
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagainin 2-derived stapled peptides derived with the ability to deliver pDNA, mRNA, and siRNA into cells†
Motoharu
Hirano
ab,
Hidetomo
Yokoo
 ac,
Chihiro
Goto
ab,
Makoto
Oba
c,
Takashi
Misawa
*a and
Yosuke
Demizu
*abd
ac,
Chihiro
Goto
ab,
Makoto
Oba
c,
Takashi
Misawa
*a and
Yosuke
Demizu
*abd
aDivision of Organic Chemistry, National Institute of Health Sciences, 3-25-26 Tonomachi, Kawasaki, Kanagawa 210-9501, Japan. E-mail: misawa@nihs.go.jp; demizu@nihs.go.jp
bGraduate School of Medical Life Science, Yokohama City University, 1-7-29 Yokohama, Kanagawa 230-0045, Japan
cMedical Chemistry, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, Kyoto 606-0823, Japan
dGraduate School of Medicine, Dentistry and Pharmaceutical Sciences, Division of Pharmaceutical Science of Okayama University, 1-1-1 Tsushimanaka, Kita 700-8530, Japan
First published on 14th September 2023
Abstract
We have developed cell-penetrating stapled peptides based on the amphipathic antimicrobial peptide magainin 2 for intracellular delivery of nucleic acids such as pDNA, mRNA, and siRNA. Various types of stapled peptides with a cross-linked structure were synthesised in the hydrophobic region of the amphipathic structure, and their efficacy in intracellular delivery of pDNA was evaluated. The results showed that the stapled peptide st7-5 could deliver pDNA into cells. To improve the deliverability of st7-5, we further designed st7-5_R, in which the Lys residues were replaced by Arg residues. The peptide st7-5_R formed compact and stable complexes with pDNA and was able to efficiently transfer pDNA into the cell. In addition to pDNA, st7-5_R was also able to deliver mRNA and siRNA into the cell. Thus, st7-5_R is a novel peptide that can achieve efficient intracellular delivery of three different nucleic acids.
Introduction
Nucleic acid therapeutics, such as antisense and small interfering RNA (siRNA) targeting DNA or RNA, are attracting attention as the next generation of therapeutics. These nucleic acid therapeutics can in principle extend the range of drug target molecules, unlike protein-targeted small molecule or antibody drugs.1 Additionally, messenger RNA (mRNA) drugs, in which mRNA encoding a target protein is administered to express the protein in the target cell, have also been actively developed.2 An important limitation of these therapeutics is that nucleic acids are generally water-soluble and negatively charged, which results in a poor ability to permeate cell membranes and thus they are difficult to transfer into cells. Therefore, it is important to develop drug delivery tools to efficiently transport nucleic acids into cells.One drug delivery system (DDS) that has attracted much attention is a non-viral technology using nanoparticles such as cell-penetrating peptides (CPPs).3,4 CPPs function as carrier molecules to transport cargo that is difficult to translocate into the cell, such as proteins and nucleic acids. Furthermore, compared with other technologies, they are generally less cytotoxic, easier to synthesize and modify, and can be mixed with nucleic acids to form nanoparticles. Consequentially, they are being actively developed as drug delivery tools.
Many reported CPPs are rich in the cationic amino acids arginine (Arg) and lysine (Lys) and can be classified into either cationic CPPs, such as Tat5,6 and oligoarginine,7,8 or amphiphilic CPPs, such as transportan9 and penetratin.10 Various CPP derivatives have been developed to improve their cell membrane permeability and effective delivery.11 The correlation between the secondary structures of cationic CPPs and their cell membrane permeability has revealed that stabilizing their helical structures improves their permeability. For example, introducing side-chain stapling on CPPs promotes helical formation and improves cell membrane permeability.12,13 In fact, the side-chain stapling of natural CPPs, such as Tat and oligoarginine, has exhibited higher cell membrane permeability than natural CPPs.14 The development of other cell membrane-permeable molecules based on controlling their helical conformational has also been reported.15–17
We have also reported novel CPPs using α,α-disubstituted amino acids (dAAs), which are non-proteinogenic amino acids that stabilize the CPPs' helical structure and improved their cell-membrane penetration.18 In particular, nona-arginine (R9)-based CPPs containing cyclic dAAs with the side-chain guanidino group could form more stable helical structures and exhibit higher cell membrane permeability to deliver plasmid DNA (pDNA) more efficiently than CPPs with nonhelical R9.19 We have also reported that a helix-stabilized block-type peptide consisting of a hydrophobic segment and R9 can deliver siRNA into the cell and induce RNA interference (RNAi).20
Recently, antimicrobial peptides (AMPs) have attracted attention as a new class of CPPs. As representative amphipathic peptides, AMPs are one of the innate host defence mechanisms of a wide range of species, including humans, and are the primary means of protecting the host from pathogenic microorganisms.21 Membrane damage and intracellular activity have been shown to be involved in the antimicrobial action of many AMPs, and that interaction with the cell membrane and subsequent intracellular entry are considered important for their functional activity.22 AMPs exhibit cell membrane permeability in human cells,23 and have been successful in their intracellular delivery of proteins and nucleic acids.24,25 A relatively short (23 residues) AMP called magainin 2 forms a typical linear α-helical structure and is also permeable to human cells.26
Recently, we developed helix-stabilized AMPs from the magainin 2 sequence. By identifying sequences essential for antimicrobial activity and stabilizing the helix structure of those sequences, we were able to enhance antimicrobial activity against Gram-positive and Gram-negative bacteria, including drug-resistant bacteria.27,28 Given the relationship between helical structure and cell permeability of CPPs, we hypothesized that our AMPs could also serve as intracellular delivery tools for nucleic acids. In this study, we aimed to develop novel intracellular delivery molecules using the essential sequence of magainin 2 identified above to stabilize its helical structure. We designed various stapled peptides based on this magainin 2 derivative and evaluated their secondary structures, cell membrane permeability, enzyme resistance, and intracellular delivery of pDNA, mRNA, and siRNA to determine their potential as drug delivery tools (Fig. 1).
