
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrifying synthesis of organosilicon compounds – from electrosynthesis to electrocatalysis
Krzysztof
Kuciński

Adam Mickiewicz University, Uniwersytetu Poznanskiego St. 8, 61-614 Poznan, Poland. E-mail: kucinski.k@amu.edu.pl
First published on 2nd February 2023
Abstract
Occasionally, a synthetic strategy is developed that seems to be a game changer. Not long ago, mechanochemistry or ionic liquids were intensively explored to afford organosilicon compounds. These and other approaches still offer benefits and influence the development of this field. Electrochemistry is currently a new synthetic strategy. Despite its long history, electrosynthesis or electrocatalysis have not become routine procedures in the synthesis of organosilicons. One may justifiably ask why. Accessibility and reproducibility are one of the challenges of electrochemical approaches. However, recent developments in the field of electrified organic synthesis have provided new insights and allowed electrochemistry to become an approachable tool. In this perspective article, several notable contributions to the electrochemical synthesis of organosilicon compounds are highlighted.
Looking at organic chemistry we notice a tremendous growth of interest in the development of novel electrochemical methodologies. Ackermann,1–4 Baran,5–8 Lin,9,10 Mei,11–13 Waldvogel14–16 and many other eminent scientists have proverbially brought electrosynthesis/electrocatalysis to light by turning it into a perfectly working synthetic tool.17–32 This, in turn, has many implications for the development of materials chemistry. By linking these two factors – synthesis and applications – one can bring to mind organosilicon chemistry.33–35 Especially, the synthetically produced silicones (siloxanes), which can be considered milestones for modern-day industry. Moreover, the organosilicons themselves are useful intermediates allowing the synthesis of very complex molecules.36–39
Until we look at the recent development in the field, one should always pay attention to previous studies (Fig. 1).40–43 Most of the pioneering electrochemical transformations of organosilicons were developed in the 1970s and 1980s. However, despite valuable conclusions, the above-mentioned investigations were not noticed with due diligence by the audience. In 1976, Litscher and Hengge developed a new method for the facile synthesis of disilanes from chlorosilanes (Fig. 1, A).44 It is worth noting, that the formation of Si–Si bonds is challenging by catalytic methods (due to the strong thermodynamic force for Si–O–Si bond formation).45 In 1985, Shono et al. described an electroreductive silylation of allylic and benzylic halides (Fig. 1, B).46 At the same time, Yoshida and coworkers reported a series of electrochemical studies giving insights into the formation of the C–Si bond (Fig. 1, C), and its further cleavage.47–49
 | ||
| Fig. 1 Previous developments concerning electrochemical transformations of organosilicons. | ||
In general, the generation of silyl radical intermediates seems to play an important role during the synthesis of targeted products. Speaking of which, the silyl radicals can be generated in several ways (Fig. 2).50–56
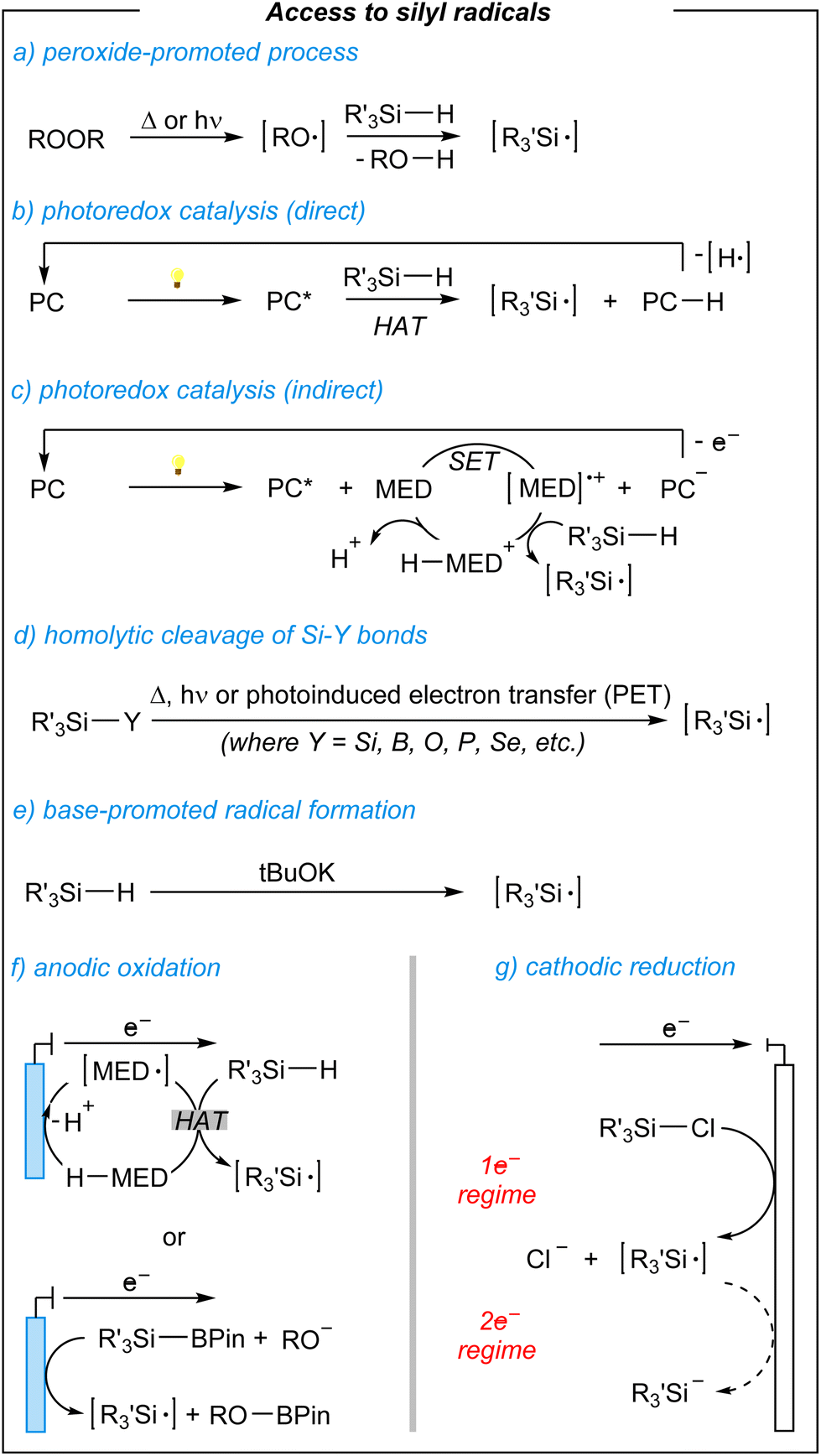 | ||
| Fig. 2 Overview of general ways for the generation of silyl radicals. | ||
The most obvious one is the peroxide-promoted radical formation via the homolyses process (thermal or photochemical), and subsequent hydrogen-atom abstraction from hydrosilanes (Fig. 2, a).50 On the other hand, hydrogen-atom transfer (HAT) can be achieved by using photoredox catalysis.51 Here, two main operative modes are involved based on the use of direct HAT photocatalysts (Fig. 2, b) or indirect photocatalysis requiring additional mediators (Fig. 2, c). Silyl radicals can be also derived from the cleavage of Si–Y bonds (where Y = Si, B, O, P, Se, etc.), via single electron transfer (SET), however, there is a limited variety of readily available species, and their preparation prolongs the whole procedure (Fig. 2, d).57,58 A few years ago, Grubbs and coworkers developed a base-promoted silylation of C–H bonds suggesting a radical addition-dehydrogenation process (Fig. 2, e).59,60 Lastly, there are electrochemically induced silyl radicals. Here, the use of redox mediators enables the generation of silyl radicals and further anodic oxidation processes (Fig. 2, f), whilst commercially available chlorosilanes can proceed with the formation of silyl radicals in the 1e− regime via cathodic reduction (Fig. 2, g). All of the above-mentioned approaches have their benefits and drawbacks. Considering a period of increasing society's awareness in terms of sustainable developments and risks arising from irresponsible resource management, scientists pay more and more attention to the development of greener synthetic solutions.61–63 Electrochemical approaches in this regard are at a forefront of such a policy, by setting the stage for molecular synthesis with unique levels of resource economy (e.g., chemicals, wastes, energy, etc.).64 The cathodic reduction, indeed, benefits from readily available chlorosilanes and the absence of additional mediators. However, the anodic oxidation process requires additional mediators, the use of limited hydrosilanes or their precursors. Nevertheless, novel electro-mediated methodologies seem to lead to fascinating new reactions, whereas the full potential of paired electrosynthetic approaches will hopefully be uncovered in the near future.22
This short perspective will mainly focus on the recent developments in the electrochemical synthesis of organosilicon compounds. It aims to explain the significance of these developments, provide a critical look at this subject, and identify further challenges. Five recent articles (2020–2022) were selected and the features of these solutions will be discussed with an appropriate description of mechanistic studies and their usefulness.
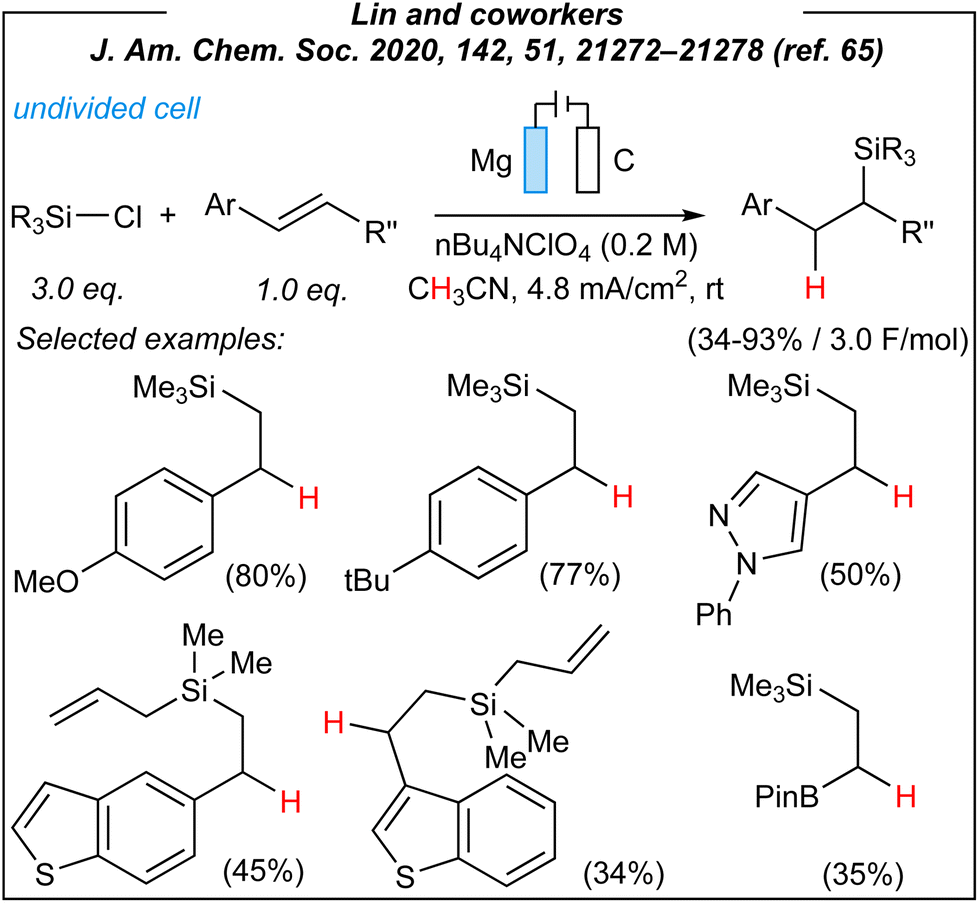
In 2020, Lin and coworkers described the synthesis of a wide range of organosilicon compounds via electrosynthesis.65 For the disilylation of alkenes (Fig. 3), the authors used the following combination: Mg anode (sacrificial), graphite cathode, CCE @ 10 mA, and TBAClO4 as the electrolyte.
