
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Group 14 metallole dianions as η5-coordinating ligands
Xiaofei
Sun
 and
Peter W.
Roesky
*
and
Peter W.
Roesky
*
Institute for Inorganic Chemistry, Karlsruhe Institute of Technology (KIT), Engesserstr.15, 76131, Karlsruhe, Germany. E-mail: roesky@kit.edu
First published on 8th September 2023
Abstract
As heavier aromatic analogs of the cyclopentadienides, group 14 dianionic metalloles exhibit more versatile reactivity and coordination modes due to the additional lone pair at the heteroatom. Compared to the well-established chemistry of monoanionic cyclopentadienide ligands, the coordination chemistry with those dianionic ligands remains underexplored. This perspective provides an overview of literature-known examples of group 14 metallole dianions (silole, germole, stannole and plumbole) adopting η5-coordinating modes. The diverse coordination modes and reactivity exhibited by these compounds highlight their potential as intriguing ligands in organometallic chemistry.
 Xiaofei Sun (right) and Peter W. Roesky (left) | Xiaofei Sun obtained her BSc in 2016 and MSc in 2018 at Karlsruhe Institute of Technology (KIT). She joined the group of Professor Peter W. Roesky in 2018 to focus on group 14 chemistry and completed her doctoral degree in 2022. In 2023, she received the Südwestmetall Award for her doctoral thesis. Currently, she continues her work in the same group to further advance low-valent group 14 chemistry research. Peter W. Roesky obtained his diploma in chemistry in 1992 from the University of Würzburg and his doctoral degree from the Technical University of Munich in 1994. After postdoctoral work at Northwestern University, USA (1995–1996), he completed his habilitation at the University of Karlsruhe in 1999. He was appointed a full professor at the Freie Universität Berlin in 2001, during which he joined the faculty of chemistry and biochemistry. In 2008, he became a full Professor of inorganic functional materials at the Karlsruhe Institute of Technology (KIT). From 2013 to 2015, he served as Dean of the Faculty of Chemistry and Biosciences at KIT. His current research interest revolves around the synthetic inorganic and organometallic chemistry of s-block metals, silicon, phosphorus, gold, and lanthanides. |
1. Introduction
Dianionic group 14 metalloles (metallacyclopentadienediides) with the general formula of [C4R4E]2− (E = Si–Pb) are isolobal to the ubiquitous cyclopentadienide (Cp−) monoanions (Fig. 1). Despite this close relationship, the coordination chemistry of such dianions has remained significantly underexplored. While the first generation of dialkalisiloles and -germoles was reported over 30 years ago,1–4 recent advances of synthetic methods have facilitated further reactivity studies of those species.5–8 Unlike the fully carbon-containing Cp− adopting the conventional η5-coordination mode, heavy group 14 dianionic compounds exhibit more versatile reactivity and coordination modes due to the additional lone pair at the heteroatom. Quantum chemical calculations and experimental investigation reveal that the dianionic group 14 metalloles also exhibit aromatic character akin to the parent Cp−.9–12 The substitution of a ring C atom by a heteroatom result in a change in terms of the electronic properties. Moreover, the two negative charges on the metallole dianions facilitate the coordination of two metals in μ–η5:η5- or μ-η1:η5-fashions, which is rarely achieved using monoanionic Cp− derivatives. As a new generation of heterocyclic species, current research is primarily focusing on utilizing those metallole dianions as ligands in coordination chemistry and uncovering novel structures by X-ray diffraction analysis. Although additional applications, for example in catalysis and as single molecular magnets (SMMs), have recently emerged, the primary emphasis remains on structural discovery.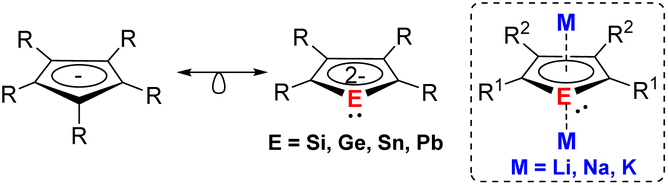 | ||
| Fig. 1 Isolobal analogy between cyclopentadienide and dianionic metallole. | ||
Several previous comprehensive review articles already exist dealing with the properties, synthesis and reactivity of those compounds.9–12 This perspective specifically summarizes the literature-known examples of η5-coordination modes of the metallole dianions.†
2. Main group compounds
In 1990, Joo and co-workers for the first time reported the synthesis of the disodium salt of tetraphenylsilole by reduction of dichlorosilole with Na metal.3 In the following decade, a handful of structurally authenticated germole and silole dianionic complexes have been reported. The X-ray structure of the lithium derivative [Li2]1 (Fig. 2) was reported for the first time by West.8[Li2]1 crystallizes as a monomer, with the two Li cations showing different coordination modes toward the silole ring (η1 and η5). Simultaneously, the tetramethylsilole dianion [K2]2 (Fig. 2) was isolated and the X-ray structure was disclosed.1,2 Compared to [Li2]1, in [K2]2 the two K cations are both coordinated by the silole ring in η5-fashion and are complexed by 18-crown-6, which resulted in an inversed sandwich structure. Interestingly, in 1996, West obtained the X-ray structures for the dilithium salt of the tetraphenylgermole [Li2]3a and [Li2]3b (Fig. 2) from dioxane.4 Depending on the crystallization temperature, the Li cations show distinct coordination modes ([Li2]3a:η5:η5, [Li2]3b:η1:η5) towards the germole ring. Also in 1996, in a comprehensive study about silole and germole compounds by Tilley, Rheingold and co-workers, the structure of the bis(germole) dianion complex [K2]4 was reported.1 Later on the tetraethylgermole dianion [Li2]5 was also structurally characterized.13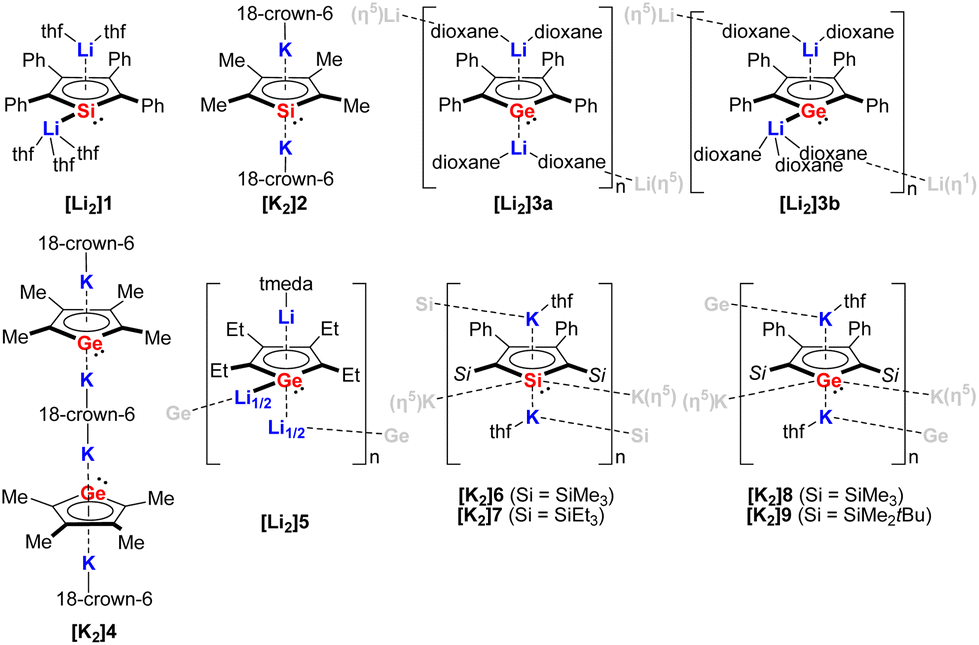 | ||
| Fig. 2 Structurally characterized silole and germole dianion complexes.1,2,4,5,8,13 | ||
[Li2]5 crystallizes as a one-dimensional coordination polymer with the germole ring being η5-coordinated by two Li atoms and η1-coordinated by one Li atom via the Ge lone pair. At that point, the scope of structurally characterized silole and germole dianions is only restricted to those possessing alkyl or aryl substituents.
