
Plant glycosyltransferases for expanding bioactive glycoside diversity
Sasilada
Sirirungruang
 abcd,
Collin R.
Barnum
e,
Sophia N.
Tang
bcf and
Patrick M.
Shih
*abcg
abcd,
Collin R.
Barnum
e,
Sophia N.
Tang
bcf and
Patrick M.
Shih
*abcg
aDepartment of Plant and Microbial Biology, University of California, Berkeley, CA, USA
bFeedstocks Division, Joint BioEnergy Institute, Emeryville, CA, USA
cEnvironmental Genomics and Systems Biology Division, Lawrence Berkeley National Laboratory, Berkeley, CA, USA
dCenter for Biomolecular Structure, Function and Application, Suranaree University of Technology, Nakhon Ratchasima, Thailand
eDepartment of Plant Biology, University of California, Davis, CA, USA
fDepartment of Molecular and Cell Biology, University of California, Berkeley, CA, USA
gInnovative Genomics Institute, University of California, Berkeley, CA, USA
First published on 28th February 2023
Abstract
Glycosylation is a successful strategy to alter the pharmacological properties of small molecules, and it has emerged as a unique approach to expand the chemical space of natural products that can be explored in drug discovery. Traditionally, most glycosylation events have been carried out chemically, often requiring many protection and deprotection steps to achieve a target molecule. Enzymatic glycosylation by glycosyltransferases could provide an alternative strategy for producing new glycosides. In particular, the glycosyltransferase family has greatly expanded in plants, representing a rich enzymatic resource to mine and expand the diversity of glycosides with novel bioactive properties. This article highlights previous and prospective uses for plant glycosyltransferases in generating bioactive glycosides and altering their pharmacological properties.
 Sasilada Sirirungruang | Sasilada Sirirungruang received her BS in chemistry and biology from Massachusetts Institute of Technology, and her PhD in molecular and cell biology from University of California, Berkeley, under the supervision of Professor Michelle Chang. She is currently a postdoctoral researcher in the laboratory of Professor Patrick Shih, focusing on developing plant biosynthetic enzyme technologies. |
 Collin R. Barnum | Collin Barnum received his BA in biology and environmental and earth sciences at Willamette University. He is a doctoral student at the University of California, Davis under the supervision of Dr Patrick Shih. His research focuses on the production of therapeutic small molecules in plants. |
 Sophia Tang | Sophia Tang received her BS in biological chemistry and chemistry from the University of Chicago. She is currently pursuing her PhD in molecular and cell biology from the University of California, Berkeley under the supervision of Patrick Shih. Her research is centered on polysaccharide engineering in plants. |
 Patrick M. Shih | Patrick Shih is an Assistant Professor at the University of California, Berkeley. His research focuses on using synthetic biology to both study and engineer biological organisms. His research group is especially interested in exploring how plant metabolism can be manipulated for novel applications in agriculture, sustainability, human health, and bioenergy. |
1. Natural product glycosides are important in drug discovery and development
Glycosylation can dramatically affect the physicochemical properties of molecules. Specifically, the pharmacokinetic and pharmacodynamic properties of glycoside natural product therapeutics can vary greatly from those of their aglycone counterparts (Fig. 1).1 Thus, controlled glycosylation is an attractive approach to modify natural products that would otherwise have poor drug-like properties, off-target effects, or high toxicity. Currently, 145 approved and experimental drugs in the US are annotated as glycosides according to https://DrugBank.com.2 They include well-known examples such as remdesivir, vancomycin, amphotericin B, and doxorubicin, which represent antiviral, antibiotic, antifungal and anti-cancer compounds, respectively. The prevalence of glycosides among successful drugs suggests that adding carbohydrate modules to drug leads is a valuable tool in drug discovery. | ||
| Fig. 1 Glycosylation can affect various properties of small molecules. (a) Solubility, which influences drug absorption, transport, and tissue and cellular distribution can be affected by glycosylation. Puerarin mono- (middle) and di-glucoside (right) are 15.6 and 100.9 times more soluble than puerarin (left).8 (b) Binding to drug targets and off targets, which governs drug efficacy and toxicity, can be modulated via glycosylation. Digitoxigenin monodigitoxoside (middle) and digitoxin (right) bind to Na+/K+-ATPase with affinity values that are 6.11 and 4.26 times that of digitoxigenin (left).12 (c) Glycosylation can affect organoleptic properties of small molecules. Rebaudioside A (middle) and D (right) have lower sweet threshold concentrations and higher bitter threshold concentrations than those of steviol (left), making them more desirable as sweeteners.52 (d) Glycosylation can also be used to generate prodrugs by masking bioactivity of bioactive molecules. The depicted prodrug combines a glucuronide trigger and a self-immolative linker to the antineoplastic agent monomethylauristatin E (MMAE). Hydrolysis of the glycosidic bond by β-glucuronidase selectively releases the active drug in the tumor microenvironment.10 | ||
The classic example of the benefits of glycosylation in drug discovery is well illustrated in the search for vancomycin derivatives against antibiotic resistance. Vancomycin has been considered an antibiotic of last resort as it prevents crosslinking of the peptidoglycan layer during cell wall synthesis.3 However, as resistance arose, vancomycin was modified in various ways to escape resistance. Notably, alterations to the number of sugars,4 the identity of sugars,5 and the modification on the sugars6 have all been reported to alter vancomycin's pharmacological properties. Other examples demonstrating the benefits of glycosylation include etoposide and teniposide, two podophyllotoxin-based anticancer drugs that are both D-glucosides and have reduced toxicity compared to the aglycone.7 In addition, glycosylation can also increase the solubility of aglycones, affecting drug absorption, transport, and distribution. Puerarin, the most abundant isoflavone from Pueraria lobata (kudzu), can be mono-, di-, or tri-O-glycosylated, which yields molecules 15.6, 100.9, and 179.1 times more soluble in water than puerarin, respectively.8 Finally, glycosylation can also be employed to mask bioactivity. Such strategy has been observed in nature in protecting reactive biosynthetic intermediates9 as well as in designing prodrugs for drug targeting purposes.10,11
Swapping different sugar moieties on a range of glycosides has also been demonstrated to affect their physicochemical properties. For instance, digitoxigenin- and ouabagenin-based cardiac glycosides have been well studied and are known to inhibit Na+/K+-ATPase pumps. Digitoxigenin-based glycosides digitoxigenin monodigitoxoside (a single digitoxose sugar on digitoxigenin) and digitoxin (three digitoxose sugars on digitoxigenin) have binding affinity values 6.11 and 4.26 times that of digitoxigenin, respectively.12 The ouabagenin-based glycosides peruvoside (thevetose on ouabagenin) and helveticoside (digitoxose on ouabagenin) have IC50 values of 0.798 ± 0.054 and 24.1 ± 0.6 times that of ouabain (rhamnose on ouabagenin).12 This shows that the identity of the sugar module plays a role in glycosides' molecular functions, particularly their interactions with drug targets.