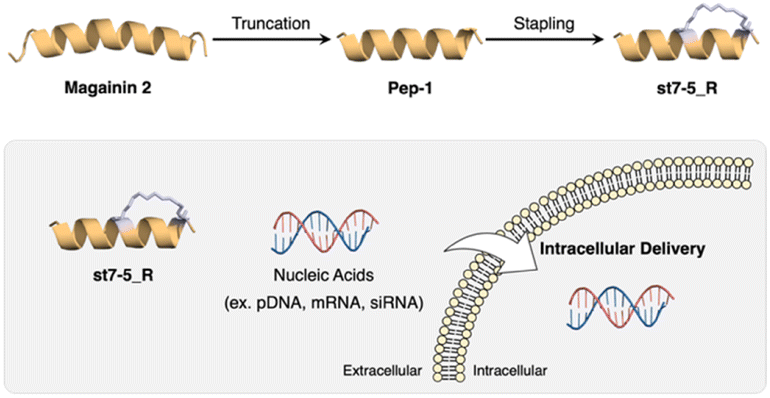 | ||
| Fig. 1 Development of CPP based on magainin 2 and intracellular delivery of nucleic acids using stapled peptides. | ||
While there have been many reports on the development of DDS by the structural expansion of CPPs, there are few reports on the development of CPPs and their delivery ability using AMPs as leads.29 In addition, most of these AMP-derived CPPs are thought to be easily degraded by proteases because they are composed of only natural amino acids. The peptides designed in this study are expected to acquire protease resistance by side-chain stapling, which should improve their delivery ability. Lastly, although intracellular delivery of nucleic acids such as pDNA, mRNA, and siRNA by CPP has been reported,30 there is only one report we are aware of that demonstrated intracellular delivery of three types of nucleic acids (pDNA, mRNA, and siRNA) with the same peptide.31 As mentioned above, while there have been reports of several CPPs based on AMPs, their application to intracellular delivery of nucleic acids is still limited, and the underlying mechanisms remain unclear. Therefore, the AMP-based CPP design method in this study is expected to guide a new type of DDS carrier design.
Results and discussion
Design and synthesis of peptides
We designed two stapled peptides based on our previously reported amphipathic peptide Pep-1,27 which is a magainin 2 derivative: (1) st4-1−6 with (S)-2-(4-pentenyl) alanine (S5) residues in the hydrophobic region at the i and i + 4 positions, and (2) st7-1–6 with (R)-2-(7-octenyl) alanine (R8) and S5 residues at the i and i + 7 positions (Table 1). The designed peptides were elongated by the Fmoc-based solid-phase method, and the side-chain stapling was conducted on resin using Grubbs catalysis.32 The peptides were then cleaved from the resin, purified by reversed-phase high-performance liquid chromatography (HPLC), and identified by liquid chromatography-mass spectrometry (LC-MS).| Peptide | Sequence |
|---|---|
| Magainin 2 | H-GIGKFLHSAKKFGKAFVGEIMNS-NH2 |
| Pep-1 | H-GIKKFLKSAKKFVKAFK-NH2 |
| st4-1 | H-S5*IKKS5*LKSAKKFVKAFK-NH2 |
| st4-2 | H-GS5*KKFS5*KSAKKFVKAFK-NH2 |
| st4-3 | H-GIKKS5*LKSS5*KKFVKAFK-NH2 |
| st4-4 | H-GIKKFLKS5*AKKS5*VKAFK-NH2 |
| st4-5 | H-GIKKFLKSS5*KKFS5*KAFK-NH2 |
| st4-6 | H-GIKKFLKSAKKS5*VKAS5*K-NH2 |
| st7-1 | H-R8*IKKFLKS5*AKKFVKAFK-NH2 |
| st7-2 | H-GR8*IKKFLKSS5*KKFVKAFK-NH2 |
| st7-3 | H-GIKKR8*LKSAKKS5*VKAFK-NH2 |
| st7-4 | H-GIKKFR8*KSAKKFS5*KAFK-NH2 |
| st7-5 | H-GIKKFLKR8*AKKFVKS5*FK-NH2 |
| st7-6 | H-GIKKFLKSR8*KKFVKAS5*K-NH2 |
| Pep-1_R | H-GIRRFLRSARRFVRAFR-NH2 |
| st7-5_R | H-GIRRFLRR8*ARRFVRS5*FR-NH2 |
| FAM-Magainin 2 | FAM-βAla-GIGKFLHSAKKFGKAFVGEIMNS-NH2 |
| FAM-Pep-1 | FAM-βAla-GIKKFLKSAKKFVKAFK-NH2 |
| FAM-st7-5 | FAM-βAla-GIKKFLKR8*AKKFVKS5*FK-NH2 |
| FAM-st7-5_R | FAM-βAla-GIRRFLRR8*ARRFVRS5*FR-NH2 |
|
|
Transfection efficiency of pDNA with stapled peptides
The transfection efficiency of pDNA with a series of synthesized stapled peptides was first evaluated by a luciferase reporter assay using pDNA encoding luciferase. The complexes were prepared so that the charge ratio (N/P ratio) of the positive charge of the cationic residues of the peptide to the negative charge of the phosphate group of pDNA was either 8 or 16. The complexes were then added to HeLa cells and incubated for 24 hours. The transfection efficiency of each peptide was determined by the level of luminescence produced by the transfected luciferase. As shown in Fig. 2A, among the stapled peptides, st7-5 had the highest transfection efficiency of pDNA. As hydrophobicity may affect cell membrane permeability, we compared the relative hydrophobicity of the peptides using ultra performance liquid chromatography retention times (Table S1†). As expected, the results showed that st7-1–6 between the i and i + 7 position stapling exhibited higher hydrophobicity than st4-1–6 between the i and i + 4 position stapling. Although there was no tendency for the hydrophobicity to change with the stapling position, st7-5 showed the slowest retention time and was the most hydrophobic. This suggests that the higher hydrophobicity of the peptides may play a role in transfection efficiency.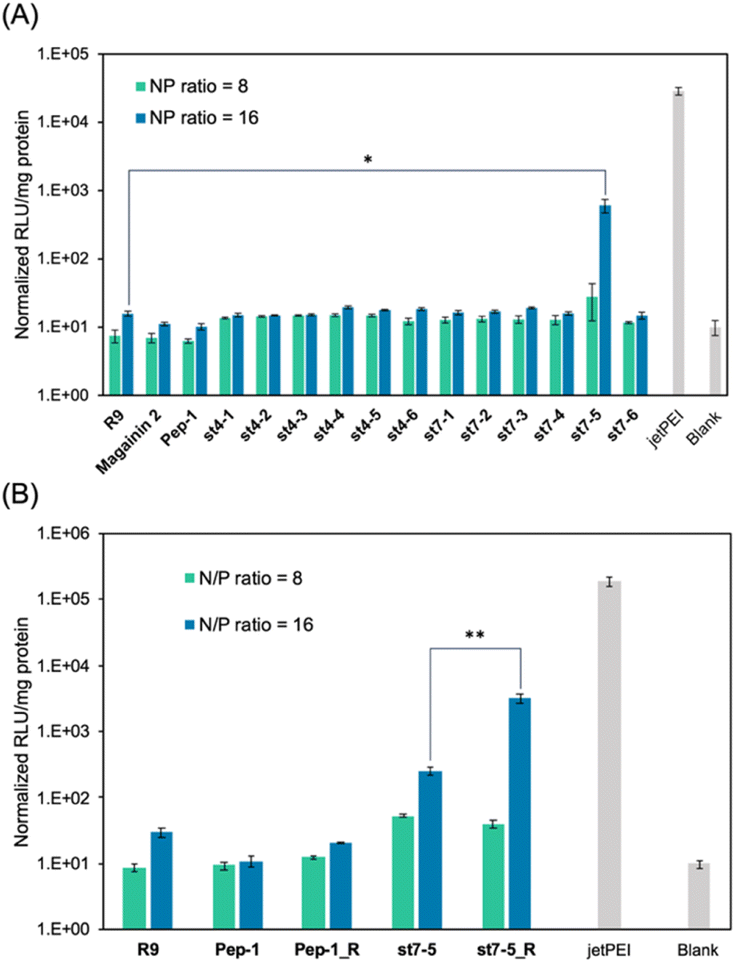 | ||
