
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Engineering ligand chemistry on Au25 nanoclusters: from unique ligand addition to precisely controllable ligand exchange†
Jiangtao
Zhao
a,
Abolfazl
Ziarati
 *a,
Arnulf
Rosspeintner
a,
Yanan
Wang
b and
Thomas
Bürgi
*a
*a,
Arnulf
Rosspeintner
a,
Yanan
Wang
b and
Thomas
Bürgi
*a
aDepartment of Physical Chemistry, University of Geneva, 30 Quai Ernest-Ansermet, 1211, Geneva 4, Switzerland. E-mail: abolfazl.ziarati@unige.ch; thomas.buergi@unige.ch
bDepartment of Chemical Engineering, University of Michigan, Ann Arbor, 2800, MI, USA
First published on 22nd June 2023
Abstract
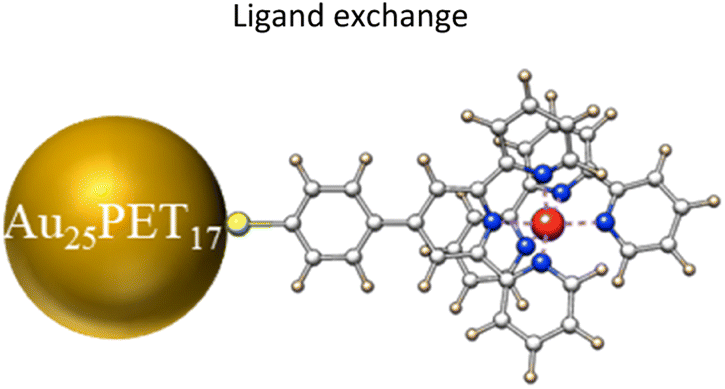
Au25 nanoclusters (NCs) protected by 18 thiol-ligands (Au25SR18, SR is a thiolate ligand) are the prototype of atomically precise thiolate-protected gold NCs. Studies concerning the alteration of the number of surface ligands for a given Au25SR18 NC are scarce. Herein we report the conversion of hydrophobic Au25PET18 (PET = 2-phenylethylthiolate) NCs to Au25SR19 [Au25PET18(metal complex)1] induced by ligand exchange reactions (LERs) with thiolated terpyridine-metal complexes (metal complex, metal = Ru, Fe, Co, Ni) under mild conditions (room temperature and low amounts of incoming ligands). Interestingly, we found that the ligand addition reaction on Au25PET18 NCs is metal dependent. Ru and Co complexes preferentially lead to the formation of Au25SR19 whereas Fe and Ni complexes favor ligand exchange reactions. High-resolution electrospray ionization mass spectrometry (HRESI-MS) was used to determine the molecular formula of Au25SR19 NCs. The photophysical properties of Au25PET18(Ru complex)1 are distinctly different from Au25PET18. The absorption spectrum is drastically changed upon addition of the extra ligand and the photoluminescence quantum yield of Au25PET18(Ru complex)1 is 14 times and 3 times higher than that of pristine Au25PET18 and Au25PET17(Ru complex)1, respectively. Interestingly, only one surface ligand (PET) could be substituted by the metal complex when neutral Au25PET18 was used for ligand exchange whereas two ligands could be exchanged when starting with negatively charged Au25PET18. This charge dependence provides a strategy to precisely control the number of exchanged ligands at the surface of NCs.
Introduction
Thiolate-protected metal nanoclusters (NCs) have attracted tremendous attention in the past few decades,1–4 and triggered extensive research interests in many emerging fields, such as biological applications,5,6 electro(photo)catalysis for energy conversion7,8 and sensing,9,10 owing to well-defined structures, unique optical and molecular-like properties.11 To date, dozens of different thiolate-protected gold NCs are known (denoted as AumSRn, m and n indicate the number of gold atoms and thiolate ligands, respectively), such as Au25SR18, Au38SR24, Au102SR44, Au144SR60, etc.12 However, to further extend the range of possible applications of NCs, the research on diversification of Au NC species with modified morphologies and properties remains important.Thiolate-ligands on the surface of the Au NCs play an important role during the synthesis of NCs,13 and influence the physicochemical properties of NCs, boost their functionalities and affect the NC size.14–16 The number of surface ligands for a given Au nuclearity is typically well-defined. For example, Au25SR18, the prototypical thiolate-protected gold NC contains exactly 18 ligands (the Au13 core is surrounded by 6 Au2SR3 staples). This NC seems the ideal candidate to study the derivatization chemistry of Au NCs. Nowadays, Au NCs are usually fabricated by a well-recognized synthetic method using neutral or anionic thiolate ligands, such as 2-phenylethanethiol (PET), glutathione, mercaptocarboxylic acids and so on.17–19 The direct synthesis of cationic-ligand-protected Au NCs is limited because of coulombic attractions between the anionic Au ion (AuCl4−) and the cationic thiol and coulombic repulsions caused by abundant cationic ligands at the surface of Au NCs.20,21 Ligand exchange reactions (LERs), in which surface ligands on Au NCs are substituted with new thiolate ligands, thus functionalizing the NCs, provide a crucial post-synthesis methodology for surface chemical modification of Au NCs.22,23 Particularly, chromophore-functionalized Au NCs were prepared and their photophysical properties studied.24–27 For instance, the optical behavior was investigated in porphyrin functionalized chiral Au38SR24 NC,28 and a directional electron transfer from the Au25 NC to the pyrene was studied in pyrene-coupled Au25 NCs.29 In addition, the ligand-exchange position controlled by steric hindrance of incoming ligands was revealed in porphyrinthiol-substituted Au25 NCs system.30 Ligand exchange with free ligand can be divided into two categories: (i) part or all surface ligands are replaced by the new ligands without changing the size and structure of the original Au NCs;31 (ii) ligand exchange induces a size/structure transformation, which was reported by Jin and co-workers.32 It provided an easy way to explore the diversity of NCs and generate new NCs with different composition and properties and to study the transformation mechanisms,33 for example, from Au25SR18 to Au28SR'20,34 from Au144SR60 to Au133SR'52, and so on.35 Some of us previously reported the transformation from Au25SR18 to Au28SR21 induced by LER with a chiral ligand under mild conditions in organic phase.36 Xie and co-workers recently discovered a new hydrophilic Au25SR19 NC by adding excess thiolate ligands (MHA = 6-mercaptohexanoic acid) to Au25MHA18 NCs,37 and the transformation mechanism was studied and proposed to occur through an oxidative etching process. In that work, the additional ligand was identical to the one protecting the original NC and the ligand addition process involving different ligands was not investigated.
In this work, we successfully obtained hydrophobic Au25SR19 by precisely adding one metal complex as ligand on the Au25PET18 surface. We demonstrated that the single ligand addition on Au25PET18 is metal dependent (metal = Ru, Fe, Co, Ni), so that the ligand addition reaction can be precisely controlled. Time-resolved high resolution ESI and UV-vis spectra were recorded to monitor the transformation process of Au25SR19, and a structure of the Au25SR19 NC is also proposed. Furthermore, it is worth to mention that neither excess thiolate ligand nor high temperature was needed to trigger this transformation. More interestingly, the number of surface ligands that can be replaced by metal complexes during LER depended on the charge of parent Au NCs.
