
Engineering the HK97 virus-like particle as a nanoplatform for biotechnology applications†
Michael D.
Woods
 a,
Matthew
Cali
a,
Bubacarr
Ceesay
a,
Shandis
Fancher
b,
Gaini
Ibrasheva
a,
Suefian
Kandeel
a,
Manon
Nassar
a,
Ali
Azghani
b,
Brent
Bill
b and
Dustin P.
Patterson
*a
a,
Matthew
Cali
a,
Bubacarr
Ceesay
a,
Shandis
Fancher
b,
Gaini
Ibrasheva
a,
Suefian
Kandeel
a,
Manon
Nassar
a,
Ali
Azghani
b,
Brent
Bill
b and
Dustin P.
Patterson
*a
aThe University of Texas at Tyler, Department of Chemistry and Biochemistry, 3900 University Dr, Tyler, TX 75799, USA. E-mail: dpatterson@uttyler.edu
bThe University of Texas at Tyler, Department of Biology, 3900 University Dr, Tyler, TX 75799, USA
First published on 14th June 2023
Abstract
The research described here looks at the development of virus-like particles (VLPs) derived from bacteriophage HK97 as versatile scaffolds for bionanomaterials construction. Based on molecular models, the Prohead I HK97 VLP was engineered to allow attachment of small molecules to the interior by introducing a reactive cysteine into the genetic sequence of the HK97 GP5 protein that self assembles to form the VLP structure. In addition, methods for entrapping large protein macromolecules were evaluated and found to produce high encapsulation numbers of green fluorescent proteins (GFP) in the internal space of the HK97 VLP. A method for modular modification of the external surface was engineered by constructing a plasmid allowing the addition of peptide sequences to the C-terminus of the GP5 protein, which was validated by appending the sortase recognition peptide sequence, LPETG, to the C-terminus of GP5 and showing the attachment of a polyglycine-GFP to the HK97 VLP through sortase mediated ligation. To demonstrate the potential for advanced applications, an HK97 VLP covalently labeled on the interior surface with fluorescein and containing an externally displayed integrin binding peptide sequence (RGD) was evaluated and found to be preferentially localized at C2C12 cells relative to the HK97 VLP lacking the RGD peptide. Together, these results support the potential of the HK97 VLP as a versatile nanoparticle platform that can be modified internally and externally in a modular fashion for the purpose of programming the VLP for desired applications.
Nanotechnology has grown exponentially in the last two decades and made great strides in the development of novel materials being exploited in applications ranging from light harvesting systems to advanced medical diagnostic and therapeutic tools. The unique size of nanomaterials makes them amenable to a wide number of applications with emergent properties not seen in bulk materials, and the near-atomic sizes make them operable in a wide range of environments, including subcellular applications. Nanomaterials have been developed from a wide range of components and can be highly diverse in their chemical make-up and composition; however, constructing structures that are homogenous in composition and structural morphology is a major challenge. One class of nanomaterials that is particularly unique in their ability to be produced homogenously in size and structural morphology are virus-like particles (VLPs), a type of protein cage structure derived from viruses. VLPs are hollow structures produced from the self-assembly of viral coat proteins that form the outer shell, called the capsid, of the virus. VLPs are unique due to their ease of production, existing knowledge of assembly and molecular structure, and biocompatible makeup, which has garnered much interest for their utilization in constructing nanomaterials.1–8 While several VLP systems have been developed, no single system provides the necessary diversity in chemical, physical, and functional properties to address all applications, and the development of new systems will help generate a diverse library of VLPs to be exploited for various applications. Here, we report the development of the VLP system based on the Hong Kong 97 (HK97) bacteriophage, which has several unique properties not found in other systems.
The HK97 bacteriophage, first identified in swine dung in 1997, naturally infects E. coli and its structure and assembly have been extensively studied. Formation of the HK97 bacteriophage begins with the assembly of 420 copies of the major capsid protein, called GP5, which forms hexamers and pentamers that assemble into the 56 nm spherical Prohead I structure (Fig. 1).9 The GP4 protease and GP3 portal proteins associate with the self-assembling GP5 major capsid protein to aid in the maturation of the HK97 phage head to its final functional morphology.10,11 The GP4 protease associates with GP5 and becomes encapsulated on the interior of Prohead I then subsequently proteolyzes GP5, removing the first 100 amino acids from the N-terminus of GP5 (the delta domain). This results in the Prohead II structure which is comprised of the truncated GP5 protein (GP5*), is largely identical to Prohead I, and preserves the internal capsid volume.10 GP3 portal proteins are optional for VLP assembly but, when included, produce a dodecameric portal that replaces a single GP5 penton in the capsid structure which allows viral DNA to be pumped into the capsid with the aid of additional accessory proteins at later stages of the maturation process.11 After formation of the Prohead II structure, the HK97 VLP can be chemically induced, in vitro by acidification or denaturation methods, to transform into the mature 66 nm Head II VLP, which results from spontaneous rearrangement of GP5* and transition through several intermediate structures.9,10,12–16 The Head II form of the HK97 VLP uniquely forms a catenane structure, in which all GP5* capsid proteins are covalently cross-linked through a lysine-aspartate side chain reaction.17 The structural transformations, omitting the various intermediates between the Prohead II and Head II, are outlined in the schematic shown in Fig. 1. The catenane structure of the HK97 Head II has been described as molecular chainmail and results in a nearly indestructible protein cage structure with high thermal stability.18
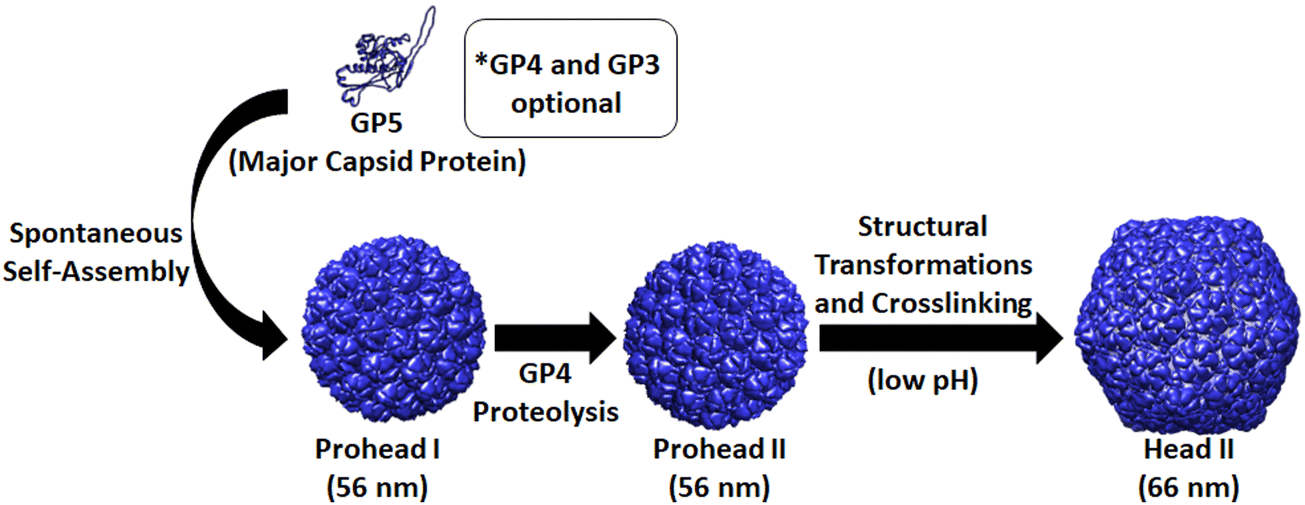 | ||
| Fig. 1 Structural morphologies of the HK97 VLP. Initial assembly of GP5 protein leads to the Prohead I structure. When assembly of GP5 occurs with the maturation protease GP4, the GP5 is proteolyzed to GP5*, with a resulting conformational change from the Prohead I to the Prohead II structure. Treatment of Prohead II with low pH leads to structural rearrangement of the GP5* subunits and their subsequent covalent crosslinking to form the Head II catenane structure. | ||
Many VLP and protein cage (PC) systems have been developed as nanoplatforms for biomaterials construction. However, bacteriophage HK97 presents many unique qualities that distinguish it from the VLPs and PCs previously described. For instance, the much larger diameter of the Prohead II (56 nm) has nearly 8 to 10 times greater internal volume compared to cowpea chlorotic mottle virus (CCMV) and cowpea mosaic virus (CPMV) (both 28 nm in diameter),19 Qbeta (24 nm), MS2 (26 nm), ferritin (12 nm), the E2 subunit pyruvate dehydrogenase (24 nm), encapsulins (20–42 nm), and other well developed systems.20 Larger PCs, such as the carboxysome (40–600 nm), do not maintain homogeneity in structural morphology,20,21 preventing the concise engineering and molecular characterization allowed by HK97. In comparison with other VLP and PC systems reported, only the P22 VLP has comparable attributes in size and dynamic properties to the bacteriophage HK97 VLPs.22 The initial P22 VLP Procapsid structure (58 nm) can be converted to a larger Expanded Shell structure (64 nm) through heating (65 degrees C) or a porous Wiffleball structure (64 nm) with 5 nm pores through heating at higher temperatures (75 degrees C). Because of its properties, the P22 VLP platform has been extensively studied and adapted for applications ranging from therapeutics to construction of nanoreactors. However, the HK97 VLP has several unique features which suggest its development could have wide ranging impacts in the VLP and PC field.
First, the HK97 VLP can assemble without need of a scaffolding protein like P22, resulting in fewer downstream treatments to prepare the VLP for encapsulation of synthetic cargo molecules. As discussed in detail later, the HK97 VLP provides the ability to encapsulate large proteins without modification to its assembly, which is significantly more challenging with a system like the P22 VLP which requires a scaffolding protein for assembly. These simplifications in producing the final product results in the HK97 VLP providing high yields and quick production times, which are invaluable for the research lab and industrial scaling alike. Additionally, the HK97 VLP provides a dynamic system that allows modifications to size and structural stability depending on the VLP form, with a highly stable catenane structure not observed in P22 or other published VLPs. The extreme stability of the catenane form expands the useful applications of the HK97 platform to include employment at high temperatures and hostile chemical environments other systems cannot withstand. Furthermore, in comparison with other VLP systems, the HK97 VLP platform has also been shown to have limited uptake into mammalian cells unless externally modified to enable entry. Huang et al. showed that wild type HK97 VLPs exhibited little interaction with mammalian cells, with negligible background interactions at only high concentrations, in contrast to CPMV, Qbeta, and the rod-shaped plant virus Potato virus X.19 This property supports the HK97 VLP as a promising candidate for therapeutic applications, such as modular cell targeting or as an inert extracellular cargo vessel, and is especially important for in vivo applications to reduce off-target effects. The unique simplicity and diverse properties of the HK97 VLP support it as a novel system that could be highly influential in bionanomaterials applications and warrants investigation. To date, relatively little has been done to develop the HK97 VLP system for advanced applications in nanotechnology. Modifications to the exterior by attachment of fluorescent fluorescein iodoisothiocyanate and the protein transferrin has been reported,19 but no further development has been reported.
