Preferential orientation of anisotropic polythiophene rods toward macroscopic chain ordering†
Received
4th October 2022
, Accepted 1st November 2022
First published on 2nd November 2022
Abstract
The orientation of polymer chains plays an important role in governing the mechanical, optical, and electronic properties of polymeric materials. Here, we report on the fabrication of anisotropic polythiophene particles with controlled lengths using a metal–organic framework template, in which the alignment of the polymer chains was retained without a host support. The formation of the rod-shaped particles with long length allowed for the facile preferential orientation of the polymer particles, and consequently resulted in the macroscopic chain alignment of unprocessable polythiophene.
Design, System, Application
Several methods have so far been developed to align polymer chains, such as mechanical stress on cast films and electron spinning. However, these strategies often require the dissolution or melting of the polymers, excluding insoluble and non-melting polymers from the conventional polymer chain alignment. Here, we disclose a versatile method for the chain alignment of an unprocessable polymer using a combination of metal−organic framework (MOF) template and rubbing methods. [In(OH)(2-bromo-1,4-benzenedicarboxylate)]n was employed as the host because the long-axis length of the MOF crystals could be controlled using the coordination modulation method. The anisotropic unsubstituted polythiophene rods were successfully obtained using the MOF as a template. We found that the long polymer rods could be easily aligned by mechanical rubbing treatment, achieving chain ordering at the macroscopic scale. In principle, this methodology can be extended to a wide range of unprocessable polymers. The ability of our designed system will facilitate the generation of a family of polymeric nanomaterials with highly anisotropic functions, beneficial for optical and electronic applications.
|
Introduction
Particle shape plays a pivotal role in determining the application of polymeric materials. Anisotropic rod-shaped polymer particles have attracted attention owing to their promising properties for optical, electronic, and biological applications.1–9 However, engineering of anisotropy at the macroscale remains challenging, because once the particles aggregate, the resulting agglomerates cannot exhibit inherent anisotropic properties. Consequently, methods involving both controlled synthesis and unidirectional alignment of the polymer rods are in high demand to exploit the full potential advantages of their functions.
One simple yet powerful methodology for controlling the morphology of polymer particles and enabling the production of novel geometrical polymer particles involves transferring the shape pattern from host materials.10–14 Metal–organic frameworks are highly crystalline materials with permanent porosity and tunable pore size at the molecular levels, making them attractive for a large scope of applications such as gas storage, sensing, drug delivery, separation, and catalysis.15–23 MOF nanochannels can be used for the polymerization of accommodated monomers and provide regulated primary and higher-order structures of polymeric nanomaterials.24–27 The MOF particle size can be tuned by the addition of capping additives that alter the coordination equilibrium at the crystal surface.28–31 Furthermore, the host materials can be easily removed under mild conditions owing to the noncovalent framework structures. Hence, MOFs are excellent porous templates for the production of well-defined polymer particles.32–37
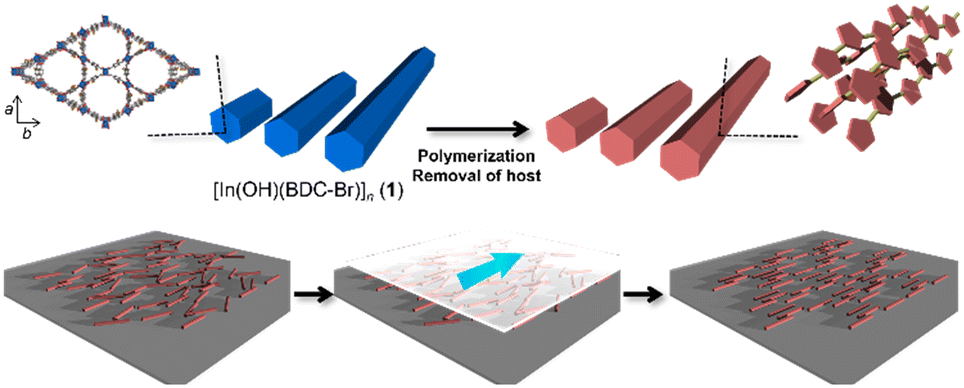
Polythiophenes have been gaining attention owing to their potential applications in nonlinear optics and electronic devices.38 Their conductivity and anisotropic optical functions can be enhanced through the alignment of the polymer chains, which can improve the device performance.39 Our group has previously reported the fabrication of unsubstituted polythiophene (PTh) rods using a MOF host with one-dimensional nanochannels, in which the orientation of the PTh chains within the pores was not disturbed after host removal.35 However, the size of the original host crystals could not be controlled, making it difficult to align the resulting PTh particles. Furthermore, the key to maintaining the chain orientation remains elusive. In this study, [In(OH)(BDC-Br)]n (1; BDC-Br = 2-bromo-1,4-benzenedicarboxylate, hexagonal channel size = 16 Å) was prepared, because the length of the host crystals can be tuned by the optimization of the synthetic conditions (e.g., heating time and capping additive).40–42 The anisotropic rod-shaped PTh particles with a controlled length was obtained using 1 as a template, in which the polymer chains were aligned along the long axis of the rods (Fig. 1). Thanks to the long length, the PTh rods could be easily aligned along a preferential direction by the mechanical rubbing treatment,43–46 achieving the macroscopic unidirectional chain orientation (Fig. 1).