In 2018, the Müller group reported the efficient multigram-synthetic protocol of the 2,5-trialkylsilyl-substituted dipotassium siloles [K2]6 and [K2]7 as well as germoles [K2]8 and [K2]9 (Fig. 2) from reduction of their corresponding neutral dihalides.5 The dipotassium salts [K2]6–[K2]9 possess very similar solid-state structures, all of them crystallize as one-dimensional coordination polymers, the silole rings are connected to each other with η1- and η5-coordinating potassium ions. This high-yield synthesis of the dianions has greatly advanced the chemistry to a new level and several fascinating novel compound classes could be approached by using [K2]6–[K2]9 as starting materials.9 For instance, Albers and Müller obtained a neutral aluminocene 10 (Scheme 1) from the reaction of [K2]8 with [Cp*AlCl2] (Cp*− = pentamethylcyclopentadienide).14 Crystallographic studies demonstrated that it adopts a classical sandwich structure with the Al center positioned between the η5-coordinating Cp* and germole ligands. Quantum chemical calculations showed that the Ge atom features an uncoordinated lone pair and a low-lying acceptor orbital, indicating its germylene character.
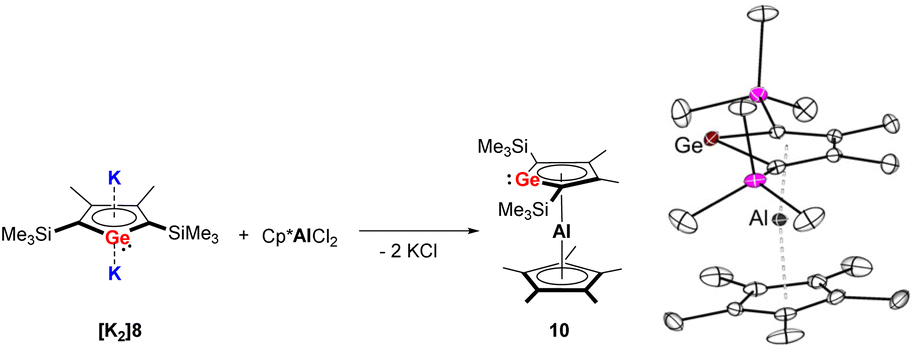 | ||
| Scheme 1 Left: Synthesis of the germaaluminocene 10; right: molecular structure of 10 in the solid state with thermal ellipsoids at 40% probability. Reproduced from the CIF file having CCDC no. 1969078.14 | ||
The dianions of the heaviest tin and lead analogs have remained elusive until the studies carried out by the Saito group. It was not until 2002 when the first synthesis of the dilithio-tetraphenylstannole [Li2]11 (Fig. 3) was reported by reduction of the neutral bi(1,1-stannole) with lithium.15 At that time, [Li2]11 could not be isolated in pure form, but trapped in the reaction with methyl iodide. In 2005, Saito succeeded in the isolation of [Li2]11 as deep-red crystals in 98% yield.16 The two Li cations are located above and below the stannole ring and each Li atom is coordinated to the stannole moiety in a η5-mode and to one Et2O molecule in η1-fashion. The aromaticity of the stannole dianion was confirmed by X-ray diffraction analysis (bond lengths, planarity), NMR spectroscopy and nucleus independent chemical shift (NICS) calculations. Shortly after this initial report, the same research group published two additional reports describing the synthesis and structure of novel stannole dianions: the dilithio-tetraethylstannole [Li2]12,17 and the dilithio-2,5-bis(silyl)-3,4-diphenylstannoles [Li2]13 and [Li2]14 (Fig. 3).6[Li2]12 was isolated as black–yellow crystals in 57% yield that crystallizes as a one-dimensional coordination polymer, wherein one Li cation is coordinated by the stannole ring an η5-fashion and one Et2O molecule an η1-fashion. The other Li cation displays η5-coordination with one stannole and η1-coordination with the Sn atom of another stannole, effectively linking the infinite polymer chain. The solid-state structures of the silyl-functionalized stannole dianions [Li2]13 and [Li2]14 bear resemblance to that of [Li2]11, exhibiting inverse-sandwich structures with two Li cations being η5-coordinated by the stannole unit and each of them coordinated by a THF molecule.