| Fig. 2 Transfection efficiency of stapled peptides and R9/pDNA complexes after a 24 h incubation in HeLa cells. The relative light unit (RLU) of (A) stapled peptides/pDNA complexes and (B) these derivatives with Lys substituted Arg, the transfected rLuc pDNA was normalized by the blank. N/P ratio = 8 (green) and 16 (blue), n = 3, mean ± standard deviation. Symbols (*) indicate statistical significance (*p < 0.05 and **p < 0.02). | ||
It has been reported that the guanidino group of Arg is important for cell-membrane permeabilization in human cells;33 therefore, replacing Lys in the peptide with Arg is expected to improve cell membrane permeability and pDNA transfection efficiency. To investigate this possibility, we designed and synthesized Pep-1_R and st7-5_R, in which all Lys residues of Pep-1 and st7-5 were replaced with Arg residues. Twenty-four hours after transfection, st7-5_R showed significantly increased luminescence compared with that seen with st7-5 transfection, but Pep-1_R did not show increased luminescence (Fig. 2B). This result suggests that not only the presence of cationic residues but also the specific positioning of side chain stapling may play a crucial role in the delivery effects observed for st7-5_R. In addition, increasing the N/P ratio from 8 to 16 also increased the luminescence intensity of both peptides. Peptides with N/P ratios of 16 and 32 exhibited the same extent of intracellular delivery efficacy (Fig. S1†). Notably, st7-5_R with an N/P ratio of 16 had the highest transfection efficiency, about 100 times higher than st7-5 and comparable with that observed with jetPEI, a transfection reagent.
Intracellular uptake of fluorescein-labelled peptides
Fluorescein-labelled peptides with 5(6)-carboxyfluorescein (FAM) at the N-terminus were synthesized and their cell membrane permeability was measured by flow cytometry (Fig. 3A). After 1 hour of compound treatment, FAM-R9, FAM-st7-5, and FAM-st7-5_R showed high fluorescence intensity in cells in a concentration-dependent manner, suggesting that they have strong membrane permeability. By contrast, FAM-magainin 2 and FAM-Pep-1 did not show cell membrane permeability. After 24 hours of compound treatment, cells incubated with FAM-st7-5 and FAM-st7-5_R had even more pronounced intracellular fluorescence intensity compared with that seen with 1 hour treatment. The results indicate that st7-5 and st7-5_R were internalized into the cells in a concentration- and time-dependent manner. This increased fluorescence was not observed, however, after 24 hour treatment with FAM-R9, which showed a 10-fold decrease in intensity at 5 μM treatment. This suggests that FAM-st7-5 and FAM-st7-5_R have higher intracellular stability likely because of the side-chain stapling insertion.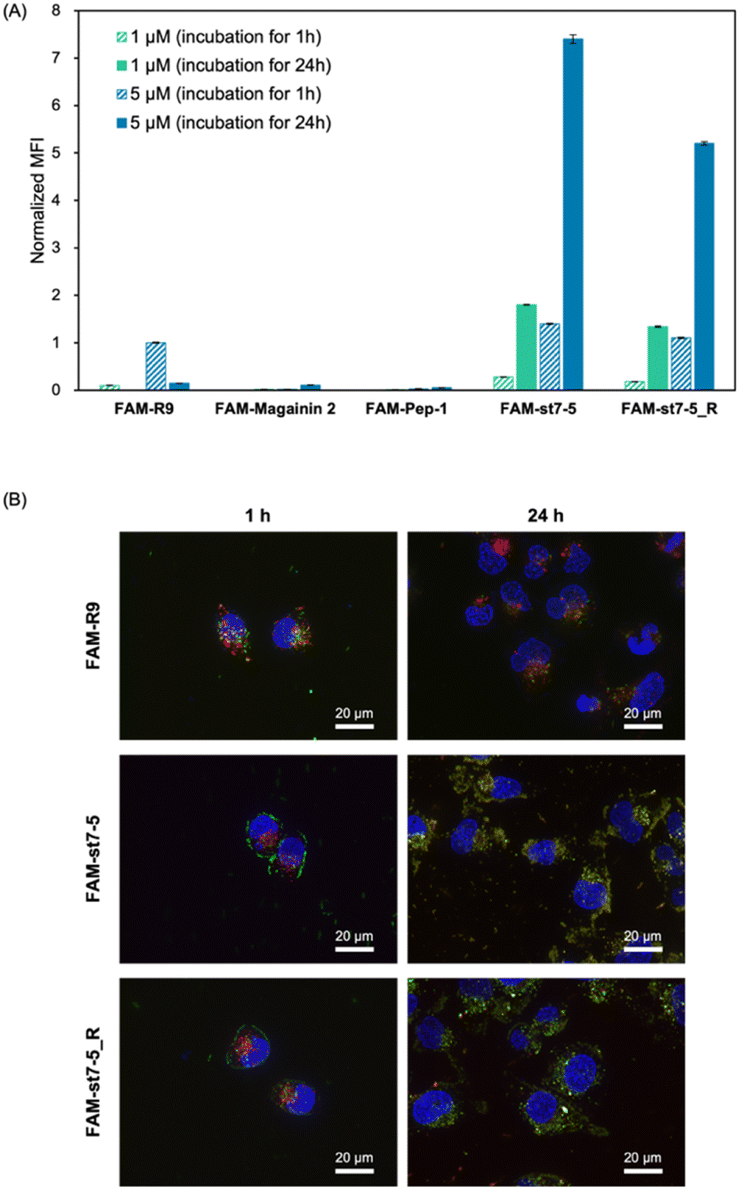 | ||
Fig. 3 (A) Intracellular uptake of fluorescein-labelled peptides into HeLa cells. Mean fluorescence intensity (MFI) of HeLa cells after incubation with 1 μM of peptide for 1 h (green, striped) and 24 h (green), and 5 μM for 1 h (blue, striped) and 24 h (blue). The MFI of each peptide was measured by flow cytometry and normalized by the MFI of FAM-R9 at 1 h n = 3, mean ± standard deviation. (B) Fluorescence microscopy images of HeLa cells treated with 5 μM FAM-R9, FAM-st7-5, and FAM-st7-5_R for 1 and 24 h at 37 °C. Late endosomes/lysosomes were stained with LysoTracker Red (red) and nuclei with Hoechst 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 342 (blue). 342 (blue). | ||
To determine if cellular uptake of these peptides depended on endocytosis, HeLa cells were pre-treated at 4 °C for 1 hour before measuring the intracellular fluorescence intensity. This pre-treatment decreased the cellular uptake of FAM-st7-5 and FAM-st7-5_R by more than 80% compared with that observed at 37 °C (Fig. S2A†), indicating that the intracellular uptake of the peptides required endocytosis.
