.4 Hz, 9-CH2), 2.65 (t, 1H, J = 2.4 Hz, 11-CH). 13C NMR (150.92 MHz, CDCl3): δ [ppm] 150.43 (s, C-2), 131.93 (d, C-3), 120.57 (s, C-4), 136.08 (s, C-5), 135.36 (d, C-6), 112.98 (d, C-7), 157.28 (s, C-8), 56.15 (t, C-9), 78.72 (s, C-10), 75.82 (d, C-11).
:1) complex solution (5.0 mL, 0.22 mmol) and stirred in the absence of light at r.t. for 5 min. To the above mixture, sodium ascorbate (83 mg, 0.42 mmol) was added, and the contents were mixed at r.t. for 19 h. The reaction mixture was then extracted into CHCl3 and washed with water. After drying the organic layer over anhydrous Na2SO4, the volatiles were removed under reduced pressure. The crude product obtained after aqueous workup was column purified on silica gel using ethanol in chloroform [from 100/0 v/v (%) to 100/7 v/v (%)] to afford 4 in a 85% yield as a purple solid. FT-IR (KBr): ν [cm−1] 3322 (w) ν(NH), 2330 (w), 1698 (s) ν(amide I), 1603 (w), 1506 (m) ν(amide II), 1455 (m), 1389 (w) δs (CH3), 1363 (m) δs (CH3), 1242 (s) ν(C–O), 1172 (s) ν(C–O), 1047 (m), 991 (s), 846 (m), 795 (s), 717 (s). MALDI MS (m/z): 1804.93 Da, 1744.02 Da. 1H NMR (600.13 MHz, CDCl3): δ [ppm] 8.85 (s, 1H, 3-CH), 8.02–8.05 (m, 1H, 6-CH), 7.25–7.30 (m, 1H, 7-CH), 5.25 (bs, 2H, 9-CH2), 7.70 (bs, 1H, 11-CH), 4.27–4.39 (m, 2H, 12-CH2), 2.3 (bs, 2H, 13-CH2), 1.53–1.62 (m, 2H, 14-CH2), 1.94-2.08 (m, 2H, 15-CH2), 3.08 (bs, 2H, 16-CH2), 1.40 (s, 9H, Boc(CH3)). 13C NMR (150.92 MHz, CDCl3): δ [ppm] 145.96 (s, C-2), a (d, C-3), 117.69 (s, C-4), 134.10 (s, C-5), a (d, C-6), a (d, C-7), 162.73 (s, C-8), 53.18 (t, C-9), 142.30 (s, C-10), 127.77 (d, C-11), 42.22 (t, C-12), 29.16 (t, C-13), 25.68 (t, C-14), 29.16 (t, C-15), 31.87 (t, C-16).
CH), 2923 (w), 2359 (w), 2342 (w), 2112 (m) ν(CC), 1605 (s), 1603 (s), 1498 (s), 1471 (s), 1381 (m) δs (CH3), 1349 (m) δs (CH3), 1288 (m) ν(C–O),1213 (s), 1173 (s) ν(C–O), 1109 (m), 1024 (s), 980 (m), 964 (s), 925 (m), 843 (m), 800 (s), 786 (s), 734 (s), 660 (m) γ (CH) (CH), 539 (s), 484 (m). MS (m/z): 831.0 [M + H]+ (ESI+, coin voltage 30 V). 1H NMR (600.13 MHz, CDCl3): δ [ppm] 8.80 (bs, 1H, 3-CH), 8.07–8.10 (m, 1H, 6-CH), 7.29–7.32 (m, 1H, 7-CH), 4.92 (d, 2H, J = 2.5 Hz, 9-CH2), 2.63 (t, 1H, J = 2.5 Hz, 11-CH), 2.83 (bs, 1H, NH). 13C NMR (150.92 MHz, CDCl3): δ [ppm] 129.4 (d, C-2), a (C-3), 119.56 (s, C-4), 135.47 (s, C-5), 135.56 (d, C-6), 113.11 (d, C-7), 157.43 (s, C-8), 56.15 (t, C-9), 78.66 (s, C-10), 75.86 (d, C-11).
CH), 2919 (w), 2359 (m), 2341 (m), 2123 (w) ν(CC), 1600 (m), 1603 (s), 1488 (s), 1375 (m) δs(CH3), 1337 (m) δs (CH3), 1280 (m) ν(C–O), 1204 (s), 1173(s) ν(C–O), 1108 (m), 1069 (m), 1026 (m), 993 (s), 928 (m), 848 (m), 796 (s), 715 (m), 668 (m) γ (CH) (CH), 634 (s), 532 (m), 424 (m). MS (m/z): 892.0 [M + H]+ (ESI+, coin voltage 25 V). 1H NMR (600.13 MHz, CDCl3): δ [ppm] 8.93 (s, 1H, 3-CH), 8.02–8.13 (m, 1H, 6-CH), 7.29–7.34 (m, 1H, 7-CH), 4.94 (d, 2H, J = 2.4 Hz, 9-CH2), 2.65 (t, 1H, J = 2.4 Hz, 11-CH). 13C NMR (150.92 MHz, CDCl3): δ [ppm] 150.43 (s, C-2), 131.93 (d, C-3), 120.57 (s, C-4), 136.08 (s, C-5), 135.36 (d, C-6), 112.98 (d, C-7), 157.28 (s, C-8), 56.15 (t, C-9), 78.72 (s, C-10), 75.82 (d, C-11).