Results and discussions
Neutral Au25PET18 (PET = 2-phenylethanethiol) was fabricated and purified according to previous reports and used as model NC in the following experiments.38 UV-vis absorption spectra (ESI, Fig. S1a†) and high-resolution electrospray ionization mass spectra (HRESI-MS) (ESI, Fig. S1b†) of as-prepared neutral Au25PET18 NC indicates successful synthesis and high purity.38,39 ESI, Fig. S2† shows 1H-NMR spectra of the Ru complex. Characteristic absorption spectra and ESI-MS of different metal complexes are displayed in ESI, Fig. S3 and Table. S1,† respectively. LER was performed by mixing Au25PET18 in dichloromethane and metal complex in acetonitrile (Fig. 1) in a glovebox at room temperature at NC/complex ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 and 1:4, respectively. The progress of the reaction was monitored by HRESI-MS (Fig. 2) at NC/complex ratio 1:2. Peak 5 at m/z = 7394.2 and peak 4 located at m/z = 6058 are assigned to the unreacted Au25PET18 and its prominent fragment.
:2 and 1:4, respectively. The progress of the reaction was monitored by HRESI-MS (Fig. 2) at NC/complex ratio 1:2. Peak 5 at m/z = 7394.2 and peak 4 located at m/z = 6058 are assigned to the unreacted Au25PET18 and its prominent fragment.
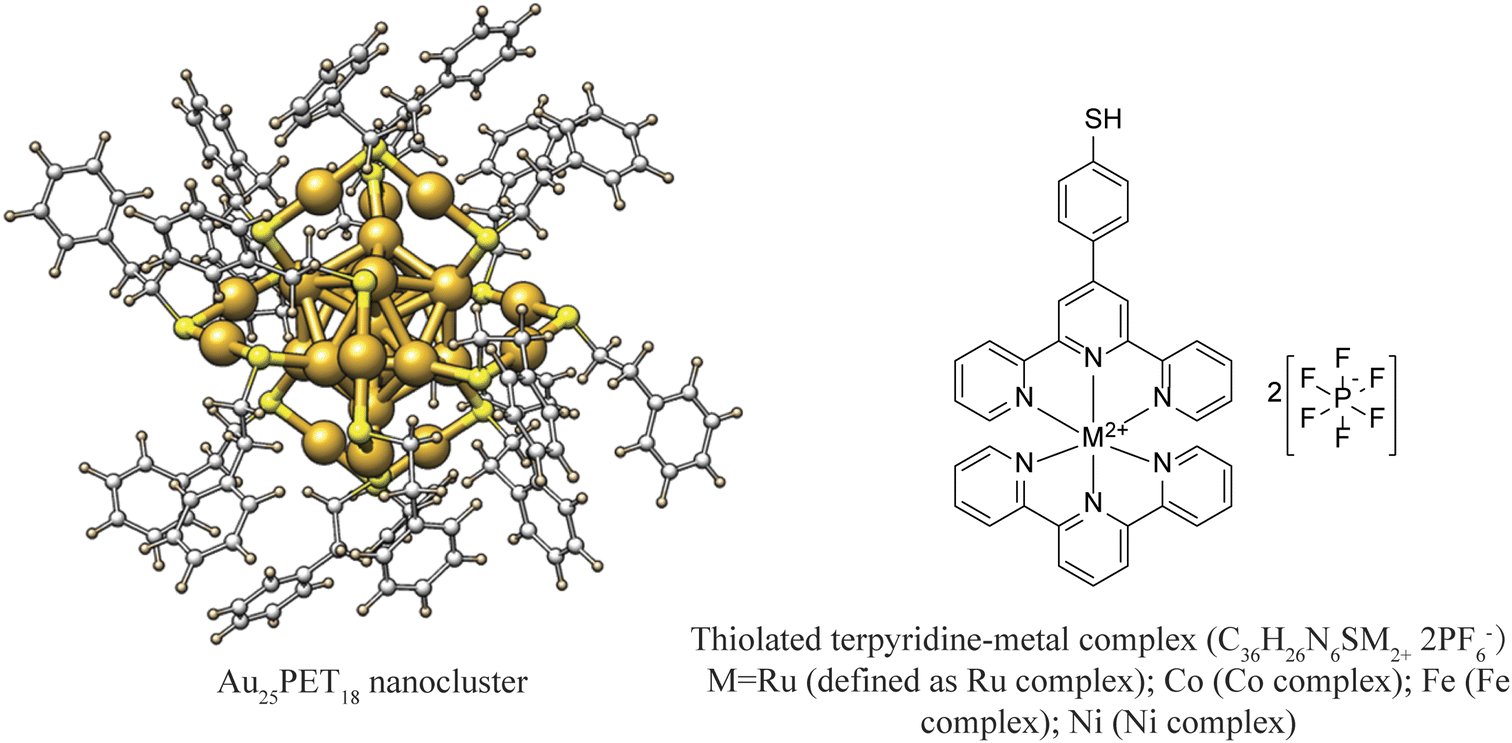 | ||
| Fig. 1 Schematic diagram of Au25PET18 NC and thiolated terpyridine-metal complex (counter ions: 2PF6−). | ||
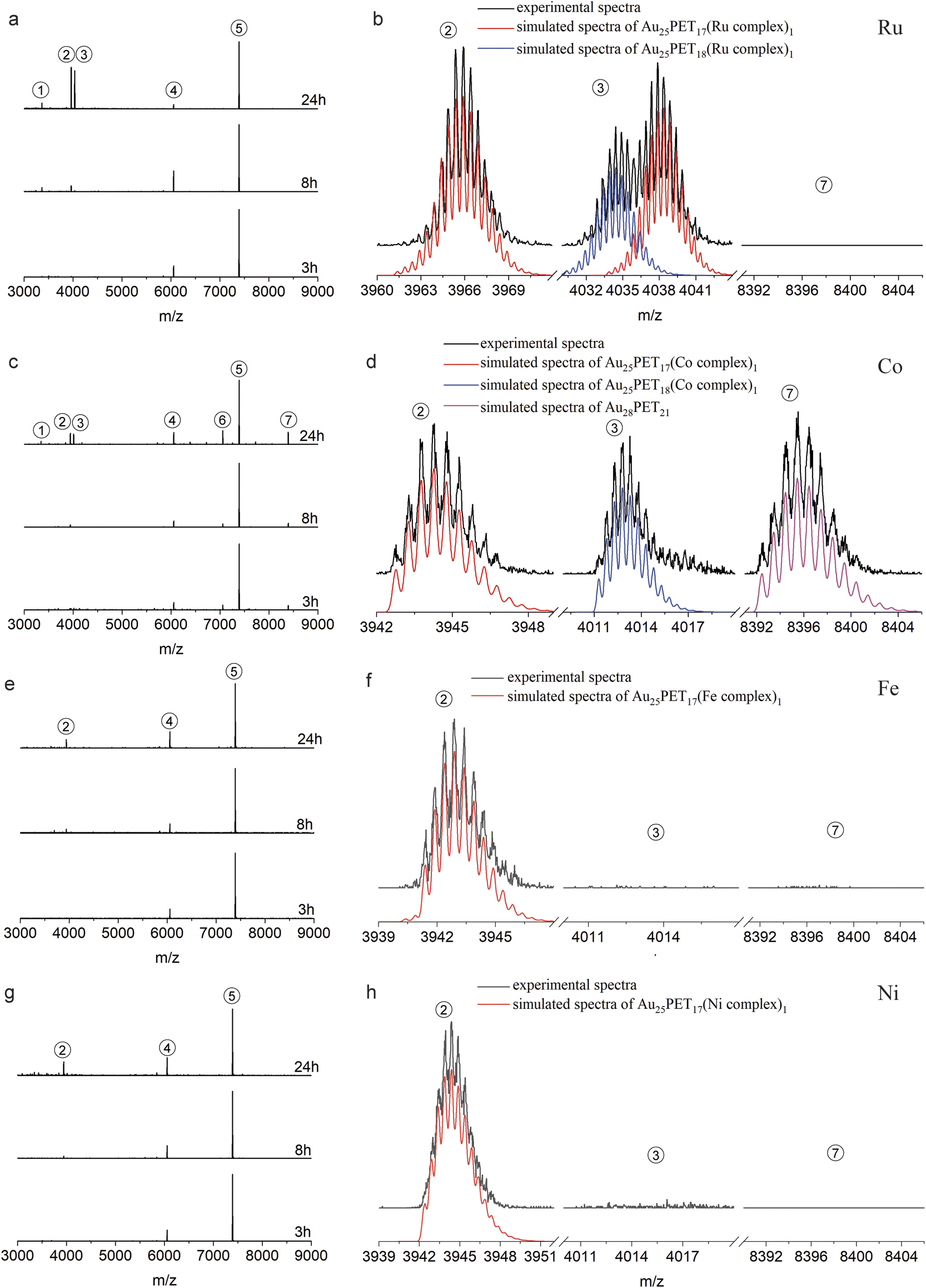 | ||
| Fig. 2 Time-dependent ESI-MS during LER with Ru complex (a and b), Co complex (c and d), Fe complex (e and f) and Ni complex (g and h) at NCs/complex ratio 1:2. (b, d, f and h) Show the comparison of experimental and simulated isotopic patterns of different species after reaction for 24 h (enlarged from a, c, e and g, respectively). (Isotopic patterns distance = 0.5 in peak 1–3 indicated the charge z = 2+, distance = 1 in peak 4–7 showed the charge z = 1+; the spectra are normalized with respect to peak 5). | ||
Fig. 2a shows the time-dependent ESI-MS during LER with the Ru complex at a ratio of 1:2, both experimental and simulated spectra of the emerging products are shown in Fig. 2b. As demonstrated in Fig. 2a and b, peak 2 and peak 3 are associated with doubly charged species because the interval between isotopic peaks is 0.5 m/z. Peak 2 at m/z = 3965.9 indicates that one PET ligand was substituted by the Ru complex, [Au25PET17(Ru complex)1]2+. The isotopic pattern of peak 3 from m/z = 4030 to m/z = 4044 demonstrates that two species overlapped in this region, the right part at m/z = 4038.3 is also consistent with one ligand exchange species, but containing one counterion PF6−. The left part located at m/z = 4034.5 is assigned to a species containing one incoming ligand (Ru complex) added to Au25PET18, [Au25PET18(Ru complex)1]2+, a species with the same nuclearity but different ligand number. Noteworthily, peak 1 at m/z = 3366.1 represents the fragment from Au25PET18(Ru complex)1, after losing Au4PET4. Such fragmentation is a common phenomenon for Au NCs during MS analysis.36