As sessile organisms, plants heavily rely on their ability to produce, modify, and perceive a range of chemicals which modulate many biological functions, including growth regulation, signaling, and defense against herbivores and pathogens. Glycosylation is arguably the predominant strategy plants utilize to store, transport, and sequester bioactive small molecules.13 Thus, plants have developed many enzymes to manipulate the carbohydrate modules of small molecules that can potentially be exploited for drug discovery, including anthraquinone, cardiac, coumarin, cyanogenic, flavonoid, glucosinolate, phenol, and saponin glycosides (Fig. 2).14 Among the many glycosides naturally produced in plants, relatively few have been studied. The structures and functions of many complex natural product glycosides remain to be discovered, due to the challenges associated with compound isolation and structural elucidation.
 | ||
| Fig. 2 Plants produce a multitude of complex glycoside natural products with pharmaceutical benefits. Sugar modules of plant glycosides (shown in blue) vary from a single simple sugar to short glycan chains and modified sugar molecules. | ||
2. Past and current glycosylation strategies
Despite having tremendous value in drug discovery, glycosides are a relatively understudied source of bioactive compounds, partly due to the limited access to these molecules. The low abundance of glycosides in complex mixtures of plant metabolites can complicate the purification process. Additionally, synthesis methods that would yield access to the pure form of glycosides of importance and their non-natural variants in large quantities are often challenging due to multiple hydroxyl groups and stereocenters on sugar moieties. Despite recent advancements in the synthesis of natural product glycosides, challenges still limit the diversity of glycosides that can be produced.Traditionally, chemical synthesis was the primary means to access low abundance glycosides found in nature. Several chemical methods to build glycosidic bonds have continually been developed over many decades. Most methods rely on the glycosyl donor contributing its anomeric carbon serving as the electrophile while the glycosyl acceptor serves as the nucleophile (Fig. 3a).15 This common strategy requires installing a leaving group at the anomeric position and protecting groups at all other hydroxyl groups of the glycosyl donor.15 Upon activation of the leaving group with a compatible electron withdrawing reagent, the anomeric carbon can be substituted by a nucleophilic glycosyl acceptor. If the glycosyl acceptor contains multiple nucleophilic groups, those not involved in the reaction also need to be protected. Many leaving groups have been developed to selectively work with different activating reagents and nucleophiles including phosphates, carbonates and esters, halides, and aryloxy groups.15,16 With carefully selected monosaccharide building blocks, the many leaving group chemistries developed allow one-pot synthesis of glycosides and oligosaccharides, which bypasses the tedious and time-consuming work-up and purification processes.16,17
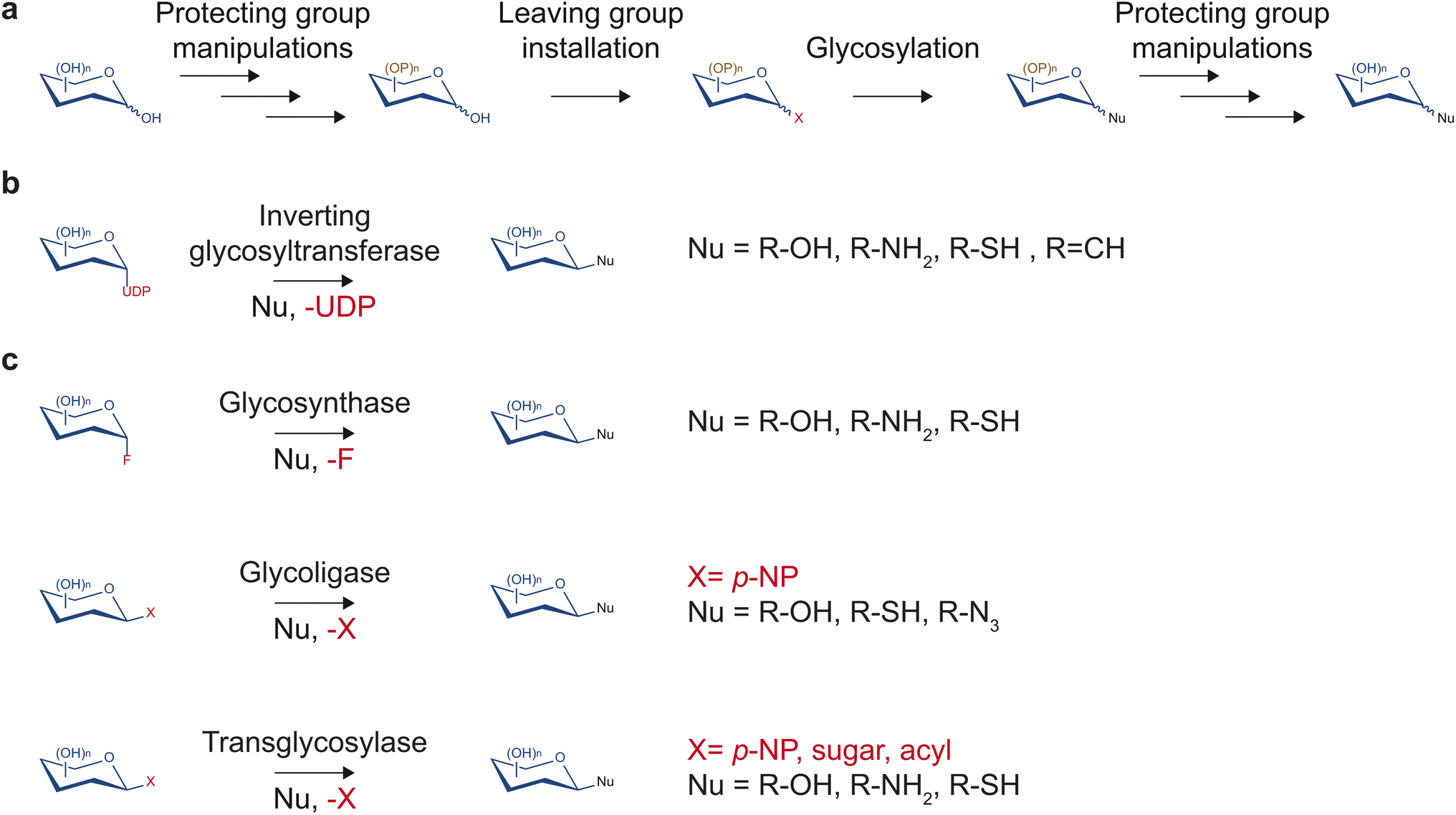 | ||
| Fig. 3 Various chemical and biological means to glycosylate small molecules. (a) Chemical glycosylation often involves nucleophilic substitution of a selectively protected sugar connected to the leaving group. (b) Glycosyltransferases transfer the sugar moiety from nucleotide sugar donors to small molecule nucleophile acceptors. (c) Engineered glycosyl hydrolases can glycosylate small molecules through the glycosynthase mechanism, glycoligase mechanism, or transglycosylase mechanism. | ||
However, due to the achiral nature of the substitution intermediate, stereochemical control of the resulting glycosidic bond requires additional attention. To achieve improved stereoselectivity, protecting groups may be installed at the C-2 position to influence the face of the sugar molecule on which nucleophilic substitution occurs. Participating substituents typically lead to 1,2-trans linkage as the major product whereas bulky nonparticipating groups generally favor to 1,2-cis linkage as the major product.18 However, the selectivity can be low and dependent on other factors such as the glycosyl acceptor and reaction conditions.