:1) complex solution (5.0 mL, 0.22 mmol) and stirred in the absence of light at r.t. for 5 min. To the above mixture, sodium ascorbate (83 mg, 0.42 mmol) was added, and the contents were mixed at r.t. for 19 h. The reaction mixture was then extracted into CHCl3 and washed with water. After drying the organic layer over anhydrous Na2SO4, the volatiles were removed under reduced pressure. The crude product obtained after aqueous workup was column purified on silica gel using ethanol in chloroform [from 100/0 v/v (%) to 100/7 v/v (%)] to afford 4 in a 85% yield as a purple solid. FT-IR (KBr): ν [cm−1] 3322 (w) ν(NH), 2330 (w), 1698 (s) ν(amide I), 1603 (w), 1506 (m) ν(amide II), 1455 (m), 1389 (w) δs (CH3), 1363 (m) δs (CH3), 1242 (s) ν(C–O), 1172 (s) ν(C–O), 1047 (m), 991 (s), 846 (m), 795 (s), 717 (s). MALDI MS (m/z): 1804.93 Da, 1744.02 Da. 1H NMR (600.13 MHz, CDCl3): δ [ppm] 8.85 (s, 1H, 3-CH), 8.02–8.05 (m, 1H, 6-CH), 7.25–7.30 (m, 1H, 7-CH), 5.25 (bs, 2H, 9-CH2), 7.70 (bs, 1H, 11-CH), 4.27–4.39 (m, 2H, 12-CH2), 2.3 (bs, 2H, 13-CH2), 1.53–1.62 (m, 2H, 14-CH2), 1.94-2.08 (m, 2H, 15-CH2), 3.08 (bs, 2H, 16-CH2), 1.40 (s, 9H, Boc(CH3)). 13C NMR (150.92 MHz, CDCl3): δ [ppm] 145.96 (s, C-2), a (d, C-3), 117.69 (s, C-4), 134.10 (s, C-5), a (d, C-6), a (d, C-7), 162.73 (s, C-8), 53.18 (t, C-9), 142.30 (s, C-10), 127.77 (d, C-11), 42.22 (t, C-12), 29.16 (t, C-13), 25.68 (t, C-14), 29.16 (t, C-15), 31.87 (t, C-16).
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) OOC(CH3)3), 79.29 (s, –COO(CH3)3), 28.27 (q, –COOC(H3)3).
N), 1693 (s) ν(amide I), 1519 (m) ν(amide II), 1445 (m), 1390 (m), 1366 (m) δ(CH3), 1250 (m), 1175 (s) ν(C–O), 766 (s). MS (m/z): 251.1 [M + Na]+ (ESI+, coin voltage 20 V), 255.1 (ESI−, coin voltage 20 V). 1H NMR (600.13 MHz, CDCl3): δ [ppm] 3.24 (bdt, 2H, J = 4.7, 5.4, 5.4 Hz, 1-CH2), 1.41–1.45 (m, 2H, 2-CH2), 1.26–1.33 (m, 2H, 3-CH2), 1.50–1.58 (m, 2H, 4-CH2), 3.35 (t, 2H, 5-CH2), 1.38 (s, 3H, Boc (CH3)). 13C NMR (150.90 MHz, CDCl3): δ [ppm] 40.03 (t, C-1), 29.64 (t, C-2), 23.91 (t, C-3), 28.39 (t, C-4), 51.23 (t, C-5), 155.74 (s, –OOC(CH3)3), 79.81 (s, –COO(CH3)3), 28.32 (q, –COOC(H3)3), 4.77 (bs, 1H, NH).
OOC(CH3)3), 79.29 (s, –COO(CH3)3), 28.27 (q, –COOC(H3)3).
N), 1693 (s) ν(amide I), 1519 (m) ν(amide II), 1445 (m), 1390 (m), 1366 (m) δ(CH3), 1250 (m), 1175 (s) ν(C–O), 766 (s). MS (m/z): 251.1 [M + Na]+ (ESI+, coin voltage 20 V), 255.1 (ESI−, coin voltage 20 V). 1H NMR (600.13 MHz, CDCl3): δ [ppm] 3.24 (bdt, 2H, J = 4.7, 5.4, 5.4 Hz, 1-CH2), 1.41–1.45 (m, 2H, 2-CH2), 1.26–1.33 (m, 2H, 3-CH2), 1.50–1.58 (m, 2H, 4-CH2), 3.35 (t, 2H, 5-CH2), 1.38 (s, 3H, Boc (CH3)). 13C NMR (150.90 MHz, CDCl3): δ [ppm] 40.03 (t, C-1), 29.64 (t, C-2), 23.91 (t, C-3), 28.39 (t, C-4), 51.23 (t, C-5), 155.74 (s, –OOC(CH3)3), 79.81 (s, –COO(CH3)3), 28.32 (q, –COOC(H3)3), 4.77 (bs, 1H, NH).