Fig. 2c and d show the time-dependent HRESI-MS for the reaction with Co complex including detail views of some selected peaks. Peak 2 at m/z = 3944.8 in Fig. 2c and its isotopic pattern in Fig. 2d indicates a NC where one surface ligand was replaced by a Co complex, [Au25PET17(Co complex)1]2+. In addition, comparison of the experimental isotope pattern of peak 3 at m/z = 4013.4 and the simulated curve of Au25PET18(Co complex)1 in Fig. 2d, also confirmed ligand addition. Peak 1 at m/z = 3344.8 is a fragment from the Au25PET18 (Co complex)1 NC after loss of Au4PET4. More interestingly, the perfect matching of simulated isotope pattern and the experimental curve for the species at m/z = 8396.8 indicates the formation of Au28PET21 (peak 7 in Fig. 2c and d). Peak 6 at m/z = 7059.4 could be due to the fragment from unreacted Au25PET18 after loss of Au1PET1 and Au28PET21 after loss of Au4PET4. ESI-MS results after reaction for 3 h, 8 h and 24 h with Fe complex and Ni complex are shown in Fig. 2e, f, g and h, respectively. Both peak 2 in Fig. 2e and f at m/z = 3943.2 and peak 2 in Fig. 2g and h at m/z = 3944.7 correspond to ligand substitution with Fe complex and Ni complex, [Au25PET17(Fe complex)1 and Au25PET17(Ni complex)1], respectively. Thus, the ligand-exchange species is the dominant product when reacting Au25PET18 with Fe and Ni complex at a ratio of 1:2. Neither ligand addition nor size transformation was observed.
Table 1 summarizes the metal-dependent products after LER for 24 h at a NC/complex ratio of 1:2. It clearly shows that Ru and Co complexes can induce both ligand exchange and ligand addition on Au25PET18, and the transformation from Au25 to Au28 also appeared in the case of the Co complex. Nevertheless, only ligand-exchange products were observed when reacting with Fe and Ni complexes.
:2. (✓ means the reaction can take place, ✗ indicates that the reaction was not observed)
| Ru | Co | Fe | Ni | |
|---|---|---|---|---|
| Ligand exchange reaction | ✓ | ✓ | ✓ | ✓ |
| Ligand addition reaction | ✓ | ✓ | ✗ | ✗ |
| Au28PET21 | ✗ | ✓ | ✗ | ✗ |
The reactions were also studied at a 1:4 NC: complex ratio (Fig. 3). Compared with the reactions at 1:2 ratio, Fig. 3a (reaction with Ru complex) and Fig. 3b (reaction with Co complex) show that a higher amount of metal complex is more favorable to induce the ligand addition on Au25PET18, forming Au25SR19. Noticeably, peak 3 in Fig. 3a and b after 24 h reaction, located at m/z = 4034.5 and m/z = 4013.4, are still assigned to Au25SR19, [Au25PET18(Ru complex)1]2+ and [Au25PET18(Co complex)1]2+, respectively. Moreover, peak 8 in Fig. 3b appearing at m/z = 4014.6 (z = 2+) indicates a new NC species corresponding to the addition of one Co complex to Au28PET21, [Au28PET21(Co complex)1]2+, also demonstrating the ligand addition reaction for another NC. Concerning LER with Fe and Ni complex at 1:4 ratio, ligand-exchange species Au25PET17(Metal complex)1 is still the main product (shown in Fig. 3c and d). Peak 3 in Fig. 3d at m/z = 4013.3 confirmed that a small amount of Au25SR19, [Au25PET18(Ni complex)1]2+, formed during the reaction with Ni complex at high ratio.