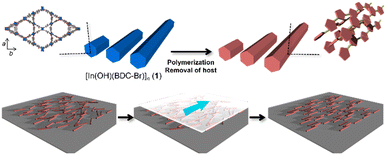 |
| | Fig. 1 Schematic image for synthesis of anisotropic PTh particles using a MOF as a template and long-range alignment of polymer rods for macroscopic chain ordering. | |
Experimental
Materials
All chemical reagents were of analytical grade, obtained from commercial suppliers (FUJIFILM Wako Pure Chemical Co., Sigma-Aldrich, Wako Chemical Co., and Tokyo Chemical Industry Co.) and used as received without further purification, unless otherwise noted. 2,2′-Azobis (isobutyronitrile) (AIBN) was recrystallized from methanol, and styrene (St) was purified by vacuum distillation before use.
Synthesis of 1
The template was synthesized and upscaled according to a previously reported protocol.40,41 2-Bromo-1,4-benzenedicarboxylic acid (441 mg) was dissolved in dimethylformamide (DMF, 86 mL). An In(NO3)3·3H2O (710 mg) was dissolved in DMF (4 mL) and then added to the organic linker solution. The longer rod particles were obtained upon heating at 100 °C for 16 h. The particles with shorter lengths were prepared by heating the solution at 100 °C for 10 min in the presence of pyridine (158 mg) as a modulator. The obtained products were isolated by filtration and washed several times with DMF and methanol before drying.
Polymerization of terthiophene (TTh) in 1
Dried host 1 was prepared by heating at 150 °C for 12 h in vacuo (<0.2 kPa) in a Pyrex reaction tube. TTh (50 mg) was mixed with the degassed MOF (100 mg) in a flask. This flask was evacuated, sealed, and heated prior to heating at 180 °C for 1 h, followed by heating at 180 °C under vacuum (<0.2 kPa) for 2 h to obtain the nanocomposite of 1 with TTh. The composite was mixed with I2 (108 mg) in a flask. The flask was then evacuated (<0.2 kPa), sealed, and subsequently heated at 90 °C for 12 h to perform the oxidative polymerization of TTh and obtain the 1-PTh composite (1 ⊃ PTh). After cooling to room temperature, 1 ⊃ PTh was dispersed in methanol (50 mL) and acetone (50 mL) and then dried in a Schlenk tube at 150 °C for 12 h under vacuum.
Polymerization of styrene (St) in 1
1 was heated in a Pyrex reaction tube at 150 °C for 12 h under vacuum (<0.2 kPa) to fully degas the sample. A mixture of St (1 mL) and AIBN (2 mg) added to the flask at room temperature. The mixture was left to stand for 60 min to insertion of the monomer and the initiator into 1, after which excess external St was removed under reduced pressure (0.2 kPa) for 1.5 h at room temperature. The 1-St nanocomposite was heated with AIBN at 95 °C for 48 h. The resulting 1 and polystyrene (PSt) composite (1 ⊃ PSt) was obtained after washing with methanol and drying under vacuum.
Liberation of the polymers from the template
The MOF/polymer composites (100 mg) were stirred overnight in a 0.05 M aqueous solution of sodium ethylenediaminetetraacetate (Na-EDTA) for the dissolution of 1. The collected polymers (PTh: 30 mg, PSt: 27 mg) were washed with water and methanol and dried under reduced pressure at room temperature.
Vortex flowing method
A suspension of the polymer particles in isopropanol (0.5 mg mL−1, 500 μL) was added dropwise to the surface of water in a beaker (100 mL) under constant stirring at 150 rpm. After the evaporation of isopropanol, the particles were transferred using tweezers onto glass substrates.
Mechanical rubbing of the polymer particles
The PTh particle suspension in isopropanol (0.5 mg mL−1, 20 μl) was dropped onto the glass substrates and dried for 30 min at room temperature. The PTh particles on the substrate was rubbed 20 times in one direction with cover glass (18 × 18 mm, Matsunami Glass Co.).
Visual orientation analysis
The orientation of the polymer particles was evaluated using the OrientationJ Java plugin of ImageJ (Biomedical Imaging Group, EPFL, Switzerland).47 The structure tensor for each pixel was calculated by computing the pixel intensity gradients in the x- and y-directions within a Gaussian-shaped window.
Physical measurements
X-ray powder diffraction (XRPD) data were collected using a Rigaku SmartLab diffractometer equipped with a Cu anode. Solid-state nuclear magnetic resonance (NMR) measurements were conducted using a 9.4 T Bruker solid-state NMR instrument with an ADVANCE III 400 MHz spectrometer. 13C cross-polarization magic angle spinning spectra were obtained using a double-resonance magic angle spinning probe (4 mm) with a recycle delay and spinning rate of 2 s and 6 kHz, respectively. The 1H-NMR spectrum was obtained using a JEOL A-500 spectrometer operating at 500 MHz. UV-vis spectra were recorded on a JASCO V-670 spectrometer. The adsorption isotherms of N2 were obtained at 77 K using BELSORP-mini equipment. Before the adsorption measurements, the samples were treated under reduced pressure (<10−2 Pa) at 100 °C for 5 h. Matrix-assisted laser deposition/ionization time-of-flight mass spectrometry (MALDI-TOF MS) spectra were recorded on an Ultraflex instrument (Bruker Daltonics) using TTh as the matrix. The thermogravimetric (TG) analysis was carried out from room temperature to 500 °C at a heating rate of 10 °C min−1 using a Rigaku Instrument Thermo plus TG 8120 in a nitrogen atmosphere. Field-emission scanning electron microscopy (FE-SEM) images were obtained using a Hitachi SU5000 FE-SEM unit. The samples were placed on a conducting carbon tape attached to an SEM grid. Energy-dispersive X-ray spectroscopy (EDX) was performed at 28 kV and at a working distance of 10 mm. Gel permeation chromatography (GPC) measurements of the PSt were performed in CHCl3 at 40 °C on three linear PSt gel columns (Shodex K-805L) connected to a JASCO PU-980 precision pump, JASCO RI-930 refractive index detector, and JASCO UV-970 UV/vis detector set at 256 nm. Transmission electron microscopy (TEM) images and selected-area electron diffraction patterns were obtained using a transmission electron microscope (JEOL, JEM-2100). The PTh dispersion in methanol was dropped and dried on a holey plastic film with a Cu mesh.