With careful planning and execution, chemical glycosylation can yield large quantities of glycosides and oligosaccharides. However, stereo- and regioselectivity remain challenging, and each reaction step requires a separate optimization. Moreover, any planned synthetic route may require customized, selectively protected glycosyl donors and acceptors, which could lengthen the synthesis process.
In contrast to synthetic routes, enzyme-based glycosylation typically exhibits high stereo- and regioselectivity. Over the years, multiple enzyme families have been discovered and engineered to facilitate a diversity of glycosidic bond formations. In particular, the glycosyltransferase family is one of the largest protein families in plants. This enzyme class utilizes nucleotide sugars to decorate proteins, glycans, and small molecules with a monosaccharide (Fig. 3b). While there are diverse families of glycosyltransferases, the GT1 family in the CAZy classification (http://www.cazy.org/)19 is of particular interest regarding drug discovery. Glycosyltransferases in the GT1 family are termed uridine diphosphate (UDP)-dependent glycosyltransferases (UGTs) as they glycosylate small molecules using UDP-sugars through an SN2-like mechanism, resulting in an inversion of configuration of the anomeric carbon.13 They contain a conserved 44-amino acid-long motif called the plant secondary product glycosylation (PSPG) box, are inverting Leloir-type glycosyltransferases, and adopt the GT-B fold.13 UGTs have been found to facilitate the formation of O-, N-, S-, and C-glycosides of a large repertoire of sugar acceptor substrates, including flavonoids, alkaloids, terpenoids, polyphenols, glycosides, as well as synthetic compounds. Notably, UGTs are involved in the biosynthesis of a range of medicinally relevant molecules such as anti-diabetics, anti-cancers, and antioxidants, and will be the focus of this report.
Outside of the glycosyltransferase family, glycosyl hydrolases, which typically catalyze hydrolysis or transfer of glycosidic bonds, have been engineered to enable glycosidic bond formation as glycosynthases, glycoligases, and transglycosylases (Fig. 3c). The majority of glycosyl hydrolases catalyze two subsequent displacement reactions at the anomeric carbon. The first is a nucleophilic attack by a catalytic sidechain leading to the glycosyl-enzyme intermediate. The second is a disruption of the intermediate by water (hydrolysis) or another acceptor (transglycosylation) that is activated by enzymatic deprotonation. Glycosynthases lack a catalytic nucleophile and use glycosyl donors with a strong leaving group such as glycosylfluoride.20 They perform glycosylation with the resulting glycosidic bond in the opposite configuration of the fluoride leaving group.20 Glycoligases have mutated catalytic acid/base residues and, thus, require strong nucleophilic sugar acceptor such as thiols in cases of thioglycoligases and hydroxyls in cases of O-glycoligases.21 Transglycosylases are natural or engineered glycosyl hydrolases that selectively reduce the stability of the hydrolysis transition state relative to that of transglycosylation to increase synthetic yield.22 Notably, recent work demonstrated that transglycosylation/hydrolysis ratio of transglycosylases from six glycosyl hydrolase families can be improved by rational protein design without extensive structural knowledge.23 Transglycosylases can utilize a wide variety of sugar donors including para-nitrophenyl(p-NP)-sugar conjugates, disaccharides, and oligosaccharides depending on their family and native functions. Engineered glycosyl hydrolases have been utilized to generate glycosides,24 defined oligosaccharides,25,26 and glycoproteins.27 However, glycosyl hydrolase-mediated glycosylation reactions either require extensive protein engineering, apply to restricted substrate ranges, are thermodynamically unfavorable, or suffer from subsequent hydrolysis of the glycoside products.
Glycosylation expands the chemical space available for drug discovery exploration. Thus, identifying viable strategies to increase glycoside diversity – altering the sugar identity, adding modifications to sugars, and changing the number of sugars decorating an aglycone – underpins the ability to find new drugs. Enzymatic glycosylation methods make possible glycorandomization, which involves the semi-random addition of sugars to the aglycone part of naturally existing glycosides to create a library of related glycosides.28 Thus, a thorough understanding of the enzymes that catalyze glycosidic bond formation is essential to facilitating drug discovery. The rest of this report will focus on plant UGTs as they are naturally biosynthetic and do not require engineering, thereby providing the most straightforward way towards synthesizing natural product glycosides.
3. Mining plant glycosyltransferases as a resource to diversify glycosides
Plants produce a wealth of glycosides (Fig. 2). Plant metabolism is especially well suited for enzymatically synthesizing a wide range of natural product glycosides because it harbors an abundant and diverse pool of sugar donors, enabling decoration of aglycones with diverse sugar moieties by UGTs. Roughly 30 nucleotide sugars have been discovered in plants,29 which leads to many permutations of potential glycoside modules. For example, almost 180 glycosides of quercetin have been described in nature including singly and multi-glycosylated compounds with and without glycan chains.30 Their presence suggests a large diversity of sugar donors and enzymatic strategies plants have to decorate a single common flavonoid molecule. In addition, more than 50 glycosidically bound volatiles have been reported in blackberries, raspberries, and grapes,31 highlighting the broad sugar acceptor range and the large number of plant UGT enzymes.3.1. Rich natural diversity of plant UGTs
The majority of plant genomes encode over 100 UGT genes, representing a rich resource to identify novel enzymes that can catalyze a wide range of reactions.32 Given the large diversity of UGTs present in the plant kingdom, it is possible that many small molecule drug leads, natural and synthetic, can be glycosylated by existing plant UGTs. A recent report (January 2022) stated that only 211 of 2983 predicted plant UGTs in the CAZy database, which only include a subset of model plants, have been functionally characterized.33 Although this represents a large increase from the year 2000 when the first plant genome was reported and only 15 plant UGTs were reportedly characterized,34 with advances in genome and transcriptome sequencing, the number of putative UGTs will continue to exponentially grow. Thus, there remains an enormous diversity of unexplored UGTs with unknown activities and substrate specificities. Characterizing these glycosyltransferases might open up a wealth of UGTs that could be harnessed for use in pharmaceutical discovery.35Characterizing individual UGTs through traditional biochemistry, genetics, and transcriptomics can be time and resource consuming. Hence, a reliable method to predict functions and characteristics of newly annotated UGTs to inform experimental efforts would be highly valuable. A few recent studies attempted to systematically characterize functions and substrate promiscuity of plant UGT enzymes. A notable large-scale study that analyzed 54 UGTs from A. thaliana on 13 sugar donors and 91 sugar acceptors found widespread substrate promiscuity among the enzymes.36 In addition, the authors reported that while primary sequence information of UGTs alone failed to capture and predict their activities, the generated dataset was sufficient to construct a decision tree model of enzyme functions based on sequence similarity to a characterized enzyme.36 However, the limited substrate scope covered in the study still leaves the vast majority of pharmaceutically relevant chemical space unexplored.
