
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible-light-induced organocatalytic enantioselective N–H insertion of α-diazoesters enabled by indirect free carbene capture†
Wengang
Guo
 *a,
Ying
Zhou
a,
Hongling
Xie
a,
Xin
Yue
a,
Feng
Jiang
a,
Hai
Huang
a,
Zhengyu
Han
a and
Jianwei
Sun
*abc
*a,
Ying
Zhou
a,
Hongling
Xie
a,
Xin
Yue
a,
Feng
Jiang
a,
Hai
Huang
a,
Zhengyu
Han
a and
Jianwei
Sun
*abc
aAdvanced Catalysis and Green Manufacturing Collaborative Innovation Center, School of Petrochemical Engineering, Changzhou University, Changzhou, 213164, China. E-mail: sunjw@ust.hk; wgguo@cczu.edu.cn
bDepartment of Chemistry, The Hong Kong Branch of Chinese National Engineering Research Centre for Tissue Restoration & Reconstruction, The Hong Kong University of Science and Technology (HKUST), Clear Water Bay, Kowloon, Hong Kong SAR, China
cShenzhen Research Institute, HKUST, No. 9 Yuexing 1st Rd, Shenzhen 518057, China
First published on 29th November 2022
Abstract
While asymmetric insertion of metal carbenes into H–X (X = C, N, O, etc.) bonds has been well-established, asymmetric control over free carbenes is challenging due to the presence of strong background reactions and lack of any anchor for a catalyst interaction. Here we have achieved the first photo-induced metal-free asymmetric H–X bond insertion of this type. With visible light used as a promoter and a chiral phosphoric acid used as a catalyst, α-diazoesters and aryl amines underwent smooth N–H bond insertion to form enantioenriched α-aminoesters with high efficiency and good enantioselectivity under mild conditions. Key to the success was the use of DMSO as an additive, which served to rapidly capture the highly reactive free carbene intermediate to form a domesticated sulfoxonium ylide.
Introduction
Insertion of a carbene into an H–X bond (X = C, N, O, etc.) is a well-known reaction that provides expedient access to functionalized organic molecules.1,2 However, free carbenes, conventionally generated from diazo compounds under hash conditions (e.g., high temperature, UV irradiation), are highly reactive and hard to control regarding chemo- and stereoselectivity and functional group compatibility (Scheme 1a).1,3 In contrast, taming their reactivity by combining them with metals to form metal carbenes has allowed these insertion reactions to proceed not only under mild conditions with good functional group tolerance, but also with straightforward enantiocontrol via metal–ligand interactions (Scheme 1b).2 Recently, free carbenes have been generated under mild conditions by deploying visible light irradiation of the corresponding diazo compounds, which allowed a wide range of insertion processes to take place without the aid of a metal catalyst.4–6 However, the leap toward a mild metal-free catalytic enantioselective insertion of a photo-generated free carbene into an H–X bond has yet to be achieved (Scheme 1c).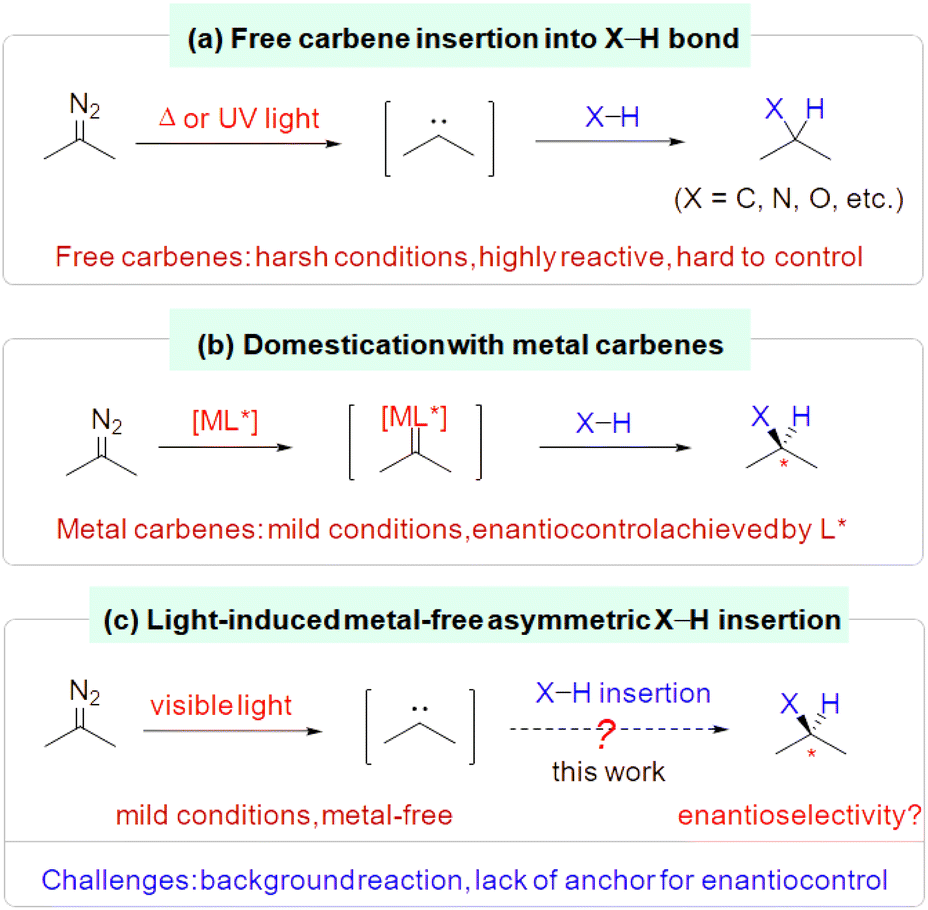 | ||
| Scheme 1 Introduction to insertion of carbenes into H–X bonds. | ||
The key to tackling this formidable challenge is thought to involve finding a way (1) to outcompete or inhibit the facile racemic background reaction once the free carbene is generated and (2) to enable a metal-free chiral inducer to interact with the reaction partners to achieve effective enantiocontrol. Typically, free carbenes and the H–X bond insertion partners (such as amines, alcohols) either lack obvious anchors for catalyst interaction or have flexible conformations that make it difficult to achieve asymmetric induction.1–3 Moreover, many chiral organocatalysts have polar H–X bonds that may not be compatible with free carbenes.7,8 In this context, here we report our progress using an indirect approach.
Results and discussion
Reaction development
We started our preliminary study with α-diazoester 1a as the carbene precursor and para-methoxy aniline as the reaction partner, aiming to achieve an asymmetric N–H insertion reaction (Scheme 2).9,10 Initially, we evaluated some typical chiral organocatalysts, including the cinchona alkaloid quinine, thiourea, urea, and squaramide. With blue light-emitting diode (LED) irradiation, these reactions proceeded smoothly at room temperature to form the desired N–H insertion product 3a in excellent yields, but unfortunately, all in racemic forms (see Fig. S1 in ESI† for details). Next, we examined a few chiral phosphoric acids (CPAs, Scheme 2).11 To our delight, these reactions showed some enantioselectivity. Of the CPAs tested, CPA E3 led to the best enantioselectivity (39.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :60.5 e.r.). Notably, the reaction did not proceed without light. Further considerable efforts to screen other conditions, including solvents and temperature, did not improve the outcome.
:60.5 e.r.). Notably, the reaction did not proceed without light. Further considerable efforts to screen other conditions, including solvents and temperature, did not improve the outcome.
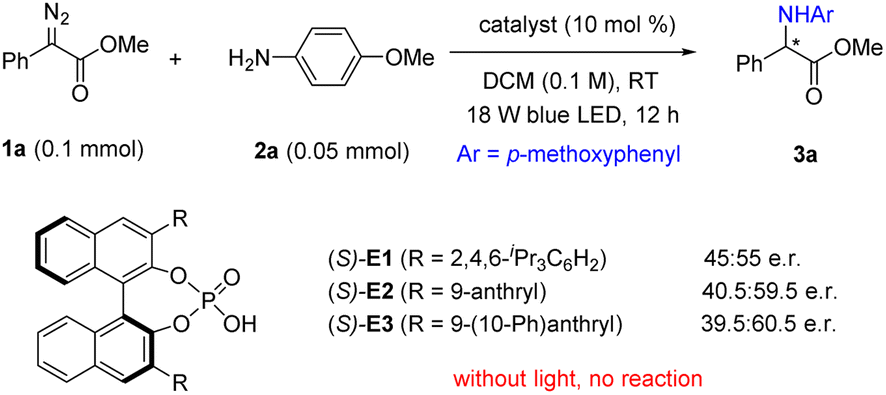 | ||
| Scheme 2 Preliminary results. | ||
The low enantioselectivity was believed to be primarily due to the facile background reactions of the free carbene. We hypothesized that the enantioselectivity would be improved if the free carbene, once generated, could be immediately captured by an additive Y to in situ generate a less-reactive intermediary carbenoid species (e.g., ylide),12 which would react with the amine in a more controlled manner (Scheme 3). However, several requirements need to be fulfilled in this design. First, the additive should be strong enough to outcompete the amine so that it is able to alter the normal reactivity (k2 ≫ k1) and thus shut down any background reaction. In addition, this intermediary carbenoid should have sufficient reactivity toward the amine partner in the presence of the organocatalyst, which in the meantime should be able to control the enantioselectivity during N–H bond insertion. Furthermore, the additive should not contain labile groups that can react with the free carbene to form side products (e.g., rearrangement products).13 Finally, with its likely use in a stoichiometric amount, the additive should be compatible with the organocatalyst and not affect the subsequent stereocontrol. Overall, these requirements have apparently made this design quite difficult to implement.
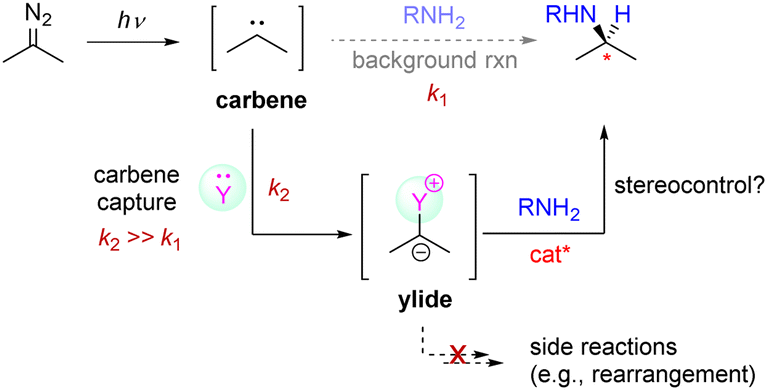 | ||
| Scheme 3 Design strategy involving carrying out carbene capture with an additive in order to inhibit any background reaction from occurring. | ||
We hypothesized that the lone pair on sulfur, phosphorus, or halogen might be able to trap the free carbene, and the resulting ylides from these species could be both stable and reactive.12 In particular, sulfur ylides have been used for asymmetric H–X bond insertion reactions.14,15 Thus, we examined PPh3, halobenzenes and sulfides, but they did not lead to any obvious improvement.16 However, use of DMSO resulted in a dramatic increase in enantioselectivity (to 78:22 e.r.). Other sulfoxides, including cyclic and aryl-substituted ones, were inferior (Fig. 1).
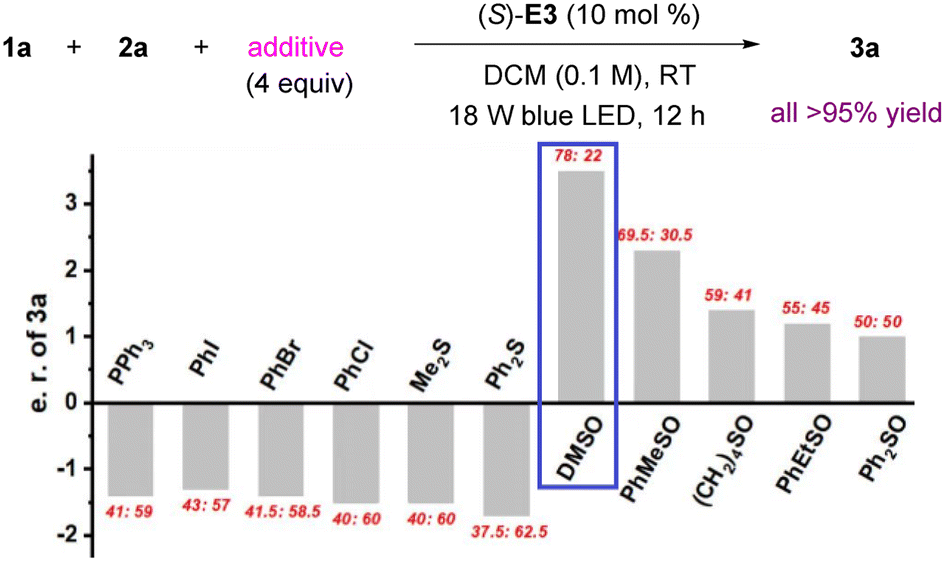 | ||
| Fig. 1 Effect of additives. | ||
With DMSO, we next varied other parameters, including solvent, temperature, and concentration as well as the ester group used. Unfortunately, no improvement was obtained. We then turned our attention to any effect of the amount of DMSO on enantioselectivity. Since its main function in this reaction was to trap the free carbene, increasing its loading was thought to perhaps facilitate carbene capture, thus diminishing any racemic background reaction. However, DMSO is not a typical solvent for organocatalysis, as its high polarity may interrupt the catalyst–substrate interaction. Our initial use of four equivalents of DMSO (Fig. 1) was arbitrary and may not have been optimal due to the above two factors that lead to opposite effects to stereocontrol. Thus, we systematically evaluated the influence of DMSO loading (Fig. 2). As the DMSO loading was increased, the enantioselectivity initially increased significantly, but then gradually decreased. The highest enantioselectivity was obtained with 32 equivalents of DMSO (90:10 e.r.). Notably, decreasing the temperature or using LEDs with different wavelengths did not improve the enantioselectivity (see Table S2† for details).
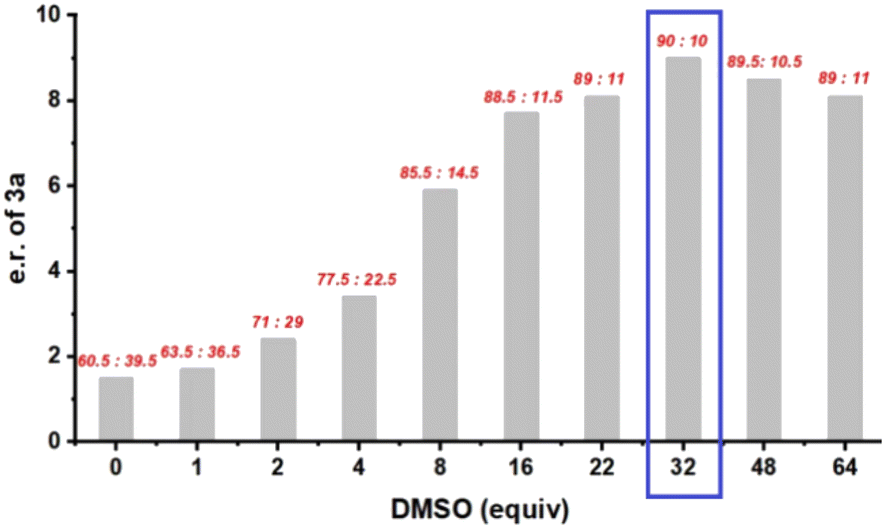 | ||
| Fig. 2 Effect of DMSO loading. | ||
With the above optimized conditions, we next investigated the generality of this mild organocatalytic asymmetric carbene insertion protocol (Scheme 4). A wide range of α-aryl diazoacetates bearing different substituents reacted smoothly with para-methoxyaniline to give the desired α-aryl glycines in excellent yields with good enantioselectivities. The incorporation of a thiophene heterocycle at the α-position (3h) did not affect the reactivity. Various arylamines were also amenable to this protocol. Unfortunately, aliphatic amines were not compatible, as use of these compounds led to racemic products. This result can be explained by a strong background reaction occurring with the more basic aliphatic amines, and their high basicity levels may have also deactivated the CPA catalyst. Late-stage functionalization of some natural product derivatives was also feasible with this protocol (3q–3s). Some potentially reactive functional groups were well tolerated, and the desired products were all formed with good efficiency and stereocontrol. Note that substrate-directed stereocontrol of these molecules under metal-catalyzed or catalyst-free conditions have typically been difficult to achieve.5j Finally, synthesis of 3a on a scale of 2 mmol was equally efficient.
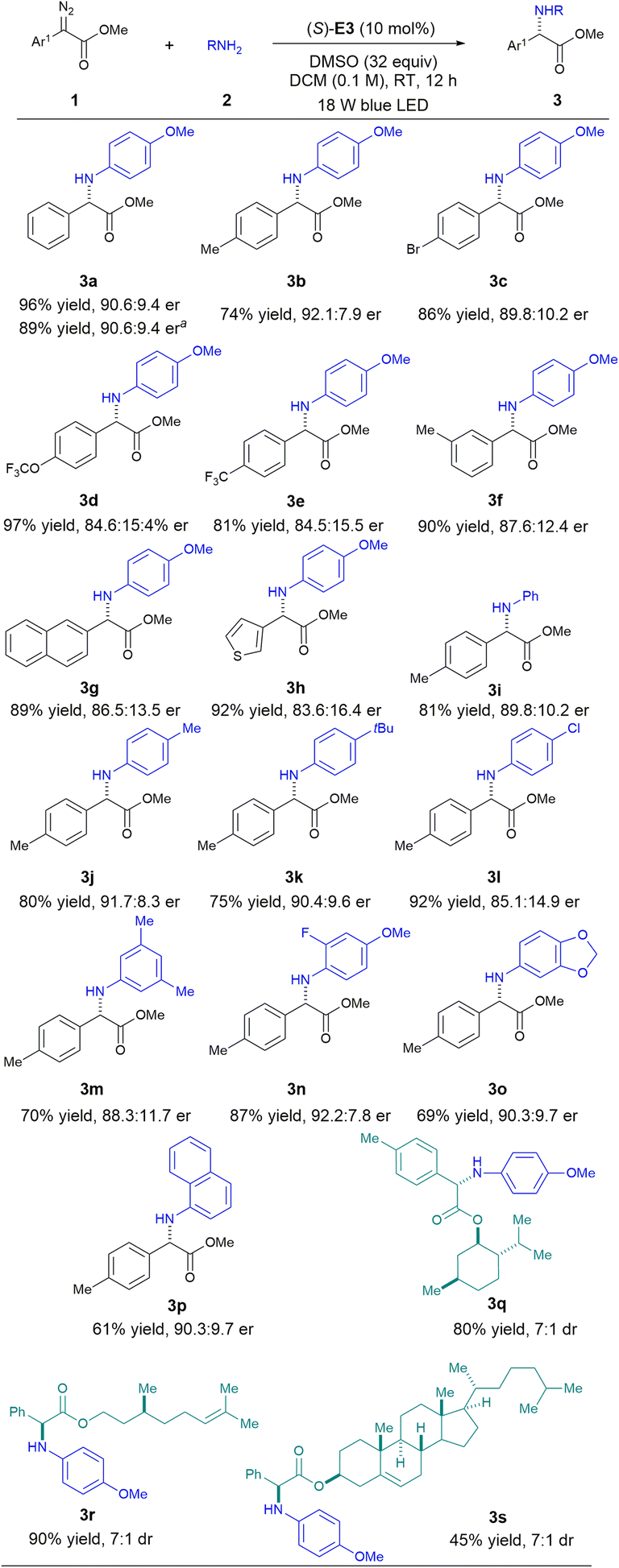 | ||
| Scheme 4 Reaction scope: 1 (0.2 mmol), 2 (0.1 mmol), catalyst (0.01 mmol), DMSO (3.2 mmol), DCM (1.0 mL). aRun at a 2.0 mmol scale. | ||
To determine more about the reaction mechanism, particularly the role of DMSO, we carried out some control experiments (Scheme 5). In the absence of amine and (S)-E3, the free carbene generated from diazoester 1a was successfully trapped by DMSO to form the sulfoxonium ylide 4a in 75% yield (Scheme 5a).12b,c Without light, this step did not proceed, suggesting that light-induced free carbene generation was likely involved. Next, this ylide was treated with amine 2a and catalyst (S)-E3, which rapidly reacted to form 3a in 90% yield and 94.5:5.5 e.r. (Scheme 5b). These results confirmed the chemical and kinetic competence of the sulfoxonium ylide 4a and also supported our design, shown in Scheme 2. We were also interested in probing any possible role of light in the C–N bond-forming step. Indeed, the reaction of 4a with 2a was very slow with the same light irradiation (Scheme 5c). This result suggested a possible lack of a significant role played by light in the second step of the standard protocol. Instead, the CPA catalyst was considered to be essential for activation and enantiocontrol of the sulfoxonium ylides.15 Finally, TEMPO was added to the standard reaction of 1a with 2a; this addition had a very minor influence on the outcome (Scheme 5d), consistent with the involvement of singlet carbenes rather than of radicals or triplet carbenes in the reaction.6g
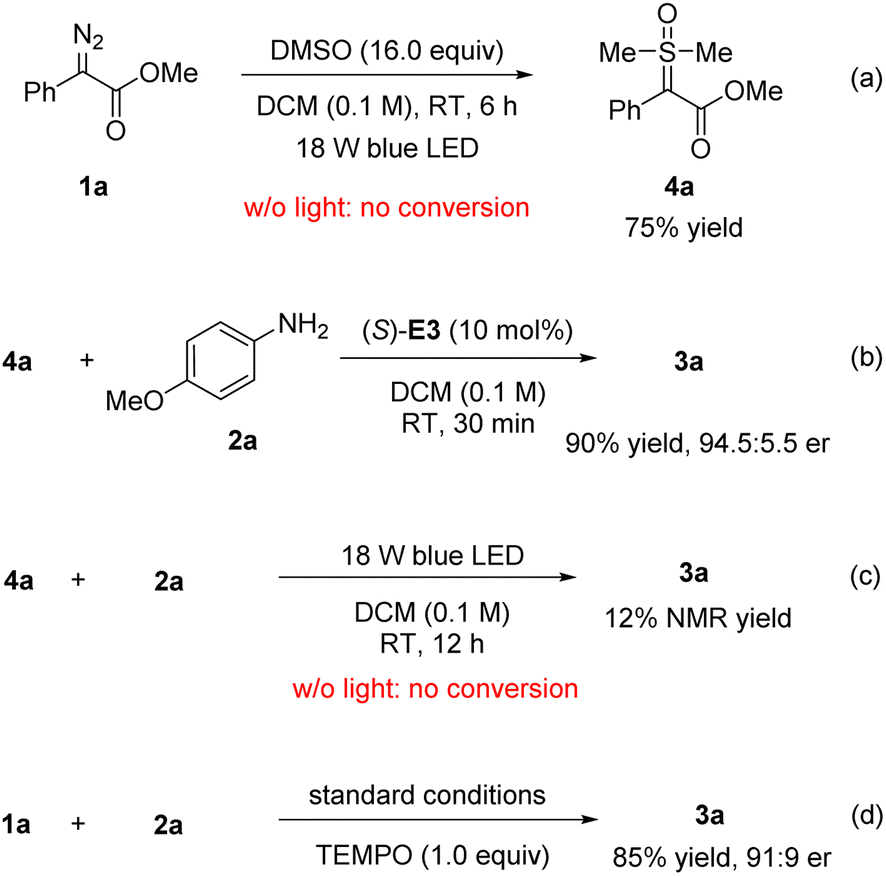 | ||
| Scheme 5 Control experiments. | ||
A possible mechanism is proposed in Scheme 6. The reaction begins with photo-initiated free carbene formation and capture by DMSO to form sulfoxonium ylide 4. Next, the chiral acid catalyst protonates the ylide to form sulfoxonium ions IM1 and IM2, which are diastereometers and rapidly interconvert with each other. And finally, according to the mechanism, the chiral anion then directs the amine to react preferentially with one of the two diastereomers to form the enantioenriched product 3via dynamic kinetic resolution.15b
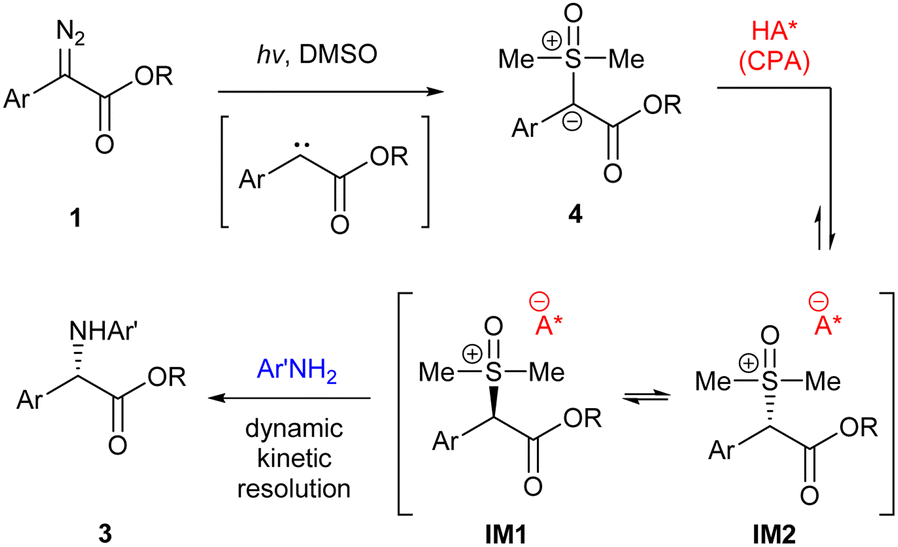 | ||
| Scheme 6 Proposed mechanism. | ||
Conclusions
In conclusion, we have developed a photo-induced metal-free enantioselective X–H insertion reaction of diazo compounds under mild conditions. Due to facile racemic background reactions and the lack of a robust anchor for catalyst interaction, the development of direct asymmetric insertion of free carbenes into X–H bonds remains a challenge, though highly desirable. Here, we have made progress indirectly in this regard by employing visible light as a promoter, a chiral phosphoric acid as a catalyst, and most importantly, a suitable additive. A range of α-diazoesters and arylamines successfully underwent one-pot asymmetric N–H insertion to form the desired enantioenriched α-aminoesters with high efficiency and good enantioselectivity. Key to the success was the use of a suitable amount of DMSO as an additive, which captured the free carbene in the form of a domesticated carbenoid, namely a sulfoxonium ylide, for the stereocontrolled N–H bond insertion. Control experiments provided solid evidence for the role of DMSO. The development of more direct and efficient metal-free asymmetric X–H insertion reactions of free carbenes is ongoing in our laboratory.Data availability
All experimental data is available within the article and the ESI.†Author contributions
W. Guo: conceptualization, methodology, investigation, data curation, and writing of the original draft. Y. Zhou, H. Xie, X. Yue, and F. Jiang: resources. H. Huang, and Z. Han: writing-review. J. Sun: conceptualization, supervision, writing-review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the Hong Kong Research Grants Council (16303420, 16309321), the Innovation and Technology Commission (ITC-CNERC14SC01), the Jiangsu Key Laboratory of Advanced Catalytic Materials and Technology (BM2012110), the Shenzhen Science and Technology Innovation Committee (JCYJ20200109141408054), and the start-up funding to W. G. by Changzhou University (ZMF21020030). We also thank the Analysis and Testing Center, NERC Biomass of Changzhou University for the assistance in NMR analysis. This work is dedicated to the 45th anniversary of the founding of Changzhou University.Notes and references
- (a) H. Zollinger, Diazo Chemistry II: Aliphatic, Inorganic and Organometallic Compounds, Wiley-VCH, New York, 1995 CrossRef; (b) R. A. Moss, M. S. Platz and M. Jones Jr, Reactive Intermediate Chemistry, Wiley-Interscience, New York, 2004 Search PubMed; (c) J. March, Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley-Interscience, New York, 6th edn, 2007 Search PubMed.
- Reviews on metal carbenes: (a) H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861–2903 CrossRef CAS; (b) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704–724 CrossRef CAS; (c) S.-F. Zhu and Q.-L. Zhou, Acc. Chem. Res., 2012, 45, 1365–1377 CrossRef CAS; (d) S.-F. Zhu and Q.-L. Zhou, Natl. Sci. Rev., 2014, 1, 580–603 CrossRef CAS; (e) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981–10080 CrossRef CAS; (f) N. J. Thumar, Q. H. Wei and W. H. Hu, in Advances in Organometallic Chemistry, ed. P. J. Perez, Academic Press, 2016, vol. 66, pp. 33–91 Search PubMed; (g) Y. Xia, D. Qiu and J. Wang, Chem. Rev., 2017, 117, 13810–13889 CrossRef CAS; (h) B. Ma, L. Liu and J. Zhang, Asian J. Org. Chem., 2018, 7, 2015–2025 CrossRef CAS.
- For a review and selected pioneering examples with free carbenes: (a) E. Buchner and T. Curtius, Ber. Dtsch. Chem. Ges., 1885, 18, 2377–2379 CrossRef; (b) W. v. E. Doering, R. G. Buttery, R. G. Laughlin and N. Chaudhuri, J. Am. Chem. Soc., 1956, 78, 3224 CrossRef CAS; (c) N. R. Candeias and C. A. M. Afonso, Curr. Org. Chem., 2009, 13, 763–787 CrossRef CAS.
- For reviews on recent developments of visible-light-induced reactions of diazo compounds: (a) L. W. Ciszewski, K. Rybicka-Jasinska and D. Gryko, Org. Biomol. Chem., 2019, 17, 432–448 RSC; (b) Z. Yang, M. L. Stivanin, I. D. Jurberg and R. M. Koenigs, Chem. Soc. Rev., 2020, 49, 6833–6847 RSC; (c) J. Durka, J. Turkowska and D. Gryko, ACS Sustainable Chem. Eng., 2021, 9, 8895–8918 CrossRef CAS.
- For selected examples of visible-light-induced H–X bond insertion with diazo compounds: (a) I. Jurberg and H. M. L. Davies, Chem. Sci., 2018, 9, 5112–5118 RSC; (b) Z. Zhang, D. Yadagiri and V. Gevorgyan, Chem. Sci., 2019, 10, 8399–8404 RSC; (c) C. Empel, F. W. Patureau and R. M. Koenigs, J. Org. Chem., 2019, 84, 11316–11322 CrossRef CAS; (d) S. Jana, Z. Yang, F. Li, C. Empel, J. Ho and R. M. Koenigs, Angew. Chem., Int. Ed., 2020, 59, 5562–5566 CrossRef CAS PubMed; (e) S. Jana, F. Li, C. Empel, D. Verspeek, P. Aseeva and R. M. Koenigs, Chem.–Eur. J., 2020, 26, 2586–2591 CrossRef CAS PubMed; (f) F. He, F. Li and R. M. Koenigs, J. Org. Chem., 2020, 85, 1240–1246 CrossRef CAS PubMed; (g) J. Chen, S. Liu, X. Lv, K. Hong, J. Lei, X. Xu and W. Hu, J. Org. Chem., 2020, 85, 13920–13928 CrossRef CAS; (h) M. L. Stivanin, A. A. G. Fernandes, A. F. da Silva, C. Y. Okada Jr and I. D. Jurberg, Adv. Synth. Catal., 2020, 362, 1106–1111 CrossRef CAS; (i) S. Jana, C. Pei, C. Empel and R. M. Koenigs, Angew. Chem., Int. Ed., 2021, 60, 13271–13279 CrossRef CAS; (j) S. Zhou, B. Cai, C. Hu, X. Cheng, L. Li and J. Xuan, Chin. Chem. Lett., 2021, 32, 2577–2581 CrossRef CAS; (k) Z.-L. Chen, C. Empel, K. Wang, P.-P. Wu, B.-G. Cai, L. Li, R. M. Koenigs and J. Xuan, Org. Lett., 2022, 24, 2232–2237 CrossRef CAS PubMed; (l) K. Yan, H. He, J. Li, Y. Luo, R. Lai, L. Guo and Y. Wu, Chin. Chem. Lett., 2021, 32, 3984–3987 CrossRef CAS.
- For other types of reactions of diazo compounds promoted by visible light: (a) Z. Wang, A. G. Herraiz, A. M. del Hoyo and M. G. Suero, Nature, 2018, 554, 86–91 CrossRef CAS PubMed; (b) T. Xiao, M. Mei, Y. He and L. Zhou, Chem. Commun., 2018, 54, 8865–8868 RSC; (c) S. Jana, Z. Yang, C. Pei, X. Xu and R. M. Koenigs, Chem. Sci., 2019, 10, 10129–10134 RSC; (d) B.-G. Cai, S.-S. Luo, L. Li, L. Li, J. Xuan and W.-J. Xiao, CCS Chem., 2020, 2, 2764–2771 Search PubMed; (e) X. Cheng, B.-G. Cai, H. Mao, J. Lu, L. Li, K. Wang and J. Xuan, Org. Lett., 2021, 23, 4109–4114 CrossRef CAS; (f) R. D. C. Gallo, M. Duarte, A. F. da Silva, C. Y. Okada, V. M. Deflon and I. D. Jurberg, Org. Lett., 2021, 23, 8916–8920 CrossRef CAS PubMed . For a recent CPA-catalyzed asymmetric variant of this reaction:; (g) H. Zhang, Z. Wang, Z. Wang, Y. Chu, S. Wang and X.-P. Hui, ACS Catal., 2022, 12, 5510–5516 CrossRef CAS.
- (a) A. Berkessel and H. Gröger, Asymmetric Organocatalysis: From Biomimetic Concepts to Applications in Asymmetric Synthesis, Wiley-VCH, 2006 Search PubMed; (b) Ł. Albrecht, A. Albrecht and L. Dell'Amico, Asymmetric Organocatalysis: New Strategies, Catalysts, and Opportunities, Wiley-VCH, 2022 Search PubMed.
- For an example: Y. Zhang, X. Zhang, J. Zhao and J. Jiang, Org. Biomol. Chem., 2021, 19, 5772–5776 RSC.
- For metal-catalyzed asymmetric N–H insertions with α-diazoesters: (a) S. Bachmann, D. Fielenbach and K. A. Jørgensen, Org. Biomol. Chem., 2004, 2, 3044–3049 RSC; (b) B. Liu, S.-F. Zhu, W. Zhang, C. Chen and Q.-L. Zhou, J. Am. Chem. Soc., 2007, 129, 5834–5835 CrossRef CAS PubMed; (c) E. C. Lee and G. C. Fu, J. Am. Chem. Soc., 2007, 129, 12066–12067 CrossRef CAS PubMed; (d) X. Xu, P. Y. Zavalij and M. P. Doyle, Angew. Chem., Int. Ed., 2012, 51, 9829–9833 CrossRef CAS PubMed; (e) M.-L. Li, J.-H. Yu, Y.-H. Li, S.-F. Zhu and Q.-L. Zhou, Science, 2019, 366, 990–994 CrossRef CAS PubMed; (f) M.-L. Li, J.-B. Pan and Q.-L. Zhou, Nat. Catal., 2022, 5, 571–577 CrossRef CAS.
- For a thermal organocatalytic example: H. Saito, D. Morita, T. Uchiyama, M. Miyake and S. Miyairi, Tetrahedron Lett., 2012, 53, 6662–6664 CrossRef CAS.
- (a) T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566–1568 CrossRef CAS; (b) D. Uraguchi and M. Terada, J. Am. Chem. Soc., 2004, 126, 5356–5357 CrossRef CAS; (c) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed; (d) T. Akiyama and K. Mori, Chem. Rev., 2015, 115, 9277–9306 CrossRef CAS PubMed; (e) T. James, M. van Gemmeren and B. List, Chem. Rev., 2015, 115, 9388–9409 CrossRef CAS.
- For selected examples of carbene capture by a lone pair to form carbenoid species: (a) R. M. Moriarty, B. R. Bailey III, O. Prakash and I. Prakash, J. Am. Chem. Soc., 1985, 107, 1375–1378 CrossRef CAS; (b) J. Lu, L. Li, X.-K. He, G.-Y. Xu and J. Xuan, Chin. J. Chem., 2021, 39, 1646–1650 CrossRef CAS; (c) L. S. Munaretto, C. Y. dos Santos, R. D. C. Gallo, C. Y. Okada, V. M. Deflon and I. D. Jurberg, Org. Lett., 2021, 23, 9292–9296 CrossRef CAS PubMed; (d) V. V. Khade, A. S. Thube, P. K. Warghude and R. G. Bhat, Tetrahedron Lett., 2021, 77, 153258 CrossRef CAS.
- For some examples, see: (a) R. Hommelsheim, Y. Guo, Z. Yang, C. Empel and R. M. Koenigs, Angew. Chem., Int. Ed., 2019, 58, 1203–1207 CrossRef CAS; (b) Z. Yang, Y. Guo and R. M. Koenigs, Chem.–Eur. J., 2019, 25, 6703–6706 CrossRef CAS; (c) S. Yan, J. Rao and C.-Y. Zhou, Org. Lett., 2020, 22, 9091–9096 CrossRef CAS PubMed; (d) K. Orłowska, K. Rybicka-Jasinska, P. Krajewski and D. Gryko, Org. Lett., 2020, 22, 1018–1021 CrossRef.
- For selected reviews on asymmetric transformations from sulfoxonium ylides: (a) G. D. Bisag, S. Ruggieri, M. Fochi and L. Bernardi, Org. Biomol. Chem., 2020, 18, 8793–8809 RSC; (b) C. A. D. Caiuby, L. G. Furniel and A. C. B. Burtoloso, Chem. Sci., 2022, 13, 1192–1209 RSC; (c) D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Chem. Rev., 2019, 119, 8701–8780 CrossRef CAS PubMed; (d) J. D. Neuhaus, R. Oost, J. Merad and N. Maulide, Top. Curr. Chem., 2018, 376, 15–62 CrossRef; (e) L.-Q. Lu, T.-R. Li, Q. Wang and W.-J. Xiao, Chem. Soc. Rev., 2017, 46, 4135–4149 RSC.
- For selected recent examples of asymmetric X–H insertion reactions of sulfur ylides: (a) W. Guo, Y. Luo, H. H.-Y. Sung, I. D. Williams, P. Li and J. Sun, J. Am. Chem. Soc., 2020, 142, 14384–14390 CrossRef CAS; (b) W. Guo, M. Wang, Z. Han, H. Huang and J. Sun, Chem. Sci., 2021, 12, 11191–11196 RSC; (c) W. Guo, F. Jiang, S. Li and J. Sun, Chem. Sci., 2022, 13, 11648–11655 RSC; (d) L. G. Furniel, R. Echemendía and A. C. B. Burtoloso, Chem. Sci., 2021, 12, 7453–7459 RSC; (e) A. N. Leveille, R. Echemendía, A. E. Mattson and A. C. B. Burtoloso, Org. Lett., 2021, 23, 9446–9450 CrossRef CAS PubMed; (f) P. B. Momo, A. N. Leveille, E. H. E. Farrar, M. N. Grayson, A. E. Mattson and A. C. B. Burtoloso, Angew. Chem., Int. Ed., 2020, 59, 15554–15559 CrossRef CAS PubMed.
- Notably, opposite chiral induction was observed between the cases with sulfoxides and other additives. Indeed, the latter cases were the same as the situation without additive. Further studies indicated that under such conditions, non-sulfoxide additives did not efficiently trap the free carbene. Therefore, their asymmetric induction was similar to that of free carbene. In contrast, sulfoxides were able to trap the free carbene, and thus asymmetric induction was via sulfoxonium ions and thus different from the others.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05149d |
| This journal is © The Royal Society of Chemistry 2023 |