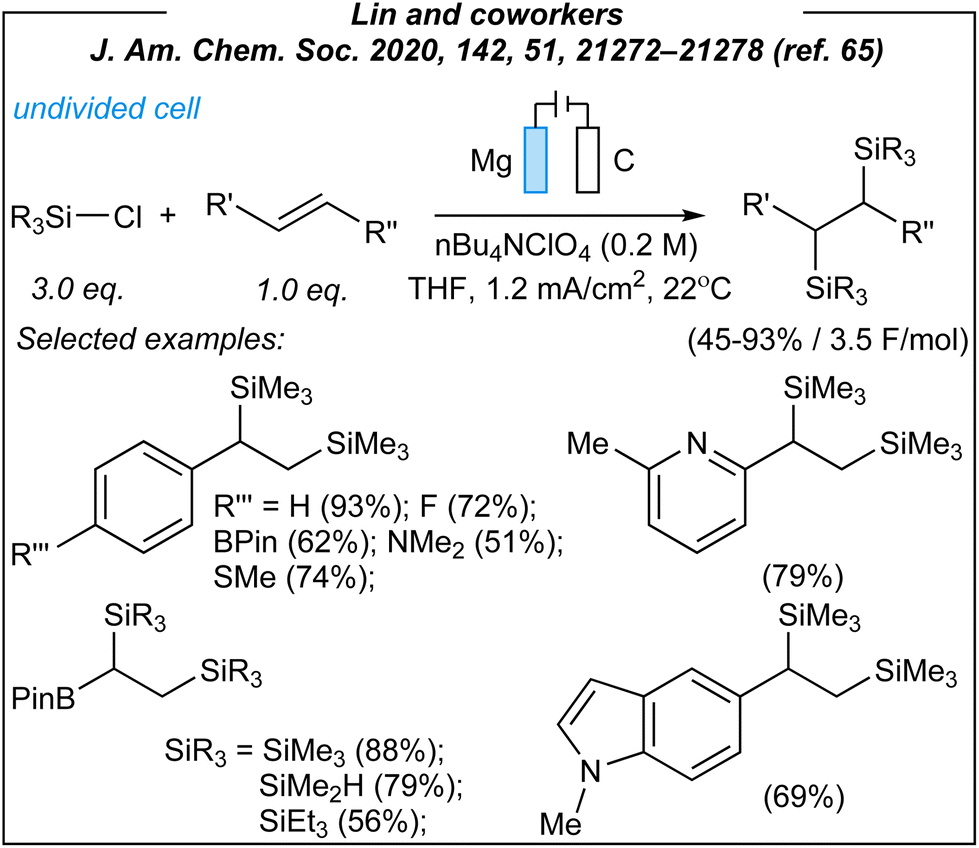 | ||
| Fig. 3 Disilylation of alkenes via electroreductive activation. | ||
Upon the investigation of the reaction scope, several styrenes, vinylboronates, and vinyl-N-heterocycles were efficiently disilylated at rt, in moderate to very good yields (45–93%). Moreover, the authors performed analogues digermylation, and used other chlorosilanes than chlorotrimethylsilane such as chlorodimethylsilane, chloropentamethyldisilane and chlorotriethylsilane. Among the unreactive or less reactive (and less selective) reagents, one can mention β-methylstyrene, styrenes with strong electron-withdrawing groups, 3-vinylpyridine, allylbenzene, cyclooctene (alkene substrates), as well as dichlorosilanes, chlorotert-butyldimethylsilane and chlorodimethylphenylsilane (chlorosilanes). Meanwhile, a silyl radical pathway was proposed. The authors conducted detailed mechanistic studies by performing several electroanalytical experiments. The reaction system involved three species that could be reduced at the cathode – chlorosilane, alkene, and magnesium cation (Mg2+). The last one was excluded since magnesium (Mg0, electrogenerated or as a powder) gave no product under the optimized conditions. Moreover, the electrolysis performed in a divided cell led to the desired product. Then, cyclic voltammetry experiments suggested the reduction of chlorosilane prior to the alkene. The plausible mechanism is depicted in Fig. 4.
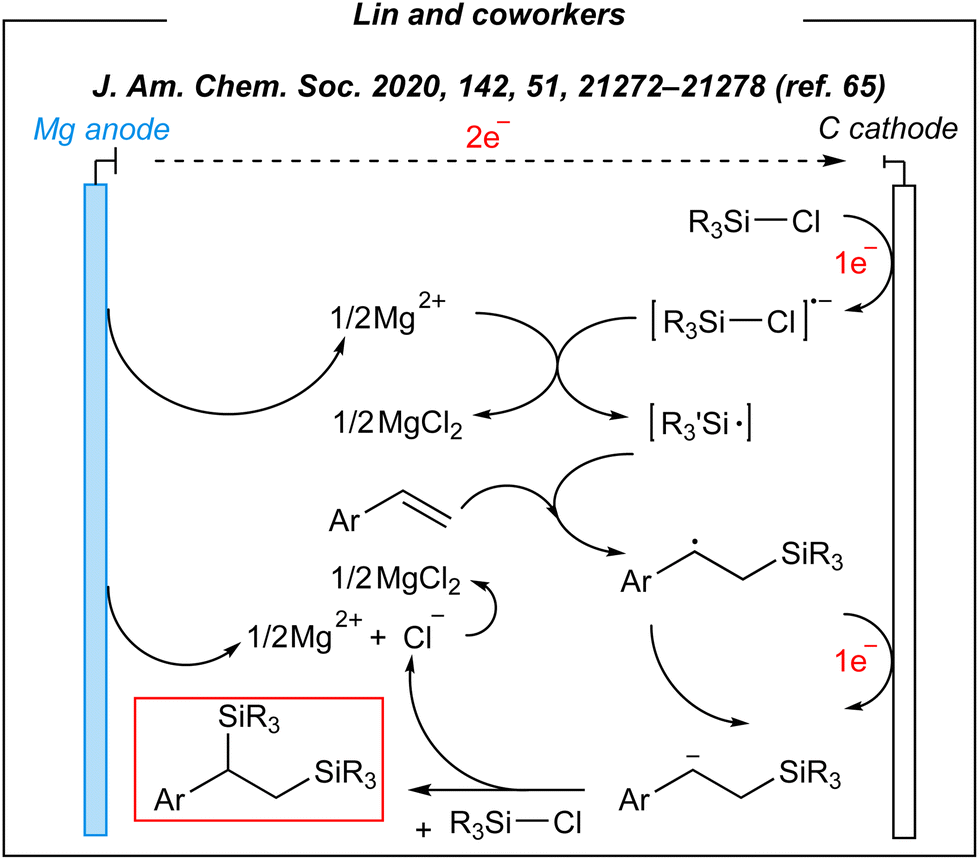 | ||
| Fig. 4 Plausible mechanism for the disilylation of alkenes under electrochemical conditions. | ||
Next, considering the plausible mechanism of this electroreductive process, the authors envisaged a completely novel methodology for the formation of silacycles (Fig. 5). Thus, several tethered silanes including dichlorodi-, tri- and tetrasilanes, as well as dichlorodisiloxane and 1,2-bis(chlorodimethylsilyl)ethane were utilized as the substrates under the same conditions. As the result, seven new silacycles (five- and six-membered ones) were obtained in good yields up to 92%. Subsequently, the authors again showed how important is to understand the nature of the process. After the synthesis of the above-mentioned silacycles, the portfolio of the products was extended to monosilylated alkanes (via formal hydrosilylation) and allylsilanes. In the case of the former, acetonitrile was used as both solvent and the hydrogen source (Fig. 6). In the latter case, the introduction of a leaving group in β-position to the carbanion intermediate was required.
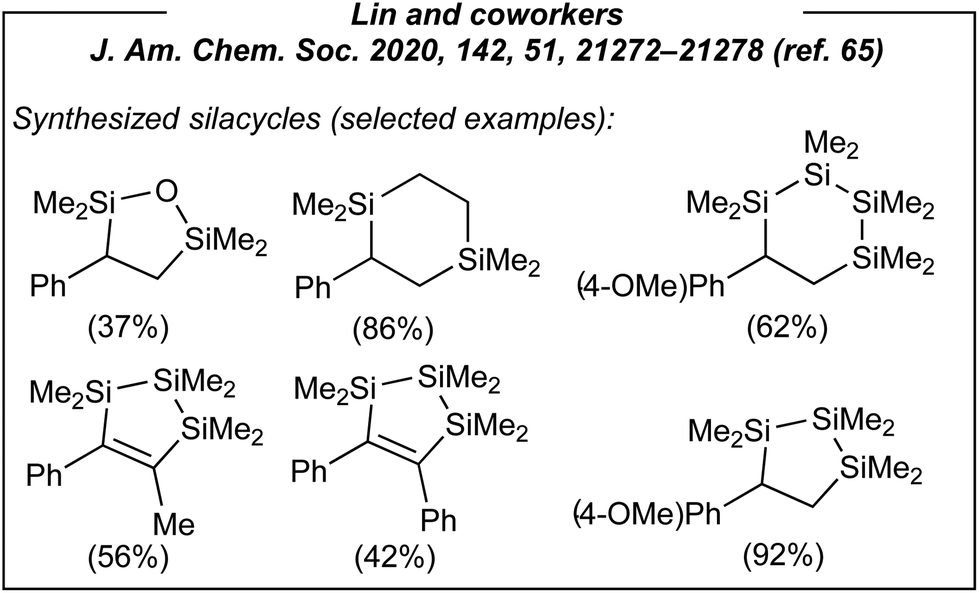 | ||
| Fig. 5 Selected silacycles obtained via electroinduced disilylation of alkenes with tethered dichlorosilyl derivative. | ||
 | ||
| Fig. 6 Hydrosilylation of alkenes via electroreductive activation. | ||
Finally, Lin demonstrated the derivatization of the obtained organosilicon products to a variety of structurally diverse compounds via known transformations including fluorination, oxidation, and Fleming–Tamao oxidation.66
To sum up, Lin and coworkers reported a novel methodology for the construction of sp3 C–Si moieties, induced by electricity. Firstly, they used chlorosilanes. This group of silicon compounds is the most numerous on the commercial market, thus their prices are typically lower than corresponding hydrosilanes. Of the major drawbacks should be noted moisture sensitivity and corrosive character of SiCl-containing derivatives. And all this implies the necessity for an inert atmosphere during the process. Moreover, the authors used tetrabutylammonium perchlorate (TBAClO4) as the electrolyte. This is a strong oxidizer, which is potentially explosive.67,68 However, a more user-friendly electrolyte such as tetrabutylammonium bis(trifluoromethanesulfonyl)imide (TBATFSI) can also be utilized giving only slightly inferior conversion. The electrochemical system consists of an anode (Mg) and graphite cathode (carbon-based electrodes are typically inexpensive). Whilst the former one is sacrificial, extended reaction time and delivered charge can lead to the deposition of combustible Mg0 layer on the cathode surface. Next, the authors used standardized IKA® equipment. This is a commercialized setup, which gives an appropriate level of reproducibility. This is a very important aspect, whereas the electrode arrangement can lead to different efficiencies. Finally, the novelty and significance of this approach will be evaluated. This is a novel methodology giving a wide range of fascinating insights. It may lead to further developments in the field. Furthermore, hitherto methodologies leading to the same products were limited. In the case of disilylation reaction, previous methods were mainly based on the use of strong reducing agents,69 expensive Pt/Pd catalysts,70–75 or silylium-ion catalysis.76,77 The latter one, despite its high scientific significance, is still in its infancy and in this particular case is mostly limited to one disilane (hexamethyldisilane). The Lin group showed also the possibility to perform transition metal-free hydrosilylation using chlorosilanes (however, it should be noted that preparation of carbon electrodes may introduce traces of metal impurities78). In the vast majority of cases, the hydrosilylation of unsaturated bonds requires the use of more expensive hydrosilanes, and radical initiators or transition metal complexes (mostly Pt or Ru).34,79–82 Thus, the utilization of chlorosilanes instead of hydrosilanes or (limited in number) transfer hydrosilylation agents83 seems to be an intriguing idea. There were only two examples, where chlorosilanes were used to give formal hydrosilylation products (magnesium-promoted reductive silylation reported by Maekawa84 and electrochemically-induced silylation of styrenes by Jouikov85). In conclusion, the electrochemical disilylation reported by Lin and coworkers is a sustainable alternative to conventional stoichiometric or catalytic methods, and despite some limitations in scope (e.g., mostly an introduction of alkylsilyl group), it can be considered as useful synthetic approach.
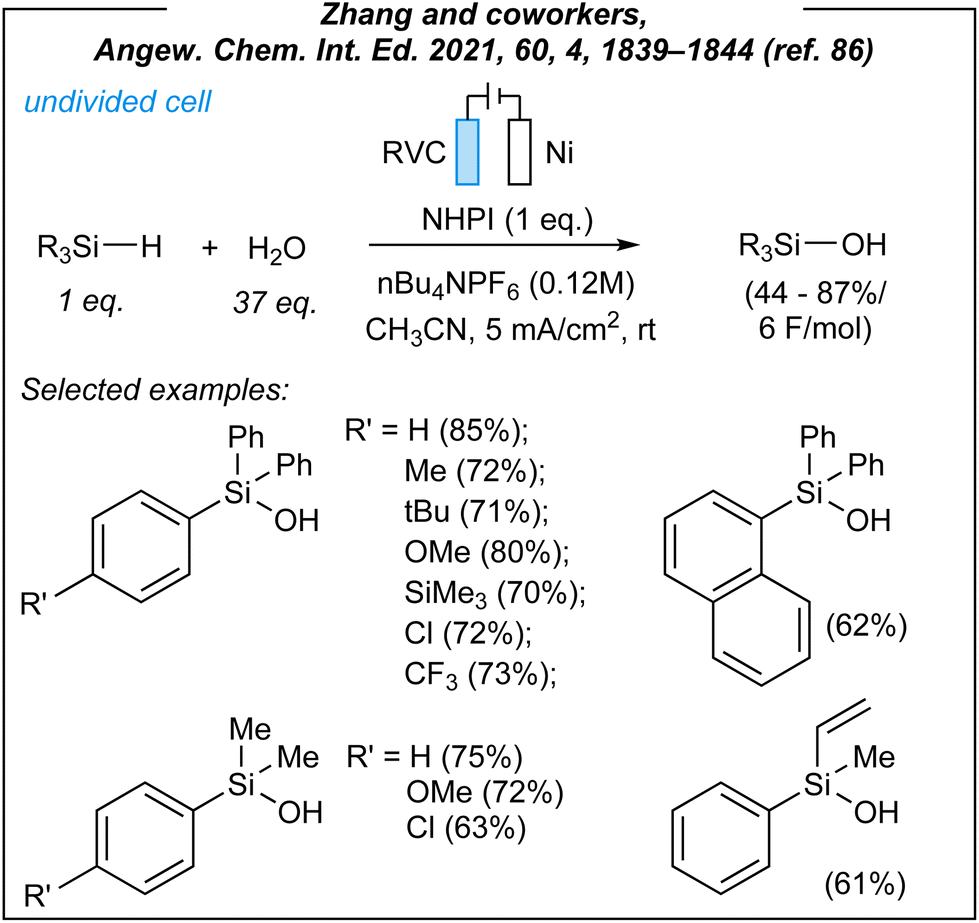
In 2021, Zhang and coworkers described the synthesis of a wide range of organosilanols via electrosynthesis.86 For the hydrolysis of hydrosilanes (Fig. 7), the authors used the following combination: RVC anode (RVC = reticulated vitreous carbon), nickel foam cathode, CCE @ 12 mA, and nBu4NPF6 as the electrolyte. During the optimization studies, several HAT mediators were tested including NHPI (selected, N-hydroxyphthalimide), NHSI (N-hydroxysuccinimide), TEMPO [(2,2,6,6-tetramethylpiperi-din-1-yl)oxyl], and DABCO (1,4-diazabicyclo[2.2.2]octane). The mediator-free attempt was also carried out and proved the essential role of the HAT-step. Next, the authors explored the substrate scope concerning several hydrosilanes. A wide range of triphenylsilane derivatives worked efficiently under the reaction conditions, as well as naphthyl-, alkyl-, alkenyl-, alkynyl, and heteroaryl-substituted substrates. Moreover, the synthetically useful dihydro derivatives such as 1,4-bis(diphenylsilyl)benzene and diphenylsilane were also well tolerated, and led to their silanol analogues in moderate yields (56% and 62%, respectively).
