
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multiscale QM/MM modelling of catalytic systems with ChemShell†
You
Lu
a,
Kakali
Sen
 a,
Chin
Yong
a,
David S. D.
Gunn
a,
John A.
Purton
a,
Jingcheng
Guan
b,
Alec
Desmoutier
b,
Jamal
Abdul Nasir
b,
Xingfan
Zhang
b,
Lei
Zhu
b,
Qing
Hou
b,
Joe
Jackson-Masters
c,
Sam
Watts
c,
Rowan
Hanson
c,
Harry N.
Thomas
c,
Omal
Jayawardena
c,
Andrew J.
Logsdail
c,
Scott M.
Woodley
b,
Hans M.
Senn
d,
Paul
Sherwood
e,
C. Richard A.
Catlow
bc,
Alexey A.
Sokol
b and
Thomas W.
Keal
*a
a,
Chin
Yong
a,
David S. D.
Gunn
a,
John A.
Purton
a,
Jingcheng
Guan
b,
Alec
Desmoutier
b,
Jamal
Abdul Nasir
b,
Xingfan
Zhang
b,
Lei
Zhu
b,
Qing
Hou
b,
Joe
Jackson-Masters
c,
Sam
Watts
c,
Rowan
Hanson
c,
Harry N.
Thomas
c,
Omal
Jayawardena
c,
Andrew J.
Logsdail
c,
Scott M.
Woodley
b,
Hans M.
Senn
d,
Paul
Sherwood
e,
C. Richard A.
Catlow
bc,
Alexey A.
Sokol
b and
Thomas W.
Keal
*a
aSTFC Scientific Computing, Daresbury Laboratory, Keckwick Lane, Daresbury, Warrington, WA4 4AD, UK. E-mail: thomas.keal@stfc.ac.uk
bKathleen Lonsdale Materials Chemistry, Department of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK
cCardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff CF10 3AT, UK
dSchool of Chemistry, University of Glasgow, Joseph Black Building, Glasgow G12 8QQ, UK
eDepartment of Chemistry, Lancaster University, Lancaster, LA1 4YB, UK
First published on 20th April 2023
Abstract
Hybrid quantum mechanical/molecular mechanical (QM/MM) methods are a powerful computational tool for the investigation of all forms of catalysis, as they allow for an accurate description of reactions occurring at catalytic sites in the context of a complicated electrostatic environment. The scriptable computational chemistry environment ChemShell is a leading software package for QM/MM calculations, providing a flexible, high performance framework for modelling both biomolecular and materials catalysis. We present an overview of recent applications of ChemShell to problems in catalysis and review new functionality introduced into the redeveloped Python-based version of ChemShell to support catalytic modelling. These include a fully guided workflow for biomolecular QM/MM modelling, starting from an experimental structure, a periodic QM/MM embedding scheme to support modelling of metallic materials, and a comprehensive set of tutorials for biomolecular and materials modelling.
Introduction
Modelling catalytic processes is a challenge for computational chemistry, as it requires an accurate characterisation of reactivity influenced by a usually complex chemical environment. This type of problem is, however, well suited to a hybrid multiscale approach, where the reaction site is treated using an accurate quantum mechanical (QM) description of the electronic structure, while the environment is accounted for through a more approximate, but less computationally expensive method. A major advantage of the hybrid approach is the ability to use a high level of theory in the central QM region, as illustrated by the work of Piccini et al.,1 who achieved chemical accuracy in the calculation of rate constants for catalytic reactions in zeolites. The environment can be described either using a relatively inexpensive level of QM theory, or with a classical molecular mechanics (MM) forcefield.Hybrid QM/MM methods offer a good balance of computational efficiency and modelling accuracy and have become established as a powerful tool for catalytic science. QM/MM methods are available in a range of computational chemistry software packages, for which the implementations broadly fall into two categories. In the first category are extensions to existing electronic structure or classical molecular dynamics (MD) packages, with examples including AMS,2 AMBER,3,4 CHARMM,5 CP2K,6 deMon2k,7 Gaussian,8,9 GROMACS,10 NAMD,11 and ORCA,12 among others. QM/MM methods are implemented in these packages either through incorporating the required additional features directly into the software, or through interfacing with other software that can perform the additional calculations. Advantages of this category include the potential for efficient integration with a specific codebase, and a familiar starting point for existing users of the software, but at the cost of being unable to use features from alternative codes which might be relevant to a given scientific problem.
The second category of QM/MM implementations is exemplified by the ChemShell computational chemistry environment,13–15 which is well established as a flexible scripting platform for chemical calculations with a focus on hybrid multiscale methods. Recognising that most computational chemistry software is specialised in either QM or MM calculations, ChemShell implements a code-coupling framework that acts as a wrapper around external QM and MM packages and combines their outputs into a full QM/MM simulation. The user is therefore able to mix and match their preferred external codes according to their practical experience and specific features they wish to use. This attractive feature has made the second category an increasingly popular option for QM/MM methods development. Other computational chemistry programs taking a similar approach include ASE,16 Cuby,17 Fromage,18 Janus,19 LICHEM,20 MiMiC,21 PUPIL,22 QMCube,23 QMMM,24 and Spicy.25
ChemShell has a modular design to ensure that it is robust when working with a potentially large number of different combinations of external packages. The modular framework in the latest release of the code is illustrated in Fig. 1. At the lowest level in the framework are objects defined in ChemShell for storing chemical data and methods for manipulating the data. Molecular and materials systems are defined using Fragment objects, which contain details of the system's geometry, periodicity, charges and other atomic attributes. This information can either be specified directly via Python commands or imported from common structural data formats such as XYZ or PDB. The output of chemical calculations, such as energies and gradients of a system, are stored in Result objects.
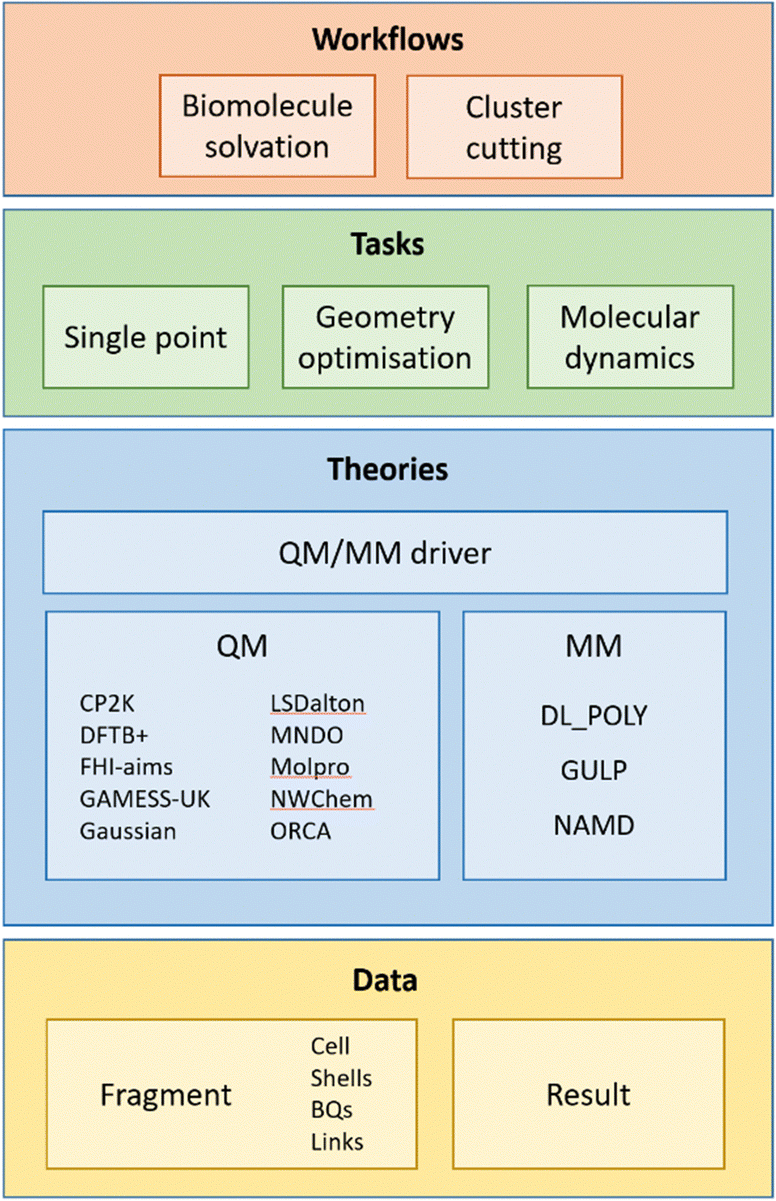 | ||
| Fig. 1 Overview of ChemShell's modular framework. Chemical systems are defined by Fragment objects. Theories define QM, MM or QM/MM calculations on the Fragment data, which in turn can be invoked by high-level chemical Tasks such as geometry optimisations. At the top level, Workflows are complex combinations of Tasks to support specific forms of modelling. | ||
At the next level, calculations are defined using Theory objects. The nature of the calculation is defined by specifying either an external QM or MM package, with relevant additional options to describe the calculation parameters (for example, the functional and basis set for a density functional theory (DFT) calculation using a QM package, or the forcefield definition for an MM package). Hybrid methods can also be specified using ChemShell's QM/MM driver, which combines the user's choice of QM and MM theories. In the hybrid case the QM/MM driver will then also handle the boundary between the QM and MM regions of the system, based on a user-provided list of atoms that define the QM region. Full details of the standard QM/MM schemes implemented in ChemShell can be found in previous articles.13–15
Once a Theory has been defined, it can then be used for one or more high-level chemical tasks, which are also defined as Python objects. The simplest Task is a single-point calculation, where the energy, and optionally also the gradient, will be returned. More complicated Tasks include geometry optimisation (using the DL-FIND geometry optimisation library26 distributed with ChemShell) and molecular dynamics. These Tasks invoke the chosen Theory whenever an energy or gradient is required, and so are completely flexible as to how the underlying Theory has been defined.
Finally, at the top level, specialised guided Workflows have been implemented to provide functionality that combines multiple Tasks and other features in the code to achieve a specific goal. Examples of ChemShell Workflows include the cluster cutting process to set up solid-state embedding models, and the new biomolecular solvation workflow, which we describe later in this article.
All ChemShell calculations are controlled via an input script. These scripts were originally written in the Tcl programming language, but in recent years a major redevelopment of ChemShell was undertaken,15 and the new software is scriptable using the Python programming language, which has become established as the most widely used scripting language for scientific applications. The open source, Python-based version of ChemShell (“Py-ChemShell”) is intended to succeed the original Tcl-based package and is the only version now under active development, although Tcl-ChemShell continues to be maintained as a legacy software resource.
A strong focus of the ChemShell project is on scalability to high performance computing (HPC) systems, including through task-farming parallelisation.27 An advanced task-farming framework is provided, which allows many chemical tasks to be performed in parallel, in addition to making use of parallelism in the external codes it calls, resulting in highly scalable QM/MM calculations.15 The Py-ChemShell build system is also designed with HPC-specific builds in mind, with automated build configurations provided for a range of HPC platforms including ARCHER2, the UK's national supercomputing service. These configuration scripts can be easily adapted to support other HPC systems.
In this review, we present a survey of recent ChemShell applications and describe new software developments in Py-ChemShell up to and including version 21.0, released in December 2021. Over this period there have been many underpinning improvements to the code, as well as new interfaces to the quantum-mechanical packages ORCA,28 FHI-aims,29 Molpro,30 Gaussian,31 and CP2K,32 the semi-empirical QM codes MNDO33 and DFTB+,34 and the classical molecular dynamics package NAMD.35 There have also been significant new high-level functionalities, which we describe in the next sections, including a comprehensive guided workflow for biomolecular QM/MM calculations, further refinements to the covalent and ionic solid-state embedding models, and a new periodic QM/MM embedding scheme targeting metallic systems, each accompanied by an illustrative example system.
QM/MM simulations of biomolecules
ChemShell is widely used for modelling complex biomolecular systems, especially in the investigation of enzyme-catalysed reaction mechanisms, where a quantum-mechanical description of the active site is necessary to characterise the catalytic processes, while a hybrid approach is needed to efficiently calculate the full protein system. Metalloenzymes are particularly well-suited to QM/MM calculations, as they usually require at least a DFT level of theory to adequately describe their reactivity at the metal site. The versatile cytochrome P450 superfamily of haem enzymes (Fig. 2) are among the most studied systems using QM/MM simulations with ChemShell, and continue to be a very active topic of research.36–51 In recent work Kalita et al. used a combination of molecular dynamics simulations and QM/MM to explore oxygen binding and the protonation steps leading to formation of compound I in two archetypal P450 systems.52 The team used these techniques also to understand the behaviour of a bioengineered CYP450BME variant that catalyses a reaction (C–H amination) that natural P450s do not.53 Lu et al. used ChemShell to illuminate the key role of a neighbouring residue in the oxidation of ethanol by CYP2E1, which results in a preferred mechanism involving an unusual initial hydrogen abstraction from the ethanol O–H bond.54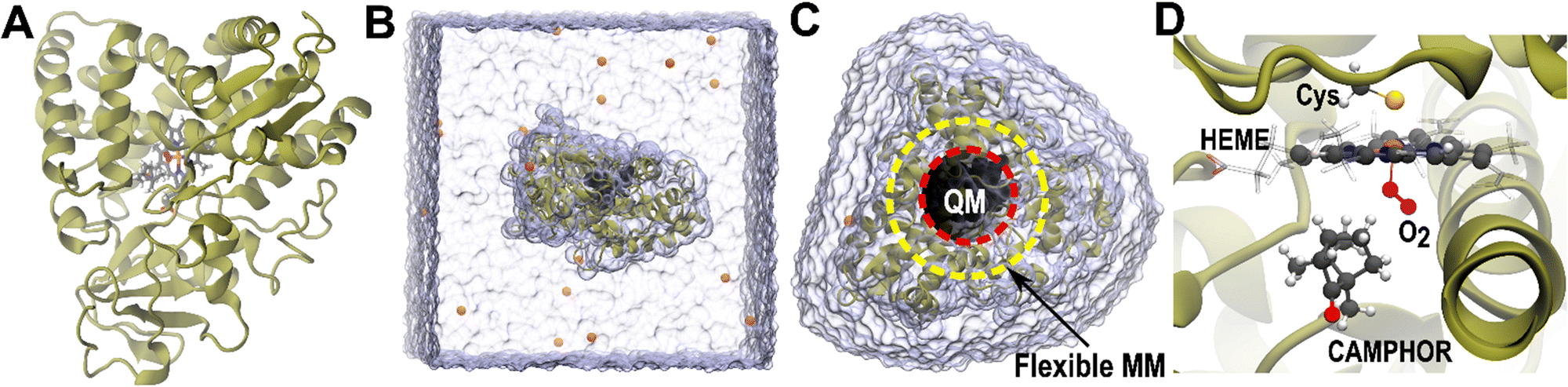 | ||
| Fig. 2 Stages in the process of the biomolecular workflow for P450cam: (A) the original PDB structure 1DZ8, (B) the smallest repeat cell of the periodic, solvated protein model at the end of the solvation workflow, ready for equilibration using molecular dynamics, (C) a non-periodic protein and water shell model cut out from an MD snapshot which forms the starting point for QM/MM calculations, (D) the QM region of the QM/MM model, containing haem unit, bound oxygen, camphor and cysteine side chain, using a ball and stick representation. | ||
ChemShell has been used to investigate many other haem-containing proteins, including recent studies by Liu and co-workers on the latex clearing protein LcpK30, a b-type cytochrome,55 the inactivation mechanism of neuronal nitric oxide synthase,56 and a proton-coupled electron transfer process in coprohaem decarboxylase, which is involved in haem biosynthesis.57 Other recent works have looked at catalase-peroxidases (KatGs),58 indoleamine-2,3-dioxygenase,59 and YfeX, a promising candidate for carbine transfer biocatalysis.60
The wider field of iron-containing enzymes also continues to be a fruitful target for QM/MM studies.61–73 In recent work Tian et al. have studied the unique non-haem iron enzyme ergothioneine synthase, using QM/MM to identify oxygen binding modes, determine the preferred spin state for reactivity, and clarify the sequence of sulfoxidation and C–S bond formation in its catalytic cycle.74 The 2-oxoglutarate (2OG) dependent non-haem iron oxygenases catalyse a wide variety of oxidative processes and have been the target of multiple recent studies.75–86 Chaturvedi et al. used ChemShell QM/MM calculations to investigate the reaction mechanism of PHF8, which catalyses demethylation of methylated lysine in histones, and is a target for cancer treatments.87 The simulations revealed that rearrangement of 2OG at the iron centre is an important part of the mechanism, which may help inform the design of inhibitors.