Due to its unique properties in structural changes, stability, and selective cell entry, we set out to engineer a modular HK97 VLP platform, with a focus on the Prohead I morphology, which could be easily adapted to alter the interior and/or exterior of the VLP as necessary for a wide range of applications (Fig. 2). Conceptually, the HK97 VLP was divided into three main domains for development: the internal surface, internal volume, and external surface. For modification of the interior, we wanted to develop the HK97 VLP so that we could either localize small molecules or trap large macromolecular species on the interior. For small molecule entrapment, we examined the molecular structure of the internal surface to see if we could introduce a cysteine residue that would act as a reaction site for covalent attachment and incorporation of synthetic molecules in the protected confines of the HK97 VLP interior. Cysteine residues provide a reactive thiol that shows selective reactivity toward functional groups, such as alpha-haloamides or maleimides, that can be easily appended to small molecules allowing localization and covalent attachment to the exposed cysteine. For entrapment of large macromolecules, we focused on the encapsulation of proteins as our target molecule because proteins are biologically active and have a wide range of applications where protein stabilization and delivery are desired. For encapsulation, we looked at exploiting the targeting peptide sequence from GP4 as a way of trafficking proteins to the interior of the HK97 VLP during assembly.10 It has been previously shown that a small decorator protein could be integrated on the interior of the HK97 VLP this way,10 but larger proteins have not been examined. Therefore, we looked to examine the encapsulation of green fluorescent protein (GFP) via this strategy to confirm that large functional proteins, such as enzymes, could effectively be encapsulated without disruption of the assembly process. By developing the HK97 VLP to have both a reactive cysteine and exploiting the targeting peptide sequence from GP4, we looked to develop a versatile encapsulation system for a wide range of guest molecules that could take advantage of the protective environment provided by the HK97 VLP cage structure.
 | ||
| Fig. 2 Developing the HK97 VLP as a versatile nanoparticle platform for bionanomaterials construction. The HK97 VLP has an internal surface, interior hollow space, and external surface that can be modified to develop designer VLP nanoparticles. Mutation of amino acids exposed to the interior (alanine 302, orange, and serine 305, magenta, highlighted) to allow site specific chemical labeling would allow modification of internal surface. Modification of large macromolecular cargos with localization tags that interact with the GP5 protein during assembly would enable encapsulation of the macromolecules on the VLP interior. Genetic engineering of the external surface (C-terminus residue, red) with peptide tags would enable chemical programming of the HK97 VLP exterior for advanced applications. Combining the ability to modify the inside and outside of the HK97 VLP should allow designer VLPs with tailored properties for advanced applications (bottom scheme). | ||
Additionally, we looked at developing the exterior so that it could be chemically programed to further extend the HK97 VLP's usefulness and diversity of future applications. Applications such as therapeutic delivery vehicles and enzyme immobilization on surfaces require external modification for attaching targeting moieties or for surface adherence. Methods that could be general, easily installed, would allow addition of small and large functional groups, and would not interfere with the previously described internal modification of the HK97 VLP with a cysteine residue were desired. To meet these constraints, we observed that the C-terminus of the HK97 VLP is positioned on the exterior (Fig. 2) and could serve as an anchoring point for the attachment of various functional groups. To this end, we examined a modular genetic approach whereby desired peptides could be added genetically via recombinant DNA technology. The versatility of peptides includes the ability to integrate cell targeting functions through targeting sequences such as the integrin binding RGD, to associate with specific surface substrates through surface binding peptides, to attach large proteins through systems such as sortase or Snap/Snare, and attachment of chemically and biochemically active peptides identified through processes such as phage display and biopanning. To this end, we developed an expression vector that enabled quick integration of new peptide sequences to the GP5 gene that would be directly attached to the HK97 VLP upon expression and displayed on the surface. This approach, which is only possible given the unique protein sequence and assembly of the HK97 VLP, allows convenient and reproducible adaptation of the surface-exposed peptides to optimize the HK97 VLP for a given application.
The research presented here examines the engineering of the HK97 VLP to produce a modular nanoparticle system that can be modified both externally and internally. The results show that the internal surface can be easily modified via a site mutated cysteine residue that yields high labeling rates. In addition, we show that large protein cargoes can be encapsulated on the interior by genetically fusing a portion of the GP4 protein sequence containing the targeting peptide (TP) portion. GFP was utilized for studying the encapsulation and two approaches were utilized, a co-expression strategy that simultaneously expressed both GP5 coat protein and GFP-TP and a sequential expression strategy in which GFP-TP expression was induced first followed by GP5 induction and expression to assemble the HK97 VLP particle. For the exterior, a plasmid that allows rapid incorporation of new peptides was constructed and two peptide sequences were examined, the LPETG sortase recognition peptide sequence and the RGD integrin binding sequence. The incorporation of the LPETG sequence to the C-terminus allows for attachment of proteins containing N-terminal polyglycines (2 or more) through a covalent bond generated between the T residue and the N-terminal glycine on the other protein, which is catalyzed by the enzyme sortase. The sortase method has been previously shown to be useful for other VLP systems for attachment of various proteins,23 and GFP containing an N-terminal poly-glycine was examined in our study. The integrin binding sequence, RGD, was examined to see if the attachment would allow cell targeting, which was investigated by coupling the RGD modification with the HK97 containing the internal cysteine to permit fluorescent molecule attachment and tracking of the HK97 VLP in in vitro cell culture studies. Together, the results show the development of a versatile scaffold that can be chemically programmed on the outside, inside, or both outside and inside through modular methods that make the HK97 VLP a highly adaptable system for nanomaterials construction.
Materials and methods
Materials
DNA modifying enzymes were purchased from New England Biolabs (Ipswich, MA). The pRSFDuet-1 plasmid was purchased from Life Technologies (Grand Island, NY). Genes encoding the bacteriophage HK97 GP5 and GP4 were purchased from GenScript (Piscataway, NJ). DNA primers were purchased from Eurofins MWG Operon (Huntsville, AL) or Invitrogen (Waltham, MA). E. coli ClearColi BL21(DE3) chemically competent and 10G ECloni electrocompotent cells were purchased from Lucigen (Middleton, WI). MinElute Reaction Cleanup and QIAprep Spin Miniprep kits were purchased from Qiagen (Valencia, CA). All other chemical reagents were from Fisher Scientific (Pittsburgh, PA) or Sigma Aldrich (St. Louis, MO).Molecular biology
For constructing expression vectors for producing HK97 VLPs, the pRSFDuet-1 plasmid was utilized. Initially, the genes encoding GP5 and GP4 were purchased and obtained in the pUC57 plasmid (GP4/GP5-pUC57). The gene encoding GP5 was transferred to pRSFDuet-1 by NdeI and XhoI double digestion of the pRSFDuet-1 plasmid and the GP4/GP5-pUC57 plasmid, separately. Double digests were purified using the MinElute Reaction Cleanup Kit (Qiagen), the resulting products were mixed, and the GP5 gene was ligated into multiple cloning site 2 of the pRSFDuet-1 plasmid with T4 DNA ligase. The ligation reaction mixture was subsequently transformed into 10G ECloni electrocompetent cells and plated out on a LB-Agar plate containing kanamycin (50 μg mL−1) for selection of the pRSFDuet-1 plasmid. After overnight growth of media plates, colonies containing the GP5 gene were identified by colony PCR using OneTaq Master Mix and primers for the GP5 gene. Colonies indicating hits for the GP5 gene were used to inoculate 5 mL of kanamycin-selective LB broth and grown up overnight for harvesting and extraction of DNA using the QIAprep Spin Miniprep kit. Nucleic acid sequencing was performed by Eurofins to confirm the proper gene sequence. The pRSFDuet-1 plasmid containing GP5 in multiple cloning site 2 (GP5-pRSF) was subsequently used to construct several other plasmids.For construction of a plasmid that could be utilized for producing the mature Head II form of the HK97 VLP, the GP5-pRSF and GP4/GP5-pUC57 plasmid were double digested with NcoI and BamHI to enable insertion of GP4 into multiple cloning site 1 of GP5-pRSF. Subsequent purification, ligation, transformation, and analysis for obtaining GP5-pRSF were performed to obtain the plasmid containing GP4 in site 1 and GP5 in site 2 of the multiple cloning sites of pRSFDuet-1 to yield the GP4/GP5-pRSF plasmid.
The published crystal structure of each HK97 VLP form (PDB ID: 3QPR for Prohead I, 3E8K for Prohead II, and 1OHG for Head II) was evaluated to identify conservative mutation sites for the addition of exposed internal cysteine moieties. Cysteine mutants of the GP5 protein used in this study were produced by PCR using the PfuTurbo DNA polymerase (Agilent) according to the manufacturers protocol and the following primers:
A302C_forward 5′-ccacatgatgttactggtgaaacactgaggaccaccgaagatata-3′
A302C_reverse 5′-tatatcttcggtggtcctcagtgtttcaccagtaacatcatgtgg-3′
S305C_forward 5′-ccccacatgatgttacaggtgaatgcctgagga-3′
S305C_reverse 5′-tcctcaggcattcacctgtaacatcatgtgggg-3′
To construct plasmids for producing GP5 containing modified C-termini, the GP5 gene was amplified from the GP4/GP5-pUC57 plasmid using the forward primer 5-aaaccatggcatatgtctgaactcgctctc-3′ and a reverse primer 5′-ttttgccg![[c with combining low line]](https://www.rsc.org/images/entities/char_0063_0332.gif)
![[t with combining low line]](https://www.rsc.org/images/entities/char_0074_0332.gif)
![[g with combining low line]](https://www.rsc.org/images/entities/char_0067_0332.gif)
![[a with combining low line]](https://www.rsc.org/images/entities/char_0061_0332.gif) ttagctaccgccggtttccggcagtcctccggcacctccgccgcctgctgagccagaagagaaggtgcccttg-3′ encoding a flexible polyglycine sequence and the sortase recognition sequence (amino acid sequence LPETG). Restriction sites (underlined) were included in the reverse primer to allow removal, modification, or replacement of either the flexible polypeptide sequence (BglII/KpnI) or the LPETG encoding peptide (KpnI/XhoI). The PCR-amplified gene containing GP5 and the C-terminal LPETG gene sequence (GP5-LPETG) was purified with a MinElute Reaction Cleanup kit (Qiagen) and subsequently inserted into multiple cloning site 2 of the pRSFDuet-1 plasmid as described previously for the GP5-pRSF plasmid. Using the resulting GP5-LPETG-pRSF plasmid, the GP5 gene containing the RGDS sequence at the C-terminus was constructed by digesting the plasmid with KpnI and XhoI, purifying the digested product with a MinElute Reaction Cleanup kit, and performing a ligation with annealed oligonucleotides 5′-ccgtggcgatagctaac-3′ and 5′-tcgagttagctatcgccacgggtac-3′ encoding the amino acid sequence RGDS with correct sticky ends for insertion into the plasmid, yielding the GP5-RGDS-pRSF plasmid. To produce the GP5-S305C-RGDS-pRSF plasmid, GP5-RGDS-pRSF plasmid was utilized as a template and PCR site directed mutatgenesis was performed using the S305C primers and same methodology noted earlier.
ttagctaccgccggtttccggcagtcctccggcacctccgccgcctgctgagccagaagagaaggtgcccttg-3′ encoding a flexible polyglycine sequence and the sortase recognition sequence (amino acid sequence LPETG). Restriction sites (underlined) were included in the reverse primer to allow removal, modification, or replacement of either the flexible polypeptide sequence (BglII/KpnI) or the LPETG encoding peptide (KpnI/XhoI). The PCR-amplified gene containing GP5 and the C-terminal LPETG gene sequence (GP5-LPETG) was purified with a MinElute Reaction Cleanup kit (Qiagen) and subsequently inserted into multiple cloning site 2 of the pRSFDuet-1 plasmid as described previously for the GP5-pRSF plasmid. Using the resulting GP5-LPETG-pRSF plasmid, the GP5 gene containing the RGDS sequence at the C-terminus was constructed by digesting the plasmid with KpnI and XhoI, purifying the digested product with a MinElute Reaction Cleanup kit, and performing a ligation with annealed oligonucleotides 5′-ccgtggcgatagctaac-3′ and 5′-tcgagttagctatcgccacgggtac-3′ encoding the amino acid sequence RGDS with correct sticky ends for insertion into the plasmid, yielding the GP5-RGDS-pRSF plasmid. To produce the GP5-S305C-RGDS-pRSF plasmid, GP5-RGDS-pRSF plasmid was utilized as a template and PCR site directed mutatgenesis was performed using the S305C primers and same methodology noted earlier.