Results and discussion
Fabrication of the anisotropic polymer particles
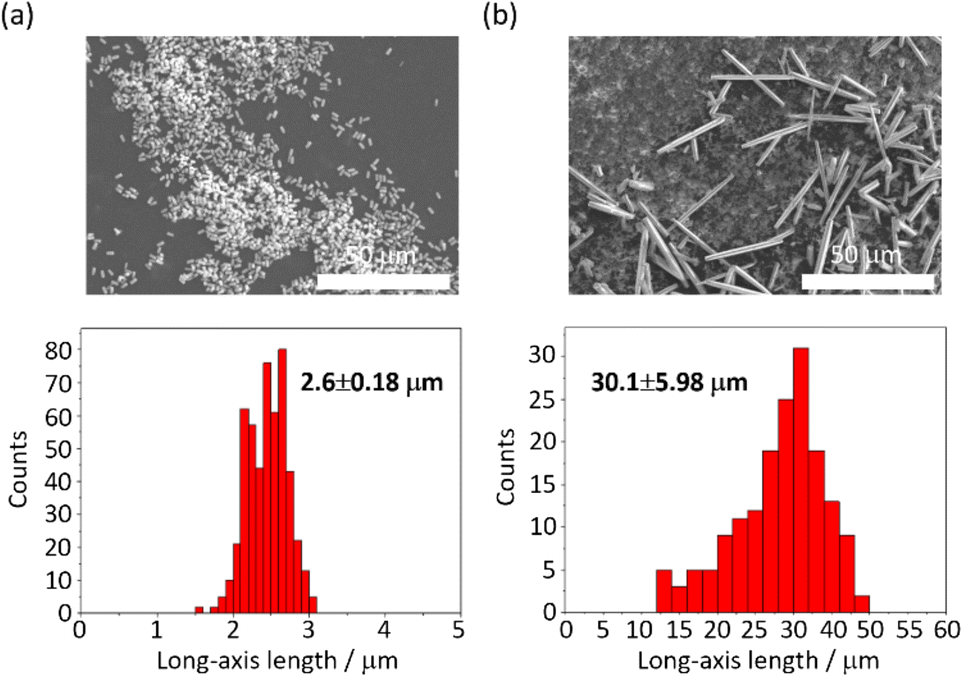
The MOF 1 is built up by the connection of metal-centered octahedral unit InO4(OH)2 with BDC-Br to form the Kagome net (Fig. 1).40 The addition of pyridine blocked the c-axis-oriented growth of the crystals, as pyridine reversibly coordinated to the In center exposed on the hexagonal facet of 1.48 The SEM images revealed that the long-axis length of the MOF crystals was successfully controlled using the modulation method (Fig. 2; average long-axis length: 2.6 ± 0.18, 30.1 ± 5.98 μm).