More complex active sites containing multiple metal centres are also amenable to QM/MM simulations. Several recent studies have investigated reactivity of non-haem diiron enzymes.88–91 Song et al. used ChemShell to model PhnZ, a mixed-valence diiron oxygenase, which catalyses the conversion of organophosphonic acids to inorganic phosphate via a mechanism involving the substrate bonding to one Fe(III) site and oxygen to the other.92 [NiFe]-hydrogenases have also been explored,93,94 which are of great interest in the development of the hydrogen economy due to their ability to catalyse H2 oxidation at ambient conditions. The radical S-adenosylmethionine (SAM) enzyme family, which are dependent on a [4Fe–4S] cofactor, also continue to attract attention for their ability to catalyse a wide range of redox reactions.95 Feng et al. used QM/MM calculations to provide insight into how the reactivity of the SAM enzyme Dph2 is affected by the electronic state of the [4Fe–4S] cluster.96 More complicated still are the double-cubane cluster protein,97 featuring an [Fe8S9] complex, and the iron-molybdenum cofactor (FeMoco) in molybdenum-dependent nitrogenase98–100 and iron–vanadium cofactor in vanadium-dependent nitrogenase.101 Thorhallson and Bjornsson have used QM/MM calculations with ChemShell to explore the energy surface of reduced states of FeMoco, showing how the electronic structure changes along a proposed redox-state cycle.102 The electronic structure of such systems calculated by broken-symmetry DFT is highly dependent on the choice of exchange–correlation functional.103
Copper enzymes catalyse a wide variety of reactions, often utilising multinuclear copper centres.104 Wu et al. used QM/MM calculations in ChemShell to investigate the form of the active species in two binuclear copper monooxygenases, which catalyse stereospecific C–H hydroxylation of precursors to hormones and neurotransmitters, resulting in a full description of the catalytic cycle.105 Sen et al. have investigated copper-containing nitrite reductases,106 which reduce nitrite to nitric acid as part of the global nitrogen cycle. These enzymes contain two copper centres, one of which catalyses the reduction reaction while the other is involved in electron transfer. A combination of ChemShell QM/MM simulations and serial crystallography, which provides a “movie” of the reduction process in a protein crystal, yield complementary information on the system, resulting in a full description of the reaction mechanism.107
The flexibility of QM/MM approaches to describe a wide range of metal cofactors is demonstrated by recent ChemShell applications on magnesium-dependent taxadiene synthase108 and ala-glu-epimerase,109 xyloside α-1,3-xylosyltransferase (where magnesium was modelled in place of manganese),110 nickel-containing quercetinases,111,112 tungsten-dependent benzoyl-coenzyme A reductase113 and zinc-dependent leukotriene A4 hydrolase,114 matrix metalloproteinase-1115 and matrix metalloproteinase-9.116 Zhao et al. used ChemShell to find the most favourable pathway for catalysis of N–N bond formation by a family of zinc-binding cupin proteins.117
While most recent ChemShell protein studies have targeted metalloenzymes, QM/MM simulations are equally applicable to other types of enzymes.118–126 Cantú Reinhard et al. investigated the case of nogalamycin monoxygenase (NMO), which, unlike most other monooxygenases, is able to catalyse the oxidation of a substrate without either a metal or organic cofactor. QM/MM calculations revealed paths for activation of the substrate by triplet O2, giving insight into the NMO reaction mechanism.127 Enzymes can also contain a mixture of metal and non-metal active sites, as in the case of cyclooxygenase-2, which has a haem-containing peroxidase active site as well as a cyclooxygenase active site. The peroxidase active site is responsible for generating a tyrosyl radical, which initiates the reaction at the cyclooxygenase active site. The subsequent deoxygenation of the substrate arachidonic acid at the cyclooxygenase active site can be efficiently characterised using QM/MM methods as the peroxidase site does not need to be included in the QM region at this stage.128–130
Environmental chemistry is an increasingly prominent topic for QM/MM simulations, which have been used to investigate biodegradation of pesticides,131–133 plastics,134,135 and other hazardous pollutants.136–142 Feng et al. used ChemShell to study the degradation of polyethylene terephthalate (PET),143 comparing the common features of reactions catalysed by the enzymes IsPETase and IsMHETase and analysing the structural characteristics that affect catalytic activity, pointing the way to the design of more efficient enzymes for plastic recycling.
It is well-established that intrinsic and external electric fields can significantly affect catalytic processes, and these are the focus of several recent studies using ChemShell QM/MM calculations in combination with electric fields modelled using the TITAN code.144–147 Yadav et al. used these methods to show that a single-site difference between two cytochrome P450 peroxygenases creates a local electric field that changes the stability of intermediate species in the catalytic cycle, resulting in a different reaction mechanism and products.148 Ramanan et al. looked at the effect of applying external electric fields on arginine demethylation catalysed by a 2OG-oxygenase, revealing that they promote C–H activation and inhibit N–H activation pathways.149
QM/MM simulations of biomolecules are not limited to proteins, and several recent ChemShell studies have focussed on DNA, RNA, and their components.150–157 Biocatalysis applications involving DNA repair enzymes are an active topic in this area.158–161 Naydenova et al. used ChemShell to investigate the catalytic mechanism of uracil DNA glycosylase, which initiates excision of uracil DNA lesions.162 QM/MM simulations revealed the key role of an active-site histidine residue and alternative reaction pathways that explain why mutation reduces, but does not completely suppress, enzymatic activity.
Other complex biomolecular targets in recent work include membrane enzymes,163 protein–protein interactions,164 and the bacterial ribosome.165
QM/MM methods are now widely applied to the study of excited electronic states, in tandem with electronic structure software packages capable of time-dependent density functional theory or wavefunction-based excited state calculations. Light-sensitive proteins that have been simulated with ChemShell in recent studies include a wide range of rhodopsins,166–173 photosystem II,174–176 calcium-regulated photoproteins,177 green fluorescent protein,178 the phototoxic protein KillerRed,179 bacterial phytochromes,180,181 and cyanobacteriochromes.182–184 Excited state calculations are typically used to simulate experimental UV-vis spectra,185 and so are useful for characterising any molecular system containing a chromophore, including catalytic intermediates.
The vast majority of biomolecular applications use a form of DFT for the QM region, but QM/MM approaches can also accommodate higher-level methods where necessary.186 Nutho et al. used ChemShell QM/MM calculations to generate potential energy profiles up to the LCCSD(T)/MM level to help establish the reaction mechanism for inhibition of zika virus protease by peptidyl aldehydes.187,188 Douglas-Gallardo et al. used ChemShell QM/MM calculations and high-level projector-embedding calculations with Molpro to provide corrections to free energy barriers for carbon dioxide formation by the RuBisCO enzyme, calculated using semi-empirical QM/MM molecular dynamics, to increase the accuracy of the reaction profiles obtained for different protonation states of key amino acid residues.189 Free energies may also be estimated at the DFT/MM level using QM/MM free energy perturbation, as in the study of Zheng et al. on HCV protease.190
Biomolecular workflows in Py-ChemShell
The 21.0 release of Py-ChemShell introduces QM/MM functionality for biomolecular modelling into the redeveloped version of ChemShell for the first time. In the original Tcl-based version of ChemShell, it was expected that the user would set up and equilibrate the biomolecular system of interest using a classical MD package before importing it into ChemShell to perform QM/MM calculations. It is still possible to follow this workflow in Py-ChemShell, but this requires a high level of familiarity with modelling best practices. We have therefore implemented a more comprehensive guided workflow within ChemShell, starting from the beginning with an experimental crystal structure, and guiding the user with a set of best practices for system setup, solvation and equilibration, resulting in a model that is ready for QM/MM simulations. The aim of this workflow is to reduce the prior knowledge required for new users while also offering sufficient flexibility to experienced users, who may also benefit from the efficiency of a more automated setup process. In the following sections we summarise the steps in the workflow with reference to the example of P450cam, a well-studied cytochrome P450 enzyme that catalyses the hydroxylation of camphor, as shown in Fig. 2. The workflow we describe is illustrated by the flowchart in Fig. 3.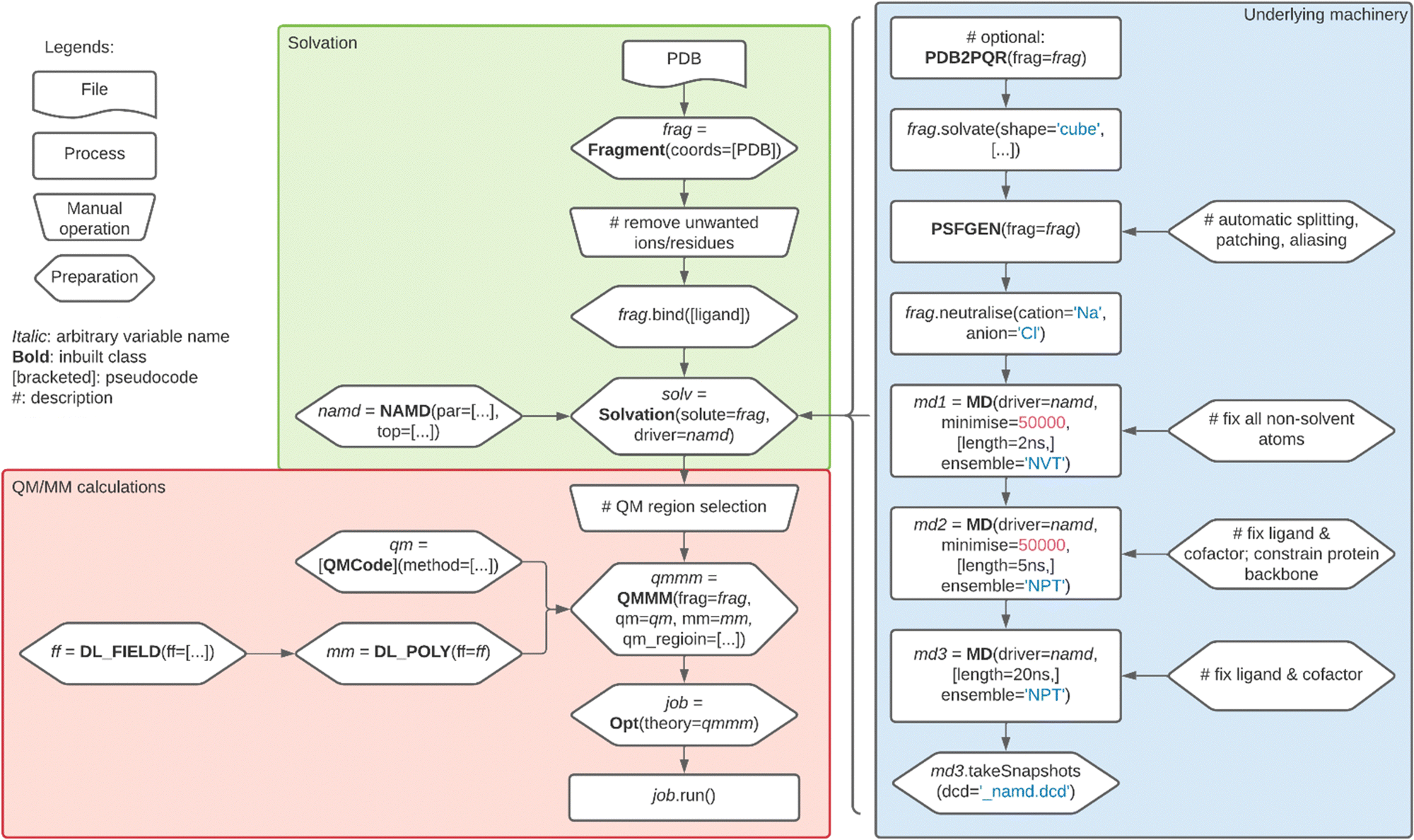 | ||
| Fig. 3 Flowchart of the workflow for biomolecular solvation and QM/MM calculations in Py-ChemShell. The underlying machinery shown in the blue box is automated, but may be controlled by the user via setting of options at each stage. | ||
System preparation
The preparation of a protein for QM/MM calculations begins with a structure obtained using X-ray crystallography or other experimental techniques. The structural data may be read from a PDB file on disk, or ChemShell can download a structure directly from the Protein Data Bank if the user supplies a PDB ID. For P450cam the initial structure (PDB reference 1DZ8)191 is shown in Fig. 2A.When ChemShell imports a raw PDB file, it performs some initial processing, such as choosing a single atomic position if alternatives are given (based on a consideration of mean occupancy and, if that is not conclusive, B-factors, where a lower B-factor indicates reduced thermal motion and so less uncertainty in the specified atomic coordinates). It does not, however, attempt to remediate any missing amino acid residues, and the user should make sure that the protein chain to be modelled is complete before proceeding.
Following the import of the PDB file, the structure usually needs to be manipulated further into a form suitable for simulation. This may include selection of a specific chain to be simulated, removal of unwanted ions or molecules, and addition of relevant ligands if they are not already present in the structure. ChemShell provides a number of powerful atom selection tools to assist with these steps.
At this point, the protein model is ready for ChemShell's guided solvation workflow. This routine automates the remaining setup stages, solvation of the system, and equilibration using classical MD through an interface to NAMD.35 The first task in the workflow is to add hydrogen atoms to the protein model, which are not observed by X-ray crystallography, and so not included in the PDB structure. To address this, ChemShell provides a wrapper to call the PDB2PQR192,193 utility program, which automates the addition of hydrogen atoms. PDB2PQR also assigns partial charges to the model using values from the forcefield selected by the user, and calls PROPKA194,195 to predict the pKa values of titratable groups in the protein, so that protonation states can be assigned.
Once the missing hydrogen atoms, protonation states, and partial charges have been assigned, the next stage is to solvate the protein model. By default, ChemShell will place the protein model in a pre-equilibrated cubic TIP3P water box, with an appropriate cavity created for the protein by deleting water molecules that occupy the same volume. A cubic box is used in preparation for the periodic MD simulations that will be run to equilibrate the full protein system. The size of the box can be adjusted as required.
In order to run the MD simulations with NAMD, a Protein Structure File (PSF) must be generated. ChemShell provides a wrapper for the PSFGEN utility in NAMD to create the PSF file. To improve the automation of this stage the ChemShell wrapper will automatically apply patches corresponding to common additional species, such as the haem group in P450cam. The user may also supply their own patches for more unusual species, as they would with a standalone PSFGEN run.
Finally, before running the MD, the workflow neutralises the solvated system through addition of counterions to the solvent environment. By default, sodium ions are added to neutralise a negatively charged system, or chloride ions to neutralise a positively charged system. The final periodic solvated P450cam system ready for equilibration is shown in Fig. 2B.
MD equilibration
The MD stage of the workflow automates a three-step equilibration process for the solvated protein system. In the initial implementation of the workflow, NAMD is used as the classical MD driver.In the first step of the process, the solvent environment and neutralising counterions are allowed to relax around the protein and any ligands or cofactors, which are fixed at their initial positions. The aim of this MD step is to remove any solvent inhomogeneities around the protein created by the initial solvation. The relaxation is accomplished through an initial partial energy minimisation, followed by an MD simulation under NVT conditions at 300 K. Default values for the minimisation and MD timescales are provided based on previous experience (as given in Fig. 3), but can be adjusted by the user as desired.
In the second step, the protein is allowed to relax in the solvent environment, albeit with backbone constraints in place to retain the overall structure at this stage. Again, an initial energy minimisation is performed, to enable the protein to swiftly reconfigure, followed by an MD simulation. In this step, the MD simulation is run using an NPT ensemble, so that the water density can equilibrate to natural conditions.
At this stage the user must make a choice about whether species such as cofactors or ligands are fixed or free to move in the rest of the MD simulations. This may be an enforced decision, most commonly because a full set of forcefield parameters is not readily available for the species at hand, which therefore needs to be kept fixed. It may also be desirable to fix the species in an initial configuration if it is known to be the correct initial configuration for a reaction to take place. In our P450cam example, this is the case for the camphor molecule, whose binding position and orientation should be kept during MD as they are correct for the subsequent H-abstraction reaction. While fixing parts of the system introduces a degree of artificiality into the MD equilibration, it should be noted that if the fixed species are within the QM region in the subsequent QM/MM calculations they will be fully optimised at that stage.
The third step is a final “production” MD simulation, again under NPT conditions but without any constraints on the protein backbone. This simulation provides the trajectory from which snapshots will be taken as a starting point for QM/MM calculations.
Py-ChemShell provides a set of analysis tools for MD trajectories that can be used to check if equilibration has completed, for example by looking at whether the RMSD of the protein backbone has converged. The temperature and energies along the trajectory can also be plotted. Once the MD simulation stage has been successfully completed, a separate ChemShell routine takes snapshots at regular intervals along the trajectory for the following QM/MM stage.
QM/MM calculations
Snapshots from the final MD simulation are the starting point for biomolecular QM/MM calculations in ChemShell. However, once the initial equilibration is complete, it is preferable to avoid the use of periodic boundary conditions as this would restrict the range of QM codes that could be used and is relatively computationally expensive, as well as potentially introducing artefacts due to periodic protein–protein interactions. Instead, ChemShell provides routines to cut the protein and a shell of solvent molecules out of the MD snapshots, resulting in a non-periodic model that is suitable for subsequent QM/MM calculations, as illustrated for the example of P450cam in Fig. 2C. This model will be saved to a PQR format file, along with the assigned partial charges from the initial setup stages.At this point, the model is ready for QM/MM calculations. The QM region now needs to be defined, and ChemShell's atom selection tools can again be used to build the QM atom list from the relevant chemical groups. An example minimal QM region for P450cam is shown in Fig. 2D, consisting of the core haem group, cysteinate side chain, oxygen ligand, and camphor molecule. The chosen QM theory should also be defined in the usual way for QM/MM calculations. An integer value must be specified for the charge of the QM region, which is most commonly obtained by summing the MM partial charges corresponding to the QM region.