For investigating the encapsulation of proteins on the interior of the HK97 VLP, a plasmid was constructed using the GP5-pRSF plasmid with an empty multiple cloning site 1. The C-terminal portion of the GP4 gene encompassing the previously identified targeting peptide (TP) sequence, which promotes encapsulation of the GP4 protein, was amplified from the GP4/GP5-pUC57 plasmid using the forward primer 5′-aaaccatgggatccggcggtgatggcattgaaacgatccgtgatg-3′ and the reverse primer 5′-aaagagctcggatccttatttacctaag-3′. The amplified TP gene and the GP5-pRSF plasmid were then double digested with BamHI and SacI, separately. The digest products were subsequently purified with a MinElute Reaction Cleanup kit, ligated, transformed, and screened as described above for the other constructs. After DNA sequencing (Eurofins) confirmed incorporation of the TP gene in multiple cloning site 1 of the GP5-pRSF plasmid, yielding the TP/GP5-pRSF plasmid, the green fluorescent protein (GFP) gene was inserted upstream of the TP by performing a NcoI and BamHI double digest of a plasmid containing the GFP gene and the TP/GP5-pRSF plasmid. These products were subsequently purified, ligated, transformed, and screened as described for other constructs to obtain a plasmid containing a GFP-TP gene fusion in multiple cloning site 1 and the GP5 gene in multiple cloning site 2 (yielding the GFP-TP/GP5-pRSF plasmid). Transfer of GFP-TP into the pBAD plasmid was accomplished by NcoI and SacI double digestion of the GFP-TP/GP5-pRSF plasmid and pBAD plasmids and subsequently performing purification, ligation, transformation, and verification of sequence as noted for the other constructs to obtain a GFP-TP-pBAD plasmid. All other plasmids for producing proteins, such as sortase and N-terminal polyglycine-GFP, were obtained and have been described in a previous publication.23
Prohead HK97 VLP expression and purification
All media used was sterile, kanamycin-selective LB broth (50 mg/L kanamycin, 10 g/L sodium chloride, 10 g/L tryptone, 5 g/L yeast extract). Glycerol stocks of Escherichia coli ClearColi BL21 containing plasmids for GP5 or GP4/GP5 co-expression and mutant variants (i.e. GP5 A302C, GP5 S305C, LPETG-GP5, and RGDS-GP5) were used to inoculate 15 mL of selective media shaken overnight at 250 rpm and 37.0 °C. These cultures were then used to inoculate 1 L of selective media in a 2.8 L Fernbach flask and shaken at 250 rpm at 37 °C. Once an optical density (OD) of 0.4–0.8 at 600 nm was measured, the cell solution was cooled at 4 °C for 15–30 minutes and isopropyl-beta-D-1-thiogalactopyranoside (IPTG) was added to yield a final concentration of 0.5 mM. The cell solution was shaken overnight at 250 rpm at 28 °C and then the cells were pelleted by centrifugation at 3000 g for 20 min at 4 °C. All cell pellets were stored at −20 °C until needed (typically overnight). Wet cell pellets were thawed and resuspended in 0.22 μm filtered Tris Lysis Buffer (50 mM Tris, 175 mM sodium chloride, pH 7.4) for lysis.Lysis was performed on ice using a Qsonica Sonicator (Qsonica, LLC, Newtown, CT) and achieved with 5 min of total sonication on time, 1 second on and 2 seconds off cycle, at 70 amplitude, with careful care taken to ensure the sample remained cool throughout. The lysate was centrifuged on a Beckman Coulter Allegra at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g for 40 min at 4 °C and the soluble lysate was subsequently decanted. Initial purification of Prohead I or II from the soluble lysate was achieved with ultracentrifugation on a Sorvall wX+ Ultra Series centrifuge (Thermoscientific) at 38000 rpm using a Fiberlite F50L-8 × 39 rotor for 50 min at 4 °C over a sucrose cushion (35% sucrose in phosphate buffered saline (PBS; 10 mM NaPO4 175 mM sodium chloride pH 7.40)). The VLP pellet was then resuspended in PBS with gentle rocking overnight at 4 °C. The resuspended solution was then centrifuged on a Sorvall Legend Micro 17 microcentrifuge (Thermoscientific) at 17000 g for 5 min and the supernatant was filtered through a Millex-GP 0.22 μm syringe filter. The filtrate was then purified with size-exclusion chromatography (SEC) down a GE Healthcare HiPrep 16/60 Sephacryl S-500 HR column with a 1.0 mL min−1 flowrate of PBS using a Bio-Rad NGC Chromatography system. Eluent absorbance was measured at 280 nm and fractions were collected and stored at 4 °C. Clean fractions were conservatively combined and stored at 4 °C.
000 g for 40 min at 4 °C and the soluble lysate was subsequently decanted. Initial purification of Prohead I or II from the soluble lysate was achieved with ultracentrifugation on a Sorvall wX+ Ultra Series centrifuge (Thermoscientific) at 38000 rpm using a Fiberlite F50L-8 × 39 rotor for 50 min at 4 °C over a sucrose cushion (35% sucrose in phosphate buffered saline (PBS; 10 mM NaPO4 175 mM sodium chloride pH 7.40)). The VLP pellet was then resuspended in PBS with gentle rocking overnight at 4 °C. The resuspended solution was then centrifuged on a Sorvall Legend Micro 17 microcentrifuge (Thermoscientific) at 17000 g for 5 min and the supernatant was filtered through a Millex-GP 0.22 μm syringe filter. The filtrate was then purified with size-exclusion chromatography (SEC) down a GE Healthcare HiPrep 16/60 Sephacryl S-500 HR column with a 1.0 mL min−1 flowrate of PBS using a Bio-Rad NGC Chromatography system. Eluent absorbance was measured at 280 nm and fractions were collected and stored at 4 °C. Clean fractions were conservatively combined and stored at 4 °C.
Head II HK97 VLP production
For production of the mature Head II HK97 VLPs, GP4/GP5 Prohead II HK97 VLPs were produced as described above. GP4/GP5 Prohead II HK97 VLPs were diluted 20-fold in Maturation Buffer (100 mM sodium citrate, 200 mM sodium chloride, pH 4.0), mixed thoroughly by inversion, and incubated at room temperature for 5 hours with gentle shaking on a tabletop shaker. Upon completion of the incubation period, the HK97 VLPs were pelleted from solution by ultracentrifugation and then resuspended in Neutralization Buffer (200 mM sodium phosphate, 40 mM sodium chloride, pH 7.0) by gentle rocking at 4 °C overnight. Mature HK97 VLPs were filtered through a Millex-GP 0.22 μm syringe filter and subsequently evaluated by SDS-PAGE, native agarose gel electrophoresis, and dynamic light scattering (DLS) as described in sections below.Agarose gel shift assay
For comparing the relative size and morphology changes of Prohead I, II, and Head II, an agarose gel shift assay was performed. Samples contained a total of 20 μg of protein, as determined by UV-Vis spectroscopy, in buffer mixed with 6X loading dye. The gel shift assays were run on a 1% agarose gel in TAE buffer (40 mM Tris, 20 mM acetic acid, 0.4 mM EDTA, pH 8.0) at 65 volts for 3 hours. Agarose gels were stained with Coomassie stain for 30 minutes and de-stained until good resolution of the protein bands was observed.Cysteine labeling
For labeling of GP5 cysteine mutants, 20 mM 5-(Iodoacetamido)fluorescein (5-IAF) in DMSO was diluted to 0.1 mM in HK97 VLPs, HK97 A302C VLPs, HK97 S305C VLPs, or RGDS-HK97 S305C VLPs (1 mg mL−1) in PBS to give an approximately 4-fold excess of 5-IAF to GP5 subunits. Cysteine residues contain a reactive thiol functional group that selectively reacts with the iodoacetamide functional group found in 5-IAF. Thiols found in cysteine can also be used to selectively react with maleimide functional groups used in fluorescent labeling. The reaction was shaken at 250 rpm at room temperature for 2 hours. To stop the reaction and quench excess 5-IAF, β-mercaptoethanol was added in excess, the reaction mixture was diluted in PBS, and then centrifuged on a Sorvall wX+ Ultra Series centrifuge (Thermoscientific) at 38000 rpm using a Fiberlite F50L-8 × 39 rotor for 50 min at 4 °C. The VLP pellets were washed by resuspending in PBS using gentle rocking and re-centrifuged on the ultracentrifuge for a total of three times to wash away any residual labeling agent. The final pellets were resuspended in PBS with gentle rocking at 4 °C overnight to attain the desired concentration. 5-IAF labeled HK97 VLPs were analyzed by UV-Vis Spectroscopy on a Cary 300 Bio UV-Visible spectrophotometer by denaturation in 6 M guanidine hydrochloride and collection of absorbance values at 280 nm for the GP5 protein (ε280 = 37930 M−1 cm−1) and 492 nm for fluorescein (ε492 = 78000 M−1 cm−1). The amount of labeling was determined based on the ratio of protein-to-fluorescein concentrations found, assuming labeling of a single cysteine site per GP5 protein.
SDS page gel analysis
Protein samples were mixed with 4× loading buffer containing DTT, heated in a boiling water bath for 10 minutes, and subsequently spun down on a bench top centrifuge. Samples were separated on a gel containing a 4% polyacrylamide stacking gel and a 16% polyacrylamide running/separating gel using a constant current of 40 mA until dyed protein standards were sufficiently resolved. Gels were stained with InstantBlue Stain (Expedeon) until sufficiently visible (approximately 1 hour). Gels for samples labeled with 5-IAF were first imaged under UV light prior to Coomassie staining to visualize bands with bound 5-IAF, then subsequently stained and imaged. Images were taken on an AlphaImager Mini (Protein Simple) and analyzed using the AlphaView SA software.Dynamic light scattering analysis
VLP solution homogeneity and particle size were determined using DLS on a Malvern Zetasizer Nano Series. Samples were measured post-SEC and diluted with Millipore Milli-Q water to a final protein concentration of 0.25 mg mL−1. Water was used as the sample dispersant (RI = 1.330, η = 0.8872 cP) and protein was used as the material (RI = 1.450, absorbance = 0.001). The sample temperature was maintained at 25.0 °C and 173° light backscattering and analyzed using Malvern's proprietary instrument software.TEM imaging
Samples (10 μL, 0.1 mg mL−1 protein) were applied to glow-discharged formvar coated grids, incubated for 30 seconds, and excess liquid was removed with filter paper. Grids were then washed with 10 μL of distilled water, liquid was removed with filter paper shortly after addition, and then stained with 5 μL 2% uranyl acetate after which excess stain was removed with filter paper. Samples were imaged on a Tecnai G2 Spirit transmission electron microscope equipped with a LaB6 source using a voltage of 120 kV (UTSW Medical School Electron Microscopy Core facility).Cargo encapsulation
To examine encapsulation of protein cargos on the inside of the HK97 VLP, GFP-TP/GP5-pRSF and GFP-TP-pBAD plasmids were constructed, as described above, which targets GFP to be encapsulated inside the GP5 capsid in place of GP4. Two approaches of encapsulation were examined: a simultaneous co-expression of the GFP-TP fusion protein with GP5 using the GFP-TP/GP5-pRSF plasmid and a temporally controlled expression of GFP-TP first with subsequent expression of GP5 for encapsulation by using GFP-TP-pBAD co-transformed with the GP5-pRSF plasmid. For the simultaneous co-expression, GFP-TP/GP5-pRSF plasmid was transformed into E. coli BL21(DE3) (Lucigen) and grown up in LB medium at 37 °C in the presence of kanamycin (0.05 mg mL−1) to maintain selection for the plasmid. Once the cell culture reached the mid log phase (OD600 = 0.8), the flasks containing the bacterial cultures were removed and chilled at 4 °C for 30 minutes. Then, the cultures were returned to the incubator/shaker, induced with IPTG to a final concentration of 0.5 mM, and allowed to incubate overnight at 28 °C before harvesting of cells by centrifugation. For the temporally controlled approach, the GFP-TP-pBAD and GP5-pRSF plasmids were co-transformed into E. coli BL21(DE3) and grown in LB medium selective for ampicillin (0.1 mg mL−1) and kanamycin (0.05 mg mL−1) at 37 °C. Once the cells reached mid log phase (OD600 = 0.8), L-arabinose was added to a final concentration of 33.3 mM to induce GFP-TP fusion protein expression and the cultures were incubated overnight (∼16 hours) at 28 °C, after which IPTG was added to a final concentration of 0.5 mM to induce expression of GP5 expression. After 6 hours following IPTG induction, cells were harvested by centrifugation and cell pellets were stored at −20 °C until purification. Purification of both constructs was carried out as described above for Prohead HK97 VLPs. Resulting HK97-GFP VLPs were analyzed by SDS-PAGE, DLS, and ultracentrifugation through a cesium chloride gradient (0.2–0.4 mg mL−1 cesium chloride in PBS). Densitometry of SDS-PAGE gels was performed using the AlphaView and a background correction (Local Background). Sum value of pixel density was used to determine relative abundances of protein bands used in characterization of VLP loading as previously reported for other VLPs.24 For calculation of the abundance of the HK97 GP5 protein, the sum value pixel densities of the major coat protein band at 42 kDa and its polymer bands at higher molecular weights were added together to determine the total abundance of GP5 major coat protein. The ratio of GP5 to GFP-TP was determined by dividing the total sum value pixel density of the GP5 bands by the total sum value pixel density of GFP-TP. The total number of GP5 major coat proteins per VLP particle is known to be 420, which was assumed to be maintained based on the molecular characterization of the GFP-TP/HK97 VLPs. The number of GFP-TP proteins encapsulated per HK97 VLP was determined by dividing the number of GP5 major coat proteins (420) by the GP5:GFP-TP ratio (example shown in Fig. S13, ESI†). Three independent preparations were evaluated to determine final average and standard deviations for loadings of GFP-TP inside the HK97 VLP by the different strategies.C-terminal modification with GFP
Ligation reactions were set up based on previously reported reaction conditions from our lab for the conjugation of proteins to other VLP platforms.23 Briefly, LPETG-HK97 VLPs and polyG-GFP were mixed at a 1:10 ratio of the GP5-LPETG to polyG-GFP protein subunit concentrations to a final concentration of 60 μM protein in Tris Ligation Buffer (50 mM Tris, 150 mM sodium chloride, 6 mM calcium chloride, pH 8.0). Sortase was added to mixtures to give a 1:1 ratio of sortase to GP5. Reactions were incubated at 42 °C for 4 hours, quenched with EDTA, and stored at 4 °C for SDS-PAGE or ultracentrifugation analysis.