In another report, 29 UGTs chosen to represent the overall phylogenetic diversity of plant UGT enzymes were investigated for their promiscuity.33 Zhang et al. found that promiscuous UGTs cluster together phylogenetically and highlighted a particular UGT enzyme FiGT2 to be especially promiscuous as it accepts 10 of 29 sugar acceptors and 10 of 15 sugar donors tested.33 The high promiscuity prevalent among plant UGTs found in these systematic studies suggests that there are possibly many acceptable enzyme substrates, natural and synthetic, that remain to be discovered. In addition, the enzyme versatility suggests that they are amenable to engineering and optimization for a specific, desired activity. Future structure–function studies of UGTs will yield important insights into functional prediction and rational engineering efforts of UGTs.
3.2. Engineering plant UGTs
In addition to the enormous diversity of natural plant UGT enzymes and their substrates, enzyme engineering can further expand the scope of glycosylation activity. Recent rational design-based UGT engineering efforts have achieved remarkable results including altering enzyme chemoselectivity37,38 and regioselectivity.38,39Teze et al. investigated the mechanism underlying chemoselectivity of a tri-functional UGT.37 The authors found that while O-glycosylation occurs through the deprotonation of a hydroxyl nucleophile and requires a His–Asp catalytic dyad, N- and S-glycosylation can occur independently of the presence of a catalytic base as long as the nucleophile can be properly positioned in relation to the orientation of the donor.37 With that revelation, the authors succeeded at engineering singly functional mutant enzymes.37
Li et al. demonstrated the use of phylogenetic analysis, structural information, and Rosetta design to engineer highly regioselective UGT variants for silybin glycosylation from a non-regioselective parental enzyme.39 The authors engineered double mutants that achieve 94%, >99% and >99% regioselectivity on the 3-OH, 7-OH and 3,7-O-diglycoside of silybin A respectively, all with a total of fewer than 100 mutants generated in the study.39
Together, these studies demonstrate that UGTs are amenable to engineering for both improved chemoselectivity and regioselectivity. Similar principles employed in them can be adapted to other non-specific UGTs to engineer enzymes with targeted characteristics for drug discovery applications.
3.3. C-glycosyltransferases
Another significant feature of enzymatic glycosylation is the ability to catalyze carbon–carbon bond formation between the aglycone and the carbohydrate module whereas chemical processes to produce the same glycoside require a challenging, multistep process. C-glycosides are of interest in drug discovery because they are less susceptible to enzymatic and chemical degradation compared to O-glycosides due to the non-polar, carbon–carbon bond. C-glycosyltransferases most commonly act upon natural products, but some also catalyze the formation of C-pentosides like pseudouridine in RNA and post-translational tryptophan mannosylation.40,41Thus far, most discovered plant C-glycosyltransferases act on structurally related sugar acceptors.42,43 Besides six UGTs from groups 73 and 74 that have been reported to yield C-glycosides of hydroxylaminodinitrotoluene,44 most other discovered C-glycosyltransferases work on hydroxylated aromatic rings.45–47 Expanding the substrate scope of C-glycosyltransferases via new discovery can greatly expand the relevant chemical space available for drug discovery. Towards such goal, He et al. found a promiscuous C-glycosyltransferase TcCGT1 to regiospecifically catalyze the 8-C-glycosylation of 36 flavones and other flavonoids, as well as the O-glycosylation of various phenolics.48 Similarly, the GgCGT from Glycyrrhiza glabra was revealed to mono-C-glycosylate 27 phenolics and di-C-glycosylate 6 substrates containing a flopropione unit.49 Both enzymes were found to accept several types of nucleotide sugars.
Moreover, C-glycosyltransferases have also been engineered to expand their substrate scope. Chen et al. described a UGT able to catalyze di-C-glycosylation of aromatic compounds and, through comparative biochemistry, were able to engineer the enzyme to increase its donor and acceptor scope.50 The resulting enzyme is able to perform mono-C-glycosylation using 11 different sugar donors and di-C-glycosylation of monoglycosides containing glucosyl, galactosyl, rhamnosyl, arabinosyl, acetyl-glucosaminyl, xylosyl, and glucuronyl moiety using UDP-glucose. Finding or generating additional UGTs capable of performing C-glycosylation with broad donor and acceptor scope will prove invaluable in drug discovery.
Taken together, the diversity, promiscuity, specificity, versatility, and plasticity of plant UGTs make them a set of practical tools that can be exploited for targeted production of glycoside pharmaceuticals. The enzymes may simplify processes that are otherwise long and inefficient to one or a few steps. In addition, the family of enzymes may also be utilized to generate a large repertoire of novel glycoside analogues that may have unique or improved properties but were previously inaccessible through nature or chemical synthesis, including a variety of novel sugars on known glycosides, glycan chains, and C-glycosides.
4. Enzymatic glycosylation expands the repertoire of bioactive plant glycosides
UGT-mediated glycosylation has started to be applied to the production of medicinally relevant molecules. Although a substantial number of UGTs have been discovered and characterized, there are few examples that showcase the full potential of this enzyme technology. Here, we explore three specific case studies of plant glycoside therapeutics, whose functional investigations demonstrate the strength of enzymatic glycosylation as a useful strategy in drug discovery. These examples highlight that UGTs can be manipulated in various ways in vitro and in vivo to achieve desired results in terms of increased product titers, heterologous production, or the generation of novel glycoside variants with improved properties. In all cases presented here, the findings would be difficult or impossible to achieve with chemical glycosylation due to the complexity of the aglycone structures, demonstrating the strength of enzymatic glycosylation in natural product drug discovery.4.1. Steviol glycosides
Optimizing glycoside biosynthetic pathways is a useful strategy to enhance the formation of valuable glycosides while limiting the production of off-target compounds. Steviol glycosides are glycoconjugates commonly used as non-nutritive sweeteners produced by a small number of plants, namely Stevia rebaudiana. They are composed of a diterpenoid core decorated with glucose, rhamnose, and/or xylose moieties via various glycosidic linkages. The variety of sugars and linkages found among steviol glycosides, which determine their organoleptic properties by binding to target receptors, produce over 30 structures with unique properties.51 Stevioside is the major steviol glycoside found in S. rebaudiana and is 210–300 times sweeter than sucrose; however, it has a notable bitter aftertaste compared to other steviol glycosides, prompting efforts to produce alternative steviol glycosides with higher sweetness and lower bitterness.51Overexpression of native UGTs can improve the abundance of valuable glycosides. Rebaudioside A, D, and M (Reb A, D, and M) share the core structure with stevioside but contain more elaborate sugar modules (Fig. 2). All three molecules contain β-1,3-glucose moieties installed by UGT76G1 and are sweeter and less bitter than stevioside.52,53 While more valuable, Reb A, D, and M are all less abundant than stevioside, making methods to improve their yields desirable. In particular, Kim et al. overexpressed UGT76G1 in S. rebaudiana to examine the effects on Reb A yields.54 Upon the overexpression of UGT76G1, the Reb A to stevioside ratio improved from 0.30 to 1.55 without any changes to total steviol glycoside content.54
Improvements in product yield of high value steviol glycosides can also be achieved through UGT engineering. UGT76G1 naturally catalyzes the production of Reb M through two consecutive additions of glucose molecules. The first glycosylation event produces the intermediate Reb D, which is then converted to Reb M in the subsequent glycosylation. However, the enzyme has a low affinity for Reb D, limiting the yields of Reb M.55,56 The structure of UGT76G1 was recently determined, enabling engineering efforts to optimize the enzyme.57–59 To alleviate the bottleneck, Yu et al. used structural investigation, docking, and saturation mutagenesis to engineer UGT76G1 to increase its affinity for Reb D. The authors found S195Q mutant to have double the catalytic efficiency of the wildtype enzyme and increase the production of Reb M by up to over 60% in in vitro reactions using E. coli lysate.56 In another study, a UGT76G1 T284S/M88L/L200A mutant was found to have an increased catalytic activity towards Reb D and was able to convert Reb D to Reb M at up to 90.50% yield in fed-batch fermentation culture of E. coli.60
Steviol, Reb A, D, and M showcase the importance of the number and identity of sugars in determining the properties of glycosides. The production of Reb A, D, and M shows that alterations to UGT expression level or specificity can serve as a tool in enhancing the yield of desired glycosides. It is a prime example of how some glycoside variants in a complex mixture can be selectively enhanced for various applications. Applying this concept to other families of therapeutic molecules found in a complex matrix in nature may facilitate the production of a single variant with desired properties.