Results and discussion
pH dependence of building blocks
The morpheein behaviour of human peroxiredoxin 3 (Prx, Fig. 1a) was characterized by means of TEM. Solutions of 0.1 mg mL−1 of protein were diluted from a stock solution in Milli-Q water into a final buffer of HEPES (20 mM) and a reducing agent tris((2-carboxyethyl)phosphine) (TCEP, 2 mM) with varying pH. At pH 8, Prx forms toroidal dodecamers (Fig. 1a and b) of 12 nm in diameter, with internal pores of 6 nm.52,53 Upon acidification, dimers and oligomers formed by co-facial stacking are found (pH 6, Fig. 1c), with the length increasing to tubules at pH 4 (Fig. 1d and e). The stacking is likely consequence of burying the hydrophobic patches between dodecamers, as well as polar interactions.54 A closer look to the calculated electrostatic surface potential at pH 8 shows an overall negative charge (in red) distributed both on the toroidal surface and the rim (Fig. 1f, left, top and bottom respectively). Close to the pI (5.9 for Prx), an even distribution of positive and negative charges are found over all surfaces. As expected, at pH 4 a major component of positive charges (blue) is found. However, it is worth mentioning that upon ring stacking, only the outer rim is available to interact, and it shows apical patches of negative charges.A tetracationic Por (1) was synthesized (Fig. 1g, see the ESI† for detailed protocol and characterization). Briefly, the (C8)-OH groups on the commercially available P ring were first substituted with propargyl chloride in the presence of K2CO3 to furnish 2. Compound 2 was then reacted with Zn(OAc)2 to obtain the metalated Zn(II)Por 3. The chelation step with Zn(II) was necessary, to protect the inner ring from sequestering Cu(II) from the Cu(I)-catalyzed Huisgen 1,3-dipolar cycloaddition reaction (CuAAC). Accordingly, 4 was prepared by appending the tert-butoxy (Boc)-protected azide derivative of cadaverine (5) using the previously described CuAAC method (Scheme S2, ESI†).55 Acid hydrolysis of 4 using 1.0 M HCl (gas) in ethyl acetate removed Zn(II) and Boc-protecting groups simultaneously, and 1 was obtained. All the intermediates and final products were characterized by NMR, mass and FTIR (see ESI†).
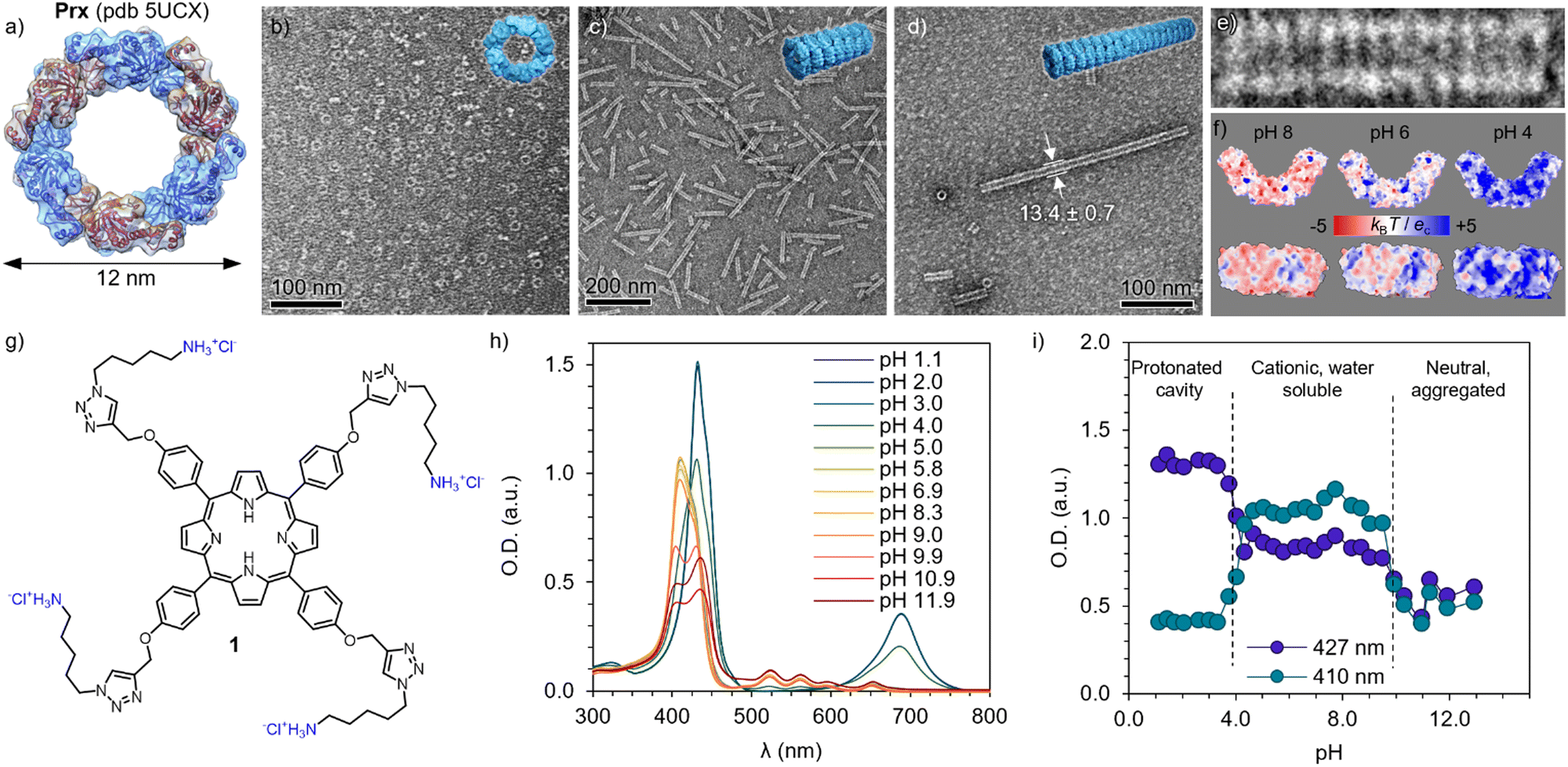 | ||
