
Steering CO2 electroreduction selectivity towards CH4 and C2H4 on a tannic acid-modified Cu electrode†
Keqiang
Xu
a,
Jinhan
Li
a,
Fangming
Liu
a,
Wence
Xu
a,
Tete
Zhao
a and
Fangyi
Cheng
 *ab
*ab
aKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Renewable Energy Conversion and Storage Center, College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: fycheng@nankai.edu.cn
bEngineering Research Center of High-efficiency Energy Storage (Ministry of Education), Tianjin 300071, China
First published on 31st January 2023
Abstract
CO2 electroreduction (CO2RR) offers a promising way to address CO2 emission and high-value utilization but it remains challenging to steer the selectivity of products due to complicated reaction pathways. Herein, tannic acid (TA) is reported as a modifier to regulate the selectivity of C2H4 and CH4 over the Cu catalyst. With an optimized TA amount, the maximal Faradaic efficiency of C2H4 and CH4 increases from 35.46% and 18.56% to 53.00% and 53.27%, respectively. In situ attenuated total reflection surface-enhanced infrared absorption spectra demonstrate that TA modification stabilizes the adsorbed CO and CHO intermediate and strengthens the interaction of hydrogen bonds with H2O. Kinetic isotope effect analysis of H2O/D2O reveals that TA-modified Cu could activate H2O dissociation to accelerate the proton-coupled electron transfer. Theoretical calculations further indicate the decrease of the energy barrier from *CO hydrogenation to *CHO by TA modification. The results evidence the importance of molecule modification to tailor the C2/C1 product selectivity in the CO2RR via concurrently stabilizing the intermediate and promoting proton transfer.
Introduction
Recently, the electrocatalytic CO2 reduction reaction (CO2RR) powered by renewable energy has attracted much attention, as it not only reduces CO2 emission but also produces chemicals.1,2 Among the catalysts reported for the CO2RR, copper-based nanomaterials are promising in generating multi-electron (>2e−) products due to the positive adsorption energy for *H and negative adsorption energy for *CO.3–8 Compared to the two electron products (e.g., CO and HCOOH) in the CO2RR,9–12 the multi-electron products (e.g., CH4, C2H4, CH3CH2OH, CH3CH2CH2OH, etc.) are more desired with higher economic value since hydrocarbons such as CH4 and C2H4 are attractive due to their easy transport, convenient storage and high energy value.13 However, it is difficult to tailor the selectivity of hydrocarbons because of multiple reaction pathways and the competing hydrogen evolution reaction (HER) in the CO2RR. In the process of the CO2RR on Cu catalysts, there are two significant factors having crucial effects on the selectivity of hydrocarbons.14,15 On the one hand, the intermediate (i.e., *CO) would be stabilized on the Cu surface to prevent CO release. On the other hand, the formation of hydrocarbon involves multiple proton-coupled electron transfer (PCET) processes, making it essential to accelerate the PCET to improve the sluggish kinetics. However, the high local pH near the surface of the electrode retards the dissociation of water to provide protons.16,17 Moreover, the *CHO intermediate is a key species to generate hydrocarbons in the CO2RR.18,19 Therefore, stabilizing *CO species and activating water are of paramount importance for the hydrogenation of *CO to *CHO to form hydrocarbons.Molecular modification of electrocatalysts exerts a significant effect on the CO2RR.20,21 Molecules such as amino acid,22 poly(acrylamide),23 polypyrrole24 and polyaniline25 can stabilize intermediates to facilitate the selectivity of hydrocarbons through the interaction of hydrogen bonds with *CHO or CO dimer intermediates. According to the density functional theory (DFT) results, hydrogen bonds could decrease the energy barrier for *CO protonation to the *CHO intermediate.26 In addition to stabilizing the intermediate, hydrogen bonds could function as a “proton pump” to promote the transfer of protons by stabilizing OH− of H2O or act as a “bridge” to promote proton transportation in electrochemical reactions.27–30 Thus, we expect that molecules that can construct hydrogen bonds would stabilize the intermediate and boost proton transfer to enhance the selectivity of hydrocarbon in the CO2RR. Among the reported functional groups, –OH or –NH2 is shown to form hydrogen bonds with intermediates.25,31 Moreover, molecules should be firmly modified on the surface of electrodes to maintain stability during electrocatalysis. Taking the above factors into consideration, we select tannic acid (TA, structurally shown in Fig. S1, ESI†) to investigate its effect on hydrocarbon generation in the CO2RR. Note that TA, which has been found widely in plants, features abundant phenolic hydroxyl and carbonyl groups32 and could be coated on nanoparticles steadily via covalent or non-covalent interactions.33 Besides, TA has also been reported to improve the performance of zinc ion batteries and electrocatalysis as a result of hydrogen interaction and adsorption effects.34,35 It is attractive and worth investigating the functionalization effect of TA as a molecular modifier to modulate the selectivity of the Cu electrode, which has not been reported to the best of our knowledge.
In this work, we modify tannic acid molecules on the surface of Cu and investigate their effects on the selectivity of hydrocarbons in the CO2RR. We demonstrate surface TA functionalization to regulate the selectivity over the Cu catalyst and the selectivity of hydrocarbon transformation from C2H4 to CH4 with an increasing amount of modified TA. In situ attenuated total reflection surface-enhanced infrared absorption spectra reveal that TA modification could stabilize *CO and *CHO intermediates and enhance hydrogen bond interaction with H2O near the electrode surface. Kinetic isotope effect experiments verify facilitated dissociation of H2O assisted by TA to promote *CO hydrogenation. DFT calculations further validate that the TA-functionalized Cu catalyst would stabilize the *CO intermediate and reduce the energy barrier for the *CO to *CHO process. This study suggests the promising use of TA as an efficient modifier to enhance the selectivity of hydrocarbons in the CO2RR on Cu catalysts.