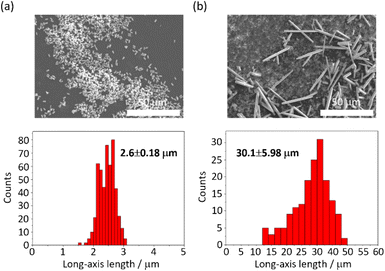 |
| | Fig. 2 SEM images and size distributions (± their standard deviations) of 1 (a) with and (b) without pyridine. | |
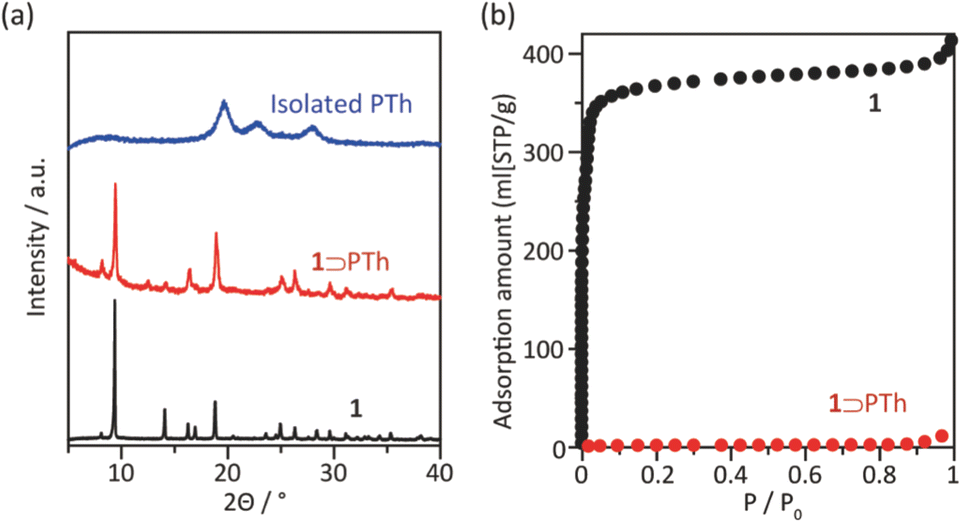
The synthesis of PTh was conducted by the oxidative polymerization of TTh as a monomer in the nanochannels of 1.35 TTh was introduced into 1 by the sublimation method, affording 1 and TTh composites (1 ⊃ TTh). 1 ⊃ TTh was then exposed to I2 vapor for the oxidative polymerization of TTh within the nanochannels. The XRPD measurements of the nanocomposite of 1, including PTh (1 ⊃ PTh), indicated that the crystal structures were maintained during polymerization (Fig. 3a). The N2 adsorption isotherm of 1 ⊃ PTh indicated a drastic decrease in the amount of adsorption compared to that of 1 (Fig. 3b). The morphology (size, shape, and surface) of 1 did not change after polymerization (Fig. 4). SEM-EDX elemental mapping measurements demonstrated the homogeneous distribution of the PTh chains in the MOF crystals (Fig. S1†). These results indicated that the polymerization of TTh proceeded inside the nanochannels of 1.
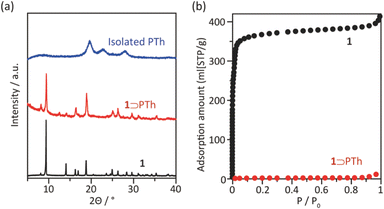 |
| | Fig. 3 (a) XRPD patterns of 1, 1 ⊃ PTh, and PTh liberated from 1. (b) N2 adsorption isotherms of 1 and 1 ⊃ PTh at 77 K (1; SBET = 1447 m2 g−1, 1 ⊃ PTh; SBET = 4 m2 g−1). | |
 |
| | Fig. 4 SEM images of (a and b) 1 with different lengths obtained by the coordination modulation method, (c and d) 1 ⊃ PTh, and (e and f) PTh isolated from 1. The morphologies and size of the recovered PTh objects retained those of the original hosts. | |
PTh was liberated from 1 by the removal of the host in an aqueous solution of Na-EDTA. Solid-state 13C-NMR and MALDI-TOF-MS analyses confirmed the formation of PTh without branching (Fig. S2 and S3†).49 SEM-EDX analysis demonstrated the absence of the In element, thus indicating the complete removal of 1 (Fig. S1†), which was further confirmed by the absence of the diffraction peaks corresponding to residues of 1 in the XRPD pattern (Fig. 3a). The SEM images of the isolated PTh revealed that the polymer particles retained the morphology and size of the original host template, despite the removal of a large fraction of the composite mass (Fig. 4). The length of the PTh particles would have a profound effect on not only their orientation degree but also anisotropic functions (ex. conductivity, luminescence, and nonlinear optical effect).50 The crystal growth of the MOF particles was regulated by the modulation method (Fig. 2), which allowed for the fabrication of the PTh rods with a controlled length via replication of the host crystals (Fig. 4e and f).
Microstructure of the polymer chains
Further characterization of the polymer particles revealed the microstructures of the polymer chains. The XRPD analysis of the isolated PTh showed characteristic peaks of crystalline PTh with a herringbone packing structure, thus indicating its reorganization into a crystalline configuration (Fig. 3a). As the direction of crystal growth in 1 was parallel to the c-axis, the polymer chains were predicted to be aligned along the long axis of the obtained polymer rod particles, as previously reported.35 The selected-area electron diffraction patterns via the TEM images were analyzed, and the diffraction spots indexed as (xy0) and (002) in the directions perpendicular and parallel to the long axis of the polymer particle, respectively, were visualized (Fig. 5). The (002) diffraction peak corresponded to the lattice plane perpendicular to the axis of the polymer backbone, indicating that the orientation of the PTh chains was not disturbed after digesting the host matrices even though PTh is a linear polymer without cross-linking. The UV-vis spectra revealed that the PTh chains formed well-ordered conjugated segments, unlike the PTh prepared by solution polymerization (Fig. S4†).35,51
 |
| | Fig. 5 (a) TEM image and (b) selected area electron diffraction patterns of PTh from 1. | |