| Fig. 1 Building blocks and their pH-dependent properties. (a) Structure of the human peroxiredoxin III ((Prx), PDB ID: 5UCX). Protein dimers are shown in alternating blue and magenta colours, forming a dodecamer. TEM images of Prx in 20 mM HEPES, 2 mM TCEP at pH 8 (b), pH 6 (c) and pH 4 (d), showing discrete rings, short, and long tubular stacks, respectively. (e) Detailed micrograph of fourteen stacked rings at pH 4, showing co-facial aggregation. (f) Isoelectric surfaces calculated at pH 8, 6 and 4 (left to right) of Prx. (g) Chemical structure of cationic Por 1, showing four positive charges in its hydrochloric form. (h) Absorption spectra of 1 at varying pH, showing clear changes in the Soret band (350–420 nm) and Q band(s) (520–700 nm). (i) pH dependence of the Soret band at 410 and 427 nm, showing clear transitions between protonated cavity (pH < 4), acidic form (4 < pH < 10), and basic form inducing aggregation (pH > 10). | ||
Bearing four primary amines, the overall charge and therefore aqueous solubility is expected to vary with the pH. Therefore, the absorption spectra was recorded at varying pH (Fig. 1h). At high pH, broad peaks and features are found in the Soret band (400–450 nm), consistent with aggregation and loss in solubility upon deprotonation of the primary amines (pKa 8.9). Upon acidification, the Soret band exhibits a blue-shift and remains constant upon pH 4, where it undergoes a sharp red-shift and, more clearly, a Q-band appears at 680 nm, indicating a protonation of the inner cavity of the Por.56 The trend is clearly observed by plotting the absorbance at 410 and 427 nm depending on the pH (Fig. 1i), depicting the three regimes. As expected, 1 shows good and stable aqueous solubility in the pH regime studied for the Prx. This assignment is in good agreement with previously reported spermine-substituted Por.54
Electrostatic assembly of hybrids
The electrostatic self-assembly of Prx and Por was characterized by dynamic light scattering (DLS), in the range of pH suitable for 1 (4 < pH < 8). At pH 8, Prx shows an apparent hydrodynamic diameter (Dh) of 12 ± 3 nm, corresponding well with the monomeric toroidal protein (Fig. 2a and Table S1, ESI†). Upon titration with 1, a sharp decrease of scattering from monomeric toroid is observed, followed up by an increase in >1000 nm species, which corresponds to large complexes. This is likely consequence of the strong electrostatic interaction between highly charged Prx and 1. At pH 6 (Fig. 2b), the monomer peak slowly shifts to higher Dh, showing at the same time the appearance of large complexes. The more stepwise interaction can be explained by a hindered electrostatic interaction. Last, at pH 4, only medium size aggregates can be found (ca. 50 nm) together with larger-sized complexes. Upon titration with 1, a minor decrease in the medium size complexes is observed, hinting a limited interaction between the now