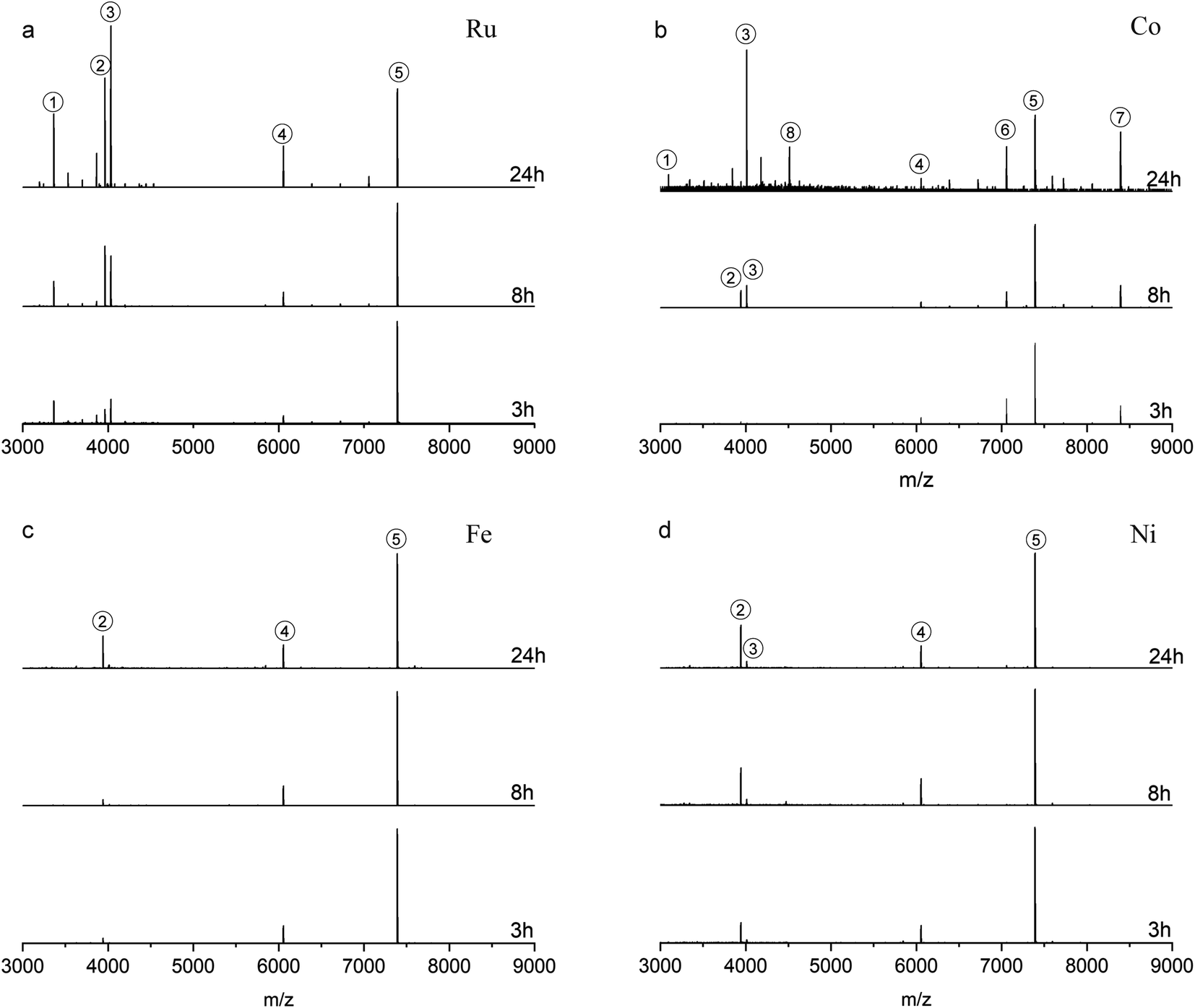 | ||
| Fig. 3 ESI-MS after reaction with Ru complex (a), Co complex (b), Fe complex (c), Ni complex (d) at NCs/complex ratio 1:4 (the spectra are normalized with respect to peak 5). | ||
Since the Ru complex can induce both ligand exchange and ligand addition reactions, it was selected to further investigate the ligand addition process during LER at a 1:10 NC: complex ratio. HRESI-MS (ESI, Fig. S4†) indicates that most of the initial Au25PET18 was converted to Au25SR19 (peak 1 at 3366.1 and peak 3 at 4034.5) after 24 h, demonstrating that excess ligand is beneficial for the formation of Au25SR19, and the addition of a second complex was not observed.
In order to make sure that the ESI-MS signal of Au25SR19 is not due to non-covalent interaction (π–π stacking) between the surface ligand of Au25PET18 and was prepared and mixed with Au25PET18. After reaction for 24 h, the sample was analyzed by HRESI-MS (ESI, Fig. S5†). The ESI mass spectra do not show the peak at m/z = 3959.8 which is assigned to the ligand addition. This indicates that the ligand is added through covalent bonding and not through π–π interaction.
Table 2 summarizes the proposed species and their corresponding m/z peaks based on the results in Fig. 2 and 3. Ru and Co complexes favor ligand addition (peak 3) reaction. The Co complex can also induce the transformation from Au25 to Au28 (peak 7) during LER. Nevertheless, with Fe and Ni complexes, ligand exchange (peak 2) is the preferred reaction.
| Species | m/z |
|---|---|
|
(z = 2+), M = Ru: 4034.5, M = Co: 4013.4, peak 3 |
|
(z = 2+), M = Ru: 3965.9, M = Co: 3944.8, M = Fe: 3943.2, M = Ni: 3944.7, peak 2 |

|
(z = 2+), 8396.8, peak 7 |
| Ligand addition-Au4PET4 | (z = 2+), M = Ru: 3366.1, M = Co: 3344.8, peak 1 |
A change of the ligand number while keeping the number of gold atoms constant will unavoidably affect the structure of the ligand shell and thus the properties of the whole NC. To study the photophysical properties, Au25SR19 [SR19 = PET18(Ru complex)1] was prepared at a NC/Ru complex ratio of 1:10 and purified by silica column chromatography with eluents dichloromethane and acetonitrile (3:1). HRESI-MS indicated good purity (Fig. 4a). The isotope patterns located at m/z = 4034.5 and m/z = 3366.1 (Fig. 4b) match perfectly with simulated spectra of Au25SR19 and its fragment after losing Au4PET4, which confirmed the molecular formula of Au25PET18(Ru complex)1. Fig. 4c shows some fragment peaks from Au25SR19 and