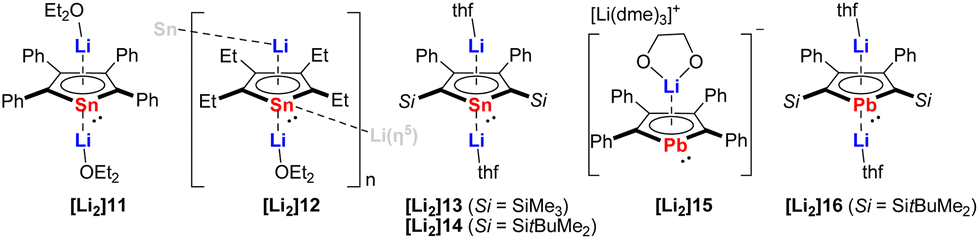 | ||
| Fig. 3 Dilithiostannole and -plumboles from Saito.6,7,15–18 | ||
Five years after the isolation of the first stannole dianion,16 Saito reported the synthesis and structure of the heaviest group 14 congener of the metallole dianions, namely, the dilithiotetraphenylplumbole [Li2]15 (Fig. 3).18 The synthesis of [Li2]15 was accomplished analogously to that of the lighter analogs by reduction of the neutral metallole ring with elemental Li. Complex [Li2]15 adopts a charge-separated structure upon crystallization from DME. In the anionic part, the Li atom is η5-coordinated by the planar plumbole ring while the cationic part is composed of a DME-solvated Li atom. The authors further introduced bulky silyl groups in the 2,5-positions of the ring and synthesized another dilithioplumbole [Li2]16 (Fig. 3)7 which has a similar monomeric solid-state structure as the dilithiostannoles [Li2]11, [Li2]13 and [Li2]15.
In recent work by Murugesu, the scope of the plumbole dianion 162− was expanded to its disodium and dipotassium salts, that are formed by reduction of the neutral plumbole with elemental Na or K (Scheme 2).19 Depending on the solvent used for crystallization, the solid-state structures are either monomeric ([Na2]16a) or polymeric ([Na2]16b, [K2]16a, [K2]16b). This breakthrough clearly showed that the stabilization of the dianionic plumbole ring does not necessarily rely on the η5-coordination of lithium.
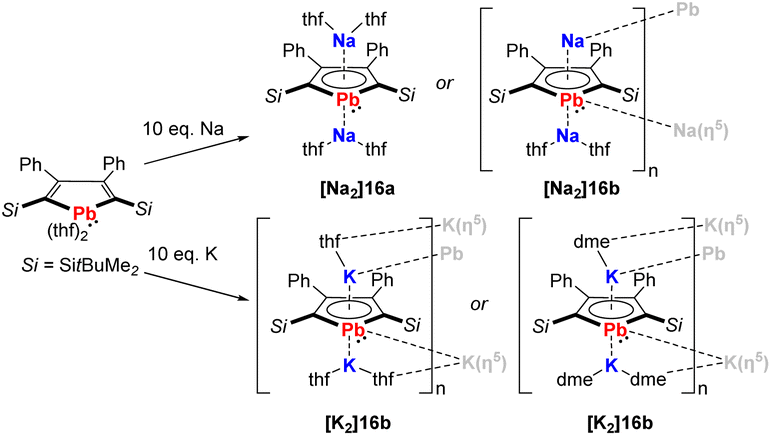 | ||
| Scheme 2 Synthesis of the disodium and dipotassium plumboles.19 | ||
A very nice example illustrating the high reducing ability of the metallole dianions is the reduction of pentaphosphaferrocene and pentaarsaferrocene by the dilithioplumbole [Li2]16 (Scheme 3).20 As products, the heterometallic Fe–Li triple-decker compounds 17 and 18 were isolated, which are composed of Cp* and plumbole as end-decks and envelope-shaped cyclo-E5 (E = P, As) units as middle decks. The plumbole rings maintained their planarity but the C–C bond lengths displayed alternating single and double bond character, therefore they are best described as plumbacyclopentadienylidenes.
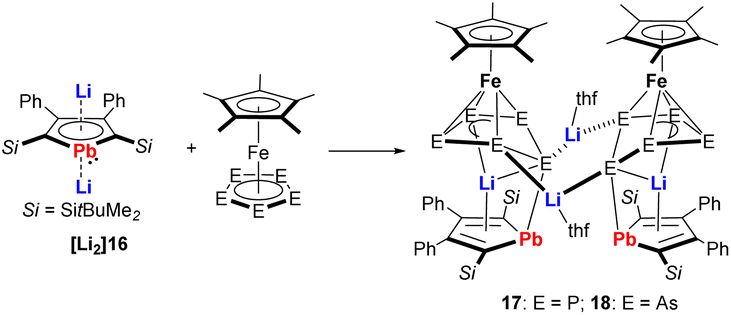 | ||
| Scheme 3 Reaction of dilithioplumbole [Li2]16 with pentaphospha- and pentaarsaferrocene.20 | ||
3. Transition metal compounds
Starting from the lithium salt of the germole monoanion, the first transition metal complex 19 (Scheme 4) comprising a germole dianion was prepared unexpectedly by reaction with [Cp*HfMe2Cl] via elimination of Me3SiCl. The latter compound further reacts with one more equivalent of the germole monoanion to form the Hf(IV) germole complex 19 and bis(trimethlylsilyl) germole as side product.21 The Hf(IV) germole complex 19 crystallized as a dimer with one of the two Li cations being sandwiched between the two germole dianions in η5-fashion while the other one is coordinated with both Ge atoms of the two germole rings.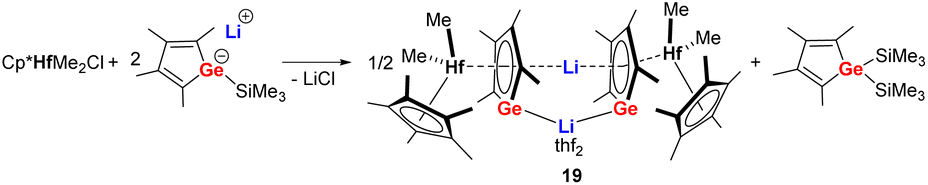 | ||
| Scheme 4 Synthesis of the germole Hf(IV) complex 19.21 | ||
Surprisingly, after this very initial report from the Tilley group in 2000, for a long time, no transition metal compounds with dianionic group 14 metallole ligand was reported, despite the fact that dialkalimetalloles should be ideal building blocks to construct double- or multi-decker complexes by simple salt metathesis reaction with suitable metal halide precursors.