Results and discussion
Fig. 1 schematically shows the process of CO2RR on a Cu catalyst with or without TA modification. In the absence of the TA molecule, the primary products of CO2RR on Cu are H2 and C2H4. In the presence of TA on a Cu surface, the adsorbed CO (denoted as *CO), which is generated from CO2 reduction and acts as the intermediate for further reduction, would be stabilized by hydrogen bonds constructed by TA molecules. Besides, H2O can be activated to accelerate dissociation to produce protons (*H) with the assistance of TA molecules. The *CO could then combine with *H to produce *CHO that is an important intermediate for the formation of hydrocarbons. Furthermore, the modification of more TA would activate more H2O to produce more *H, leading to more *CHO intermediate generation. Accordingly, the product selectivity of C2H4 and CH4 is expected to be tailorable by adjusting the amount of modified TA.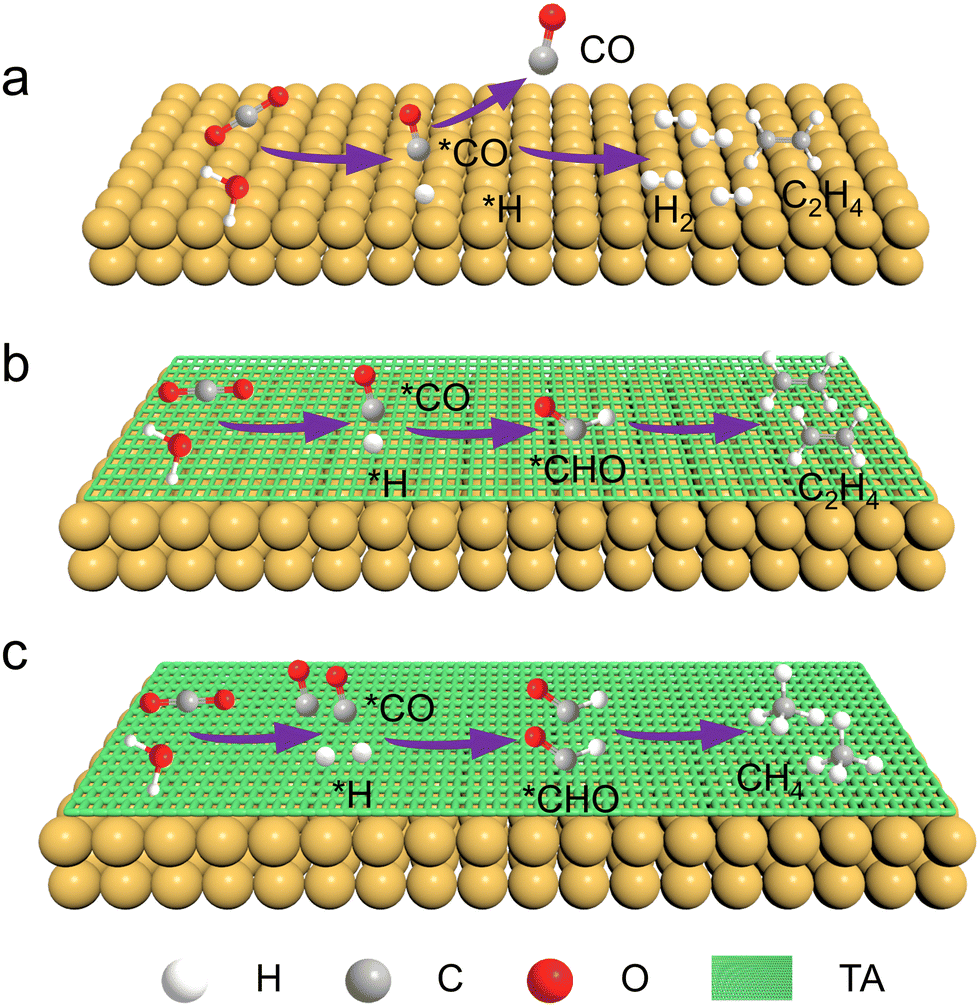 | ||
| Fig. 1 Schematic illustration of CO2RR on the surfaces of (a) Cu, (b) Cu–1TA and (c) Cu–3TA catalysts. | ||
Fig. S2 (ESI†) illustrates the process of the preparation of Cu and Cu–TA nanoparticles (NPs). Firstly, Cu NPs were synthesized by heating a mixture of Cu(OAc)2·H2O and sodium ascorbate at 100 °C for 3 h.36 Then, the obtained Cu NPs were mixed with a desired amount of TA in ethanol solution and ultrasonicated for 0.5 h to prepare Cu–xTA (x represented the designated mass ratio of TA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu). The actual mass fraction of TA in Cu–xTA (x from 0.5 to 4) samples is 22.45%, 31.38%, 48.99%, 58.48% and 65.89%, which is determined by thermogravimetric analysis (TGA, Fig. S3, ESI†). The prepared samples were characterized by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). As shown in Fig. S4 (ESI†) and Fig. 2a, Cu NPs present a uniform spherical shape. From the high-resolution TEM (HRTEM) image (Fig. 2b), the lattice distance is 0.20 nm, which belongs to the (111) crystal plane of Cu. Additionally, selected area electron diffraction (SAED, Fig. 2c) and X-ray diffraction (XRD, Fig. S5, ESI†) indicates the polycrystalline feature of the prepared Cu NPs with the preferentially exposed (111) crystal plane.
:Cu). The actual mass fraction of TA in Cu–xTA (x from 0.5 to 4) samples is 22.45%, 31.38%, 48.99%, 58.48% and 65.89%, which is determined by thermogravimetric analysis (TGA, Fig. S3, ESI†). The prepared samples were characterized by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). As shown in Fig. S4 (ESI†) and Fig. 2a, Cu NPs present a uniform spherical shape. From the high-resolution TEM (HRTEM) image (Fig. 2b), the lattice distance is 0.20 nm, which belongs to the (111) crystal plane of Cu. Additionally, selected area electron diffraction (SAED, Fig. 2c) and X-ray diffraction (XRD, Fig. S5, ESI†) indicates the polycrystalline feature of the prepared Cu NPs with the preferentially exposed (111) crystal plane.
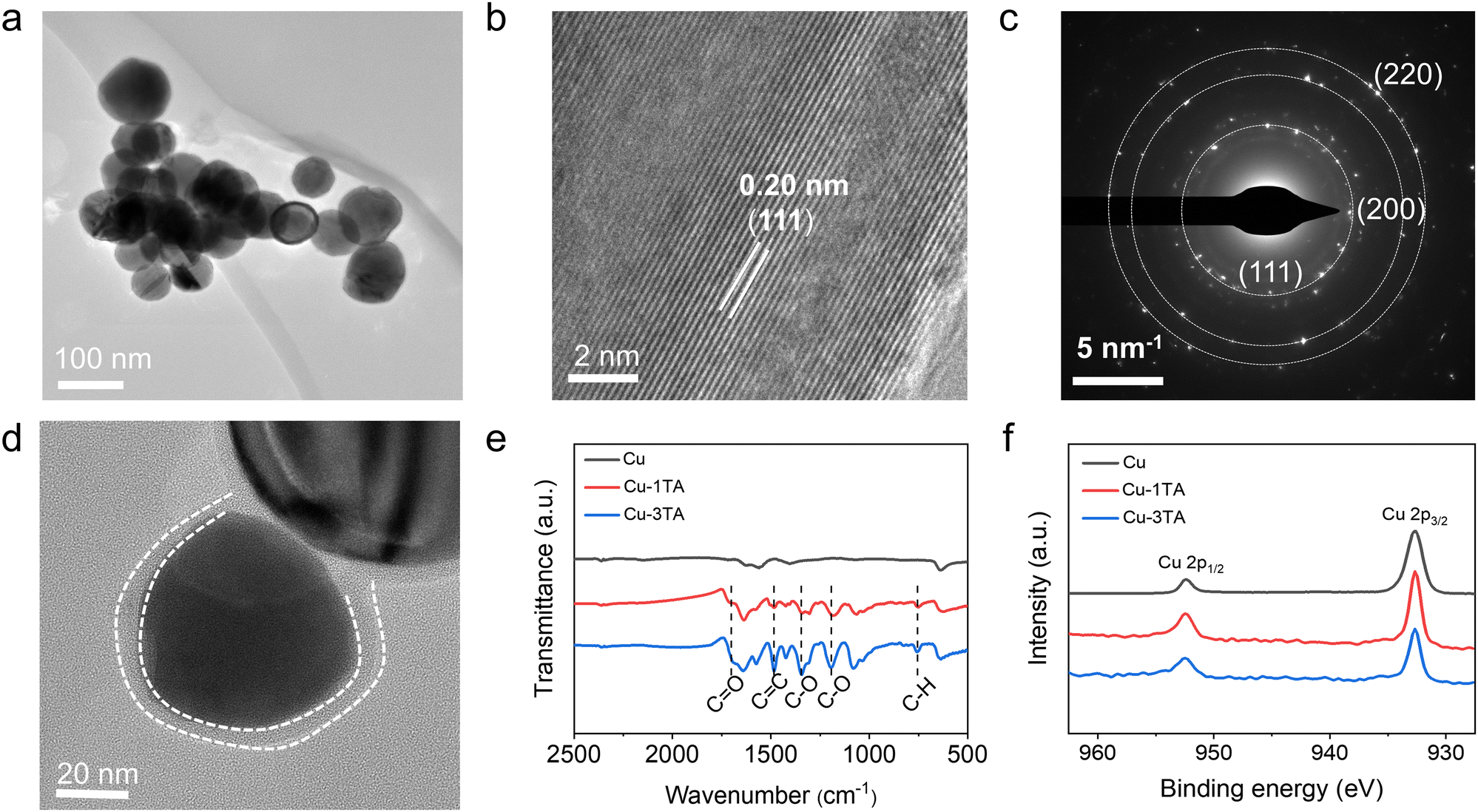 | ||
| Fig. 2 Typical (a) TEM, (b) HRTEM and (c) SAED images of Cu NPs. (d) TEM image of Cu–1TA NPs. (e) FTIR and (f) XPS spectra of Cu, Cu–1TA and Cu–3TA NPs. | ||
Microscopy and spectroscopies were performed to analyse TA modified Cu NPs. From TEM images (Fig. 2d and Fig. S6, ESI†), Cu–1TA and Cu–3TA show a spherical core–shell structure with a coating layer thickness of 4.5 and 12 nm, respectively. Fig. 2e comparatively displays the Fourier transform infrared (FTIR) spectra of Cu, Cu–1TA and Cu–3TA NPs. Compared with neat Cu NPs, the FTIR curves of Cu–TA NPs exhibit characteristic peaks of TA molecules at a wavenumber of 1700 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration), 1483 cm−1 (aromatic CC stretching vibration), 1342 and 1195 cm−1, (C–O stretching), and 759 cm−1 (C–H out-plane bending), confirming the modification of TA on Cu. In addition, we carried out the X-ray photoelectron spectroscopy (XPS) measurements. The full spectrum and high-resolution Cu 2p spectra are shown in Fig. S7 (ESI†) and Fig. 2f, respectively. For Cu and Cu–TA NPs, there are two peaks at 932.5 and 952.5 eV, which could be assigned to signals of Cu 2p3/2 and Cu 2p1/2, respectively. Satellite peaks of Cu2+ are not discernible, indicating good preservation of the metallic state of Cu NPs. The XRD analysis further suggests no apparent difference of the crystal phase between Cu, Cu–1TA and Cu–3TA NPs (Fig. S5, ESI†).