4.2. Montbretin A
In addition to making direct interactions with protein targets, sugars can play structural roles in positioning the bioactive functional groups of therapeutic compounds. Montbretin A is a low abundance, heavily glycosylated, acylated flavonol glycoside found in the corms of Crocosmia x crocosmiiflora.61 Montbretin A is a drug candidate in treating diabetes as it inhibits the human pancreatic amylase (HPA) with high specificity, thereby limiting the degradation of starch in the gastrointestinal tract.62Montbretin A consists of a myricetin core, a caffeoyl moiety, and five sugars, which are installed by five UGT enzymes (Fig. 2).63,64 Although the myricetin core and the caffeoyl modification have the most substantial effects on Ki, the peripheral sugar moieties play roles in contributing weak but significant interactions with its HPA target.62,65 The importance of these sugar moieties was demonstrated by inhibition assays showing increases in Ki as additional sugars were removed from montbretin A.62,65 For example, the removal of xylose, terminal rhamnose, and terminal glucose of montbretin A increased Ki from 8.1 nM to 93.3–400 nM.62,65 The effects of the sugar moieties on the Ki of montbretin A derivatives suggests that altering their identities could further influence the HPA-inhibiting property of montbretin A. While UGT703H1, which is responsible for installing xylose, was shown to be selective against other sugar donors, UGT729A2, responsible for installing terminal rhamnose, was demonstrated to accept UDP-xylose in addition to UDP-rhamnose.63 Utilizing the donor substrate promiscuity inherent to UGT792A2 in heterologous production may make montbretin A derivatives with varying terminal sugars available for functional screening.
Additionally, the rhamnose-glucose disaccharide that links the myricetin core to the caffeoyl group has crucial roles in positioning those two groups. The disaccharide positions the caffeoyl group to form a π-stacking interaction with the myricetin moiety, enabling effective inhibition of HPA. Thus, modifying the disaccharide linker can be a way to fine-tune the binding affinity of montbretin A. While synthetic analogues of montbretin A that replaced the rhamnose-glucose disaccharide with a non-sugar linker had a substantially higher Ki compared to that of montbretin A,62 more subtle changes such as changing the sugars of the disaccharide linker have not been explored. Such modifications may be achieved as UGT77B2 and UGT709G2, which install rhamnose and glucose moieties respectively, have been demonstrated to accept nucleotide sugar donors other than their native substrates.35
The effects of selectively removing the sugar decorations of montbretin A display the diverse roles that sugars can play in the bioactive properties of glycosides and emphasize the possibilities in altering pharmacological properties of drug leads by modifying their sugar module. Identification and reconstitution of the complete biosynthetic pathway of montbretin A, including five UGT enzymes, heterologously in Nicotiana benthamiana yielded up to 7 μg g−1 fresh weight.63 With detailed studies into the UGT enzymes in the pathway and engineering them, it is possible to employ this heterologous platform to increase production titers and elaborate novel montbretin A analogues currently inaccessible.
4.3. Cardiotonic steroids
Cardiotonic steroids have been a useful therapy for patients with heart failure and/or atrial fibrillation for centuries. They consist of a steroid core, a five- or six-membered lactone ring, and in some cases a glycan chain (Fig. 2).66 This class of compounds inhibits the Na+/K+-ATPase pumps, resulting in the improvement of cardiac contraction force, increasing left ventricular systolic function.67 Due to the prevalence of their molecular target in various tissues, cardiotonic steroids have also been proposed as a possible treatment for cancer,68 reducing cellular prion protein,69 and intraocular pressure;70 however, they also exhibit many potential systemic off-target effects. Thus, cardiotonic steroids' applications in therapeutics is challenging and limited to a small therapeutic window. In fact, high doses of cardiotonic steroids lead to potentially lethal toxicity. As such, there is much interest in optimizing these molecules to be more suitable for therapeutics.Altering the sugar moiety of cardiotonic steroids has the potential to affect their properties. By measuring binding affinities of 37 cardiotonic steroids including compound series that systematically vary the number of sugars, Paula et al. determined that the presence of the α-sugar moiety positively affected binding, whereas that of the γ-sugar exhibited negative effects.12 Evomonoside (rhamnose attached to digitoxigenin) and neriifolin (thevetose attached to digitoxigenin) have 2.8 and 2.6 times higher affinity than digitoxigenin monodigitoxide respectively, suggesting that the interaction between rhamnose and the ATPase contributes positively to affinity.12,71 This observation is supported by evidence that digitoxose does not form hydrogen bonds with the protein in a similar manner that rhamnose does.72
One strategy to circumvent systemic toxicity is to alter binding affinity and to develop isoform-specific derivatives as Na+/K+-ATPases are present in multiple isoforms, which are distributed unevenly in different cell types.70,73 While the α1 isoform is the most prevalent isoform, the α2 isoform is thought to be functionally more important for cardiac muscle contraction.74 As such, derivatives of digoxin with modifications to the oligosaccharide chains have been synthesized to improve their specificity for the α2 isoform.70,73 Katz et al. found that C4 modification to the sugar module of digoxin drastically increases their selectivity for the α2 isoform, which appears to differ from the α1 isoform by the size of the opening of the binding cavity. This study demonstrates that exploring the sugar diversity of cardiotonic steroids is a promising approach to optimizing their pharmacological properties.