Next, to identify which endocytic pathway was being used, HeLa cells were co-treated with each endocytosis inhibitor (nystatin, a caveolae-mediated endocytosis inhibitor; sucrose, a clathrin-mediated endocytosis inhibitor; and amiloride, a macropinocytosis inhibitor) (Fig. S2B†). Interestingly, the endocytosis inhibitors had differential effects on the two peptides. While sucrose decreased the intracellular fluorescence intensity of both FAM-st7-5 and FAM-st7-5_R, nystatin only affected FAM-st7-5. Amiloride did not affect either peptide. This suggests that both peptides require clathrin-mediated endocytosis, but FAM-st7-5 also might use caveolae-mediated endocytosis.
The intracellular localization of FAM-labelled peptides after 1 hour of treatment was examined by fluorescence microscopy (Fig. 3B) using Hoechst 33342 (blue) and LysoTracker (red) to label nuclei and late endosomes/lysosomes, respectively. As expected, FAM-R9 co-localized with late endosomes/lysosomes, suggesting that it is incorporated into the cell via endocytosis. While the stapled FAM-st7-5 and FAM-st7-5_R peptides also co-localized with late endosomes/lysosomes, they also localized at the plasma membrane. Moreover, as a result, the fluorescence of FAM-R9 was decreased after 24 h incubation, while that of FAM-st7-5 and FAM-st7-5_R significantly increased and the peptides were localized in cytosol. These results suggest that FAM-st7-5 and FAM-st7-5_R interact with the cell membrane.
Cytotoxicity of peptides
The cytotoxicity of magainin 2, Pep-1, st7-5, and st7-5_R in HeLa cells was evaluated using WST-8 (Fig. S3†). Magainin 2 and Pep-1 did not exhibit cytotoxicity even at 30 μM. While lower concentrations (10 μM) of the stapled st7-5 and st7-5_R peptides did not show significant cytotoxicity, they were cytotoxic at 30 μM (25% cell viability for st7-5, 54% cell viability for st7-5_R), which corresponds to the N/P ratio of 16 used in the evaluation of pDNA delivery (Fig. S1†). Importantly, a concentration of 5 μM of the st7-5 and st7-5_R peptides is sufficient for efficient intracellular delivery, which suggests that they will not be toxic to cells under these conditions.Protease resistance of peptides
Many peptides that are composed entirely of proteinogenic amino acids are not protease-resistant and are easily degraded in cellulo and in vivo. Because side-chain stapling and the introduction of non-proteinogenic amino acids are known to improve protease resistance,34 it is expected that the stapled peptides synthesized in this study will be resistant to proteases. To evaluate this, we explored the resistance of st7-5 and st7-5_R to trypsin, which is a representative protease and targets the cationic amino acids Arg and Lys. Compared with magainin 2, Pep-1, and R9, st7-5 and st7-5_R exhibited higher resistance to trypsin (Fig. S4†). After 24 hours of incubation, while magainin 2, Pep-1, and R9 6.4% had completely degraded, 70–80% of st7-5 and st7-5_R still remained. Even after 48 hours, more than 40% of st7-5 and st7-5_R did not degrade.Intracellular uptake of the peptide/Cy5-pDNA complex
Cy5 labelled pDNA (Cy5-pDNA) was prepared and the subcellular localization of Cy5-pDNA was examined by fluorescence microscopy (Fig. 4A). We used Hoechst 33342 (blue) as a nuclear marker and LysoPrime Green as a lysosomal marker. Cy5-pDNA complexed with R9, st7-5, and st7-5_R was localized in the cytoplasm or the nucleus. This suggests that these complexes might escape from the endosomes and transport pDNA into other intracellular compartments. Moreover, intracellular uptake of the peptide/Cy5-pDNA complex in HeLa cells was observed by flow cytometry. Analysis of intracellular fluorescence intensity showed that while R9-incubated cells exhibited only slight fluorescence, cells treated with R9, st7-5, and st7-5_R was localized in the cytoplasm or the nucleus, but lysosome. Additionally, st7-5_R-treated cells showed the highest fluorescence intensity, about 10 times higher than that observed after st7-5 treatment (Fig. 4B). Furthermore, the intracellular uptake pathway of pDNA/peptides complex using endocytosis inhibitors were investigated. As shown in Fig. 4C, the inhibition of each endocytosis pathway decreased the intracellular uptake, suggesting that the pDNA/peptide complex was delivered through endocytosis pathway. These results suggest that st7-5 and st7-5_R can be efficiently taken up into the cell and escape from endosomes via the endocytosis pathway.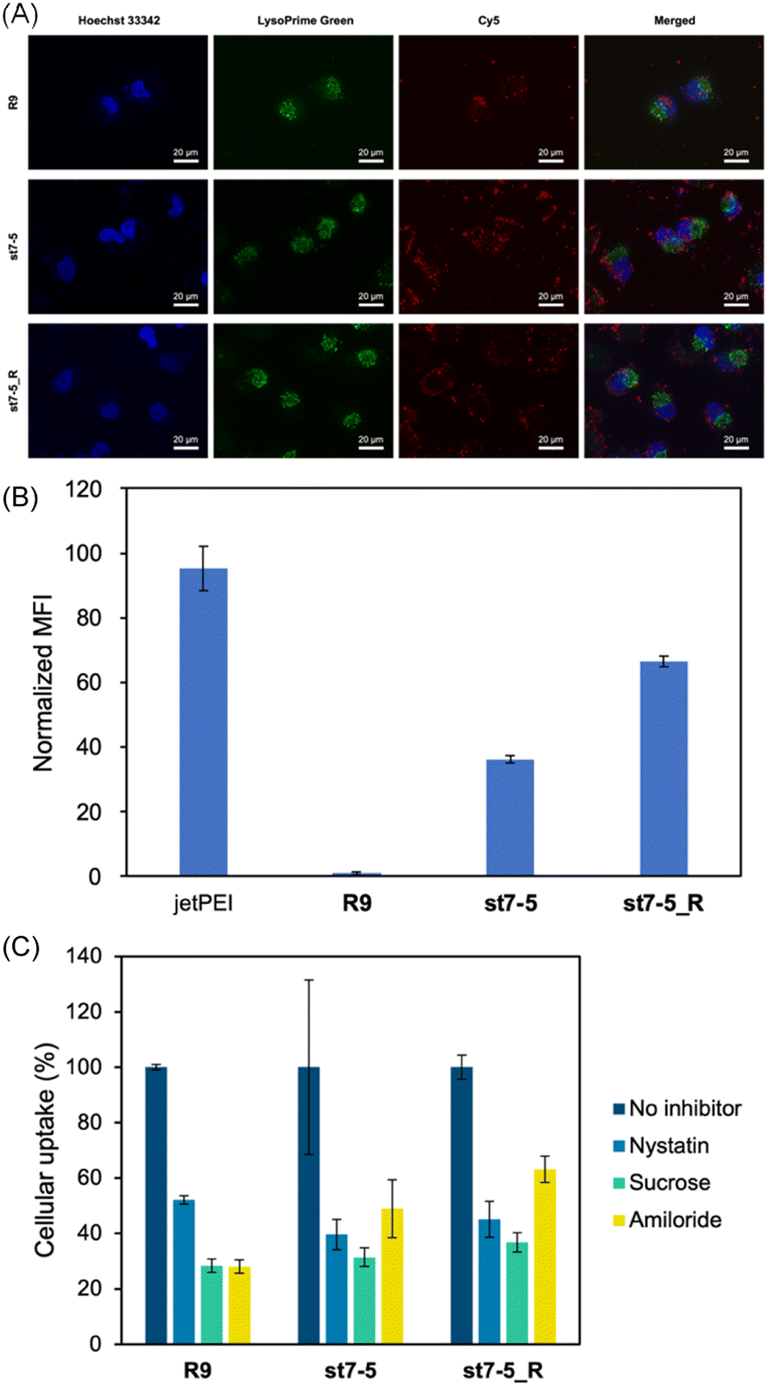 | ||