the isotope patterns. The peak at m/z = 406.0 originates from the fragment PET + Ru complex ([C8H9S-SC36H25N6Ru]2+). In addition, the peak located at m/z = 937.1 is assigned to [Au4PET3(Ru complex)1]2+. Comparing the simulated isotopic patterns of Au25SR19 for covalent and noncovalent attachment of the metal complex with the experimental mass spectra (ESI, Fig. S10†) shows that the simulated isotopic patterns for the covalent attachment matches well the experiment, thus indicating a covalent binding of the metal complex to the cluster. The observed Au4SR4 fragment in ESI-MS may originate from staples (SR-Au-SR-Au-SR) of the NC e.g. from multistep rearrangement of two or more staples on the surface of the NC prior to fragmentation.40,41 A possible structure of Au25SR19 was already proposed by Xie et al., which is characterized by the removal of one Au atom from the Au13 core and the formation of a Au3SR4 staple.37
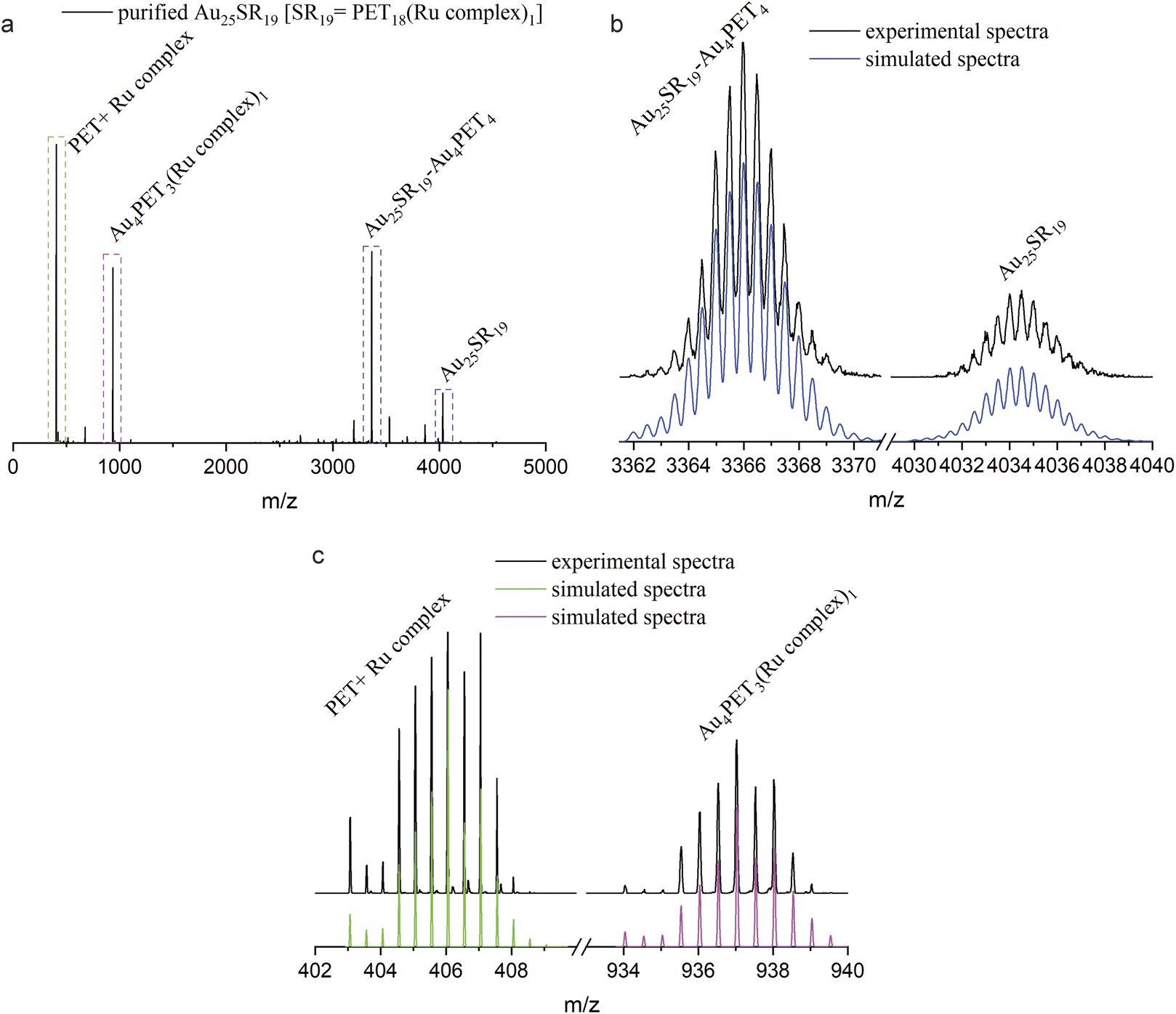 | ||
| Fig. 4 (a) ESI-MS of Au25SR19 [SR19 = PET18(Ru complex)1] after purification; (b) and (c): detailed isotopic peaks (in the square in a) and simulated isotopic patterns of labeled species, peak distance = 0.5. | ||
The molar decadic extinction coefficient and differential luminescence quantum yield for Au25PET18, Au25PET17(Ru complex)1, and Au25SR19 [SR19 = PET18(Ru complex)1] are studied in Fig. 5a and b, respectively. As shown in Fig. 5a, the characteristic peaks from Au25SR18 still remained in the ligand-exchange product [Au25PET17(Ru complex)1], but disappeared after ligand addition [Au25PET18(Ru complex)1]. The intense peak at around 484.5 nm (2.56 eV) due to the Ru complex appeared in both Au25PET17(Ru complex)1 and Au25PET18(Ru complex)1, confirming that the Ru complex was associated with the NC.
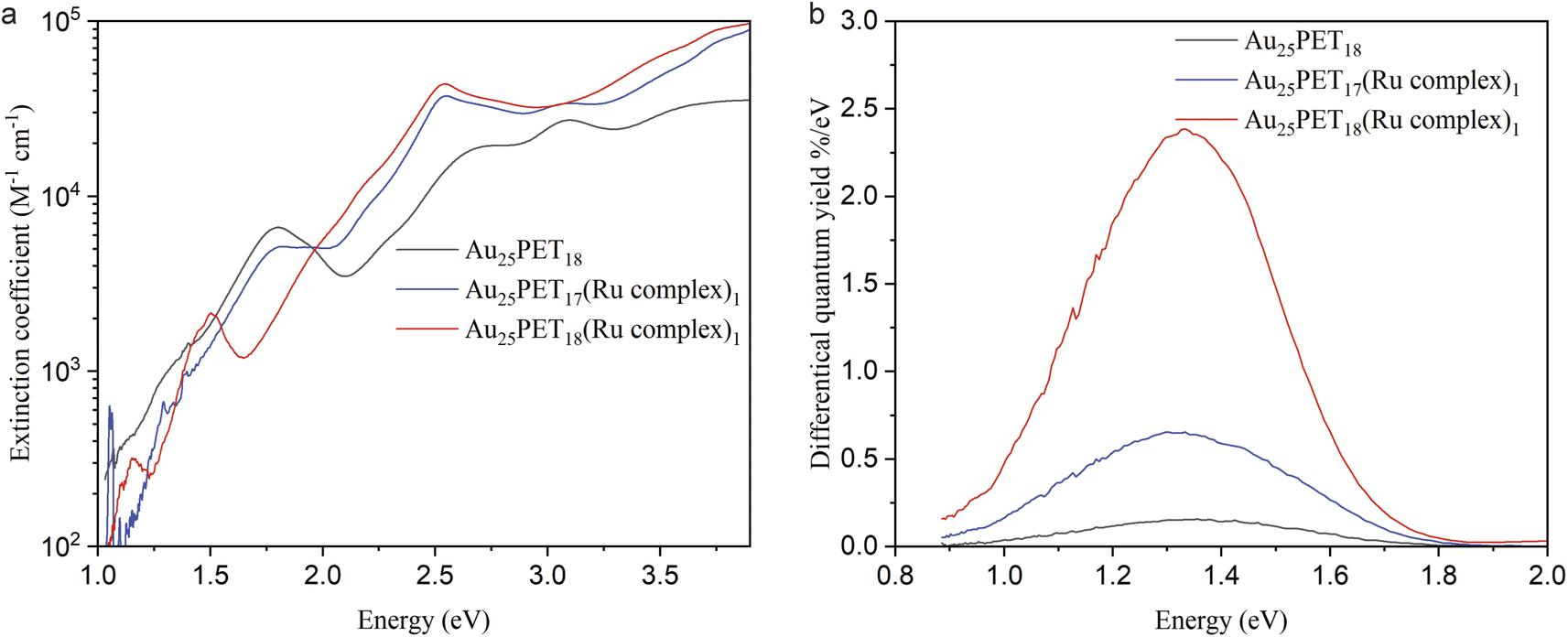 | ||
| Fig. 5 (a) The extinction coefficient for Au25PET18, Au25PET17(Ru complex)1, and Au25SR19 [SR19 = PET18(Ru complex)1]; (b) spectra of the differential luminescence quantum yield for Au25PET18, Au25PET17(Ru complex)1 and Au25PET18(Ru complex)1. | ||
The extinction coefficient for Au25PET17(Ru complex)1 and Au25PET18(Ru complex)1 after subtraction of the contribution from the Ru complex are shown in ESI, Fig. S11.† Au25PET17(Ru complex)1 exhibited very similar features compared with parent Au25PET18, while the spectrum of Au25PET18(Ru complex)1 completely changed, indicating that the electronic structure of Au25PET18(Ru complex)1 changed drastically compared to Au25PET18.