For the MM part of a biomolecular QM/MM calculation, DL_POLY196 is used to evaluate MM energies and gradients, as it provides a flexible, general purpose forcefield engine for systems set up either using the guided workflow or imported from elsewhere. The Python-based version of ChemShell interfaces to the open-source version 5 release of DL_POLY, which scales well to very large QM/MM systems.15 For biomolecular calculations using CHARMM forcefields,197 the forcefield parameters are managed by a wrapper to the DL_FIELD program,198 which is used to convert CHARMM forcefield data automatically into DL_POLY format. User-defined parameter sets outside the standard CHARMM forcefields can also be provided to DL_FIELD, e.g. for the camphor molecule in the case of P450cam.
Once the biomolecular QM/MM model has been fully specified, the calculations are run as with any other QM/MM system using the modular ChemShell framework. Tasks can now be defined and run, making use of high-level ChemShell features, such as the range of geometry optimisation techniques provided by DL-FIND to characterise reaction mechanisms,26 including the nudged elastic band and dimer methods.
In the present release of Py-ChemShell (v21.0), the full workflow is designed to be used with CHARMM forcefields. However, the user may not wish to use the complete workflow: the import mechanism via DL_FIELD is separate to the solvation stage, and if the initial CHARMM MD runs have been run elsewhere, these snapshots can be directly imported to QM/MM calculations in ChemShell. An alternative workflow is supported for AMBER forcefields,199,200 which can be imported directly into Py-ChemShell for QM/MM calculations. In this case, it is assumed that the system has been set up and equilibrated externally, for example in AMBER, and snapshots taken from there can be imported into ChemShell. Conversion tools are also provided for importing existing QM/MM setups that have been built in Tcl-ChemShell. An online case study tutorial, which goes step-by-step through the ChemShell commands used to set up the P450cam model and run QM/MM calculations on it, is available on the ChemShell website.201
Future releases of Py-ChemShell will provide a full guided workflow for AMBER forcefields, following the same lines as for CHARMM forcefields. The workflow will also be extended to support solvation and equilibration of DNA strands in a similar manner to proteins.
Solid state embedding
Although QM/MM methods have been more commonly used in the area of biocatalysis, they are equally applicable to solid state modelling. ChemShell provides both covalent and ionic embedded cluster models for heterogeneous catalysis applications. The covalent embedding model is extensively used for studies of porous catalytic materials, with a number of recent ChemShell investigations targeting microporous zeolites202–205 and mesoporous MCM-41.206,207 Nastase et al. used QM/MM simulations of the zeolites H-ZSM-5 and H-Y to study a crucial step in the conversion of methanol to hydrocarbons where the intermediate dimethyl ether is formed.208 The results highlighted the importance of the strength of the Brønsted acidic character at the active site on the stability of dimethyl ether, which may help to explain the superior catalytic performance of H-ZSM-5. Abdul Nasir et al. investigated the effect of water and ammonia solvent molecules on the selective catalytic reduction of NOx species by the copper-exchanged zeolite Cu-CHA.209 The results showed that solvation of the active sites has a strong influence on the energy profile of the catalytic cycle, with differing effects on the oxidative and reductive parts. Liu et al. used QM/MM methods to investigate the structure and stability of vanadium-doped active sites in Al-MCM-41, and their potential use as heterogeneous catalysts for the oxidation of furans.210 Besides catalysis applications, covalent QM/MM embedding methods in ChemShell have also been recently applied to the excited state properties of organic materials211–216 and the adsorption of hydrogen onto amorphous solid water.217The ionic QM/MM embedded cluster approach can be used to model both bulk defects and surface reactivity. In recent ChemShell studies it has been used to characterise native point defects in GaN,218 to create optimised models of rutile TiO2 surfaces,219 to study oxygen vacancies in TiO2 using DFT and high-level wavefunction methods,220 to improve interatomic potential descriptions of CeO2,221 to investigate bulk and surface vacancies in MnO with potential catalytic applications to the transformation of CO2,222 and to study the defect properties of Cu in ZnO, an important industrial catalyst for the synthesis of methanol.223 Sainna et al. used ChemShell to study the reaction of glycerol over a catalytic MgO surface to form methanol.224 The results indicated that the most favourable route to methanol production involves a disproportionation reaction on the surface of MgO, while water is unlikely to play an important part in the process.
The nature of the QM/MM model employed changes significantly depending on whether a covalent or ionic embedded cluster approach is required, and we describe below separately the capabilities of ChemShell for each approach. Both approaches, however, start from the construction of a finite cluster model cut from a periodic bulk or slab model of the system. The finite cluster QM/MM model has several significant advantages over a periodic representation of the system, including avoidance of unphysical periodic interactions of the local defect with itself, straightforward handling of charged systems, and the ability to use a wide range of non-periodic QM software packages, improving the computational efficiency of the simulations. However, the creation of a cluster model necessarily involves the loss of the periodic electrostatic environment. To compensate for this, a set of point charges is arranged outside the cluster and fitted to reproduce the bulk electrostatic potential and field within the active region of the cluster. ChemShell provides a guided workflow for automated construction of the clusters and point charge fitting, following the procedure detailed in ref. 13.
Covalent embedded cluster model
For covalent or semi-covalent systems such as zeolites, which can be described using forcefields with covalent bonding terms, a similar QM/MM approach can be used as in the modelling of biomolecules.225 Link atoms are added to the QM region where bonds are broken at the boundary with the MM region, and the charge shift scheme is applied to avoid over-polarisation of the QM region. In the case of zeolites, an additional charge adjustment to the MM region is required because the natural choice of the QM region, groups of silicon or aluminium atoms and their connected oxygen atoms, will not have a zero charge.13 This is an automated procedure in all versions of ChemShell for the case of aluminosilicate systems. In the Python-based version of ChemShell, the adjustment routine has been generalised so that other systems can be treated in the same way, with charge modifications calculated using user-defined bond contributions from each pair of atoms at the boundary.A typical embedded cluster model of a zeolite is shown in Fig. 4. This example, with a cluster radius of 21.2 Å, consists of 2157 atoms. In ChemShell the standard additive electrostatic embedding scheme is generally used for zeolite calculations. As with electrostatic embedding calculations on biomolecules, any QM software package capable of supporting point charges in the QM Hamiltonian may be used. Typically calculations are performed using DFT with a hybrid exchange–correlation functional. In Py-ChemShell, the MM region is usually calculated with GULP,226 using the Hill–Sauer aluminosilicate forcefield.227,228
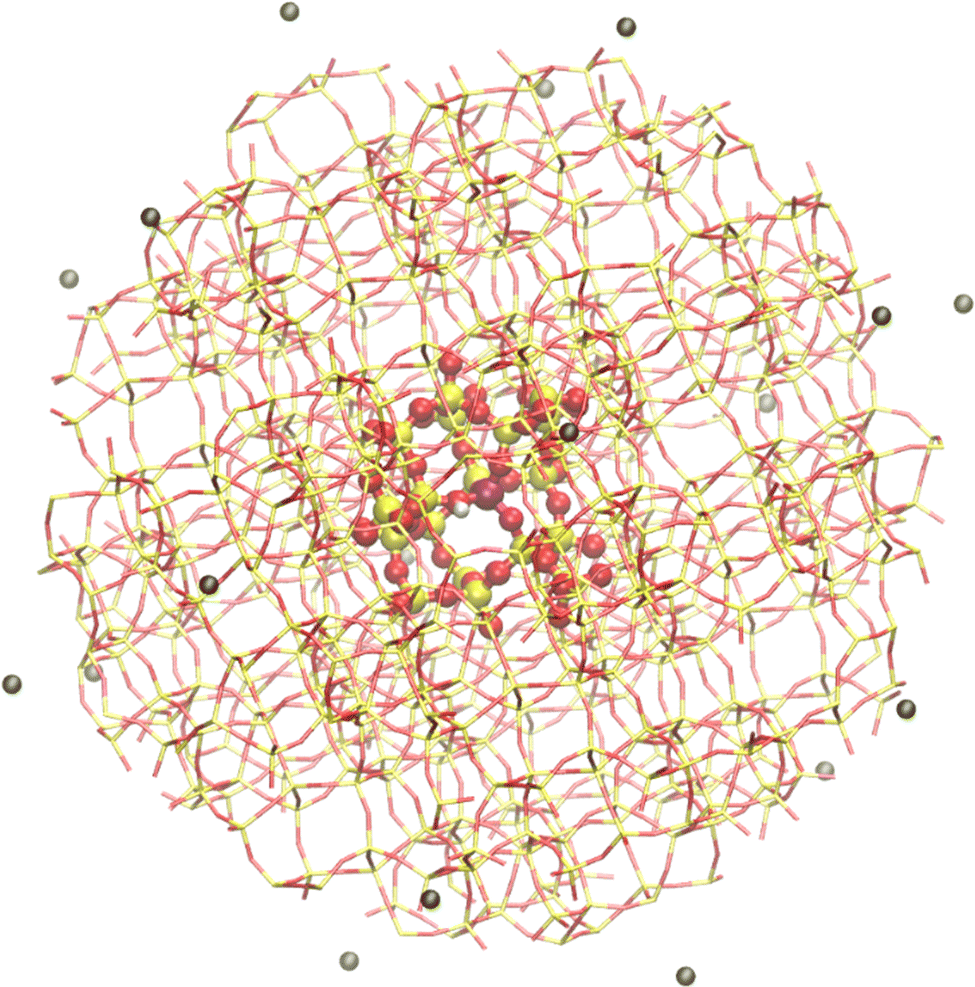 | ||
| Fig. 4 ChemShell embedded cluster model of the zeolite H-ZSM-5. Yellow atoms = Si, red = O, purple = Al, white = H, black = external point charges. A typical QM region of 67 atoms is shown with a ball and stick representation. | ||
A zeolite case study tutorial is available on the ChemShell website,229 detailing the setup of siliceous and aluminosilicate embedded cluster models, geometry optimisation of the zeolite system, calculation of ionisation potentials and deprotonation energies, and calculation of vibrational frequencies when methanol is adsorbed in the zeolite framework.
Ionic embedded cluster model
The QM/MM embedding model must be modified for ionically bonded systems such as transition metal oxides, where a link atom approach at the boundary is no longer appropriate. In Py-ChemShell, we follow the ionic embedding procedure established in the original Tcl-based version of ChemShell,230 where pseudopotentials are applied to atoms in a boundary region to localise the electron density within the inner QM region.QM/MM calculations using the ionic embedding model typically use polarisable embedding, where the QM and MM parts of the model are polarised by each other. In ChemShell, the polarisable MM region is represented by shell model potentials, where shell positions are relaxed under the electrostatic influence of the QM region. As with electrostatic embedding, the QM region is in turn polarised by the MM environment, so the calculation must be iterated self-consistently until the polarisation effects have converged.
In v21.0 of Py-ChemShell, the ionic embedding model is supported by NWChem,231 GAMESS-UK232 and FHI-aims, which contain the necessary mechanisms for handling pseudopotentials at the boundary sites. The polarisable MM region is calculated using shell model potentials implemented in GULP.
A typical example ionic QM/MM embedded cluster model of a CO2 molecule adsorbed onto MgO is shown in Fig. 5. In order to model the surface, a hemispherical cluster of radius 30 Å has been cut from a periodic slab model of MgO, with external point charges fitted to the electrostatic potential and electric field in a central sampling region of radius 15 Å, which is also the region allowed to relax when the cluster is optimised. Typically, 20–30 atoms around the central adsorption site are included in the QM region. The QM region is usually modelled using DFT with a hybrid exchange–correlation functional. Large-core effective core potentials are placed on the Mg2+ cations in the boundary between the QM and MM regions. The MM region is calculated using a shell model potential parameterised for MgO.233
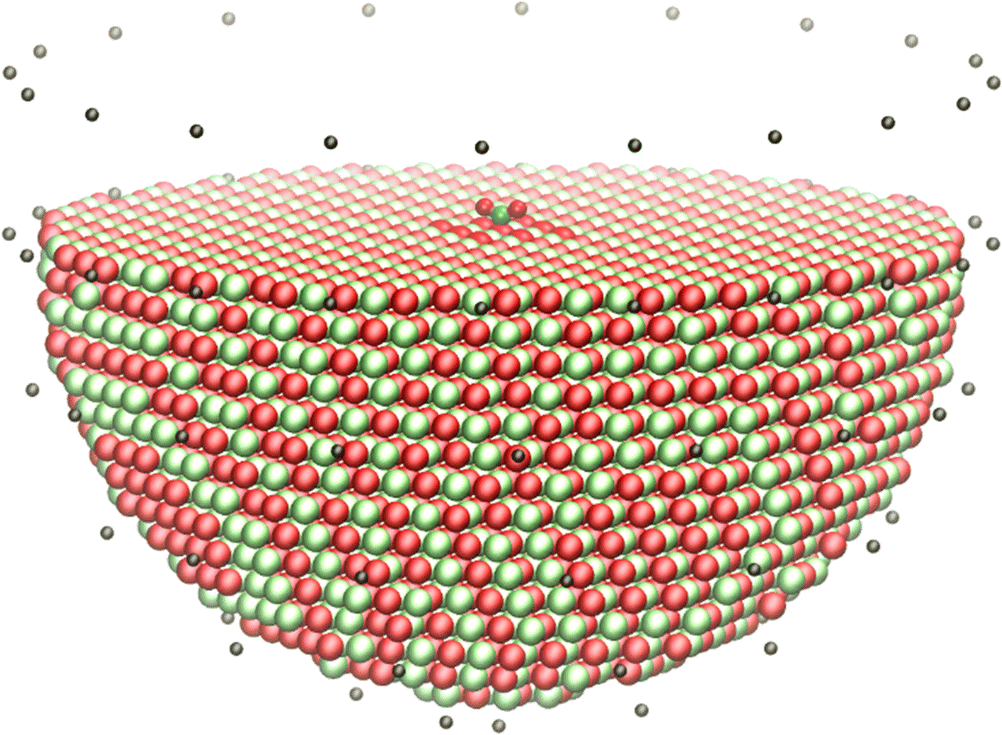 | ||
| Fig. 5 ChemShell embedded cluster model of CO2 adsorbed onto an MgO surface. Red spheres = O, green = Mg, cyan = C, black = external point charges. | ||
Two ionic embedding case study tutorials are available on the ChemShell website. The first takes the user through the process of setting up a cluster representing an MgO surface to study surface reactivity, including addition of an adsorbed CO2 molecule, and calculating vibrational frequencies and ionisation potentials of the system.234 The second demonstrates how a bulk MgO cluster can be set up and doped with Li, with the aim of calculating the barrier for electron migration, calculation of vibrational frequencies of the transition state, and calculation of electron paramagnetic resonance properties.235
Periodic QM/MM embedding
The non-periodic models described above can be applied to a wide range of systems across biomolecular and materials modelling, but there are some problems for which they are not suitable. Adsorption on metallic surfaces is one example of this: while taking a QM/MM approach would offer the same advantages in principle over pure QM methods as for other surface studies, the delocalised electronic structure of the metal surface is not properly described by a finite QM cluster, and so the standard non-periodic embedding models described above cannot be applied.To enable systems of this kind to be modelled, we have implemented the “QM/Me” embedding scheme of Meyer and Reuter236 in Py-ChemShell. The QM/Me method is fully described in the original paper so here we only highlight the differences with the non-periodic embedded cluster model implemented in ChemShell.
Like the previous methods, QM/Me follows a hybrid embedding approach, with the QM region containing part of the metal surface and the adsorbate in order to capture adsorbate–adsorbate and substrate–adsorbate interactions accurately. Importantly, in QM/Me the QM region is calculated with periodic boundary conditions (using a slab model) so that the band structure of the metallic system is correctly described. This region is embedded in a much larger bath-like MM region, which is used to describe longer range elastic substrate–substrate interactions, and is again calculated with periodic boundary conditions. However, the two calculations cannot simply be added together, as the QM calculation would also describe substrate–substrate interactions to some extent and so these will be overcounted. To avoid this problem, the QM region is calculated twice: once including both surface and adsorbate and once for the surface alone. The second QM calculations is then subtracted from the first, removing the surface–surface interactions while retaining the desired description of the adsorption. The system is then fully described as:
| EQM/Me(R) = [EQM(Rslab ∪ Rads) − EQM(Rslab)] + EMe(Rslab ∪ Renv) |
The ChemShell implementation of QM/Me follows the original scheme, while also being fully integrated into the modular ChemShell framework so that in principle any interfaced QM and MM packages can be used, providing they are capable of calculations with periodic boundary conditions. Our initial implementation released in Py-ChemShell v21.0 is intended for use with the interface to CP2K32 for the periodic QM calculations, and GULP for the MM region.