O stretching vibration), 1483 cm−1 (aromatic CC stretching vibration), 1342 and 1195 cm−1, (C–O stretching), and 759 cm−1 (C–H out-plane bending), confirming the modification of TA on Cu. In addition, we carried out the X-ray photoelectron spectroscopy (XPS) measurements. The full spectrum and high-resolution Cu 2p spectra are shown in Fig. S7 (ESI†) and Fig. 2f, respectively. For Cu and Cu–TA NPs, there are two peaks at 932.5 and 952.5 eV, which could be assigned to signals of Cu 2p3/2 and Cu 2p1/2, respectively. Satellite peaks of Cu2+ are not discernible, indicating good preservation of the metallic state of Cu NPs. The XRD analysis further suggests no apparent difference of the crystal phase between Cu, Cu–1TA and Cu–3TA NPs (Fig. S5, ESI†).
The CO2RR activity of Cu and Cu–TA catalysts was evaluated in a gas-tight H-Cell using a Nafion-117 proton exchange membrane (Fig. S8, ESI†). Linear sweep voltammetry (LSV) curves were conducted in Ar or CO2-saturated 0.1 M aqueous KHCO3 solution. The higher current density in CO2-saturated solution than in Ar-saturated solution (Fig. S9, ESI†) indicates the CO2RR activity of prepared catalysts. The electrochemical double layer capacitance, which correlates with the electrochemical surface area (ECSA) is 39.73, 38.64 and 33.67 μF for Cu, Cu–1TA and Cu–3TA, respectively (Fig. S10, ESI†). Thus, the surface modification of TA exerts only a slight impact on the exposed active sites of Cu catalysts. Meanwhile, there is a slight increase of charge transfer resistance (Rct) after TA modification, as shown in the electrochemical impedance spectroscopy (EIS) spectra (Fig. S11, ESI†). Besides, we measured OH− adsorption and desorption behaviour to investigate the influence of TA on Cu NPs. The OH− adsorption potential is related to the crystal planes of Cu NPs37 and the redox potential around 0.54 V in the polarization curves indicates OH− adsorption on the Cu(111) facet.38 As viewed from the discernible positive shift of the anodic peak potential (Fig. S12, ESI†), TA modification retards OH− adsorption on the Cu catalyst. Furthermore, the lower OH− adsorption current again indicates that the OH− adsorption is suppressed on Cu–TA catalysts, which could be explained by the covering of TA on the surface of Cu NPs.
To further evaluate the product distribution for catalysts, on-line gas chromatography (GC) and ex situ nuclear magnetic resonance (NMR) spectroscopy were conducted to determine gas and liquid products (Fig. S13, ESI†), respectively. Fig. S14 (ESI†) shows the faradaic efficiency (FE) of all products and chronoamperometry curves for the Cu catalyst. Main products are H2 and C2H4 and the FE of H2 and C2H4 reach up to 48.01% and 35.46% at −1.2 V, respectively. The high selectivity of H2 decreases the CO2RR activity of the Cu catalyst, which is adverse for the CO2RR. We then tested different Cu–xTA (x from 0.5 to 4) catalysts to determine the optimized TA modification amount. Fig. 3a and b show FE distribution curves of C2H4 and CH4 at −1.2 V and −1.4 V, respectively. Interestingly, the FE of C2H4 decreases and CH4 increases with an increasing amount of modified TA. For the Cu–1TA catalyst, the FE of C2H4 increases to 53.00% from 35.46% at −1.2 V, which shows the highest C2H4 selectivity among the Cu–TA catalysts. Moreover, our results show a superior performance in comparison with reported results among molecule modified Cu–based catalysts (Table S1, ESI†). For the Cu–3TA catalyst, the FE of CH4 reaches 36.32% at −1.4 V, which is about 11 times as large as the unmodified Cu catalyst (FE of CH4 is 3.31% at −1.4 V). The above results indicate the selectivity of C2H4 and CH4 could be steered via adjusting the TA modification amount. Thus, we focus on Cu–1TA and Cu–3TA catalysts to explore the effect of TA modification. The FE of all products and chronoamperometry curves of Cu–1TA and Cu–3TA catalysts at different potentials are shown in Fig. S15 (ESI†) in detail. Fig. 3c and d show FE of C2H4 and CH4 for Cu, Cu–1TA and Cu–3TA catalysts under different potentials, respectively. The results clearly showcase the transformation of selectivity from C2H4 to CH4 with increasing amounts of modified TA and the FE of C2H4 decreases and CH4 increases from the Cu–1TA to the Cu–3TA catalyst. We also performed stability tests and FE of C2H4 still maintains 50.06% after lasting for 14000 s for the Cu–1TA catalyst, revealing that Cu–1TA has considerable stability (Fig. S16, ESI†). Moreover, from the SEM and TEM images collected after electrolysis, there is no apparent morphological evolution for the Cu–1TA catalyst (Fig. S17 and S18, ESI†). The FTIR spectrum further evidences the presence of TA on the Cu surface (Fig. S19a, ESI†) while the XRD pattern reveals the preservation of the metallic Cu phase (Fig. S19b, ESI†).