Interestingly, the luminescence spectra show very similar shape and peak position (Fig. 5b, ESI, Fig. S12† also shows emission spectra after normalization) for all three species Au25PET18, Au25PET17(Ru complex)1 and Au25PET18(Ru complex)1. The emission peak for pure Ru complex appears at 670 nm (1.85 eV, ESI, Fig. S12†). However, after binding to Au25 NCs, quenching of the chromophore fluorescence is observed, which can be due to energy transfer from the metal complex to the NC.28 The luminescence peak at 1.33 eV for Au25PET18, Au25PET17(Ru complex)1 and Au25PET18(Ru complex)1 probably stems from the core of the Au NCs, indicating that the core is not changing after ligand addition. However, Au25PET18(Ru complex)1 shows a drastic enhancement of quantum yield (Fig. 5b). Table 3 summarizes the luminescence quantum yields for these NCs. An increase of the luminescence quantum yield is observed for Au25PET18(Ru complex)1 compared to the initial Au25PET18. All samples exhibit a violation of Kasha's rule, as the luminescence shows up at higher energy than the lowest energy absorption band.
| Quantum yield | |
|---|---|
| Au25PET18 | 0.075% |
| Au PET17(Ru complex)1 | 0.32% |
| Au25PET18(Ru complex)1 | 1.05% |
To further investigate the mechanism for the ligand addition reaction, unreacted Au25PET18 was separated from the reaction mixture, as shown in Fig. 6a, and studied. Fig. 6b shows UV-vis spectra of the top and bottom parts of fraction 1. According to these spectra, the bottom part can be assigned to neutral Au25PET18 (written as [Au25PET18]0), but in the UV-vis spectra of the top part, both peaks at around 400 nm and 700 nm were bleached, which indicates positively charged Au25PET18 (written as [Au25PET18]+).42 ESI-MS (Fig. 6c) was also applied to confirm the unreacted Au25PET18 of fraction 1 (top part). Since neutral Au25PET18 was used as the starting point for all ligand exchange reactions, however, both neutral and positively charged Au25PET18 were observed after ligand addition, we propose that [Au25PET18]+ should be the intermediate in the ligand addition reaction. Noteworthily, neutral Au25PET18 cannot be oxidized to [Au25PET18]+ on the silica column. Therefore, the transformation process from Au25SR18 to Au25SR19 is proposed as below: [Au25PET18]0 is oxidized to [Au25PET18]+, then [Au25PET18]+ NCs react with an external thiolate ligand to form a new isoelectric species, [Au25SR19]0, realizing the addition of one thiolate ligand to the original NC. The transformation mechanism is also consistent with the oxidization process proposed by Xie et al.37 In our work, the ligand addition reaction is dependent on the metal cations in the complexes, which indicates that metals play an important role in the process of ligand addition. We hypothesize that Ru and Co complexes are able to oxidize neutral Au25PET18 NCs to [Au25PET18]+ (in contrast to Fe and Ni complexes), so that the ligand addition reaction can take place easily. Moreover, the reported reduction potentials for the addition of the first electron to [M(tpy)2]2+ (M = Co, Ni, Fe, Ru; tpy = 2,2′:6′,2′′-terpyridine)43,44 indicate the following oxidant power of [M(tpy)2]2+: [Co(tpy)2]2+ > [Ru(tpy)2]2+ >[Fe(tpy)2]2+ >[Ni(tpy)2]2+. This observation is also consistent with our hypothesis that Co and Ru complexes have stronger tendency to oxidize the NC.
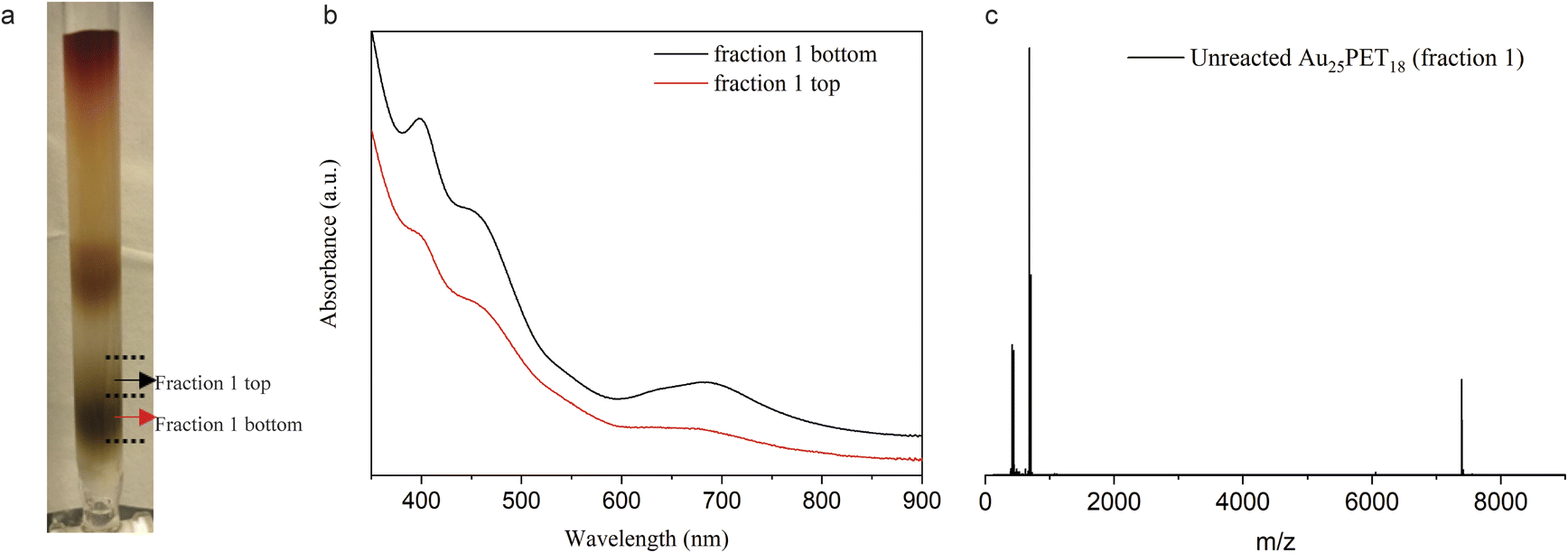 | ||
| Fig. 6 (a) Photograph of silica column after LER of Au25PET18 with Ru complex for 24 h at NC/complex ratio 1:4, fraction 2 contains both ligand exchange and ligand addition products; (b) UV-vis spectra of fraction 1 (top part and bottom part, as indicated in a); (c) ESI-MS of fraction 1 top part. | ||
Furthermore, we found that only one ligand at the surface could be replaced by metal complexes when neutral [Au25PET18]0 was used for LERs. In order to shed light on this interesting phenomenon, we performed LER starting from negatively charged Au25PET18 (written as [Au25PET18]−1) at a NC/complex ratio of 1:2. Fig. 7 shows HRESI-MS and detailed isotopic patterns after mixing either [Au25PET18]0 or [Au25PET18]−1 with the Ru complex and reacting for 24 h. As shown in Fig. 7a and b, products related with one ligand exchange (peak 2 and right part of peak 3) and addition of only one ligand (left part of peak 3) were observed when LER started form [Au25PET18]0. Interestingly, when [Au25PET18]−1 was used for LER with the Ru complex, the product of two ligand exchanges, Au25PET16(Ru complex)2, (peak X in Fig. 7c and d), was observed in addition to species resulting from one ligand exchange and ligand addition, respectively. The emerging peak X at m/z = 2117 (charge z = 4+) is assigned to [Au25PET16(Ru complex)2]4+ (molecular formula: (Au25C200H194N12S18Ru2)4+). The measured and simulated isotope patterns shown in Fig. 7b match very well. Another prominent peak at m/z = 2643.9 matches a NC with one ligand exchanged but with z = 3+, [Au25PET17(Ru complex)1]3+. Based on these interesting results, we hypothesize that the number of the metal complexes that can be exchanged to the NCs is strongly related to the charge of parent NCs. The Au25SR18 NC may be oxidized during LER with Ru complex, and easily accessible charge states for Au25SR18 are −1, 0, +1. Therefore, only one ligand can be substituted by metal complex on initially neutral Au25PET18, but two on initially negatively charged [Au25PET18]−. This could provide a promising method to precisely control the ligand-exchange number at the surface of NCs.