overall positively charged protein and dye. While this information is a clear sign of electrostatic recognition leading to aggregation, the calculated Dh can only be interpreted qualitatively given the large size as well as the expected non-spherical (rod-like) shape at pH 6 and 4. Additionally, the derived count rate gives a calculated count of photons scattered by the sample. Following the Stokes–Einstein equation, and assuming no significant dilution of protein or variation in the refractive index or viscosity through the titration, an increase of the derived count rate is consequence of the increase in diffusion coefficient and, therefore, particle size. In the Prx titration with 1 (Fig. 2d) we observe at pH 8 the derived count rate to sharply increase around 3 × 10−6 M, saturating at 2 × 10−5 M.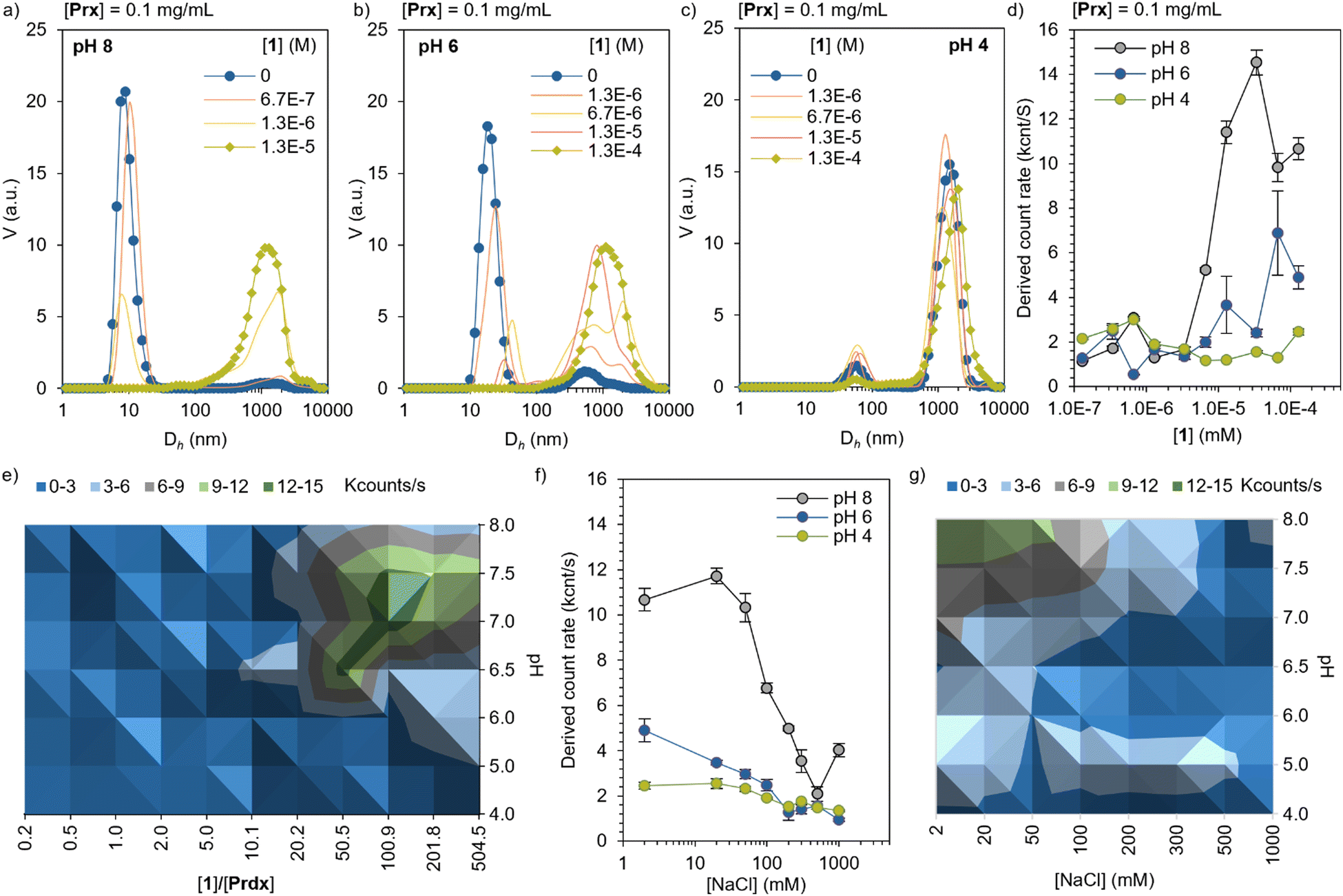 | ||
| Fig. 2 Self-assembly of Prx–1 complexes by dynamic light scattering (DLS). Titration of [Prx] = 0.1 mg mL−1 with increasing concentrations of 1, given as volume-averaged size distributions at (a) pH 8, (b) pH 6, and (c) pH 4. (d) Derived count rate, showing a sharp increase upon formation of large complexes. (e) Diagram of complex formation at different pH and 1 ratios. (f) Complex disassembly upon increase of the ionic strength of the media, achieved by NaCl concentration. (g) Phase diagram of disassembly at different pH and NaCl concentration. | ||
At pH 6, a more modest and milder increase is observed. At pH 4, no noticeable increase was found in the range explored. A diagram prepared by measuring the derived count rate of a constant Prx concentration (0.1 mg mL−1) as a function of pH and ratio of 1, shows that the regions with highest binding affinity are located between pH 7.5 and 6.5, and [1] between 50 to 200 equivalents (Fig. 2e, green area). In order to show the electrostatic character of the interaction, Prx–1 complexes of constant concentration ([Prx] = 0.1 mg mL−1, [1] = 5 × 10−5 M) were titrated with NaCl. The increase of the ionic strength screens the electrostatic interactions, effectively disassembling the complexes. At pH 8, the strong interaction is not fully reversed until 500 mM of NaCl. At pH 6, the interaction is screened at 200 mM, confirming the weaker electrostatic interaction. At pH 4 the same trend is observed, with a modest yet noticeable drop at the same NaCl concentration. Like the binding experiment, a diagram of the disassembly mediated by ionic strength was prepared at different pH values (Fig. 2g). The data shows the expected trend of stronger electrostatic interaction at higher pH.