This situation changed after Saito succeeded in the synthesis and isolation of the dilithium salts of stannole and plumbole. Using the dilithiostannoles [Li2]13 and [Li2]14 bearing silyl and phenyl substituents, Saito and co-workers prepared the neutral triple-decker ruthenocenes 20 and 21 (Fig. 4) with Cp* ligands as end-decks and the middle stannole dianion coordinating the two Ru(II) metals in μ-η5:η5 mode.22 This is a remarkable example since similar triple-decker species [(Cp-M)(μ-η5:η5-Cp)(M-Cp)]+ (M = Fe, Ru, Ni)28,29 composed of three Cp ligands are cationic and the introduction of such heterocycles with two negative charges enabled for the first time the isolation of a neutral analog. By changing the amount of [Cp*RuCl]4, one Cp*Ru unit could also be incorporated to form the anionic stannole–Cp* double decker 22 (Fig. 4). More recently, the anionic ruthenocene 23 (Fig. 4) comprising the heaviest group 14 derivate, the plumbole dianion, was also reported.23 NMR studies reveal that upon coordination of the plumbole to the Ru(II) center, the 207Pb NMR signal appears at lower frequencies (1552 ppm) compared to that of [Li2]16 (2573 ppm). Calculations suggest that the difference is mainly attributed to the back-donation from Ru to the plumbole ring, which means an increase of electron density in the latter moiety is observed that leads to magnetic shielding on the nuclei in the plumbole ring.
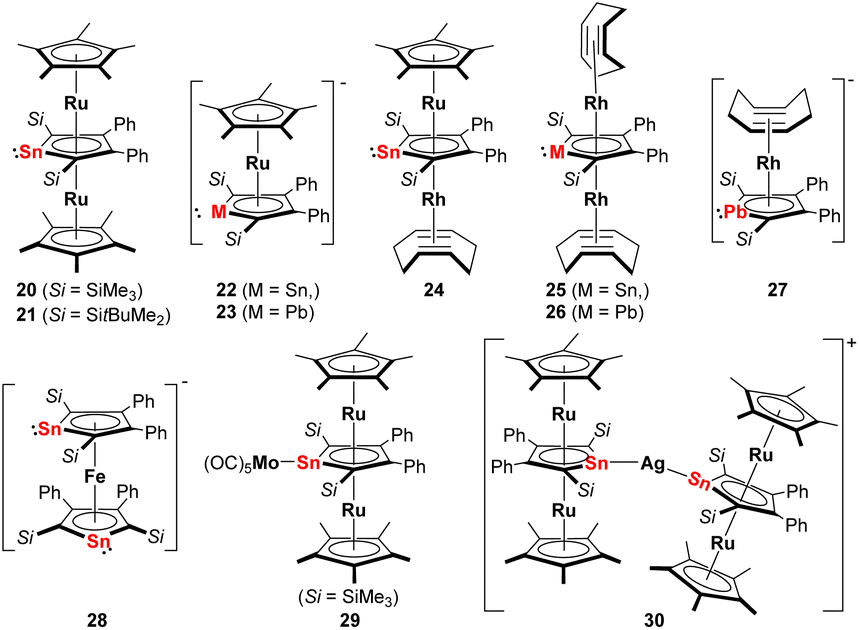 | ||
| Fig. 4 Late transition metal complexes with η5-coordinating dianionic stannoles or plumboles; Si = SitBuMe2, unless otherwise specified.22–27 | ||
Using the anionic ruthenocene 22 as precursor, the heterobimetallic Ru–Rh stannole triple-decker complex 24 (Fig. 4) could be accomplished.25 By decomposition of 24 in ionic liquids [BMIm][BF4] or [BMIm][NTf2] (BMIm = 1-butyl-3-methyl-imidazolium; NTf2 = bis(trifluoromethylsulfonyl)imide), the corresponding metal nanoparticles were prepared. The ionic liquids enabled the formation of non-agglomerated metal nanoparticle dispersion without the addition of capping ligands, polymers, or surfactants. For better comparison, the same group also synthesized the birhodium triple-decker species 25 (Fig. 4) by reacting [Li2]14 with [Rh(cod)Cl]2 (cod = cyclooctadiene).25 Analogously, the birhodium plumbole triple-decker complex 26 (Fig. 4) was prepared one year later.27 In all these complexes, the stannole or plumbole rings coordinate the Ru(II) or Rh(I) atoms in an η5-fashion. Due to the bulky substituents on the metallole rings, the two η4-coordinating cod ligands in 25 and 26 are in a staggered arrangement. When the amount of [Rh(cod)Cl]2 in the reaction with [Li2]16 was reduced, the anionic Rh(I) complex 27 (Fig. 4) has also been achieved. Among those, the neutral plumbole complex 26 revealed high catalytic activity in the [2 + 2 + 2] cycloaddition of triynes, enabling the synthesis of benzene derivatives. Notably, the catalytic activity of the plumbole complex 26 surpasses that of [CpRh(cod)] which highlights the potential application of dianionic group 14 metalloles as an alternative to conventional Cp ligands in catalytic application.
Another intriguing example of applying metallole dianions in transition metal chemistry is the synthesis of the anionic stannaferrocene 28 which could be obtained from reaction of the respective dilithiostannole [Li2]13 with [Fe(acac)3] (acac = acetylacetonate).26 Complex 28 was isolated from a THF/n-pentane mixture as dark purple crystals in 15% yield. The molecular structure (Fig. 5) clearly demonstrates that the central Fe ion is sandwiched between the two planar η5-stannole rings, therefore exhibiting great structural resemblance to the archetypical sandwich compound ferrocene. To minimize steric hindrance, the two nearly identical stannole moieties adopt gauche conformation. Due to the strong reducing nature of the dilithiostannole, the formal oxidation state of the iron center should be +1, as evidenced by Mössbauer spectroscopy and quantum chemical calculations, resulting in formally mono anionic ligands.
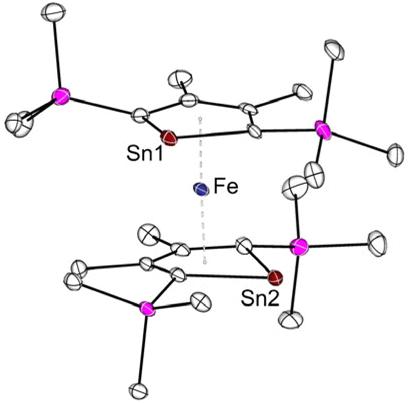 | ||
| Fig. 5 Molecular structure of the anion of Saito's stannaferrocene 28 in the solid state with thermal ellipsoids at 40% probability. Reproduced from the CIF file having CCDC No. 1859348.26 | ||
More recently, the Saito group employed the triple-decker ruthenocene compounds 20 and 21 as novel stannylene ligands for other transition metals.24 The reaction of 20 with [Mo(CO)6] furnished the triple-decker Mo(CO)5 complex 29 (Fig. 4) in which the stannole lone pair coordinates the Mo(0) moiety. Upon complexation, the geometric parameters of the triple-decker fragment remain almost unchanged. Alternatively, the Ag complex 30 that is linearly coordinated by two triple-decker units, could also be synthesized by reacting two equivalents of 21 (Fig. 5) with a Ag(I) carborane. The two triple-decker motifs in 30 are oriented perpendicularly to each other due to steric congestion arising from the bulky silyl substituents. This efficiently showcased that multi-decker metallole complexes can act as strongly electron-donating tetrylene ligands by coordinating the lone pair toward a second metal fragment in η1-fashion.
The reactivity of the metallole dianions is influenced not solely by the heteroatom but also by the ring substituents and the solvent used in the reaction. As discussed before, sandwich-type Ru(II) stannole complexes 20, 21 and 22 were obtained when the silyl-functionalized stannole dianion was employed. However, using the tetraethyl stannole dianion and the same Ru(II) starting material [Cp*RuCl]4, dinuclear Ru(II) species with butterfly of inverse-sandwich structures were isolated.30 During the investigation of the reactivity of the dilithiostannoles with the early transition metal precursor [Cp2HfCl2], it turned out that the outcome of the reaction depends on the solvent which was used.31,32 For instance, when the bulky silyl-functionalized dilithiostannole [Li2]14 was employed, the salt elimination reaction in toluene led to the formation of a dinuclear Hf(IV) complex in which the two Hf(IV) fragments are both coordinated by the stannole in η1-fashion.32 In sharp contrast, the same reaction in THF resulted in the formation of a pyramid-like Hf(IV) complex.31 The analogous reaction of the tetraethylstannole dianion [Li2]12 with [Cp2TiCl2] gave the six-membered Sn4Ti2 ring complex 31 or the three-membered Sn2Ti ring complex 32 depending on the molar ratio of the two starting materials (Scheme 5).33 During the reaction some redox processes were taking place leading additionally to unidentified low-valent Ti(III) compounds. 31 can be regarded as the reduced version of 32 and was alternatively obtained quantitatively by the reduction of 32 with dilithiostannole [Li2]12. In complex 31, the two lithocene moieties are connected with each other via the two bridging Cp2Ti fragments.
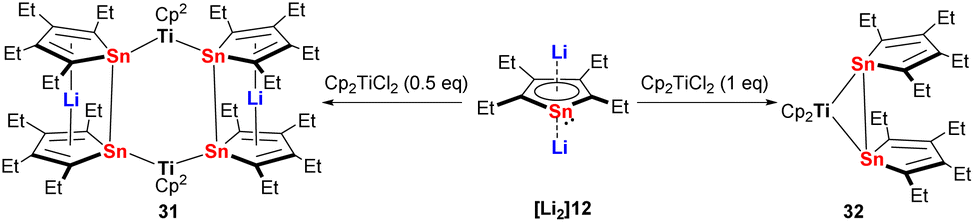 | ||
| Scheme 5 Reaction of tetraethyldilithiostannole [Li2]12 with [Cp2TiCl2], leading to the formation of 31 and 32.33 | ||
When the germole dianion [K2]8 was reacted in a similar procedure with group 4 metallocene dichlorides [Cp2MCl2] (M = Ti, Zr, Hf) (Scheme 6), the outcome of the reaction is different compared to the heavier Sn analog.34–36 In the case of Hf, the bicyclic germylene 34 was formed via anionic hafnocene germylene intermediate [K]33c.36 For the lighter metals Ti and Zr, the dimeric Zr(IV) germole complex 36 or Ti(III) complex 37 were isolated (Scheme 6).35 The formation of the latter compound proceeds via the intermediate 35, which was isolated in small amounts. When the Hf compounds [K]33c or 34 were further used in the reactions with the tetramethylimidazolylidene or [PPh3AuCl], the ligand underwent rearrangement.34
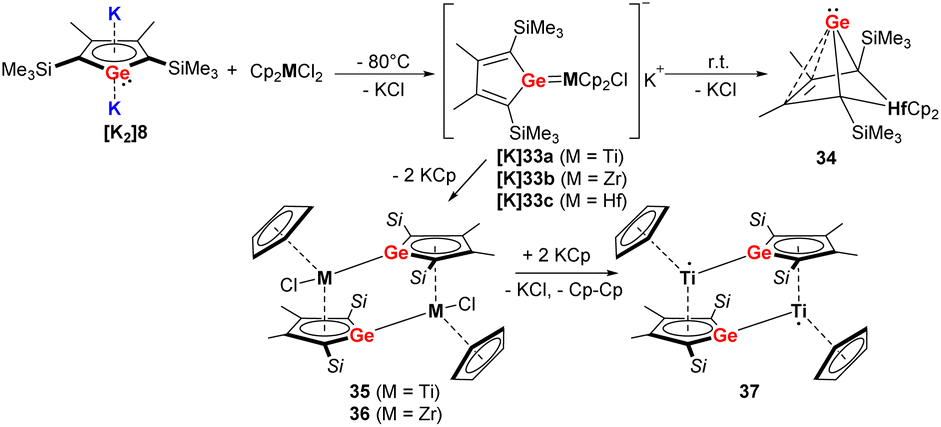 | ||
| Scheme 6 Reaction of Müller's dipotassiogermole [K2]8 towards group 4 metallocene dichlorides.35,36 | ||
4. Rare-earth compounds
The reactivity of dianionic metalloles has already been investigated to some extent, particularly in relation to transition metals. However, in comparison, rare-earth complexes comprising group 14 metallole dianions are extremely scarce. Progress has been made in this area since 2021 when Tan and co-workers expanded the coordination chemistry of the metallole dianions to rare-earth elements and isolated a series of yttrium germole complexes 38–40 (Fig. 6).37 The reaction between the dipotassium germole [K2]8 with YCl3 in equimolar ratio in THF did not lead to the conventional salt elimination product. Instead, the dianionic yttrium germole salt [(η5–8)YCl3][K(2.2.2-crypt)]2 (38) was isolated as yellow crystals after the addition of 2.2.2-cryptand to the reaction mixture. The Y(III) atom in 38 is coordinated by the germole dianion in an η5-fashion with a Y–CtGermole distance of 2.331 Å. When the molecular ratio between [K2]8 and [YCl3(thf)3.5] was changed to 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and the reaction was performed in toluene at 80 °C, the salt metathesis proceeded and complex 39 could be isolated as dark brown crystals in 24% yield. Complex 39 crystallizes as a dimer with each Y center being η5-coordinated to two trans-oriented germole dianions. In addition, each Y(III) atom exhibits an additional η1-coordination with the neighboring germole ring. Moreover, the two K atoms are coordinated in an η5-fashion to the germole ring and in an η6-fashion to a toluene molecule. A similar dimeric dinuclear Y species 40 was obtained by the reaction of [K2]8 with [Cp*YCl2], the two yttrium centers are both coordinated by one germole moiety and one Cp* moiety in η5-interaction modes.
:1 and the reaction was performed in toluene at 80 °C, the salt metathesis proceeded and complex 39 could be isolated as dark brown crystals in 24% yield. Complex 39 crystallizes as a dimer with each Y center being η5-coordinated to two trans-oriented germole dianions. In addition, each Y(III) atom exhibits an additional η1-coordination with the neighboring germole ring. Moreover, the two K atoms are coordinated in an η5-fashion to the germole ring and in an η6-fashion to a toluene molecule. A similar dimeric dinuclear Y species 40 was obtained by the reaction of [K2]8 with [Cp*YCl2], the two yttrium centers are both coordinated by one germole moiety and one Cp* moiety in η5-interaction modes.
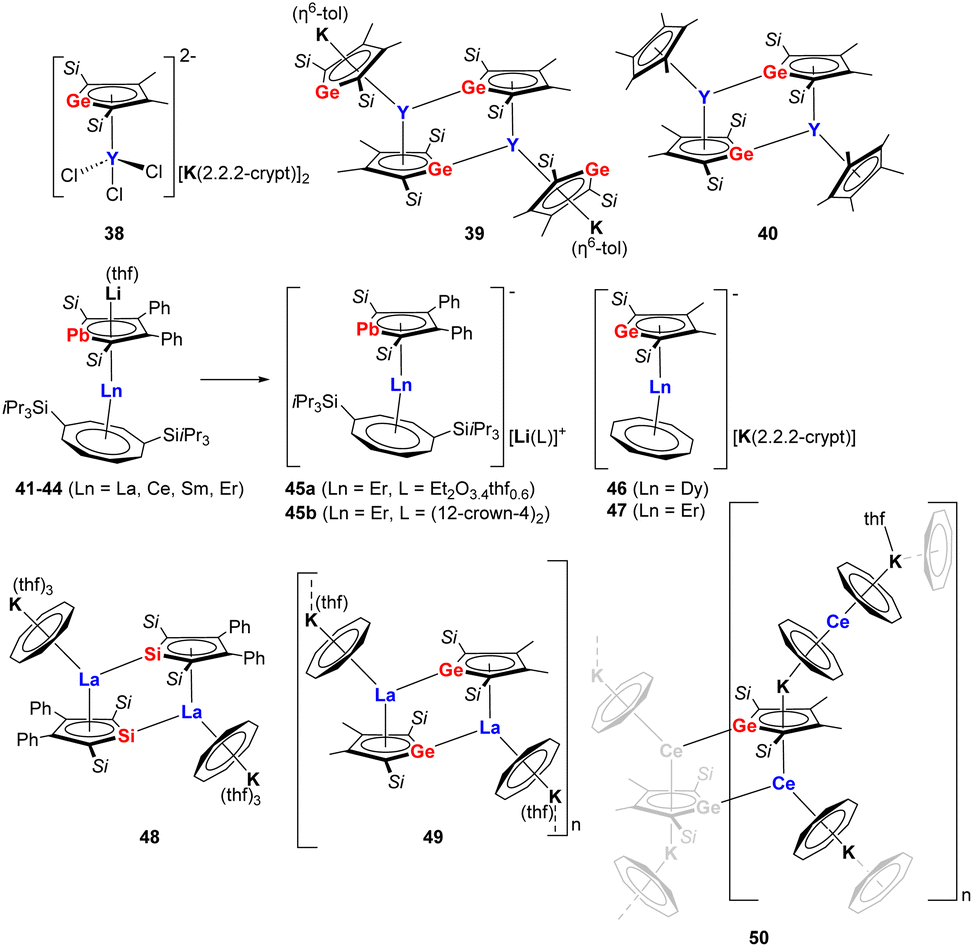 | ||
| Fig. 6 Rare-earth compounds with η5-coordinating metallole dianions.37–40 | ||
It is interesting to note that the Roesky group achieved the isolation of a series of monomeric lanthanide sandwich compounds comprising the heaviest 14 metallole dianion (41–45b, Fig. 6) upon treating the dilithioplumbole [Li2]16 with suitable lanthanide borohydride precursors [Ln(η8-COTTIPS)BH4] (COTTIPS = 1,4-bis-triisopropylsilyl-cyclooctatetraendiide).38 Four isostructural sandwich species 41–44 (Ln = La, Ce, Sm, Er) were obtained through crystallization from n-heptane (41, 42, 44) or toluene (43), among which the erbium congener 44 exhibits magnetic hysteresis up to 5 K, providing unequivocal evidence of single molecule magnet behavior. Interestingly, the Li cation could be trapped by crystallization from Et2O (45a) or addition of 12-crown-4 (45b), which has led to modulation of the magnetic properties of the Er complex due to slight changes in the bonding metrics of the central [(η5-16)Er(η8-COTTIPS)]− fragment.
Another noteworthy example of near-linear (approx. 175°) lanthanide group 14 metallole sandwich complexes (46–47, Fig. 6) was described by the Layfield group in 2023.39 In this case, the germole-ligated [K(2.2.2-crypt)][(η5-8)Dy(η8-COT)] (46) and [K(2.2.2-crypt)][(η5-8)Er(η8-COT)] (47) complexes were synthesized by reacting [K2]8 with [(η8-COT)Ln(BH4)(thf)2] in 1:1 THF and toluene mixture. The Ln–CtGermole distances are 2.3063(2) (46) and 2.2693(10) Å (47). Remarkably, the axial geometry provided by germole is maintained as the lone pair of Ge do not engage in bonding interactions with nearby Ln(III) ions. Based on magnetic measurements, the Dy(III) complex (46) did not feature SMM behavior, while for the Er(III) analog (47) a magnetic hysteresis loop up to 10 K was observed.
Roesky et al. also highlighted that different combinations of the dianionic silole [K2]6/germole [K2]8 and early Ln(III) ions (La, Ce) in combination with the cyclooctatetraendiide ligand can lead to different dimensionality and intermolecular interactions.40 These includes a dimeric La–silole complex (48, Fig. 6), a La(III)–germole ladder-type 2D polymer (49) and a Ce(III)–germole 3D coordination polymer (50).
5. Conclusion and perspectives
The coordination chemistry of group 14 metallole dianions has already yielded significant findings in both in p- and d-block chemistry and recently this chemistry was extended to f-block metals. This expansion of the field highlights the versatility and potential of metallole dianions in diverse areas of chemistry. To cater to more specific applications, the incorporation of ring heteroatoms offers electronic fine-tuning capabilities that cannot be achieved with solely carbon-based ring systems. In particular, the synthesis of dianionic group 14 metalloles has become more straightforward and advanced, which plays pivotal role in their further application and investigation. Despite the significant progress made in this area in the past twenty years, the periodic table of elements still offers abundant opportunities for future investigations of the reactivity of group 14 metalolle dianions. There remains significant untapped potential for utilizing such intriguing heterocycles as ligands to stabilize novel structure motifs. The combination of metallole dianions with diverse metal centers opens up great possibilities for discovering new catalytic processes, designing advanced luminescent materials, and developing high-performance magnetic compounds. Thus, it was already shown that the heteroatoms have an influence on the SMM properties of the corresponding lanthanide complexes and that the magnetic properties can be modulated. Present investigations of the catalytic properties are mainly limited to Rh(I) complexes. However, since the classical Cp complexes of various d- and f-elements have a wide range of catalytic applications depending on the metal used, it can be assumed that the potential of group 14 metallol complexes in catalysis is far from being fully explored. As research in this area continues to progress, we anticipate that in the near future investigations will give more exciting results.Author contributions
X. S. – investigation and writing. P. W. R. – writing, reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Deutsche Forschungsgemeinschaft (DFG) is acknowledged for financial support within the Reinhart Koselleck-Projekt 440644676, RO 2008/19-1.References
- W. P. Freeman, T. D. Tilley, L. M. Liable-Sands and A. L. Rheingold, Synthesis and Study of Cyclic π-Systems Containing Silicon and Germanium. The Question of Aromaticity in Cyclopentadienyl Analogues, J. Am. Chem. Soc., 1996, 118, 10457–10468 CrossRef CAS.
- W. P. Freeman, T. D. Tilley, G. P. A. Yap and A. L. Rheingold, Silolyl Anions and Silole Dianions: Structure of [K([18]crown-6)+]2[C4Me4Si2−], Angew. Chem., Int. Ed. Engl., 1996, 35, 882–884 CrossRef CAS.
- W.-C. Joo, J.-H. Hong, S.-B. Choi, H.-E. Son and C. Hwan Kim, Synthesis and reactivity of 1,1-disodio-2,3,4,5-tetraphenyl-1-silacyclopentadiene, J. Organomet. Chem., 1990, 391, 27–36 CrossRef CAS.
- R. West, H. Sohn, D. R. Powell, T. Müller and Y. Apeloig, The Dianion of Tetraphenylgermole is Aromatic, Angew. Chem., Int. Ed. Engl., 1996, 35, 1002–1004 CrossRef CAS.
- Z. Dong, C. R. W. Reinhold, M. Schmidtmann and T. Müller, Trialkylsilyl-Substituted Silole and Germole Dianions, Organometallics, 2018, 37, 4736–4743 CrossRef CAS.
- T. Kuwabara, J.-D. Guo, S. Nagase, M. Minoura, R. H. Herber and M. Saito, Enhancement of Stannylene Character in Stannole Dianion Equivalents Evidenced by NMR and Mössbauer Spectroscopy and Theoretical Studies of Newly Synthesized Silyl-Substituted Dilithiostannoles, Organometallics, 2014, 33, 2910–2913 CrossRef CAS.
- M. Saito, M. Nakada, T. Kuwabara and M. Minoura, A reversible two-electron redox system involving a divalent lead species, Chem. Commun., 2015, 51, 4674–4676 RSC.
- R. West, H. Sohn, U. Bankwitz, J. Calabrese, Y. Apeloig and T. Müller, Dilithium Derivative of Tetraphenylsilole: An η1 - η5 Dilithium Structure, J. Am. Chem. Soc., 1995, 117, 11608–11609 CrossRef CAS.
- Z. Dong, L. Albers and T. Müller, Trialkylsilyl-Substituted Silole and Germole Dianions as Precursors for Unusual Silicon and Germanium Compounds, Acc. Chem. Res., 2020, 53, 532–543 CrossRef CAS PubMed.
- T. Kuwabara and M. Saito, in Comprehensive Heterocyclic Chemistry IV, ed. D. S. Black, J. Cossy and C. V. Stevens, Elsevier, Oxford, 2022, pp. 798–832, DOI:10.1016/B978-0-12-409547-2.14789-4.
- M. Saito, Transition-Metal Complexes Featuring Dianionic Heavy Group 14 Element Aromatic Ligands, Acc. Chem. Res., 2018, 51, 160–169 CrossRef CAS PubMed.
- J. Wei, W.-X. Zhang and Z. Xi, The aromatic dianion metalloles, Chem. Sci., 2018, 9, 560–568 RSC.
- S.-B. Choi, P. Boudjouk and J.-H. Hong, Unique Bis-η5/η1 Bonding in a Dianionic Germole. Synthesis and Structural Characterization of the Dilithium Salt of the 2,3,4,5-Tetraethyl Germole Dianion, Organometallics, 1999, 18, 2919–2921 CrossRef CAS.
- L. Albers, P. Tholen, M. Schmidtmann and T. Müller, A germaaluminocene, Chem. Sci., 2020, 11, 2982–2986 RSC.
- M. Saito, R. Haga and M. Yoshioka, Formation of the first monoanion and dianion of stannole, Chem. Commun., 2002, 1002–1003 RSC.
- M. Saito, R. Haga, M. Yoshioka, K. Ishimura and S. Nagase, The Aromaticity of the Stannole Dianion, Angew. Chem., Int. Ed., 2005, 44, 6553–6556 CrossRef CAS PubMed.
- S. Masaichi, K. Takuya, K. Chika, Y. Michikazu, I. Kazuya and N. Shigeru, Synthesis, Structure, and Reaction of Tetraethyldilithiostannole, Chem. Lett., 2010, 39, 700–701 CrossRef.
- M. Saito, M. Sakaguchi, T. Tajima, K. Ishimura, S. Nagase and M. Hada, Dilithioplumbole: A Lead-Bearing Aromatic Cyclopentadienyl Analog, Science, 2010, 328, 339–342 CrossRef CAS PubMed.