| Fig. 4 (A) Fluorescence microscopy of HeLa cells treated with peptide/Cy5 labelled pDNA (Cy5-pDNA) complex for 24 hours at 37 °C. R9, st7-5, and st7-5_R were mixed with Cy5-pDNA to prepare a complex with N/P ratio = 16. Lysosomes were stained with LysoPrime Green and nuclei with Hoechst 33342. (B) The intracellular uptake of Cy5-labeled pDNA into HeLa cells. The MFI of cells after incubation of HeLa cells with each peptide/pDNA complex at NP ratio = 16 for 24 h (1 μg of Cy5-labeled pDNA/well). The MFI of each peptide was measured by flowcytometry and normalized by the MFI of R9/pDNA complex. n = 3, mean ± standard deviation. (C) Effects of endocytosis inhibitors (nystatin: a caveolae-mediated endocytosis inhibitor; sucrose: a clathrin-mediated endocytosis inhibitor; amiloride: a macropinocytosis inhibitor) on intracellular uptake of Cy5-labeled pDNA on HeLa cells. n = 3, mean ± standard deviation. | ||
Structural properties of the peptide/pDNA complex
Fluorescence measurements of peptide/pDNA complex solutions prepared at various charge ratios (Fig. S5†) confirmed the formation of peptide/pDNA complexes using FAM-labeled peptides. The presence of FAM labels led to fluorescence quenching due to self-quenching upon complex formation. However, all peptide/pDNA complexes showed comparable curves, with fluorescence intensity increasing above N/P ratio = 1.0, indicating a stoichiometric N/P ratio in these complexes.The ability to form stable complexes with pDNA was compared with those of R9 and st7-5 by an agarose gel shift assay (Fig. S6A†). For pDNA alone, three bands were identified (relaxed circular, linear, and supercoiled). However, for all peptide/pDNA complexes (N/P ratios of 4, 8, and 16), the pDNA-derived bands disappeared. This indicates that R9, st7-5 and st7-5_R are completely complexed with pDNA when the N/P ratio is 4 or higher.
We then measured the size and zeta potential of the peptide/pDNA complexes (Table 2). As the N/P ratio increased, the particle size of the composite and the particle size distribution became smaller, indicating that a uniform complex was formed. In particular, st7-5_R formed the compact complex with a particle size of 125.1 ± 0.2 nm at an NP ratio of 16. This is considerably smaller the complexes formed by R9 and st7-5, which had particle sizes of 188.7 ± 0.7 and 234.0 ± 2.1 nm, respectively. Furthermore, zeta potential measurements revealed that st7-5_R exhibited the highest zeta potential (27.0 ± 0.3 mV) when the N/P ratio was 16, suggesting that it forms particles with excellent dispersion stability with pDNA. st7-5_R's formation of a more compact complex than that formed by st7-5 could be attributed to the fact that, compared with Lys, Arg has a stronger interaction with DNA and RNA.35,36 Because the size of the complex strongly influences the intracellular transport of nucleic acids,37 this may explain why st7-5_R has the most efficient transfection. However, particle size alone may not explain transfection efficiency because st7-5, which formed a larger complex than R9, transfected pDNA more efficiently than R9. This suggests that amphiphilicity likely also contributes to intracellular translocation as hydrophobic amino acid residues of amphipathic peptides, which have been reported to be important for intracellular translocation, interact with the cell membrane.38
| Peptide | N/P ratio | Size (nm) | PDI (μ/Γ2) | Zeta-potential (mV) |
|---|---|---|---|---|
| R9 | 8 | 357.5 ± 0.3 | 0.21 ± 0.01 | 22.1 ± 0.3 |
| 16 | 188.7 ± 0.7 | 0.19 ± 0.01 | 25.8 ± 0.2 | |
| st7-5 | 8 | 1522.0 ± 39.0 | 0.61 ± 0.02 | 23.4 ± 0.1 |
| 16 | 234.0 ± 2.1 | 0.27 ± 0.02 | 25.6 ± 0.1 | |
| st7-5_R | 8 | 352.2 ± 4.1 | 0.28 ± 0.00 | 22.3 ± 0.3 |
| 16 | 125.1 ± 0.2 | 0.20 ± 0.01 | 27.0 ± 0.3 |
mRNA delivery with stapled peptides
Vaccines for the prevention of infectious diseases as well as in cancer treatment, regenerative medicine, and genome editing therapy.39 However, a key limitation is that mRNA is unstable in vivo and easily degraded. Therefore, there is a need for stable and effective delivery of mRNA into cells to enhance protein expression. Because we showed that st7-5_R improves complex formation and transfection of pDNA, we explored if it could also improve mRNA transfection as well.We investigated the mRNA transfection efficiency of the peptide/mRNA complex in HeLa cells using a luciferase-encoded mRNA in the luciferase reporter assay. After preparation of the peptide/mRNA complexes with an N/P ratio of 16, the complexes were added to HeLa cells and incubated for 48 hours. Similar to what was observed with pDNA, cells transfected with mRNA complexed with either st7-5 and st7-5_R exhibited luminescence, suggesting that mRNA was introduced into the cells (Fig. 5). In particular, st7-5_R showed the strongest luminescence at N/P ratio = 16, demonstrating it to be the most efficient complex. By contrast, treatment of R9 with mRNA showed little to no luminescence, suggesting that the introduction of side-chain stapling and amphipathic properties is important for efficient mRNA delivery.