Fig. 6 shows an example of a QM/Me model used to optimise O2 adsorbed onto a Pd(100) surface. In this case, the MM bath contained 500 Pd atoms and was calculated with the modified embedded atom method with second nearest neighbour (MEAM-2nn237). DFT with the PBE exchange–correlation functional238 has been used for the QM region. The basis set employed was TZV2P-MOLOPT-GTH for oxygen and SZV-MOLOPT-SR-GTH for palladium239,240 and the pseudopotential was GTH-PBE.241 In this demonstrative case the QM region contained a 3-layer slab of 48 Pd atoms for which a 2 × 4 × 1 Monkhorst–Pack mesh was used for K-points. The QM/Me scheme, including definition of the two QM calculations, is fully automated using the “QM/Me” option in Py-ChemShell.
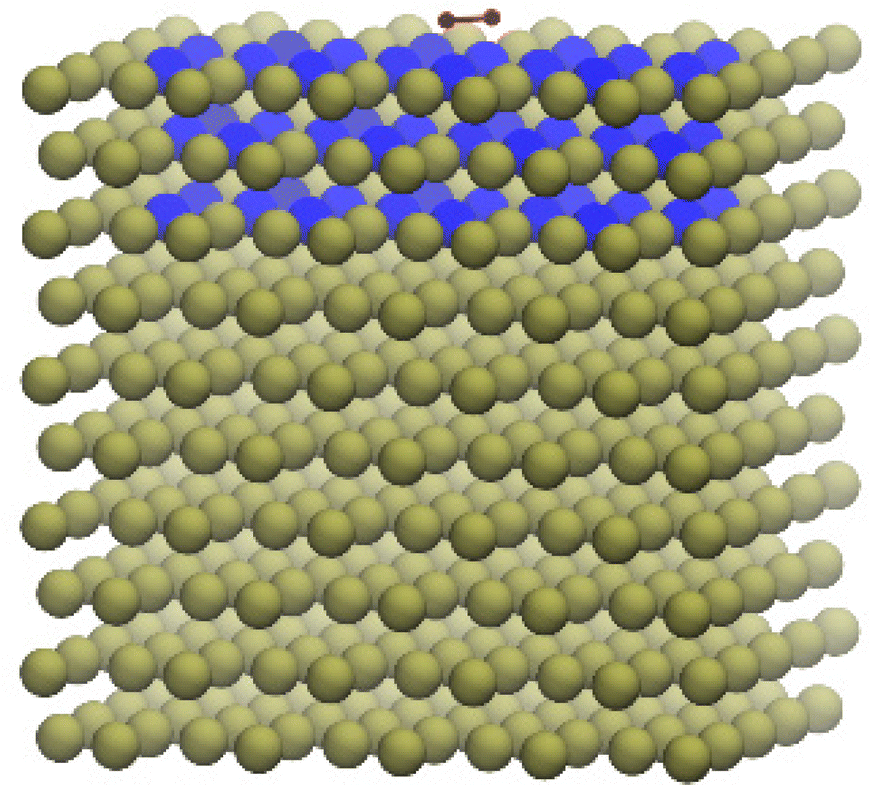 | ||
| Fig. 6 O2 (red) absorbed on the Pd surface optimised using the QM/Me embedding scheme. The 3-layer QM slab is displayed in blue colour. | ||
In addition to the QM/Me method, Py-ChemShell v21.0 has new functionality to support periodic calculations in general, in particular with the ability to optimise unit cells with DL-FIND. Future releases of ChemShell will extend the QM/Me method to other QM codes (FHI-aims, CASTEP and CRYSTAL), and implement other periodic QM/MM embedding schemes within the same framework.
Conclusions and outlook
In this review we have surveyed the wide range of biomolecular and materials QM/MM applications in biomolecular and solid state catalysis that have recently been studied using ChemShell and outlined the capabilities of the latest release of the redeveloped Python-based version of the software. The 2021 release of Py-ChemShell includes the full functionality required for biomolecular QM/MM workflows, guiding users from the initial download of an experimental protein structure, through setup and equilibration with classical molecular dynamics, and then on to full QM/MM calculations using the flexible, modular ChemShell framework. The latest release of Py-ChemShell also implements the periodic QM/Me scheme for surface-adsorbate systems, complementing the established finite cluster embedding models for materials modelling. The addition of QM/Me extends the range of materials that can be studied to include metallic systems, where a delocalised QM description is required.The Python-based version of ChemShell is under very active development. The next release of the software is planned for mid-2023 and will include a generic molecular dynamics module, enabling MD with any of the QM/MM schemes described in this paper and any combination of QM and MM codes supported by ChemShell. It will also feature routines for harmonic IR and Raman spectroscopic signatures242 and anharmonic corrections based on the vibrational self-consistent field (VSCF) approach, as well as additional interfaces to external QM packages.
Looking further ahead, several new development projects are underway, targeting different aspects of QM/MM functionality in ChemShell. A major scientific focus of the method developments is the emerging research area of hybrid catalysis, which involves catalysts that combine elements of the usual categories of heterogeneous, homogeneous and biomolecular catalysts. New features in development targeting these systems include advanced multilayer QM/MM models, including high-level wavefunction-in-DFT embedding and solvent boundary potentials, and enhanced sampling methods to fully characterise the behaviour of solvated catalysts. Support for free energy methods in Py-ChemShell will be enabled through a planned interface to the PLUMED library.243 Free energy methods previously available in Tcl-ChemShell (such as umbrella sampling and QM/MM free energy perturbation) will be supported together with additional methods such as metadynamics, to provide the user with a range of options for obtaining free energies. Beyond this, adaptive QM/MM methods will be introduced to allow solvent molecules to enter or leave the QM region, which will provide a dynamic description of solvent around a catalytic centre.
Another key area for future development will be the integration of machine learning (ML) approaches for interatomic potentials into embedding models for materials modelling. ML methods have also been recently introduced into DL-FIND to accelerate geometry optimisation by Kästner and co-workers.244–246
Complementing the scientific method developments, the underlying ChemShell software infrastructure is being enhanced to meet the needs of new computing platforms. As part of the UK's ExCALIBUR exascale computing initiative, ChemShell is being retooled for the exascale computing era, with a particular focus on complex QM/MM workflows coupling to highly scalable molecular and periodic DFT software to target problems in computational catalysis.247 ChemShell is also being developed to manage multiscale embedding workflows involving emerging quantum computing technologies in combination with conventional high performance computing.
Together these developments provide a flexible, scalable toolkit for QM/MM simulations that is applicable to all types of catalysis: heterogeneous, homogeneous and bio-catalysis. To download ChemShell and access the online documentation, tutorials and forum, please visit chemshell.org.
Author contributions
The software developments described in this review are the result of several projects. The biomolecular QM/MM features in Py-ChemShell v21.0 were developed and validated by YL, KS, CY, HMS, PS and TWK. Solid state embedding model enhancements and validation in Py-ChemShell v19.0–21.0 were carried out by YL, DSDG, JG, AD, JAN, XZ, LZ, QH, AJL, CRAC, AAS and TWK. The covalent and ionic embedding case study tutorials were created by JJ-M, SW, RH, HNT, OJ, TWK and AJL. Periodic QM/MM embedding and related development work in Py-ChemShell v21.0 was carried out by YL, DSDG, JAP and TWK as part of the SAINT project led by SMW. The manuscript was drafted by TWK, YL and KS, and was reviewed and edited by all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
TWK, CY, HMS, PS, CRAC and AAS acknowledge funding for the biomolecular and solid state embedding developments described in this review from EPSRC grant EP/R001847/1. SMW, CRAC and TWK acknowledge funding for the periodic QM/MM developments from EPSRC grant EP/P022235/1. The SAINT project team are grateful to Jörg Meyer and Matt Watkins for discussions concerning the QM/Me method and CP2K. Additional support for solid state embedding developments was provided by CoSeC, the Computational Science Centre for Research Communities, in collaboration with the Materials Chemistry Consortium, one of the UK's High End Computing consortia, and the UK's Collaborative Computational Project 5 (CCP5), which are funded by EPSRC grants EP/R029431/1 and EP/M022617/1. We acknowledge use of computing resources provided by STFC Scientific Computing Department's SCARF cluster and the ARCHER2 UK National Supercomputing Service (https://www.archer2.ac.uk). AJL acknowledges funding by the UKRI Future Leaders Fellowship program (MR/T018372/1). AJL and TWK thank CCP5 for supporting several student summer projects at Cardiff University to create online case study tutorials for Py-ChemShell.Notes and references
- G. Piccini, M. Alessio and J. Sauer, Ab Initio Calculation of Rate Constants for Molecule–Surface Reactions with Chemical Accuracy, Angew. Chem., Int. Ed., 2016, 55, 5235–5237 CrossRef CAS PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, Chemistry with ADF, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- R. Salomon-Ferrer, D. A. Case and R. C. Walker, An overview of the Amber biomolecular simulation package, WIREs Comput. Mol. Sci., 2013, 3, 198–210 CrossRef CAS.
- A. W. Götz, M. A. Clark and R. C. Walker, An extensible interface for QM/MM molecular dynamics simulations with AMBER, J. Comput. Chem., 2014, 35, 95–108 CrossRef PubMed.
- B. R. Brooks, C. L. Brooks III, A. D. Mackerell Jr., L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, CHARMM: The biomolecular simulation program, J. Comput. Chem., 2009, 30, 1545–1614 CrossRef CAS PubMed.
- T. Laino, F. Mohamed, A. Laio and M. Parrinello, An Efficient Real Space Multigrid QM/MM Electrostatic Coupling, J. Chem. Theory Comput., 2005, 1, 1176–1184 CrossRef CAS PubMed.
- A. de la Lande, A. Alvarez-Ibarra, K. Hasnaoui, F. Cailliez, X. Wu, T. Mineva, J. Cuny, P. Calaminici, L. López-Sosa, G. Geudtner, I. Navizet, C. Garcia Iriepa, D. R. Salahub and A. M. Köster, Molecular Simulations with in-deMon2k QM/MM, a Tutorial-Review, Molecules, 2019, 24, 1653 CrossRef CAS PubMed.
- T. Vreven, K. Morokuma, Ö. Farkas, H. B. Schlegel and M. J. Frisch, Geometry optimization with QM/MM, ONIOM, and other combined methods. I. Microiterations and constraints, J. Comput. Chem., 2003, 24, 760–769 CrossRef CAS PubMed.
- T. Vreven, M. J. Frisch, K. N. Kudin, H. B. Schlegel and K. Morokuma, Geometry optimization with QM/MM methods II: Explicit quadratic coupling, Mol. Phys., 2006, 104, 701–714 CrossRef CAS.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, GROMACS: Fast, flexible, and free, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- M. C. R. Melo, R. C. Bernardi, T. Rudack, M. Scheurer, C. Riplinger, J. C. Phillips, J. D. C. Maia, G. B. Rocha, J. V. Ribeiro, J. E. Stone, F. Neese, K. Schulten and Z. Luthey-Schulten, NAMD goes quantum: an integrative suite for hybrid simulations, Nat. Methods, 2018, 15, 351–354 CrossRef CAS PubMed.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, The ORCA quantum chemistry program package, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- P. Sherwood, A. H. de Vries, M. F. Guest, G. Schreckenbach, C. R. A. Catlow, S. A. French, A. A. Sokol, S. T. Bromley, W. Thiel, A. J. Turner, S. Billeter, F. Terstegen, S. Thiel, J. Kendrick, S. C. Rogers, J. Casci, M. Watson, F. King, E. Karlsen, M. Sjøvoll, A. Fahmi, A. Schäfer and C. Lennartz, QUASI: A general purpose implementation of the QM/MM approach and its application to problems in catalysis, THEOCHEM, 2003, 632, 1–28 CrossRef CAS.
- S. Metz, J. Kästner, A. A. Sokol, T. W. Keal and P. Sherwood, ChemShell—a modular software package for QM/MM simulations, WIREs Comput. Mol. Sci., 2014, 4, 101–110 CrossRef CAS.
- Y. Lu, M. R. Farrow, P. Fayon, A. J. Logsdail, A. A. Sokol, C. R. A. Catlow, P. Sherwood and T. W. Keal, Open-Source, Python-Based Redevelopment of the ChemShell Multiscale QM/MM Environment, J. Chem. Theory Comput., 2019, 15, 1317–1328 CrossRef CAS PubMed.
- A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. B. Jensen, J. Kermode, J. R. Kitchin, E. L. Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. B. Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, The atomic simulation environment—a Python library for working with atoms, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef PubMed.
- J. Řezáč, Cuby: An integrative framework for computational chemistry, J. Comput. Chem., 2016, 37, 1230–1237 CrossRef PubMed.
- M. Rivera, M. Dommett, A. Sidat, W. Rahim and R. Crespo-Otero, fromage: A library for the study of molecular crystal excited states at the aggregate scale, J. Comput. Chem., 2020, 41, 1045–1058 CrossRef CAS PubMed.
- B. Zhang, D. Altarawy, T. Barnes, J. M. Turney and H. F. I. Schaefer, Janus: An Extensible Open-Source Software Package for Adaptive QM/MM Methods, J. Chem. Theory Comput., 2019, 15, 4362–4373 CrossRef CAS PubMed.
- E. G. Kratz, A. R. Walker, L. Lagardère, F. Lipparini, J.-P. Piquemal and G. Andrés Cisneros, LICHEM: A QM/MM program for simulations with multipolar and polarizable force fields, J. Comput. Chem., 2016, 37, 1019–1029 CrossRef CAS PubMed.
- J. M. H. Olsen, V. Bolnykh, S. Meloni, E. Ippoliti, M. P. Bircher, P. Carloni and U. Rothlisberger, MiMiC: A Novel Framework for Multiscale Modeling in Computational Chemistry, J. Chem. Theory Comput., 2019, 15, 3810–3823 CrossRef CAS PubMed.
- J. Torras, E. Deumens and S. B. Trickey, Software Integration in Multi-scale Simulations: the PUPIL System, J. Comput.-Aided Mater. Des., 2006, 13, 201–212 CrossRef CAS.
- S. Martí, QMCube (QM3): An all-purpose suite for multiscale QM/MM calculations, J. Comput. Chem., 2021, 42, 447–457 CrossRef PubMed.
- H. Lin, Y. Zhang, S. Pezeshki, A. W. Duster, B. Wang, X.-P. Wu, L. Gagliardi and D. G. Truhlar, QMMM 2018, https://comp.chem.umn.edu/qmmm/.
- P. Seeber, S. Seidenath, J. Steinmetzer and S. Gräfe, Growing Spicy ONIOMs: Extending and generalizing concepts of ONIOM and many body expansions, WIREs Comput. Mol. Sci., 2022, e1644 Search PubMed.
- J. Kästner, J. M. Carr, T. W. Keal, W. Thiel, A. Wander and P. Sherwood, DL-FIND: An Open-Source Geometry Optimizer for Atomistic Simulations, J. Phys. Chem. A, 2009, 113, 11856–11865 CrossRef PubMed.
- T. W. Keal, P. Sherwood, G. Dutta, A. A. Sokol and C. R. A. Catlow, Characterization of hydrogen dissociation over aluminium-doped zinc oxide using an efficient massively parallel framework for QM/MM calculations, Proc. R. Soc. A, 2011, 467, 1900–1924 CrossRef CAS.
- F. Neese, The ORCA program system, WIREs Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter and M. Scheffler, Ab initio molecular simulations with numeric atom-centered orbitals, Comput. Phys. Commun., 2009, 180, 2175–2196 CrossRef CAS.
- H.-J. Werner, P. J. Knowles, F. R. Manby, J. A. Black, K. Doll, A. Heßelmann, D. Kats, A. Köhn, T. Korona, D. A. Kreplin, Q. Ma, T. F. Miller, A. Mitrushchenkov, K. A. Peterson, I. Polyak, G. Rauhut and M. Sibaev, The Molpro quantum chemistry package, J. Chem. Phys., 2020, 152, 144107 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Rev. C.01, 2009 Search PubMed.
- T. D. Kühne, M. Iannuzzi, M. Del Ben, V. V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schütt, F. Schiffmann, D. Golze, J. Wilhelm, S. Chulkov, M. H. Bani-Hashemian, V. Weber, U. Borštnik, M. Taillefumier, A. S. Jakobovits, A. Lazzaro, H. Pabst, T. Müller, R. Schade, M. Guidon, S. Andermatt, N. Holmberg, G. K. Schenter, A. Hehn, A. Bussy, F. Belleflamme, G. Tabacchi, A. Glöβ, M. Lass, I. Bethune, C. J. Mundy, C. Plessl, M. Watkins, J. VandeVondele, M. Krack and J. Hutter, CP2K: An electronic structure and molecular dynamics software package - Quickstep: Efficient and accurate electronic structure calculations, J. Chem. Phys., 2020, 152, 194103 CrossRef PubMed.