- R. M. Diaz-Rodriguez, A. A. Kitos and M. Murugesu, Expanding the series of alkali metal plumbolyl complexes to Na and K, Dalton Trans., 2022, 51, 14420–14428 RSC.
- X. Sun, A. K. Singh, R. Yadav, D. Jin, M. Haimerl, M. Scheer and P. W. Roesky, Triple-decker complexes incorporating three distinct deck architectures, Chem. Commun., 2022, 58, 673–676 RSC.
- J. M. Dysard and T. D. Tilley, Synthesis and Reactivity of η5-Silolyl, η5-Germolyl, and η5-Germole Dianion Complexes of Zirconium and Hafnium, J. Am. Chem. Soc., 2000, 122, 3097–3105 CrossRef CAS.
- T. Kuwabara, J.-D. Guo, S. Nagase, T. Sasamori, N. Tokitoh and M. Saito, Synthesis, Structures, and Electronic Properties of Triple- and Double-Decker Ruthenocenes Incorporated by a Group 14 Metallole Dianion Ligand, J. Am. Chem. Soc., 2014, 136, 13059–13064 CrossRef CAS PubMed.
- M. Nakada, T. Kuwabara, S. Furukawa, M. Hada, M. Minoura and M. Saito, Synthesis and reactivity of a ruthenocene-type complex bearing an aromatic π-ligand with the heaviest group 14 element, Chem. Sci., 2017, 8, 3092–3097 RSC.
- M. Saito, J. Hamada, S. Furukawa, M. Hada, L. Dostál and A. Růžička, Transition-Metal Capping to Suppress Back-Donation to Enhance Donor Ability, Organometallics, 2020, 39, 4191–4194 CrossRef CAS.
- M. Saito, N. Matsunaga, J. Hamada, S. Furukawa, M. Minoura, S. Wegner, J. Barthel and C. Janiak, Heterobimetallic triple-decker complexes derived from a dianionic aromatic stannole ligand, Dalton Trans., 2018, 47, 8892–8896 RSC.
- M. Saito, N. Matsunaga, J. Hamada, S. Furukawa, T. Tada and R. H. Herber, Anionic Stannaferrocene and Its Unique Electronic State, Chem. Lett., 2019, 48, 163–165 CrossRef CAS.
- M. Saito, M. Nakada, T. Kuwabara, R. Owada, S. Furukawa, R. Narayanan, M. Abe, M. Hada, K. Tanaka and Y. Yamamoto, Inverted Sandwich Rh Complex Bearing a Plumbole Ligand and Its Catalytic Activity, Organometallics, 2019, 38, 3099–3103 CrossRef CAS.
- S. K. Ghag, M. L. Tarlton, E. A. Henle, E. M. Ochoa, A. W. Watson, L. N. Zakharov and E. J. Watson, Synthesis and Structures of Triple-Decker Complexes with a Bridging Tetramethylcyclopentadienyl Ligand, Organometallics, 2013, 32, 1851–1857 CrossRef CAS.
- A. Salzer and H. Werner, A New Route to Triple-Decker Sandwich Compounds, Angew. Chem., Int. Ed. Engl., 1972, 11, 930–932 CrossRef CAS.
- T. Kuwabara, M. Saito, J.-D. Guo and S. Nagase, Unexpected Formation of Ru2Sn2 Bicyclic Four-Membered Ring Complexes with Butterfly and Inverse-Sandwich Structures, Inorg. Chem., 2013, 52, 3585–3587 CrossRef CAS PubMed.
- T. Kuwabara, M. Nakada, J. Hamada, J. D. Guo, S. Nagase and M. Saito, (η4-Butadiene)Sn(0) Complexes: A New Approach for Zero-Valent p-Block Elements Utilizing a Butadiene as a 4π-Electron Donor, J. Am. Chem. Soc., 2016, 138, 11378–11382 CrossRef CAS PubMed.
- T. Kuwabara and M. Saito, Synthesis of a Stannole Dianion Complex Bearing a μ-η1;η1-Coordination Mode: Different Electronic State of Stannole Dianion Ligands Depending on Their Hapticity, Organometallics, 2015, 34, 4202–4204 CrossRef CAS.
- T. Kuwabara, J. D. Guo, S. Nagase and M. Saito, Diversity of the Structures in a Distannene Complex and its Reduction to Generate a Six-Membered Ti2Sn4 Ring Complex, Angew. Chem., Int. Ed., 2014, 53, 434–438 CrossRef CAS PubMed.
- Z. Dong, K. Bedbur, M. Schmidtmann and T. Müller, Hafnocene-based Bicyclo[2.1.1]hexene Germylenes – Formation, Reactivity, and Structural Flexibility, J. Am. Chem. Soc., 2018, 140, 3052–3060 CrossRef CAS PubMed.
- Z. Dong, O. Janka, J. Kösters, M. Schmidtmann and T. Müller, A Dimeric η1,η5-Germole Dianion Bridged Titanium(III) Complex with a Multicenter Ti–Ge–Ge–Ti Bond, Angew. Chem., Int. Ed., 2018, 57, 8634–8638 CrossRef CAS PubMed.
- Z. Dong, C. R. W. Reinhold, M. Schmidtmann and T. Müller, A Germylene Stabilized by Homoconjugation, Angew. Chem., Int. Ed., 2016, 55, 15899–15904 CrossRef CAS PubMed.
- J. Liu, K. Singh, S. Dutta, Z. Feng, D. Koley, G. Tan and X. Wang, Yttrium germole dianion complexes with Y–Ge bonds, Dalton Trans., 2021, 50, 5552–5556 RSC.
- L. Münzfeld, X. Sun, S. Schlittenhardt, C. Schoo, A. Hauser, S. Gillhuber, F. Weigend, M. Ruben and P. W. Roesky, Introduction of plumbole to f-element chemistry, Chem. Sci., 2022, 13, 945–954 RSC.
- S. De, A. Mondal, Z.-Y. Ruan, M.-L. Tong and R. A. Layfield, Dynamic Magnetic Properties of Germole-ligated Lanthanide Sandwich Complexes, Chem. – Eur. J., 2023, e202300567 CrossRef CAS.
- X. Sun, L. Münzfeld, D. Jin, A. Hauser and P. W. Roesky, Silole and germole complexes of lanthanum and cerium, Chem. Commun., 2022, 58, 7976–7979 RSC.
Footnote |
| † Benzannulated dianionic metalloles will not be discussed in this perspective. |
| This journal is © the Partner Organisations 2023 |