To reveal the effects of the structural characteristics, such as backbone rigidity and flexibility, on the chain reorganization process upon host removal, particles of PSt, a nonconjugated polymer, were fabricated using 1 as a template (see the Experimental section), and their microstructures were characterized. XRPD, TG, 1H-NMR, and GPC results confirmed the formation of PSt (Mn = 39![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Mw/Mn = 2.73) (Fig. S5–S8†). The SEM images revealed that the PSt rod particles were successfully obtained upon removing the framework (Fig. S9†). Unlike in the case of PTh, a diffraction peak was not observed in the XRPD pattern of the PSt isolated from 1; instead, diffuse scattering was observed, which is typical for amorphous atactic PSt (Fig. S5†). The electron diffraction method cannot be applied to evaluate the macromolecular orientation due to its amorphous state. The orientation of the PSt chains was examined using polarized Raman spectroscopy.52,53 The rod samples were placed in such a manner that the long axis was parallel and perpendicular to the direction of the polarized excitation laser. The intensity of the Raman band at 623 cm−1 was attributed to the antisymmetric skeletal vibration of the benzene ring, which is known to vary with the polarization direction.52 The PSt particles showed similar Raman band intensities between the parallel and perpendicular directions at 623 cm−1, indicating that the polymer chains were not oriented along the long-axis direction (Fig. S10†). In contrast, it should be noted that the alignment of the PTh chains was supported by the Raman spectroscopy measurements, in which the intensity of the peak at 1436 cm−1, ascribable to the C
000, Mw/Mn = 2.73) (Fig. S5–S8†). The SEM images revealed that the PSt rod particles were successfully obtained upon removing the framework (Fig. S9†). Unlike in the case of PTh, a diffraction peak was not observed in the XRPD pattern of the PSt isolated from 1; instead, diffuse scattering was observed, which is typical for amorphous atactic PSt (Fig. S5†). The electron diffraction method cannot be applied to evaluate the macromolecular orientation due to its amorphous state. The orientation of the PSt chains was examined using polarized Raman spectroscopy.52,53 The rod samples were placed in such a manner that the long axis was parallel and perpendicular to the direction of the polarized excitation laser. The intensity of the Raman band at 623 cm−1 was attributed to the antisymmetric skeletal vibration of the benzene ring, which is known to vary with the polarization direction.52 The PSt particles showed similar Raman band intensities between the parallel and perpendicular directions at 623 cm−1, indicating that the polymer chains were not oriented along the long-axis direction (Fig. S10†). In contrast, it should be noted that the alignment of the PTh chains was supported by the Raman spectroscopy measurements, in which the intensity of the peak at 1436 cm−1, ascribable to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C in-phase stretching mode of thiophene, drastically changed, depending on the polarization direction of the excitation laser (Fig. S11†).54 These results revealed that, unlike in the case of PTh, the chain orientation of PSt was not preserved after the removal of the MOF matrix. PSt had a much larger molecular weight than that of PTh (Fig. S2 and S8†), and the loading amount of PSt was almost the same as that of PTh (see the Experimental section). Consequently, it is likely that the rigidity of the polymer backbones and effective interchain interactions are essential for the orientation memory effect.
C in-phase stretching mode of thiophene, drastically changed, depending on the polarization direction of the excitation laser (Fig. S11†).54 These results revealed that, unlike in the case of PTh, the chain orientation of PSt was not preserved after the removal of the MOF matrix. PSt had a much larger molecular weight than that of PTh (Fig. S2 and S8†), and the loading amount of PSt was almost the same as that of PTh (see the Experimental section). Consequently, it is likely that the rigidity of the polymer backbones and effective interchain interactions are essential for the orientation memory effect.