C-terminal RGDS and targeting
Immortalized mouse skeletal myoblast cell line C2C12 (ATCC, USA) was utilized and maintained in Dulbecco Modified Eagle Medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin complex at 37 °C and humidified 5% carbon dioxide. For transformation and follow up imaging experiments, the cells were transferred to image grade, cell-culture Petri dishes with a growth medium designed to induce skeletal muscle differentiation. The elongation and multinucleation skeletal muscle cells were treated and processed for imaging at about 70% confluency. Procapsid I HK97 S305C VLPs with and without a C-terminal RGDS sequence were produced and labeled with 5-IAF as described above for HK97 cysteine mutant labeling. After characterization, 5-IAF labeled HK97 S305C or RGDS-HK97 S305C VLPs (50 ng mL−1) were placed in 5 mL culture grade Petri dishes and incubated with differentiated C2C12 cells for 60 minutes. C2C12 cells were rinsed 3 times with sterile Hanks Buffered Saline Solution (HBSS) to remove excess/free HK97 VLPs. Cells were fixed with 2% paraformaldehyde (10 minutes) and permeabilized with 0.1% TritonX-100 in PBS (5 minutes). Fixed and permeabilized C2C12 cells were incubated with 1 μg mL−1 4′,6-diamidino-2-phenylindole (DAPI) fluorescent probe (5 minutes) then rinsed with HBSS 7 times. Cells were imaged on a Zeiss LSM8 confocal microscope with Aryscan to determine localization of HK97 VLPs.Results and discussion
Production of the Prohead I, Prohead II, and Head II HK97 VLPs
Our initial investigations looked at the construction of plasmids for the expression and production of either the Prohead I or Prohead II HK97 VLP for subsequent production of the Head II form of the HK97 VLP. For Prohead I, the gene of GP5 was inserted into multiple cloning site 2 of the pRSFDuet-1 plasmid (GP5-pRSF), which would result in the in vivo assembly of GP5 to the Prohead I HK97 VLP (Fig. 1) without GP4-induced proteolytic cleavage and the resulting formation of Prohead II. For Head II production, the GP4 maturation protease gene was included in multiple cloning site 1 of the pRSFDuet-1 plasmid containing GP5 (GP4/GP5-pRSF), which allows co-expression of both GP4 and GP5, leading to Prohead II formation upon assembly and gaining the ability for chemical transformation to the Head II form after purification (Fig. 1). After construction of the plasmids, expression, purification, and characterization of the different constructs was carried out. Results showed that the GP5-pRSF system produced Prohead I HK97 VLPs with relatively high yields of 20 mg L−1 of media or more after conservative selection of fractions after the final purification by SEC. The purification process for Prohead I HK97 VLPs only required two centrifugation steps and a final SEC clean-up, a very efficient approach relative to the previously described methodology for HK97 VLPs production.19 Purification of the Prohead II VLPs, produced by co-expression of GP4 and GP5, using the same purification methods as for Prohead I was observed to produce large VLP pellets after ultracentrifugation, similar to those of Prohead I and resulting in a similar yield. Transformation of purified Prohead II HK97 VLPs to Head II by acidification led to marginal yields of less than 1 mg L−1 of media, with the lower yield resulting from aggregation of HK97 VLPs as indicated by size exclusion chromatograms showing a greater abundance of aggregates near the void volume of the column (Fig. S1, ESI†).Prohead I and Prohead II HK97 VLP samples were analyzed by SDS-PAGE and bands of ∼42 kDa, corresponding to uncleaved GP5, were observed in all samples (Fig. 3A and B). An additional band was observed for Prohead I between 170 and 130 kDa, which has been previously reported as GP5 crosslinked product (poly-GP5) resulting as an artifact from the SDS-PAGE preparation methods utilized.16 The poly-GP5 band observed for Prohead I was shown to be removed (Fig. S2, ESI†) by performing a more laborious TCA precipitation preparation method that has been previously reported16 but was not utilized further after confirming the origins of the band. Prohead II HK97 VLP samples showed a reduced molecular weight band of ∼31 kDa corresponding to proteolyzed GP5 (GP5*), consistent with previous reports.10,12,16 A faint band was observed near the expected 25 kDa for GP4 in both Prohead II and Head II samples.10 The presence of uncleaved GP5 (42 kDa band) in Prohead II and Head II samples indicates that GP4 proteolysis was not complete, which is speculated to be due to reduced expression levels of the protease GP4, although this has not been investigated. SDS-PAGE analysis of Head II HK97 VLP samples showed results similar to the Prohead II, retaining both the 42 kDa GP5 and 31 kDa GP5* band, but with more intense staining of larger poly-GP5 bands and slightly more staining in the well area of the gel (Fig. 3B).
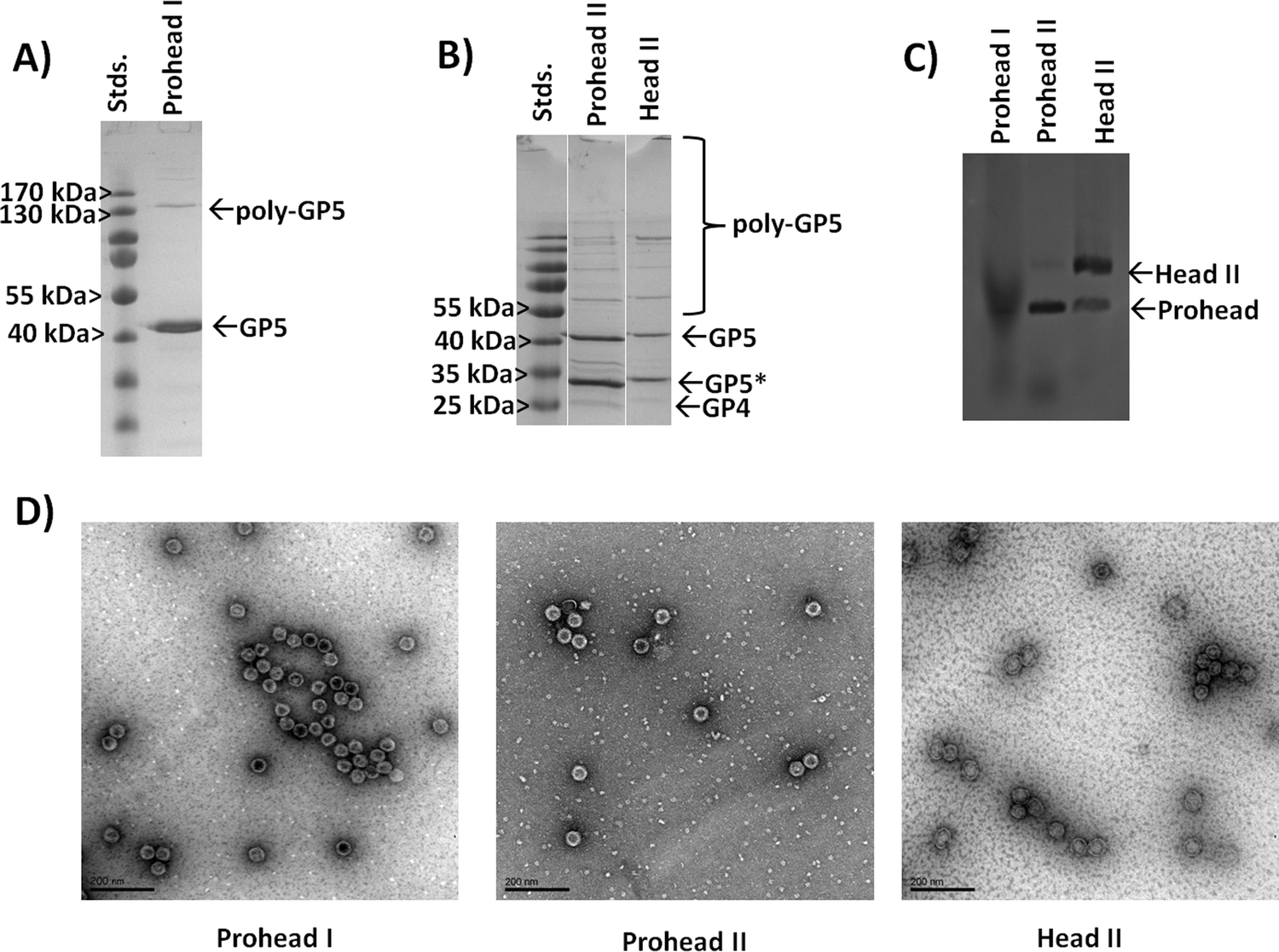 | ||
| Fig. 3 Characterization of Prohead I, Prohead II, and Head II HK97 VLPs produced through recombinant expression in E. coli. (A) SDS-PAGE analysis of Prohead I HK97 VLPs assembled from the GP5 protein (expected 42 kDa). (B) Analysis of Prohead II and transformed Head II HK97 VLPs, which show the expected GP5* band (31 kDa) resulting from proteolysis by GP4 (25 kDa). Higher molecular weight bands indicate crosslinking by the GP5* protein. (C) Agarose gel shift assay analysis of Prohead I, Prohead II, and Head II Hk97 VLPs. The larger diameter Head II morphology is found to migrate more slowly, consistent with its larger diameter. (D) Transmission electron microscopy (TEM) images of the Prohead I, Prohead II, and Head II HK97 VLPs show particles consistent with the expected morphologies. Scale bars are 200 nm in all images. | ||
To further investigate the expected size expansion of the Head II HK97 VLP samples, an agarose gel shift assay was performed to examine Prohead I, Prohead II and Head II HK97 VLP samples (Fig. 3C). Prohead I samples showed a more diffuse band that ran furthest on the gel, consistent with a smaller diameter particle which is less impeded by the gel during separation. The Prohead II sample showed a more defined band running equal to the Prohead I, as expected, but also contained a faint band that ran more slowly, indicating a larger diameter. The faint band in Prohead II corresponds with an intense band seen in the Head II sample, corresponding to the Head II HK97 VLP, and is consistent with the findings by others that a small population of the Head form is found in Prohead II samples, which forms during purification or the preparation process.11,13 A population of the smaller diameter Prohead I/II VLP structures also remained in the Head II sample as well, indicated by the minor band with reduced impedance in the gel that aligned with the major bands of Prohead I and II. HK97 VLPs were further analyzed by dynamic light scattering (DLS) to examine their solution diameters. Prohead I particles were found to have diameters of 55.5 ± 1.8 nm, consistent with the 56 nm diameter expected for these structures, and average polydispersity index (PDI) values of 0.039 ± 0.027 (Fig. S3, ESI†), indicating that the samples were homogeneous in size (values less than 0.08 are considered homogeneous). SEC-purified Prohead II samples were found to have similar average diameters by DLS of 53.2 ± 1.0 nm and PDI values of 0.012 ± 0.002 (Fig. S4, ESI†), showing little variation in external diameter. DLS further confirmed the increase in size of the matured HK97 VLP to 64.2 ± 4.5 nm, consistent with the diameter of 66 nm expected for the Head II form, and an average PDI of 0.128 ± 0.099 (Fig. S5, ESI†). The higher PDI suggests some heterogeneity within the Head II sample, likely a small amount of untransformed Prohead and aggregates from the transformation process, supported by the observations in the gel shift assay and SEC chromatogram (Fig. S1, ESI†). A comparison of DLS data for the different samples is provided in Table 1. Taken together, the results show that Head II is not completely pure from Prohead II and SEC does not provide sufficient resolution to separate the different species. Transmission electron microscopy (TEM) was used to further examine the Prohead I, Prohead II, and Head II HK97 VLPs, with samples showing results consistent with DLS measurements and the structural morphology and size expected for Prohead I, Prohead II, and Head II (Fig. 3D). These results show the ability to produce the different structural forms of HK97 VLPs, albeit with incomplete transformation of Prohead II to Head II, with excellent results for the Prohead I form that was investigated further as an adaptable nanoparticle platform.