- W. Thiel, MNDO2020 semiempirical electronic structure package, https://mndo.kofo.mpg.de/.
- B. Hourahine, B. Aradi, V. Blum, F. Bonafé, A. Buccheri, C. Camacho, C. Cevallos, M. Y. Deshaye, T. Dumitrică, A. Dominguez, S. Ehlert, M. Elstner, T. van der Heide, J. Hermann, S. Irle, J. J. Kranz, C. Köhler, T. Kowalczyk, T. Kubař, I. S. Lee, V. Lutsker, R. J. Maurer, S. K. Min, I. Mitchell, C. Negre, T. A. Niehaus, A. M. N. Niklasson, A. J. Page, A. Pecchia, G. Penazzi, M. P. Persson, J. Řezáč, C. G. Sánchez, M. Sternberg, M. Stöhr, F. Stuckenberg, A. Tkatchenko, V. W.-Z. Yu and T. Frauenheim, DFTB+, a software package for efficient approximate density functional theory based atomistic simulations, J. Chem. Phys., 2020, 152, 124101 CrossRef CAS PubMed.
- J. C. Phillips, D. J. Hardy, J. D. C. Maia, J. E. Stone, J. V. Ribeiro, R. C. Bernardi, R. Buch, G. Fiorin, J. Hénin, W. Jiang, R. McGreevy, M. C. R. Melo, B. K. Radak, R. D. Skeel, A. Singharoy, Y. Wang, B. Roux, A. Aksimentiev, Z. Luthey-Schulten, L. V. Kalé, K. Schulten, C. Chipot and E. Tajkhorshid, Scalable molecular dynamics on CPU and GPU architectures with NAMD, J. Chem. Phys., 2020, 153, 044130 CrossRef CAS PubMed.
- K. D. Dubey and S. Shaik, Choreography of the Reductase and P450BM3 Domains Toward Electron Transfer Is Instigated by the Substrate, J. Am. Chem. Soc., 2018, 140, 683–690 CrossRef CAS PubMed.
- V. Postils, M. Saint-André, A. Timmins, X.-X. Li, Y. Wang, J. M. Luis, M. Solà and S. P. De Visser, Quantum Mechanics/Molecular Mechanics Studies on the Relative Reactivities of Compound I and II in Cytochrome P450 Enzymes, Int. J. Mol. Sci., 2018, 19, 1974 CrossRef PubMed.
- H. Su, G. Ma and Y. Liu, Theoretical Insights into the Mechanism and Stereoselectivity of Olefin Cyclopropanation Catalyzed by Two Engineered Cytochrome P450 Enzymes, Inorg. Chem., 2018, 57, 11738–11745 CrossRef CAS PubMed.
- X. Wang, J. Shi and Y. Liu, Oxidative Rearrangement Mechanism of Pentalenolactone F Catalyzed by Cytochrome P450 CYP161C2 (PntM), Inorg. Chem., 2018, 57, 8933–8941 CrossRef CAS PubMed.
- L. Bao, W. Liu, Y. Li, X. Wang, F. Xu, Z. Yang, Y. Yue, C. Zuo, Q. Zhang and W. Wang, Carcinogenic Metabolic Activation Process of Naphthalene by the Cytochrome P450 Enzyme 1B1: A Computational Study, Chem. Res. Toxicol., 2019, 32, 603–612 Search PubMed.
- H. Su, B. Wang and S. Shaik, Quantum-Mechanical/Molecular-Mechanical Studies of CYP11A1-Catalyzed Biosynthesis of Pregnenolone from Cholesterol Reveal a C–C Bond Cleavage Reaction That Occurs by a Compound I-Mediated Electron Transfer, J. Am. Chem. Soc., 2019, 141, 20079–20088 CrossRef CAS PubMed.
- F. G. Cantú Reinhard, Y.-T. Lin, A. Stańczak and S. P. de Visser, Bioengineering of Cytochrome P450 OleTJE: How Does Substrate Positioning Affect the Product Distributions?, Molecules, 2020, 25, 2675 CrossRef PubMed.
- A. S. Chowdhury, H. S. Ali, A. S. Faponle and S. P. de Visser, How external perturbations affect the chemoselectivity of substrate activation by cytochrome P450 OleTJE, Phys. Chem. Chem. Phys., 2020, 22, 27178–27190 RSC.
- C. Hui, W. Singh, D. Quinn, C. Li, T. S. Moody and M. Huang, Regio- and stereoselectivity in the CYP450BM3-catalyzed hydroxylation of complex terpenoids: a QM/MM study, Phys. Chem. Chem. Phys., 2020, 22, 21696–21706 RSC.
- C. Wang, P. Wu, Z. Wang and B. Wang, The molecular mechanism of P450-catalyzed amination of the pyrrolidine derivative of lidocaine: insights from multiscale simulations, RSC Adv., 2021, 11, 27674–27680 RSC.
- J. Wei and Y. Liu, Mechanistic Insights into the P450 TleB-Catalyzed Unusual Intramolecular C–N Bond Formation Involved in the Biosynthesis of Indolactam V, J. Chem. Inf. Model., 2021, 61, 3638–3648 CrossRef CAS PubMed.
- X. Zhang, Y. Jiang, Q. Chen, S. Dong, Y. Feng, Z. Cong, S. Shaik and B. Wang, H-Bonding Networks Dictate the Molecular Mechanism of H2O2 Activation by P450, ACS Catal., 2021, 11, 8774–8785 CrossRef CAS.
- S. Kalita, S. Shaik and K. D. Dubey, Mechanistic Conundrum of C–C Bond Cleavage by CYP51, ACS Catal., 2022, 12, 5673–5683 CrossRef CAS.
- W. Singh, S. F. G. Santos, P. James, G. W. Black, M. Huang and K. D. Dubey, Single-Site Mutation Induces Water-Mediated Promiscuity in Lignin Breaking Cytochrome P450GcoA, ACS Omega, 2022, 7, 21109–21118 CrossRef CAS PubMed.
- S. Yadav, V. Kardam, A. Tripathi, T. G. Shruti and K. D. Dubey, The Performance of Different Water Models on the Structure and Function of Cytochrome P450 Enzymes, J. Chem. Inf. Model., 2022, 62, 6679–6690 CrossRef CAS PubMed.
- J. Zhou, X. Zhang, Y. Li, S. Feng, Q. Zhang and W. Wang, Endocrine-disrupting metabolic activation of 2-nitrofluorene catalyzed by human cytochrome P450 1A1: A QM/MM approach, Environ. Int., 2022, 166, 107355 CrossRef CAS PubMed.
- S. Kalita, S. Shaik, H. K. Kisan and K. D. Dubey, A Paradigm Shift in the Catalytic Cycle of P450: The Preparatory Choreography during O2 Binding and Origins of the Necessity for Two Protonation Pathways, ACS Catal., 2020, 10, 11481–11492 CrossRef CAS.
- S. Kalita, S. Shaik and K. D. Dubey, MD simulations and QM/MM calculations reveal the key mechanistic elements which are responsible for the efficient C–H amination reaction performed by a bioengineered P450 enzyme, Chem. Sci., 2021, 12, 14507–14518 RSC.
- Q. Lu, J. Song, P. Wu, C. Li and W. Thiel, Mechanistic Insights into the Directing Effect of Thr303 in Ethanol Oxidation by Cytochrome P450 2E1, ACS Catal., 2019, 9, 4892–4901 CrossRef CAS.
- S. Zhang and Y. Liu, Mechanical Insights into the Enzymatic Cleavage of Double C–C Bond in Poly(cis-1,4-isoprene) by the Latex Clearing Protein, Inorg. Chem., 2020, 59, 9627–9637 CrossRef CAS PubMed.
- L. Yan, Y. Wang, S. Zhang, X. Li, J. Wei, Z. Wang and Y. Liu, Inactivation Mechanism of Neuronal Nitric Oxide Synthase by (S)-2-Amino-5-(2-(methylthio)acetimidamido)pentanoic Acid: Chemical Conversion of the Inactivator in the Active Site, Inorg. Chem., 2021, 60, 9345–9358 CrossRef CAS PubMed.
- G. Tian, G. Hao, X. Chen and Y. Liu, Tyrosyl Radical-Mediated Sequential Oxidative Decarboxylation of Coproporphyrinogen III through PCET: Theoretical Insights into the Mechanism of Coproheme Decarboxylase ChdC, Inorg. Chem., 2021, 60, 13539–13549 CrossRef CAS PubMed.
- B. Wang, I. Fita and C. Rovira, Theory Uncovers the Role of the Methionine–Tyrosine–Tryptophan Radical Adduct in the Catalase Reaction of KatGs: O2 Release Mediated by Proton-Coupled Electron Transfer, Chem. – Eur. J., 2018, 24, 5388–5395 CrossRef CAS PubMed.
- J. Chauhan, S. R. Maddi, K. D. Dubey and S. Sen, Developing C2-Aroyl Indoles as Novel Inhibitors of IDO1 and Understanding Their Mechanism of Inhibition via Mass Spectroscopy, QM/MM Calculations and Molecular Dynamics Simulation, Front. Chem., 2021, 9, 691319 CrossRef CAS PubMed.
- V. Sosa Alfaro, S. O. Waheed, H. Palomino, A. Knorrscheidt, M. Weissenborn, C. Z. Christov and N. Lehnert, YfeX – A New Platform for Carbene Transferase Development with High Intrinsic Reactivity, Chem. – Eur. J., 2022, 28, e202201474 CrossRef CAS PubMed.
- Y. Li, X. Wang, R. Zhang, J. Wang, Z. Yang, L. Du, X. Tang, Q. Zhang and W. Wang, Computational Evidence for the Enzymatic Transformation of 2-Hydroxypropylphosphonate to Methylphosphonate, ACS Earth Space Chem., 2018, 2, 888–894 CrossRef CAS.
- P. Saura, I. Kaganer, D. Heydeck, J. M. Lluch, H. Kühn and À. González-Lafont, Mutagenesis of Sequence Determinants of Truncated Porcine ALOX15 Induces Changes in the Reaction Specificity by Altering the Catalytic Mechanism of Initial Hydrogen Abstraction, Chem. – Eur. J., 2018, 24, 962–973 CrossRef CAS PubMed.
- A. Timmins, N. J. Fowler, J. Warwicker, G. D. Straganz and S. P. de Visser, Does Substrate Positioning Affect the Selectivity and Reactivity in the Hectochlorin Biosynthesis Halogenase?, Front. Chem., 2018, 6, 513 CrossRef CAS PubMed.
- J. Bai, Q. Hou, W. Zhu and Y. Liu, Mechanical insights into the oxidative cleavage of resveratrol catalyzed by dioxygenase NOV1 from Novosphingobium aromaticivorans: confirmation of dioxygenase mechanism by QM/MM calculations, Catal. Sci. Technol., 2019, 9, 444–455 RSC.
- S. Li, J. Lu and W. Lai, Mechanistic insights into ring cleavage of hydroquinone by PnpCD from quantum mechanical/molecular mechanical calculations, Org. Biomol. Chem., 2019, 17, 8194–8205 RSC.
- J. Lu and W. Lai, Mechanistic Insights into a Stibene Cleavage Oxygenase NOV1 from Quantum Mechanical/Molecular Mechanical Calculations, ChemistryOpen, 2019, 8, 228–235 CrossRef CAS PubMed.
- S. Zhang and Y. Liu, Mechanism of fatty acid decarboxylation catalyzed by a non-heme iron oxidase (UndA): a QM/MM study, Org. Biomol. Chem., 2019, 17, 9808–9818 RSC.
- I. Ivanov, A. B. Golovanov, C. Ferretti, M. Canyelles-Niño, D. Heydeck, S. Stehling, J. M. Lluch, À. González-Lafont and H. Kühn, Mutations of Triad Determinants Changes the Substrate Alignment at the Catalytic Center of Human ALOX5, ACS Chem. Biol., 2019, 14, 2768–2782 CrossRef CAS PubMed.
- X. Li, W. Zhu and Y. Liu, Mechanistic Insights into the Oxidative Rearrangement Catalyzed by the Unprecedented Dioxygenase ChaP Involved in Chartreusin Biosynthesis, Inorg. Chem., 2020, 59, 13988–13999 CrossRef CAS PubMed.
- J. Lu, B. Wang, S. Shaik and W. Lai, QM/MM Calculations Reveal the Important Role of α-Heteroatom Substituents in Controlling Selectivity of Mononuclear Nonheme HppE-Catalyzed Reactions, ACS Catal., 2020, 10, 9521–9532 CrossRef CAS.
- A. Cruz, À. González-Lafont and J. M. Lluch, Deciphering the Molecular Details of the Lipoxin Formation Mechanism in the 5(S),15(S)-DiHpETE Biosynthetic Pathway Catalyzed by Reticulocyte 15-Lipoxygenase-1, J. Phys. Chem. B, 2020, 124, 11406–11418 CrossRef CAS PubMed.
- Y. Wang, L. Yan, X. Li, S. Zhang, J. Wei and Y. Liu, Formation Mechanism of Cofactor Cys–Tyr in the Cysteine Dioxygenases (CDO and F2-CDO) and Its Influence on Catalysis: A QM/MM Study, Inorg. Chem., 2021, 60, 7844–7856 CrossRef CAS PubMed.
- Y. Liu and Y. Liu, Computational Study of Aromatic Hydroxylation Catalyzed by the Iron-Dependent Hydroxylase PqqB Involved in the Biosynthesis of Redox Cofactor Pyrroloquinoline Quinone, Inorg. Chem., 2022, 61, 5943–5956 CrossRef CAS PubMed.
- G. Tian, H. Su and Y. Liu, Mechanism of Sulfoxidation and C–S Bond Formation Involved in the Biosynthesis of Ergothioneine Catalyzed by Ergothioneine Synthase (EgtB), ACS Catal., 2018, 8, 5875–5889 CrossRef CAS.
- H. Li, W. Zhu and Y. Liu, Mechanism of Uncoupled Carbocyclization and Epimerization Catalyzed by Two Non-Heme Iron/α-Ketoglutarate Dependent Enzymes, J. Chem. Inf. Model., 2019, 59, 5086–5098 CrossRef CAS PubMed.
- J. Xue, J. Lu and W. Lai, Mechanistic insights into a non-heme 2-oxoglutarate-dependent ethylene-forming enzyme: selectivity of ethylene-formation versusL-Arg hydroxylation, Phys. Chem. Chem. Phys., 2019, 21, 9957–9968 RSC.
- L. Yan and Y. Liu, Insights into the Mechanism and Enantioselectivity in the Biosynthesis of Ergot Alkaloid Cycloclavine Catalyzed by Aj_EasH from Aspergillus japonicus, Inorg. Chem., 2019, 58, 13771–13781 CrossRef CAS PubMed.
- J. Bai, L. Yan and Y. Liu, Catalytic mechanism of the PrhA (V150L/A232S) double mutant involved in the fungal meroterpenoid biosynthetic pathway: a QM/MM study, Phys. Chem. Chem. Phys., 2019, 21, 25658–25668 RSC.
- H. Li and Y. Liu, Mechanistic Investigation of Isonitrile Formation Catalyzed by the Nonheme Iron/α-KG-Dependent Decarboxylase (ScoE), ACS Catal., 2020, 10, 2942–2957 CrossRef CAS.
- R. Ramanan, S. S. Chaturvedi, N. Lehnert, C. J. Schofield, T. G. Karabencheva-Christova and C. Z. Christov, Catalysis by the JmjC histone demethylase KDM4A integrates substrate dynamics, correlated motions and molecular orbital control, Chem. Sci., 2020, 11, 9950–9961 RSC.
- X. Zhang, Z. Wang, J. Gao and W. Liu, Chlorination versus hydroxylation selectivity mediated by the non-heme iron halogenase WelO5, Phys. Chem. Chem. Phys., 2020, 22, 8699–8712 RSC.
- Y. Liu, J. Shi and Y. Liu, Mechanistic Insights into the Oxidative Ring Expansion from Penicillin N to Deacetoxycephalosporin C Catalyzed by a Nonheme Iron(II) and α-KG-Dependent Oxygenase, Inorg. Chem., 2020, 59, 12218–12231 CrossRef CAS PubMed.
- W. Singh, C. Hui, C. Li and M. Huang, Thebaine is Selectively Demethylated by Thebaine 6-O-Demethylase and Codeine-3-O-demethylase at Distinct Binding Sites: A Computational Study, Inorg. Chem., 2021, 60, 10199–10214 CrossRef CAS PubMed.
- S. S. Chaturvedi, R. Ramanan, J. Hu, R. P. Hausinger and C. Z. Christov, Atomic and Electronic Structure Determinants Distinguish between Ethylene Formation and l-Arginine Hydroxylation Reaction Mechanisms in the Ethylene-Forming Enzyme, ACS Catal., 2021, 11, 1578–1592 CrossRef CAS.
- S. S. Chaturvedi, S. B. Jaber Sathik Rifayee, S. O. Waheed, J. Wildey, C. Warner, C. J. Schofield, T. G. Karabencheva-Christova and C. Z. Christov, Can Second Coordination Sphere and Long-Range Interactions Modulate Hydrogen Atom Transfer in a Non-Heme Fe(II)-Dependent Histone Demethylase?, JACS Au, 2022, 2, 2169–2186 CrossRef CAS PubMed.