 | ||
| Fig. 5 Luciferase-encoding mRNA transfection into HeLa cells. HeLa cells were incubated for 48 hours after addition of each peptide/mRNA complex at N/P ratio of 8 (green) or 16 (blue). n = 5, mean ± standard deviation. | ||
siRNA delivery with stapled peptides
Lastly, we investigated the transfection efficiency of siRNA with the peptides. Peptides complexed with siRNA against luciferase or control were transfected into Huh-7 cells stably expressing luciferase (Huh-7-Luc cells). The transfection efficiency of the peptide/siRNA complex was measured by a loss of luminescence in the luciferase reporter assay (Fig. 6). The siRNAs used were Luc-siRNA, which binds to the mRNA of the target luciferase protein and causes RNAi, and Scr-siRNA, which was designed not to bind to the mRNA by randomizing the sequence. As expected, the luminescence intensity of luciferase did not decrease in the st7-5_R/Scr-siRNA complex. However, the luminescence intensity decreased significantly in the st7-5_R/Luc-siRNA complex, suggesting that st7-5_R efficiently delivers siRNA into the cell. Treatment of siRNA/peptide complex with the N/P ratio of 16 resulted in a tendency to decrease the luminescent intensity compared to the N/P ratio of 8. Especially at N/P ratio = 16, st7-5_R reduced the luminescence signal by nearly 70%. This reduction is comparable with what was observed with the transfection reagent jetPEI. While the luminescence intensity of luciferase was reduced when treated with the st7-5/Luc-siRNA complex, it was similarly reduced when treated with the st7-5/Scr-siRNA complex. This suggests that this may be related to cytotoxicity of the complex rather than interference with the luciferase gene. The complex with R9 did not result in a decrease in luminescence intensity, indicating that the siRNA was not transfected.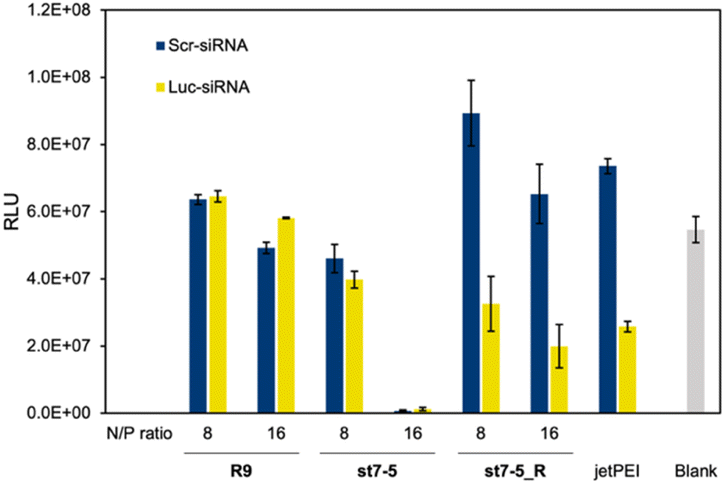 | ||
| Fig. 6 Transfection efficiency of siRNA into cultured cells. Luciferase activity of Huh-7-Luc cells treated with each peptide/siRNA complex at an N/P ratio of 8 or 16 and incubated for 48 h. Blue bars: scrambled sequence siRNA (Scr-siRNA). Yellow bars: firefly GL3 luciferase siRNA (Luc-siRNA). n = 5, mean ± standard deviation. | ||
Structural properties of the peptide/mRNA and siRNA complex
The ability of each peptide to form complexes with mRNA and siRNA was assessed using an agarose gel shift assay, similar to the procedure employed for pDNA. In the case of st7-5_R complexes with mRNA and siRNA at N/P ratios of 4, 8, and 16, the bands corresponding to nucleic acid disappeared (Fig. S6B and C†). This observation indicates that st7-5_R fully complexes with both mRNA and siRNA when the N/P ratio is 4 or higher. Conversely, in the complexes of R9 and st7-5 with mRNA at N/P ratios of 4, 8, and 16, the bands originating from mRNA vanished (Fig. S6B†), indicating that R9 and st7-5 fully complexed with mRNA at these N/P ratios. However, in the case of siRNA complexes with R9 and st7-5 at N/P ratios of 4, 8, and 16, the bands corresponding to siRNA were still visible (Fig. S6C†), suggesting that R9 and st7-5 did not fully complex with siRNA at these N/P ratios.The particle size and zeta potential of the peptide/mRNA and siRNA complexes were measured and are presented in Table S2.† Notably, the particle size of the formed complexes decreased proportionally with an increase in the N/P ratio. When preparing complexes with peptides at N/P ratio = 16, the sizes of the complexes with mRNA were as follows: R9 formed a complex of 177.7 ± 28.8 nm, st7-5 formed a complex of 211.3 ± 3.2 nm, and st7-5_R formed a compact complex of 158.3 ± 47.4 nm (Table S2A†). Similarly, for siRNA complexes, R9 formed a complex of 690.6 ± 8.7 nm, st7-5 formed a complex of 284.8 ± 21.1 nm, and st7-5_R formed a compact complex of 304.0 ± 5.7 nm (Table S2B†). These results clearly demonstrate that st7-5_R exhibits the ability to form complete complexes with both mRNA and siRNA at an N/P ratio greater than 4, and at an N/P ratio of 16, it forms compact complexes. The high complexing ability of st7-5_R for both mRNA and siRNA is expected to significantly enhance the efficiency of intracellular delivery for these nucleic acids.