 | ||
| Fig. 7 Silanol-oriented hydrolysis of hydrosilanes under electrochemical conditions. | ||
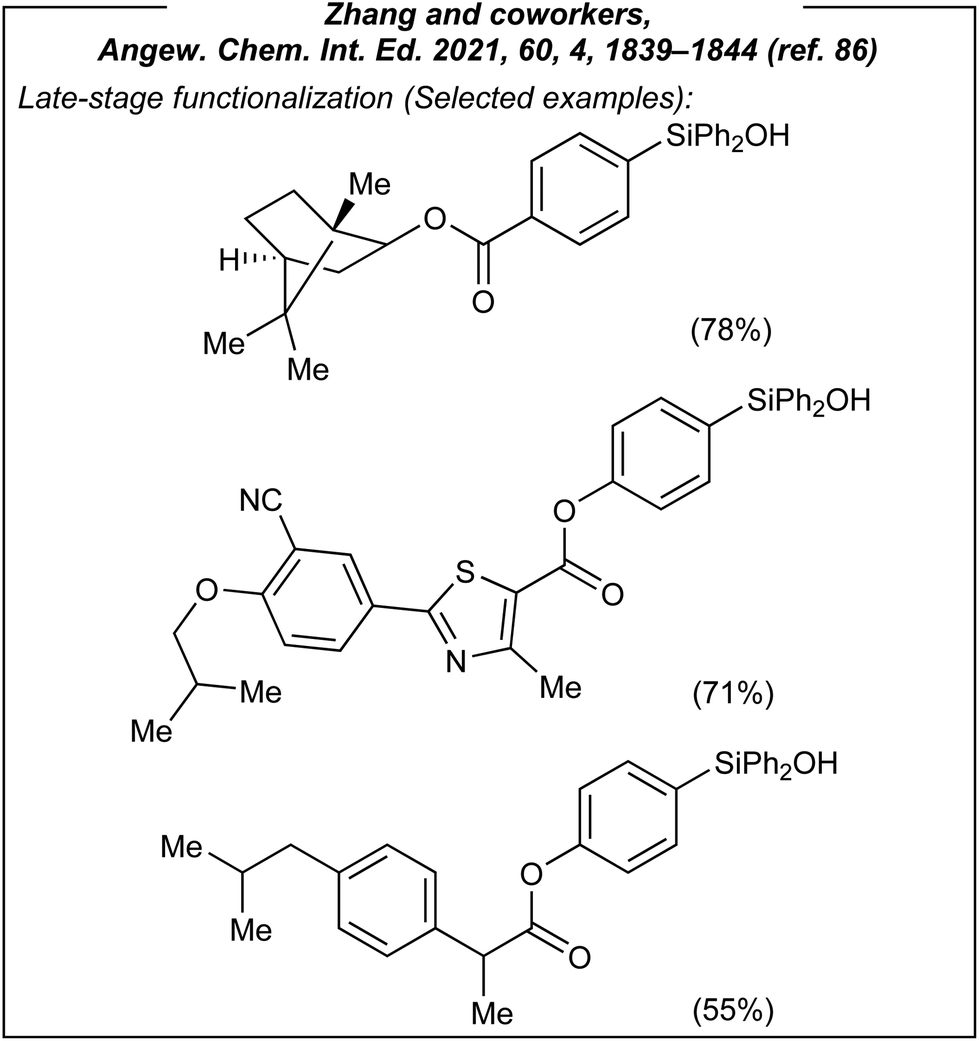
Encouraged by these results, Zhang investigated also the use of silyl-derived natural products and drugs, which are biorelevant scaffolds (Fig. 8). All of them afforded the expected products in moderate to good yields (44–78%).
 | ||
| Fig. 8 Selected biorelevant compounds obtained via electroinduced hydrolysis of Si–H bonds. | ||
Furthermore, the authors replaced water with alcohol to check the possible alcoholysis of hydrosilanes under electrochemical conditions. Using quinuclidine as the HAT mediator (3.0 eq.), the alcohols including methanol, 1-adamantanol, L-menthol, and (−)-nopol provided four silyl ethers in moderate to very good yields (51–89%).
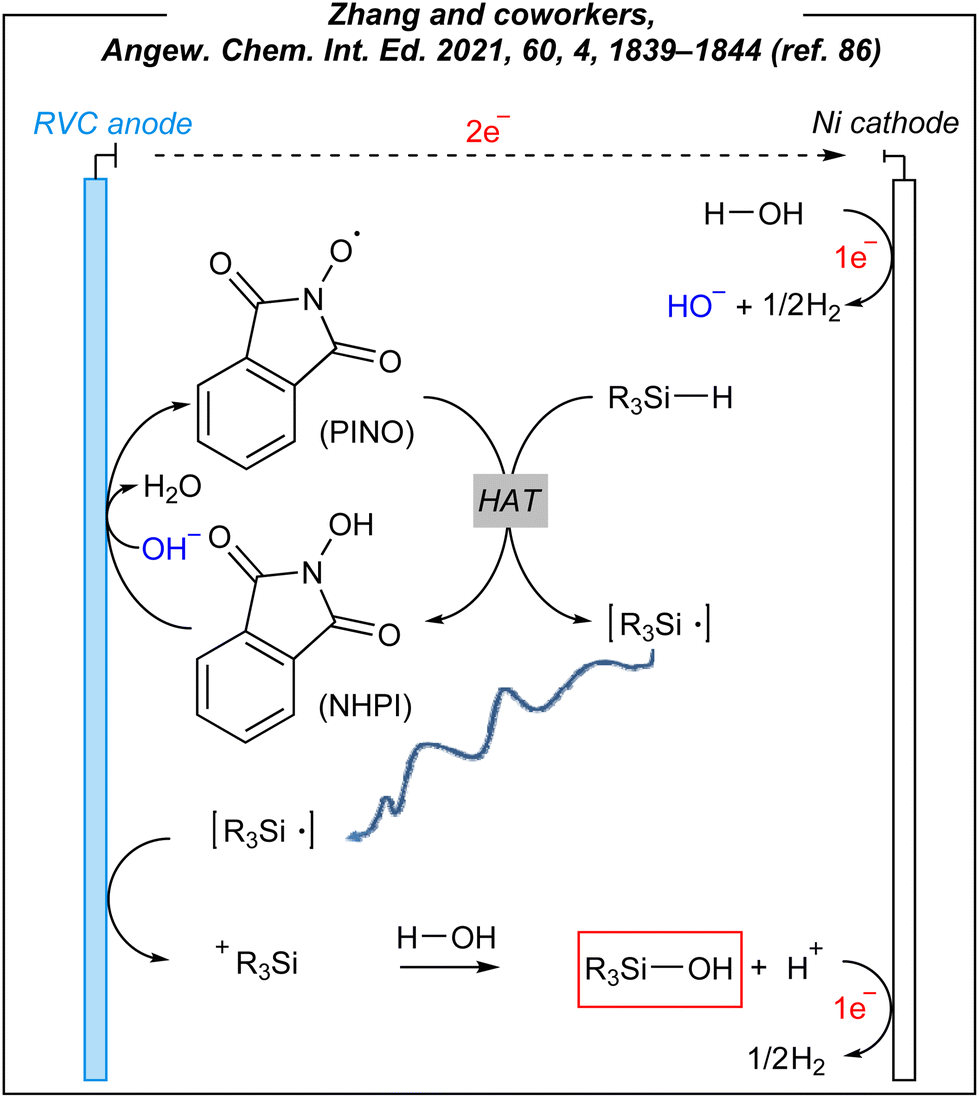
To gain some mechanistic insights into this electroinduced transformation, the authors conducted several electrochemical experiments. The reaction system involved a HAT mediator – NHPI, which undergoes an anodic oxidation process facilitated by cathodically-generated hydroxides. As the result, the PINO radical (phthalimide N-oxyl) is afforded and it abstracts the hydrogen atom from the Si–H bond of hydrosilanes. The formed silyl radicals are subsequently oxidized to silyl cations. The formed R3Si+ species are instantaneously trapped by O–nucleophiles (hydroxide anion or water can quench the cation, but an excess of water favors the latter one) to give the corresponding silanols. It is worth noting that other mediums such as dichloromethane and dimethylformamide were ineffective solvents in this process, whilst acetonitrile, known for stabilizing silyl cations,87 ensured the best conversion. The plausible mechanism is depicted in Fig. 9.
 | ||
| Fig. 9 Plausible mechanism for the hydrolysis of hydrosilanes under electrochemical conditions. | ||
To sum up, Zhang and coworkers reported a novel methodology for the construction of O–Si moieties, induced by electricity. First of all, the authors used hydrosilanes as the substrates. Unlike chlorosilanes, SiH-containing molecules are less sensitive and lead to non-corrosive byproducts. However, they are not as readily available as halosilanes, which serve as the first-choice substrates in conventional methods.88 Indeed, chlorosilanes readily undergo hydrolysis to give corrosive HCl (usually trapped by amine bases) and silanols. Unfortunately, the chemoselectivity of these transformations is generally low, whereas silanols can easily undergo condensation to disiloxanes. Zhang's approach gives access to both types of silanols, these easily available from chlorosilanes (e.g., triisopropylsilanol), and those difficult to be conventionally produced with good selectivity (e.g., SiMe2-containing silanols). What is more, the authors performed the challenging hydrolysis of silyl-derived biorelevant compounds, which contain sensitive functional groups. Furthermore, in the vast majority of cases, the conversion of Si–H to Si–OH moieties requires the use of stoichiometric oxidants and/or expensive transition metal catalysts.39,89 Thus, the electrosynthetic approach seems to be a very significant alternative to known procedures utilizing hydrosilanes as the substrates. The authors performed also pioneering alcoholysis reactions leading to alkoxysilanes under electrochemical conditions. In the past, several sustainable methods were developed,39,90–93 but this electroinduced approach might be interesting in the case of the compounds with sensitive groups. Furthermore, the electrochemical system consists of an RVC anode and a nickel cathode. Both are classified as not so expensive. Similarly to Lin,65 the Zhang group used an IKA® ElectraSyn 2.0, which ensures the reproducibility of the obtained results. In addition, what is equally important, the authors tested the reaction under electricity-free (as well as mediator-free and catalyst-free) conditions to show the necessity of electroinduced synthesis. This is a very important aspect, whereas some processes can be performed via uncatalyzed reactions or without additional terms and conditions.94–96 In conclusion, the electrochemical hydrolysis reported by Zhang and coworkers is an interesting alternative to conventional stoichiometric or catalytic methods, and despite some limitations (e.g., stoichiometric amounts of HAT mediator, moderate yields, etc.), it can be considered as a useful synthetic approach.