Alignment of the PTh rod particles
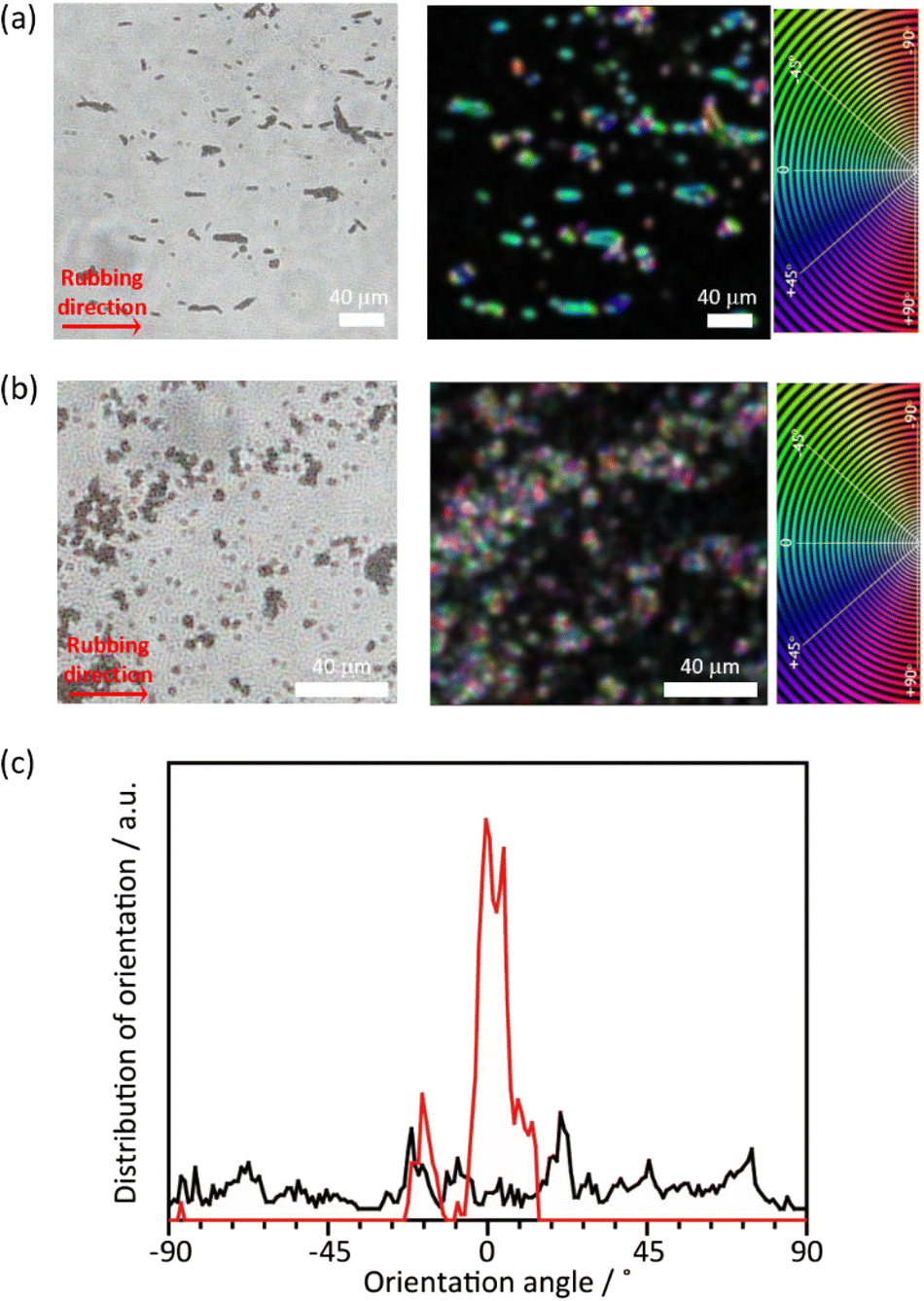
Although the PTh chains are oriented within each particle, their anisotropy is lost at the macroscopic scale when the particles aggregate. For uniaxial orientation of the polymer particles, a vortex Langmuir–Blodgett (vortex-LB) method was employed, whereby the vortex motion caused by the mechanical stirring of a subphase medium directs the floating particles to align at the liquid interface.55 The dispersion of PTh particles was added dropwise to the water surface under stirring, and the resulting film was transferred onto a glass substrate. However, the PTh particles were not fully oriented, and the strong hydrophobicity of PTh resulted in the formation of random agglomerates on the surface (Fig. S12†). The PTh dispersion was then dropped onto glass substrates, and the particles were aligned using the mechanical rubbing method.43–46 The film of the PTh rods on the substrate were rubbed using a cover glass. Strikingly, this simple method allowed for the preferential alignment of the particles. The orientation degree of the particles was analyzed before and after the rubbing treatment using the OrientationJ plugin (see the Experimental section for details). An unimodal and narrow angular distribution was obtained with longer-length PTh rods, demonstrating the orientation of the rod particles, while the shorter rod particles provided a nearly isotropic orientational distribution, presumably due to the reduced shear orientation effects (Fig. 6 and S13†).56 These results clearly underlined the necessity to control the particle length for achieving the alignment of the particles. Several techniques have been employed to align polymer chains, such as applying mechanical stress on cast films and electrospinning;57,58 however, these strategies mostly require the melting or dissolution of the polymers. These methods cannot be applied to PTh because strong interchain interactions render PTh nonfusible and insoluble with a nonmelting behavior. The results of this study demonstrated that the combination of the MOF template with simple rubbing methods can result in the macroscopic alignment of unprocessable polymers.
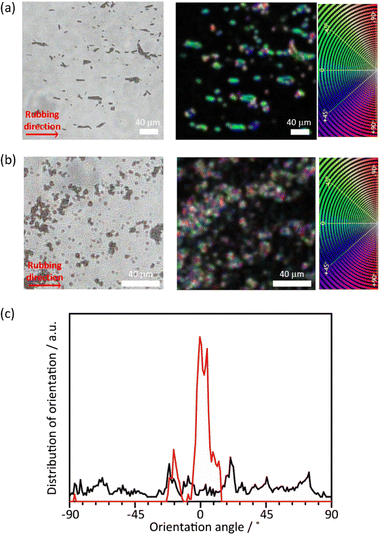 |
| | Fig. 6 (a and b) Microscope images and orientation analyses of (a) long and (b) short PTh rod particles after mechanical rubbing treatment. Orientation analyses were color-coded according to the orientation color map from −90° to +90°. (c) Angle orientation of the PTh rods with long (red) and short (black) length. | |
Conclusions
In conclusion, this paper reports the fabrication of anisotropic PTh particles with tunable lengths using a MOF template. The microstructure of the polymer chains was explored by XRPD, selected-area electron diffraction, and polarized Raman spectroscopy measurements, and the results indicated that the inherent rigidity of the polymer chains as well as the inter-chain interactions were an essential prerequisite for maintaining the polymer chain alignment during recovery. Tuning the length of the rod particles allowed for the unidirectional ordering of the PTh particles by a mechanical rubbing treatment, which led to the alignment of the PTh chains at the macroscopic scale. This system can contribute to the preparation of a variety of polymeric materials with highly anisotropic functional properties for their application in advanced optoelectronic devices.59
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by JST-CREST (JPMJCR20T3) and JST-PRESTO (JPMJPR21A7) programs, and a Grant-in-Aid for Scientific Research (JP16H06517, JP21H01738, and JP21H05473) from the Ministry of Education, Culture, Sports, Science and Technology, Government of Japan. This work was carried out by the joint research in the Institute for Solid State Physics, the University of Tokyo.