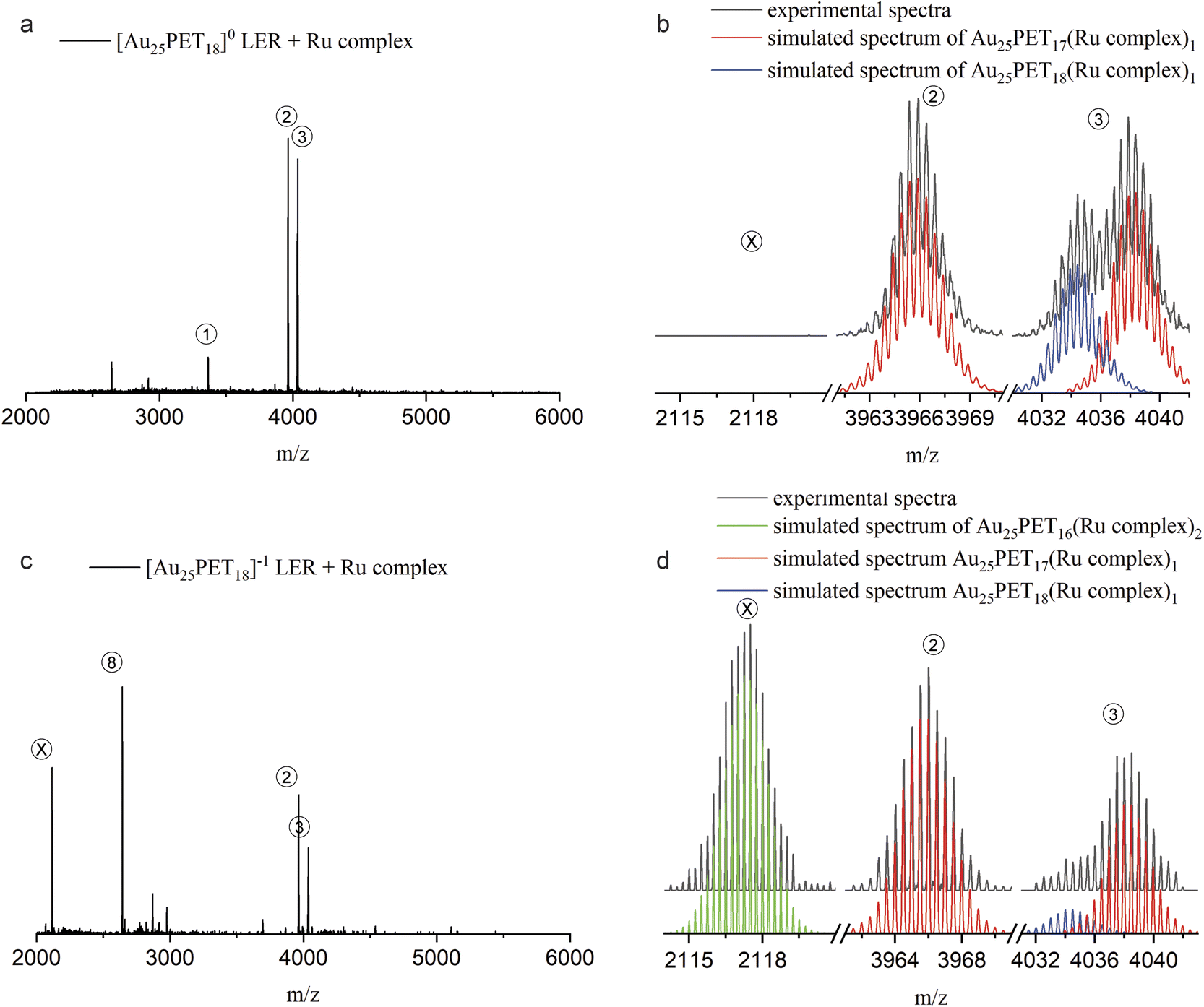 | ||
| Fig. 7 ESI-MS after LER started from neutral Au25PET18 (a and b) and negatively charged Au25PET18 (c and d) with Ru complex at NC/complex ratio 1:2 for 24 h. | ||
Conclusions
In conclusion, Au25SR19 was successfully obtained by precisely adding one thiolated terpyridine-metal complex to Au25PET18 NCs during ligand exchange reaction at mild conditions. Moreover, we demonstrated that the reaction is metal dependent. Ru and Co complexes were favorable for ligand addition leading to Au25SR19, whereas Fe and Ni complexes promoted the exchange of one ligand. Also, NC/complex ratio and reaction time were studied, revealing that more metal complex and longer reaction time were advantageous for ligand addition on Au25. Therefore, the ligand addition process can be easily and precisely controlled. HRESI-MS also gave some hints concerning the structure of Au25SR19. The new NC showed different electronic structure and strongly enhanced luminescence compared with pristine Au25PET18. The manipulation of the number of ligands at a given Au NCs provides additional possibilities for the diversification of NCs and to change their physiochemical properties, thus boosting the application of Au NCs. More interestingly, only one ligand can be replaced on neutral Au25PET18 but two on negatively charged Au25PET18 during LER with metal complexes, which provides us with a novel methodology to precisely control the ligand-exchange number at the surface of NCs.Experimental section shown in ESI.†
Data availability
The original data leading to this publication are available at https://doi.org/10.5281/zenodo.8079142.Author contributions
T. B. and A. Z. conceived the idea of the research. J. Z. performed all of the experiments, analysed data and wrote the manuscript with the help of Y. W., A. Z. and T. B. The photophysical experiment was conducted with the help of A. R. All authors contributed to editing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. B. acknowledges the generous support of the Swiss National Science Foundation (grant 200020_192232) and the University of Geneva. The authors acknowledge the Mass Spectrometry core facility (MZ ChemBio). We thank Luis Llanes Montesino for help with the photoluminescence experiments. J. Z. would like to thank China Scholarship Council (CSC201807040048) for the financial supporting.Notes and references
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- A. W. Cook and T. W. Hayton, Acc. Chem. Res., 2018, 51, 2456–2464 CrossRef CAS PubMed.
- Q. Yao, X. Yuan, V. Fung, Y. Yu, D. T. Leong, D.-e. Jiang and J. Xie, Nat. Commun., 2017, 8, 927 CrossRef PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- Y. Su, T. Xue, Y. Liu, J. Qi, R. Jin and Z. Lin, Nano Res., 2019, 12, 1251–1265 CrossRef CAS.