Morphology of the complexes
A fine control on the binding affinity is a key parameter to obtain highly ordered assemblies: high binding affinities lead to low order/amorphous aggregates, and low affinities to no recognition, moderate affinities are desirable to yield well ordered structures.57 In high binding affinity regime (pH 8), the strong interaction leads to clusters of toroidal proteins with no apparent orientation as shown by TEM images (Fig. 3a and Fig. S1, ESI†). This is in agreement with the overall negative charge of Prx (see Fig. 1f), that is well accessible in the monomer and upon interaction with 1. Therefore, since the protein shows no preferential binding direction, it leads to random aggregation. At pH 6, showing a milder binding affinity, clusters of short and partially aligned tubules are observed (Fig. 3b and Fig. S2, ESI†). Last, at pH 4, large structures of well-aligned tubes of Prx–1 hybrids are found (Fig. 3c and Fig. S3, ESI†). With typical widths of ca. 100 nm and lengths at the micron range, the tightly packed bundles consist of approximately eight Prx proteins in diameter, and hundreds stack. The envisioned mechanism of formation has two steps: first tubule formation is controlled by pH, followed by bundling effect caused by electrostatic interaction with 1. This highly ordered structure is likely a consequence of two factors: (i) the tubule stacking masks the more positively charged surface, and (ii) the remaining negative charge remains on the toroidal rim, enabling a side-by-side interaction of the tubes. The orientation of 1 at the protein interphase is likely random in the pH range explored, given the small size of 1 compared to Prx and the high degree of freedom of the charged side chains. It is worthy to mention that these morphologies are the result of a delicate interplay of forces and, subsequently, small deviations in ionic strength or the absence of reducing agents as TCEP yields amorphous aggregation. | ||
| Fig. 3 Morphology of the Prx–1 complexes. TEM micrographs of the hybrids at (a) pH 8, (b) pH 6, and (c) pH 4. Inset: Close-up of the bundle. Samples prepared by pH adjustment in HEPES 20 mM, TCEP 2 mM. [Prx] = 0.1 mg mL−1 and [1] = 0.2 mg mL−1. (d) Schematic representation of the two step process. | ||
Conclusions
In this manuscript, we report an orthogonal approach towards highly ordered biohybrid materials. We exploit the biological versatility of morpheein proteins to direct the assembly in one direction as tubular stacks, to subsequently tune the electrostatic interactions of the rim to direct the assembly orthogonally. First, we delimited the pH functional range for both moieties, then explore their interaction, based on electrostatic interaction. Last, the morphology of the hybrids was characterized by microscopy techniques. At high pH, strong binding affinities are found, although it leads to poor control and amorphous aggregation. Close to the isoelectric point of the protein, the interaction is moderate, and some order in form of clumped tubules is found. Finally, at pH 4, highly ordered structures are found, directed by the mildest interactions. Our approach, based on the ubiquitous electrostatic interaction to post-functionalize protein cage stacks, shows tremendous potential to be applied in other 1D protein stacks. This opens the toolbox of interactions for protein cage self-assembly, facilitating the often challenging preparation of anisotropic biohybrid materials. These types of materials have the advantage of high directionality and biocompatibility, rendering unique candidates for their exploitation in tissue engineering (e.g. aligning cell growth) or, when doped with conducting (nano)materials, suitable for sensors and wearable electronics.15Author contributions
The authors contributed the following: E. A.-P. biohybrid formation and characterization, project design, funding acquisition, drafting. Z. Ö. Synthesis and characterization. W. Z. Drafting. M. A. K.: project design, drafting.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the funding from the Marie Sklodowska-Curie grant (794536), Academy of Finland (projects 308578, 303804, 267497, 341057), Jane and Aatos Erkko Foundation. We acknowledge the provision of facilities and technical support by Aalto University Bioeconomy Facilities and OtaNano-Nanomicroscopy Center (Aalto-NMC).Notes and references
- A. F. Dishman and B. F. Volkman, ACS Chem. Biol., 2018, 13, 1438–1446 CrossRef CAS PubMed.
- Z. A. Wood, E. Schröder, J. R. Harris and L. B. Poole, Trends Biochem. Sci., 2003, 28, 32–40 CrossRef CAS PubMed.
- M. Aran, D. Ferrero, A. Wolosiuk, S. Mora-García and R. A. Wolosiuk, J. Biol. Chem., 2011, 286, 23441–23451 CrossRef CAS PubMed.
- M. E. Conway and C. Lee, Biomol. Concepts, 2015, 6, 269–284 CAS.
- F. Angelucci, A. Bellelli, M. Ardini, R. Ippoliti, F. Saccoccia and V. Morea, FEBS J., 2015, 282, 2827–2845 CrossRef CAS PubMed.
- A. Korpi, E. Anaya-Plaza, S. Välimäki and M. Kostiainen, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2020, 12, e1578 CAS.
- Q. Luo, C. Hou, Y. Bai, R. Wang and J. Liu, Chem. Rev., 2016, 116, 13571–13632 CrossRef CAS PubMed.
- J. R. McMillan, O. G. Hayes, P. H. Winegar and C. A. Mirkin, Acc. Chem. Res., 2019, 52, 1939–1948 CrossRef CAS PubMed.
- N. P. King, J. B. Bale, W. Sheffler, D. E. McNamara, S. Gonen, T. Gonen, T. O. Yeates and D. Baker, Nature, 2014, 510, 103–108 CrossRef CAS PubMed.
- J. Sun, C. DuFort, M. C. Daniel, A. Murali, C. Chen, K. Gopinath, B. Stein, M. De, V. M. Rotello, A. Holzenburg, C. C. Kao and B. Dragnea, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1354–1359 CrossRef CAS PubMed.