References
- N. J. Halas, Nano Lett., 2010, 10, 3816–3822 CrossRef CAS.
- S. C. Glotzer and M. J. Solomon, Nat. Mater., 2007, 6, 557–562 CrossRef PubMed.
- S. Lee, C. M. Yoon, J. Y. Hong and J. Jang, J. Mater. Chem. C, 2014, 2, 6010–6016 RSC.
- C. W. Liu, Y. C. Wei, C. C. Liu and K. W. Wang, J. Mater. Chem., 2012, 22, 4641–4644 RSC.
- H. Wang, R. Zhao, Y. Li, H. Liu, F. Li, Y. Zhao and G. Nie, Nanomedicine, 2016, 12, 439–448 CrossRef CAS PubMed.
- D. Muenmart, A. B. Foster, A. Harvey, M. T. Chen, O. Navarro, V. Promarak, M. C. McCairn, J. M. Behrendt and M. L. Turner, Macromolecules, 2014, 47, 6531–6539 CrossRef CAS.
- T. Adachi, L. Tong, J. Kuwabara, T. Kanbara, A. Saeki, S. Seki and Y. Yamamoto, J. Am. Chem. Soc., 2013, 135, 870–876 CrossRef CAS PubMed.
- A. J. C. Kuehne, M. C. Gather and J. Sprakel, Nat. Commun., 2012, 3, 1088 CrossRef PubMed.
- S. I. Moon and T. J. McCarthy, Macromolecules, 2003, 36, 4253–4255 CrossRef CAS.
- P. Sozzani, S. Bracco, A. Comotti, R. Simonutti, P. Valsesia, Y. Sakamoto and O. Terasaki, Nat. Mater., 2006, 5, 545–551 CrossRef CAS PubMed.
- G. Distefano, A. Comotti, S. Bracco, M. Beretta and P. Sozzani, Angew. Chem., Int. Ed., 2012, 51, 9258–9262 CrossRef CAS.
- J. F. Hulvat and S. I. Stupp, Angew. Chem., Int. Ed., 2003, 42, 778–781 CrossRef CAS.
- D. Park, B. Kim, M. Jang, K. Bae and J. Joo, Appl. Phys. Lett., 2005, 86, 113116 CrossRef.
- J. I. Lee, S. H. Cho, S.-M. Park, J. K. Kim, J. K. Kim, J.-W. Yu, Y. C. Kim and T. P. Russell, Nano Lett., 2008, 8, 2315–2320 CrossRef CAS PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2011, 112, 724–781 CrossRef.
- S. Kitagawa, R. Kitaura and S. I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS.
- R. J. Kuppler, D. J. Timmons, Q.-R. Fang, J.-R. Li, T. A. Makal, M. D. Young, D. Yuan, D. Zhao, W. Zhuang and H.-C. Zhou, Coord. Chem. Rev., 2009, 253, 3042–3066 CrossRef CAS.
- G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- X. Zhao, X. Song, Y. Li, Z. Chang and L. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 5633–5640 CrossRef CAS.
- H. Miyasaka, Bull. Chem. Soc. Jpn., 2021, 94, 2929–2955 CrossRef CAS.
- N. Hosono, Bull. Chem. Soc. Jpn., 2021, 94, 60–69 CrossRef CAS.
- M. Takahashi, Bull. Chem. Soc. Jpn., 2021, 94, 2602–2612 CrossRef CAS.
- N. Ding, H. Li, X. Feng, Q. Wang, S. Wang, L. Ma, J. Zhou and B. Wang, J. Am. Chem. Soc., 2016, 138, 10100–10103 CrossRef CAS PubMed.
- T. Kitao, Y. Zhang, S. Kitagawa, B. Wang and T. Uemura, Chem. Soc. Rev., 2017, 46, 3108–3133 RSC.
- H.-C. Lee, M. Antonietti and B. V. Schmidt, Polym. Chem., 2016, 7, 7199–7203 RSC.
- N. Hosono and T. Uemura, Bull. Chem. Soc. Jpn., 2021, 94, 2139–2148 CrossRef CAS.
- S. Furukawa, J. Reboul, S. Diring, K. Sumida and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5700–5734 RSC.
- T. Tsuruoka, S. Furukawa, Y. Takashima, K. Yoshida, S. Isoda and S. Kitagawa, Angew. Chem., Int. Ed., 2009, 48, 4739–4743 CrossRef CAS.
- A. Umemura, S. Diring, S. Furukawa, H. Uehara, T. Tsuruoka and S. Kitagawa, J. Am. Chem. Soc., 2011, 133, 15506–15513 CrossRef CAS.
- S. Diring, S. Furukawa, Y. Takashima, T. Tsuruoka and S. Kitagawa, Chem. Mater., 2010, 22, 4531–4538 CrossRef CAS.
- T. Ishiwata, Y. Furukawa, K. Sugikawa, K. Kokado and K. Sada, J. Am. Chem. Soc., 2013, 135, 5427–5432 CrossRef CAS PubMed.
- M. Tsotsalas, J. Liu, B. Tettmann, S. Grosjean, A. Shahnas, Z. Wang, C. Azucena, M. Addicoat, T. Heine, J. Lahann, J. Overhage, S. Bräse, H. Gliemann and C. Wöll, J. Am. Chem. Soc., 2014, 136, 8–11 CrossRef CAS.