- T.-Y. Chen, Z. Zheng, X. Zhang, J. Chen, L. Cha, Y. Tang, Y. Guo, J. Zhou, B. Wang, H. Liu and W. Chang, Deciphering the Reaction Pathway of Mononuclear Iron Enzyme-Catalyzed N ≡ C Triple Bond Formation in Isocyanide Lipopeptide and Polyketide Biosynthesis, ACS Catal., 2022, 12, 2270–2279 CrossRef CAS PubMed.
- S. S. Chaturvedi, R. Ramanan, N. Lehnert, C. J. Schofield, T. G. Karabencheva-Christova and C. Z. Christov, Catalysis by the Non-Heme Iron(II) Histone Demethylase PHF8 Involves Iron Center Rearrangement and Conformational Modulation of Substrate Orientation, ACS Catal., 2020, 10, 1195–1209 CrossRef CAS PubMed.
- S. Zhang, X. Li, Y. Wang, L. Yan, J. Wei and Y. Liu, Computational Study of the C5-Hydroxylation Mechanism Catalyzed by the Diiron Monooxygenase PtmU3 as Part of the Platensimycin Biosynthesis, Inorg. Chem., 2021, 60, 17783–17796 CrossRef CAS PubMed.
- J. Liu, P. Wu, S. Yan, Y. Li, Z. Cao and B. Wang, Spin-Regulated Inner-Sphere Electron Transfer Enables Efficient O—O Bond Activation in Nonheme Diiron Monooxygenase MIOX, ACS Catal., 2021, 11, 6141–6152 CrossRef CAS.
- Y. Wang, L. Dong, H. Su and Y. Liu, Dioxygen Activation and Nδ,Nε-Dihydroxylation Mechanism Involved in the Formation of N-Nitrosourea Pharmacophore in Streptozotocin Catalyzed by Nonheme Diiron Enzyme SznF, Inorg. Chem., 2022, 61, 15721–15734 CrossRef CAS PubMed.
- T.-P. Zhou, W.-H. Deng, Y. Wu and R.-Z. Liao, QM/MM Calculations Suggest Concerted O−O Bond Cleavage and Substrate Oxidation by Nonheme Diiron Toluene/o-Xylene Monooxygenase, Chem. – Asian J., 2022, 17, e202200490 CAS.
- X. Song, J. Liu and B. Wang, Emergence of Function from Nonheme Diiron Oxygenases: A Quantum Mechanical/Molecular Mechanical Study of Oxygen Activation and Organophosphonate Catabolism Mechanisms by PhnZ, ACS Catal., 2022, 12, 2009–2022 CrossRef CAS.
- A. M. Escorcia and M. Stein, QM/MM Investigation of the Role of a Second Coordination Shell Arginine in [NiFe]-Hydrogenases, Front. Chem., 2018, 6, 164 CrossRef PubMed.
- S. Moubarak, N. Elghobashi-Meinhardt, D. Tombolelli and M. A. Mroginski, Probing the Structure of [NiFeSe] Hydrogenase with QM/MM Computations, Appl. Sci., 2020, 10, 781 CrossRef CAS.
- J. Feng and B. Wang, Super-exchange and exchange-enhanced reactivity in Fe4S4-mediated activation of SAM by radical SAM enzymes, Chin. J. Chem. Phys., 2021, 34, 532–540 CrossRef CAS.
- J. Feng, S. Shaik and B. Wang, Spin-Regulated Electron Transfer and Exchange-Enhanced Reactivity in Fe4S4-Mediated Redox Reaction of the Dph2 Enzyme During the Biosynthesis of Diphthamide, Angew. Chem., Int. Ed., 2021, 60, 20430–20436 CrossRef CAS PubMed.
- N. Elghobashi-Meinhardt, D. Tombolelli and M. A. Mroginski, Electronic and Structural Properties of the Double Cubane Iron-Sulfur Cluster, Catalysts, 2021, 11, 245 CrossRef CAS.
- C. V. Stappen, A. T. Thorhallsson, L. Decamps, R. Bjornsson and S. DeBeer, Resolving the structure of the E1 state of Mo nitrogenase through Mo and Fe K-edge EXAFS and QM/MM calculations, Chem. Sci., 2019, 10, 9807–9821 RSC.
- A. T. Thorhallsson, B. Benediktsson and R. Bjornsson, A model for dinitrogen binding in the E4 state of nitrogenase, Chem. Sci., 2019, 10, 11110–11124 RSC.
- N. Spiller, R. Bjornsson, S. DeBeer and F. Neese, Carbon Monoxide Binding to the Iron–Molybdenum Cofactor of Nitrogenase: a Detailed Quantum Mechanics/Molecular Mechanics Investigation, Inorg. Chem., 2021, 60, 18031–18047 CrossRef CAS PubMed.
- B. Benediktsson and R. Bjornsson, Quantum Mechanics/Molecular Mechanics Study of Resting-State Vanadium Nitrogenase: Molecular and Electronic Structure of the Iron–Vanadium Cofactor, Inorg. Chem., 2020, 59, 11514–11527 CrossRef CAS PubMed.
- A. T. Thorhallsson and R. Bjornsson, The E2 state of FeMoco: Hydride Formation versus Fe Reduction and a Mechanism for H2 Evolution, Chem. – Eur. J., 2021, 27, 16788–16800 CrossRef CAS PubMed.
- B. Benediktsson and R. Bjornsson, Analysis of the Geometric and Electronic Structure of Spin-Coupled Iron–Sulfur Dimers with Broken-Symmetry DFT: Implications for FeMoco, J. Chem. Theory Comput., 2022, 18, 1437–1457 CrossRef CAS PubMed.
- H. Jiang and W. Lai, Monophenolase and catecholase activity of Aspergillus oryzae catechol oxidase: insights from hybrid QM/MM calculations, Org. Biomol. Chem., 2020, 18, 5192–5202 RSC.
- P. Wu, F. Fan, J. Song, W. Peng, J. Liu, C. Li, Z. Cao and B. Wang, Theory Demonstrated a “Coupled” Mechanism for O2 Activation and Substrate Hydroxylation by Binuclear Copper Monooxygenases, J. Am. Chem. Soc., 2019, 141, 19776–19789 CrossRef CAS PubMed.
- K. Sen, M. A. Hough, R. W. Strange, C. W. Yong and T. W. Keal, A QM/MM Study of Nitrite Binding Modes in a Three-Domain Heme-Cu Nitrite Reductase, Molecules, 2018, 23, 2997 CrossRef PubMed.
- K. Sen, M. A. Hough, R. W. Strange, C. Yong and T. W. Keal, QM/MM Simulations of Protein Crystal Reactivity Guided by MSOX Crystallography: A Copper Nitrite Reductase Case Study, J. Phys. Chem. B, 2021, 125, 9102–9114 CrossRef CAS PubMed.
- J. P. M. van Rijn, A. M. Escorcia and W. Thiel, QM/MM study of the taxadiene synthase mechanism, J. Comput. Chem., 2019, 40, 1902–1910 CrossRef CAS PubMed.
- A. Tripathi and K. Dutta Dubey, Combined MD and QM/MM Calculations Reveal Allostery-Driven Promiscuity in Dipeptide Epimerases of Enolase Family, Chem. – Asian J., 2022, 17, e202200528 CAS.
- L. Yan and Y. Liu, The Retaining Mechanism of Xylose Transfer Catalyzed by Xyloside α-1,3-Xylosyltransferase (XXYLT1): a Quantum Mechanics/Molecular Mechanics Study, J. Chem. Inf. Model., 2020, 60, 1585–1594 CrossRef CAS PubMed.
- H. Li, X. Wang, G. Tian and Y. Liu, Insights into the dioxygen activation and catalytic mechanism of the nickel-containing quercetinase, Catal. Sci. Technol., 2018, 8, 2340–2351 RSC.
- W.-J. Wang, W.-J. Wei and R.-Z. Liao, Deciphering the chemoselectivity of nickel-dependent quercetin 2,4-dioxygenase, Phys. Chem. Chem. Phys., 2018, 20, 15784–15794 RSC.
- H.-X. Qian and R.-Z. Liao, QM/MM Study of Tungsten-Dependent Benzoyl-Coenzyme A Reductase: Rationalization of Regioselectivity and Predication of W vs Mo Selectivity, Inorg. Chem., 2018, 57, 10667–10678 CrossRef CAS PubMed.
- M. Canyelles-Niño, À. González-Lafont and J. M. Lluch, Theoretical Characterization of the Step-by-Step Mechanism of Conversion of Leukotriene A4 to Leukotriene B4 Catalysed by the Enzyme Leukotriene A4 Hydrolase, Int. J. Mol. Sci., 2022, 23, 3140 CrossRef PubMed.
- A. Varghese, S. S. Chaturvedi, B. DiCastri, E. Mehler, G. B. Fields and T. G. Karabencheva-Christova, Effects of the Nature of Metal Ion, Protein and Substrate on the Catalytic Center in Matrix Metalloproteinase-1: Insights from Multilevel MD, QM/MM and QM Studies, ChemPhysChem, 2022, 23, e202100680 CrossRef CAS.
- B. Chen, Z. Kang, E. Zheng, Y. Liu, J. W. Gauld and Q. Wang, Hydrolysis Mechanism of the Linkers by Matrix Metalloproteinase-9 Using QM/MM Calculations, J. Chem. Inf. Model., 2021, 61, 5203–5211 CrossRef CAS PubMed.
- G. Zhao, W. Peng, K. Song, J. Shi, X. Lu, B. Wang and Y.-L. Du, Molecular basis of enzymatic nitrogen-nitrogen formation by a family of zinc-binding cupin enzymes, Nat. Commun., 2021, 12, 7205 CrossRef CAS PubMed.
- B. Ling, X. Wang, H. Su, R. Liu and Y. Liu, Protonation state and fine structure of the active site determine the reactivity of dehydratase: hydration and isomerization of β-myrcene catalyzed by linalool dehydratase/isomerase from Castellaniella defragrans, Phys. Chem. Chem. Phys., 2018, 20, 17342–17352 RSC.
- N. Mohamed-Raseek, H. D. Duan, P. Hildebrandt, M. A. Mroginski and A.-F. Miller, Spectroscopic, thermodynamic and computational evidence of the locations of the FADs in the nitrogen fixation-associated electron transfer flavoprotein, Chem. Sci., 2019, 10, 7762–7772 RSC.
- S. Romero-Téllez, J. M. Lluch, À. González-Lafont and L. Masgrau, Comparing Hydrolysis and Transglycosylation Reactions Catalyzed by Thermus thermophilus β-Glycosidase. A Combined MD and QM/MM Study, Front. Chem., 2019, 7, 200 CrossRef PubMed.
- B. Ling, H. Li, L. Yan, R. Liu and Y. Liu, Conversion mechanism of enoyl thioesters into acyl thioesters catalyzed by 2-enoyl-thioester reductases from Candida Tropicalis, Phys. Chem. Chem. Phys., 2019, 21, 10105–10113 RSC.
- W.-H. Deng, Y. Lu and R.-Z. Liao, Revealing the Mechanism of Isethionate Sulfite-Lyase by QM/MM Calculations, J. Chem. Inf. Model., 2021, 61, 5871–5882 CrossRef CAS PubMed.
- K. D. Dubey and W. Singh, Simulations reveal the key role of Arg15 in the promiscuous activity in the HisA enzyme, Org. Biomol. Chem., 2021, 19, 10652–10661 RSC.
- S. Miranda-Rojas, K. Blanco-Esperguez, I. Tuñón, J. Kästner and F. Mendizábal, Exploration of the Activation Mechanism of the Epigenetic Regulator MLL3: A QM/MM Study, Biomolecules, 2021, 11, 1051 CrossRef CAS PubMed.
- K. Blanco-Esperguez, I. Tuñón, J. Kästner, F. Mendizábal and S. Miranda-Rojas, Unraveling the Role of the Tyrosine Tetrad from the Binding Site of the Epigenetic Writer MLL3 in the Catalytic Mechanism and Methylation Multiplicity, Int. J. Mol. Sci., 2022, 23, 10339 CrossRef CAS PubMed.
- Y. Wang, X. Li, J. Wei, X. Zhang and Y. Liu, Mechanism of Sugar Ring Contraction and Closure Catalyzed by UDP-d-apiose/UDP-d-xylose Synthase (UAXS), J. Chem. Inf. Model., 2022, 62, 632–646 CrossRef CAS PubMed.
- F. G. Cantú Reinhard, J. L. DuBois and S. P. de Visser, Catalytic Mechanism of Nogalamycin Monoxygenase: How Does Nature Synthesize Antibiotics without a Metal Cofactor?, J. Phys. Chem. B, 2018, 122, 10841–10854 CrossRef PubMed.
- A. Cebrián-Prats, À. González-Lafont and J. M. Lluch, Unraveling the Molecular Details of the Complete Mechanism That Governs the Synthesis of Prostaglandin G2 Catalyzed by Cyclooxygenase-2, ACS Omega, 2019, 4, 2063–2074 CrossRef.
- A. Cebrián-Prats, À. González-Lafont and J. M. Lluch, Understanding the Molecular Details of the Mechanism That Governs the Oxidation of Arachidonic Acid Catalyzed by Aspirin-Acetylated Cyclooxygenase-2, ACS Catal., 2020, 10, 138–153 CrossRef.
- A. Suñer-Rubio, A. Cebrián-Prats, À. González-Lafont and J. M. Lluch, Unraveling how the Gly526Ser mutation arrests prostaglandin formation from arachidonic acid catalyzed by cyclooxygenase-2: a combined molecular dynamics and QM/MM study, RSC Adv., 2020, 10, 986–997 RSC.
- S. Zhang, X. Li, Y. Wang, J. Wei, X. Zhang and Y. Liu, Computational Study of the Peroxygenase Mechanism Catalyzed by Hemoglobin Dehaloperoxidase Involved in the Degradation of Chlorophenols, Inorg. Chem., 2022, 61, 2628–2639 CrossRef CAS PubMed.
- Y. Fu, Y. Zhang, F. Fan, B. Wang and Z. Cao, Degradation of pesticides diazinon and diazoxon by phosphotriesterase: insight into divergent mechanisms from QM/MM and MD simulations, Phys. Chem. Chem. Phys., 2022, 24, 687–696 RSC.
- Y. Fu, F. Fan, B. Wang and Z. Cao, Water-Regulated Mechanisms for Degradation of Pesticides Paraoxon and Parathion by Phosphotriesterase: Insight from QM/MM and MD Simulations, Chem. – Asian J., 2022, 17, e202200439 CAS.
- S. Feng, Y. Yue, J. Chen, J. Zhou, Y. Li and Q. Zhang, Biodegradation mechanism of polycaprolactone by a novel esterase MGS0156: a QM/MM approach, Environ. Sci.: Processes Impacts, 2020, 22, 2332–2344 RSC.
- M. Zheng, Y. Li, W. Dong, W. Zhang, S. Feng, Q. Zhang and W. Wang, Depolymerase-Catalyzed Polyethylene Terephthalate Hydrolysis: A Unified Mechanism Revealed by Quantum Mechanics/Molecular Mechanics Analysis, ACS Sustainable Chem. Eng., 2022, 10, 7341–7348 CrossRef CAS.
- J. Wang, X. Tang, Y. Li, R. Zhang, L. Zhu, J. Chen, Y. Sun, Q. Zhang and W. Wang, Computational evidence for the degradation mechanism of haloalkane dehalogenase LinB and mutants of Leu248 to 1-chlorobutane, Phys. Chem. Chem. Phys., 2018, 20, 20540–20547 RSC.
- R. Zhang, T. Zhuang, Q. Zhang and W. Wang, Effect of F, Cl, Br and I substitution on the BphB enzyme for the degradation of halogenated biphenyls, revealed by quantum and molecular mechanics, Environ. Chem. Lett., 2019, 17, 1167–1173 CrossRef CAS.
- Y. Li, Y. Yue, H. Zhang, Z. Yang, H. Wang, S. Tian, J. Wang, Q. Zhang and W. Wang, Harnessing fluoroacetate dehalogenase for defluorination of fluorocarboxylic acids: in silico and in vitro approach, Environ. Int., 2019, 131, 104999 CrossRef CAS PubMed.
- J. Wang, J. Chen, X. Tang, Y. Li, R. Zhang, L. Zhu, Y. Sun, Q. Zhang and W. Wang, Catalytic Mechanism for 2,3-Dihydroxybiphenyl Ring Cleavage by Nonheme Extradiol Dioxygenases BphC: Insights from QM/MM Analysis, J. Phys. Chem. B, 2019, 123, 2244–2253 CrossRef CAS PubMed.
- J. Wang, X. Tang, Y. Zhang, Y. Li, L. Zhu, Q. Zhang and W. Wang, How to complete the tautomerization and substrate-assisted activation prior to C–C bond fission by meta-cleavage product hydrolase LigY?, Catal. Sci. Technol., 2020, 10, 5856–5869 RSC.
- Y. Yue, J. Fan, G. Xin, Q. Huang, J. Wang, Y. Li, Q. Zhang and W. Wang, Comprehensive Understanding of Fluoroacetate Dehalogenase-Catalyzed Degradation of Fluorocarboxylic Acids: A QM/MM Approach, Environ. Sci. Technol., 2021, 55, 9817–9825 CrossRef CAS PubMed.
- J. Wei, Y. Wang, X. Li, X. Zhang and Y. Liu, Mechanistic Insights into Pyridine Ring Degradation Catalyzed by 2,5-Dihydroxypyridine Dioxygenase NicX, Inorg. Chem., 2022, 61, 2517–2529 CrossRef CAS PubMed.
- S. Feng, Y. Yue, M. Zheng, Y. Li, Q. Zhang and W. Wang, IsPETase- and IsMHETase-Catalyzed Cascade Degradation Mechanism toward Polyethylene Terephthalate, ACS Sustainable Chem. Eng., 2021, 9, 9823–9832 CrossRef CAS.
- T. Stuyver, J. Huang, D. Mallick, D. Danovich and S. Shaik, TITAN: A Code for Modeling and Generating Electric Fields—Features and Applications to Enzymatic Reactivity, J. Comput. Chem., 2020, 41, 74–82 CrossRef CAS PubMed.
- K. Dutta Dubey, T. Stuyver, S. Kalita and S. Shaik, Solvent Organization and Rate Regulation of a Menshutkin Reaction by Oriented External Electric Fields are Revealed by Combined MD and QM/MM Calculations, J. Am. Chem. Soc., 2020, 142, 9955–9965 CrossRef CAS PubMed.
- S. O. Waheed, S. S. Chaturvedi, T. G. Karabencheva-Christova and C. Z. Christov, Catalytic Mechanism of Human Ten-Eleven Translocation-2 (TET2) Enzyme: Effects of Conformational Changes, Electric Field, and Mutations, ACS Catal., 2021, 11, 3877–3890 CrossRef CAS.
- S. A. Siddiqui and K. D. Dubey, Can the local electric field be a descriptor of catalytic activity? A case study on chorismate mutase, Phys. Chem. Chem. Phys., 2022, 24, 1974–1981 RSC.
- S. Yadav, S. Shaik, S. A. Siddiqui, S. Kalita and K. D. Dubey, Local Electric Fields Dictate Function: The Different Product Selectivities Observed for Fatty Acid Oxidation by Two Deceptively Very Similar P450-Peroxygenases OleT and BSβ, J. Chem. Inf. Model., 2022, 62, 1025–1035 CrossRef CAS PubMed.
- R. Ramanan, S. O. Waheed, C. J. Schofield and C. Z. Christov, What Is the Catalytic Mechanism of Enzymatic Histone N-Methyl Arginine Demethylation and Can It Be Influenced by an External Electric Field?, Chem. – Eur. J., 2021, 27, 11827–11836 CrossRef CAS PubMed.
- S. Reiter, D. Keefer and R. de Vivie-Riedle, RNA Environment Is Responsible for Decreased Photostability of Uracil, J. Am. Chem. Soc., 2018, 140, 8714–8720 CrossRef CAS PubMed.
- W. Lee and S. Matsika, Role of charge transfer states into the formation of cyclobutane pyrimidine dimers in DNA, Faraday Discuss., 2019, 216, 507–519 RSC.
- Y. Zhang, J. Wang and S. Yang, Notable effect of water on excess electron attachment to aqueous DNA deoxyribonucleosides, Phys. Chem. Chem. Phys., 2019, 21, 8925–8932 RSC.
- Y. Zhang, P. Xie, S. Yang and K. Han, Ionization and Electron Attachment for Nucleobases in Water, J. Phys. Chem. B, 2019, 123, 1237–1247 CrossRef CAS PubMed.
- A. Gheorghiu, P. V. Coveney and A. A. Arabi, The influence of base pair tautomerism on single point mutations in aqueous DNA, Interface Focus, 2020, 10, 20190120 CrossRef CAS PubMed.
- S. Chandorkar, S. Raghunathan, T. Jaganade and U. D. Priyakumar, Multiscale Modeling of Wobble to Watson–Crick-Like Guanine–Uracil Tautomerization Pathways in RNA, Int. J. Mol. Sci., 2021, 22, 5411 CrossRef CAS PubMed.
- A. Gheorghiu, P. V. Coveney and A. A. Arabi, The influence of external electric fields on proton transfer tautomerism in the guanine–cytosine base pair, Phys. Chem. Chem. Phys., 2021, 23, 6252–6265 RSC.
- M. I. Sorour, A. H. Marcus and S. Matsika, Modeling the Electronic Absorption Spectra of the Indocarbocyanine Cy3, Molecules, 2022, 27, 4062 CrossRef CAS PubMed.
- E. Naydenova, J. C. B. Dietschreit and C. Ochsenfeld, Reaction Mechanism for the N-Glycosidic Bond Cleavage of 5-Formylcytosine by Thymine DNA Glycosylase, J. Phys. Chem. B, 2019, 123, 4173–4179 CrossRef CAS PubMed.
- S. O. Waheed, R. Ramanan, S. S. Chaturvedi, N. Lehnert, C. J. Schofield, C. Z. Christov and T. G. Karabencheva-Christova, Role of Structural Dynamics in Selectivity and Mechanism of Non-heme Fe(II) and 2-Oxoglutarate-Dependent Oxygenases Involved in DNA Repair, ACS Cent. Sci., 2020, 6, 795–814 CrossRef CAS PubMed.
- A. Kreppel and C. Ochsenfeld, The Enzymatic Decarboxylation Mechanism of 5-Carboxy Uracil: A Comprehensive Quantum Chemical Study, J. Chem. Theory Comput., 2021, 17, 96–104 CrossRef CAS PubMed.
- S. O. Waheed, A. Varghese, S. S. Chaturvedi, T. G. Karabencheva-Christova and C. Z. Christov, How Human TET2 Enzyme Catalyzes the Oxidation of Unnatural Cytosine Modifications in Double-Stranded DNA, ACS Catal., 2022, 12, 5327–5344 CrossRef CAS PubMed.
- E. Naydenova, S. Roßbach and C. Ochsenfeld, QM/MM Study of the Uracil DNA Glycosylase Reaction Mechanism: A Competition between Asp145 and His148, J. Chem. Theory Comput., 2019, 15, 4344–4350 CrossRef CAS PubMed.
- S. Olatunji, K. Bowen, C.-Y. Huang, D. Weichert, W. Singh, I. G. Tikhonova, E. M. Scanlan, V. Olieric and M. Caffrey, Structural basis of the membrane intramolecular transacylase reaction responsible for lyso-form lipoprotein synthesis, Nat. Commun., 2021, 12, 4254 CrossRef CAS PubMed.
- A. Meiners, S. Bäcker, I. Hadrović, C. Heid, C. Beuck, Y. B. Ruiz-Blanco, J. Mieres-Perez, M. Pörschke, J.-N. Grad, C. Vallet, D. Hoffmann, P. Bayer, E. Sánchez-García, T. Schrader and S. K. Knauer, Specific inhibition of the Survivin–CRM1 interaction by peptide-modified molecular tweezers, Nat. Commun., 2021, 12, 1505 CrossRef CAS PubMed.
- G. König, P. Sokkar, N. Pryk, S. Heinrich, D. Möller, G. Cimicata, D. Matzov, P. Dietze, W. Thiel, A. Bashan, J. E. Bandow, J. Zuegg, A. Yonath, F. Schulz and E. Sanchez-Garcia, Rational prioritization strategy allows the design of macrolide derivatives that overcome antibiotic resistance, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2113632118 CrossRef PubMed.
- D. Ehrenberg, N. Krause, M. Saita, C. Bamann, R. K. Kar, K. Hoffmann, D. Heinrich, I. Schapiro, J. Heberle and R. Schlesinger, Atomistic Insight into the Role of Threonine 127 in the Functional Mechanism of Channelrhodopsin-2, Appl. Sci., 2019, 9, 4905 CrossRef CAS.
- E. W. Hernández-Rodríguez, A. M. Escorcia, M. W. van der Kamp, A. L. Montero-Alejo and J. Caballero, Multi-scale simulation reveals that an amino acid substitution increases photosensitizing reaction inputs in Rhodopsins, J. Comput. Chem., 2020, 41, 2278–2295 CrossRef PubMed.
- M. Broser, A. Spreen, P. E. Konold, E. Peter, S. Adam, V. Borin, I. Schapiro, R. Seifert, J. T. M. Kennis, Y. A. Bernal Sierra and P. Hegemann, NeoR, a near-infrared absorbing rhodopsin, Nat. Commun., 2020, 11, 5682 CrossRef CAS PubMed.
- T. Stensitzki, S. Adam, R. Schlesinger, I. Schapiro and K. Heyne, Ultrafast Backbone Protonation in Channelrhodopsin-1 Captured by Polarization Resolved Fs Vis-pump—IR-Probe Spectroscopy and Computational Methods, Molecules, 2020, 25, 848 CrossRef CAS PubMed.
- P. Skopintsev, D. Ehrenberg, T. Weinert, D. James, R. K. Kar, P. J. M. Johnson, D. Ozerov, A. Furrer, I. Martiel, F. Dworkowski, K. Nass, G. Knopp, C. Cirelli, C. Arrell, D. Gashi, S. Mous, M. Wranik, T. Gruhl, D. Kekilli, S. Brünle, X. Deupi, G. F. X. Schertler, R. M. Benoit, V. Panneels, P. Nogly, I. Schapiro, C. Milne, J. Heberle and J. Standfuss, Femtosecond-to-millisecond structural changes in a light-driven sodium pump, Nature, 2020, 583, 314–318 CrossRef CAS PubMed.
- J. F. Bada Juarez, P. J. Judge, S. Adam, D. Axford, J. Vinals, J. Birch, T. O. C. Kwan, K. K. Hoi, H.-Y. Yen, A. Vial, P.-E. Milhiet, C. V. Robinson, I. Schapiro, I. Moraes and A. Watts, Structures of the archaerhodopsin-3 transporter reveal that disordering of internal water networks underpins receptor sensitization, Nat. Commun., 2021, 12, 629 CrossRef CAS PubMed.
- A. Rozenberg, I. Kaczmarczyk, D. Matzov, J. Vierock, T. Nagata, M. Sugiura, K. Katayama, Y. Kawasaki, M. Konno, Y. Nagasaka, M. Aoyama, I. Das, E. Pahima, J. Church, S. Adam, V. A. Borin, A. Chazan, S. Augustin, J. Wietek, J. Dine, Y. Peleg, A. Kawanabe, Y. Fujiwara, O. Yizhar, M. Sheves, I. Schapiro, Y. Furutani, H. Kandori, K. Inoue, P. Hegemann, O. Béjà and M. Shalev-Benami, Rhodopsin-bestrophin fusion proteins from unicellular algae form gigantic pentameric ion channels, Nat. Struct. Mol. Biol., 2022, 29, 592–603 CrossRef CAS PubMed.
- S. Mous, G. Gotthard, D. Ehrenberg, S. Sen, T. Weinert, P. J. M. Johnson, D. James, K. Nass, A. Furrer, D. Kekilli, P. Ma, S. Brünle, C. M. Casadei, I. Martiel, F. Dworkowski, D. Gashi, P. Skopintsev, M. Wranik, G. Knopp, E. Panepucci, V. Panneels, C. Cirelli, D. Ozerov, G. F. X. Schertler, M. Wang, C. Milne, J. Standfuss, I. Schapiro, J. Heberle and P. Nogly, Dynamics and mechanism of a light-driven chloride pump, Science, 2022, 375, 845–851 CrossRef CAS PubMed.
- A. Sirohiwal, F. Neese and D. A. Pantazis, Protein Matrix Control of Reaction Center Excitation in Photosystem II, J. Am. Chem. Soc., 2020, 142, 18174–18190 CrossRef CAS PubMed.
- A. Sirohiwal, F. Neese and D. A. Pantazis, How Can We Predict Accurate Electrochromic Shifts for Biochromophores? A Case Study on the Photosynthetic Reaction Center, J. Chem. Theory Comput., 2021, 17, 1858–1873 CrossRef CAS PubMed.
- A. Sirohiwal, F. Neese and D. A. Pantazis, Chlorophyll excitation energies and structural stability of the CP47 antenna of photosystem II: a case study in the first-principles simulation of light-harvesting complexes, Chem. Sci., 2021, 12, 4463–4476 RSC.
- B.-W. Ding, E. V. Eremeeva, E. S. Vysotski and Y.-J. Liu, Luminescence Activity Decreases When v-coelenterazine Replaces Coelenterazine in Calcium-Regulated Photoprotein—A Theoretical and Experimental Study, Photochem. Photobiol., 2020, 96, 1047–1060 CrossRef CAS PubMed.
- P. Armengol, L. Spörkel, R. Gelabert, M. Moreno, W. Thiel and J. M. Lluch, Ultrafast action chemistry in slow motion: atomistic description of the excitation and fluorescence processes in an archetypal fluorescent protein, Phys. Chem. Chem. Phys., 2018, 20, 11067–11080 RSC.
- W. Lee, I. Kim and Y. M. Rhee, A proton transfer network that generates deprotonated tyrosine is a key to producing reactive oxygen species in phototoxic KillerRed protein, Phys. Chem. Chem. Phys., 2018, 20, 22342–22350 RSC.
- M. Fernandez Lopez, A. D. Nguyen, F. Velazquez Escobar, R. González, N. Michael, Ż. Nogacz, P. Piwowarski, F. Bartl, F. Siebert, I. Heise, P. Scheerer, W. Gärtner, M. A. Mroginski and P. Hildebrandt, Role of the Propionic Side Chains for the Photoconversion of Bacterial Phytochromes, Biochemistry, 2019, 58, 3504–3519 CrossRef CAS PubMed.
- Y. Hontani, M. Baloban, F. V. Escobar, S. A. Jansen, D. M. Shcherbakova, J. Weißenborn, M. Kloz, M. A. Mroginski, V. V. Verkhusha and J. T. M. Kennis, Real-time observation of tetrapyrrole binding to an engineered bacterial phytochrome, Commun. Chem., 2021, 4, 1–11 CrossRef PubMed.
- C. Wiebeler and I. Schapiro, QM/MM Benchmarking of Cyanobacteriochrome Slr1393g3 Absorption Spectra, Molecules, 2019, 24, 1720 CrossRef CAS PubMed.
- S. G. Sokolovski, E. A. Zherebtsov, R. K. Kar, D. Golonka, R. Stabel, N. B. Chichkov, A. Gorodetsky, I. Schapiro, A. Möglich and E. U. Rafailov, Two-photon conversion of a bacterial phytochrome, Biophys. J., 2021, 120, 964–974 CrossRef CAS PubMed.
- S. Altmayer, L. Köhler, P. Bielytskyi, W. Gärtner, J. Matysik, C. Wiebeler and C. Song, Light- and pH-dependent structural changes in cyanobacteriochrome AnPixJg2, Photochem. Photobiol. Sci., 2022, 21, 447–469 CrossRef CAS PubMed.
- M. Paulikat, R. A. Mata and R. Gelabert, A high-throughput computational approach to UV-Vis spectra in protein mutants, Phys. Chem. Chem. Phys., 2019, 21, 20678–20692 RSC.
- G. Bistoni, I. Polyak, M. Sparta, W. Thiel and F. Neese, Toward Accurate QM/MM Reaction Barriers with Large QM Regions Using Domain Based Pair Natural Orbital Coupled Cluster Theory, J. Chem. Theory Comput., 2018, 14, 3524–3531 CrossRef CAS PubMed.
- B. Nutho, A. J. Mulholland and T. Rungrotmongkol, Quantum Mechanics/Molecular Mechanics (QM/MM) Calculations Support a Concerted Reaction Mechanism for the Zika Virus NS2B/NS3 Serine Protease with Its Substrate, J. Phys. Chem. B, 2019, 123, 2889–2903 CrossRef CAS PubMed.
- B. Nutho, A. J. Mulholland and T. Rungrotmongkol, The reaction mechanism of Zika virus NS2B/NS3 serine protease inhibition by dipeptidyl aldehyde: a QM/MM study, Phys. Chem. Chem. Phys., 2019, 21, 14945–14956 RSC.
- O. A. Douglas-Gallardo, J. A. Murillo-López, J. Oller, A. J. Mulholland and E. Vöhringer-Martinez, Carbon Dioxide Fixation in RuBisCO Is Protonation-State-Dependent and Irreversible, ACS Catal., 2022, 12, 9418–9429 CrossRef CAS.
- C. Zheng, M. Schneider, A. Marion and I. Antes, The Q41R mutation in the HCV-protease enhances the reactivity towards MAVS by suppressing non-reactive pathways, Phys. Chem. Chem. Phys., 2022, 24, 2126–2138 RSC.