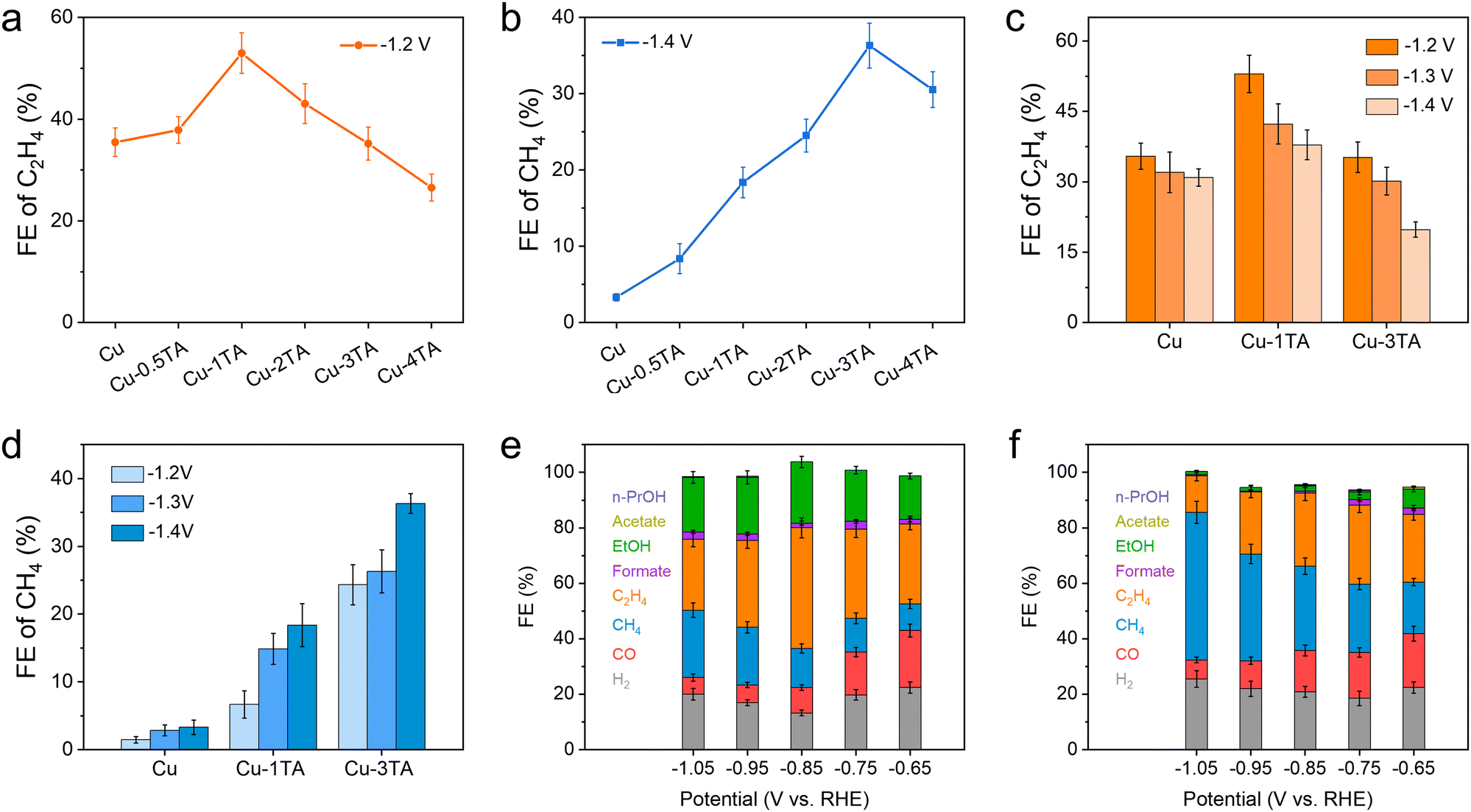 | ||
| Fig. 3 (a and b) Faradaic efficiency (FE) of C2H4 and CH4 on Cu and Cu–xTA (x from 0.5 to 4) catalysts in CO2-saturated 0.1 M KHCO3 solution in H-Cell. (c and d) FE of C2H4 and CH4 on Cu, Cu–1TA and Cu–3TA catalysts under different potentials in H-Cells. (e and f) FE of products distribution on Cu–1TA and Cu–3TA catalysts at different applied potential in 1 M KHCO3 solution in flow cells. | ||
To overcome the limit of CO2 solubility in aqueous solution and achieve higher current density, we evaluated the performance of catalysts in a home-made flow cell in 1 M KHCO3 solution. The working mechanism and EIS spectra of the flow cell are proposed and shown in Fig. S20 and S21 (ESI†), respectively. The chronoamperometry curves of Cu, Cu–1TA and Cu–3TA catalyst under different potentials are displayed in Fig. S22 (ESI†), where all current density surpasses −100 mA cm−2 at applied potentials. The FE of C2H4 is 43.54% at −0.85 V for the Cu–1TA catalyst (Fig. 3e), which is higher than that for the Cu catalyst (the FE of C2H4 is 27.07% at −0.85 V in Fig. S23, ESI†). The sum of FE for C2+ is 65.57% for the Cu–1TA catalyst at −0.85 V. Moreover, the current density of the Cu–1TA catalyst could attain −340 mA cm−2 at −0.85 V and partial current density for the C2+ products is −222.94 mA cm−2. Surprisingly, for the Cu–3TA catalyst (Fig. 3f), the FE of CH4 reaches up to 53.27% and the corresponding current density is about 1000 mA cm−2 at −1.05 V, the partial current density for CH4 can reach up to 532.70 mA cm−2. The result shows the highest partial current density of methane in the reported literature (Table S2, ESI†). The higher FE of CH4 for the Cu–3TA catalyst in a flow cell might be attributed to the stronger buffering ability of 1 M KHCO3 than 0.1 M KHCO3, which is conducive to CH4 formation.39 Fig. S24 and S25 (ESI†) show the representative GC profiles of gaseous products and the corresponding concentration of Cu–1TA and Cu–3TA catalysts, clearly showing the transformation of selectivity between C2H4 and CH4. Fig. S26 (ESI†) shows the standard curves of CH4 and C2H4 components for GC analysis. The product distribution in a flow cell again indicates that the selectivity of C2H4 and CH4 could be tailored with increasing amounts of modified TA.