- J. Yang, Y. Peng, S. Li, J. Mu, Z. Huang, J. Ma, Z. Shi and Q. Jia, Coord. Chem. Rev., 2022, 456, 214391 CrossRef CAS.
- X. Cui, J. Wang, B. Liu, S. Ling, R. Long and Y. Xiong, J. Am. Chem. Soc., 2018, 140, 16514–16520 CrossRef CAS PubMed.
- S. Li, A. V. Nagarajan, D. R. Alfonso, M. Sun, D. R. Kauffman, G. Mpourmpakis and R. Jin, Angew. Chem., 2021, 133, 6421–6426 CrossRef.
- L. Shang, J. Xu and G. U. Nienhaus, Nano Today, 2019, 28, 100767 CrossRef.
- J. Sun and Y. Jin, J. Mater. Chem. C, 2014, 2, 8000–8011 RSC.
- Y. Cao, T. Chen, Q. Yao and J. Xie, Acc. Chem. Res., 2021, 54, 4142–4153 CrossRef CAS PubMed.
- B. Kumar, T. Kawawaki, N. Shimizu, Y. Imai, D. Suzuki, S. Hossain, L. V. Nair and Y. Negishi, Nanoscale, 2020, 12, 9969–9979 RSC.
- Q. Yao, T. Chen, X. Yuan and J. Xie, Acc. Chem. Res., 2018, 51, 1338–1348 CrossRef CAS PubMed.
- Z. Gan, J. Chen, J. Wang, C. Wang, M.-B. Li, C. Yao, S. Zhuang, A. Xu, L. Li and Z. Wu, Nat. Commun., 2017, 8, 14739 CrossRef CAS PubMed.
- C. Zeng, Y. Chen, A. Das and R. Jin, J. Phys. Chem. Lett., 2015, 6, 2976–2986 CrossRef CAS PubMed.
- Y. Niihori, Y. Kikuchi, A. Kato, M. Matsuzaki and Y. Negishi, ACS Nano, 2015, 9, 9347–9356 CrossRef CAS PubMed.
- H. Häkkinen, Nat. Chem., 2012, 4, 443–455 CrossRef PubMed.
- X. Yuan, B. Zhang, Z. Luo, Q. Yao, D. T. Leong, N. Yan and J. Xie, Angew. Chem., 2014, 126, 4711–4715 CrossRef.
- T. Bürgi, Nanoscale, 2015, 7, 15553–15567 RSC.
- Y. Ishida, K. Narita, T. Yonezawa and R. L. Whetten, J. Phys. Chem. Lett., 2016, 7, 3718–3722 CrossRef CAS PubMed.
- Z. Huang, Y. Ishida, K. Narita and T. Yonezawa, J. Phys. Chem. C, 2018, 122, 18142–18150 CrossRef CAS.
- C. L. Heinecke, T. W. Ni, S. Malola, V. Mäkinen, O. A. Wong, H. Häkkinen and C. J. Ackerson, J. Am. Chem. Soc., 2012, 134, 13316–13322 CrossRef CAS PubMed.
- Y. Wang and T. Bürgi, Nanoscale Adv., 2021, 3, 2710–2727 RSC.
- A. Dominguez-Castro, C. R. Lien-Medrano, K. Maghrebi, S. Messaoudi, T. Frauenheim and A. Fihey, Nanoscale, 2021, 13, 6786–6797 RSC.
- E. Hulkko, T. Lahtinen, V. Marjomaki, E. Pohjolainen, V. Saarnio, K. Sokolowska, A. Ajitha, M. Kuisma, L. Lehtovaara, G. Groenhof, H. Hakkinen and M. Pettersson, Nanoscale Adv., 2021, 3, 6649–6658 RSC.
- K. Pyo, N. H. Ly, S. M. Han, M. B. Hatshan, A. Abuhagr, G. Wiederrecht, S. W. Joo, G. Ramakrishna and D. Lee, J. Phys. Chem. Lett., 2018, 9, 5303–5310 CrossRef CAS PubMed.
- R. K. Pandey, C. Chakraborty, U. Rana, S. Moriyama and M. Higuchi, J. Mater. Chem. A, 2016, 4, 4398–4401 RSC.
- B. Varnholt, R. Letrun, J. J. Bergkamp, Y. Fu, O. Yushchenko, S. Decurtins, E. Vauthey, S. X. Liu and T. Burgi, Phys. Chem. Chem. Phys., 2015, 17, 14788–14795 RSC.
- M. S. Devadas, K. Kwak, J.-W. Park, J.-H. Choi, C.-H. Jun, E. Sinn, G. Ramakrishna and D. Lee, J. Phys. Chem. Lett., 2010, 1, 1497–1503 CrossRef CAS.
- W. Suzuki, R. Takahata, Y. Chiga, S. Kikkawa, S. Yamazoe, Y. Mizuhata, N. Tokitoh and T. Teranishi, J. Am. Chem. Soc., 2022, 144, 12310–12320 CrossRef CAS PubMed.
- G. H. Woehrle, L. O. Brown and J. E. Hutchison, J. Am. Chem. Soc., 2005, 127, 2172–2183 CrossRef CAS PubMed.
- C. Zeng, C. Liu, Y. Pei and R. Jin, ACS Nano, 2013, 7, 6138–6145 CrossRef CAS PubMed.
- M. P. Maman, A. S. Nair, H. Cheraparambil, B. Pathak and S. Mandal, J. Phys. Chem. Lett., 2020, 11, 1781–1788 CrossRef CAS PubMed.
- C. Zeng, T. Li, A. Das, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2013, 135, 10011–10013 CrossRef CAS PubMed.
- X. Kang and M. Zhu, Chem. Mater., 2019, 31, 9939–9969 CrossRef CAS.
- Y. Wang, B. Nieto-Ortega and T. Bürgi, Chem. Commun., 2019, 55, 14914–14917 RSC.
- Y. Cao, V. Fung, Q. Yao, T. Chen, S. Zang, D.-e. Jiang and J. Xie, Nat. Commun., 2020, 11, 5498 CrossRef CAS.
- X. Kang, H. Chong and M. Zhu, Nanoscale, 2018, 10, 10758–10834 RSC.
- C. Comby-Zerbino, F. Bertorelle, P. Dugourd, R. Antoine and F. Chirot, J. Phys. Chem. A, 2020, 124, 5840–5848 CrossRef CAS.
- R. Hamouda, F. Bertorelle, D. Rayane, R. Antoine, M. Broyer and P. Dugourd, Int. J. Mass Spectrom., 2013, 335, 1–6 CrossRef CAS.
- L. A. Angel, L. T. Majors, A. C. Dharmaratne and A. Dass, ACS Nano, 2010, 4, 4691–4700 CrossRef CAS.
- D. R. Kauffman, D. Alfonso, C. Matranga, P. Ohodnicki, X. Deng, R. C. Siva, C. Zeng and R. Jin, Chem. Sci., 2014, 5, 3151–3157 RSC.
- D. Ghosh, T. Kajiwara, S. Kitagawa and K. Tanaka, Eur. J. Inorg. Chem., 2020, 2020, 1814–1818 CrossRef CAS.
- N. Elgrishi, M. B. Chambers, V. Artero and M. Fontecave, Phys. Chem. Chem. Phys., 2014, 16, 13635–13644 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc01177a |
| This journal is © The Royal Society of Chemistry 2023 |