| HK97 VLP construct | Z-average diameter ± std. dev. (nm) | Average polydispersity index (PDI) ± std. dev. |
|---|---|---|
| Prohead I | 55.5 ± 1.8 | 0.039 ± 0.027 |
| Prohead II | 53.2 ± 1.0 nm | 0.012 ± 0.002 |
| Head II | 64.2 ± 4.5 | 0.128 ± 0.099 |
| HK97 S305C | 55.2 ± 0.5 | 0.029 ± 0.007 |
| HK97 S305C 5-IAF labeled | 55.2 ± 0.7 | 0.047 ± 0.028 |
| GFP-TP/HK97 Co-Express | 52.7 ± 1.59 | 0.018 ± 0.014 |
| GFP-TP/HK97 Seq-Express | 59.0 ± 2.91 | 0.036 ± 0.011 |
| LPETG-HK97 | 55.3 ± 1.7 | 0.033 ± 0.011 |
| RGDS-HK97 | 56.7 ± 2.7 | 0.078 ± 0.044 |
| RGDS-HK97 5-IAF labeled | 54.8 ± 1.9 | 0.073 ± 0.019 |
Design and production of HK97 allowing internal chemical modification
After verifying the ability to produce HK97 VLPs, we went about designing the VLP so that we could selectively trap small molecules on the interior (Fig. 4A). Although all subsequent studies would be performed on the Prohead I HK97 VLP morphology, molecular models for both the Prohead and Head II forms were examined to find residues that were only exposed on the internal surface in both morphologies. We also looked for amino acid residues where mutation to a cysteine residue would be a relatively conservative change to preserve the wild type protein folding dynamics required for proper assembly. From the structures, it was observed that alanine 302 (A302) and serine 305 (S305) residues of the GP5 protein were located on the internal surface with exposed functional groups to the internal volume of both the Prohead (Fig. 2 and Fig. S6, ESI†) and Head forms of the HK97 VLP (Fig. S7, ESI†). PCR was used to create two different mutant GP5 proteins that would yield HK97 VLPs with cysteine situated on the interior; the GP5 A302C, which has the alanine at position 302 mutated to cysteine, and GP5 S305C, which has the serine at position 305 mutated to a cysteine. Subsequent Prohead I variants are referred to based on the GP5 mutant they are comprised from. Initial expression and purification studies of the HK97 GP5 A302C VLP showed cross-linking of GP5 by SDS-PAGE (Fig. S8, ESI†) and larger structural formation by DLS, with diameters of 65.2 ± 2.6 nm and PDI values of 0.080 ± 0.011 (Fig. S9, ESI†). These results were more consistent with those seen for mature Head II VLPs instead of the expected Prohead I structure being investigated, suggesting the A302C mutation altered the structural properties of the Prohead I HK97 VLP. However, it was observed that the GP5 S305C mutant was able to form regular HK97 Prohead I VLPs with final production levels similar to that of the wild type GP5. Analysis of the HK97 S305C VLPs by SDS-PAGE showed a band consistent with the molecular weight of GP5 (Fig. 4B) and DLS analysis of the particles produced diameters of 55.2 ± 0.5 nm and PDI values of 0.029 ± 0.007 (Fig. S10, ESI†), similar to what was observed for Prohead I HK97 VLPs produced from wild type GP5 (Fig. 4C and Table 1). TEM imaging of HK97 S305C VLPs showed particle sizes consistent with those expected for the Prohead I (Fig. 4D) and in agreement with DLS. HK97 S305C VLPs were further analyzed for labeling with 5-iodoacetamidefluorescein (5-IAF), containing the fluorescein fluorphore, in comparison with wild type HK97 GP5 VLPs treated with 5-IAF. Ultracentrifugation of HK97 S305C VLPs after labeling showed a distinct orange/yellow color that was not observed for the wild type HK97 VLPs (Fig. 4E). Analysis by SDS-PAGE showed intense labeling of bands corresponding to the GP5 S305C mutant when viewed under UV irradiation that overlapped with the same bands found upon staining with Coomassie. This was not observed for wild type GP5, which showed only minimal intensity from UV, even though the Coomassie stained gel showed similar protein band intensity and abundance to that of the S305C sample (Fig. 4F). In addition, no significant change was observed to the diameter by DLS after labeling (Fig. S11, ESI† and Table 1), with average diameters of 55.2 ± 0.7 nm and PDI values of 0.047 ± 0.028, indicating that the attachment of molecules to the HK97 VLP interior did not have any adverse effect on the structure. Investigations of the number of cysteine sites labeled per GP5 subunit in the Hk97 S305C VLP, assuming only one site per subunit (420 subunits make up the HK97 VLP), using the protein-to-fluorophore absorption ratio found labeling levels nearing 50 percent (43.8% or 184 sites per HK97 VLP). Although complete labeling (i.e. 100%) was not observed, the overall labeling levels suggest that the HK97 VLP enables sufficient sites for attachment and localization of small molecules to the molecular interior for practical applications.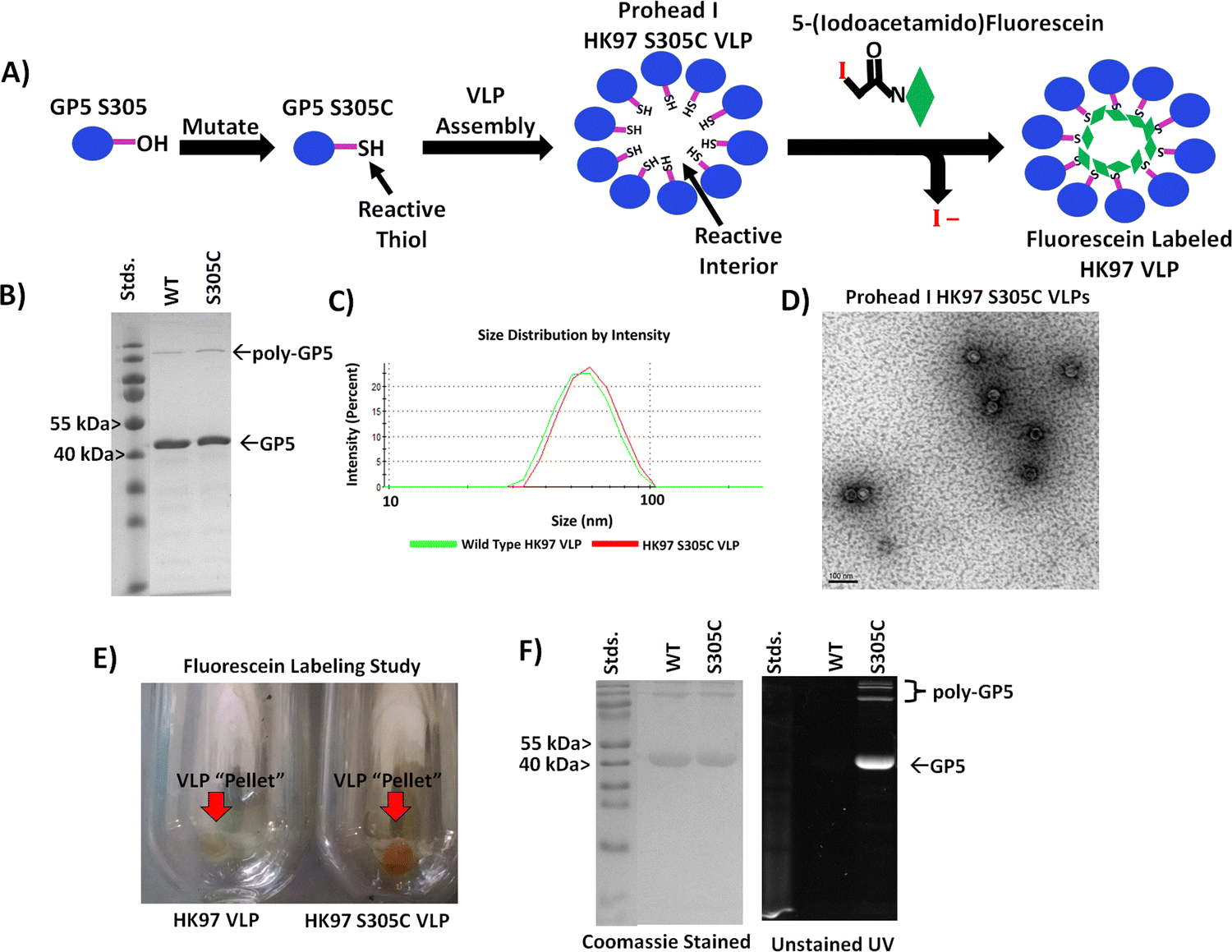 | ||
| Fig. 4 Evaluation of the HK97 S305 VLP for internal chemical modification. (A) Schematic showing the strategy for producing an HK97 VLP that can be chemically labeled by incorporating a cysteine at position 305 in the GP5 sequence (normally containing serine). (B) SDS-PAGE analysis of the HK97 VLP wild type (WT) GP5 and the GP5 S305C variant (S305C). (C) Comparison of the DLS spectra for wild type Prohead (green trace) vs. the GP5 S305C variant (red trace), which produces the same size HK97 VLP. (D) TEM imaging of HK97 S305C VLPs showed particles identical in size and morphology of those seen for WT Prohead I HK97 VLPs (Fig. 3D for comparison). (E) Ultracentrifugation after labeling reactions produced VLP “pellets” of WT (HK97 VLP) and S305C HK97 VLPs that showed only strong staining for the S305C variant. (F) SDS-PAGE analysis of samples from 5-IAF labeling studies indicated that only the S305C GP5 protein labeled sufficiently, as indicated by UV images taken before staining with Coomassie (right), with only a little labeling observable for the WT GP5 HK97 VLPs, even though the same amount of protein was loaded onto the SDS-PAGE gel, as indicated by Coomassie staining (left). | ||
Entrapment of large macromolecules within the Prohead I HK97 VLP interior
While small molecule encapsulation is important for any nanoparticle platform, the entrapment and encapsulation of larger macromolecular molecules and structures is also desirable. The large size of the HK97 VLP Prohead and Head morphologies (56 or 66 nm outer diameter) make it an ideal platform for the encapsulation of larger macromolecular structures. For entrapment of large macromolecules inside the HK97 VLP, focus was put on the encapsulation of proteins as the target molecule, since proteins are biologically active and there are a wide range of applications in which protein stabilization and delivery are desired. For encapsulation of proteins, we looked at exploiting the previously reported targeting peptide (TP) sequence from the C-terminus of the GP4 maturation protease as a way of trafficking proteins to the interior of the HK97 VLP during assembly. It has been previously shown that a small decorator protein could be integrated on the interior by creating a fusion protein of the decorator with the TP, but larger proteins have not been examined.10 We constructed a plasmid containing the gene for amino acids 163 through 226 from GP4, comprising the TP portion of GP4, which was placed in the multiple cloning site 1 of pRSFDuet-1 plasmid containing GP5 in multiple cloning site 2 (TP/GP5-pRSF). To facilitate insertion of large protein genes on the N-terminal side of the TP, NcoI/BamHI restriction sites were situated upstream of the TP gene to allow in-line insertion of desired protein genes.To investigate the ability of the TP to traffic large proteins to the interior of the HK97 VLP, the gene encoding green fluorescent protein (GFP) was inserted into the plasmid to construct a GFP-TP gene fusion (GFP-TP/GP5-pRSF) (Fig. 5A). GFP was chosen as a model protein because of the ability to easily visualize and confirm encapsulation through its fluorescent properties. Expression and purification were performed in the same manner as the other Prohead HK97 VLPs, with the resulting ultracentrifugation pellets and SEC fractions containing VLPs showing a green color (Fig. 5B), indicating co-purification of the GFP with the HK97 VLP. An additional ultracentrifugation run of the SEC purified GFP-TP/HK97 VLPs through a cesium chloride gradient was compared with “wild type” Prohead I HK97 VLPs, with GFP-TP/HK97 VLPs showing green VLP bands, consistent with the GFP being trapped inside the HK97 VLP, in comparison with the typical white bands found for the wild type sample (Fig. 5C). SDS-PAGE analysis of purified samples showed bands corresponding to both GFP-TP (∼38 kDa) and GP5 (Fig. 5D), consistent with encapsulation and not covalent bonding of the GFP-TP to GP5. DLS results indicated that the particles were unchanged in their size, with average diameter of 52.7 ± 1.59 nm and PDI of 0.018 ± 0.014 observed (Fig. S12, ESI†), further supporting the encapsulation of GFP-TP instead of bonding to the exterior surface. Densitometry results of SDS-PAGE gels (Fig. S13, ESI†) indicated an encapsulation of 68 ± 19 GFP-TP proteins per HK97 VLP for the GFP-TP/GP5-pRSF co-expression plasmid, consistent with approximately 60 GP4 proteins normally encapsulated inside wild type Prohead I.9
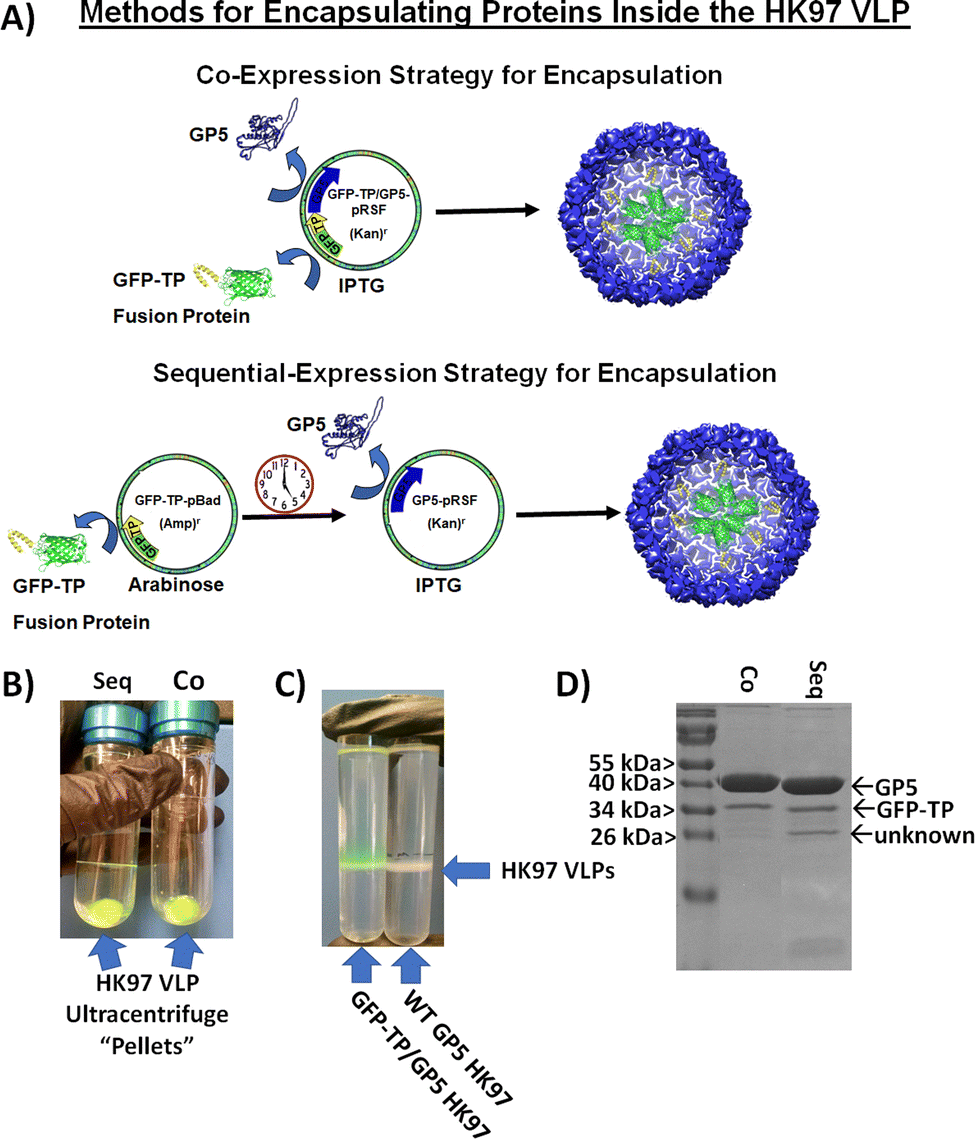 | ||
| Fig. 5 Encapsulation of GFP on the interior of the HK97 VLP to show the ability to encapsulate large macromolecular cargoes on the interior of the HK97 VLP. (A) Assembly strategies for encapsulating GFP-targeting peptide fusion proteins (GFP-TP) on the interior of the HK97 VLP through either co-expression (upper scheme) or a temporally controlled sequential expression (lower scheme). (B) Purified HK97 VLP pellets produced by sequential (Seq) and co-expression (Co) showed green color, indicating co-purification of GFP-TP and HK97 VLPs, suggesting encapsulation of GFP-TP. (C) Comparative analysis of HK97/GFP-TP VLPs with wild type (WT) HK97 VLPs through a cesium chloride gradient after ultracentrifugation. HK97-GFP VLPs ran the same as WT HK97 VLPs, but have green color due to the encapsulation of GFP-TP. (D) Representative SDS-PAGE analysis of the two strategies for encapsulation of GFP-TP inside the HK97 VLP, which showed insignificant difference in the amount of the GFP-TP encapsulated by densitometry. | ||
Unlike VLP systems which requires a scaffolding protein for assembly, such as the P22 VLP, HK97 VLP assembly is independent of the TP moiety. Therefore, TP fusion protein mediated encapsulation may be limited if TP availability is scarce, and we hypothesized higher amounts of GFP-TP available for packaging before VLP assembly would result in higher packaging efficiency. To that end, we examined a two-plasmid approach that would instead allow temporal control of the GFP-TP expression relative to the GP5 coat protein (Fig. 5A). The gene for GFP-TP was transferred to the pBAD plasmid (GFP-TP-pBAD), inducible by arabinose, and was co-transformed with the pRSFDuet-1 plasmid, inducible by IPTG, containing only GP5 (GP5-pRSF). The separation of GFP-TP and GP5 on separate plasmids that are controlled by different inducers enables GFP-TP, or other future cargo proteins, to be expressed first to build-up GFP-TP in the cytosol before initiation of GP5 expression and subsequent assembly into the HK97 VLP. Expression of GFP-TP was induced for overnight expression (∼16 hours), to create a high intracellular concentration of GFP-TP available for encapsulation upon GP5 induction, which was subsequently induced for 6 hours. Overall, analysis of the sequential expression showed no difference in encapsulation levels relative to the simultaneous co-expression approach (Fig. 5B-D, Fig. S13 and S14, ESI†), with 68 ± 18 GFP-TPs encapsulated per HK97 VLP for the sequential method. An additional band of lower molecular weight of approximately 26 kDa was sometimes observed in either co-expression or sequential expression trials, although more prevalent in the sequential expression, as seen and noted in the sequential expression sample in Fig. 5D. This band has a molecular weight similar to GFP lacking the TP (26 kDa), which theoretically could be trafficked to the interior of the HK97 VLP without the TP if it were to form a dimer with a GFP-TP (GFP is known to form dimers25). However, we omitted the band from our analysis because it was inconsistent, its contribution would not drastically alter the loading number, and future proteins of interest may not exhibit the same dimerization properties of GFP; therefore, the loading of the GFP-TP fusion protein was of sole interest. DLS values for sequential expression did show a slightly higher average diameter of 59.0 ± 2.91 nm and PDI of 0.036 ± 0.011, but not drastically different from the co-expression samples. Overall, both methods produced HK97 VLPs that showed similar purification and retention properties by SEC to those of Prohead I HK97 VLPs (Fig. S15, ESI†). The co-expression method may have yielded similar results as the sequential method because of GFP's exceedingly high expression levels in E. coli, allowing substantial GFP production even in the co-expression setting. Proteins expressed at lower levels than GFP or proteins that require special folding or maturation processes before encapsulation inside the VLP structure, as has been reported recently for the P22 system,24 might derive greater benefit from the sequential approach, and each protein should be evaluated on a case-by-case basis to determine which method provides optimal results. The results present options for producing HK97 VLPs that allow researchers to adapt expression methods to the unique cargo proteins that are being utilized.
Modular approach for external modification
The exterior of a VLP serves as a key foundation for integrating VLPs into a variety of applications. The ability to rapidly modify the surface of a VLP in a modular fashion is highly desirable to allow adaptation for many applications. To achieve a general approach with the HK97 VLP platform, we examined the molecular model to determine a strategy which would not interfere with our thiol reactive approach for internal chemical modification, removing the prospects of utilizing reactive cysteine residues on the exterior, as has been described in the past.19 In addition, the introduction of cysteine on the exterior would likely cause undesirable aggregation and disulfide cross-linking between VLPs without including reductants to prevent crosslinking, which might interfere with other downstream chemical modification desired. In our examination, it was found that the GP5 C-terminus of the HK97 VLP in both Prohead and Head II morphologies points to the exterior (Fig. 2 and Fig. S7, ESI†), although not all the amino acids could be observed in the structural models which is likely due to them being unstructured. This positioning of the GP5 C-terminus suggested that it could serve as an anchoring point for the attachment of various functional moieties onto the exterior of the HK97 VLP.To this end, we explored a modular genetic approach whereby peptides of various types could be added genetically via recombinant DNA technology. Peptides can provide wide ranging functionalities including the ability to: integrate cell targeting functions; associate with certain surface substrates through surface binding peptides; serve as attachment sites for large proteins through ligation systems, such as Sortase or SNAP/SNARE; or provide attachment sites for a number of chemically and biochemically active peptides identified through processes like combinatorial chemistry, phage display, and biopanning. Therefore, we developed an expression plasmid that enabled quick integration of new peptide sequences to the C-terminus of the GP5 gene that would be directly attached to the HK97 VLP upon expression and, we hypothesized, displayed on the surface (Fig. 6A). In our plasmid design, we introduced DNA restriction enzyme sites at the C-terminal end of the GP5 gene sequence that allowed cassette removal and insertion of genes utilizing KpnI and XhoI restriction enzymes to make modification rapid. To ensure adequate separation of the peptides from the VLP surface, we incorporated a modular flexible polyglycine peptide spacer between the GP5 C-terminus and where the peptide fragment could be inserted. Due to reliance on genetic programming, the overall design of the plasmid ensured modification of the HK97 VLPs produced would occur at every GP5 C-terminus and that modification could be easy adapted in a modular fashion requiring only standard molecular biology techniques.
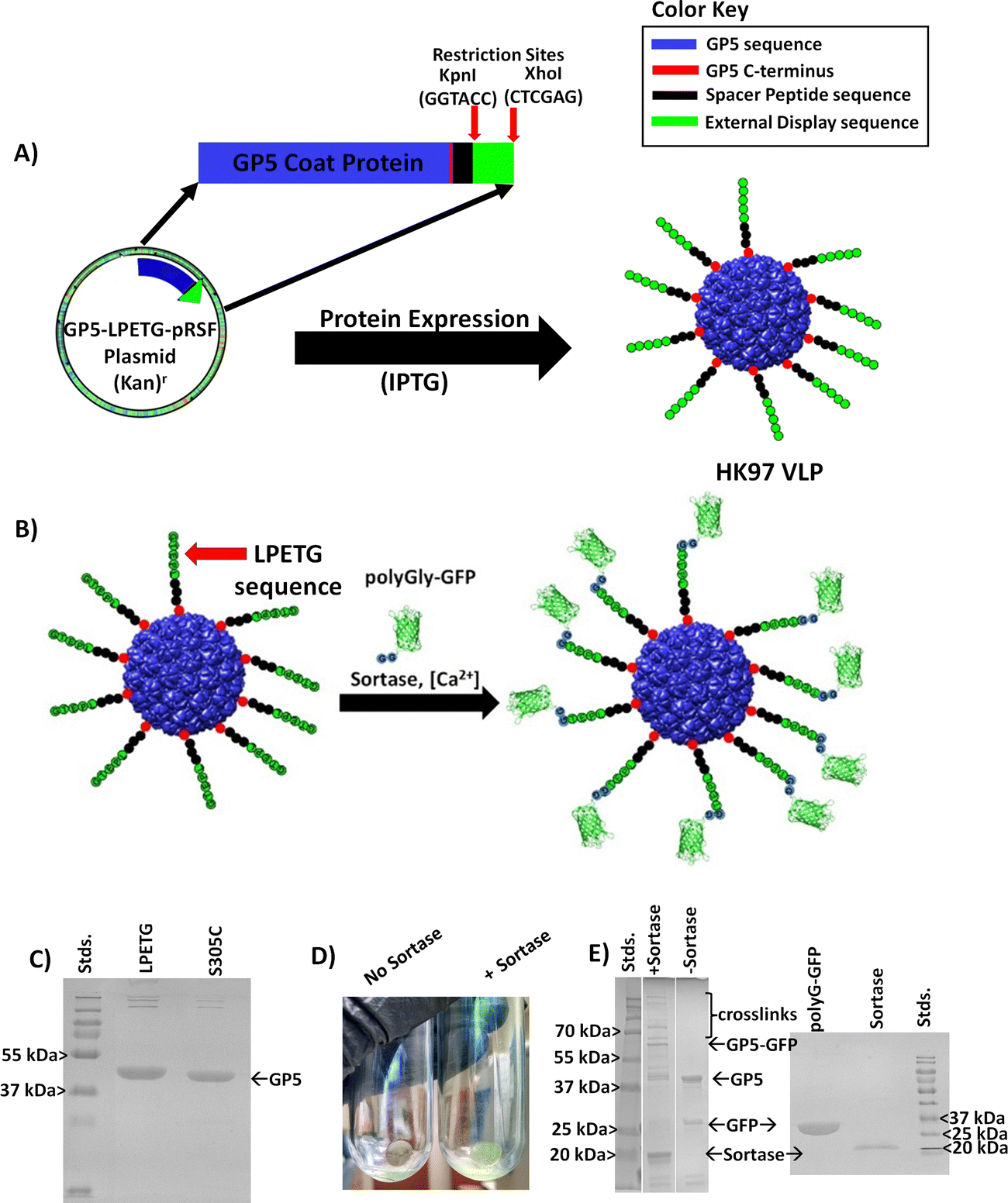 | ||
| Fig. 6 Design and examination of the external modification of the HK97 VLP via the GP5 C-terminus. (A) Design of a plasmid for the addition of amino acid peptide sequences (green) separated by a flexible peptide spacer (black) to the C-terminus (red) of the GP5 protein (blue) that assembles into the HK97 VLP with the peptide sequence displayed on the exterior. (B) Schematic showing the proof of concept approach to determine the accessibility of C-terminally modified HK97 VLPs using an HK97 VLP containing GP5 with the LPETG amino acid sequence attached to the C-terminus, enabling sortase mediated ligation (calcium dependent) of an N-terminus polyglycine (polyGly) containing GFP. The sortase reaction is expected to covalently attach polyG-GFP to the exterior of the LPETG-HK97 VLP if it is exposed and accessible. (C) SDS-PAGE analysis of HK97 VLPs produced from GP5-LPETG (LPETG) in comparison with GP5 S305C (S305C), with GP5-LPETG showing a slight shift higher in molecular weight, consistent with an increase in amino acids and molecular weight. (D) Comparison of results for the ultracentrifugation of LPETG-HK97 VLP/polyG-GFP crosslinking reactions with either no sortase (left) or addition of sortase (right) indicating that co-pelleting of the polyG-GFP and LPETG-HK97 VLPs was only seen when sortase was added, suggesting crosslinking to the exterior. (E) SDS-PAGE analysis of sortase-mediated ligation reactions, showing addition of sortase (+Sortase) led to higher molecular bands consistent with covalent attachment of polyG-GFP to GP5-LPETG. SDS-PAGE gels containing individual components used for sortase ligation is provided for comparison. | ||
To investigate the designed plasmid for expression of C-terminally displayed peptides and to verify that the C-terminus was truly exposed on the exterior, we genetically inserted the sortase recognition sequence Leucine-Proline-Glutamate-Threonine-Glycine (LPETG) to the C-terminus (GP5-LPETG-pRSF resulting plasmid). The enzyme sortase catalyzes peptide bond formation between the threonine (T) of the LPETG sequence and a molecule containing an N-terminal polyglycine, permitting selection of a desired protein to conjugate to the modified VLP as shown schematically in Fig. 6B. The LPETG-HK97 VLP was expressed and purified as was carried out for the other HK97 VLPs and showed the expected characteristics of the Prohead I HK97 VLP. SDS-PAGE showed a band of slightly higher molecular weight from GP5 (Fig. 6C), consistent with the fusion of the spacer and LPETG peptide to the GP5 protein. Analysis of the LPETG-HK97 VLP by DLS showed average diameter of 55.3 ± 1.7 nm and PDI of 0.033 ± 0.011 (Fig. S16, ESI† and Table 1), showing no significant change, as expected with the addition of a small and external flexible peptide sequence.
To further analyze and confirm the external display of the peptide on the surface, a sortase-mediated ligation was carried out using GFP containing an N-terminal polyglycine (polyG-GFP, Fig. 6B). The reaction produced green LPETG-HK97 VLP pellets after ultracentrifugation, consistent with sortase-mediated attachment of polyG-GFP to the LPETG-HK97 VLP, which was not seen in controls which omitted sortase (Fig. 6D). The SDS-PAGE verified peptide bond formation of the polyG-GFP to GP5-LPETG by the presence of a band corresponding to the ligated molecular weight of the two proteins (∼68 kDa). Additional bands of much higher molecular weight were also observed in the sortase mediated reaction but not found in the control. Banding patterns like this are not without precedent, as a previous sortase mediated ligation study involving the P22 VLP showed similar banding, which was attributed to additional cross-linking resulting from the broader specificity of sortase when using high substrate concentrations and extended incubation times,23 as was used here to prove the accessibility of the C-terminus on the exterior of the HK97 VLP. DLS of reaction products showed increased diameter of 98.1 ± 0.23 nm and PDI of 0.330 ± 0.006, indicating the VLPs had increased in size and heterogeneity due to external GFP attachment, which also had contributions from remaining components interacting in the mixture (Fig. S17, ESI†). These results verified the external exposure of the C-terminus and that peptides added as an extension are fully accessible on the exterior for molecular interactions, even those requiring large enzyme and protein-protein interactions as demonstrated by the sortase ligation. Furthermore, the results indicate that functional moieties can be added to the exterior in vitro, which suggests that this method may also allow the attachment of non-protein molecules as well.
Examining internal/external co-modification of the HK97 VLP platform for programming bioactivity
To further examine the display of peptides from the surface and integrating external modification with internal modification, we looked at evaluating a peptide sequence with known bioactivity for cellular localization to support the use of the HK97 VLP as a potential cell delivery vehicle. For external modification, we chose to attach the integrin-binding RGD sequence for attachment to the VLP surface,26,27 to see if it would enable increased localization of HK97 VLPs to mammalian cells. To visualize the location of the HK97 VLPs, we looked to integrate the capacity to fluorescently label the HK97 VLP by using the GP5 S305 mutant combined with the integrin binding RGD. A schematic of our strategy is shown in Fig. 7A. For the study, we constructed a plasmid (GP5-S305C-RGDS-pRSF) that combined the internal S305C mutation with an external display of the RGDS peptide sequence on the VLP to enable fluorescent tracking of the HK97 VLPs with 5-IAF. The RGDS-HK97 S305C VLPs were expressed and purified, and the resulting particles were analyzed by SDS-PAGE and DLS. SDS-PAGE showed a band consistent with GP5 (Fig. 7B), since only minimal amino acids were appended which did not significantly alter the molecular weight, and DLS showed particles with average diameter of 56.7 ± 2.7 nm and PDI of 0.078 ± 0.044 (Fig. S18, ESI† and Table 1), indicating the Prohead I particles were properly formed and homogeneous. TEM images of RGDS-HK97 S305C VLPs (Fig. 7C) showed particles with morphologies consistent with Prohead I and sizes observed by DLS (Table 1). RGDS-HK97 S305C VLPs were labeled with 5-IAF as carried out for HK97 S305C VLPs, with similar labeling levels observed for the RGDS-HK97 S305C VLP (50.65% or 213 sites labeled) as was found for the HK97 S305C VLP. DLS was performed after labeling, which indicated that there was no change to the size of the RGDS-HK97 S305C VLPs upon labeling after purification, with average diameter of 54.8 ± 1.9 nm and PDI of 0.073 ± 0.019 (Fig. S19, ESI† and Table 1). To assess in vitro cellular localization, we compared binding of HK97 S305C VLPs and RGDS-HK97 S305C VLPs labeled with 5-IAF to C2C12 cells, a mouse myoblast cell line that is commonly used to examine cell differentiation and mechanistic biochemical pathways. Confocal microscope imaging of C2C12 cells treated with RGDS-HK97 S305C-5-IAF VLPs were shown to have increased green fluorescent labeling (from fluorescein found in 5-IAF) in comparison to those treated with the HK97 S305C-5-IAF VLPs which lack the RGDS peptide (Fig. 7D). These results support that external modification of the HK97 VLP platform allows modification of bioactivity and binding interactions with biological targets, such as cell surfaces, in addition to attachment of macromolecules. Future investigations will look to evaluate if these interactions lead to cell surface binding only or internalization of the HK97 VLPs into cells. In addition, the results show the ability to combine modifications to the platform, enabling the user to modify both the inside and outside to chemically manipulate the HK97 VLP platform using chemically orthogonal strategies.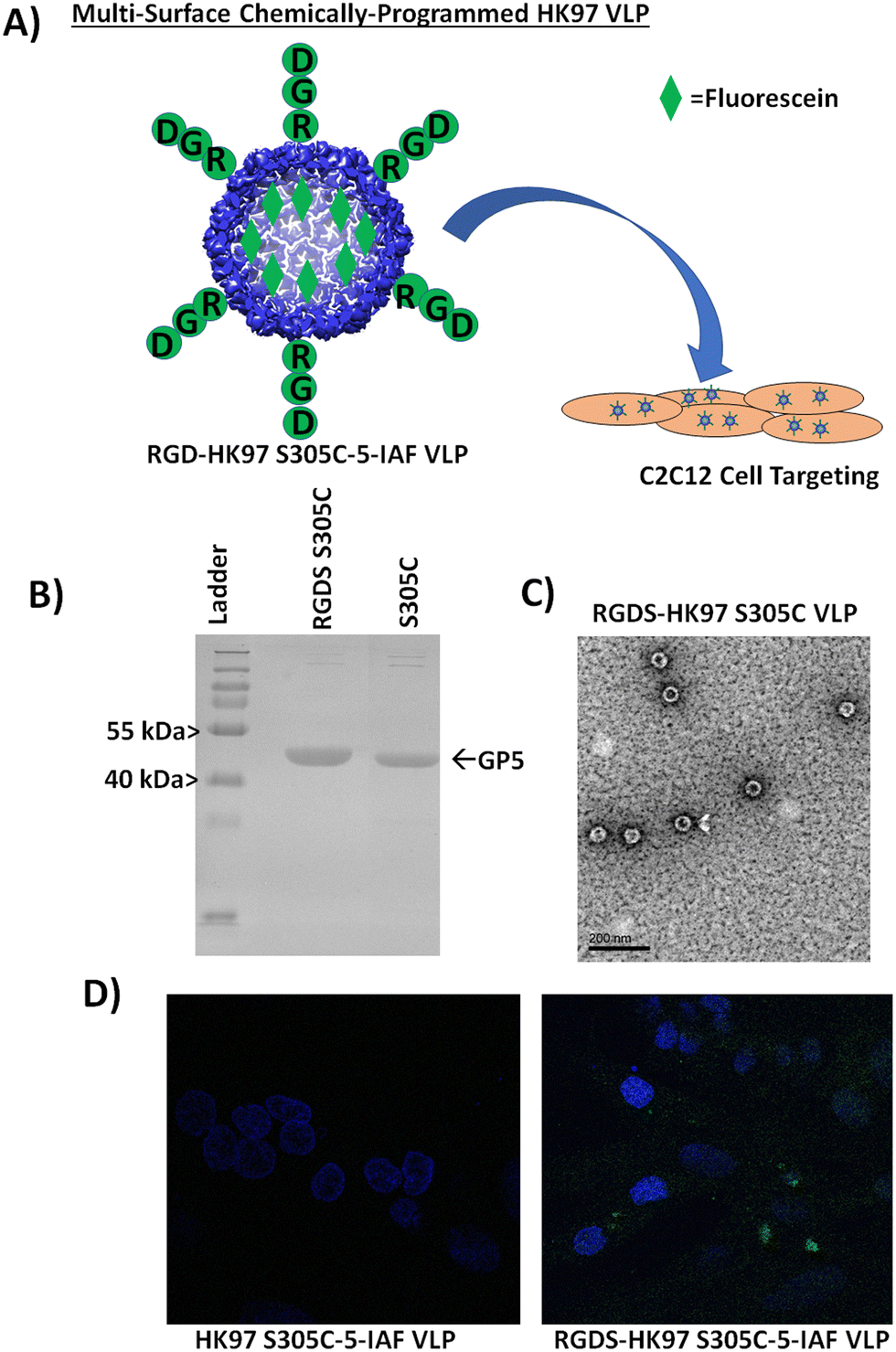 | ||
| Fig. 7 Characterization of a cell targeting HK97 VLP containing the RGDS peptide sequence (RGDS-HK97 VLP). (A) Schematic representation of developing an HK97 VLP capable of targeting cells and tracking cell adhesion by fluorescence. Combining an HK97 VLP containing an RGD on the C-terminus with the GP5 S305 mutation would allow chemical labeling and tracking of the VLP with fluorescein (5-IAF) that would be trafficked to cells containing integrin receptors through the RGD peptide (RGDS was used). (B) SDS-PAGE characterization of HK97 S305C VLP containing the RGDS peptide on the exterior (RGDS-HK97 S305C VLP) vs. HK97 S305C VLP. (C) TEM image of the RGDS-HK97 S305C VLPs after purification (scale bar is 200 nm). (D) Comparison of C2C12 cell localization of HK97 S305C (left) and RGDS-HK97 S305C VLPs (right) labeled with 5-iodoacetamidefluorescein (5-IAF). Fluorescein is observed as green over the cells stained with 4′,6-diamidino-2-phenylindole (blue). Only VLPs with the genetic-appended RGDS peptide were found to localize at the cells. | ||
Conclusion
The HK97 VLP presents a new versatile platform for the construction of bionanomaterials. Our results show the Prohead I HK97 VLP can be readily modified on its interior or exterior without changing its overall structural morphology or purification properties (Fig. S20, ESI†). Internal surface modifications can be made via a site mutated cysteine residue that yields high labeling rates when used with cysteine-selective reagents like iodoacetamides. In addition, we showed that large protein cargoes can be encapsulated on the interior by genetically fusing the targeting peptide (TP) portion of the GP4 protein to GFP to make the GFP-TP fusion protein. Two strategies for encapsulation of GFP-TP were examined: a co-expression strategy that simultaneously expressed both GP5 coat protein and GFP-TP and a sequential expression strategy in which GFP-TP expression was induced first, followed by GP5 expression induction to assemble the Prohead I HK97 VLP particle. While minimal differences were observed between the two encapsulation methods applied to GFP encapsulation, the alternate approaches provide flexibility for adaptation for other types of encapsulated protein cargos, particularly poorly expressed proteins or those requiring additional maturation after folding, which are not seen in the model GFP protein examined. For the exterior, a plasmid that allows rapid incorporation of new peptides was constructed and two peptide sequences were examined: the LPETG sortase recognition peptide sequence and the RGDS integrin binding sequence. The incorporation of the LPETG sequence to the GP5 C-terminus allowed the sortase-catalyzed attachment of the modified GFP protein containing an N-terminal polyglycine to the exterior of the HK97 VLP, verifying the external exposure of the C-terminus and also providing a strategy for addition of large protein and non-protein components to the exterior of the HK97 VLP. Attachment of the integrin binding sequence, RGDS, to the exterior of HK97 VLPs enabled their selective localization to C2C12 cells that was not observed in HK97 VLPs lacking the peptide. Together, our results demonstrate the ability to modify the HK97 VLP in a robust, modular fashion and provide a foundation for future use of the HK97 platform as a scaffold for developing new bionanomaterials for diverse applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research reported in this publication was supported by The University of Texas at Tyler internal interdisciplinary grant to BB, AA & DPP. Support for MDW, BC, MC, SK, MN, and GI was provided by The Welch Foundation Grant BP-0037. TEM imaging was performed by the University of Texas Southwestern Medical Center Electron Microscopy Core Facility.References
- A. Shahrivarkevishahi,
et al., Virus-like particles: a self-assembled toolbox for cancer therapy, Mater. Today Chem., 2022, 24, 100808 CrossRef CAS
.
- J. He,
et al., Virus-like Particles as Nanocarriers for Intracellular Delivery of Biomolecules and Compounds, Viruses, 2022, 14(9), 1905, DOI:10.3390/v14091905
- J. L. Mejía-Méndez, R. Vazquez-Duhalt, L. R. Hernández, E. Sánchez-Arreola and H. Bach, Virus-like Particles: Fundamentals and Biomedical Applications, Int. J. Mol. Sci., 2022, 23(15), 8579, DOI:10.3390/ijms23158579
- D. McNeale, N. Dashti, L. C. Cheah and F. Sainsbury, Protein cargo encapsulation by virus-like particles: Strategies and applications, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2022, e1869, DOI:10.1002/wnan.1869
- L. Li and G. Chen, Precise Assembly of Proteins and Carbohydrates for Next-Generation Biomaterials, J. Am. Chem. Soc., 2022, 144, 16232–16251 CrossRef CAS PubMed
- E. Selivanovitch and T. Douglas, Virus capsid assembly across different length scales inspire the development of virus-based biomaterials, Curr. Opin. Virol., 2019, 36, 38–46 CrossRef CAS PubMed
- B. Ikwuagwu and D. Tullman-Ercek, Virus-like particles for drug delivery: a review of methods and applications, Curr. Opin. Biotechnol, 2022, 78, 102785 CrossRef CAS PubMed
- C. Hou,
et al., Virus-Based Supramolecular Structure and Materials: Concept and Prospects, ACS Appl. Bio Mater., 2021, 4, 5961–5974 CrossRef CAS PubMed
- R. K. Huang,
et al., The Prohead-I structure of bacteriophage HK97: implications for scaffold-mediated control of particle assembly and maturation, J. Mol. Biol., 2011, 408, 541–554 CrossRef CAS PubMed
- R. L. Duda, B. Oh and R. W. Hendrix, Functional domains of the HK97 capsid maturation protease and the mechanisms of protein encapsidation, J. Mol. Biol., 2013, 425, 2765–2781 CrossRef CAS PubMed
- J. B. Maurer, B. Oh, C. L. Moyer and R. L. Duda, Capsids and portals influence each other's conformation during assembly and maturation, J. Mol. Biol., 2020, 432, 2015–2029 CrossRef CAS PubMed
- L. Gan,
et al., Control of crosslinking by quaternary structure changes during bacteriophage HK97 maturation, Mol. Cell, 2004, 14, 559–569 CrossRef CAS PubMed
- D. Tso, R. W. Hendrix and R. L. Duda, Transient contacts on the exterior of the HK97 procapsid that are essential for capsid assembly, J. Mol. Biol., 2014, 426, 2112–2129 CrossRef CAS PubMed
- D. Veesler,
et al., Architecture of a dsDNA viral capsid in complex with its maturation protease, Structure, 2014, 22, 230–237 CrossRef CAS PubMed
- P. D. Ross,
et al., Crosslinking renders bacteriophage HK97 capsid maturation irreversible and effects an essential stabilization, EMBO J., 2005, 24, 1352–1363 CrossRef CAS PubMed
- R. L. Duda,
et al., Structural transitions during bacteriophage HK97 head assembly, J. Mol. Biol., 1995, 247, 618–635 CAS
- W. R. Wikoff,
et al., Topologically linked protein rings in the bacteriophage HK97 capsid, Science, 2000, 289, 2129–2133 CrossRef CAS PubMed
- R. L. Duda, Protein chainmail: catenated protein in viral capsids, Cell, 1998, 94, 55–60 CrossRef CAS PubMed
- R. K. Huang, N. F. Steinmetz, C.-Y. Fu, M. Manchester and J. E. Johnson, Transferrin-mediated targeting of bacteriophage HK97 nanoparticles into tumor cells, Nanomedicine, 2011, 6, 55–68 CrossRef CAS PubMed
- D. Diaz, A. Care and A. Sunna, Bioengineering Strategies for Protein-Based Nanoparticles, Genes, 2018, 9(7), 370, DOI:10.3390/genes9070370
- L.-N. Liu, M. Yang, Y. Sun and J. Yang, Protein stoichiometry, structural plasticity and regulation of bacterial microcompartments, Curr. Opin. Microbiol., 2021, 63, 133–141 CrossRef CAS PubMed
- V. A. Essus,
et al., Bacteriophage P22 capsid as a pluripotent nanotechnology tool, Viruses, 2023, 15(2), 516, DOI:10.3390/v15020516
- D. Patterson,
et al., Sortase-Mediated Ligation as a Modular Approach for the Covalent Attachment of Proteins to the Exterior of the Bacteriophage P22 Virus-like Particle, Bioconjugate Chem., 2017, 28, 2114–2124 CrossRef CAS PubMed
- D.
P. Patterson, C. Hjorth, A. Hernandez Irias, N. Hewagama and J. Bird, Delayed In Vivo Encapsulation of Enzymes Alters the Catalytic Activity of Virus-Like Particle Nanoreactors, ACS Synth. Biol., 2022, 11, 2956–2968 CrossRef PubMed
-
M. Chalfie and S. Kain, Green Fluorescent Protein: Properties, Applications and Protocols (Methods of Biochemical Analysis), John Wiley and Sons, 2005 Search PubMed
- F. Wang,
et al., The functions and applications of RGD in tumor therapy and tissue engineering, Int. J. Mol. Sci., 2013, 14, 13447–13462 CrossRef PubMed
- S. Sani,
et al., Biological Relevance of RGD-Integrin Subtype-Specific Ligands in Cancer, ChemBioChem, 2021, 22, 1151–1160 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb00318c |
| This journal is © The Royal Society of Chemistry 2023 |