To clarify the effect of the TA molecule and the possible reaction mechanism of CO2RR in our work, we conducted in situ attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIRS) spectra to monitor intermediate behaviour on Cu, Cu–1TA and Cu–3TA catalysts. The photo and working mechanism of in situ ATR-SEIRS are shown in Fig. S27 (ESI†). From the spectra in Fig. 4a and c, we could see that there are two downward bands located at ∼1738 and ∼2085 cm−1 in the Cu–3TA catalyst, respectively. The band at ∼1738 cm−1 can be indexed as the stretching of *CHO,40,41 gradually increasing as the potentials become more negative. The band at ∼2085 cm−1 is attributed to the linear-bond CO adsorption on the Cu surface.42,43 The *CO intermediate on the Cu–3TA catalyst possesses a stronger signal than the Cu catalyst, which demonstrates that TA modification can stabilize *CO on the Cu surface, which is beneficial for further reaction to form the *CHO intermediate. Thus, the Cu–3TA catalyst has a stronger signal about the *CHO intermediate than the Cu catalyst. Fig. S28a (ESI†) shows the in situ ATR-SEIRS spectra, which reveal the presence of *CO and *CHO intermediates on the Cu–1TA catalyst.
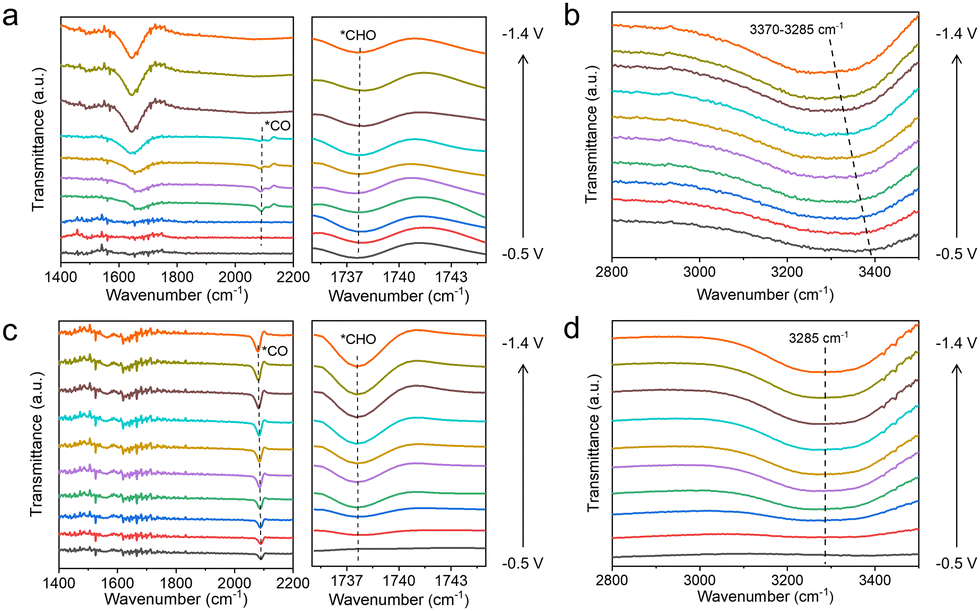 | ||
| Fig. 4 In situ attenuated total reflection surface-enhanced infrared absorption spectroscopy spectra of (a and b) Cu and (c and d) Cu–3TA catalysts in CO2-saturated 0.1 M KHCO3 solution. | ||
The proton, participating in the formation of CO2RR products, comes from the dissociation of H2O44 and it has been reported that hydrogen bonds could promote the proton transfer.45 We then performed in situ ATR-SEIRS spectroscopy to investigate the change of hydrogen bonds after TA modification. For Cu–1TA and Cu–3TA catalysts, the stretching vibration of –OH in H2O shifts to a lower wavenumber at an earlier potential (Fig. 4b, d and Fig. S28b, ESI†). We consider that Cu–TA catalysts could enhance hydrogen bond interaction with H2O and promote proton transfer near the electrode surface. Next, we carried out kinetic isotope effect (KIE) tests to explore how TA effects H2O dissociation on the Cu electrode.46,47 We replace H2O with D2O as the solvent in 1 M KHCO3 solution. The formation rate of C2H4 and CH4 drops more significantly for the Cu catalyst, whereas the formation rates of C2H4 and CH4 decline less for Cu–1TA and Cu–3TA catalysts (Fig. S29, ESI†), respectively. Fig. 5a and b show the calculated KIE values for C2H4 and CH4. The KIE value of C2H4 and CH4 is 2.10 and 2.37 for the Cu catalyst, respectively, suggesting that the proton transfer is a rate-determining process, and the KIE value for Cu–1TA and Cu–3TA catalysts is determined to be 1.44 and 1.10, respectively. The smaller KIE value of Cu–TA catalyst than the Cu catalyst proves that the H2O dissociation process is not the rate-determining step, manifesting that TA-modified Cu could promote H2O to dissociate. The smallest KIE value for Cu–3TA reveals that the modification of more TA can activate more H2O and it could supply more protons for *CO hydrogenation to form the *CHO intermediate. Thus, the selectivity of CH4 is enhanced with increasing amount of modified TA.