- V. Liljeström, A. Ora, J. Hassinen, H. T. Rekola, N. Nonappa, M. Heilala, V. Hynninen, J. J. Joensuu, R. H. A. Ras, P. Törmä, O. Ikkala and M. A. Kostiainen, Nat. Commun., 2017, 8, 671 CrossRef PubMed.
- M. Künzle, T. Eckert and T. Beck, J. Am. Chem. Soc., 2016, 138, 12731–12734 CrossRef PubMed.
- M. Lach, C. Strelow, A. Meyer, A. Mews and T. Beck, ACS Appl. Mater. Interfaces, 2022, 14, 10656–10668 CrossRef CAS PubMed.
- N. P. King, W. Sheffler, M. R. Sawaya, B. S. Vollmar, J. P. Sumida, I. André, T. Gonen, T. O. Yeates and D. Baker, Science, 2012, 336, 1171–1174 CrossRef CAS PubMed.
- J. B. Bale, S. Gonen, Y. Liu, W. Sheffler, D. Ellis, C. Thomas, D. Cascio, T. O. Yeates, T. Gonen, N. P. King and D. Baker, Science, 2016, 353, 389–394 CrossRef CAS PubMed.
- J. Laniado, K. A. Cannon, J. E. Miller, M. R. Sawaya, D. E. McNamara and T. O. Yeates, ACS Nano, 2021, 15, 4277–4286 CrossRef CAS PubMed.
- E. Selivanovitch, M. Uchida, B. Lee and T. Douglas, ACS Nano, 2021, 15, 15687–15699 CrossRef CAS PubMed.
- M. Lach, M. Künzle and T. Beck, Chem. – Eur. J., 2017, 23, 17482–17486 CrossRef CAS PubMed.
- A. Korpi and M. A. Kostiainen, ChemNanoMat, 2022, 8, e202100458 CrossRef CAS.
- Y. T. Lai, E. Reading, G. L. Hura, K. L. Tsai, A. Laganowsky, F. J. Asturias, J. A. Tainer, C. V. Robinson and T. O. Yeates, Nat. Chem., 2014, 6, 1065–1071 CrossRef CAS PubMed.
- E. Anaya-Plaza, A. Shaukat, I. Lehtonen and M. A. Kostiainen, Adv. Healthcare Mater., 2021, 10, 2001162 CrossRef CAS PubMed.
- A. Shaukat, E. Anaya-Plaza, N. K. Beyeh and M. A. Kostiainen, Chem. – Eur. J., 2022, 28, e202104341 CrossRef CAS PubMed.
- M. Ardini, A. Bellelli, D. L. Williams, L. Di Leandro, F. Giansanti, A. Cimini, R. Ippoliti and F. Angelucci, Bioconjugate Chem., 2021, 32, 43–62 CrossRef CAS PubMed.
- E. Miceli, M. Kar and M. Calderón, J. Mater. Chem. B, 2017, 5, 4393–4405 RSC.
- G. Giovannini, M. Ardini, N. Maccaferri, X. Zambrana-Puyalto, G. Panella, F. Angelucci, R. Ippoliti, D. Garoli and F. De Angelis, Adv. Opt. Mater., 2020, 8, 1901583 CrossRef CAS.
- S. Manuguri, K. Webster, N. A. Yewdall, Y. An, H. Venugopal, V. Bhugra, A. Turner, L. J. Domigan, J. A. Gerrard, D. E. Williams and J. Malmström, Nano Lett., 2018, 18, 5138–5145 CrossRef CAS PubMed.
- M. Ardini, F. Giansanti, L. Di Leandro, G. Pitari, A. Cimini, L. Ottaviano, M. Donarelli, S. Santucci, F. Angelucci and R. Ippoliti, Nanoscale, 2014, 6, 8052–8061 RSC.
- M. Ardini, J. A. Huang, V. Caprettini, F. De Angelis, F. Fata, I. Silvestri, A. Cimini, F. Giansanti, F. Angelucci and R. Ippoliti, Biochim. Biophys. Acta, Gen. Subj., 2020, 1864, 129617 CrossRef CAS PubMed.
- H. Sun, X. Zhang, L. Miao, L. Zhao, Q. Luo, J. Xu and J. Liu, ACS Nano, 2016, 10, 421–428 CrossRef CAS PubMed.
- L. Miao, J. Han, H. Zhang, L. Zhao, C. Si, X. Zhang, C. Hou, Q. Luo, J. Xu and J. Liu, ACS Nano, 2014, 8, 3743–3751 CrossRef CAS PubMed.
- L. Zhao, H. Zou, H. Zhang, H. Sun, T. Wang, T. Pan, X. Li, Y. Bai, S. Qiao, Q. Luo, J. Xu, C. Hou and J. Liu, ACS Nano, 2017, 11, 938–945 CrossRef CAS PubMed.
- S. Sim, T. Niwa, H. Taguchi and T. Aida, J. Am. Chem. Soc., 2016, 138, 11152–11155 CrossRef CAS PubMed.
- T. Sendai, S. Biswas and T. Aida, J. Am. Chem. Soc., 2013, 135, 11509–11512 CrossRef CAS PubMed.