In 2021, He and coworkers reported the radical 1,2-silylfunctionalization of alkenes under electrochemical conditions.97 For the silyl-oxygenation process (Fig. 10), the authors used the following combination: graphite anode, platinum cathode, CCE @ 3 mA, NHPI as HAT mediator (1.2 eq.), and LiClO4 as the electrolyte. Upon the investigation of the reaction scope, several hydrosilanes (including secondary and tertiary ones) were matched with electron-deficient alkenes and different N-oxyl species. In most cases, that was a combination of triphenylsilanes, acrylonitrile, and NHPI. As the result, several β-silyl-cyanohydrines were obtained with moderate to good yields. Moreover, He demonstrated that acrylates, vinyl phenyl sulfone, and vinyl phenyl ketone are also suitable alkene partners, whilst styrene and vinyl ether more or less gave no product. Furthermore, tertiary and secondary germanes were also reactive under these electrochemical conditions, giving β-germyl-cyanohydrines in moderate yields (33–60%). Finally, the described protocol was scaled up to a 5 mmol scale yielding 72% (1.71 g) of 2-((1,3-dioxoisoindolin-2-yl)oxy)-3-(triphenylsilyl)propanenitrile. This once again makes it clear that the proposed methodology has significant application potential. Next, the authors demonstrated the derivatization of the obtained β-silyl-cyanohydrines to a variety of structurally diverse compounds via transformations of all three functional groups found in the mentioned scaffold.
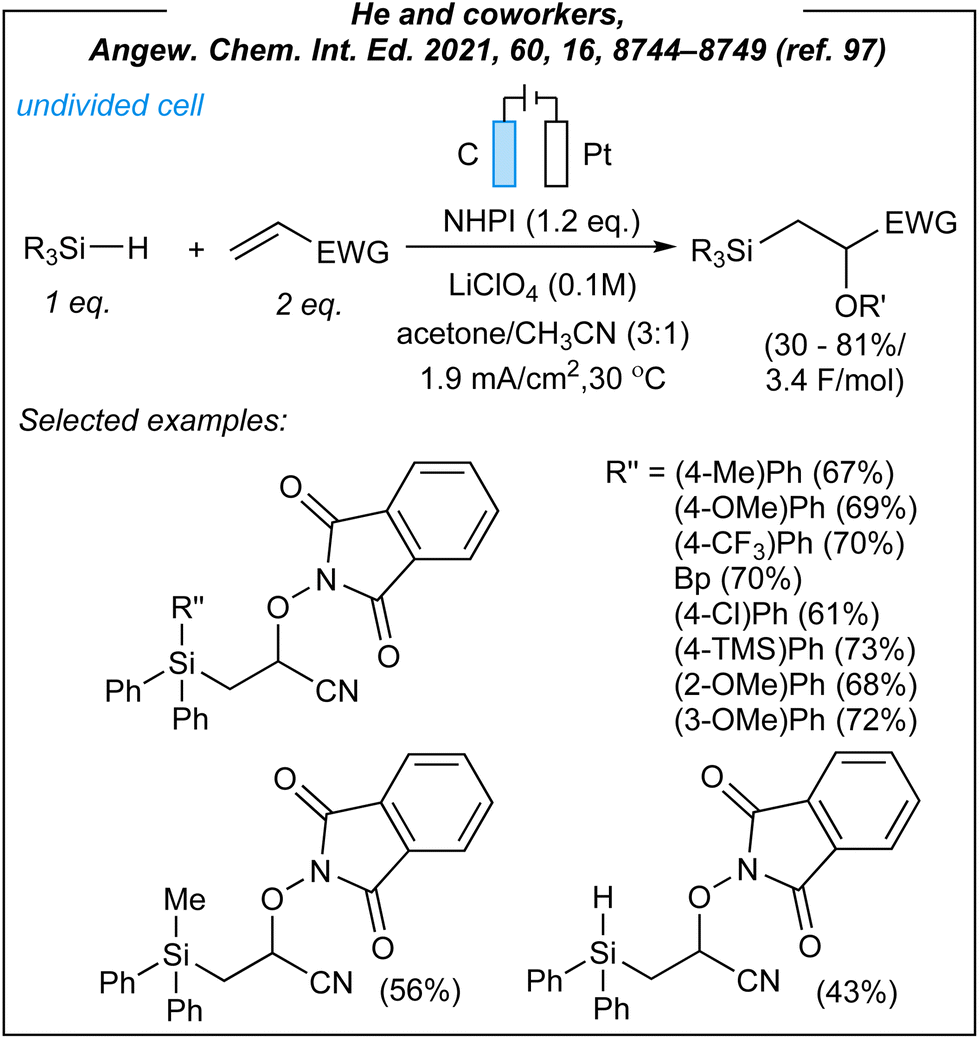 | ||
| Fig. 10 Radical 1,2-silylfunctionalization of alkenes under electrochemical conditions. | ||
Thus, it was shown that Si–H group can be efficiently converted to Si–OH moiety in the presence of oxidant and rhenium catalyst (however, it would be interesting to check previously mentioned Zhang's electrochemical conditions to perform this reaction under catalyst-free and external oxidant-free conditions). Moreover, it was demonstrated that nitrile as well as N-oxyl groups can be efficiently transformed into other functions such as tetrazoles and α-aminoxy derivatives respectively.
To gain mechanistic insights into these electroinduced reactions, the authors conducted some preliminary experiments and suggested a plausible mechanism (Fig. 11). A radical clock experiment was carried out giving the ring-opening product in 28% yield. This experiment implied that the radical pathway is operative. It is well-known the direct anodic oxidation of hydrosilanes is very difficult due to the high redox potential of Si–H bonds. The CV studies confirmed these presumptions, showing that oxidation of NHPI to PINO should be favoured. As the result, the PINO radical abstracts the hydrogen atom from the Si–H bond of hydrosilanes. The formed silyl radicals are subsequently added to the alkene substrate giving silyl alkyl radical, which is followed by the radical–radical coupling between the latter ones with N-oxyl species to give 1,2-silyl-oxygenation products (Fig. 11).
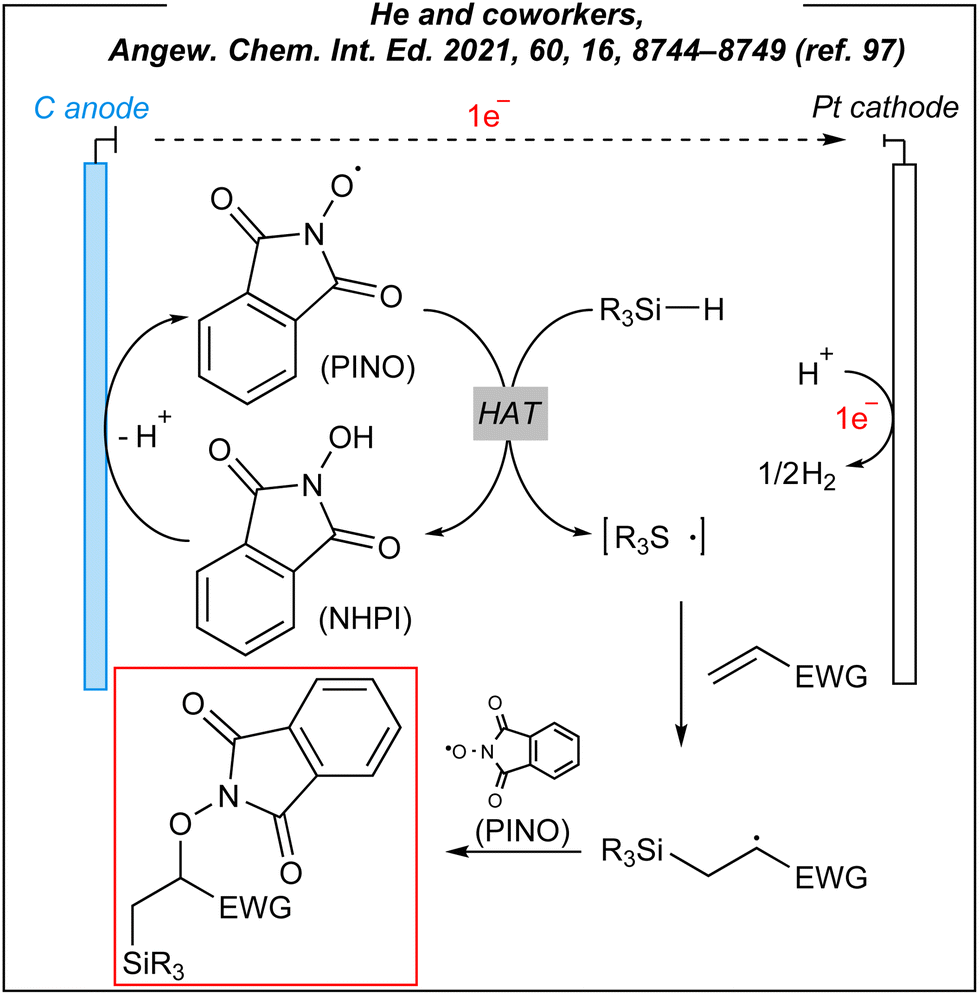 | ||
| Fig. 11 Plausible mechanism for the 1,2-silylfunctionalization. | ||
To sum up, He and coworkers reported a novel methodology for the silyl-oxygenation of activated alkenes, induced by electricity. First of all, the authors used hydrosilanes as the substrates, which is a typical choice for this type of reaction. However, usually, the scope of hydrosilane is limited to triethylsilane or tris(trimethylsilyl)silane, and the conversion of Si–H moieties requires the use of stoichiometric peroxides, traditional transition-metal catalysis or metallaphotocata-lysis.98–107 Despite some disadvantages, the mentioned approaches are more or less a sustainable way to obtain the desired 1,2-silylfunctionalized products. Here, He's method stands out from others in terms of a wide range of silicon substrates. Furthermore, the electrochemical system consists of a graphite anode and a platinum cathode. The latter is classified as an expensive electrode but also reusable. Similarly to Lin65 and Zhang,86 He and coworkers again used an IKA® ElectraSyn 2.0, which ensures the reproducibility of the obtained results. In conclusion, the electrochemical 1,2-silylfunctionalization of alkenes reported by He and coworkers is an interesting alternative to other synthetic methods. Despite some limitations in the alkene scope, it can be considered a useful synthetic approach.
In 2021, Han and Jing achieved the synthesis of dibenzosiloles through electrocatalytic sila-Friedel–Craftss reaction.108 For this process (Fig. 12), the authors used the following combination: RVC anode, platinum cathode, CCE @ 5 mA, NHPI as HAT mediator (1.2 eq.), pyridine as an additive (2.0 eq.) and nBu4NPF6 as the electrolyte. During the optimization studies, several HAT mediators were tested including NHPI (selected), NHSI, quinuclidine, and DABCO. The mediator-free attempt was also carried out and proved the essential role of the HAT-step. Han also demonstrated the necessity of electricity, and that pyridine was the additive of choice. After considerable experimentation, it was also found that CH3CN/HFIP mixture (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) is crucial, and the absence of HFIP (hexafluoroiso-propanol) gave inferior results. Upon the investigation of the reaction scope, it was revealed that biaryl-2-yldiphenylsilanes with both electron-donating groups and electron-withdrawing substituents at the phenyl rings are compatible with the process conditions, leading to the desired products in moderate to excellent yields (32–96%).
:1) is crucial, and the absence of HFIP (hexafluoroiso-propanol) gave inferior results. Upon the investigation of the reaction scope, it was revealed that biaryl-2-yldiphenylsilanes with both electron-donating groups and electron-withdrawing substituents at the phenyl rings are compatible with the process conditions, leading to the desired products in moderate to excellent yields (32–96%).
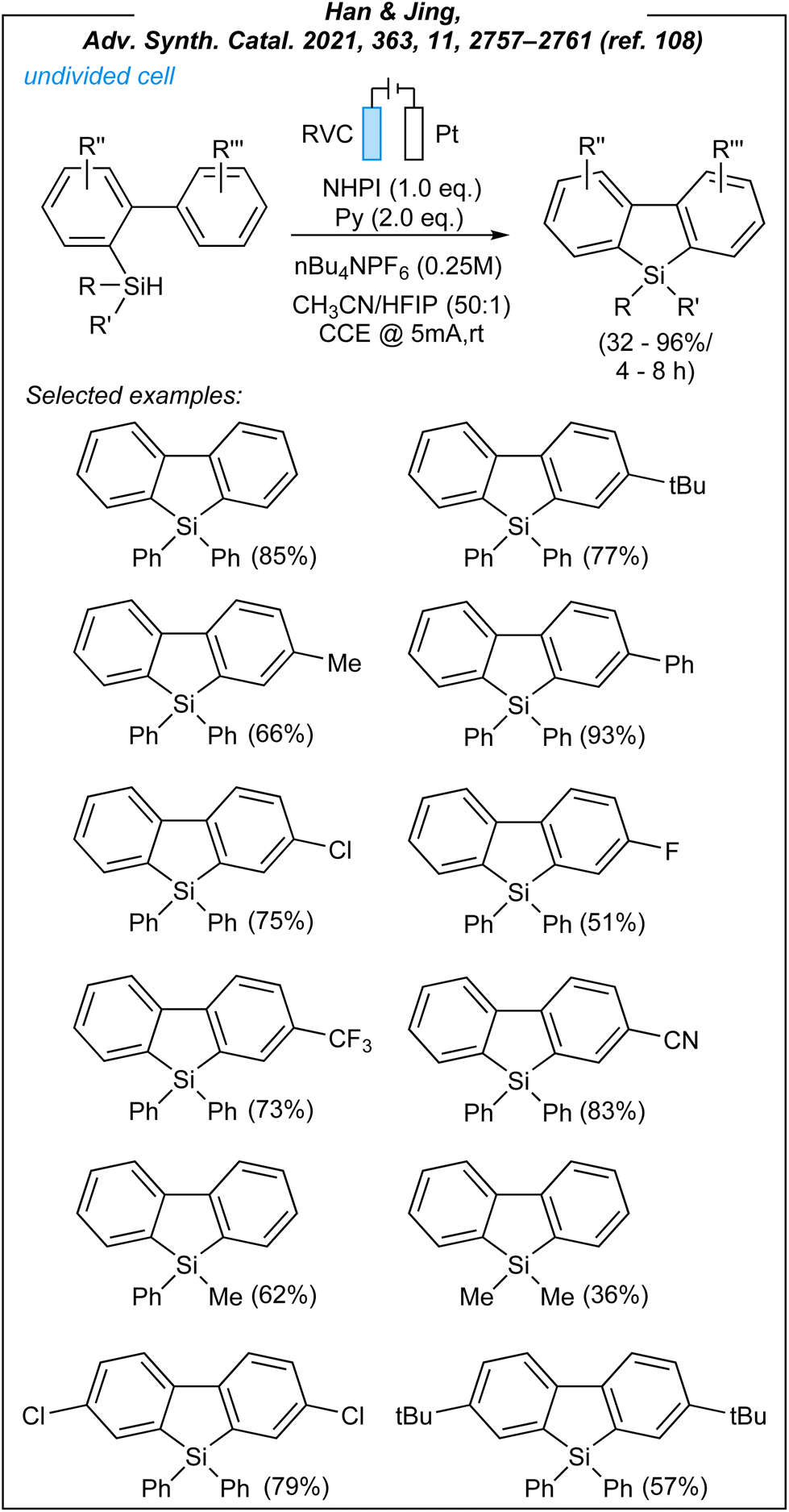 | ||
| Fig. 12 Electroinduced sila-Friedel–Craftss reaction leading to dibenzosiloles. | ||
However, in the vast majority of cases, the substrates with activated rings afforded higher yields. It is worth noting that the nature of substituents at silicon atoms is crucial, probably due to stabilization effects. Thus, the best results were obtained for biaryl-2-yldiphenylsilanes, whilst biaryl-2-ylmethylphenylsilane and biaryl-2-yldimethylsilane gave inferior results (62% and 36%, respectively).