- I. Schlichting, J. Berendzen, K. Chu, A. M. Stock, S. A. Maves, D. E. Benson, R. M. Sweet, D. Ringe, G. A. Petsko and S. G. Sligar, The Catalytic Pathway of Cytochrome P450cam at Atomic Resolution, Science, 2000, 287, 1615–1622 CrossRef CAS PubMed.
- T. J. Dolinsky, J. E. Nielsen, J. A. McCammon and N. A. Baker, PDB2PQR: an automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations, Nucleic Acids Res., 2004, 32, W665–W667 CrossRef CAS PubMed.
- T. J. Dolinsky, P. Czodrowski, H. Li, J. E. Nielsen, J. H. Jensen, G. Klebe and N. A. Baker, PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations, Nucleic Acids Res., 2007, 35, W522–W525 CrossRef PubMed.
- M. H. M. Olsson, C. R. Søndergaard, M. Rostkowski and J. H. Jensen, PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions, J. Chem. Theory Comput., 2011, 7, 525–537 CrossRef CAS PubMed.
- C. R. Søndergaard, M. H. M. Olsson, M. Rostkowski and J. H. Jensen, Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values, J. Chem. Theory Comput., 2011, 7, 2284–2295 CrossRef PubMed.
- I. T. Todorov, W. Smith, K. Trachenko and M. T. Dove, DL_POLY_3: new dimensions in molecular dynamics simulations via massive parallelism, J. Mater. Chem., 2006, 16, 1911–1918 RSC.
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig, B. L. de Groot, H. Grubmüller and A. D. MacKerell, CHARMM36m: an improved force field for folded and intrinsically disordered proteins, Nat. Methods, 2017, 14, 71–73 CrossRef CAS PubMed.
- C. W. Yong, Descriptions and Implementations of DL_F Notation: A Natural Chemical Expression System of Atom Types for Molecular Simulations, J. Chem. Inf. Model., 2016, 56, 1405–1409 CrossRef CAS PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- C. Tian, K. Kasavajhala, K. A. A. Belfon, L. Raguette, H. Huang, A. N. Migues, J. Bickel, Y. Wang, J. Pincay, Q. Wu and C. Simmerling, ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution, J. Chem. Theory Comput., 2020, 16, 528–552 CrossRef PubMed.
- Modelling a P450 enyzme—Py-ChemShell 21.0.1 documentation, https://chemshell.org/static_files/py-chemshell/tutorial/build/html/p450_enzyme/index.html, (accessed January 27, 2023).
- S. a F. Nastase, A. J. O’Malley, C. R. A. Catlow and A. J. Logsdail, Computational QM/MM investigation of the adsorption of MTH active species in H-Y and H-ZSM-5, Phys. Chem. Chem. Phys., 2019, 21, 2639–2650 RSC.
- S. K. Matam, S. A. F. Nastase, A. J. Logsdail and C. R. A. Catlow, Methanol loading dependent methoxylation in zeolite H-ZSM-5, Chem. Sci., 2020, 11, 6805–6814 RSC.
- S. A. F. Nastase, A. J. Logsdail and C. R. A. Catlow, QM/MM study of the reactivity of zeolite bound methoxy and carbene groups, Phys. Chem. Chem. Phys., 2021, 23, 17634–17644 RSC.
- L. Negahdar, N. E. Omori, M. G. Quesne, M. D. Frogley, F. Cacho-Nerin, W. Jones, S. W. T. Price, C. R. A. Catlow and A. M. Beale, Elucidating the Significance of Copper and Nitrate Speciation in Cu-SSZ-13 for N2O Formation during NH3-SCR, ACS Catal., 2021, 11, 13091–13101 CrossRef CAS.
- Z.-M. Wang, L.-J. Liu, B. Xiang, Y. Wang, Y.-J. Lyu, T. Qi, Z.-B. Si, H.-Q. Yang and C.-W. Hu, The design and catalytic performance of molybdenum active sites on an MCM-41 framework for the aerobic oxidation of 5-hydroxymethylfurfural to 2,5-diformylfuran, Catal. Sci. Technol., 2019, 9, 811–821 RSC.
- L.-J. Liu, Z.-M. Wang, S. Fu, Z.-B. Si, Z. Huang, T.-H. Liu, H.-Q. Yang and C.-W. Hu, Catalytic mechanism for the isomerization of glucose into fructose over an aluminium-MCM-41 framework, Catal. Sci. Technol., 2021, 11, 1537–1543 RSC.
- S. A. F. Nastase, C. R. A. Catlow and A. J. Logsdail, QM/MM study of the stability of dimethyl ether in zeolites H-ZSM-5 and H-Y, Phys. Chem. Chem. Phys., 2021, 23, 2088–2096 RSC.
- J. Abdul Nasir, J. Guan, T. W. Keal, A. W. Desmoutier, Y. Lu, A. M. Beale, C. R. A. Catlow and A. A. Sokol, Influence of Solvent on Selective Catalytic Reduction of Nitrogen Oxides with Ammonia over Cu-CHA Zeolite, J. Am. Chem. Soc., 2023, 145, 247–259 CrossRef CAS PubMed.
- L.-J. Liu, Z.-M. Wang, Y.-J. Lyu, J.-F. Zhang, Z. Huang, T. Qi, Z.-B. Si, H.-Q. Yang and C.-W. Hu, Catalytic mechanisms of oxygen-containing groups over vanadium active sites in an Al-MCM-41 framework for production of 2,5-diformylfuran from 5-hydroxymethylfurfural, Catal. Sci. Technol., 2020, 10, 278–290 RSC.
- Y. Jiang, Z. Shuai and M. Liu, Roles of Long-Range Hopping, Quantum Nuclear Effect, and Exciton Delocalization in Exciton Transport in Organic Semiconductors: A Multiscale Study, J. Phys. Chem. C, 2018, 122, 18365–18375 CrossRef CAS.
- H. Ma, Q. Peng, Z. An, W. Huang and Z. Shuai, Efficient and Long-Lived Room-Temperature Organic Phosphorescence: Theoretical Descriptors for Molecular Designs, J. Am. Chem. Soc., 2019, 141, 1010–1015 CrossRef CAS PubMed.
- T. Zhang, W. Shi, D. Wang, S. Zhuo, Q. Peng and Z. Shuai, Pressure-induced emission enhancement in hexaphenylsilole: a computational study, J. Mater. Chem. C, 2019, 7, 1388–1398 RSC.
- G. Gong, H. Wu, T. Zhang, Z. Wang, X. Li and Y. Xie, Aggregation-enhanced emission in tetraphenylpyrazine-based luminogens: theoretical modulation and experimental validation, Mater. Chem. Front., 2021, 5, 5012–5023 RSC.
- Q. Peng, H. Ma and Z. Shuai, Theory of Long-Lived Room-Temperature Phosphorescence in Organic Aggregates, Acc. Chem. Res., 2021, 54, 940–949 CrossRef CAS PubMed.
- K. Qin, W. Gong, J. Gao, D. Hu, H. Shi, W. Yao, Z. An and H. Ma, Theoretical Insight Into the Ultralong Room-Temperature Phosphorescence of Nonplanar Aromatic Hydrocarbon, Front. Chem., 2021, 9, 740018 CrossRef CAS PubMed.
- G. Molpeceres and J. Kästner, Adsorption of H2 on amorphous solid water studied with molecular dynamics simulations, Phys. Chem. Chem. Phys., 2020, 22, 7552–7563 RSC.
- Z. Xie, Y. Sui, J. Buckeridge, C. R. A. Catlow, T. W. Keal, P. Sherwood, A. Walsh, M. R. Farrow, D. O. Scanlon, S. M. Woodley and A. A. Sokol, Donor and acceptor characteristics of native point defects in GaN, J. Phys. Appl. Phys., 2019, 52, 335104 CrossRef CAS.
- M. Kick and H. Oberhofer, Towards a transferable design of solid-state embedding models on the example of a rutile TiO2 (110) surface, J. Chem. Phys., 2019, 151, 184114 CrossRef CAS PubMed.
- J. Chen, N. A. Bogdanov, D. Usvyat, W. Fang, A. Michaelides and A. Alavi, The color center singlet state of oxygen vacancies in TiO2, J. Chem. Phys., 2020, 153, 204704 CrossRef CAS PubMed.
- X. Zhang, L. Zhu, Q. Hou, J. Guan, Y. Lu, T. W. Keal, J. Buckeridge, C. R. A. Catlow and A. A. Sokol, Toward a Consistent Prediction of Defect Chemistry in CeO2, Chem. Mater., 2023, 35, 207–227 CrossRef CAS PubMed.
- A. J. Logsdail, C. A. Downing, T. W. Keal, P. Sherwood, A. A. Sokol and C. R. A. Catlow, Hybrid-DFT Modeling of Lattice and Surface Vacancies in MnO, J. Phys. Chem. C, 2019, 123, 8133–8144 CrossRef CAS.
- Q. Hou, J. Buckeridge, A. Walsh, Z. Xie, Y. Lu, T. W. Keal, J. Guan, S. M. Woodley, C. R. A. Catlow and A. A. Sokol, The Interplay of Interstitial and Substitutional Copper in Zinc Oxide, Front. Chem., 2021, 9, 780935 CrossRef CAS PubMed.
- M. A. Sainna, S. Nanavati, C. Black, L. Smith, K. Mugford, H. Jenkins, M. Douthwaite, N. F. Dummer, C. R. A. Catlow, G. J. Hutchings, S. H. Taylor, A. J. Logsdail and D. J. Willock, A combined periodic DFT and QM/MM approach to understand the radical mechanism of the catalytic production of methanol from glycerol, Faraday Discuss., 2021, 229, 108–130 RSC.
- P. Sherwood, A. H. de Vries, S. J. Collins, S. P. Greatbanks, N. A. Burton, M. A. Vincent and I. H. Hillier, Computer simulation of zeolite structure and reactivity using embedded cluster methods, Faraday Discuss., 1997, 106, 79–92 RSC.
- J. D. Gale and A. L. Rohl, The General Utility Lattice Program (GULP), Mol. Simul., 2003, 29, 291–341 CrossRef CAS.
- J. R. Hill and J. Sauer, Molecular mechanics potential for silica and zeolite catalysts based on ab initio calculations. 1. Dense and microporous silica, J. Phys. Chem., 1994, 98, 1238–1244 CrossRef CAS.
- J.-R. Hill and J. Sauer, Molecular Mechanics Potential for Silica and Zeolite Catalysts Based on ab Initio Calculations. 2. Aluminosilicates, J. Phys. Chem., 1995, 99, 9536–9550 CrossRef CAS.
- Modelling zeolites—Py-ChemShell 21.0.1 documentation, https://chemshell.org/static_files/py-chemshell/tutorial/build/html/zeol_covalent/index.html, (accessed January 27, 2023).
- A. A. Sokol, S. T. Bromley, S. A. French, C. R. A. Catlow and P. Sherwood, Hybrid QM/MM embedding approach for the treatment of localized surface states in ionic materials, Int. J. Quantum Chem., 2004, 99, 695–712 CrossRef CAS.
- E. Aprà, E. J. Bylaska, W. A. de Jong, N. Govind, K. Kowalski, T. P. Straatsma, M. Valiev, H. J. J. van Dam, Y. Alexeev, J. Anchell, V. Anisimov, F. W. Aquino, R. Atta-Fynn, J. Autschbach, N. P. Bauman, J. C. Becca, D. E. Bernholdt, K. Bhaskaran-Nair, S. Bogatko, P. Borowski, J. Boschen, J. Brabec, A. Brunauer, E. Cauët, Y. Chen, G. N. Chuev, C. J. Cramer, J. Daily, M. J. O. Deegan, T. H. Dunning, M. Dupuis, K. G. Dyall, G. I. Fann, S. A. Fischer, A. Fonari, H. Früchtl, L. Gagliardi, J. Garza, N. Gawande, S. Ghosh, K. Glaesemann, A. W. Götz, J. Hammond, V. Helms, E. D. Hermes, K. Hirao, S. Hirata, M. Jacquelin, L. Jensen, B. G. Johnson, H. Jónsson, R. A. Kendall, M. Klemm, R. Kobayashi, V. Konkov, S. Krishnamoorthy, M. Krishnan, Z. Lin, R. D. Lins, R. J. Littlefield, A. J. Logsdail, K. Lopata, W. Ma, A. V. Marenich, J. Martin del Campo, D. Mejia-Rodriguez, J. E. Moore, J. M. Mullin, T. Nakajima, D. R. Nascimento, J. A. Nichols, P. J. Nichols, J. Nieplocha, A. Otero-de-la-Roza, B. Palmer, A. Panyala, T. Pirojsirikul, B. Peng, R. Peverati, J. Pittner, L. Pollack, R. M. Richard, P. Sadayappan, G. C. Schatz, W. A. Shelton, D. W. Silverstein, D. M. A. Smith, T. A. Soares, D. Song, M. Swart, H. L. Taylor, G. S. Thomas, V. Tipparaju, D. G. Truhlar, K. Tsemekhman, T. Van Voorhis, Á. Vázquez-Mayagoitia, P. Verma, O. Villa, A. Vishnu, K. D. Vogiatzis, D. Wang, J. H. Weare, M. J. Williamson, T. L. Windus, K. Woliński, A. T. Wong, Q. Wu, C. Yang, Q. Yu, M. Zacharias, Z. Zhang, Y. Zhao and R. J. Harrison, NWChem: Past, present, and future, J. Chem. Phys., 2020, 152, 184102 CrossRef PubMed.
- M. F. Guest, I. J. Bush, H. J. J. Van Dam, P. Sherwood, J. M. H. Thomas, J. H. Van Lenthe, R. W. A. Havenith and J. Kendrick, The GAMESS-UK electronic structure package: algorithms, developments and applications, Mol. Phys., 2005, 103, 719–747 CrossRef CAS.
- A. J. Logsdail, C. A. Downing, T. W. Keal, P. Sherwood, A. A. Sokol and C. R. A. Catlow, Modelling the chemistry of Mn-doped MgO for bulk and (100) surfaces, Phys. Chem. Chem. Phys., 2016, 18, 28648–28660 RSC.
- Modelling an MgO surface—Py-ChemShell 21.0.1 documentation, https://chemshell.org/static_files/py-chemshell/tutorial/build/html/mgo_surface/index.html, (accessed January 27, 2023).
- Diffusion of Li in MgO—Py-ChemShell 21.0.1 documentation, https://chemshell.org/static_files/py-chemshell/tutorial/build/html/li_mgo/index.html, (accessed January 27, 2023).
- J. Meyer and K. Reuter, Modeling Heat Dissipation at the Nanoscale: An Embedding Approach for Chemical Reaction Dynamics on Metal Surfaces, Angew. Chem., Int. Ed., 2014, 53, 4721–4724 CrossRef CAS PubMed.
- M. I. Baskes, Modified embedded-atom potentials for cubic materials and impurities, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 2727–2742 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Lippert, J. Hutter and M. Parrinello, A hybrid Gaussian and plane wave density functional scheme, Mol. Phys., 1997, 92, 477–488 CrossRef CAS.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- S. Goedecker, M. Teter and J. Hutter, Separable dual-space Gaussian pseudopotentials, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703–1710 CrossRef CAS PubMed.
- J. Guan, Y. Lu, K. Sen, J. Abdul Nasir, A. W. Desmoutier, Q. Hou, X. Zhang, A. J. Logsdail, G. Dutta, A. M. Beale, R. W. Strange, C. Yong, P. Sherwood, H. M. Senn, C. R. A. Catlow, T. W. Keal and A. A. Sokol, Philos. Trans. R. Soc., A, 2023 DOI:10.1098/rsta.2022.0234.
- G. A. Tribello, M. Bonomi, D. Branduardi, C. Camilloni and G. Bussi, PLUMED 2: New feathers for an old bird, Comput. Phys. Commun., 2014, 185, 604–613 CrossRef CAS.
- A. Denzel and J. Kästner, Gaussian process regression for geometry optimization, J. Chem. Phys., 2018, 148, 094114 CrossRef.
- A. Denzel and J. Kästner, Gaussian Process Regression for Transition State Search, J. Chem. Theory Comput., 2018, 14, 5777–5786 CrossRef CAS PubMed.
- D. Born and J. Kästner, Geometry Optimization in Internal Coordinates Based on Gaussian Process Regression: Comparison of Two Approaches, J. Chem. Theory Comput., 2021, 17, 5955–5967 CrossRef CAS PubMed.
- T. W. Keal, A.-M. Elena, A. A. Sokol, K. Stoneham, M. I. J. Probert, C. S. Cucinotta, D. J. Willock, A. J. Logsdail, A. Zen, P. J. Hasnip, I. J. Bush, M. Watkins, D. Alfè, C.-K. Skylaris, B. F. E. Curchod, Q. Cai and S. M. Woodley, Materials and Molecular Modeling at the Exascale, Comput. Sci. Eng., 2022, 24, 36–45 CAS.
Footnote |
| † The authors would like to dedicate this review to the memory of our late colleague, friend and mentor Prof. Walter Thiel, who did so much to support and advance the ChemShell project. |
| This journal is © the Owner Societies 2023 |