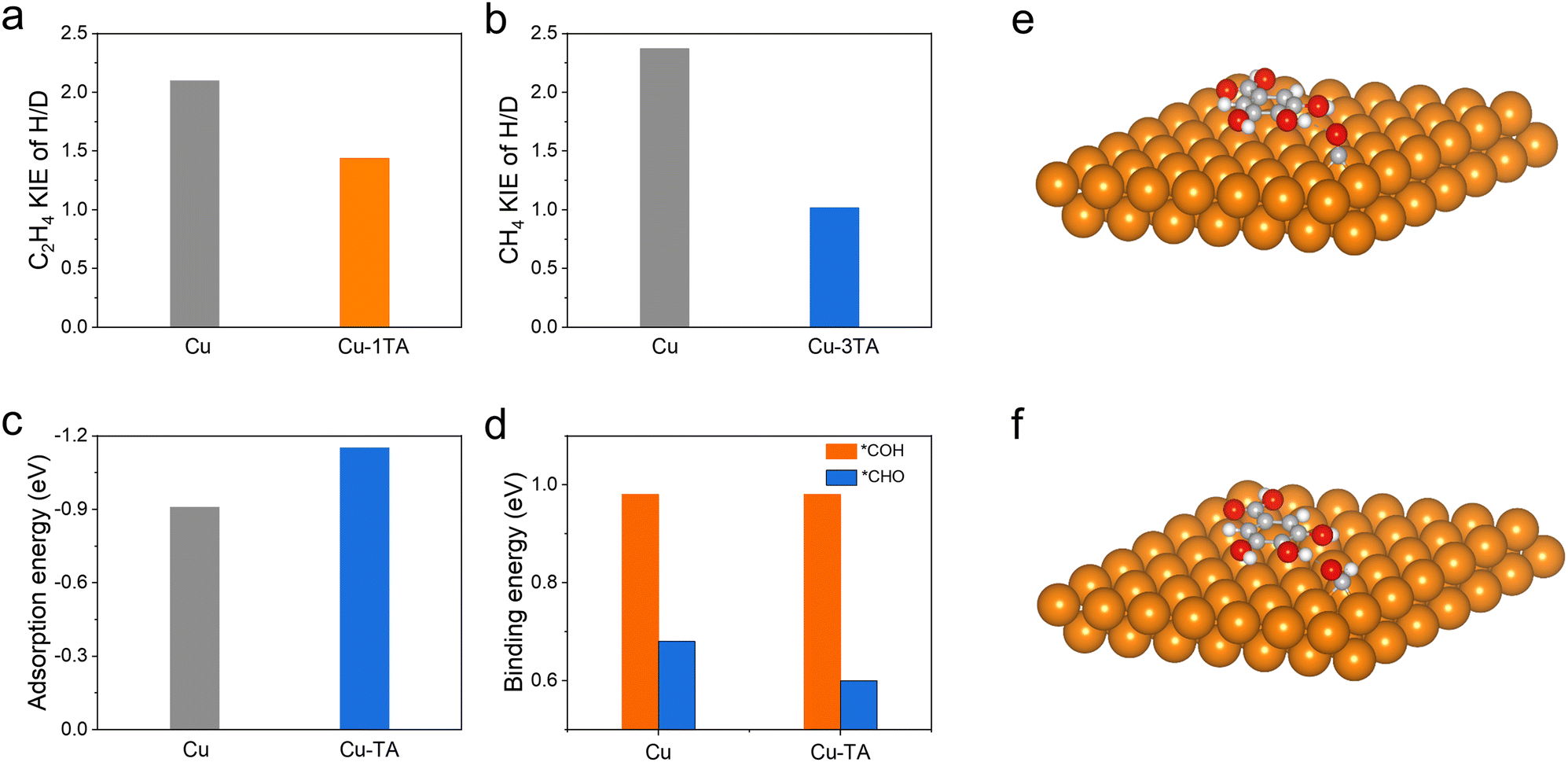 | ||
| Fig. 5 (a) Kinetic isotope effect (KIE) value of C2H4 on Cu and Cu–1TA catalysts at −0.85 V in a flow cell. (b) KIE value of CH4 on Cu and Cu–3TA catalysts at −1.05 V in a flow cell. (c) Calculated adsorption energy of *CO on Cu and Cu–TA catalysts. (d) Binding energy from *CO to *COH or *CHO intermediate on Cu and Cu–TA catalysts. (e and f) Optimized adsorption configuration of *CO and *CHO intermediates on the Cu–TA catalyst. | ||
From the results of in situ ATR-SEIRS and KIE experiments, the TA-functionalized Cu electrode could not only stabilize the adsorbed *CO intermediate, but also activate water dissociation to promote the PCET process. We propose that the *CHO intermediate is prone to form on Cu–TA catalysts as compared to pristine Cu. In the case of less TA modification (i.e., from Cu–0.5TA to Cu–2TA in Cu–xTA), *CO would couple with *CHO to form C2H4 and the Cu–1TA catalyst exhibits the highest C2H4 selectivity. However, more TA modification (i.e., Cu–3TA and Cu–4TA) favours the formation of a hydrogen bond network, which facilitates proton transfer and water dissociation.27,28 More protons would combine with *CO to form more *CHO intermediate while more *CHO species benefits CH4 generation. Therefore, the selectivity between CH4 and C2H4 could be steered via altering the amount of modified TA. Then, we carried out DFT calculations to further unravel the origin of TA-modified Cu for tailoring the selectivity of hydrocarbons in CO2RR. To simplify the TA molecule structure, we selected 3,4,5-trihydroxybenzoic acid as a molecular model because it is the monomer of TA hydrolysis. First, the possible adsorption configuration of TA on the (111) crystal plane of Cu NPs is optimized (Fig. S30, ESI†). Then, we calculated the adsorption free energies of the *CO intermediate on Cu and Cu–TA NPs. The *CO adsorption energy is found to be more negative on Cu–TA NPs (Fig. 5c and e), demonstrating that the *CO intermediate could be stabilized on the Cu surface with TA modification. Next, we compared the binding energy of the *CO to *CHO or *COH process. The optimized adsorption configurations are shown in Fig. 5f, Fig. S31 and S32 (ESI†), respectively. The binding energy from *CO to *COH is identical for Cu and Cu–TA NPs, while it decreases for Cu–TA NPs (Fig. 5d). The result indicates that the protonation process of *CO to *CHO is favoured for TA-modified Cu. From the above theoretical analysis, we confirm TA-modified Cu could stabilize *CO and boost the *CHO formation.
Conclusions
In summary, we report the preparation of TA-modified Cu catalysts by simply mixing and ultrasound preparation for CO2 reduction. By controlling the amount of TA modification, we could achieve a considerable FE of C2H4 (53.00%) for the Cu–1TA catalyst and FE of CH4 (53.27%) for the Cu–3TA catalyst, respectively. In situ ATR-SEIRS spectra show that TA modification could stabilize *CO and *CHO intermediates and enhance the hydrogen bond interaction with H2O near the electrode surface. Kinetic tests further illustrate that TA modification can accelerate H2O dissociation to supply indispensable protons to promote PCET. Furthermore, computational investigation indicates stabilization of the *CO intermediate and lowering of the energy barrier for *CO protonation to *CHO after TA decoration. This study highlights an effective molecule modification strategy to tailor the C2/C1 product selectivity via collectively stabilizing the *CO intermediate and activating H2O dissociation.Author contributions
K. X. and F. C. designed the study. K. X. prepared and characterized the materials, performed measurements, and drafted the manuscript. J. L. carried out DFT computations. F. C. supervised the research and co-wrote the paper. All authors contributed to data analysis and results discussion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by NSFC (21835004, 21925503, 21871149, and 52001172), MOE (B12015), and the Fundamental Research Funds for the Central Universities. Computations were carried out on TianHe-1(A) at National Supercomputer Center in Tianjin.References
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- X. Tan, C. Yu, Y. Ren, S. Cui, W. Li and J. Qiu, Recent advances in innovative strategies for the CO2 electroreduction reaction, Energy Environ. Sci., 2021, 14, 765–780 RSC.
- Y. Yan, L. W. Ke, Y. Ding, Y. Zhang, K. Rui, H. J. Lin and J. X. Zhu, Recent advances in Cu-based catalysts for electroreduction of carbon dioxide, Mater. Chem. Front., 2021, 5, 2668–2683 RSC.
- Y. L. Shi, C. F. Wen, X. F. Wu, J. Y. Zhao, F. X. Mao, P. F. Liu and H. G. Yang, In situ reconstruction of vegetable sponge-like Bi2O3 for efficient CO2 electroreduction to formate, Mater. Chem. Front., 2022, 6, 1091–1097 RSC.