The presented reaction system involved a HAT mediator – NHPI, which undergoes deprotonation facilitated by pyridine, and a subsequent anodic oxidation process. As the result, the PINO radical is afforded and it abstracts the hydrogen atom from the Si–H bond of hydrosilane. The formed silyl radical is subsequently oxidized to a silyl cation. The formation of R3Si+ species was indirectly confirmed by quenching the cations with water to afford the corresponding silanols (similar to Zhang's procedure). However, under standard conditions, the silyl cations cyclize through sila-Friedel–Crafts reaction. The plausible mechanism is depicted in Fig. 13.
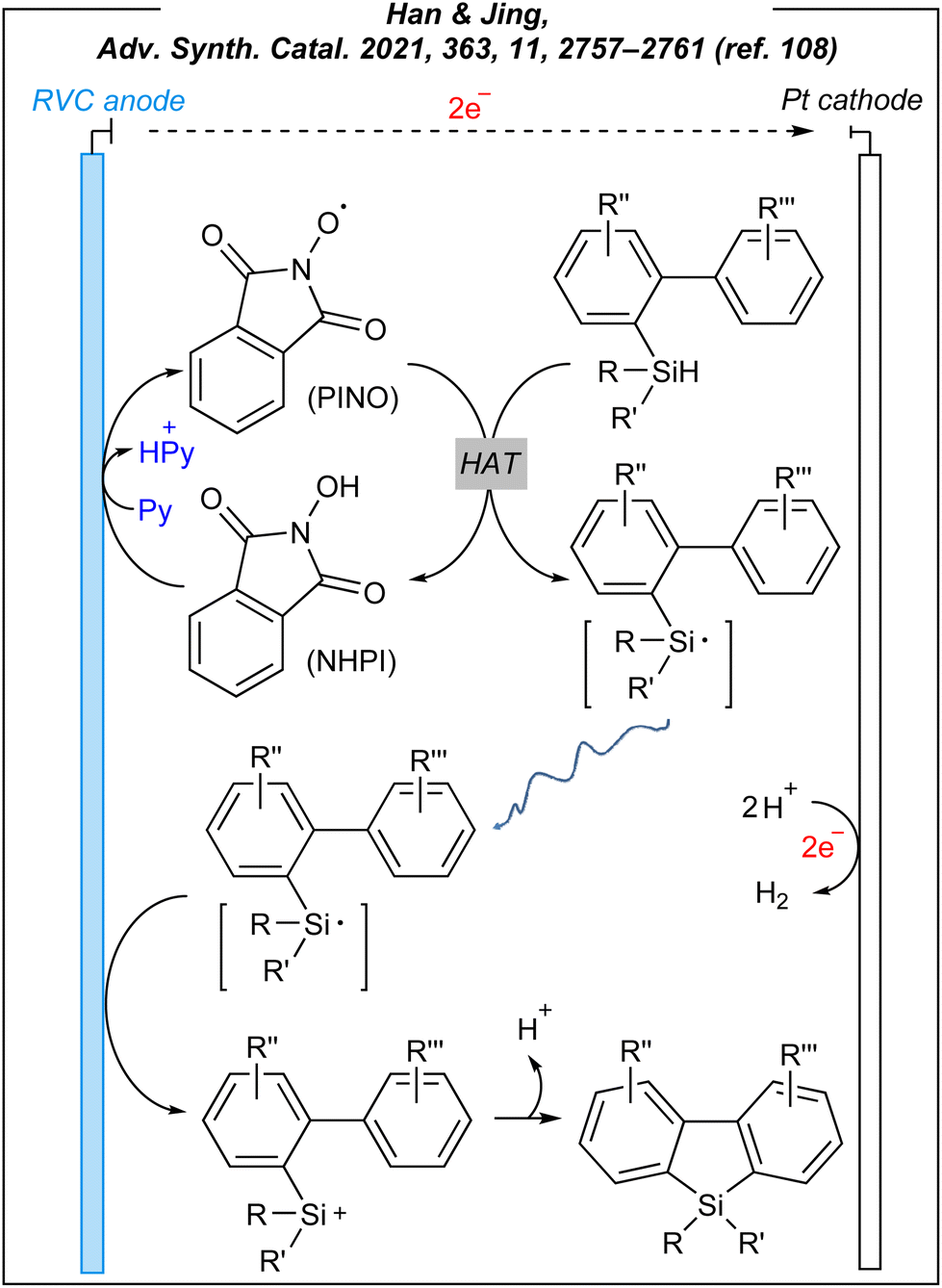 | ||
| Fig. 13 Plausible mechanism for the electrocatalytic sila-Friedel–Craftss reaction. | ||
To sum up, Han and coworkers reported a novel methodology for the synthesis of dibenzosiloles, induced by electricity. The reaction proceeds via cyclization of in situ generated silyl cations (via sequence: hydrosilane – silyl radical – silyl cation). The SiH-substituted biaryls are a typical choice for this type of reaction. The authors showed that triarylsilanes give the best results, with a wide range of substituents at biaryl rings. To date, there were four major ways to obtain dibenzosiloles. The conventional coupling between readily available chlorosilane with organometallic derivative has a limited scope and gives the desired products with low yields.109,110 Secondly, there are radical approaches usually suffering from harsh conditions and stoichiometric amounts of oxidants.111,112 There are also classical dehydrogenative coupling processes employing very expensive transition metal complexes (such as Rh).113–115 Finally, the sila-Friedel–Crafts reactions can be performed in the presence of trityl cation116 or cationic Ru–S complexes.117,118 Thus, from the synthetic point of view, the electrosynthetic approach seems to be a sustainable alternative to known procedures (e.g., no TM-catalysts, no external oxidants, etc.) Furthermore, the electrochemical system consists of an inexpensive graphite anode and an expensive platinum cathode. The authors used a homemade reactor providing no crucial information about the anode dimensions and the current density. In conclusion, the electrochemical sila-Friedel–Crafts reaction reported by Han and coworkers is an interesting alternative to known synthetic protocols, but it has some limitations in the scope (e.g., mostly the utilization of triarylsilyl derivatives) and supporting information for the users.
Recently, Poisson and coworkers disclosed a novel strategy to carry out hydrosilylation reaction by using the Suginome reagent (PhMe2Si–Bpin) and alkynes under electrochemical conditions (Fig. 14).119
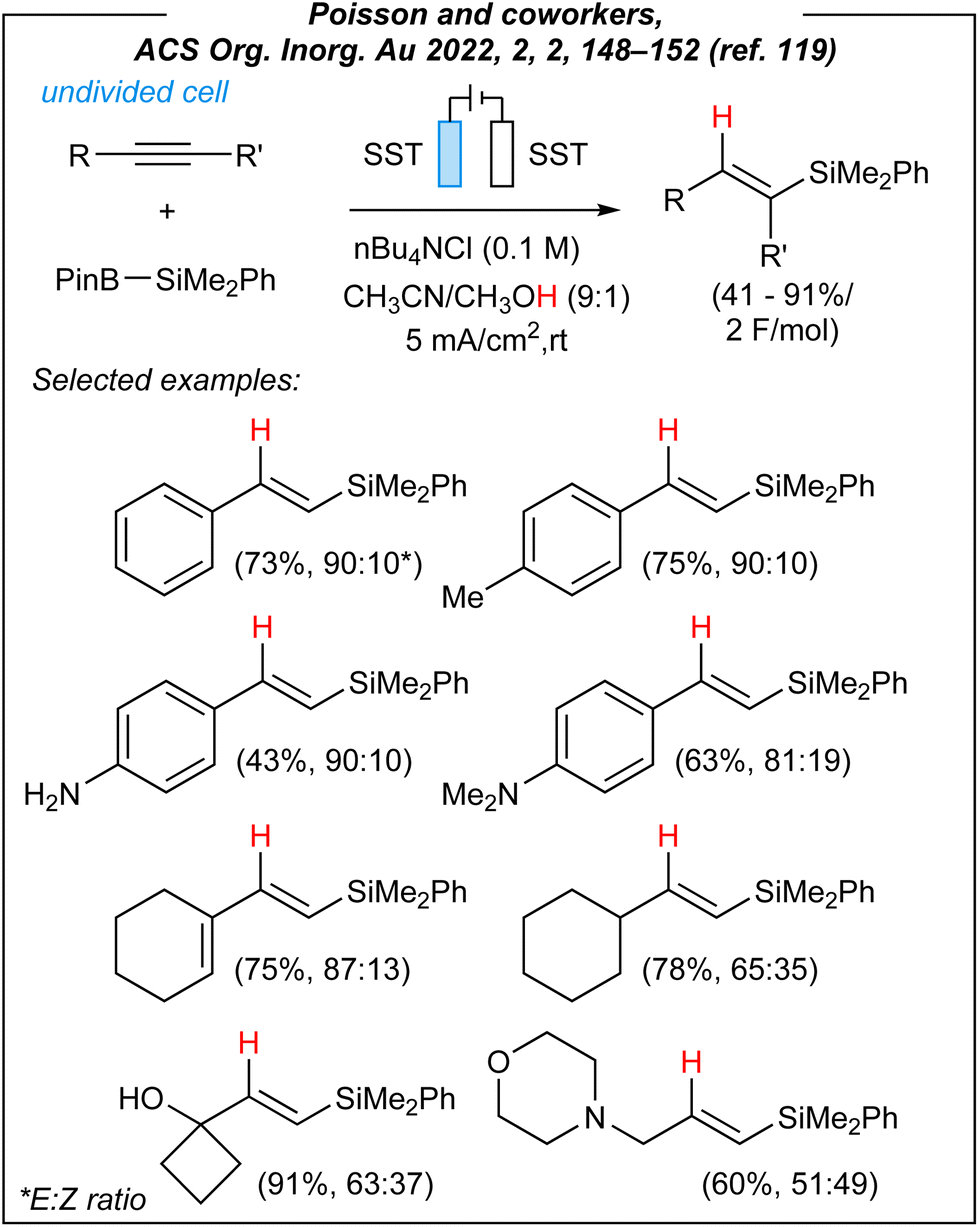 | ||
| Fig. 14 Hydrosilylation of alkynes under electrochemical conditions. | ||
This article clearly shows how important recent developments in the field were. For the formal hydrosilylation process, the authors used the following combination: stainless steel electrodes (both anode and cathode), CCE @ 10 mA, and nBu4NCl as the electrolyte. Control experiments showed that a mixture of CH3CN/CH3OH (9:1) is crucial to obtain the desired products, and the essential role of electricity was also confirmed. The switch from nBu4NCl to nBu4NBF4 led to lower conversion but slightly better selectivity. Subsequently, the authors investigated the scope of this hydrosilylation approach. A wide range of phenylacetylene derivatives worked efficiently under the reaction conditions. Furthermore, commercial ene–yne derivative also participated effectively in this process, while preserving the ene-functionality untouched. Finally, the authors were able to convert a few biorelevant complex molecules, as well as four internal alkynes.
To gain some mechanistic insights into this electroinduced transformation, the authors conducted some experiments. Firstly, the origin of the hydrogen atom (red colour in Fig. 14) added to the vinyl radicals was examined. Unlike Lin, and despite a lower bond dissociation energy of the C–H bond in CH3CN molecule, Poisson proposed that CH3OH serves as the hydrogen source (not CH3CN). It was confirmed via labeling experiments; however, the true nature of this process is still unknown. The plausible mechanism is depicted in Fig. 15.
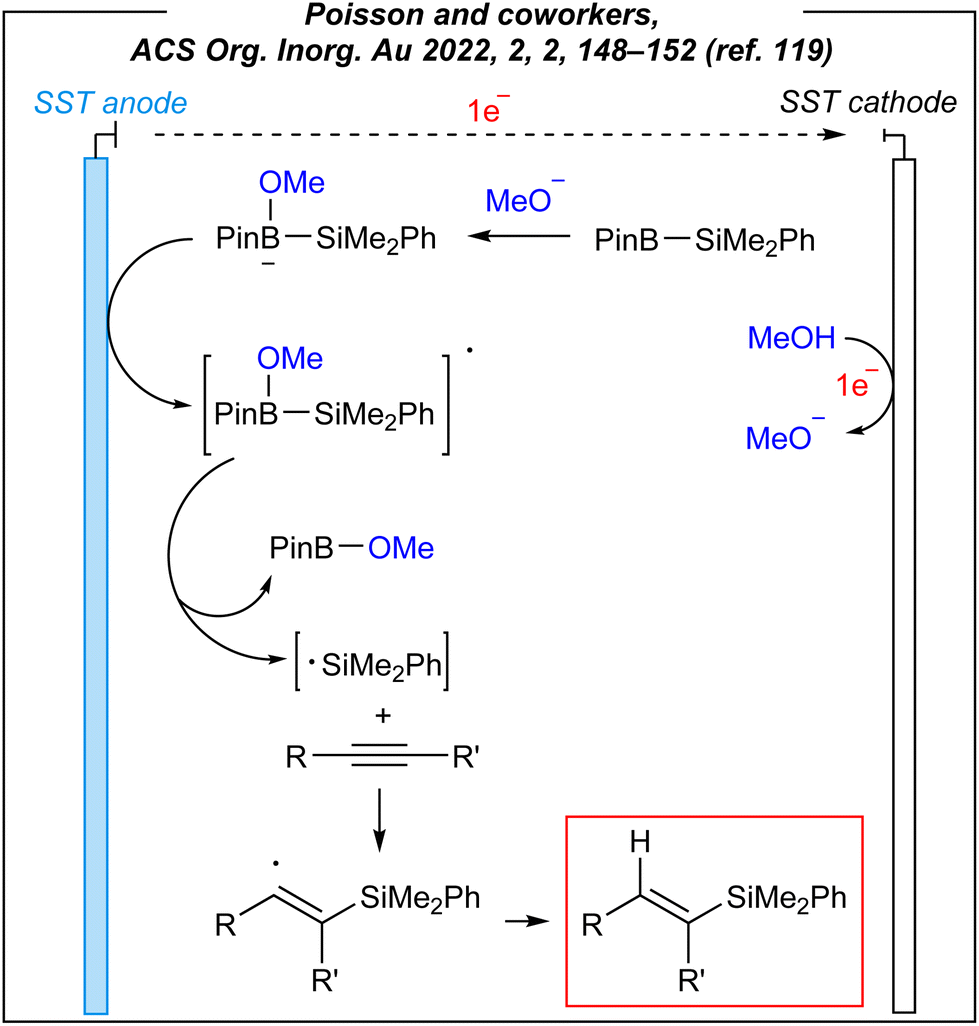 | ||
| Fig. 15 Plausible mechanism for the electroinduced hydrosilylation. | ||
It assumes, that firstly, the methoxide anion reacts with the Suginome reagent to form anionic borate species, which is followed by the anodic oxidation of the latter to form borate radical species. Their decomposition leads to methoxyboronic acid pinacol ester and silyl radicals. The next step involves a nucleophilic silyl radical addition to the alkyne to generate a Si−C bond. The highly reactive vinyl radicals undergo the radical–radical coupling or they abstract the proton from tetracoordinated boron species (formed from methanol and Suginome reagent).
To sum up, Poisson and coworkers reported a novel methodology for the hydrosilylation of alkynes, induced by electricity. First of all, the authors used only Suginome reagent as the source of silicon (it is worth noting, that recently, the authors disclosed also another interesting application of this compound under electrochemical conditions120). The mentioned 2-(dimethylphenylsilyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane is commercially available but quite expensive (Merck – 104€ per 1 g; Gelest – 462.00$ per 2.5 g). The future expansion of the scope (for other B–Si species) would imply an additional preparation of such precursors. Fortunately, the scope of alkynes includes a wide range of terminal and internal derivatives, giving access to several hydrosilylation products. Of course, there are plenty of methods to achieve Si–H addition to unsaturated bonds.34,121–123 However, they involve the use of radical generators or different catalysts (mainly TM-complexes). Therefore, a very important step is searching for an alternative to highly expensive oxidants, as well as catalysts based on platinum or rhodium.124–129 Furthermore, in terms of the electrochemical system, the authors used readily available stainless steel electrodes. Similarly to Lin,65 Zhang,86 and He,97 also Poisson and coworkers used an IKA® ElectraSyn 2.0, which ensures the reproducibility of the obtained results. In conclusion, the presented electrochemical hydrosilylation of alkynes is a fresh look at the process of Si–H addition to unsaturated bonds. Despite some limitations (e.g., all reactions limited to the Suginome reagent, the mixture of E/Z isomers, etc.), it can be considered as one of the pioneering achievements, which should stimulate further development of this field.
The main goal of this perspective was to showcase the diverse synthetic approaches associated with the electroinduced cleavage of Si–H, Si–Cl, and Si–B bonds enabling the synthesis of several organosilicon compounds. These, in turn, create a platform for both known and completely new materials. During the last two years, a significant recent impetus has been gained by the use of electrosynthesis. Notably, this also involved the use of knowledge regarding previous investigations dating back to the 1970s. The major advances had been achieved with the reproducibility and accessibility of the electrochemical approaches. Commercial, standardized equipment such as IKA® ElectraSyn 2.0 may revolutionize the electrochemical synthesis itself. Given the importance of organosilicon compounds for material chemistry, further exciting advances are expected in this constantly-evolving research area. These should address the use of less expensive precursors and additives, the issue of selectivity, and probably the development of electrocatalytic approaches for more challenging transformations. Finally, it is hoped that further achievements will meet the criteria of broadly understood green synthesis, which will ensure the sustainable development of our societies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a National Science Centre Grant UMO-2021/43/D/ST4/00132.References
- R. Zhao, Z. Lin, I. Maksso, J. Struwe and L. Ackermann, Electrochemical Cross-Electrophile-Coupling for Transition-Metal-Free Allylic Carboxylation with Ambient CO2, ChemElectroChem, 2022, 9, e202200989 CAS.
- M. Stangier, A. Scheremetjew and L. Ackermann, Chemo- and Site-Selective Electro-Oxidative Alkane Fluorination by C(sp3)−H Cleavage, Chem. – Eur. J., 2022, 28, e202201654 CrossRef CAS PubMed.
- K. Kuciński, H. Simon and L. Ackermann, Rhoda-Electrocatalyzed C−H Methylation and Paired Electrocatalyzed C−H Ethylation and Propylation, Chem. – Eur. J., 2022, 28, e202103837 CrossRef PubMed.
- B. Sadowski, B. Yuan, Z. Lin and L. Ackermann, Rhodaelectro-Catalyzed peri-Selective Direct Alkenylations with Weak O-Coordination Enabled by the Hydrogen Evolution Reaction (HER), Angew. Chem., Int. Ed., 2022, 61, e202117188 CrossRef CAS.
- T. S.-B. Lou, Y. Kawamata, T. Ewing, G. A. Correa-Otero, M. R. Collins and P. S. Baran, Scalable, Chemoselective Nickel Electrocatalytic Sulfinylation of Aryl Halides with SO2, Angew. Chem., Int. Ed., 2022, 61, e202208080 CAS.
- S. J. Harwood, M. D. Palkowitz, C. N. Gannett, P. Perez, Z. Yao, L. Sun, H. D. Abruña, S. L. Anderson and P. S. Baran, Modular terpene synthesis enabled by mild electrochemical couplings, Science, 2022, 375, 745–752 CrossRef CAS PubMed.
- B. Zhang, Y. Gao, Y. Hioki, M. S. Oderinde, J. X. Qiao, K. X. Rodriguez, H.-J. Zhang, Y. Kawamata and P. S. Baran, Ni-electrocatalytic Csp3−Csp3 doubly decarboxylative coupling, Nature, 2022, 606, 313–318 CrossRef CAS PubMed.
- S. Gnaim, A. Bauer, H.-J. Zhang, L. Chen, C. Gannett, C. A. Malapit, D. E. Hill, D. Vogt, T. Tang, R. A. Daley, W. Hao, R. Zeng, M. Quertenmont, W. D. Beck, E. Kandahari, J. C. Vantourout, P.-G. Echeverria, H. D. Abruna, D. G. Blackmond, S. D. Minteer, S. E. Reisman, M. S. Sigman and P. S. Baran, Cobalt-electrocatalytic HAT for functionalization of unsaturated C–C bonds, Nature, 2022, 605, 687–695 CrossRef CAS PubMed.
- L. F. T. Novaes, Y. Wang, J. Liu, X. Riart-Ferrer, W.-C. Cindy Lee, N. Fu, J. S. K. Ho, X. P. Zhang and S. Lin, Electrochemical Diazidation of Alkenes Catalyzed by Manganese Porphyrin Complexes with Second-Sphere Hydrogen-Bond Donors, ACS Catal., 2022, 12, 14106–14112 CrossRef CAS.
- W. Zhang, L. Lu, W. Zhang, Y. Wang, S. D. Ware, J. Mondragon, J. Rein, N. Strotman, D. Lehnherr, K. A. See and S. Lin, Electrochemically driven cross-electrophile coupling of alkyl halides, Nature, 2022, 604, 292–297 CrossRef CAS.
- D. Liu, Z.-R. Liu, Z.-H. Wang, C. Ma, S. Herbert, H. Schirok and T.-S. Mei, Paired electrolysis-enabled nickel-catalyzed enantioselective reductive cross-coupling between α-chloroesters and aryl bromides, Nat. Commun., 2022, 13, 7318 CrossRef CAS PubMed.
- Z.-H. Wang, L. Wei, K.-J. Jiao, C. Ma and T.-S. Mei, Nickel-catalyzed decarboxylative cross-coupling of indole-3-acetic acids with aryl bromides by convergent paired electrolysis, Chem. Commun., 2022, 58, 8202–8205 RSC.
- Y.-K. Xing, X.-R. Chen, Q.-L. Yang, S.-Q. Zhang, H.-M. Guo, X. Hong and T.-S. Mei, Divergent rhodium-catalyzed electrochemical vinylic C–H annulation of acrylamides with alkynes, Nat. Commun., 2021, 12, 930 CrossRef CAS PubMed.
- L. G. Gombos, L. Werner, D. Schollmeyer, C. A. Martínez-Huitle and S. R. Waldvogel, Selective Electrochemical Dibromination of Terpenes and Naturally Derived Olefins, Eur. J. Org. Chem., 2022, 2022, e202200857 CAS.
- D. Pollok, L. M. Großmann, T. Behrendt, T. Opatz and S. R. Waldvogel, A General Electro-Synthesis Approach to Amaryllidaceae Alkaloids, Chem. – Eur. J., 2022, 28, e202201523 CrossRef CAS PubMed.
- A. D. Beck, S. Haufe, J. Tillmann and S. R. Waldvogel, Challenges in the Electrochemical Synthesis of Si2Cl6 Starting from Tetrachlorosilane and Trichlorosilane, ChemElectroChem, 2022, 9, e202101374 CAS.
- R. D. Little, A Perspective on Organic Electrochemistry, J. Org. Chem., 2020, 85, 13375–13390 CrossRef CAS PubMed.
- P. Gandeepan, L. H. Finger, T. H. Meyer and L. Ackermann, 3d metallaelectrocatalysis for resource economical syntheses, Chem. Soc. Rev., 2020, 49, 4254–4272 RSC.
- L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Electrocatalysis as an enabling technology for organic synthesis, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC.
- M. C. Leech and K. Lam, A practical guide to electrosynthesis, Nat. Rev. Chem., 2022, 6, 275–286 CrossRef.
- M. C. Leech, A. D. Garcia, A. Petti, A. P. Dobbs and K. Lam, Organic electrosynthesis: from academia to industry, React. Chem. Eng., 2022, 5, 977–990 RSC.
- F. Marken, A. J. Cresswell and S. D. Bull, Recent Advances in Paired Electrosynthesis, Chem. Rec., 2021, 21, 2585–2600 CrossRef CAS PubMed.
- D. Pletcher, Organic electrosynthesis – A road to greater application. A mini review, Electrochem. Commun., 2018, 88, 1–4 CrossRef CAS.
- A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Electrifying Organic Synthesis, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed.
- D. Pollok and S. R. Waldvogel, Electro-organic synthesis – a 21st century technique, Chem. Sci., 2020, 11, 12386–12400 RSC.
- K.-J. Jiao, Z.-H. Wang, C. Ma, H.-L. Liu, B. Cheng and T.-S. Mei, The applications of electrochemical synthesis in asymmetric catalysis, Chem Catal., 2022, 2, 3019–3047 CrossRef.
- X. Cheng, A. Lei, T.-S. Mei, H.-C. Xu, K. Xu and C. Zeng, Recent Applications of Homogeneous Catalysis in Electrochemical Organic Synthesis, CCS Chem., 2022, 4, 1120–1152 CrossRef CAS.
- L. Ackermann, Metalla-electrocatalyzed C–H Activation by Earth-Abundant 3d Metals and Beyond, Acc. Chem. Res., 2020, 53, 84–104 CrossRef CAS PubMed.
- Z. N. Gafurov, A. O. Kantyukov, A. A. Kagilev, O. G. Sinyashin and D. G. Yakhvarov, Electrochemical methods for synthesis and in situ generation of organometallic compounds, Coord. Chem. Rev., 2021, 442, 213986 CrossRef CAS.
- C. Ma, P. Fang, D. Liu, K.-J. Jiao, P.-S. Gao, H. Qiu and T.-S. Mei, Transition metal-catalyzed organic reactions in undivided electrochemical cells, Chem. Sci., 2021, 12, 12866–12873 RSC.
- S. B. Beil, D. Pollok and S. R. Waldvogel, Reproducibility in Electroorganic Synthesis—Myths and Misunderstandings, Angew. Chem., Int. Ed., 2021, 60, 14750–14759 CrossRef CAS PubMed.
- Y. Zhang, Z. Cai, S. Warratz, C. Ma and L. Ackermann, Recent advances in electrooxidative radical transformations of alkynes, Sci. China: Chem., 2023 DOI:10.1007/s11426-022-1438-0 accepted article.
- M. A. Brook, Silicon in organic, organometallic, and polymer chemistry, Wiley, 2000 Search PubMed.
- H. Maciejewski, C. Pietraszuk, P. Pawluć and B. Marciniec, Hydrosilylation – A Comprehensive Review on Recent Advances, Springer, 2009 Search PubMed.
- D. B. Cordes, P. D. Lickiss and F. Rataboul, Recent developments in the chemistry of cubic polyhedral oligosilsesquioxanes, Chem. Rev., 2010, 110, 2081–2173 CrossRef CAS PubMed.
- K. Kuciński and G. Hreczycho, Catalytic Formation of Silicon–Heteroatom (N, P, O, S) Bonds, ChemCatChem, 2017, 9, 1868–1885 CrossRef.
- Q. Bu, E. Gońka, K. Kuciński and L. Ackermann, Cobalt-Catalyzed Hiyama-Type C−H Activation with Arylsiloxanes: Versatile Access to Highly ortho-Decorated Biaryls, Chem. – Eur. J., 2019, 25, 2213–2216 CAS.
- H. F. T. Klare, L. Albers, L. Süsse, S. Keess, T. Müller and M. Oestreich, Silylium Ions: From Elusive Reactive Intermediates to Potent Catalysts, Chem. Rev., 2021, 121, 5889–5985 CrossRef CAS PubMed.
- K. Kuciński, H. Stachowiak-Dłużyńska and G. Hreczycho, Catalytic silylation of O–nucleophiles via Si–H or Si–C bond cleavage: A route to silyl ethers, silanols and siloxanes, Coord. Chem. Rev., 2022, 459, 214456 CrossRef.
- R. E. Dessy, W. Kitching and T. Chivers, Organometallic Electrochemistry. I. Derivatives, of Group IV-B Elements, J. Am. Chem. Soc., 1966, 88, 453–459 CrossRef CAS.
- J. Yoshida, Electrochemical reactions of organosilicon compounds, in Electrochemistry V, ed. E. Steckhan, Springer, Berlin, Heidelberg, 1994, pp. 39–81 Search PubMed.
- V. V. Jouikov, Electrochemical reactions of organosilicon compounds, Russ. Chem. Rev., 1997, 66, 509 CrossRef.
- V. Jouikov and G. Salaheev, Electrochemical silylation of phenylacetylene, Electrochim. Acta, 1996, 41, 2623–2629 CrossRef CAS.
- E. Hengge and G. Litscher, A New Electrochemical Method of Forming SiSi Bonds, Angew. Chem., Int. Ed. Engl., 1976, 15, 370–370 CrossRef.
- H. K. Sharma and K. H. Pannell, The Photochemical Irradiation of R3SiH in the Presence of [(η5-C5H5)Fe(CO)2CH3] in DMF Leads to Disiloxanes not Disilanes, Angew. Chem., Int. Ed., 2009, 48, 7052–7054 CrossRef CAS PubMed.
- T. Shono, Y. Matsumura, S. Katoh and N. Kise, An electroreductive synthesis of allylsilanes and benzylsilanes, Chem. Lett., 1985, 14, 463–466 CrossRef.
- J. Yoshida, K. Muraki, H. Funahashi and N. Kawabata, Electrochemical synthesis of organosilicon compounds, J. Organomet. Chem., 1985, 284, C33–C35 CrossRef CAS.
- J. Yoshida, K. Muraki, H. Funahashi and N. Kawabata, Electrochemical synthesis of organosilicon compounds, J. Org. Chem., 1986, 51, 3996–4000 CrossRef CAS.
- J. Yoshida, T. Murata and S. Isoe, Electrochemical oxidation of organosilicon compounds I. Oxidative cleavage of carbon-silicon bond in allylsilanes and benzylsilanes, Tetrahedron Lett., 1986, 27, 3373–3376 CrossRef CAS.
- X. Shang and Z.-Q. Liu, Recent developments in free-radical-promoted C–Si formation via selective C–H/Si–H functionalization, Org. Biomol. Chem., 2016, 14, 7829–7831 RSC.
- J.-S. Li and J. Wu, Recent Developments in the Photo-Mediated Generation of Silyl Radicals and Their Application in Organic Synthesis, ChemPhotoChem, 2018, 2, 839–846 CrossRef CAS.
- I. D. Jenkins and E. H. Krenske, Mechanistic Aspects of Hydrosilane/Potassium tert-Butoxide (HSiR3/KOtBu)-Mediated Reactions, ACS Omega, 2020, 5, 7053–7058 CrossRef CAS PubMed.
- S. Ghosh, D. Lai and A. Hajra, Visible-light-induced silylation: an update, Org. Biomol. Chem., 2021, 19, 2399–2415 RSC.
- X. Zhang, J. Fang, C. Cai and G. Lu, Recent advances in synthesis of organosilicons via radical strategies, Chin. Chem. Lett., 2021, 32, 1280–1292 CrossRef CAS.
- L.-Q. Ren, N. Li, J. Ke and C. He, Recent advances in photo- and electro-enabled radical silylation, Org. Chem. Front., 2022, 9, 6400–6415 RSC.
- M. Khatravath, R. K. Maurya, A. Dey, A. G. Burra, R. Chatterjee and R. Dandela, Recent Advancements in Development of Radical Silylation Reactions, Curr. Org. Chem., 2022, 26, 920–960 CrossRef.
- X. Yu, M. Lübbesmeyer and A. Studer, Oligosilanes as Silyl Radical Precursors through Oxidative Si−Si Bond Cleavage Using Redox Catalysis, Angew. Chem., Int. Ed., 2021, 60, 675–679 CrossRef CAS PubMed.
- N.-X. Xu, B.-X. Li, C. Wang and M. Uchiyama, Sila- and Germacarboxylic Acids: Precursors for the Corresponding Silyl and Germyl Radicals, Angew. Chem., Int. Ed., 2020, 59, 10639–10644 CrossRef CAS PubMed.
- A. A. Toutov, W. B. Liu, K. N. Betz, A. Fedorov, B. M. Stoltz and R. H. Grubbs, Silylation of C-H bonds in aromatic heterocycles by an Earth-abundant metal catalyst, Nature, 2015, 518, 80–84 CrossRef CAS PubMed.
- W. B. Liu, D. P. Schuman, Y. F. Yang, A. A. Toutov, Y. Liang, H. F. T. Klare, N. Nesnas, M. Oestreich, D. G. Blackmond, S. C. Virgil, S. Banerjee, R. N. Zare, R. H. Grubbs, K. N. Houk and B. M. Stoltz, Potassium tert-Butoxide-Catalyzed Dehydrogenative C-H Silylation of Heteroaromatics: A Combined Experimental and Computational Mechanistic Study, J. Am. Chem. Soc., 2017, 139, 6867–6879 CrossRef CAS PubMed.
- P. Anastas and N. Eghbali, Green Chemistry: Principles and Practice, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- M. B. Gawande, V. D. B. Bonifacio, R. Luque, P. S. Branco and R. S. Varma, Solvent-Free and Catalysts-Free Chemistry: A Benign Pathway to Sustainability, ChemSusChem, 2014, 7, 24–44 CrossRef CAS PubMed.
- A. Sarkar, S. Santra, K. Kundu, A. Hajra, G. V. Zyryanov, O. N. Chupakhin, V. N. Charushin and A. Majee, A decade update on solvent and catalyst-free neat organic reactions: a step forward towards sustainability, Green Chem., 2016, 18, 4475–4525 RSC.
- N. Sbei, T. Hardwick and N. Ahmed, Green Chemistry: Electrochemical Organic Transformations via Paired Electrolysis, ACS Sustainable Chem. Eng., 2021, 9, 6148–6169 CrossRef CAS.
- L. Lu, J. C. Siu, Y. Lai and S. Lin, An Electroreductive Approach to Radical Silylation via the Activation of Strong Si–Cl Bond, J. Am. Chem. Soc., 2020, 142, 21272–21278 CrossRef CAS PubMed.
- G. R. Jones and Y. Landais, The oxidation of the carbon-silicon bond, Tetrahedron, 1996, 52, 7599–7662 CrossRef CAS.
- K. L. Hervert, in Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, 2013 Search PubMed.
- Y. Yuan and A. Lei, Is electrosynthesis always green and advantageous compared to traditional methods?, Nat. Commun., 2020, 11, 802 CrossRef CAS PubMed.
- M. Yus, P. Martínez and D. Guijarro, DTBB-Catalysed dilithiation of styrene and its methyl-derivatives: introduction of two electrophilic reagents, Tetrahedron, 2021, 57, 10119–10124 CrossRef.
- T. Hayashi, T. Kobayashi, A. M. Kawamoto, H. Yamashita and M. Tanaka, Platinum complex catalyzed double silylation of ethylene and norbornene with disilanes, Organometallics, 1990, 9, 280–281 CrossRef CAS.
- Y. Ito, M. Suginome and M. Murakami, Palladium(II) acetate-tert-alkyl isocyanide as a highly efficient catalyst for the inter- and intramolecular bis-silylation of carbon-carbon triple bonds, J. Org. Chem., 1991, 56, 1948–1951 CrossRef CAS.
- M. Suginome and Y. Ito, Transition-Metal-Catalyzed Additions of Silicon−Silicon and Silicon−Heteroatom Bonds to Unsaturated Organic Molecules, Chem. Rev., 2000, 100, 3221–3256 CrossRef CAS PubMed.
- J. Terao, A. Oda and N. Kambe, Palladium-Catalyzed Dimerization Disilylation of 1,3-Butadiene with Chlorosilanes, Org. Lett., 2004, 6, 3341–3344 CrossRef CAS PubMed.
- M. Wollenburg, J. Bajohr, A. D. Marchese, A. Whyte, F. Glorius and M. Lautens, Palladium-Catalyzed Disilylation and Digermanylation of Alkene Tethered Aryl Halides: Direct Access to Versatile Silylated and Germanylated Heterocycles, Org. Lett., 2020, 22, 3679–3683 CrossRef CAS PubMed.
- Y. Zhang, X.-C. Wang, C.-W. Ju and D. Zhao, Bis-silylation of internal alkynes enabled by Ni(0) catalysis, Nat. Commun., 2021, 12, 68 CrossRef CAS PubMed.
- Q. Wu, A. Roy, E. Irran, Z.-W. Qu, S. Grimme, H. F. T. Klare and M. Oestreich, Catalytic Difunctionalization of Unactivated Alkenes with Unreactive Hexamethyldisilane through Regeneration of Silylium Ions, Angew. Chem., Int. Ed., 2019, 58, 17307–17311 CrossRef CAS PubMed.
- A. Roy, V. Bonetti, G. Wang, Q. Wu, H. F. T. Klare and M. Oestreich, Silylium-Ion-Promoted Ring-Opening Hydrosilylation and Disilylation of Unactivated Cyclopropanes, Org. Lett., 2020, 22, 1213–1216 CrossRef CAS PubMed.
- V. Natarajan, N. K. Porwal, Y. Babu, B. Rajeswari, B. A. Dhawale, M. Kumar, S. V. Godbole and V. K. Manchanda, Direct determination of metallic impurities in graphite by EDXRF, Appl. Radiat. Isot., 2010, 68, 1128–1131 CrossRef CAS PubMed.
- Y. Nakajima and S. Shimada, Hydrosilylation Reaction of Olefins: Recent Advances and Perspective, RSC Adv., 2015, 5, 20603–20616 RSC.
- R. Zhou, Y. Y. Goh, H. Liu, H. Tao, L. Li and J. Wu, Visible-Light-Mediated Metal-Free Hydrosilylation of Alkenes through Selective Hydrogen Atom Transfer for Si−H Activation, Angew. Chem., Int. Ed., 2017, 56, 16621–16625 CrossRef CAS PubMed.
- M. Zaranek, B. Marciniec and P. Pawluć, Ruthenium-catalysed hydrosilylation of carbon–carbon multiple bonds, Org. Chem. Front., 2016, 3, 1337–1344 RSC.
- T. K. Meister, K. Riener, P. Gigler, J. Stohrer, W. A. Herrmann and F. E. Kühn, Platinum Catalysis Revisited—Unraveling Principles of Catalytic Olefin Hydrosilylation, ACS Catal., 2016, 6, 1274–1284 CrossRef CAS.
- S. Amrein, A. Timmermann and A. Studer, Radical Transfer Hydrosilylation/Cyclization Using Silylated Cyclohexadienes, Org. Lett., 2001, 3, 2357–2360 CrossRef CAS PubMed.
- T. Zhang, Z. Zhang, Y. Nishiyama and H. Maekawa, Facile and highly selective silylation of vinylpyridines at the β-olefinic carbon by magnesium-promoted reduction, Tetrahedron, 2016, 72, 2293–2299 CrossRef CAS.
- V. Jouikov and L. Grigorieva, Electrochemically induced silylation of unsaturated compounds, Electrochim. Acta, 1996, 41, 469–470 CrossRef CAS.
- H. Liang, L.-J. Wang, Y.-X. Ji, H. Wang and B. Zhang, Selective Electrochemical Hydrolysis of Hydrosilanes to Silanols via Anodically Generated Silyl Cations, Angew. Chem., Int. Ed., 2021, 60, 1839–1844 CrossRef CAS PubMed.
- K. Kuciński, H. Stachowiak and G. Hreczycho, Silylation of Silanols with Hydrosilanes via Main-Group Catalysis: The Synthesis of Unsymmetrical Siloxanes and Hydrosiloxanes, Inorg. Chem. Front., 2020, 7, 4190–4196 RSC.
- J. A. Cella and J. C. Carpenter, Procedures for the preparation of silanols, J. Organomet. Chem., 1994, 480, 23–26 CrossRef CAS.
- M. Jeon, J. Han and J. Park, Catalytic synthesis of silanols from hydrosilanes and applications, ACS Catal., 2012, 2, 1539–1549 CrossRef CAS.
- A. Harinath, J. Bhattacharjee, S. Anga and T. K. Panda, Dehydrogenative Coupling of Hydrosilanes and Alcohols by Alkali Metal Catalysts for Facile Synthesis of Silyl Ethers, Aust. J. Chem., 2017, 70, 724–730 CrossRef CAS.
- M. Skrodzki, M. Zaranek, S. Witomska and P. Pawluc, Direct dehydrogenative coupling of alcohols with hydrosilanes promoted by sodium tri(Sec-butyl)borohydride, Catalysts, 2018, 8, 618–624 CrossRef.
- K. Kuciński, H. Stachowiak and G. Hreczycho, Silylation of Alcohols, Phenols, and Silanols with Alkynylsilanes - an Efficient Route to Silyl Ethers and Unsymmetrical Siloxanes, Eur. J. Org. Chem., 2020, 2020, 4042–4049 CrossRef.
- K. Pypowski and M. Mojzych, Reactivity of heterocyclic α-aminomethylsilanes with alcohols, Chem. Heterocycl. Compd., 2021, 57, 320–324 CrossRef CAS.
- H. Stachowiak, J. Kaźmierczak, K. Kuciński and G. Hreczycho, Catalyst-free and solvent-free hydroboration of aldehydes, Green Chem., 2018, 20, 1738–1742 RSC.
- E. O. Pentsak, D. B. Eremin, E. G. Gordeev and V. P. Ananikov, Phantom Reactivity in Organic and Catalytic Reactions as a Consequence of Microscale Destruction and Contamination-Trapping Effects of Magnetic Stir Bars, ACS Catal., 2019, 9, 3070–3081 CrossRef CAS.
- S. Sau, M. Pramanik, A. Bal and P. Mal, Reported Catalytic Hydrofunctionalizations that Proceed in the Absence of Catalysts: The Importance of Control Experiments, Chem. Rec., 2022, 22, e202100208 CrossRef CAS PubMed.
- J. Ke, W. Liu, X. Zhu, X. Tan and C. He, Electrochemical Radical Silyl-Oxygenation of Activated Alkenes, Angew. Chem., Int. Ed., 2021, 60, 8744–8749 CrossRef CAS PubMed.
- O. Tayama, T. Iwahama, S. Sakaguchi and Y. Ishii, The First Hydroxysilylation of Alkenes with Triethylsilane under Dioxygen Catalyzed by N-Hydroxyphthalimide, Eur. J. Org. Chem., 2003, 2003, 2286–2289 CrossRef.
- Y. Lan, X.-H. Chang, P. Fan, C.-C. Shan, Z.-B. Liu, T.-P. Loh and Y.-H. Xu, Copper-Catalyzed Silylperoxidation Reaction of α,β-Unsaturated Ketones, Esters, Amides, and Conjugated Enynes, ACS Catal., 2017, 7, 7120–7125 CrossRef CAS.
- Y. Yang, R.-J. Song, X.-H. Ouyang, C.-Y. Wang, J.-H. Li and S. Luo, Iron-Catalyzed Intermolecular 1,2-Difunctionalization of Styrenes and Conjugated Alkenes with Silanes and Nucleophiles, Angew. Chem., Int. Ed., 2017, 56, 7916–7919 CrossRef CAS PubMed.
- J. Hou, A. Ee, H. Cao, H.-W. Ong, J.-H. Xu and J. Wu, Visible-Light-Mediated Metal-Free Difunctionalization of Alkenes with CO2 and Silanes or C(sp3)−H Alkanes, Angew. Chem., Int. Ed., 2018, 57, 17220–17224 CrossRef CAS PubMed.
- K. Nozawa-Kumada, T. Ojima, M. Inagi, M. Shigeno and Y. Kondo, Di-tert-butyl Peroxide (DTBP)-Mediated Oxysilylation of Unsaturated Carboxylic Acids for the Synthesis of Silyl Lactones, Org. Lett., 2020, 22, 9591–9596 CrossRef CAS PubMed.
- Z. Zhang and X. Hu, Arylsilylation of Electron-Deficient Alkenes via Cooperative Photoredox and Nickel Catalysis, ACS Catal., 2020, 10, 777–782 CrossRef CAS.
- M. Zheng, J. Hou, L. Hua, W.-Y. Tang, L.-W. Zhan and B.-D. Li, Visible-Light-Mediated Divergent Silylfunctionalization of Alkenes, Org. Lett., 2021, 23, 5128–5132 CrossRef CAS PubMed.
- S. Neogi, A. Kumar Ghosh, S. Mandal, D. Ghosh, S. Ghosh and A. Hajra, Three-Component Carbosilylation of Alkenes by Merging Iron and Visible-Light Photocatalysis, Org. Lett., 2021, 23, 6510–6514 CrossRef CAS PubMed.
- W. Zheng, Y. Xu, H. Luo, Y. Feng, J. Zhang and L. Lin, Light-Promoted Arylsilylation of Alkenes with Hydrosilanes, Org. Lett., 2022, 24, 7145–7150 CrossRef CAS PubMed.
- S. Plöger and A. Studer, Visible-Light-Mediated Radical Silyl-Oximation of Activated Alkenes Using tert-Butyl Nitrite and Silanes, Org. Lett., 2022, 24, 8568–8572 CrossRef PubMed.
- P. Han, M. Yin, H. Li, J. Yi, L. Jing and B. Wei, Synthesis of Dibenzosiloles through Electrocatalytic Sila-Friedel-Crafts Reaction, Adv. Synth. Catal., 2021, 363, 2757–2761 CrossRef CAS.
- L. Li, J. Xiang and C. Xu, Synthesis of Novel Ladder Bis-Silicon-Bridged p-Terphenyls, Org. Lett., 2007, 9, 4877–4879 CrossRef CAS PubMed.
- A. Kaga, H. Iida, S. Tsuchiya, H. Saito, K. Nakano and H. Yorimitsu, Aromatic Metamorphosis of Thiophenes by Means of Desulfurative Dilithiation, Chem. – Eur. J., 2021, 27, 4567–4572 CrossRef CAS PubMed.
- D. Leifert and A. Studer, 9-Silafluorenes via Base-Promoted Homolytic Aromatic Substitution (BHAS) – The Electron as a Catalyst, Org. Lett., 2015, 17, 386–389 CrossRef CAS PubMed.
- C. Yang, J. Wang, J. Li, W. Ma, K. An, W. He and C. Jiang, Visible-Light Induced Radical Silylation for the Synthesis of Dibenzosiloles via Dehydrogenative Cyclization, Adv. Synth. Catal., 2018, 360, 3049–3054 CrossRef CAS.
- W. Ma, L.-C. Liu, K. An, T. He and W. He, Rhodium-Catalyzed Synthesis of Chiral Monohydrosilanes by Intramolecular C−H Functionalization of Dihydrosilanes, Angew. Chem., Int. Ed., 2021, 60, 4245–4251 CrossRef CAS PubMed.
- Y. Xu, W. Xu, X. Chen, X. Luo, H. Lu, M. Zhang, X. Yang, G. Deng, Y. Liang and Y. Yang, Me3SiSiMe2(OnBu): a disilane reagent for the synthesis of diverse silacycles via Brook- and retro-Brook-type rearrangement, Chem. Sci., 2021, 12, 11756–11761 RSC.
- L. Zhang, K. An, Y. Wang, Y.-D. Wu, X. Zhang, Z.-X. Yu and W. He, A Combined Computational and Experimental Study of Rh-Catalyzed C–H Silylation with Silacyclobutanes: Insights Leading to a More Efficient Catalyst System, J. Am. Chem. Soc., 2021, 143, 3571–3582 CrossRef CAS PubMed.
- S. Furukawa, J. Kobayashi and T. Kawashima, Development of a Sila-Friedel−Crafts Reaction and Its Application to the Synthesis of Dibenzosilole Derivatives, J. Am. Chem. Soc., 2009, 131, 14192–14193 CrossRef CAS PubMed.
- L. Omann and M. Oestreich, A Catalytic SEAr Approach to Dibenzosiloles Functionalized at Both Benzene, Angew. Chem., Int. Ed., 2015, 54, 10276–10279 CrossRef CAS PubMed.
- L. Omann and M. Oestreich, Catalytic Access to Indole-Fused Benzosiloles by 2-Fold Electrophilic C–H Silylation with Dihydrosilanes, Organometallics, 2017, 36, 767–776 CrossRef CAS.
- T. Biremond, P. Jubault and T. Poisson, Electrochemical Hydrosilylation of Alkynes, ACS Org. Inorg. Au, 2022, 2, 148–152 CrossRef CAS.
- M. Aelterman, T. Biremond, P. Jubault and T. Poisson, Electrochemical Synthesis of gem-Difluoro- and γ-Fluoro-Allyl Boronates and Silanes, Chem. – Eur. J., 2022, 28, e202202194 CrossRef CAS PubMed.
- J. V. Obligacion and P. J. Chirik, Earth-abundant transition metal catalysts for alkene hydrosilylation and hydroboration, Nat. Rev. Chem., 2018, 2, 15–34 CrossRef CAS PubMed.
- S. Harder, Early Main Group Metal-Catalyzed Hydrosilylation of Unsaturated Bonds, in Early Main Group Metal Catalysis, John Wiley & Sons, Ltd, 2020, pp. 151–173 Search PubMed.
- J. Walkowiak, J. Szyling, A. Franczyk and R. L. Melen, Hydroelementation of diynes, Chem. Soc. Rev., 2022, 51, 869–994 RSC.
- A. Franczyk, K. Stefanowska, M. Dutkiewicz, D. Frąckowiak and B. Marciniec, A highly selective synthesis of new alkenylsilsesquioxanes by hydrosilylation of alkynes, Dalton Trans., 2017, 46, 158–164 RSC.
- D. Brząkalski, M. Rzonsowska and B. Dudziec, Regioselective formation of β-(E)- and α-alkenylsilane moiety attached to silsesquioxane core via Pt- and Ru-based hydrosilylation, Appl. Organomet. Chem., 2018, 32, e4267 CrossRef.
- P. Żak, M. Bołt, M. Kubicki and C. Pietraszuk, Highly selective hydrosilylation of olefins and acetylenes by platinum(0) complexes bearing bulky N-heterocyclic carbene ligands, Dalton Trans., 2018, 47, 1903–1910 RSC.
- J. Szyling, R. Januszewski, K. Jankowska, J. Walkowiak, I. Kownacki and A. Franczyk, Synthesis of bifunctional disiloxanes via subsequent hydrosilylation of alkenes and alkynes, Chem. Commun., 2021, 57, 4504–4507 RSC.
- K. Stefanowska, T. Sokolnicki, J. Walkowiak, A. Czapik and A. Franczyk, Directed cis-hydrosilylation of borylalkynes to borylsilylalkenes, Chem. Commun., 2022, 58, 12046–12049 RSC.
- M. Bołt, A. Mermela and P. Żak, Influence of Bis-NHC Ligand on Platinum-Catalyzed Hydrosilylation of Internal Alkynes, Eur. J. Inorg. Chem., 2023, 26, e202200622 CrossRef.
| This journal is © the Partner Organisations 2023 |