Conclusions
In this study, we designed stapled peptides from the magainin 2 derivative Pep-1 for the intracellular delivery of three different types of nucleic acids, pDNA, mRNA, and siRNA. We showed that st7-5, which was stapled by replacing amino acids in the hydrophobic region of Pep-1, had greater cell membrane permeability than magainin 2 and Pep-1, and that was comparable to or higher than R9. Moreover, st7-5_R, in which Lys residues of st7-5 are replaced by Arg residues, had a cell membrane permeability comparable to that of st7-5 and its intracellular pDNA delivery was more efficient than that of st7-5. Furthermore, a physiochemical property analysis revealed that st7-5_R formed a more stable and compact complex with pDNA compared with that observed with st7-5. This indicates that both cell membrane permeability and nucleic acid complex formation are important for an effective CPP design. In addition to pDNA, st7-5_R also efficiently transfected mRNA and siRNA. The stapled peptide st7-5_R enabled stable protein expression even 48 hours after mRNA transfection and induced gene silencing by about 70% after siRNA delivery. Future research should explore the detailed mechanisms of how these various complexes escape from endosomes and release nucleic acids, as this remains unclear.Together, these results demonstrate the usefulness of AMPs in CPP development. These stapled peptides are broadly applicable because they can be simply mixed with nucleic acids for cellular transfection. Additionally, they could be applied in vivo by controlling surface charge and complex size through molecular modification.40,41 To the best of our knowledge, this is the first example of intracellular delivery of three different nucleic acids, pDNA, mRNA, and siRNA, using amphipathic CPPs, and it is expected to be useful in the development of novel CPPs for the therapeutic delivery of mRNA and siRNA.
Data availability
Additional experimental data supporting this article are included in the ESI.† Reasonable requests for additional information can be made to the corresponding authors.Author contributions
Y. D. designed the research, and M. H., T. M., and Y. D wrote the paper. M. H., H. Y., C. G., M. O., T. M., and Y. D. performed the experiments and analysed results. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors express their deepest appreciation to Dr Yuki Takeuchi-Haraya and Dr Yasuhiro Abe (National Institute of Health. Sciences) for supporting the DLS measurements. This study was supported in part by grants from the Japan Agency for Medical Research and Development (Grant numbers: 22mk0101197j0002, 22fk0210110j0401, 22ak0101185j0301, and 22fk0310506j0701 to Y. D.; 21fk08622j0001 and 22fk0108622j0002 to T. M.), the Japan Society for the Promotion of Science and the Ministry of Education, Culture, Sports, Science and Technology (JSPS/MEXT KAKENHI Grant umbers JP18H05502 and 21K05320 to Y. D., 20K06958 to T. M.), Chugai Foundation for Innovative Drug Discovery Science: C-FINDs (to Y. D. and H. Y.), JSPS KAKENHI Grant Number JP22J23111 (to M. H.), the Public Promoting Association Asano Foundation for Studies on Medicine (to H. Y.). We thank Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.Notes and references
- A. Khvorova and J. K. Watts, The chemical evolution of oligonucleotide therapies of clinical utility, Nat. Biotechnol., 2017, 35, 238–248 CrossRef CAS PubMed
.
- U. Sahin, K. Kariko and O. Tureci, mRNA-based therapeutics–developing a new class of drugs, Nat. Rev. Drug Discovery, 2014, 13, 759–780 CrossRef CAS PubMed
- S. C. De Smedt, J. Demeester and W. E. Hennink, Cationic polymer based gene delivery systems, Pharm. Res., 2000, 17, 113–126 CrossRef CAS PubMed
- X. Hou, T. Zaks, R. Langer and Y. Dong, Lipid nanoparticles for mRNA delivery, Nat. Rev. Mater., 2021, 6, 1078–1094 CrossRef CAS PubMed
- A. D. Frankel and C. O. Pabo, Cellular uptake of the tat protein from human immunodeficiency virus, Cell, 1988, 55, 1189–1193 CrossRef CAS PubMed
- E. Vives, P. Brodin and B. Lebleu, A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus, J. Biol. Chem., 1997, 272, 16010–16017 CrossRef CAS PubMed
- S. Futaki, T. Suzuki, W. Ohashi, T. Yagami, S. Tanaka, K. Ueda and Y. Sugiura, Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery, J. Biol. Chem., 2001, 276, 5836–5840 CrossRef CAS PubMed
- P. A. Wender, D. J. Mitchell, K. Pattabiraman, E. T. Pelkey, L. Steinman and J. B. Rothbard, The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13003–13008 CrossRef CAS PubMed
- M. Pooga, M. Hällbrink, M. Zorko and Ü. Langel, Cell penetration by transportan, Faseb. J., 1998, 12, 67–77 CrossRef CAS PubMed
- D. Derossi, A. H. Joliot, G. Chassaing and A. Prochiantz, The third helix of the Antennapedia homeodomain translocates through biological membranes, J. Biol. Chem., 1994, 269, 10444–10450 CrossRef CAS PubMed
- F. Heitz, M. C. Morris and G. Divita, Twenty years of cell-penetrating peptides: from molecular mechanisms to therapeutics, Br. J. Pharmacol., 2009, 157, 195–206 CrossRef CAS PubMed
- S. Hyun, Y. Choi, H. N. Lee, C. Lee, D. Oh, D. K. Lee, C. Lee, Y. Lee and J. Yu, Construction of histidine-containing hydrocarbon stapled cell penetrating peptides for in vitro and in vivo delivery of siRNAs, Chem. Sci., 2018, 9, 3820–3827 RSC
- L. D. Walensky and G. H. Bird, Hydrocarbon-stapled peptides: principles, practice, and progress, J. Med. Chem., 2014, 57, 6275–6288 CrossRef CAS PubMed
- Q. Chu, R. E. Moellering, G. J. Hilinski, Y.-W. Kim, T. N. Grossmann, J. T. H. Yeh and G. L. Verdine, Towards understanding cell penetration by stapled peptides, MedChemComm, 2015, 6, 111–119 RSC
- N. Umezawa, M. A. Gelman, M. C. Haigis, R. T. Raines and S. H. Gellman, Translocation of a beta-peptide across cell membranes, J. Am. Chem. Soc., 2002, 124, 368–369 CrossRef CAS PubMed
- J. Iriondo-Alberdi, K. Laxmi-Reddy, B. Bouguerne, C. Staedel and I. Huc, Cellular internalization of water-soluble helical aromatic amide foldamers, Chembiochem, 2010, 11, 1679–1685 CrossRef CAS PubMed
- M. Bornerie, A. Brion, G. Guichard, A. Kichler and C. Douat, Delivery of siRNA by tailored cell-penetrating urea-based foldamers, Chem. Commun., 2021, 57, 1458–1461 RSC
- H. Yamashita, T. Kato, M. Oba, T. Misawa, T. Hattori, N. Ohoka, M. Tanaka, M. Naito, M. Kurihara and Y. Demizu, Development of a Cell-penetrating Peptide that Exhibits Responsive Changes in its Secondary Structure in the Cellular Environment, Sci. Rep., 2016, 6, 33003 CrossRef CAS PubMed
- H. Yamashita, M. Oba, T. Misawa, M. Tanaka, T. Hattori, M. Naito, M. Kurihara and Y. Demizu, A Helix-Stabilized Cell-Penetrating Peptide as an Intracellular Delivery Tool, Chembiochem, 2016, 17, 137–140 CrossRef CAS PubMed
- T. Misawa, N. Ohoka, M. Oba, H. Yamashita, M. Tanaka, M. Naito and Y. Demizu, Development of 2-aminoisobutyric acid (Aib)-rich cell-penetrating foldamers for efficient siRNA delivery, Chem. Commun., 2019, 55, 7792–7795 RSC
- A. A. Bahar and D. Ren, Antimicrobial peptides, Pharmaceuticals, 2013, 6, 1543–1575 CrossRef PubMed
- C. D. Fjell, J. A. Hiss, R. E. Hancock and G. Schneider, Designing antimicrobial peptides: form follows function, Nat. Rev. Drug Discovery, 2011, 11, 37–51 CrossRef PubMed
- S. T. Henriques, M. N. Melo and M. A. Castanho, Cell-penetrating peptides and antimicrobial peptides: how different are they?, Biochem. J., 2006, 399, 1–7 CrossRef CAS PubMed
- X. Zhang, K. Oglecka, S. Sandgren, M. Belting, E. K. Esbjorner, B. Norden and A. Graslund, Dual functions of the human antimicrobial peptide LL-37-target membrane perturbation and host cell cargo delivery, Biochim. Biophys. Acta, 2010, 1798, 2201–2208 CrossRef CAS PubMed
- W. W. Hu, Z. W. Lin, R. C. Ruaan, W. Y. Chen, S. L. Jin and Y. Chang, A novel application of indolicidin for gene delivery, Int. J. Pharm., 2013, 456, 293–300 CrossRef CAS PubMed
- K. Takeshima, A. Chikushi, K. K. Lee, S. Yonehara and K. Matsuzaki, Translocation of analogues of the antimicrobial peptides magainin and buforin across human cell membranes, J. Biol. Chem., 2003, 278, 1310–1315 CrossRef CAS PubMed
- C. Goto, M. Hirano, K. Hayashi, Y. Kikuchi, Y. Hara-Kudo, T. Misawa and Y. Demizu, Development of Amphipathic Antimicrobial Peptide Foldamers Based on Magainin 2 Sequence, ChemMedChem, 2019, 14, 1911–1916 CrossRef CAS PubMed
- M. Hirano, C. Saito, H. Yokoo, C. Goto, R. Kawano, T. Misawa and Y. Demizu, Development of Antimicrobial Stapled Peptides Based on Magainin 2 Sequence, Molecules, 2021, 26, 444 CrossRef CAS PubMed
- K. Splith and I. Neundorf, Antimicrobial peptides with cell-penetrating peptide properties and vice versa, Eur. Biophys. J., 2011, 40, 387–397 CrossRef CAS PubMed
- H. Yokoo, M. Oba and S. Uchida, Cell-Penetrating Peptides: Emerging Tools for mRNA Delivery, Pharmaceutics, 2021, 14, 78 CrossRef PubMed
- S. Ali, C. Dussouillez, B. Padilla, B. Frisch, A. J. Mason and A. Kichler, Design of a new cell penetrating peptide for DNA, siRNA and mRNA delivery, J. Gene Med., 2022, 24, e3401 CrossRef CAS PubMed
- Y. W. Kim, T. N. Grossmann and G. L. Verdine, Synthesis of all-hydrocarbon stapled alpha-helical peptides by ring-closing olefin metathesis, Nat. Protoc., 2011, 6, 761–771 CrossRef CAS PubMed
- S. Futaki and I. Nakase, Cell-Surface Interactions on Arginine-Rich Cell-Penetrating Peptides Allow for Multiplex Modes of Internalization, Acc. Chem. Res., 2017, 50, 2449–2456 CrossRef CAS PubMed
- C. E. Schafmeister, J. Po and G. L. Verdine, An All-Hydrocarbon Cross-Linking System for Enhancing the Helicity and Metabolic Stability of Peptides, J. Am. Chem. Soc., 2000, 122, 5891–5892 CrossRef CAS
- B. Lustig, S. Arora and R. L. Jernigan, RNA base-amino acid interaction strengths derived from structures and sequences, Nucleic Acids Res., 1997, 25, 2562–2565 CrossRef CAS PubMed
- R. Sathyapriya and S. Vishveshwara, Interaction
of DNA with clusters of amino acids in proteins, Nucleic Acids Res., 2004, 32, 4109–4118 CrossRef CAS PubMed
- F. D. Ledley, Nonviral gene therapy: the promise of genes as pharmaceutical products, Hum. Gene Ther., 1995, 6, 1129–1144 CrossRef CAS PubMed
- K. Takayama, I. Nakase, H. Michiue, T. Takeuchi, K. Tomizawa, H. Matsui and S. Futaki, Enhanced intracellular delivery using arginine-rich peptides by the addition of penetration accelerating sequences (Pas), J. Controlled Release, 2009, 138, 128–133 CrossRef CAS PubMed
- N. Pardi, M. J. Hogan, F. W. Porter and D. Weissman, mRNA vaccines - a new era in vaccinology, Nat. Rev. Drug Discovery, 2018, 17, 261–279 CrossRef CAS PubMed
- M. Ogris, S. Brunner, S. Schuller, R. Kircheis and E. Wagner, PEGylated DNA/transferrin-PEI complexes: reduced interaction with blood components, extended circulation in blood and potential for systemic gene delivery, Gene Ther., 1999, 6, 595–605 CrossRef CAS PubMed
- K. L. Veiman, K. Kunnapuu, T. Lehto, K. Kiisholts, K. Parn, U. Langel and K. Kurrikoff, PEG shielded MMP sensitive CPPs for efficient and tumor specific gene delivery in vivo, J. Controlled Release, 2015, 209, 238–247 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04124g |
| This journal is © The Royal Society of Chemistry 2023 |