- H. Guo, D.-H. Si, H.-J. Zhu, Q.-X. Li, Y.-B. Huang and R. Cao, Ni single-atom sites supported on carbon aerogel for highly efficient electroreduction of carbon dioxide with industrial current densities, eScience, 2022, 2, 295–303 CrossRef.
- Z. Zhu, Z. Li, J. Wang, R. Li, H. Chen, Y. Li, J. S. Chen, R. Wu and Z. Wei, Improving NiNX and pyridinic N active sites with space-confined pyrolysis for effective CO2 electroreduction, eScience, 2022, 2, 445–452 CrossRef.
- M. B. Ross, P. De Luna, Y. Li, C.-T. Dinh, D. Kim, P. Yang and E. H. Sargent, Designing materials for electrochemical carbon dioxide recycling, Nat. Catal., 2019, 2, 648–658 CrossRef CAS.
- R. Wang, T. Li, R. Gao, J. Qin, M. Li, Y. Guo and Y. Song, Carbonized Yolk-shell Metal-Organic Frameworks for Electrochemical Conversion of CO2 into Ethylene, Chem. Res. Chin. Univ., 2022 DOI:10.1007/s40242-022-2149-z.
- F. You, J. Wan, J. Qi, D. Mao, N. Yang, Q. Zhang, L. Gu and D. Wang, Lattice Distortion in Hollow Multi-Shelled Structures for Efficient Visible-Light CO2 Reduction with a SnS2/SnO2 Junction, Angew. Chem., Int. Ed., 2020, 59, 721–724 CrossRef CAS.
- Z. Tao and H. Wang, Pb3(CO3)2(OH)2 is an Active Phase in Electrocatalytic CO2 Reduction to Formate, Chem. Res. Chin. Univ., 2020, 36, 1145–1146 CrossRef CAS.
- J. Yi, Q. Li, S. Chi, Y. Huang and R. Cao, Boron-doped Covalent Triazine Framework for Efficient CO2 Electroreduction, Chem. Res. Chin. Univ., 2022, 38, 141–146 CrossRef CAS.
- L. Wang, J. Wan, Y. Zhao, N. Yang and D. Wang, Hollow Multi-Shelled Structures of CO3O4 Dodecahedron with Unique Crystal Orientation for Enhanced Photocatalytic CO2 Reduction, J. Am. Chem. Soc., 2019, 141, 2238–2241 CrossRef CAS.
- Y. Wei, F. You, D. Zhao, J. Wan, L. Gu and D. Wang, Heterogeneous Hollow Multi-Shelled Structures with Amorphous-Crystalline Outer-Shells for Sequential Photoreduction of CO2, Angew. Chem., Int. Ed., 2022, 61, e202212049 CAS.
- W. Ma, X. He, W. Wang, S. Xie, Q. Zhang and Y. Wang, Electrocatalytic reduction of CO2 and CO to multi-carbon compounds over Cu-based catalysts, Chem. Soc. Rev., 2021, 50, 12897–12914 RSC.
- B. Talukdar, S. Mendiratta, M. H. Huang and C.-H. Kuo, Recent Advances in Bimetallic Cu-Based Nanocrystals for Electrocatalytic CO2 Conversion, Chem. – Asian J., 2021, 16, 2168–2184 CrossRef CAS.
- W. Ma, S. Xie, X.-G. Zhang, F. Sun, J. Kang, Z. Jiang, Q. Zhang, D.-Y. Wu and Y. Wang, Promoting electrocatalytic CO2 reduction to formate via sulfur-boosting water activation on indium surfaces, Nat. Commun., 2019, 10, 892 CrossRef.
- Y. Li, A. Xu, Y. Lum, X. Wang, S.-F. Hung, B. Chen, Z. Wang, Y. Xu, F. Li, J. Abed, J. E. Huang, A. S. Rasouli, J. Wicks, L. K. Sagar, T. Peng, A. H. Ip, D. Sinton, H. Jiang, C. Li and E. H. Sargent, Promoting CO2 methanation via ligand-stabilized metal oxide clusters as hydrogen-donating motifs, Nat. Commun., 2020, 11, 6190 CrossRef CAS.
- J. H. Montoya, A. A. Peterson and J. K. Nørskov, Insights into CC Coupling in CO2 Electroreduction on Copper Electrodes, ChemCatChem, 2013, 5, 737–742 CrossRef CAS.
- A. J. Garza, A. T. Bell and M. Head-Gordon, Mechanism of CO2 Reduction at Copper Surfaces: Pathways to C2 Products, ACS Catal., 2018, 8, 1490–1499 CrossRef CAS.
- F. Li, A. Thevenon, A. Rosas-Hernández, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Tao, Z.-Q. Liang, M. Luo, X. Wang, H. Li, C. P. O’Brien, C.-S. Tan, D.-H. Nam, R. Quintero-Bermudez, T.-T. Zhuang, Y. C. Li, Z. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Molecular tuning of CO2-to-ethylene conversion, Nature, 2020, 577, 509–513 CrossRef CAS PubMed.
- D.-H. Nam, P. De Luna, A. Rosas-Hernández, A. Thevenon, F. Li, T. Agapie, J. C. Peters, O. Shekhah, M. Eddaoudi and E. H. Sargent, Molecular enhancement of heterogeneous CO2 reduction, Nat. Mater., 2020, 19, 266–276 CrossRef CAS.
- M. S. Xie, B. Y. Xia, Y. Li, Y. Yan, Y. Yang, Q. Sun, S. H. Chan, A. Fisher and X. Wang, Amino acid modified copper electrodes for the enhanced selective electroreduction of carbon dioxide towards hydrocarbons, Energy Environ. Sci., 2016, 9, 1687–1695 RSC.
- S. Ahn, K. Klyukin, R. J. Wakeham, J. A. Rudd, A. R. Lewis, S. Alexander, F. Carla, V. Alexandrov and E. Andreoli, Poly-Amide Modified Copper Foam Electrodes for Enhanced Electrochemical Reduction of Carbon Dioxide, ACS Catal., 2018, 8, 4132–4142 CrossRef CAS.
- Y. Ji, C. Yang, L. Qian, L. Zhang and G. Zheng, Promoting electrocatalytic carbon monoxide reduction to ethylene on copper-polypyrrole interface, J. Colloid Interface Sci., 2021, 600, 847–853 CrossRef CAS.
- X. Wei, Z. Yin, K. Lyu, Z. Li, J. Gong, G. Wang, L. Xiao, J. Lu and L. Zhuang, Highly Selective Reduction of CO2 to C2+ Hydrocarbons at Copper/Polyaniline Interfaces, ACS Catal., 2020, 10, 4103–4111 CrossRef CAS.
- X. Bai, L. Shi, Q. Li, C. Ling, Y. Ouyang, S. Wang and J. Wang, Synergistic Effect of Metal Doping and Tethered Ligand Promoted High-Selectivity Conversion of CO2 to C2 Oxygenates at Ultra-Low Potential, Energy Environ. Mater., 2022, 5, 892–898 CrossRef CAS.