- J. R. McMillan and C. A. Mirkin, J. Am. Chem. Soc., 2018, 140, 6776–6779 CrossRef CAS PubMed.
- M. Endo, M. Fujitsuka and T. Majima, Chem. – Eur. J., 2007, 13, 8660–8666 CrossRef CAS PubMed.
- J. Zhang, X. Wang, K. Zhou, G. Chen and Q. Wang, ACS Nano, 2018, 12, 1673–1679 CrossRef CAS PubMed.
- Y. Li, C. Xia, R. Tian, L. Zhao, J. Hou, J. Wang, Q. Luo, J. Xu, L. Wang, C. Hou, B. Yang, H. Sun and J. Liu, ACS Nano, 2022, 16, 8012 CrossRef CAS PubMed.
- O. G. Hayes, B. E. Partridge and C. A. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2106808118 CrossRef CAS PubMed.
- V. Almeida-Marrero, E. van de Winckel, E. Anaya-Plaza, T. Torres and A. de la Escosura, Chem. Soc. Rev., 2018, 47, 7369–7400 RSC.
- J. Mikkilä, E. Anaya-Plaza, V. Liljeström, J. R. Caston, T. Torres, A. De La Escosura and M. A. Kostiainen, ACS Nano, 2016, 10, 1565–1571 CrossRef PubMed.
- E. Anaya-Plaza, A. Aljarilla, G. Beaune, Nonappa, J. V. I. Timonen, A. Escosura, T. Torres and M. A. Kostiainen, Adv. Mater., 2019, 31, 1902582 CrossRef PubMed.
- W. R. Osterloh, K. M. Kadish and E. Van Caemelbecke, Handbook of Porphyrin Science, World scientific, 2023, vol. 47 Search PubMed.
- G. Bottari, O. Trukhina, M. Ince and T. Torres, Coord. Chem. Rev., 2012, 256, 2453–2477 CrossRef CAS.
- G. Bottari, G. de la Torre, D. M. Guldi and T. Torres, Coord. Chem. Rev., 2021, 428, 213605 CrossRef CAS.
- J. Chakraborty, I. Nath and F. Verpoort, Coord. Chem. Rev., 2016, 326, 135–163 CrossRef CAS.
- H. Lu and N. Kobayashi, Chem. Rev., 2016, 116, 6184–6261 CrossRef CAS PubMed.
- Q. Sun, L. M. Mateo, R. Robles, P. Ruffieux, G. Bottari, T. Torres, R. Fasel and N. Lorente, Adv. Sci., 2022, 9, 2105906 CrossRef CAS PubMed.
- Q. Sun, L. M. Mateo, R. Robles, P. Ruffieux, N. Lorente, G. Bottari, T. Torres and R. Fasel, J. Am. Chem. Soc., 2020, 142, 18109–18117 CrossRef CAS PubMed.
- M. Strohalm, D. Kavan, P. Novák, M. Volný and V. Havlíček, Anal. Chem., 2010, 82, 4648–4651 CrossRef CAS PubMed.
- E. Jurrus, D. Engel, K. Star, K. Monson, J. Brandi, L. E. Felberg, D. H. Brookes, L. Wilson, J. Chen, K. Liles, M. Chun, P. Li, D. W. Gohara, T. Dolinsky, R. Konecny, D. R. Koes, J. E. Nielsen, T. Head-Gordon, W. Geng, R. Krasny, G. Wei, M. J. Holst, J. A. McCammon and N. A. Baker, Protein Sci., 2018, 27, 112–128 CrossRef CAS PubMed.
- Z. Özdemir, D. Šaman, K. Bertula, M. Lahtinen, L. Bednárová, M. Pazderková, L. Rárová, Nonappa and Z. Wimmer, Langmuir, 2021, 37, 2693–2706 CrossRef PubMed.
- N. A. Yewdall, H. Venugopal, A. Desfosses, V. Abrishami, Y. Yosaatmadja, M. B. B. Hampton, J. A. A. Gerrard, D. C. C. Goldstone, A. K. K. Mitra and M. Radjainia, Structure, 2016, 24, 1120–1129 CrossRef CAS PubMed.
- N. A. Yewdall, A. V. Peskin, M. B. Hampton, D. C. Goldstone, F. G. Pearce and J. A. Gerrard, Biochem. Biophys. Res. Commun., 2018, 497, 558–563 CrossRef CAS PubMed.
- F. Angelucci, F. Saccoccia, M. Ardini, G. Boumis, M. Brunori, L. Di Leandro, R. Ippoliti, A. E. Miele, G. Natoli, S. Scotti and A. Bellelli, J. Mol. Biol., 2013, 425, 4556–4568 CrossRef CAS PubMed.
- M. Yang, Z. Özdemir, H. Kim, S. Nah, E. Andris, X. Li, Z. Wimmer and J. Yoon, Adv. Healthcare Mater., 2022, 11, 2200529 CrossRef CAS PubMed.
- J. Sobczyński, H. H. Tønnesen and S. Kristensen, Pharmazie, 2013, 68, 100–109 Search PubMed.
- A. M. Kalsin, Science, 2012, 420, 420–425 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Chemical characterization and supplementary micrographs. See DOI: https://doi.org/10.1039/d3tb00369h |
| This journal is © The Royal Society of Chemistry 2023 |