Recently, a group of three UGT74AN enzymes from Asclepias curassavica were discovered to glycosylate cardiotonic steroids regiospecifically at the C3 position,75–77 opening the possibility to enzymatically alter the sugar decoration of cardiotonic steroids. UGT74AN1 was shown to glucosylate over 20 steroid substrates, making it the most promiscuous C3 regiospecific steroid UGT discovered to date.75 In a subsequent study, Huang et al. used UGT74AN3 in a one-pot reaction in combination with cyclodextrin glycosyltransferase to generate mono-, di-, tri-, tetra-, and penta-O-glucosides of seven bufadienolide and cardenolide aglycones.76 Moreover, a related enzyme from the same organism, UGT74AN2, was structurally characterized and engineered to expand the scope of accepted sugar donor specificity. Naturally UGT74AN1/2/3 all only accept UDP-glucose as a donor substrate, but an engineered triple mutant of UGT74AN2 has comparable activities on UDP-glucose, UDP-glucuronic acid, UDP-galactose, and UDP-rhamnose.77 These enzymes and methods form a set of practical tools to expand the sugar module diversity of cardiotonic steroids. Such development may lead to the discovery of cardiotonic glycoside variants with new binding specificity or improved in vivo properties, lessening their toxicity and expanding therapeutic windows.
5. Implications and prospects
Glycosylation is an effective approach to modify and diversify pharmacological properties of natural products. With recent advances in the understanding of glycosyltransferases, enzymatic glycosylation becomes a complementary approach to chemical glycosylation in synthesizing complex glycosides. Enzymatic glycosylation has advantages in regio-, chemo-, and stereospecificity, although each enzyme requires individual investigation to determine its characteristics, including sugar donor and acceptor scopes, kinetics, and preferred reaction conditions.While discovering novel putative UGTs is straightforward in the post-genomic era, characterizing them remains a tremendously low throughput process. Detailed structural and biochemical studies on each individual UGT will facilitate selecting and engineering them for specific applications. Such endeavors are extremely time-consuming; nevertheless, significant progress has been made in both enzyme discovery and engineering. Advances in high-throughput approaches to systematically characterize UGTs may provide the global understanding of the enzyme family to begin elucidating more basic structure–function principles to not only predict function better, but also refine engineering efforts. Currently, there are limited resources to predict UGT functions based on their sequence information alone. Further studies that aim at functional prediction of carbohydrate-related biosynthetic enzymes could greatly facilitate their applications in drug discovery, as well as in research and other industries.
While conceptually intriguing, the implementations of UGT-mediated small molecule glycosylation methods to facilitate drug discovery so far have seen few examples. A successful application of UGTs in glycoside production at industrial scale will require careful considerations. In vitro processes will have to consider sugar donor costs and recycling strategies, as well as concentrations and solubility of substrates, and completeness of reaction. On the other hand, in vivo processes will require considerations regarding substrate availability, off-targets, competing pathways, and product isolation. Recent process engineering efforts involving UDP-glucose production and UDP cycling by coupling UGTs with sucrose synthase enzymes demonstrated that enzymatic glycosylation can be optimized to meet the high efficiency demands of industrial applications.78–80 Notably, a gram scale synthesis of C-glycoside nothofagin was demonstrated in both a batch synthesis79 and a flow synthesis80 process. A means to produce large amounts of other nucleotide sugars inexpensively will further facilitate wide-ranging applications of UGTs in drug discovery.
Overall, there is a great promise in using UGTs to expand the chemical space available for drug discovery. Synthesizing new glycoside variants for functional exploration is an area in which strategic applications of UGT-mediated glycosylation can have great positive impacts on drug discovery. Moreover, UGTs may help simplify or optimize the production process of existing glycosides, increasing access to relevant drug molecules.
6. Conflicts of interest
There are no conflicts to declare.7. Acknowledgement
We thank the DOE Joint BioEnergy Institute, which is supported by the Office of Science, Office of Biological and Environmental Research, the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.8. References
- L. Xu, T. Qi, L. Xu, L. Lu and M. Xiao, J. Carbohydr. Chem., 2016, 35, 1–23 CrossRef.
- D. S. Wishart, Y. D. Feunang, A. C. Guo, E. J. Lo, A. Marcu, J. R. Grant, T. Sajed, D. Johnson, C. Li, Z. Sayeeda, N. Assempour, I. Iynkkaran, Y. Liu, A. Maciejewski, N. Gale, A. Wilson, L. Chin, R. Cummings, D. Le, A. Pon and M. Wilson, Nucleic Acids Res., 2018, 46, D1074–D1082 CrossRef CAS PubMed.
- C. Walsh, Nature, 2000, 406, 775–781 CrossRef CAS PubMed.
- N.-X. Chin and H. C. Neu, Diagn. Microbiol. Infect. Dis., 1991, 14, 181–184 CrossRef CAS PubMed.
- X. Fu, C. Albermann, J. Jiang, J. Liao, C. Zhang and J. S. Thorson, Nat. Biotechnol., 2003, 21, 1467–1469 CrossRef CAS PubMed.
- M. Ge, Z. Chen, H. R. Onishi, J. Kohler, L. L. Silver, R. Kerns, S. Fukuzawa, C. Thompson and D. Kahne, Science, 1999, 284, 507–511 CrossRef CAS PubMed.
- J. Xiao, M. Gao, Z. Sun, Q. Diao, P. Wang and F. Gao, Eur. J. Med. Chem., 2020, 208, 112830 CrossRef CAS PubMed.
- W. Huang, Q. He, Z.-R. Zhou, H.-B. He and R.-W. Jiang, ACS Omega, 2020, 5, 12251–12258 CrossRef CAS PubMed.
- L. Barleben, S. Panjikar, M. Ruppert, J. Koepke and J. Stöckigt, Plant Cell, 2007, 19, 2886–2897 CrossRef CAS PubMed.
- B. Renoux, F. Raes, T. Legigan, E. Péraudeau, B. Eddhif, P. Poinot, I. Tranoy-Opalinski, J. Alsarraf, O. Koniev, S. Kolodych, S. Lerondel, A. Le Pape, J. Clarhaut and S. Papot, Chem. Sci., 2017, 8, 3427–3433 RSC.
- A. Fernandes, A. Viterisi, F. Coutrot, S. Potok, D. A. Leigh, V. Aucagne and S. Papot, Angew. Chem., Int. Ed., 2009, 48, 6443–6447 CrossRef CAS PubMed.
- S. Paula, M. R. Tabet and W. J. Ball, Biochemistry, 2005, 44, 498–510 CrossRef CAS PubMed.
- T. Louveau and A. Osbourn, Cold Spring Harbor Perspect. Biol., 2019, 11, a034744 CrossRef CAS PubMed.
- B. Soto-Blanco, in Herbal biomolecules in healthcare applications, Elsevier, 2022, pp. 239–282 Search PubMed.
- D. P. Galonić and D. Y. Gin, Nature, 2007, 446, 1000–1007 CrossRef PubMed.
- T. J. Boltje, T. Buskas and G.-J. Boons, Nat. Chem., 2009, 1, 611–622 CrossRef CAS PubMed.