- Y.-R. Wang, M. Liu, G.-K. Gao, Y.-L. Yang, R.-X. Yang, H.-M. Ding, Y. Chen, S.-L. Li and Y.-Q. Lan, Implanting Numerous Hydrogen-Bonding Networks in a Cu-Porphyrin-Based Nanosheet to Boost CH4 Selectivity in Neutral-Media CO2 Electroreduction, Angew. Chem., Int. Ed., 2021, 60, 21952–21958 CrossRef CAS.
- X. Li, B. Lv, X.-P. Zhang, X. Jin, K. Guo, D. Zhou, H. Bian, W. Zhang, U.-P. Apfel and R. Cao, Introducing Water-Network-Assisted Proton Transfer for Boosted Electrocatalytic Hydrogen Evolution with Cobalt Corrole, Angew. Chem., Int. Ed., 2022, 61, e202114310 CAS.
- C.-F. Li, J.-W. Zhao, L.-J. Xie, J.-Q. Wu, Q. Ren, Y. Wang and G.-R. Li, Surface-Adsorbed Carboxylate Ligands on Layered Double Hydroxides/Metal–Organic Frameworks Promote the Electrocatalytic Oxygen Evolution Reaction, Angew. Chem., Int. Ed., 2021, 60, 18129–18137 CrossRef CAS.
- W. Li, F. Li, H. Yang, X. Wu, P. Zhang, Y. Shan and L. Sun, A bio-inspired coordination polymer as outstanding water oxidation catalyst via second coordination sphere engineering, Nat. Commun., 2019, 10, 5074 CrossRef CAS.
- J.-D. Yi, R. Xie, Z.-L. Xie, G.-L. Chai, T.-F. Liu, R.-P. Chen, Y.-B. Huang and R. Cao, Highly Selective CO2 Electroreduction to CH4 by In Situ Generated Cu2O Single-Type Sites on a Conductive MOF: Stabilizing Key Intermediates with Hydrogen Bonding, Angew. Chem., Int. Ed., 2020, 59, 23641–23648 CrossRef CAS.
- W. Yan, M. Shi, C. Dong, L. Liu and C. Gao, Applications of tannic acid in membrane technologies: A review, Adv. Colloid Interface Sci., 2020, 284, 102267 CrossRef CAS.
- T. S. Sileika, D. G. Barrett, R. Zhang, K. H. A. Lau and P. B. Messersmith, Colorless Multifunctional Coatings Inspired by Polyphenols Found in Tea, Chocolate, and Wine, Angew. Chem., Int. Ed., 2013, 52, 10766–10770 CrossRef CAS.
- B. Zhang, L. Qin, Y. Fang, Y. Chai, X. Xie, B. Lu, S. Liang and J. Zhou, Tuning Zn2+ coordination tunnel by hierarchical gel electrolyte for dendrite-free zinc anode, Sci. Bull., 2022, 67, 955–962 CrossRef CAS.
- H. Wang, X. Li, Y. Jiang, M. Li, Q. Xiao, T. Zhao, S. Yang, C. Qi, P. Qiu, J. Yang, Z. Jiang and W. Luo, A Universal Single-Atom Coating Strategy Based on Tannic Acid Chemistry for Multifunctional Heterogeneous Catalysis, Angew. Chem., Int. Ed., 2022, 61, e202200465 CAS.
- S. Thoka, M. Madasu, C.-F. Hsia, S.-Y. Liu and M. H. Huang, Aqueous-Phase Synthesis of Size-Tunable Copper Nanocubes for Efficient Aryl Alkyne Hydroboration, Chem. – Asian J., 2017, 12, 2318–2322 CrossRef CAS.
- G. Zhang, Z.-J. Zhao, D. Cheng, H. Li, J. Yu, Q. Wang, H. Gao, J. Guo, H. Wang, G. A. Ozin, T. Wang and J. Gong, Efficient CO2 electroreduction on facet-selective copper films with high conversion rate, Nat. Commun., 2021, 12, 5745 CrossRef CAS.
- S. Zhang, P. Kang and T. J. Meyer, Nanostructured Tin Catalysts for Selective Electrochemical Reduction of Carbon Dioxide to Formate, J. Am. Chem. Soc., 2014, 136, 1734–1737 CrossRef CAS.
- A. S. Varela, M. Kroschel, T. Reier and P. Strasser, Controlling the selectivity of CO2 electroreduction on copper: The effect of the electrolyte concentration and the importance of the local pH, Catal. Today, 2016, 260, 8–13 CrossRef CAS.
- L. Xiong, X. Zhang, H. Yuan, J. Wang, X. Yuan, Y. Lian, H. Jin, H. Sun, Z. Deng, D. Wang, J. Hu, H. Hu, J. Choi, J. Li, Y. Chen, J. Zhong, J. Guo, M. H. Rümmerli, L. Xu and Y. Peng, Breaking the Linear Scaling Relationship by Compositional and Structural Crafting of Ternary Cu–Au/Ag Nanoframes for Electrocatalytic Ethylene Production, Angew. Chem., Int. Ed., 2021, 60, 2508–2518 CrossRef CAS.
- L. Xiong, X. Zhang, L. Chen, Z. Deng, S. Han, Y. Chen, J. Zhong, H. Sun, Y. Lian, B. Yang, X. Yuan, H. Yu, Y. Liu, X. Yang, J. Guo, M. H. Rümmeli, Y. Jiao and Y. Peng, Geometric Modulation of Local CO Flux in Ag@Cu2O Nanoreactors for Steering the CO2RR Pathway toward High-Efficacy Methane Production, Adv. Mater., 2021, 33, 2101741 CrossRef CAS.
- Z. Gu, H. Shen, Z. Chen, Y. Yang, C. Yang, Y. Ji, Y. Wang, C. Zhu, J. Liu, J. Li, T.-K. Sham, X. Xu and G. Zheng, Efficient Electrocatalytic CO2 Reduction to C2+ Alcohols at Defect-Site-Rich Cu Surface, Joule, 2021, 5, 429–440 CrossRef CAS.
- S. Zhu, B. Jiang, W.-B. Cai and M. Shao, Direct Observation on Reaction Intermediates and the Role of Bicarbonate Anions in CO2 Electrochemical Reduction Reaction on Cu Surfaces, J. Am. Chem. Soc., 2017, 139, 15664–15667 CrossRef CAS.
- M. C. O. Monteiro, F. Dattila, B. Hagedoorn, R. García-Muelas, N. López and M. T. M. Koper, Absence of CO2 electroreduction on copper, gold and silver electrodes without metal cations in solution, Nat. Catal., 2021, 4, 654–662 CrossRef CAS.
- M. E. Tuckerman, D. Marx and M. Parrinello, The nature and transport mechanism of hydrated hydroxide ions in aqueous solution, Nature, 2002, 417, 925–929 CrossRef CAS.
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Electrocatalytic reduction of CO2 to ethylene and ethanol through hydrogen-assisted C–C coupling over fluorine-modified copper, Nat. Catal., 2020, 3, 478–487 CrossRef CAS.
- X. Yan, C. Chen, Y. Wu, S. Liu, Y. Chen, R. Feng, J. Zhang and B. Han, Efficient electroreduction of CO2 to C2+ products on CeO2 modified CuO, Chem. Sci., 2021, 12, 6638–6645 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2qm01259f |
| This journal is © the Partner Organisations 2023 |