- T. Uemura, T. Kaseda and S. Kitagawa, Chem. Mater., 2013, 25, 3772–3776 CrossRef CAS.
- T. Kitao, M. W. A. MacLean, B. Le Ouay, Y. Sasaki, M. Tsujimoto, S. Kitagawa and T. Uemura, Polym. Chem., 2017, 8, 5077–5081 RSC.
- Z. G. Gu, W. Q. Fu, M. Liu and J. Zhang, Chem. Commun., 2017, 53, 1470–1473 RSC.
- M. W. MacLean, T. Kitao, T. Suga, M. Mizuno, S. Seki, T. Uemura and S. Kitagawa, Angew. Chem., Int. Ed., 2016, 55, 708–713 CrossRef CAS PubMed.
- J. Roncali, Chem. Rev., 1992, 92, 711–738 CrossRef CAS.
- D. Kajiya, S. Ozawa, T. Koganezawa and K.-I. Saitow, J. Phys. Chem. C, 2015, 119, 7987–7995 CrossRef CAS.
- C. Volkringer, M. Meddouri, T. Loiseau, N. Guillou, J. Marrot, G. Férey, M. Haouas, F. Taulelle, N. Audebrand and M. Latroche, Inorg. Chem., 2008, 47, 11892–11901 CrossRef CAS PubMed.
- S. Choi, T. Kim, H. Ji, H. J. Lee and M. Oh, J. Am. Chem. Soc., 2016, 138, 14434–14440 CrossRef CAS.
- B. Hosseini Monjezi, B. Sapotta, S. Moulai, J. Zhang, R. Oestreich, B. P. Ladewig, K. Müller-Buschbaum, C. Janiak, T. Hashem and A. Knebel, Chem. Ing. Tech., 2022, 94, 135–144 CrossRef CAS.
- Y. Amit, A. Faust, I. Lieberman, L. Yedidya and U. Banin, Phys. Status Solidi A, 2012, 209, 235–242 CrossRef CAS.
- C. Park, K. Koh and U. Jeong, Sci. Rep., 2015, 5, 8340 CrossRef CAS PubMed.
- L. Hartmann, D. Djurado, I. Florea, J.-F. Legrand, A. Fiore, P. Reiss, S. Doyle, A. Vorobiev, S. Pouget, F. Chandezon, O. Ersen and M. Brinkmann, Macromolecules, 2013, 46, 6177–6186 CrossRef CAS.
- H. Eimura, A. Niwa, J. Uchida and T. Kato, Bull. Chem. Soc. Jpn., 2021, 94, 1588–1593 CrossRef CAS.
- Z. Püspöki, M. Storath, D. Sage and M. Unser, Adv. Anat., Embryol. Cell Biol., 2016, 219, 69–93 Search PubMed.
- L. Sun, R. Li, W. Zhan, Y. Yuan, X. Wang, X. Han and Y. Zhao, Nat. Commun., 2019, 10, 2270 CrossRef PubMed.
- J. Chen, J. Shu, S. Schobloch, A. Kroeger, R. Graf, R. Muñoz-Espí, K. Landfester and U. Ziener, Macromolecules, 2012, 45, 5108–5113 CrossRef.
- J. Mattu, T. Johansson and G. W. Leach, J. Phys. Chem. C, 2007, 111, 6868–6874 CrossRef CAS.
- P. J. Brown, D. S. Thomas, A. Köhler, J. S. Wilson, J. S. Kim, C. M. Ramsdale, H. Sirringhaus and R. H. Friend, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 642031–6420316 Search PubMed.
- D. Papkov, C. Pellerin and Y. A. Dzenis, Macromolecules, 2018, 51, 8746–8751 CrossRef CAS.
- B. Jasse and J. L. Koenig, J. Polym. Sci., Polym. Phys. Ed., 1980, 18, 731–738 CrossRef CAS.
- N. Kumari, M. Pandey, S. Nagamatsu, M. Nakamura and S. S. Pandey, ACS Appl. Mater. Interfaces, 2020, 12, 11876–11883 CrossRef CAS.
- V. Krishnan, Y. Kasuya, Q. Ji, M. Sathish, L. K. Shrestha, S. Ishihara, K. Minami, H. Morita, T. Yamazaki, N. Hanagata, K. Miyazawa, S. Acharya, W. Nakanishi, J. P. Hill and K. Ariga, ACS Appl. Mater. Interfaces, 2015, 7, 15667–15673 CrossRef CAS PubMed.
- M. Schlenk, E. Hofmann, S. Seibt, S. Rosenfeldt, L. Schrack, M. Drechsler, A. Rothkirch, W. Ohm, J. Breu, S. Gekle and S. Förster, Langmuir, 2018, 34, 4843–4851 CrossRef CAS PubMed.
- B. O'Connor, R. J. Kline, B. R. Conrad, L. J. Richter, D. Gundlach, M. F. Toney and D. M. DeLongchamp, Adv. Funct. Mater., 2011, 21, 3697–3705 CrossRef.
- M. Richard-Lacroix and C. Pellerin, Macromolecules, 2013, 46, 9473–9493 CrossRef CAS.
- T. Kato, M. Gupta, D. Yamaguchi, K. P. Gan and M. Nakayama, Bull. Chem. Soc. Jpn., 2021, 94, 357–376 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ab,
Alexandre
Legrand
ab,
Alexandre
Legrand