
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN-Alkylation of aromatic amines with alcohols by using a commercially available Ru complex under mild conditions†
Rita Mocci a,
Luciano Atzoria,
Walter Barattab,
Lidia De Lucac and
Andrea Porcheddu*a
a,
Luciano Atzoria,
Walter Barattab,
Lidia De Lucac and
Andrea Porcheddu*a
aDipartimento di Scienze Chimiche e Geologiche, Università degli Studi di Cagliari, Cittadella Universitaria, S.S. 554 bivio per Sestu, 09042 Monserrato (CA), Italy. E-mail: porcheddu@unica.it
bDipartimento di Scienze Agroalimentari, Ambientali e Animali, Università degli Studi di Udine, via delle Scienze 206, 33100 Udine, Italy
cDipartimento di Scienze Chimiche, FIsiche, Matematiche e Naturali, Università degli Studi di Sassari, via Vienna 2, 07100 Sassari, Italy
First published on 30th November 2023
Abstract
An N-alkylation procedure has been developed under very mild conditions using a known commercially available Ru-based catalyst. As a result, a wide range of aromatic primary amines has been selectively alkylated with several primary alcohols, yielding the corresponding secondary amines in high yields. The methodology also enables the methylation of anilines in refluxing methanol and the preparation of a set of heterocycles in a straightforward way.
Introduction
The straightforward functionalization of amines is a crucial transformation for the pharmaceutical and fine chemical industries.1,2 Among various methods enabling the synthesis of a new C–N bond, the so-called borrowing hydrogen (BH) strategy stands out for its intrinsic atom efficiency and selectivity traits.3–7 In a typical BH or hydrogen autotransfer (HT) reaction, alcohol is temporarily dehydrogenated in the presence of a suitable metal catalyst, resulting in a carbonylic compound available to condense with an appropriate amine. Lastly, the imine is reduced by the resulting metal hydride [M]H2 to give the alkylated secondary amine without the formation of waste other than water (Scheme 1). As well as avoiding alkyl halides, the catalytic system allows the in situ generation of the carbonyl source directly from alcohols, accessible from renewable feedstock.8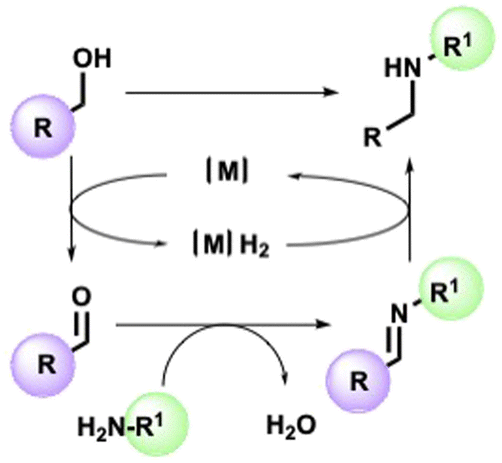 | ||
| Scheme 1 N-alkylation BH mediated general mechanism. | ||
The extraordinary utility of this reaction is demonstrated by the plethora of examples reported in the literature. Since the pioneering studies9,10 involving homogenous catalysts based on triphenylphosphine complexes of Rh, Ru, and Ir, introducing new ligands allowed the development of innovative catalytic platforms to realize more efficient processes under milder conditions.11–13
In this framework, some of these catalytic systems are represented by metal complexes requiring complex preparation and appropriate expertise. A few of the most representative ones follow: [(η6-arene)RuClL2]X or [(η6-arene)RuCl2L] where L = PR3, NHC, Py, SR2, SeR2 and X = Cl, PF6, OTf, and PNN, PNO/PNS, PNP, and NNC pincer complexes.14 Recently, some remarkable examples of BH reactions catalyzed by non-noble metals such as Fe,15 Mn,16,17 and Co18,19 have also been reported. However, Ru and Ir-based catalysts are still the most represented in this scenario.4
To this day, most N-alkylation reactions of amines with alcohols are mainly conducted at high temperatures (>100 °C).20,21 Very few examples of room-temperature BH N-alkylation are reported in the literature.16,22–25
Finding an accessible methodology to perform such a significant transformation under very mild conditions remains highly demanded and challenging. We envisioned fetching from a library of pincer Ru (and Os) complexes and bidentate amino derivatives to achieve this goal. These catalysts were selected based on their notable efficacy in facilitating both the acceptorless dehydrogenation of alcohols to ketones and the transfer hydrogenation of carbonyl compounds. Prior investigations demonstrated the proficient catalytic abilities of these ruthenium (Ru) and osmium (Os) complexes in promoting carbonyl compound/alcohol interconversion reactions, highlighting in certain instances a superior activity of Os complexes in terms of turn over frequency. Consequently, we postulated that these attributes could be harnessed for amine alkylation.26–31
Results and discussion
We started screening a set of 12 ruthenium and osmium-based catalysts by using 4-methoxy-anisidine and 1-octananol as model substrates in toluene and in the presence of 1 equivalent of tBuOK as a base (Fig. 1 and Table 1).4 Since we aimed to perform the N-alkylation under mild conditions, we tested the catalytic process only by switching the reaction temperature from 25 °C to 70 °C (Table 1). The Ru complexes performed best among the twelve catalysts examined (Table 1, entries 1–8). Regarding the Ru bidentate amino derivatives, we observe complete conversion as early as room temperature in the presence of a dppf ligand on the metal center (see entries 1 and 4 for comparison).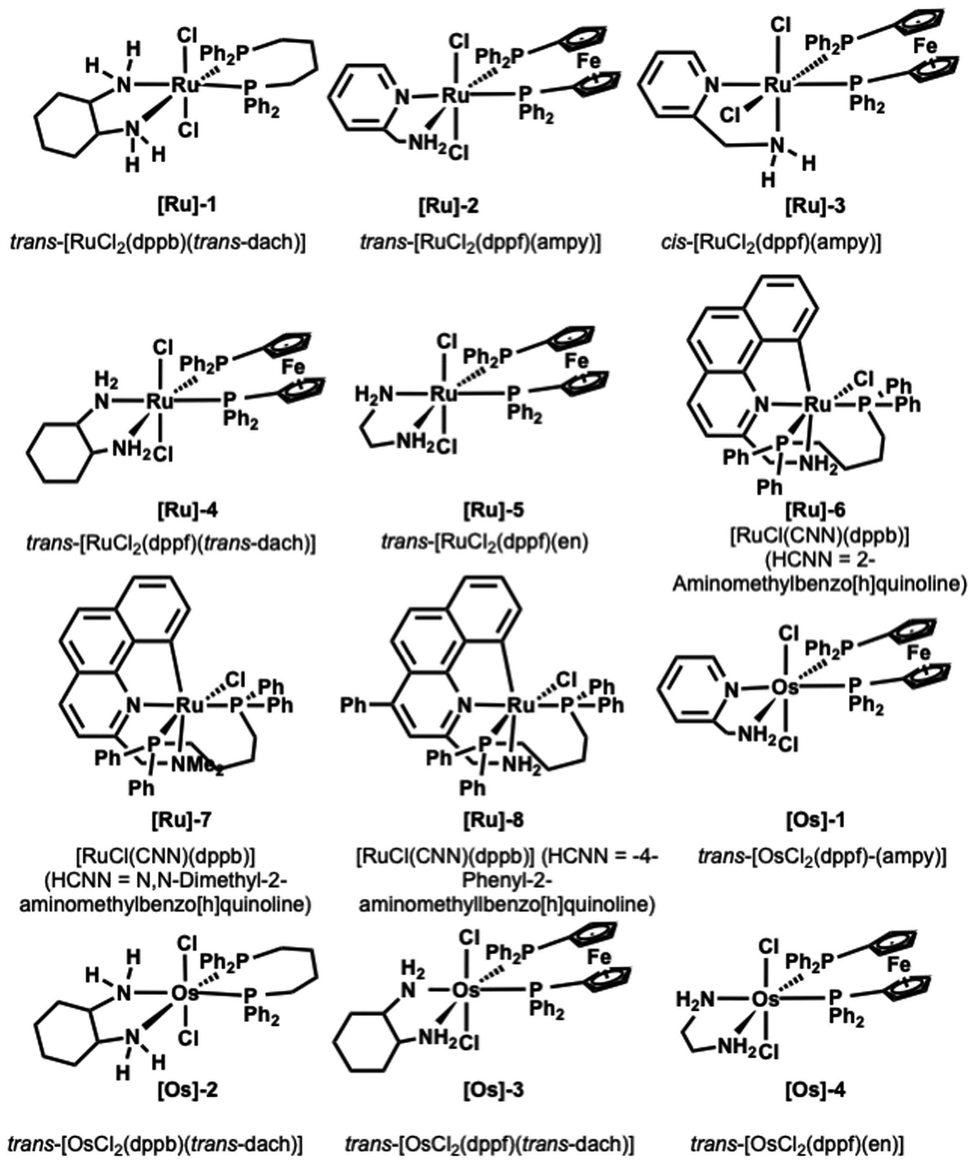 | ||
| Fig. 1 Ru and Os catalysts tested.21–25 | ||
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Time (h) | Temperature | Yield of 3b |
| a Reaction conditions: anisidine (1.0 mmol), octanol (1.0 mmol), potassium tert-butoxide (1.0 mmol), catalyst (2.5 mol%), in toluene (1.0 mL) for the given time and given temperature.b Determined by GC-MS analysis.c Catalyst loading 2 mol%. | ||||
| 1 | [Ru]-1 | 36 | 70 °C | — |
| 2 | [Ru]-2 | 12 | 70 °C | 99 |
| 3 | [Ru]-3 | 24 | 25 °C | 99 |
| 4 | [Ru]-4 | 36 | 25 °C | 97 |
| 5 | [Ru]-5 | 24 | 25 °C | 99 |
| 6 | [Ru]-6 | 12 | 25 °C | 99 |
| 7 | [Ru]-7 | 36 | 70 °C | — |
| 8 | [Ru]-8 | 2.5 | 25 °C | 99 |
| 9c | [Ru]-3 | 24 | 25 °C | 99 |
| 10 | [Os]-1 | 36 | 70 °C | — |
| 11 | [Os]-2 | 24 | 50 °C | 99% |
| 12 | [Os]-3 | 36 | 70 °C | Traces |
| 13 | [Os]-4 | 36 | 70 °C | — |
Trans and cis isomers of RuCl2(AMPY)(DPPF) displayed a sharp gap in catalytic activity ([Ru]-2 and [Ru]-3); the cis isomer proved efficient already at 25 °C after 24 hours, while the trans isomer (see ESI, Table S1† and entries 2 and 3 in Table 1) provided complete conversion into amine only at 70 °C.29 At the same time, when dppf diphosphine complexed the metal in the presence of other diamino derivatives (1,2-cyclohexanediamine and 1,2-ethylenediamine, [Ru]-4 and [Ru]-5), we obtained the quantitative formation of the desired alkylated amine 3a in 36 and 24 hours (Table 1, entries 4 and 5).
The benzo[h]quinoline derivatives ligands confer a marked increase of activity to the catalyst. Indeed, the reaction proceeds smoothly in the presence of the catalysts [Ru]-6 and [Ru]-8 (Table 1, entries 6 and 8). Conversely, the absence of a free NH2 moiety in the analogue [Ru]-7 results in diminished performance (see Table 1, entry 7).
This behavior agrees with studies on analogous pincer CNN ruthenium(II) complexes made by Baratta et al., in which the influence of NH2 on the TH and HY catalytic activity was established.32 Conversely, Martin-Matute observed an opposite trend, finding the NMe2 pincer catalyst more active in forming secondary amine concerning the NH2 analogous.31 Finally, we tested the Os complexes shown in Fig. 1 (Table 1, entries 10–13). Even though none showed catalytic activity under room temperature conditions, we were pleased to observe the formation of the targeted N-alkylamine using [Os]-2 at only 50° C (Table 1, entry 11). This result is noteworthy because, in a previous report, the BH osmium-mediated amine alkylation occurred at 200 °C.33
With this data in hand, we expanded the scope of our findings. We synthesized an array of secondary amines in high yield using catalysts [Ru]-6 and [Ru]-8 (see ESI, Table S3† for further details). Moreover, we were more intrigued by the potential feasibility of the catalyst [Ru]-3, which is commercially available.
We completed the reaction conditions tuning by reducing the catalyst loading to 2 mol% (see ESI† for further details and entry 9, Table 1). We applied the optimized conditions to an array of amines and alcohols (see Scheme 2).
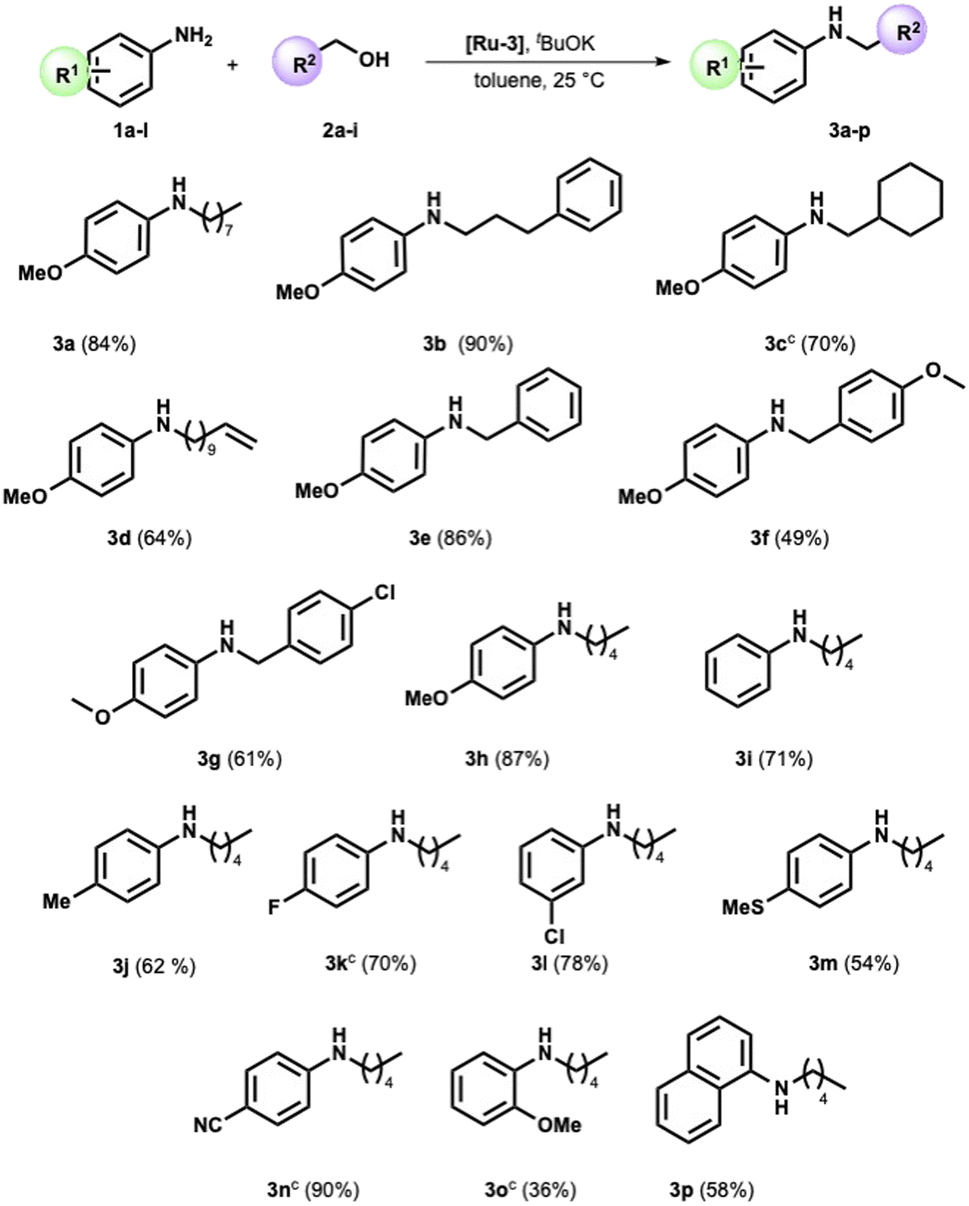 | ||
| Scheme 2 Scope of the N-alkylation reaction of aromatic amines with alcohols.a,b aReaction conditions: amine (1 mmol), alcohol (1 mmol), potassium tert-butoxide (1 mmol), [Ru]-3 (2 mol%), in toluene (1 mL) for 24 h. bIsolated yields. c36 h. | ||
Variations in the steric and electronic character of aliphatic alcohols provided the targeted compounds with high yields at a satisfactory reaction rate. Indeed, the 3-phenyl-1-propanol and cyclohexylmethanol were reacted with p-anisidine, providing the corresponding secondary amine in 90% and 70% yield, respectively (Scheme 2, compounds 3b and 3c). The reaction displayed good selectivity in the presence of the alkene moiety. The alkylamine 3d was obtained in high yield (64%), while the product of double bond hydrogenation was not detected by GC-MS analysis.
Benzyl alcohol, the archetypal starting material for the BH-mediated N-alkylation, reacted with anisidine under optimized conditions. Pleasingly, N-benzyl p-anisidine 3e was isolated in 85% yield. Compounds 3f and 3g were then successfully prepared with a yield of 49% and 61%, respectively (Scheme 2). p-Anisidine was then reacted with amyl alcohol, providing the corresponding secondary amine in high yield (Scheme 2, compound 3h).
At this stage, we verified the effect of several substituents on the aromatic ring in the amine moiety. In detail, the less nucleophilic aniline and 4-methyl aniline showed good reactivity toward the pentyl alcohol, forming products 3i and 3j in satisfying yields (71% and 62%, respectively). A halide substituent such as F or Cl is well tolerated (Scheme 2). Indeed, products 3k and 3l were selectively synthesized in high yields (Scheme 2), while the product of hydrodehalogenation was not detected. The reaction proved suitable for an aniline bearing a sulfide group, as witnessed by the formation of 3m in satisfactory yield (54%).
To our delight, when the 4-amino benzonitrile was reacted under the optimized conditions with 1-pentyl alcohol, the formation of the secondary amine 3n was almost quantitative (Scheme 2). Furthermore, the presence of the methoxy substituent in the ortho position slightly affects the outcome of the reaction (Scheme 2, compare amines 3h and 3o). Lastly, 1-naphthylamine is effectively functionalized with a yield of 58% (Scheme 2, compound 3p).
Shifting the focus to aliphatic amines yielded unfavorable outcomes. Specifically, attempts to react N-phenylethylamine with either heptanol or benzyl alcohol under the established optimized conditions proved inadequate for attaining the intended product.
Despite its value, applying BH reaction to the N-methylation is rather challenging. High temperatures are needed for the reaction, often requiring an appropriate pressure tube. However, the chance to perform this transformation, avoiding using formaldehyde or other toxic reagents such as methyl iodide or diazomethane, is of remarkable appeal.34–38
We then decided to test our catalyst in the reaction of p-anisidine and methanol. After tuning the reaction parameters (see ESI, Table S1†), we obtained the formation of the N-methyl-p-anisidine 3q in 75% yield in refluxing methanol after 48 h. The method was then extended to various anilines, providing the corresponding N-methyl functionalized product in high yield (see Scheme 3). The reaction proceeds smoothly without substituents on the aryl ring (Scheme 3, amine 3r) and with an electron-withdrawing group, such as the fluoro substituent in position 4. The 4-bromo-aniline also provided the methylated analogue in high isolated yields (see product 3t).
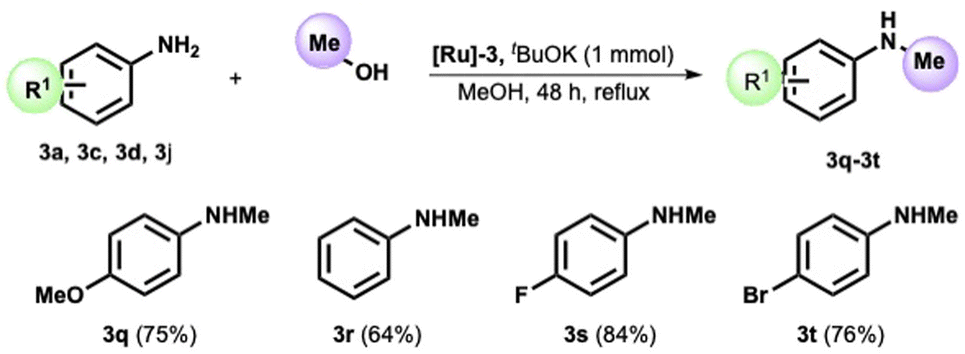 | ||
| Scheme 3 Scope of the N-methylation of aromatic amines with methanol.a,b aReaction conditions: amine (1 mmol), potassium tert-butoxide (1 mmol), [Ru]-3 (2 mol%), in refluxing methanol (2 mL) for 48 h. bIsolated yields. | ||
At last, the borrowing hydrogen and acceptorless dehydrogenation catalysis provide access to the straightforward and elegant synthesis of various relevant heterocycles.39–41 Elevated temperatures in the 100–140 °C range are typically necessary for this process to occur. This section evaluated our catalytic system for the acceptorless dehydrogenative synthesis of indole, benzimidazole, and quinoxaline.
Indole ring is perhaps the most preeminent N-heterocycle in nature and a worthy structural component in many pharmaceutical ingredients,42,43 arousing strong interest through its synthesis.44 The intramolecular cyclization of 2-aminophenethyl alcohol is an elegant strategy for this target. However, despite sporadic examples,23 operating temperatures hover around 100 °C or above. For example, in 2016, Beller reported this cyclization in the presence of an Mn pincer complex at 100 °C.16 Other examples are those written by Banerjee45 and Adhikari,46 which employ Ni catalysts for synthesizing indoles starting from aminoalcohols at 130 °C and 110 °C.
In our first trial, we tested the cyclization of 2-aminophenethyl alcohol using commercially available [Ru]-3, potassium tert-butoxide in toluene at room temperature.
Under these conditions, only 50% conversion was observed after 48 hours. However, when the temperature was increased to 70 °C for 24 hours, complete conversion of indole was achieved, as confirmed by GC-MS analysis. Indole 5a was subsequently isolated with a yield of 61%, as shown in Scheme 4.
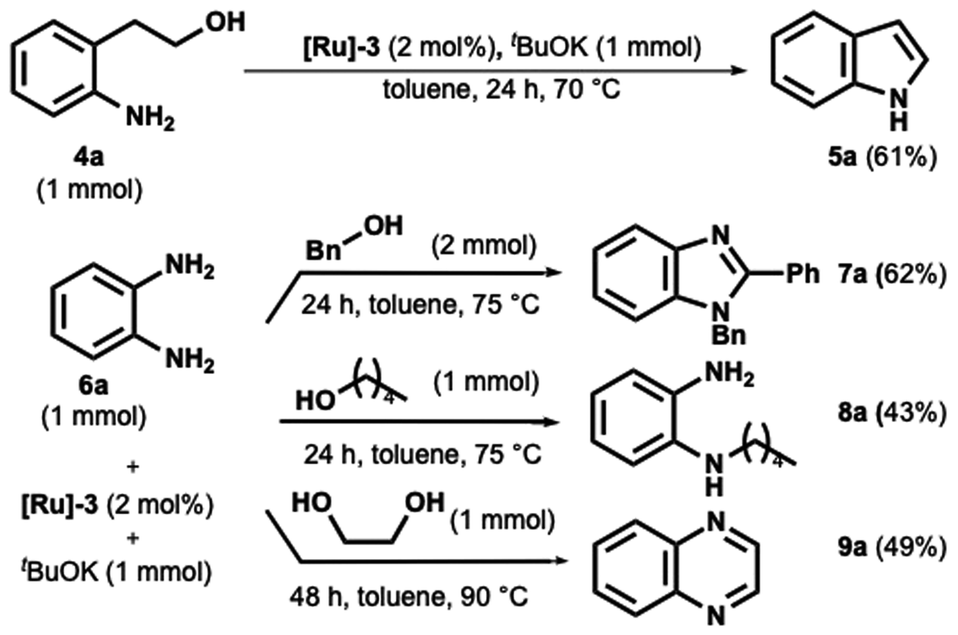 | ||
| Scheme 4 Application of the catalytic system to synthesize heterocycles. | ||
Benzimidazole derivatives synthesis has attracted significant interest due to their important biological and pharmacological properties.47,48 In this scenario, their practical, direct synthesis from primary alcohol and o-phenylenediamine has gained much attention.49–51 However, operating temperatures over 100 °C are required. We then decided to test the o-phenylenediamine in the presence of benzyl alcohol and 1-pentanol (Scheme 4). While the benzyl alcohol provided the 1,2-disubstituted benzimidazoles in satisfying yield (Scheme 4, compound 7a), the formation of the monoalkylated o-phenylenediamine was preferred to the ring closure when the diamine was coupled with 1-pentanol.
Finally, we focused on synthesizing the quinoxaline ring, a pharmaceutical occurring and key starting material nitrogen-containing heterocycle. Same here; most methods involve high temperatures up to 160 °C.52–56 The catalyst proved suitable as product 9a was obtained after 48 h at 90 °C in satisfying yield (Scheme 4).
Finally, an array of experiments for qualitative hydrogen detection were performed. For these experiments, three case substrates were reacted under an N2 atmosphere in anhydrous toluene in the presence of catalyst [Ru]-3 and tBuOK. No significative hydrogen evolution was observed during 3a, 3e, and 5a formation at room temperature after 12 hours. Conversely, indole formation from aminophenyl alcohol (see Scheme 4) at 70 °C was complemented by a consistent hydrogen evolution after only 8 hours. As a comparison, the benzylic alcohol was similarly reacted at 70 °C in the presence of anisidine, and consistent hydrogen formation was observed in this case as well.
Conclusion
In summary, we reported using a commercially available and robust [Ru]-3 catalyst for the mild N-alkylation reaction of aromatic amines with several alcohols, including methanol. The catalyst displayed good versatility when applied to the acceptorless dehydrogenative synthesis of a set of relevant heterocycles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from MIUR Italy, PNRR PE2 – NEST – Network 4 Energy Sustainable Transition – (MUR no. PE0000021 – CUP no. F53C22000770007). We acknowledge the CeSAR (Centro Servizi Ricerca d'Ateneo) core facility of the University of Cagliari, Dr Sandrina Lampis for assistance with the generation of NMR data, and Gianluigi Corrias for the technical support.References
- S. A. Lawrence, Amines: Synthesis Properties, and Applications, Cambridge University Press, 2004 Search PubMed.
- A. S. Travis, The Chemistry of Anilines, ed. Z. Rappoport, Wiley-Interscience, 2007, vol. 1, p. 717 Search PubMed.
- S. Bähn, S. Imm, L. Neubert, M. Zhang, H. Neumann and M. Beller, ChemCatChem, 2011, 3, 1853–1864 CrossRef.
- E. Podyacheva, O. I. Afanasyev, D. V. Vasilyev and D. Chusov, ACS Catal., 2022, 12, 7142–7198 CrossRef CAS.
- G. Guillena, D. J. Ramón and M. Yus, Chem. Rev., 2010, 110, 1611–1641 CrossRef CAS PubMed.
- J. Leonard, A. J. Blacker, S. P. Marsden, M. F. Jones, K. R. Mulholland and R. Newton, Org. Process Res. Dev., 2015, 19, 1400–1410 CrossRef CAS.
- A. Corma, J. Navas and M. J. Sabater, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS PubMed.
- K. Barta and P. C. Ford, Acc. Chem. Res., 2014, 47, 1503–1512 CrossRef CAS PubMed.
- R. Grigg, T. R. B. Mitchell, S. Sutthivaiyakit and N. Tongpenyai, J. Chem. Soc. Chem. Commun., 1981, 611–612 RSC.
- Y. Watanabe, Y. Tsuji and Y. Ohsugi, Tetrahedron Lett., 1981, 22, 2667–2670 CrossRef CAS.
- Y. Liu, H. Diao, G. Hong, J. Edward, T. Zhang, G. Yang, B.-M. Yang and Y. Zhao, J. Am. Chem. Soc., 2023, 145, 5007–5016 CrossRef CAS PubMed.
- A. K. Bains, A. Kundu, S. Yadav and D. Adhikari, ACS Catal., 2019, 9, 9051–9059 CrossRef CAS.
- S. Hameury, H. Bensalem and K. De Oliveira Vigier, Catalysts, 2022, 12, 1306 CrossRef CAS.
- V. Cherepakhin and T. J. Williams, ACS Catal., 2020, 10, 56–65 CrossRef CAS.
- T. Yan, B. L. Feringa and K. Barta, Nat. Commun., 2014, 5, 5602 CrossRef CAS PubMed.
- S. Elangovan, J. Neumann, J.-B. Sortais, K. Junge, C. Darcel and M. Beller, Nat. Commun., 2016, 7, 12641 CrossRef PubMed.
- M. Huang, Y. Li, Y. Li, J. Liu, S. Shu, Y. Liu and Z. Ke, Chem. Commun., 2019, 55, 6213–6216 RSC.
- G. Zhang, Z. Yin and S. Zheng, Org. Lett., 2016, 18, 300–303 CrossRef CAS PubMed.
- S. Rösler, M. Ertl, T. Irrgang and R. Kempe, Angew. Chem., Int. Ed., 2015, 54, 15046–15050 CrossRef PubMed.
- Z. Moutaoukil, E. Serrano-Díez, I. G. Collado, M. Jiménez-Tenorio and J. M. Botubol-Ares, Org. Biomol. Chem., 2022, 20, 831–839 RSC.
- B. Patel, R. Ranjan, N. R. Chauhan, S. Mukhopadhyay, A. R. Choudhury and K. M. Vyas, New J. Chem., 2023, 47, 8305–8317 RSC.
- A. B. Enyong and B. Moasser, J. Org. Chem., 2014, 79, 7553–7563 CrossRef CAS PubMed.
- J.-Q. Li and P. G. Andersson, Chem. Commun., 2013, 49, 6131–6133 RSC.
- V. R. Jumde, L. Gonsalvi, A. Guerriero, M. Peruzzini and M. Taddei, Eur. J. Org. Chem., 2015, 2015, 1829–1833 CrossRef CAS.
- R. Figliolia, S. Baldino, H. G. Nedden, A. Zanotti-Gerosa and W. Baratta, Chem.–Eur. J., 2017, 23, 14416–14419 CrossRef CAS PubMed.
- G. Chelucci, S. Baldino and W. Baratta, Acc. Chem. Res., 2015, 48, 363–379 CrossRef CAS PubMed.
- W. Baratta, L. Fanfoni, S. Magnolia, K. Siega and P. Rigo, Eur. J. Inorg. Chem., 2010, 2010, 1419–1423 CrossRef.
- E. Putignano, G. Bossi, P. Rigo and W. Baratta, Organometallics, 2012, 31, 1133–1142 CrossRef CAS.
- W. Baratta, G. Bossi, E. Putignano and P. Rigo, Chem.–Eur. J., 2011, 17, 3474–3481 CrossRef CAS PubMed.
- N. D. Schley, G. E. Dobereiner and R. H. Crabtree, Organometallics, 2011, 30, 4174–4179 CrossRef CAS.
- S. Agrawal, M. Lenormand and B. Martín-Matute, Org. Lett., 2012, 14, 1456–1459 CrossRef CAS PubMed.
- W. Baratta, M. Ballico, A. Del Zotto, E. Herdtweck, S. Magnolia, R. Peloso, K. Siega, M. Toniutti, E. Zangrando and P. Rigo, Organometallics, 2009, 28, 4421–4430 CrossRef CAS.
- M. Bertoli, A. Choualeb, D. G. Gusev, A. J. Lough, Q. Major and B. Moore, Dalton Trans., 2011, 40, 8941–8949 RSC.
- G. Yan, A. J. Borah, L. Wang and M. Yang, Adv. Synth. Catal., 2015, 357, 1333–1350 CrossRef CAS.
- T. T. Dang, B. Ramalingam and A. M. Seayad, ACS Catal., 2015, 5, 4082–4088 CrossRef CAS.
- A. Del Zotto, W. Baratta, M. Sandri, G. Verardo and P. Rigo, Eur. J. Inorg. Chem., 2004, 524–529 CrossRef CAS.
- P. Piehl, R. Amuso, A. Spannenberg, B. Gabriele, H. Neumann and M. Beller, Catal. Sci. Technol., 2021, 11, 2512–2517 RSC.
- G. T. Toyooka Akiko and K. I. Fujita, Synthesis, 2018, 50, 4617–4626 CrossRef.
- A. Mondal, R. Sharma, D. Pal and D. Srimani, Eur. J. Org. Chem., 2021, 2021, 3690–3720 CrossRef CAS.
- N. Hofmann and K. C. Hultzsch, Eur. J. Org. Chem., 2021, 2021, 6206–6223 CrossRef CAS.
- M. Maji, D. Panja, I. Borthakur and S. Kundu, Org. Chem. Front., 2021, 8, 2673–2709 RSC.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- A. Porcheddu, R. Mocci, M. Brindisi, F. Cuccu, C. Fattuoni, F. Delogu, E. Colacino and M. V. D’Auria, Green Chem., 2022, 24, 4859–4869 RSC.
- D. F. Taber and P. K. Tirunahari, Tetrahedron, 2011, 67, 7195–7210 CrossRef CAS PubMed.
- M. Vellakkaran, K. Singh and D. Banerjee, ACS Catal., 2017, 7, 8152–8158 CrossRef CAS.
- A. K. Bains, A. Biswas and D. Adhikari, Chem. Commun., 2020, 56, 15442–15445 RSC.
- J. D. C. Lambert, Bioactive heterocyclic compound classes: pharmaceuticals, 2012 Search PubMed.
- V. V Fedotov, V. L. Rusinov, E. N. Ulomsky, E. M. Mukhin, E. B. Gorbunov and O. N. Chupakhin, Chem. Heterocycl. Compd., 2021, 57, 383–409 CrossRef PubMed.
- K. Das, A. Mondal and D. Srimani, J. Org. Chem., 2018, 83, 9553–9560 CrossRef CAS PubMed.
- A. Ravindran N E, M. Yadav, M. M. Tamizh, N. Bhuvanesh, S. Sarkar and R. Karvembu, Asian J. Org. Chem., 2023, e202200675 CrossRef CAS.
- Z. Xu, D.-S. Wang, X. Yu, Y. Yang and D. Wang, Adv. Synth. Catal., 2017, 359, 3332–3340 CrossRef CAS.
- T. Hille, T. Irrgang and R. Kempe, Chem.–Eur. J., 2014, 20, 5569–5572 CrossRef CAS PubMed.
- C. S. Cho and S. G. Oh, Tetrahedron Lett., 2006, 47, 5633–5636 CrossRef CAS.
- P. Daw, A. Kumar, N. A. Espinosa-Jalapa, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2018, 8, 7734–7741 CrossRef CAS PubMed.
- A. Mondal, M. K. Sahoo, M. Subaramanian and E. Balaraman, J. Org. Chem., 2020, 85, 7181–7191 CrossRef CAS PubMed.
- R. H. Crabtree, Chem. Rev., 2017, 117, 9228–9246 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra06751c |
| This journal is © The Royal Society of Chemistry 2023 |