- S. S. Kulkarni, C.-C. Wang, N. M. Sabbavarapu, A. R. Podilapu, P.-H. Liao and S.-C. Hung, Chem. Rev., 2018, 118, 8025–8104 CrossRef CAS PubMed.
- M. Panza, S. G. Pistorio, K. J. Stine and A. V. Demchenko, Chem. Rev., 2018, 118, 8105–8150 CrossRef CAS PubMed.
- E. Drula, M.-L. Garron, S. Dogan, V. Lombard, B. Henrissat and N. Terrapon, Nucleic Acids Res., 2022, 50, D571–D577 CrossRef CAS PubMed.
- L. F. Mackenzie, Q. Wang, R. A. J. Warren and S. G. Withers, J. Am. Chem. Soc., 1998, 120, 5583–5584 CrossRef CAS.
- Y.-W. Kim, R. Zhang, H. Chen and S. G. Withers, Chem. Commun., 2010, 46, 8725–8727 RSC.
- B. Bissaro, P. Monsan, R. Fauré and M. J. O'Donohue, Biochem. J., 2015, 467, 17–35 CrossRef CAS PubMed.
- D. Teze, J. Zhao, M. Wiemann, Z. G. A. Kazi, R. Lupo, B. Zeuner, M. Vuillemin, M. E. Rønne, G. Carlström, J. Ø. Duus, Y.-H. Sanejouand, M. J. O'Donohue, E. Nordberg Karlsson, R. Fauré, H. Stålbrand and B. Svensson, Chem. – Eur. J., 2021, 27, 10323–10334 CrossRef CAS PubMed.
- Y. Malbert, S. Pizzut-Serin, S. Massou, E. Cambon, S. Laguerre, P. Monsan, F. Lefoulon, S. Morel, I. André and M. Remaud-Simeon, ChemCatChem, 2014, 6, 2282–2291 CrossRef CAS.
- L. Ruzic, J. M. Bolivar and B. Nidetzky, Biotechnol. Bioeng., 2020, 117, 1597–1602 CrossRef CAS PubMed.
- C. Possiel, M. E. Ortiz-Soto, J. Ertl, A. Münch, A. Vogel, R. Schmiedel and J. Seibel, Sci. Rep., 2019, 9, 7720 CrossRef PubMed.
- A. J. Fairbanks, Chem. Soc. Rev., 2017, 46, 5128–5146 RSC.
- B. Goel, N. Tripathi, D. Mukherjee and S. K. Jain, Eur. J. Med. Chem., 2021, 213, 113156 CrossRef CAS PubMed.
- M. Bar-Peled and M. A. O'Neill, Annu. Rev. Plant Biol., 2011, 62, 127–155 CrossRef CAS PubMed.
- M. Biesaga and K. Pyrzynska, Crit. Rev. Anal. Chem., 2009, 39, 95–107 CrossRef CAS.
- J. Pico, K. Nozadi, E. M. Gerbrandt, M. Dossett and S. D. Castellarin, Food Chem., 2022, 403, 134304 CrossRef PubMed.
- B. L. Cantarel, P. M. Coutinho, C. Rancurel, T. Bernard, V. Lombard and B. Henrissat, Nucleic Acids Res., 2009, 37, D233–D238 CrossRef CAS PubMed.
- L.-J. Zhang, D.-G. Wang, P. Zhang, C. Wu and Y.-Z. Li, ACS Synth. Biol., 2022, 11, 812–819 CrossRef CAS PubMed.
- T. Vogt and P. Jones, Transplant. Sci., 2000, 5, 360–386 Search PubMed.
- S. Irmisch, S. Jo, C. R. Roach, S. Jancsik, M. Man Saint Yuen, L. L. Madilao, M. O'Neil-Johnson, R. Williams, S. G. Withers and J. Bohlmann, Plant Cell, 2018, 30, 1864–1886 CrossRef CAS PubMed.
- M. Yang, C. Fehl, K. V. Lees, E.-K. Lim, W. A. Offen, G. J. Davies, D. J. Bowles, M. G. Davidson, S. J. Roberts and B. G. Davis, Nat. Chem. Biol., 2018, 14, 1109–1117 CrossRef CAS PubMed.
- D. Teze, J. Coines, F. Fredslund, K. D. Dubey, G. N. Bidart, P. D. Adams, J. E. Dueber, B. Svensson, C. Rovira and D. H. Welner, ACS Catal., 2021, 1810–1815 CrossRef CAS.
- Z. Wen, Z.-M. Zhang, L. Zhong, J. Fan, M. Li, Y. Ma, Y. Zhou, W. Zhang, B. Guo, B. Chen and J.-B. Wang, ACS Catal., 2021, 14781–14790 CrossRef CAS.
- J. Li, G. Qu, N. Shang, P. Chen, Y. Men, W. Liu, Z. Mei, Y. Sun and Z. Sun, Green Synth. Catal., 2021, 2, 45–53 CrossRef.
- M. Charette and M. W. Gray, IUBMB Life, 2000, 49, 341–351 CrossRef CAS PubMed.
- S. Manabe, Y. Marui and Y. Ito, Chem. – Eur. J., 2003, 9, 1435–1447 CrossRef PubMed.
- G. Tegl and B. Nidetzky, Biochem. Soc. Trans., 2020, 48, 1583–1598 CrossRef CAS PubMed.
- N. Putkaradze, D. Teze, F. Fredslund and D. H. Welner, Nat. Prod. Rep., 2021, 38, 432–443 RSC.
- F. Gandia-Herrero, A. Lorenz, T. Larson, I. A. Graham, D. J. Bowles, E. L. Rylott and N. C. Bruce, Plant J., 2008, 56, 963–974 CrossRef CAS PubMed.
- Y. Sun, Z. Chen, J. Yang, I. Mutanda, S. Li, Q. Zhang, Y. Zhang, Y. Zhang and Y. Wang, Commun. Biol., 2020, 3, 110 CrossRef CAS PubMed.
- Z.-L. Wang, H.-M. Gao, S. Wang, M. Zhang, K. Chen, Y.-Q. Zhang, H.-D. Wang, B.-Y. Han, L.-L. Xu, T.-Q. Song, C.-H. Yun, X. Qiao and M. Ye, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 30816–30823 CrossRef CAS PubMed.
- K. Mashima, M. Hatano, H. Suzuki, M. Shimosaka and G. Taguchi, Plant Cell Physiol., 2019, 60, 2733–2743 CrossRef CAS PubMed.
- J.-B. He, P. Zhao, Z.-M. Hu, S. Liu, Y. Kuang, M. Zhang, B. Li, C.-H. Yun, X. Qiao and M. Ye, Angew. Chem., Int. Ed., 2019, 58, 11513–11520 CrossRef CAS PubMed.
- M. Zhang, F.-D. Li, K. Li, Z.-L. Wang, Y.-X. Wang, J.-B. He, H.-F. Su, Z.-Y. Zhang, C.-B. Chi, X.-M. Shi, C.-H. Yun, Z.-Y. Zhang, Z.-M. Liu, L.-R. Zhang, D.-H. Yang, M. Ma, X. Qiao and M. Ye, J. Am. Chem. Soc., 2020, 142, 3506–3512 CrossRef CAS PubMed.
- D. Chen, S. Fan, R. Chen, K. Xie, S. Yin, L. Sun, J. Liu, L. Yang, J. Kong, Z. Yang and J. Dai, ACS Catal., 2018, 8, 4917–4927 CrossRef CAS.
- S. Ceunen and J. M. C. Geuns, J. Nat. Prod., 2013, 76, 1201–1228 CrossRef CAS PubMed.
- C. Hellfritsch, A. Brockhoff, F. Stähler, W. Meyerhof and T. Hofmann, J. Agric. Food Chem., 2012, 60, 6782–6793 CrossRef CAS PubMed.
- I. Prakash, A. Markosyan and C. Bunders, Foods, 2014, 3, 162–175 CrossRef PubMed.
- M. J. Kim, J. Zheng, M. H. Liao and I.-C. Jang, Plant Biotechnol. J., 2019, 17, 1037–1047 CrossRef CAS PubMed.
- K. Olsson, S. Carlsen, A. Semmler, E. Simón, M. D. Mikkelsen and B. L. Møller, Microb. Cell Fact., 2016, 15, 207 CrossRef PubMed.
- J. Yu, Y. Tao, H. Pan, L. Lin, J. Sun, R. Ma, Y. Li and H. Jia, J. Funct. Foods, 2022, 92, 105033 CrossRef CAS.
- T. Yang, J. Zhang, D. Ke, W. Yang, M. Tang, J. Jiang, G. Cheng, J. Li, W. Cheng, Y. Wei, Q. Li, J. H. Naismith and X. Zhu, Nat. Commun., 2019, 10, 3214 CrossRef PubMed.
- Z. Liu, J. Li, Y. Sun, P. Zhang and Y. Wang, Plant Commun., 2020, 1, 100004 CrossRef PubMed.
- S. G. Lee, E. Salomon, O. Yu and J. M. Jez, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 13131–13136 CrossRef CAS PubMed.
- B. Guo, Z. Deng, F. Meng, Q. Wang, Y. Zhang, Z. Yuan and Y. Rao, J. Agric. Food Chem., 2022, 70, 5088–5094 CrossRef CAS PubMed.
- Y. Asada, Y. Hirayama and T. Furuya, Phytochemistry, 1988, 27, 1497–1501 CrossRef CAS.
- L. K. Williams, X. Zhang, S. Caner, C. Tysoe, N. T. Nguyen, J. Wicki, D. E. Williams, J. Coleman, J. H. McNeill, V. Yuen, R. J. Andersen, S. G. Withers and G. D. Brayer, Nat. Chem. Biol., 2015, 11, 691–696 CrossRef CAS PubMed.
- S. Irmisch, S. Jancsik, M. Man Saint Yuen, L. L. Madilao and J. Bohlmann, Plant Physiol., 2020, 184, 97–109 CrossRef CAS PubMed.
- S. Irmisch, S. Jancsik, M. M. S. Yuen, L. L. Madilao and J. Bohlmann, Plant J., 2019, 100, 879–891 CrossRef CAS PubMed.
- C. R. Tysoe, S. Caner, M. B. Calvert, A. Win-Mason, G. D. Brayer and S. G. Withers, Chem. Sci., 2019, 10, 11073–11077 RSC.
- A. F. M. Botelho, F. Pierezan, B. Soto-Blanco and M. M. Melo, Toxicon, 2019, 158, 63–68 CrossRef CAS PubMed.
- O. J. Ziff and D. Kotecha, Trends Cardiovasc. Med., 2016, 26, 585–595 CrossRef CAS PubMed.
- F. Triana-Martínez, P. Picallos-Rabina, S. Da Silva-Álvarez, F. Pietrocola, S. Llanos, V. Rodilla, E. Soprano, P. Pedrosa, A. Ferreirós, M. Barradas, F. Hernández-González, M. Lalinde, N. Prats, C. Bernadó, P. González, M. Gómez, M. P. Ikonomopoulou, P. J. Fernández-Marcos, T. García-Caballero, P. Del Pino and M. Collado, Nat. Commun., 2019, 10, 4731 CrossRef PubMed.
- S. Eid, T. Zerbes, D. Williams, X. Wang, C. Sackmann, S. Meier, N. O. Dulin, P. Nagorny and G. Schmitt-Ulms, Int. J. Mol. Sci., 2022, 23, 14823 CrossRef CAS PubMed.
- A. Katz, D. M. Tal, D. Heller, H. Haviv, B. Rabah, Y. Barkana, A. L. Marcovich and S. J. D. Karlish, J. Biol. Chem., 2014, 289, 21153–21162 CrossRef CAS PubMed.
- A. Katz, Y. Lifshitz, E. Bab-Dinitz, E. Kapri-Pardes, R. Goldshleger, D. M. Tal and S. J. D. Karlish, J. Biol. Chem., 2010, 285, 19582–19592 CrossRef CAS PubMed.
- R. Kanai, F. Cornelius, H. Ogawa, K. Motoyama, B. Vilsen and C. Toyoshima, Proc. Natl. Acad. Sci. U. S. A., 2020, 118, e2020438118 CrossRef PubMed.
- A. Katz, D. M. Tal, D. Heller, M. Habeck, E. Ben Zeev, B. Rabah, Y. Bar Kana, A. L. Marcovich and S. J. D. Karlish, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 13723–13728 CrossRef CAS PubMed.
- M. Habeck, E. Tokhtaeva, Y. Nadav, E. Ben Zeev, S. P. Ferris, R. J. Kaufman, E. Bab-Dinitz, J. H. Kaplan, L. A. Dada, Z. Farfel, D. M. Tal, A. Katz, G. Sachs, O. Vagin and S. J. D. Karlish, J. Biol. Chem., 2016, 291, 23159–23174 CrossRef CAS PubMed.
- C. Wen, W. Huang, X.-L. Zhu, X.-S. Li, F. Zhang and R.-W. Jiang, Org. Lett., 2018, 20, 534–537 CrossRef CAS PubMed.
- W. Huang, C. Wen, Z. Zhou, Z. Fu, A. Katz, A. Plotnikov, S. J. D. Karlish and R. Jiang, Adv. Synth. Catal., 2019, 361, 3114–3119 CrossRef CAS.
- W. Huang, Y. He, R. Jiang, Z. Deng and F. Long, ACS Catal., 2022, 12, 2927–2937 CrossRef CAS.
- L. Dai, C. Liu, J. Li, C. Dong, J. Yang, Z. Dai, X. Zhang and Y. Sun, J. Agric. Food Chem., 2018, 66, 2830–2837 CrossRef CAS PubMed.
- A. Gutmann, A. Lepak, M. Diricks, T. Desmet and B. Nidetzky, Biotechnol. J., 2017, 12, 1600557 CrossRef PubMed.
- H. Liu and B. Nidetzky, Biotechnol. Bioeng., 2021, 118, 4402–4413 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |