
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic enhancement of production of solar thermochemical fuels: opportunities and limitations
Juan M.
Coronado
 * and
Alicia
Bayón
* and
Alicia
Bayón
Instituto de Catálisis y Petroleoquímica. ICP-CSIC. C/Marie Curie, 2. E-28049. Madrid, Spain. E-mail: jm.coronado@csic.es
First published on 5th June 2023
Abstract
Solar thermochemical fuels are a promising low-carbon alternative to conventional fossil fuels, which must be swiftly phased out to mitigate the consequences of climate change. Thermochemical cycles powered by concentrating solar energy at high temperatures have demonstrated efficiency in the conversion of solar to chemical energy exceeding 5% and have been assayed in pilot scale facilities of up to 50 kW. This conversion route implies the use of a solid oxygen carrier that enables CO2 and H2O splitting, generally operating in two consecutive stages. The primary product of the combined thermochemical conversion of CO2 and H2O is syngas (CO + H2), which for practical applications must be catalytically transformed into hydrocarbons or other chemicals such as methanol. This link between thermochemical cycles, involving the transformation of the whole solid used as an oxygen carrier, and catalysis, occurring only on the material surface, calls for exploitation of the synergies between these two unlike but interconnected gas–solid processes. Accordingly, in this perspective we discuss the differences and similitudes between these two transformation routes, consider the practical impact of kinetics in thermochemical solar fuel generation and explore the limits and opportunities of the catalytic promotion. With this aim, first, the potential benefits and hurdles of direct catalytic enhancement of CO2 and H2O dissociation in thermochemical cycles are discussed and then, the possibilities of improving the catalytic production of hydrocarbon fuels, basically methane, are also assessed. Finally, an outlook of the future opportunities of the catalytic promotion of thermochemical solar fuel productions is also provided.
Introduction. Sustainable solar fuels for an imminent future
The increasing awareness in the society about the risks of climate change, which is causing growing environmental and economic concerns, is placing decarbonization of energy at the focal point of challenges faced by humankind. Although global agreements such as that reached at COP26 in Glasgow in 2021 are necessary to deal with this worldwide endeavor, the scientific community must develop a broad portfolio of viable technical solutions to swiftly reduce the carbon footprint of energy.Increasing efficiency in energy consumption and promoting the use of renewable sources are the basic keystones of the new emerging energy panorama. Currently, the electrical grid is increasingly renewable, with the largest deployment of new generation plants in China, USA, Brazil, India and the European Union. In contrast, climatization, including both air-conditioning and heating systems, together with industrial applications are still globally based on fossil resources (>85% in 2019), while oil-based liquid fuels constitute more than 95% of the world's energy share in the transport sector.1
To progress in this direction, a number of routes for the production of more sustainable synthetic fuels powered by different renewable sources are currently being investigated.2,3 Solar fuel production is particularly promising because it allows storage of solar energy in chemical bonds. This can be achieved by following different transformation routes (see Fig. 1), such as photocatalysis, photoelectrochemical or thermochemical. The primary product of most of these transformations is hydrogen, which is attracting a huge deal of interest due to the versatility of this simple molecule as an energy vector, with applications in fuel cells and adapted engines.4 Furthermore, hydrogen is also an essential reagent in the chemical industry, and it can enable CO2 valorisation to produce fuels and chemicals, providing a potential way to reach neutral or even negative carbon emissions.5
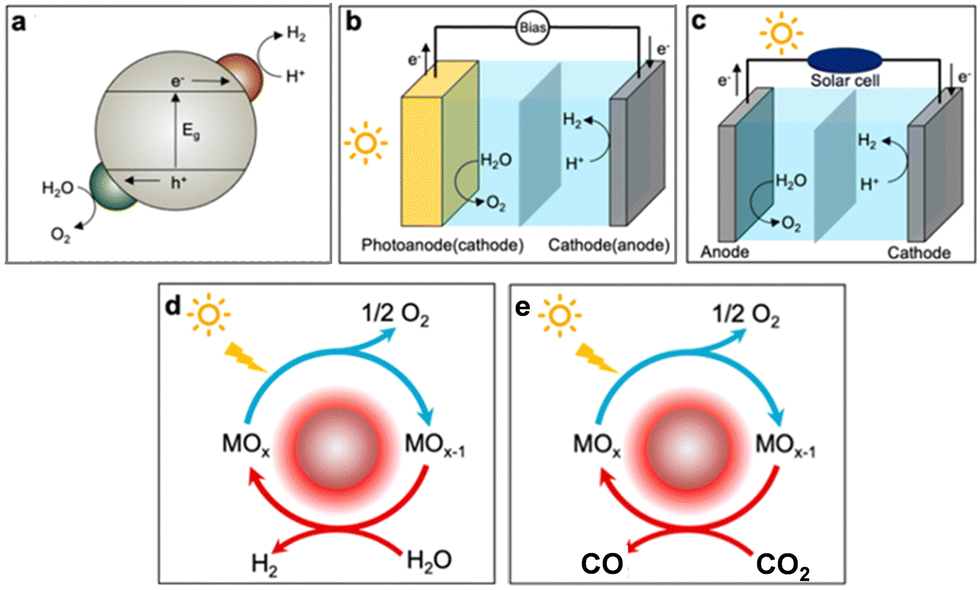 | ||
| Fig. 1 Main routes to produce solar fuels: (a) photocatalytic (PC), (b) photoelectrochemical (PEC), and (c) photovoltaic–electrochemical (PV-EC) water splitting; solar thermochemical (STC) (d) water and (e) CO2 splitting. Adapted from ref. 2 with permission from the American Chemical Society. | ||
The technical readiness levels of these technologies vary widely, but generation of green (CO2-free) hydrogen by electrochemical water splitting externally powered by wind or photovoltaic (PV) electricity is already on the verge of commercial exploitation. A usual metric to determine how much energy can be chemically stored is the solar-to-hydrogen (STH) efficiency, which is calculated as the ratio between the hydrogen rate and the solar irradiance multiplied by the exposed area of the device. For PV systems this parameter can exceed 30% in some optimized systems based on triple-junction solar cells.2,6 Yet, using commercially available crystalline Si cells coupled with optimized electrodes of Pt and WO2.72 co-doped with Co and Fe, and operating in alkaline media, an average solar-to-hydrogen efficiency of 16.9% was achieved during 50 h of operation.7 These selected examples of high yields illustrate the foremost position of this technology to commercially deliver carbon-free hydrogen. In spite of this, the price of this green hydrogen, with values expressed as the levelized cost of hydrogen (LCOH) ranging from 6 to 2 $ per kg, is still higher than that produced industrially by the steam reforming of methane (generally lower than 2 $ per kg), although economically competitive production is estimated to be achievable in few years.8
Research on alternative transformation schemes to those based on electrolysers powered by PV (Fig. 1c) and directly activated by sunlight in a single step is gathering notable interest due to their potential to achieve high energy conversion yields. Among these technologies, photoelectrochemical water splitting (see Fig. 1b) is leading the way because of its competitive efficiency for hydrogen production.2,9 Solar-to-hydrogen efficiencies higher than 19% were obtained in acidic media using a device with an Rh-based photoelectrocatalyst on a complex multi-layered semiconductor structure.10 For photocatalysis, which is exclusively activated by light (Fig. 1a), the best results were achieved using carbon nanodots dispersed STH on C3N4 as the photocatalyst. This system reaches only 2% of water splitting without using any sacrificial agent.11 Recently, an STH efficiency of 9% has been reported for water splitting using InGaN/GaN nanowires as photocatalysts with a complex co-catalyst containing Rh, Cr and Co and operating at about 70 °C under sunlight irradiation.12 This increment in the temperature appears to be instrumental to achieve higher hydrogen yields and points out the relevance of exploring photothermal approaches, based on simultaneous thermal and photonic activation, for energy and other applications.13
Thermochemical solar fuels (Fig. 1d and e) have already reached interesting energy yields that exceed 5% solar to fuel conversion,14 and they present a large theoretical potential for additional improvement. Recently, thermochemical hydrogen generation has proved to be comparable with photoelectrocatalytic hydrogen production in efficiency, providing an attractive option for those locations with high direct irradiation.15 In addition, this technology is well adapted to complement electricity generation in concentrating solar plants16 and it also allows a very selective CO2 splitting, which is not easily achieved by other technologies. However, the thermochemical route for solar fuel production is not free of drawbacks, because the requirement of operating at high temperature adds complexity to thermochemical reactors and demand thermally resistant materials that can increase the cost of the commissioning of these plants. In addition, these systems are less adapted to modular deployment than the other technologies based on direct activation by photons and need extensive heliostat fields to reach reasonable productivity.
A more detailed vision of the current state of the art of this technology, with emphasis on material development, is given in the next section.
Thermochemical solar fuels production
Currently, most of the research on solar thermochemical fuels is focussed on cycles of two stages, that involve separate and consecutive reactions that when combined lead to H2O or CO2 splitting. This is achieved with the assistance of an oxygen carrier, which generally is a redox metal oxide (see Fig. 2). The corresponding reactions of this stepwise process using a generic reducible non-stoichiometric oxide, MOx, are the following:| MOx ⇌ MOx−δ + δ/2 O2 | (1) |
| MOx−δ + δ H2O ⇌ MOx + δ H2 | (2) |
| MOx−δ + δ CO2 ⇌ MOx + δ CO | (3) |
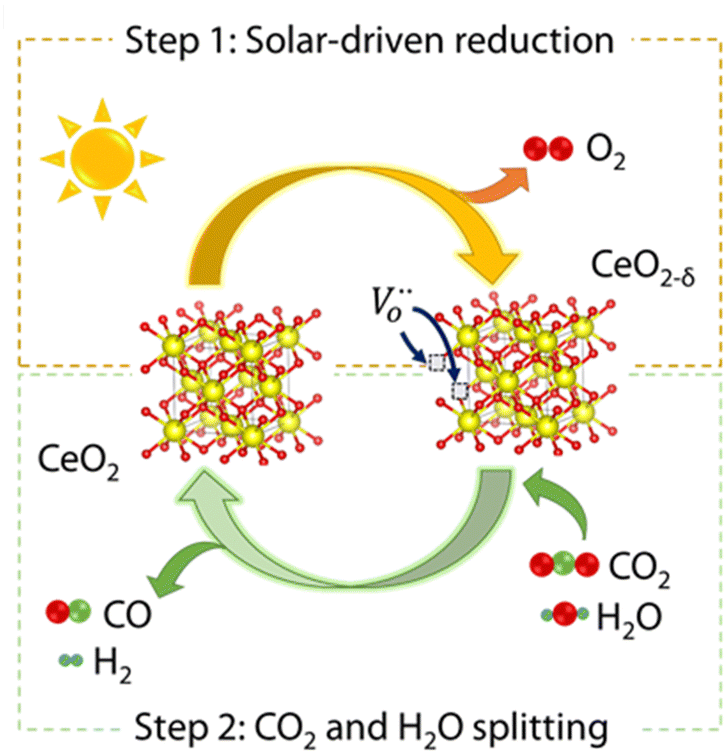 | ||
| Fig. 2 Scheme of the thermochemical cycle for CO2 and H2O splitting over CeO2. Reproduced with permission from ref. 26 with permission from RSC. | ||
In the second stage, two differentiated paths for reoxidation of the oxygen carrier can be considered as indicated in eqn (2) and (3), by using either H2O to produce H2 or CO2 to obtain CO. In practice, coprocessing of H2O and CO2 mixtures is the preferred option, because the outcome is the blend of H2 and CO, known as syngas, which can be fed to a second catalytic unit in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 proportion to produce synthetic fuels using Fischer–Tropsch technology.17 Besides, the synthesis of methanol from solar syngas with a H2/CO ratio higher than 3 has been also explored.18 Thermodynamic dictates that H2O and CO2 splitting reactions are more efficient at lower temperatures than those previously applied for vacancy formation. Therefore, efficient isothermal operation of these cycles, although preferred to avoid heat losses, is difficult to implement in practice.
:1 proportion to produce synthetic fuels using Fischer–Tropsch technology.17 Besides, the synthesis of methanol from solar syngas with a H2/CO ratio higher than 3 has been also explored.18 Thermodynamic dictates that H2O and CO2 splitting reactions are more efficient at lower temperatures than those previously applied for vacancy formation. Therefore, efficient isothermal operation of these cycles, although preferred to avoid heat losses, is difficult to implement in practice.
Thermochemical cycles with three or more stages, such as H2SO4–SOx or MnO–Na2CO3,19,20 have also been investigated due to their potential for achieving high hydrogen yields. However, the increasing complexity of the required reactors, the concerns about material corrosion as well as the challenges of efficient heat recovery caused by the significant temperature gap between the different reaction stages have limited the interest of these cycles for solar applications.
Several redox oxides have been studied as possible oxygen carriers for high temperature thermochemical cycles, although research is currently focussed on the use of perovskites and CeO2, because these materials showed promising efficiencies. In the case of CeO2, despite the initial attempts to deal with the fully reduced oxide, Ce2O3, the extreme temperatures (ca. 2000 °C) required for this transformation hindered achieving complete conversion.21 In contrast, the use of partially reduced ceria, CeO2−δ, has proved to be more successful, and the seminal article of Chueh et al. published in 2010 unveiled the remarkable performance of CeO2 as an efficient oxygen carrier due to its favourable thermodynamics.22 Typically, as it is shown in the graph of Fig. 3, thermochemical production of syngas on CeO2 occurs by reduction of the solid at 1500 °C with a pO2 lower than 0.02 bars, and subsequent reoxidation by introducing a mixture of steam and CO2 in the reactor at about 900 °C.
 | ||
| Fig. 3 Time evolution of the temperature, reactor pressure, and flow rates of the gas products (O2, CO, and H2) during a representative redox cycle using an alveolar foam of CeO2 (18.1 kg) and with a mean Psolar = 42.0 ± 6.2 kW. Reproduced with permission from ref. 17 with permission from Elsevier. | ||
The unparallel reduction entropy of CeO2, which is mostly associated with the contribution of the configurational entropy of the f electrons of the lanthanide group,23 is a key aspect to explain the notable thermochemical activity of CeO2. Subsequent investigations by the Steinfeld group, using a larger scale reactor with CeO2 shaped as reticulated foams, have set the record on sun-to-fuel efficiency of 5.25% for the thermochemical CO2 splitting at non-isothermal conditions.14 This system achieved a CO2 conversion of 83% and showed stability for 500 cycles under a concentration of 3000 suns provided by a solar simulator of 4 kW. Recently, thermochemical hydrocarbon fuel production has been demonstrated at a much larger scale reactor (50 kW) powered by a modular 500 m2 heliostat field.17 This system was fed with a mixture of CO2 and H2O vapor and, under realistic operation conditions with real sunlight, reached an efficiency of 4.1% for syngas production (see Fig.·3), which is slightly lower than that of the bench-scale prototype. This work has established that despite the complexity of operating with high solar fluxes and at elevated temperatures, deployment of plants for thermochemical fuel production is feasible with medium-sized plants with high concentrations (about 2500 suns) and located in areas with moderate solar irradiance.
Several attempts to enhance the thermochemical performance of CeO2 by introducing additional metals have been reported. Doping ceria with up to 20% mols of Zr allows doubling the hydrogen production by increasing the reducibility of the mixed Ce1−xZrxO2 oxide, which retains the fluorite structure.24 However, to fully exploit this higher hydrogen generation capacity is necessary to increment the water vapor concentration to compensate for the slower re-oxidation kinetic of the doped material. Similarly, enhancement of the CO2 splitting activity for ceria doped with 15% of Hf has also been reported but, as in the case of Zr, at the cost of sluggish hydrogen evolution.25 Morphology of the solid is also important for easing the gas–solid contact and to allow a better distribution of the incident solar radiation. Accordingly, CeO2 has been used as porous fibres or as alveolar foams in larger scale reactors.
Perovskites, with the general formula ABO3 have been proposed as potentially suitable oxygen carriers due, among other reasons, to their noteworthy chemical adaptability. The crystalline lattice of perovskites is theoretically cubic, although distortions leading to tetragonal or hexagonal symmetries are common. Perovskites are characterized by their chemical versatility that allows them to accommodate a number of different cations on both A and B sites, as long as the electroneutrality and the geometric constraints of the lattice are preserved.26,27 In this respect, the large cation A must fit in a 12-fold coordinated site, while the B element occupies an octahedral one. Furthermore, if the cation B, or less frequently that in A site, is redox active, perovskites can undergo partial oxygen release at high temperatures and low oxygen partial pressure, yielding the reduced oxide ABO3−δ.
This reactivity can be exploited in thermochemical cycles. In this way, perovskites of the family La1−xSrxMnO3 have been proved to be active for water and CO2 splitting with higher efficiency than CeO2 and at lower temperatures.28 Additional doping of the B site results in oxides such as La0.6 Sr0.4 Mn0.4 Al0.6O3 that present an attractive performance for CO2 and H2O splitting.29,30 Just to give an impression about the chemical diversity of perovskite compositions so far investigated, but without trying to be exhaustive, recent reports have also shown promising thermochemical activity of CaTi0.5Mn0.5O331 and BaCe0.25Mn0.75O332 for hydrogen production and of La0.6Sr0.4Cr1−xCoxO333 for CO generation. Furthermore, some composite systems using perovskites and CeO2 have also been investigated to take advantage of the possible synergies between the two redox components.34 Besides, ab initio calculations using Density functional theory (DFT) has emerged as a very convenient theoretical tool to determine the thermodynamic properties of a large number of perovskites and to identify chemical compositions with high potential efficiency.35 For additional information on the application of perovskites to solar thermochemical fuel production, the readers are referred to the excellent specific reviews on this topic.27,36
Mixed oxides of other structural families different than perovskites can be also applied to thermochemical water splitting. Notably, Ni ferrites, NiFe2O4, have widely been investigated for this application, and those doped with Zn were successfully used at large scale (100 kW) for solar hydrogen production, although they showed low cycling stability.37 Besides, Ni ferrites have also proved to be active for CO2 splitting at the lab scale.38 More recently, mixed oxides containing four different transition metals, Ni, Fe, Mg and Co, and consisting of a mixture of rock salt and spinel structures have demonstrated considerable water splitting activity at temperatures below 1100 °C.39 These results show a wide range of chemical compositions of metal oxides that can be explored as potential oxygen carriers. This is prompting the application of data science tools such as machine learning for the selection of appropriate candidates for oxygen carriers.40
For establishing the potential applicability of a specific material for production of solar thermochemical fuels, their thermodynamic functions for vacancy formation, both enthalpy and entropy, should be determined as a function of temperature, pressure, and non-stoichiometry. As mentioned earlier, these parameters can be obtained by DFT calculations, but validation of theoretical values requires careful, systematic and time-consuming experimental work.41 Easy reducibility of the redox oxides according to eqn (1) can be positive for achieving high non-stoichiometry, but the key aspect for the applicability of a redox material in thermochemical cycles is whether it can supply the energy necessary to drive H2O and CO2 splitting during the subsequent stage when the solid is reoxidated. Therefore, a specific material can be utilized for thermochemical fuel production under a fixed set of conditions of pressure, temperature and gas composition only if ΔG < 0 for the processes described in eqn (2) and (3). Recently, Bayón et al. have systematized these thermodynamics calculations to determine the viability of using specific perovskite compositions and considering directly the enthalpy and entropy of oxygen vacancy formation, as illustrated in the graph of Fig. 4.42 Similarly, Haile and co-workers have explored the expected limits of the solar-to-fuel efficiency that can be reached considering fixed bed reactors by using an iterative method and applying quasi-equilibrium conditions to some selected oxides.43 These results predict a solar-to fuel energy yield of 12.9% at a CeO2 reduction temperature of 1500 °C, while the estimated efficiency for other potentially promising materials under realistic operation conditions is lower, including that of the Zr-doped CeO2 (10.1%) and La0.8Sr0.2MnO3 perovskite (3.3%). So far, perovskites cannot compete with the efficiencies of CeO2 when performing a side-to-side comparison,44 although considering the immense number of possible compositions of those mixed oxides, there is significant expectation of finding novel materials that could outperform CeO2.
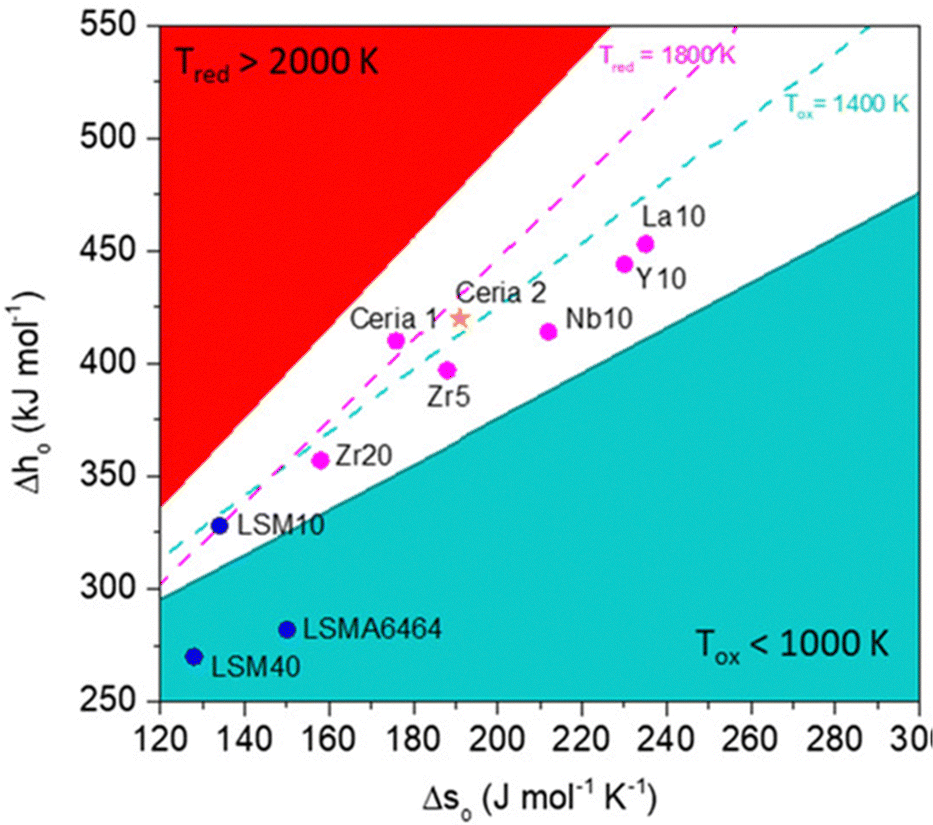 | ||
| Fig. 4 Graphs plotting the enthalpy of oxygen vacancies formation, Δho, as a function of the entropy, Δso, of selected redox oxides used in thermochemical cycles to reach Δgo = 0. Fixed oxygen partial pressure (p/p0 = 10−5) and 10% of conversion were considered for a fixed range of temperatures for the reduction (solid red and dashed pink lines) and water splitting (blue solid and dashed lines) equilibrium. The white area represents values of the thermodynamic properties with operation temperatures judged reasonable for solar thermal applications. This region is delimited for reduction below 2000 K (higher temperatures appear in the red sector) and reoxidation above 1000 K (blue area), while for reoxidation between 1800 and 1400 K is the white space between dashed lines. Dots represent values of selected materials used as oxygen carriers. Reproduced with permission from ref. 42 with permission from Wiley. | ||
Isothermal operation of both reduction and oxidation stages can provide advantages in terms of limiting heat losses, which is the main contribution to decreasing the efficiency in thermo-solar processes.15 In this respect, considering the thermodynamic functions, it has been estimated that the potential solar-to-fuel efficiency for CeO2 could reach up to 28% by implementing an optimized heat recovery system.45,46
In addition to thermodynamic considerations, experimental studies have shown that thermodynamic properties do not fully account for the suitability of redox materials to produce solar thermochemical fuels, because unfavourable kinetic may turn impractical in the use of materials with satisfactory redox properties. This is particularly relevant during the CO2 and water splitting because this stage generally occurs at lower temperatures, reducing the reaction driving force. These aspects have been highlighted in many experimental studies and partially considered in the recent model by Haile's group.43 Pure ceria presents slow reduction kinetics and relatively fast reoxidation, while perovskites and doped ceria are faster during reduction and slower during the reoxidation of the solid which takes place during the water splitting reaction.47 To cope with these constraints, the concentration of the gas reagents, CO2 and water is frequently increased to compensate for the slow reoxidation rate. However, this strategy has a negative impact on the overall efficiency of the process because the energy required for heating larger volumes of gas and the lower efficiency of separation of the products from the unreacted molecules impose an important penalty on the energy that can be accumulated on products.15 This calls for additional approaches to boost the rate of syngas production, as explored in the following sections.
The relevance of kinetics for thermochemical cycles
Due to the impact of kinetic on the productivity of solar fuels, different empirical and theoretical approaches have been used to understand the key parameters determining the rate of thermochemical cycles. This section summarizes the main research efforts in this area.The master plot method, which considers different equations for solid state kinetic in a normalized graph, has been applied to analyze the kinetic of thermochemical cycles.48 Results of this approach suggest that CO2 splitting over Zr0.25Ce0.75O2 is controlled by diffusion within the solid, while water splitting appears to follow a first-order kinetic. However, the agreement between experimental data and model predictions is far from perfect, particularly for low conversion of the redox material. In contrast, good matching of hydrogen generation over pure CeO2 was obtained by fitting experimental data with a model considering a first-order deceleration of the rate.49 Similarly, hydrogen production by water splitting over Zr-modified ceria, or co-doped with Zr and Gd or Pr, was reproduced by applying a power-law decelerating kinetic. To select the most appropriate model, an iterative algorithm was used to determine which model of solid-state kinetics better matched the experimental results.50 This approach suggests that water splitting is controlled by surface reaction rather than by diffusion in the bulk. A simple analytical model was proposed by Bulfin et al. with the aim of explaining the reduction and oxidation kinetics of ceria with O2,51 and it was lately applied to Zr-doped ceria.47 This method considers the rate of variation of the concentration of oxygen vacancies and assumes thermodynamic equilibrium to reproduce the experimental oxygen release and uptake curves. From these results, the authors concluded that doped materials showed slower kinetics than pure ceria.47 However, the reduction reaction seemed to be limited by the heating rate, despite being extremely high. These results stress the importance of considering not only the effect of the kinetic on the chemistry of the redox material but also the physical parameters affecting mass and heat transport.
In contrast with previous approaches, the so-called thermo-kinetic model poses that the rate of the two stages of water splitting can be described satisfactorily considering only the independently determined thermodynamic properties, without including any fitting parameters.52,53 This model, extensively investigated by Haile's group, stated that thermodynamic equilibrium at high enough operation temperature is reached so rapidly that the rate of the process is defined only by the flow of reagents and products at the operation conditions.54–56 This implies that both surface reaction and oxygen diffusion through the solid are considered to occur so rapidly that they do not have a measurable effect on the overall kinetics. Therefore, the only constrains to the reactivity of the redox solid are their thermodynamic properties and the effective activation energy approaches zero. Therefore, by applying a mass balance to the inlet and outlet streams, analytic equations can be obtained for water splitting that depends only on the thermodynamic properties of the oxygen carrier and selected conditions of gas composition, flow rate and temperature. Following this method, experimental results of water splitting over CeO2 and perovskite La1−xSrxMnO3 were adequately reproduced under different operation conditions. Generally, a good agreement between experimental results and the predictions of the thermo-kinetic model is obtained, but significant deviations are observed for CeO2 at the lower temperatures when the assumptions of the model do not fully hold. Similarly, an appreciable mismatch with this quasi-equilibrium model is observed for water splitting over the perovskite CaTi0.5Mn0.5O3, particularly at the beginning of the water splitting stage, due to the kinetic limitations of the material.30 In this way, experimental time-resolved studies carried out by means of the electrical conductance relaxation method show that the thermo-kinetic regime provides a good description at relatively low flow rates, high temperatures and large surface area of the oxide.51
Alternatively, detailed defect analysis considering the migration of oxygen vacancies from the bulk to the surface was used to reproduce the experimental hydrogen production over CeO2 by fitting the thermodynamic parameters of vacancy formation and the corresponding kinetic constants.57Table 1 summarizes the main kinetic studies included in this review.
| Reaction | Sample | Equipment | Methodology | E a (kJ mol−1) | Model | Ref. |
|---|---|---|---|---|---|---|
| TGA: thermogravimetric analyzer, SFR: stagnation flow reactor, XLV: Xenon Lamp in Vacuum chamber, CFR: continuous flow reactor, OR: re-oxidation in O2.a Estimated by Arrhenius plot from experimental data obtained at different temperatures.b Calculated by the multiparametric fitting of the kinetic model to the experimental data. | ||||||
| CDS | Zr0.25Ce0.75O2 | TGA | Master plot | 82–103a | Diffusion | 47 |
| WS | CeO2 | SFR | Model fitting | 29b | First order decelatory | 48 |
| WS | CeO2 doped with Zr, Gd or Pr | SFR | Model fitting | — | Power law | 49 |
| WS | CeO2 | CFR | Prediction based on thermodynamics | — | Thermo-kinetic | 52 and 53 |
| WS | La1−xSrxMnO3 | CFR | Prediction based on thermodynamics | — | Thermo-kinetic | 54 |
| OR | Ce1−xZrxO2 & CeO2 | XLV | Fitting to the experimental results | 26–30 ± 2.5 (Ce1−xZrxO2)a 36 ± 4 (CeO2)a | Based on vacancies equilibrium & diffusion/shrinking Core | 46 and 50 |
| WS | CeO2 | CFR | Fitting to the experimental results | 190 ± 50b | Based on vacancies and electrons equilibrium & transport | 56 |
In addition to the intrinsic characteristics of the redox materials, their textural properties have also an important impact on kinetic. This has been shown for CO2 splitting with CeO2, which when used as a tridimensional ordered microporous (350–50 nm) material presents a rate one order of magnitude larger than for the non-porous oxide.58
The study of the mechanism of reoxidation of the redox oxides by water vapor and CO2 can be approached by means of DFT calculations. Wolverton et al. showed that the dissociation of water on oxygen vacancies of CeO2(111) to yield surface hydroxyls presents an insignificant energy barrier.59 However, for an efficient H2 release, it is proposed that the most efficient pathway implies the reaction of Ce–H species with the Ce–OH surface group. This route proceeds with an estimated activation energy of about 110 kJ mol−1 (1.14 eV barrier). In contrast, other studies of CeO2 proposed Ce–OH species as intermediate for the desorption of H2, although with great differences in the calculated energy barrier. Thus, in one report formation of H2 present an activation energy of about 400 kJ mol−1,60 which is much larger that for water desorption, while a more moderate barrier of about 200 kJ mol−1 was determined in other DFT studies.57 More recently, the reaction of surface vacancies on Ce0.75Zr0.25O2(111) with water was also studied by DFT. The results obtained revealed that hydrogen generation occurs in two steps, first water dissociation on the surface oxygen vacancies and then the migration of a hydrogen atom to yield two hydroxyls, which subsequently react to release H2. The activation energies of these two processes are 84.5 kJ mol−1 and 169.2 kJ mol−1, respectively.61 DFT calculations for Nb-doped CeO2 suggested a similar pathway with an activation energy of 64 kJ mol−1 for hydrogen transfer and 154 kJ mol−1 for H2formation.62 Other DFT studies of Zr and Pr doped ceria also confirmed that desorption of H2 is the rate-limiting stage with energy barriers around 2 eV.50 These theoretical energy barriers are within the high-end of the values determined experimentally for water splitting, as listed in Table 1, which show a significant dispersion, with energies varying in the 80–200 kJ mol−1 range.
On the other hand, CO2 interaction on reduced Ce0.75Zr0.25O2(111) leads to the adsorption of CO2 on an oxygen vacancy and the formation of the adsorbed carbonyl with an activation energy of 72.9 kJ mol−1 (see Fig. 5). The DFT results obtained for unmodified CeO2(111) in these two processes are similar although the activation energies are slightly higher. These theoretical energy barriers are slightly higher than those experimentally determined from Arrhenius plots, which were in the 80–105 kJ mol−1 range for CO2 splitting using Zr0.25Ce0.75O2, although the accuracy of these values is limited by the small number of tests performed.48
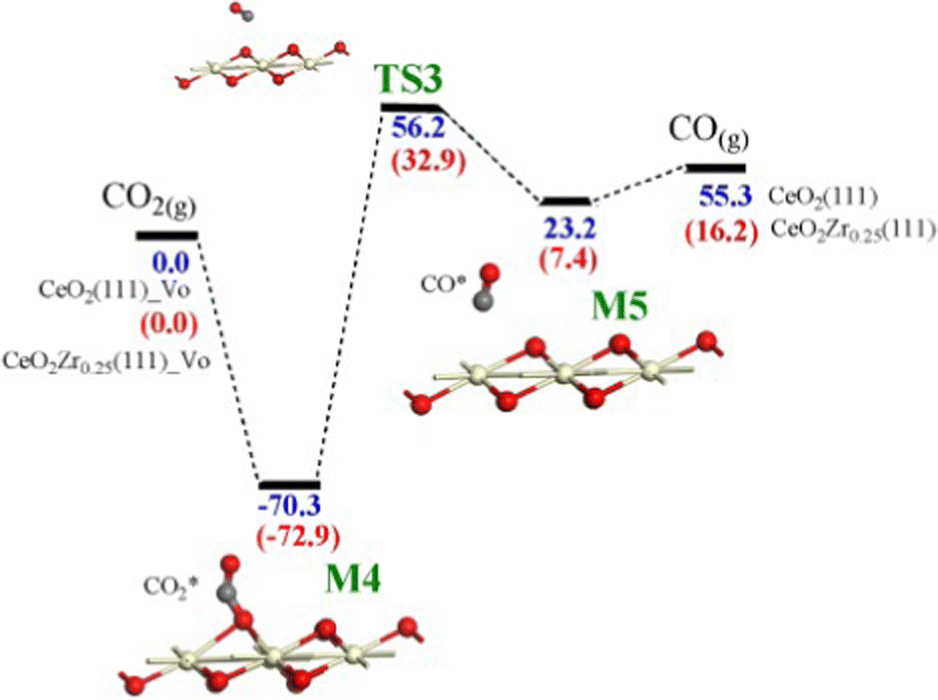 | ||
| Fig. 5 Potential energy surfaces for the successive steps of CO2 splitting calculated by DFT+U on CeO2(111) and Ce0.25Zr0.75O2(111) surfaces with an oxygen vacancy, VO. Values of the energy barriers (in kJ mol−1) are referenced to CO2(g), and those in brackets refer to Ce0.25Zr0.75O2(111). Only the three topmost layers are displayed. Reproduced from ref. 61 with permission from Elsevier. | ||
Fast kinetic implies having a very low activation energy process, which appears to be the case for oxygen release at high temperatures, but both theoretical and experimental reports indicate that dissociation of CO2 and water on reduced oxide surfaces present a significant energetic barrier. This implies that under the operation temperatures required for CO2 and H2O dissociation, the rate for the generation of both CO and H2 is lower than expected by the thermodynamics limit. Catalytic promotion can provide an opportunity for overcoming the kinetic constrains of solid re-oxidation. This is because catalysts can potentially offer alternate reaction pathways with lower activation energy, increasing the rate for the generation of H2 and CO. Therefore, catalysis holds the promise of being a game changer for materials such as many perovskites, which show kinetic limitations at the low-temperature stage of fuel production.
Critically reviewing the relevance of catalysis in thermochemical solar fuels generation, considering the reasons for the so far modest advances in this approach, exploring the possible avenues for improvement, and learning from the coupling of renewable power and catalysis are the main topics of this perspective. However, assessing the complex interplay between different active components simultaneously participating in different solid–gas reactions requires first establishing some clear definitions of what can be considered a catalyst in the context of thermochemical cycles. These fundamental aspects will be discussed in the next section.
Some working definitions: what does count as a heterogeneous catalyst in thermochemical cycles?
A catalyst can be simply described as a substance accelerating the reaction rate without modifying the thermodynamic equilibrium.63 From a kinetic point of view, the catalytic effect implies the lowering of the activation energy barrier to the process by providing an alternative reaction pathway (see Fig. 6b). In this sense, redox oxides used in the thermochemical cycles, as described by eqn (1)–(3), can be considered loosely as catalysts. In fact, although this term is seldom utilized in the recent literature, some pioneering articles about thermochemical solar production referred to the active redox oxides as catalysts.64 In this line, it can be argued that these solids provide a lower energy pathway (Fig. 6a) by interaction with the solid vacancies that allow CO2 and water splitting at a significant rate, while the redox solid is recovered unchanged after completing the process.65 Although considering an isolated cycle, the ratio between the solid oxygen carrier and the gas reagent is relatively high, and this value approaches the typical catalytic/reagent ratio when averaged in many cycles, as expected in practical applications.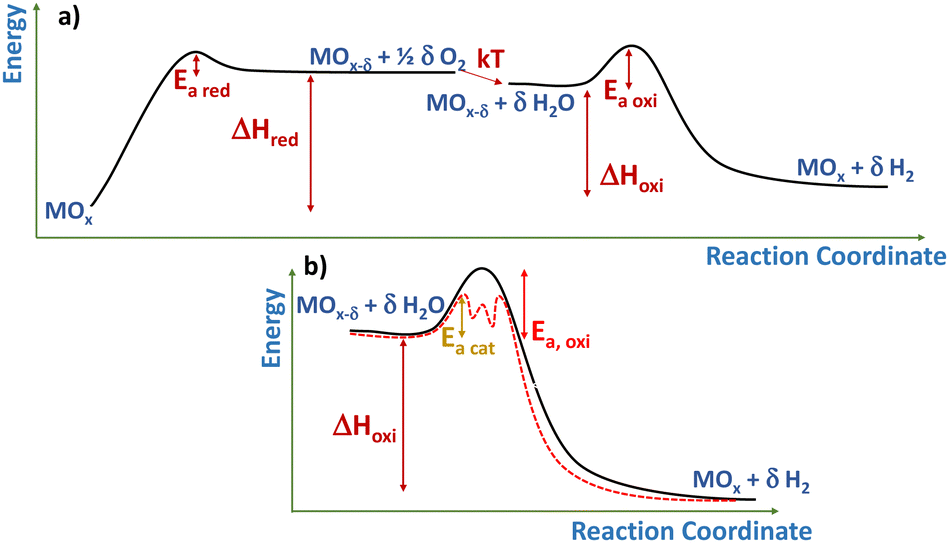 | ||
| Fig. 6 Scheme of the energy landscape of (a) a two-stage thermochemical cycle over a generic non-stoichiometric oxide, MOx and (b) detail the water splitting stage showing an alternative catalytic pathway. ΔHred and ΔHoxi represents, respectively, the reduction or oxidation enthalpy of the oxide, while Ea,red, Ea,oxi, and Ea,cat are the activation energy of, respectively, the reduction, the oxidation and the oxidation in the presence of a catalyst. | ||
The activity of catalysts can be expressed by their turnover frequency (TOF). Although specific operation conditions must be considered to accurately determine this parameter,66 TOF is usually defined as the ratio between the rate of product formation, in moles per time unit, and the number of catalytic sites, expressed in mols. Therefore, this parameter presents reciprocal time (frequency) units. Although determining the nature and concentration of the active sites can be tricky,65 this metric provides a quantitative way to compare conventional catalysis. In contrast, using TOF for establishing a direct meaningful comparison of thermochemical cycles with the equivalent catalytic process is more challenging, since the operation of chemical looping processes is intrinsically discontinuous. Therefore, the duration of a typical thermochemical cycle (from minutes to hours) is at least two orders of magnitude larger than for a typical catalytic one (seconds to milliseconds). Then, while TOF is often larger than 10 s−1 for catalytic processes, for chemical looping system, including solar thermochemical cycles, TOF is the range of ms−1.67 Thus, for example, for methane, dry reforming over NiCo/Ce0.75Zr0.25O2 at 750 °C, the TOF for CO formation is 2.5 s−1.68 In contrast, the TOF of solar thermochemical CO production using reticulated CeO2 is estimated to be about 1.1 × 10−4 s−1 (1.2 × 10−2 considering the peak rate) at 1250 °C. It is important to stress that these differences in the TOF are not necessarily due to a low instantaneous rate. In this way, it has been reported that chemical looping dry reforming of methane over La0.9Sr0.1FeO3/ZrO2 presents a higher activity for syngas production than conventional catalytic process under comparable isothermal conditions.69 In fact, the remarkably lower TOF arises from the relatively high amount of redox oxide and the longer duration of the cycle. Therefore, increasing the frequency of the cycles can be a key parameter to optimize thermochemical fuel production.
Another relevant difference between these two gas–solid processes is that catalysis is intrinsically a surface phenomenon, while transformations of the solids in thermochemical cycles imply the whole material, so diffusion in the bulk of relevant species such vacancies, oxide ions and electrons should also be considered. Accordingly, as mentioned in the previous section, some kinetic models specifically consider the diffusion of defects in the solid.57
For these two kinds of solid–gas processes a complete recovery of the solid in its initial state is expected, which allows continuous operation in both catalytic and thermochemical cycles, but in practice deactivation is an important concern. In the case of thermochemical cycles, structural and morphological degradation can arise due to the severity of the operation conditions that promote sintering and can induce structural in shaped solids during redox cycling. In fact, the durability of the oxygen carriers is an important issue when operating on large scale and under realistic conditions. For example, this is one of the main practical limitations of using Zn ferrites for solar thermochemical hydrogen productions.37 Likewise, although CeO2 has shown excellent cyclability at the lab scale,14 plant operation have revealed minor structural integrity problems of the large ceramic foam components after completing 62 cycles.17
In the context of solarization of liquid fuels production, a recent thermodynamic study has compared the production of syngas from methane steam reforming by a conventional catalytic route with isothermal chemical looping, operating in both cases under 15 bar of pressure.70 Considering the subsequent synthesis of methanol from the obtained solar syngas, the obtained results reveal that energy efficiency using CeO2 as an oxygen carrier in the chemical looping was lower (0.70) than for the catalytic conversion (0.79) due to the higher operation temperature, which in addition should be carefully controlled to avoid carbon deposition caused by methane cracking under these conditions. A direct catalytic route can be in certain cases a more convenient alternative for driving solar thermochemical processes, although the impact on the carbon footprint of each possible process should be also evaluated. Yet, one of the key advantages of the chemical looping approach is that products are obtained separately in each stage, and this allows for easily tuning the H2/CO ratio by adapting the composition of the feed.
With this background about the parallelisms and differences between the two types of processes, catalytic and thermochemical, in this perspective, we approach the use of conventional short-cycle heterogeneous catalysts to enhance longer-period thermochemical cycles. Although many of the redox oxides applied to thermochemical cycles also present relevant catalytic properties that can contribute to the overall activity, here we focus on the incorporation of additional metallic components in low amounts to promote the activity in any of the two stages. This criterion is intended to provide a clear cut definition for identifying these hybrid systems and to explore the potential synergetic effects between catalysis and thermochemical cycles. However, it should be kept in mind that as different reactions can occur simultaneously, cooperative (or competitive) interactions between components of the active solid are likely to happen. In this regard, the redox contribution of the catalytic active component to the thermochemical cycle can be generally disregarded because of its low concentration. Besides, following initial activation, catalytic metals are expected to remain in a reduced state under reaction conditions.67
Ascertaining potentially relevant interactions between components may need operando techniques, which are challenging to be applied at extremely high temperature. Accordingly, in situ studies designed to gather information at these harsh conditions are still scarce, although some recent reports have approached the in situ study of CO2 splitting over CeO2 at 800 °C.71 Furthermore, synchrotron techniques are also increasingly applied to determine mechanistic and structural aspects of thermochemical cycles.72
In the following section, we discuss the rationale for using heterogeneous catalysts at high-temperature processes, setting the limits for the feasibility of their use.
Catalytic promotion of thermochemical cycles
Although the utilization of a catalytic element has received relatively little attention in thermochemical cycles based on redox oxides, in the case of sulfur-based cycles (sulfur–iodine or hybrid), the participation of a heterogeneous catalyst is crucial for achieving efficient hydrogen production.20 This is because the key step of SO3 reduction, which takes place according to the following reaction:| SO3 (g) → SO2 (g) + 1/2 O2 (g) ΔH = 99 kJ mol−1 | (4) |
In contrast with the case of SO3 decomposition, CO2 or water splitting on reduced oxides involve a solid phase as a reactive species. This fact implies that optimization of the reaction rates should consider the contribution of both surface reactions and bulk processes such as vacancies diffusion.75 Incorporation of catalytic metals is expected to affect only surface processes, although cooperative interactions impacting the redox transformations of the solid can also be possible.
Pioneering work performed in 1980 showed an enormous boost in water splitting at a very low temperature, 200 °C, by incorporating Pd, Ni and particularly Pt on CeO2 pre-reduced in hydrogen. This last metal increased the rate of hydrogen production by three orders of magnitude.76 Even though these activation conditions are unlike those required for solar processing, this study confirmed the interest in the incorporation of catalytic active components to the redox oxide for enhancing the hydrogen yield. Likewise, addition of Rh to CeO2 also resulted in an increment of the rate for the isothermal water splitting at 1500 °C, while the effect for oxygen evolution was limited.77 However, the catalytic effect of Rh on hydrogen production is clearly enhanced (see Fig. 7) at temperatures below 1100 °C.56 Thus, the maximum rate for hydrogen generation at 800 °C is almost double over Rh/CeO2 than for CeO2.
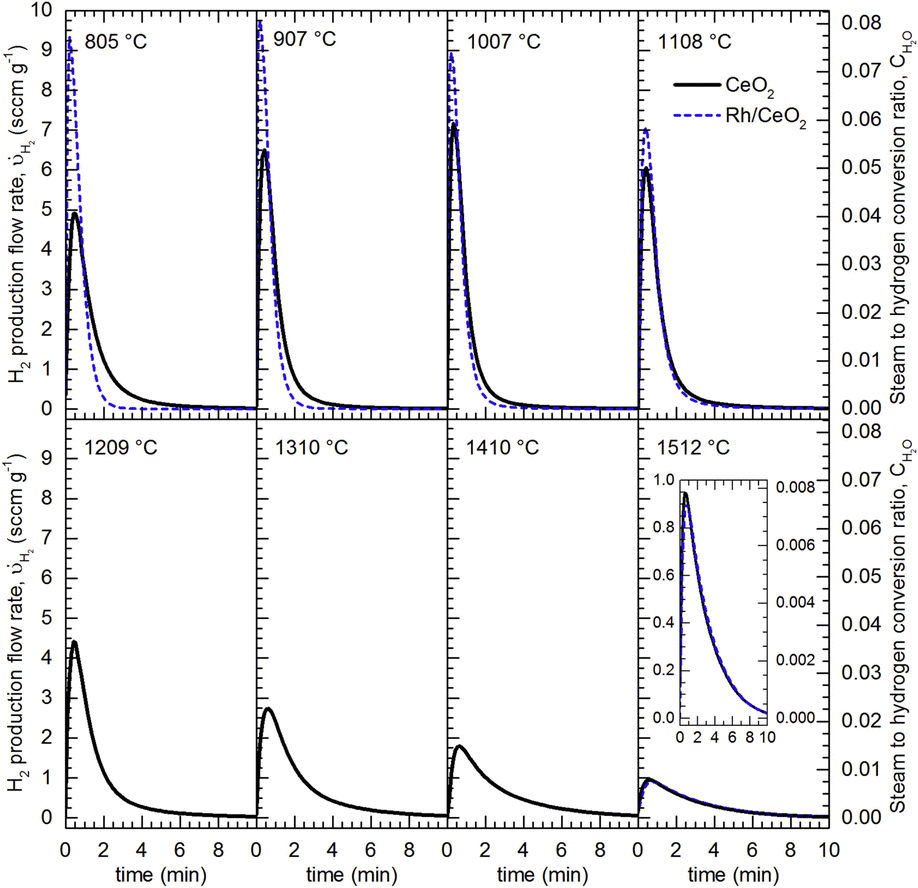 | ||
| Fig. 7 Hydrogen release profiles obtained by water splitting at different temperatures (flow of 278 sccm of 20% H2O in Ar) over CeO2 (Black lines) and Rh/CeO2 (Dashed blue lines) previously reduced in Ar at 1488 °C. Reproduced with permission from ref. 56 with permission from Elsevier. | ||
Remarkably, incorporation of Rh to CeO2 as a catalyst has been used as a way to explore the validity of the thermo-kinetic model,56 which assumes that for high enough temperatures, the rate of surface processes and bulk diffusion is fast enough to be disregarded. This is consistent with the experimentally observed overlapping of the hydrogen production curves for both Rh/CeO2 and CeO2 for assays performed above 1200 °C, as shown in Fig. 7. Therefore, this behavior suggests that there is a limited interval of temperatures for enhancing the thermochemical solar fuels production using a catalytic metal.
Recently, the impact of several noble metals, Ru, Pt and Ir, on the efficiency of CO2 splitting over Ce0.85Zr0.15O2 has been investigated by the Can Li group.78 Dispersion of these metals on Zr-doped ceria prepared via wet impregnation slightly increases oxygen release, but CO2 splitting is significantly promoted by PtOx, and in a larger extent by IrOx. Thus, for CO2 splitting at 800 °C during a 60 min period, the total CO production using a catalyst with 0.4 atom% of IrOx is two-fold that of unmodified Ce0.85Zr0.15O2. If IrOx is incorporated during the mixed oxide synthesis, using a solution combustion method, the obtained materials exceed the rate of CO production of the impregnated catalyst. Thus, the rate of CO evolution is 5.9 mL g−1 for the material loaded with 1 atom% of IrOx for the first 10 min at 800 °C, which represents almost a threefold increment with regards to unmodified Ce0.85Zr0.15O2. According to XPS results, IrOx consists mainly of Ir0 (ca. 80%) but with a significant contribution of Ir4+. Optimal catalytic effect is observed at different metal loadings for materials prepared by combustion (of 1 atom%) or impregnation (0.4 atom%). These differences have been related to variations in the metal dispersion, induced by the preparation route. The role of IrOx in boosting the activity is hypothesized to be due to an increase in the mixed (ionic and electronic) conductivity of Ce oxide, which favors oxygen vacancies formation and diffusion since these charge carriers participate actively in the process of vacancy formation and removal, which occurs through the equilibrium: 1/2O2 + VO + 2e− ↔ O2−. In addition, Ir also plays an important role in easing the rupture of the C–O bond.78 Catalytic enhancement of CO generation is significant in the 700–1000 °C interval and it reaches a maximum in the 800–900 °C range, depending on the Ir content. Although the total production of CO during the whole stage is comparable, the peak production rate is still significantly lower for IrOx/Ce0.85Zr0.15O2 than for CeO2. This is because unmodified ceria shows a more favorable kinetic than the Zr-doped oxides, and this difference is not completely compensated by the promotion effect of the noble metal catalyst. Finally, it is important to note that although the catalytic promotion of IrOx has proven to be preserved during a few cycles, it would be necessary to confirm the long-term stability of these multicomponent materials under realistic conditions.
Catalytic promotion without using any noble was verified on Cr-modified CeO2 by the van Bokhoven group.79 These materials present a remarkably faster rate for CO and hydrogen evolution than undoped oxides. The best results are obtained for the sample containing 15 atom% of Cr and prepared by incipient wetness impregnation. This material shows a rate of CO and H2 production of, respectively, 2.50 and 1.94 mL g−1 min−1 at 800 °C, while for unmodified CeO2, the corresponding rates for CO and H2 are 0.005 and 0.09 mL g−1 min−1, respectively. Furthermore, the rate of oxygen evolution up to 1500 °C in Ar atmosphere, 0.16 mL g−1 min−1, is three times more than that of CeO2. In these materials, formation of the perovskite CeCrO3 is detected in the oxidized materials in a proportion of 7–10% depending on the preparation method. This phase appears to be inert for water and CO2 splitting. However, considering the phase diagram of the Cr–O system, it is expected that metallic chromium can also be present following reduction at high temperatures and low oxygen pressure. Accordingly, the authors suggest that the promotion of CO formation can be explained by considering the catalytic role of Cr0 in favoring CO2 dissociation. In addition, these metallic centers may also play a role in easing the hydrogen release, although the promotion of water splitting with Cr-modified CeO2 is not as efficient as for CO2 activation.
Similarly to the case of CeO2, IrOx has also been applied as catalyst to enhance CO2 splitting over the perovskite LaFeO3 by the Can Li group.80 Likewise, differences in the promotion are also found depending on the preparation route. Solution-combustion method, which is expected to favor the incorporation of Ir into the perovskite lattice, is also in this system more effective than impregnation for promoting CO production. Thus, using the perovskite LaFe0.9Ir0.1O3, which shows some segregation of IrOx, the generation of CO increases about 50% at 1000 °C with regards to the unmodified ferrite during the first 10 minutes, and it is more than 30% larger, after 60 minutes of reaction, reaching a rate of 4.1 mL g−1. In contrast, using an impregnated sample containing 0.7 atom% of Ir, CO2 splitting yields only 3.8 mL g−1 in 60 minutes. Interestingly, for these perovskites, the influence of Ir on the oxygen evolution stage is much more relevant than for Ce1−xZrxO2 materials. Thus, the O2 release for LaFe0.9Ir0.1O3 is about double that for LaFeO3 and the onset temperature of this process, 800 °C, is remarkably lower than for the unmodified ferrite (1250 °C). This has been related to the well-known capacity of IrO2 of catalyzing the formation of O2, which is exploited in electrochemical devices.81 On the other hand, the stability of the CO generation on the Ir-modified materials was confirmed for 10 cycles and, although some decrease in the activity was observed, CO production remained higher than for LaFeO3.
Incorporation of noble metal catalysts for enhancing the release of oxygen from perovskites has been also investigated to enhance the oxygen storage capacity. This concept could be also potentially transferred to the first stage of solar fuels production (eqn (1)). One notable example of the potential of metals to alter the redox properties of the oxide is the promotion of reduction of thin-films of the perovskite SrCoO3 to the brownmillerite SrCoO2.5 by Pt and Ag.82 This process occurs at room temperature under vacuum, although it takes several hours. Nevertheless, in the case of high-temperature solar thermochemical processes, the application of relatively volatile metals such as Ag (melting point 961.78 °C) should be discarded.
Catalysis is very relevant for solarized processes such as chemical looping dry-reforming. This route can be considered to be in the interface of solar processes with conventional chemical looping and thermal catalysis, and it is currently attracting significant interest due to the high productivity of syngas achieved at more moderate temperatures.83 However, this process can only be considered free of fossil carbon when biogas is the only methane source.69,84–86 The use of ex-solution synthesis to anchor metal nanoparticles with catalytic activity such as Ni, Fe, Co or Ru87 into perovskites and CeO288 is currently attracting high interest for this application as a consequence of the thermal stability these metal particles anchored to the redox oxide, which prevents their sintering at high operation temperatures. However, this route of syngas production will not be further discussed here since this study is focused on the production of fully renewable thermochemical fuels.
In brief, the positive effects of the incorporation of some noble and transition metals on redox oxides applied to CO2 and water splitting have been experimentally confirmed. The most representative examples of this catalytic promotion of thermochemical fuel production are summarized in Table 2. However, results so far obtained show, in general, moderate improvements, especially at lower operation temperatures. Nevertheless, information available is still too scarce to assess the real opportunities of these bifunctional materials consisting of both redox and a catalytic component. In this regard, among other aspects, for practical utilization, it would be necessary to explore the stability of these heterogenous solids, performing more than 100 cycles, because phenomena such as encapsulation of the metal or phase segregation may limit their applicability in the long term.
| Reaction | Material | Conditions | Promotion | Ref. |
|---|---|---|---|---|
| WS: water splitting, CDS: carbon dioxide splitting, OER: oxygen evolution reaction. | ||||
| SO3 reduction | LaVO4/SiO2 | 800 °C | Catalysts are always needed in sulfur-based cycles | 74 |
| Direct. Sulfur cycle stage | ||||
| WS | Rh/CeO2 | 800–1100 °C | Double rate than CeO2 at 800 °C | 55 |
| Direct | ||||
| CDS | 1% IrOx/Ce0.85Zr0.15O2 | 700–1000 °C | Threefold increment in the rate at 800 °C with regards to unmodified Ce0.85Zr0.15O2. | 78 |
| Direct | ||||
| OER/WS/CDS | 15% Cr–CeO2 | 800–1500 °C | 500-fold increment for CO and 30 for H2 rates at 800 °C. Triple O2 production at 1500 °C | 79 |
| Direct | ||||
| OER/CDS | LaFe0.9Ir0.1O3 | 800–1250 °C | CO production rate one third larger at 1000 °C. Double production of O2 than LaFeO3 | 80 |
| Direct | ||||
| Methanation | 1% Rh/CeO2 | 350–500 °C | CH4 generation at 500 °C | 93 |
| Indirect | ||||
Catalytic upgrading of thermochemical solar fuels
In addition to its role in boosting the elementary processes of the thermochemical cycles leading to oxygen releases and syngas formation, catalytic transformations are essential for upgrading solar syngas into more valuable molecules, yielding for example kerosene or other hydrocarbon fractions.17,89 This can be uncoupled from the solar thermochemical cycle, as is the case in larger scale demonstration plants so far assayed,89 by accumulating and subsequently processing the generated solar syngas. Then, selecting the catalyst and the operation conditions, either hydrocarbons, obtained by Fischer–Tropsch synthesis, or methanol can be produced.17,89 Using the first procedure, a liquid fraction containing 16% kerosene (hydrocarbons with chain length in the C10–C15 range), and 40% diesel (C15–C20) can be obtained operating at 30 bar and 210 °C using a feed with a H2/CO ratio of 2.15. In addition, Fischer-Tropsch processing of syngas also yielded a wax phase, which has an additional 7% of kerosene and 40% of diesel. Furthermore, the Steinfeld group has shown the technical feasibility of producing fuels from just sunlight together with CO2 and H2O extracted from air in a 5 kW thermal pilot solar facility provided with a module for the capture of these gases from air, and a catalytic processing unit for liquid fuel production.89 This system yields 12.81 liters of syngas per kg of ceria, with a H2/(CO +CO2) molar ratio of 2.7. In addition, to hydrocarbon production, this solar syngas was applied to methanol synthesis using a commercial Cu–ZnO–Al2O3 catalyst. This system achieved a conversion by a single pass of 27% in a recirculation reactor operating at 230 °C and 50 bar. This plant can produce jet fuel with an estimated cost of $1.2–2 per litre, although it has an overall sun-to-liquid energy efficiency of only about 0.8%, considering the energy consumption of all the units. However, it is expected that this value can be greatly improved by optimization of the design of the plant.In order to set the limits of the economic viability, solar thermal plants to produce methanol, with separate units for thermochemical conversion, storage of the produced syngas and the synthesis of methanol have been modeled.18 Assuming that the plants are installed in Almeria (SE Spain), this simulation indicates that with a heliostat field of about 88 hectares and a tower of 220 m, about 22 kt y−1 of methanol can be produced yearly at a cost of 1.14 $ per L. Further upgrading of the model, considering the implementation of an additional unit for the capture of CO2, ideally powered by the waste heat of the plant, predicted the production of 11.8 kt y−1 with a cost of about 3€/l, despite considering a location, (Riyadh, Saudi Arabia), with higher direct irradiation.90
Despite these encouraging results for a sequential transformation, direct catalytic conversion in one step under solar thermal conditions of CO2 and H2O into simple hydrocarbons such as methane is attractive because that molecule is a more useful fuel than syngas and, in principle, a direct route can allow to increase the efficiency and simplify the system. However, one important obstacle to this end is that thermodynamics favors the production of hydrocarbons at much lower temperatures and higher pressures than those required for thermochemical CO2 and water splitting.91 However, despite these significant constraints to the maximum yield that can be achieved by direct catalytic promotion, several reports have experimentally explored the limits of this approach.
The feasibility of generating CH4 directly from CO2 and water was investigated by Haile's group by incorporating 10%wt. Ni on Sm-doped CeO2.92 Following reduction, this material allowed a significant production of CH4 at 400 °C. According to thermodynamic expectations, the activity progressively decayed with increasing temperature, so only CO and H2 are detected at 700 °C. However, in this report, the catalyst was reduced previously in hydrogen at 800 °C, and those conditions allow the achievement of higher non-stoichiometry than under usual solar conditions, and cycling stability was not investigated.
Thermochemical synthesis of CH4 from CO2 and water over Rh/CeO2 was determined to be rather effective between 350 °C and 500 °C, while unmodified ceria yields only syngas.93 In this case, the material was reduced at 1300 °C in an Ar stream, and using a H2/CO ratio of 3 and a pressure of 1 Atms, the yield of methane is about 70% at 500 °C and it drops to about 40% at 600 °C. The reason for this behavior is that as the temperature raises reverse water gas shift, which is also promoted by Rh, becomes the predominant process.91 Characterization of this material indicates that Rh is incorporated into the CeO2 lattice with the formation of oxygen vacancies and it remains stable under the assayed conditions.
In a subsequent more detailed study, it was confirmed that 1 wt% Rh/CeO2 was very effective for CH4 generation from CO2 and H2O at 500 °C following thermal reduction at 1400 °C in Ar, as shown in Fig. 8.94 In contrast, 10 wt%. Ni/CeO2 is less active than the Rh catalyst for CH4 generation at 500 °C following reduction in hydrogen at 600 °C, and it does not show any methanation activity under reduction at 1400 °C in an inert gas atmosphere. This fact has been attributed to the volatilization of Ni at these extreme temperature. In contrast, Rh/CeO2 has proven to keep the activity for methane generation for 58 successive cycles, although a progressive decline of the activity is observed after the first 10 cycles. In addition, it is important to note that the selectivity to methane is low, and hydrogen and CO are also simultaneously produced, although increasing the H2O/CO2 proportion raises the yield of methane.
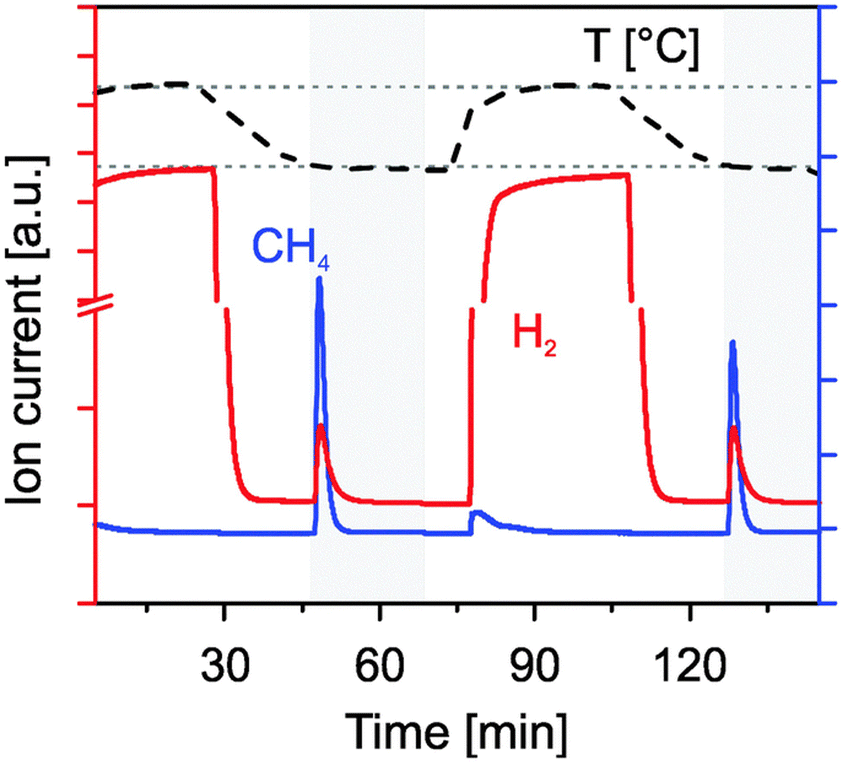 | ||
| Fig. 8 Formation of hydrogen and methane at 500 °C from water (flow of 278 sccm of 20% H2O in Ar) and carbon dioxide by reoxidation of Rh/CeO2 previously reduced in H2/Ar at 500 °C rhodium-doped ceria. Reproduced with permission from ref. 94 with permission from RSC. | ||
On the other hand, continuous catalytic production of solar fuels under the fluctuating conditions imposed by renewable sources has been evaluated in the context of power-to-gas schemes fed by electrochemically generated hydrogen.95 In this context, the effect of variable gas composition on the methanation of CO2 over Ni/CaO–Al2O3 catalyst has been investigated by operando XAS spectroscopy.96 In contrast, thermochemical solar fuel production has always relied on buffering the oscillations due to cyclic syngas production by accumulating the gas. This approach increases the reliability of the catalytic upgrading but makes less efficient the integration of the different units. Therefore, it can be worth exploring also in the case of solar thermal plants the feasibility of a more direct coupling of the solar receiver and the catalytic unit, considering the impact of the variability in the feed composition on fuel productivity.
These results, despite proving the technical feasibility of directly generating solar methane, clearly point out the technical challenges of coupling two processes that present optimal yields under very different conditions. However, the relatively low number of studies and catalyst compositions investigated, calls for additional research effort on optimizing thermochemical and catalytic upgrading processes operating in tandem.
Future outlook
Despite the promising developments in the last decade, the thermochemical route for solar fuel production has not reached yet the solar-to-fuel efficiency required for commercial development, which is estimated to be higher than 10% 2. Reaching this threshold will imply additional research efforts for the development of redox materials with enhanced properties and solar plants with optimized design. Furthermore, thermochemical production of solar fuels critically relies on catalytic processes for converting the primarily produced syngas into high energy molecules such as methanol or different hydrocarbon fractions. Accordingly, finding ways to harmonically coupling these two processes, thermochemical and catalytic, in such a way to efficiently use solar energy also for powering catalytic transformations is of high practical interest. Despite the relatively modest progress so far obtained by hybridizing these technologies, prospects for further enhancement of the catalytic production of solar thermochemical fuels can be foreseen in different areas, as discussed below.Operation at very high temperatures tends to speed-up the processes, and accordingly, direct catalytic promotion becomes irrelevant, as it has experimentally found.56 However, there are still opportunities for achieving a reduction of the redox carriers at lower temperatures by incorporating an O2 evolving catalyst and, more clearly for water and CO2 reactions, which take place at lower temperatures, where catalytic promotion is expected to be relevant. In this respect, the moderate promotion observed by noble metals such as Ir could be further improved by using ex-solution synthesis that can stabilize metal particles and maximize the interaction with the metal oxide matrix. This strategy is currently applied to SOFC and other electrochemical devices, and for chemical looping dry reforming but to our knowledge it has not been explored for the direct catalytic enhancement of CO2 or water splitting. On the other side, the use of redox active transition metals as dopants such as V or Cr among others, which present redox and catalytic properties, has already demonstrated efficacy for promoting the activity of CeO2, and it can be worth exploring in more detail their possible benefits. Obviously, many other catalytic additives, yet untested, can be potentially used for boosting thermochemical cycles.
Further development of in situ and operando studies of these catalytic systems can be of high value not only from a fundamental point of view to understand the mechanisms at the molecular scale of these processes, but likewise to rationally improve them. However, the extreme operation temperatures impose significant limitations for some spectroscopies measurements, although XAS97 and Raman71 measurements are possible for redox systems at relevant conditions. Furthermore, in situ XRD, which provides information about phase evolution, can be also of interest for investigating thermochemical processes.72
In the case of indirect, two steps transformations to upgrade solar thermochemical fuels improving the design of the solar reactors working in tandem with a catalytic one is essential to manage the flows of heat and mass efficiently. This kind of consecutive reactor has been proposed for coupling sequential catalytic reactions such as for example the production of ethanol from syngas98 or the recently proposed production of C3 oxygenates from CO2 and ethane.99 The experience gained with these tandem reactors can give insights into how to maximize yields in solar applications.
With regards to efficiently use solar power, integration of thermochemical storage systems100 can be a suitable approach to deal with the difference between the optimal operating temperatures for thermochemical and catalytic processes. However, extensive work on modeling and design will be necessary to exploit these multiunit systems efficiently. Besides, a moderate increase in the operation pressure could reduce the temperature gap between the production and the catalytic upgrading of syngas, and accordingly, this strategy can open a window for favorable operation conditions, which is worth exploring. This may also require the development of catalysts with high stability and adapted to these novel conditions. In addition, it would be necessary to further investigate how to optimize the operation under the fluctuating conditions imposed on the one hand by the thermochemical cycles and on the other hand by the inherent variability of solar flux.101,102
In summary, this panorama of the application of catalysis for promoting the production of solar thermochemical fuels has revealed that, despite the strong limitations dictated by the thermodynamics constraints, there are interesting opportunities to couple these two processes to maximize the efficiency in storing the solar energy in chemical bonds. So far, research on coupling catalysis with thermochemical processes has been relatively scarce but it seems highly possible that the technological and scientific interest in this area will increase in the near future.
Author contributions
JMC: conceptualization, writing – original draft, and funding acquisition; AB: writing – review & editing, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors appreciate the financial support from the ACES2030 program of the “Comunidad de Madrid” and European Structural Funds (P2018/EMT-4319). A. B. also thanks the “Talento” program of the “Comunidad de Madrid” for funding the project “SmartSolFuel”.References
- D. Henner, REN21, Ren21, 2017.
- H. Song, S. Luo, H. Huang, B. Deng and J. Ye, ACS Energy Lett., 2022, 7, 1043–1065 CrossRef CAS.
- Y. Zhao, W. Gao, S. Li, G. R. Williams, A. H. Mahadi and D. Ma, Joule, 2019, 3, 920–937 CrossRef CAS.
- A. M. Oliveira, R. R. Beswick and Y. Yan, Curr. Opin. Chem. Eng., 2021, 33, 100701 CrossRef.
- M. M. May and K. Rehfeld, Adv. Energy Mater., 2022, 12, 2103801 CrossRef CAS.
- J. Jia, L. C. Seitz, J. D. Benck, Y. Huo, Y. Chen, J. Wei, D. Ng, T. Bilir, J. S. Harris and T. F. Jaramillo, Nat. Commun., 2016, 7, 13237 CrossRef CAS PubMed.
- H. Chen, L. Song, S. Ouyang, J. Wang, J. Lv and J. Ye, Adv. Sci., 2019, 6, 1900465 CrossRef PubMed.
- J. Schneidewind, Adv. Energy Mater., 2022, 12, 2200342 CrossRef CAS.
- C. Ros, T. Andreu and J. R. Morante, J. Mater. Chem. A, 2020, 8, 10625–10669 RSC.
- W. H. Cheng, M. H. Richter, M. M. May, J. Ohlmann, D. Lackner, F. Dimroth, T. Hannappel, H. A. Atwater and H. J. Lewerenz, ACS Energy Lett., 2018, 3, 1795–1800 CrossRef CAS.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S. T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- P. Zhou, I. A. Navid, Y. Ma, Y. Xiao, P. Wang, Z. Ye, B. Zhou, K. Sun and Z. Mi, Nature, 2023, 613, 66–70 CrossRef CAS PubMed.
- A. Iglesias-Juez, F. Fresno, J. M. Coronado, J. Highfield, A. M. Ruppert and N. Keller, Curr. Opin. Green Sustain. Chem., 2022, 37, 100652 CrossRef CAS.
- D. Marxer, P. Furler, M. Takacs and A. Steinfeld, Energy Environ. Sci., 2017, 10, 1142–1149 RSC.
- W.-H. H. Cheng, A. De La Calle, H. A. Atwater, E. B. Stechel and C. Xiang, ACS Energy Lett., 2021, 6, 3113 Search PubMed.
- M. Romero and A. Steinfeld, Energy Environ. Sci., 2012, 5, 9234–9245 RSC.
- S. Zoller, E. Koepf, D. Nizamian, M. Stephan, A. Patané, P. Haueter, M. Romero, J. González-Aguilar, D. Lieftink, E. de Wit, S. Brendelberger, A. Sizmann and A. Steinfeld, Joule, 2022, 6, 1606–1616 CrossRef CAS PubMed.
- N. Monnerie, P. G. Gan, M. Roeb and C. Sattler, Int. J. Hydrogen Energy, 2020, 45, 26117–26125 CrossRef CAS.
- A. Bayón, V. A. De La Penã O’Shea, D. P. Serrano and J. M. Coronado, J. CO2 Util., 2020, 42, 101264 CrossRef.
- C. Sattler, M. Roeb, C. Agrafiotis and D. Thomey, Sol. Energy, 2017, 156, 30–47 CrossRef CAS.
- S. Abanades and G. Flamant, Sol. Energy, 2006, 80, 1611–1623 CrossRef CAS.
- W. C. Chueh, C. Falter, M. Abbott, D. Scipio, P. Furler, S. M. Haile and A. Steinfeld, Science, 2010, 330, 1797–1801 CrossRef CAS PubMed.
- S. S. Naghavi, A. A. Emery, H. A. Hansen, F. Zhou, V. Ozolins and C. Wolverton, Nat. Commun., 2017, 8, 1–6 CrossRef CAS PubMed.
- Y. Hao, C. K. Yang and S. M. Haile, Chem. Mater., 2014, 26, 6073–6082 CrossRef CAS.
- J. R. Scheffe, R. Jacot, G. R. Patzke and A. Steinfeld, J. Phys. Chem. C, 2013, 117, 24104–24110 CrossRef CAS.
- A. J. Carrillo, J. L. M. Rupp and J. M. Coronado, Energy Storage and Conversion Materials, The Royal Society of Chemistry, 2020, pp. 136–187 Search PubMed.
- M. Kubicek, A. H. Bork and J. L. M. Rupp, J. Mater. Chem. A, 2017, 5, 11983–12000 RSC.
- J. R. Scheffe, D. Weibel and A. Steinfeld, Energy Fuels, 2013, 27, 4250–4257 CrossRef CAS.
- M. Ezbiri, M. Takacs, D. Theiler, R. Michalsky and A. Steinfeld, J. Mater. Chem. A, 2017, 5, 4172–4182 RSC.
- A. H. McDaniel, E. C. Miller, D. Arifin, A. Ambrosini, E. N. Coker, R. O’Hayre, W. C. Chueh, J. Tong, W. C. Chueh and J. Tong, Energy Environ. Sci., 2013, 6, 2424–2428 RSC.
- X. Qian, J. He, E. Mastronardo, B. Baldassarri, W. Yuan, C. Wolverton and S. M. Haile, Matter, 2021, 4, 688–708 CrossRef CAS.
- D. R. Barcellos, M. D. Sanders, J. Tong, A. H. Mcdaniel and R. P. O’hayre, J. Energy Environ. Sci, 2018, 11, 3256 RSC.
- A. H. Bork, M. Kubicek, M. Struzik and J. L. M. Rupp, J. Mater. Chem. A, 2015, 3, 15546–15557 RSC.
- A. H. Bork, A. J. Carrillo, Z. D. Hood, B. Yildiz and J. L. M. Rupp, ACS Appl. Mater. Interfaces, 2020, 12, 32622–32632 CrossRef CAS PubMed.
- A. A. Emery, J. E. Saal, S. Kirklin, V. I. Hegde and C. Wolverton, Chem. Mater., 2016, 28, 5621–5634 CrossRef CAS.
- A. Bayon, A. de la Calle, K. K. Ghose, A. Page and R. McNaughton, Int. J. Hydrogen Energy, 2020, 45, 12653–12679 CrossRef CAS.
- M. Roeb, J. P. Säck, P. Rietbrock, C. Prahl, H. Schreiber, M. Neises, L. de Oliveira, D. Graf, M. Ebert, W. Reinalter, M. Meyer-Grünefeldt, C. Sattler, A. Lopez, A. Vidal, A. Elsberg, P. Stobbe, D. Jones, A. Steele, S. Lorentzou, C. Pagkoura, A. Zygogianni, C. Agrafiotis and A. G. Konstandopoulos, Sol. Energy, 2011, 85, 634–644 CrossRef CAS.
- R. R. Bhosale, A. Kumar, F. AlMomani, U. Ghosh, P. Sutar, G. Takalkar, A. Ashok and I. Alxneit, Ceram. Int., 2017, 43, 5150–5155 CrossRef CAS.
- S. Zhai, J. Rojas, N. Ahlborg, K. Lim, M. F. Toney, H. Jin, W. C. Chueh and A. Majumdar, Energy Environ. Sci., 2018, 11, 2172–2178 RSC.
- X. Zhai, F. Ding, Z. Zhao, A. Santomauro, F. Luo and J. Tong, Commun. Mater., 2022, 3, 42 CrossRef CAS.
- E. Mastronardo, X. Qian, J. M. Coronado and S. M. Haile, J. Mater. Chem. A, 2020, 8, 8503–8517 RSC.
- A. Bayon, A. de la Calle, E. B. Stechel and C. Muhich, Energy Technol., 2022, 10, 2100222 CrossRef CAS.
- J. Lou, Z. Tian, Y. Wu, X. Li, X. Qian, S. M. Haile and Y. Hao, Sol. Energy, 2022, 241, 504–514 CrossRef CAS.
- C. L. Muhich, S. Blaser, M. C. Hoes and A. Steinfeld, Int. J. Hydrogen Energy, 2018, 43, 18814–18831 CrossRef CAS.
- A. de la Calle, I. Ermanoski and E. B. Stechel, Int. J. Hydrogen Energy, 2022, 47, 10474–10482 CrossRef CAS.
- S. Li, V. M. Wheeler, P. B. Kreider and W. L. Lipiński, Energy Fuels, 2018, 32(10), 10838–10847 CrossRef CAS.
- B. Bulfin, F. Call, J. Vieten, M. Roeb, C. Sattler and I. V. Shvets, J. Phys. Chem. C, 2016, 120, 2027–2035 CrossRef CAS.
- A. Le Gal, S. Abanades and G. Flamant, Energy Fuels, 2011, 25, 4836–4845 CrossRef CAS.
- D. Arifin and A. W. Weimer, Sol. Energy, 2018, 160, 178–185 CrossRef CAS.
- D. Arifin, A. Ambrosini, S. A. Wilson, B. Mandal, C. L. Muhich and A. W. Weimer, Int. J. Hydrogen Energy, 2020, 45, 160–174 CrossRef CAS.
- B. Bulfin, A. J. Lowe, K. A. Keogh, B. E. Murphy, O. Lübben, S. A. Krasnikov and I. V. Shvets, J. Phys. Chem. C, 2013, 117, 24129–24137 CrossRef CAS.
- L. J. Venstrom, R. M. De Smith, R. Bala Chandran, D. B. Boman, P. T. Krenzke and J. H. Davidson, Energy Fuels, 2015, 29, 8168–8177 CrossRef CAS.
- H. Il, Ji, T. C. Davenport, M. J. Ignatowich and S. M. Haile, Phys. Chem. Chem. Phys., 2017, 19, 7420–7430 RSC.
- H. Il, Ji, T. C. Davenport, C. B. Gopal and S. M. Haile, Phys. Chem. Chem. Phys., 2016, 18, 21554–21561 RSC.
- M. J. Ignatowich, A. H. Bork, T. C. Davenport, J. L. M. Rupp, C. K. Yang, Y. Yamazaki and S. M. Haile, MRS Commun., 2017, 7, 873–878 CrossRef CAS.
- T. C. Davenport, M. Kemei, M. J. Ignatowich and S. M. Haile, Int. J. Hydrogen Energy, 2017, 42, 16932–16945 CrossRef CAS.
- Z. Zhao, M. Uddi, N. Tsvetkov, B. Yildiz and A. F. Ghoniem, J. Phys. Chem. C, 2016, 120, 16271–16289 CrossRef CAS.
- S. G. Rudisill, L. J. Venstrom, N. D. Petkovich, T. Quan, N. Hein, D. B. Boman, J. H. Davidson and A. Stein, J. Phys. Chem. C, 2013, 117, 1692–1700 CrossRef CAS.
- H. A. Hansen and C. Wolverton, J. Phys. Chem. C, 2014, 118, 27402–27414 CrossRef CAS.
- D. Marrocchelli and B. Yildiz, J. Phys. Chem. C, 2012, 116, 2411–2424 CrossRef CAS.
- S. E. Rawadieh, M. Altarawneh, I. S. Altarawneh, M. A. Batiha and L. A. Al-Makhadmeh, Mol. Catal., 2020, 498, 111256 CrossRef CAS.
- K. Razmgar, T. Shittu, I. Oluwoye, A. Khaleel, G. Senanayake and M. Altarawneh, J. CO2 Util., 2023, 67, 102339 CrossRef CAS.
- E. Roduner, Chem. Soc. Rev., 2014, 43, 8226–8239 RSC.
- A. Le Gal and S. Abanades, Int. J. Hydrogen Energy, 2011, 36, 4739–4748 CrossRef CAS.
- G. B. Marin, V. V. Galvita and G. S. Yablonsky, J. Catal., 2021, 404, 745–759 CrossRef CAS.
- S. Kozuch and J. M. L. Martin, ACS Catal., 2012, 2, 2787–2794 CrossRef CAS.
- M. S. C. Chan, E. Marek, S. A. Scott and J. S. Dennis, J. Catal., 2018, 359, 1–7 CrossRef CAS.
- M. A. Vasiliades, C. M. Damaskinos, P. Djinović, A. Pintar and A. M. Efstathiou, Catal. Commun., 2021, 149, 106237 CrossRef CAS.
- D. Sastre, C. Á. Galván, P. Pizarro and J. M. Coronado, Fuel, 2022, 309, 122122 CrossRef CAS.
- B. Bulfin, S. Ackermann, P. Furler and A. Steinfeld, Sol. Energy, 2021, 215, 169–178 CrossRef CAS.
- E. Sediva, A. J. Carrillo, C. E. Halloran and J. L. M. Rupp, ACS Appl. Energy Mater., 2021, 4, 1474–1483 CrossRef CAS.
- S. Shulda, R. T. Bell, N. A. Strange, L. Metzroth, K. N. Heinselman, S. Sainio, S. Roychoudhury, D. Prendergast, A. H. McDaniel and D. S. Ginley, Front. Energy Res., 2022, 10, 1–18 Search PubMed.
- H. A. Khan, M. I. Iqbal, A. Jaleel, I. Abbas, S. A. Abbas and K. Deog-Jung, Int. J. Hydrogen Energy, 2019, 44, 2312–2322 CrossRef CAS.
- M. Machida, A. Ikematsu, A. S. M. M. Nur and H. Yoshida, ACS Appl. Energy Mater., 2021, 4, 1696–1703 CrossRef CAS.
- H. Chang, E. Bjørgum, O. Mihai, J. Yang, H. L. Lein, T. Grande, S. Raaen, Y. A. Zhu, A. Holmen and D. Chen, ACS Catal., 2020, 10, 3707–3719 CrossRef CAS.
- K. Otsuka, M. Hatano and A. Morikawa, J. Catal., 1983, 79, 493–496 CrossRef CAS.
- Y. Hao, C. K. Yang and S. M. Haile, Phys. Chem. Chem. Phys., 2013, 15, 17084–17092 RSC.
- Z. Chen, Q. Jiang, H. An, J. Zhang, S. Hao, X. Li, L. Cai, W. Yu, K. You, X. Zhu and C. Li, ACS Catal., 2022, 12, 7719–7736 CrossRef CAS.
- S. Mostrou, R. Büchel, S. E. Pratsinis and J. A. van Bokhoven, Appl. Catal. A Gen., 2017, 537, 40–49 CrossRef CAS.
- Q. Jiang, Z. Chen, J. Tong, M. Yang, Z. Jiang and C. Li, ACS Catal., 2016, 6(2), 1172–1180 CrossRef CAS.
- V. K. Puthiyapura, S. Pasupathi, H. Su, X. Liu, B. Pollet and K. Scott, Int. J. Hydrogen Energy, 2014, 39, 1905–1913 CrossRef CAS.
- K. Wang, C. Han, Z. Shao, J. Qiu, S. Wang, S. Liu, K. Wang, C. Han, Z. P. Shao, S. M. Liu, J. S. Qiu and S. B. Wang, Adv. Funct. Mater., 2021, 31, 2102089 CrossRef CAS.
- S. Bhattar, M. A. Abedin, S. Kanitkar and J. J. Spivey, Catal. Today, 2021, 365, 2–23 CrossRef CAS.
- O. Kwon, R. Huang, T. Cao, J. M. Vohs and R. J. Gorte, Catal. Today, 2021, 382, 142–147 CrossRef CAS.
- S. Joo, A. Seong, O. Kwon, K. Kim, J. H. Lee, R. J. Gorte, J. M. Vohs, J. W. Han and G. Kim, Sci. Adv., 2020, 6, 1–9 Search PubMed.
- X. Gao, G. Liu, Y. Zhu, P. Kreider, A. Bayon, T. Gengenbach, T. Lu, Y. Liu, J. Hinkley, W. Lipiński and A. Tricoli, Nano Energy, 2018, 50, 347–358 CrossRef CAS.
- F. Schrenk, L. Lindenthal, H. Drexler, G. Urban, R. Rameshan, H. Summerer, T. Berger, T. Ruh, A. K. Opitz and C. Rameshan, Appl. Catal., B, 2022, 318, 121886 CrossRef CAS.
- A. J. Carrillo, L. Navarrete, M. Laqdiem, M. Balaguer and J. M. Serra, Mater. Adv., 2021, 2, 2924 RSC.
- R. Schäppi, D. Rutz, F. Dähler, A. Muroyama, P. Haueter, J. Lilliestam, A. Patt, P. Furler and A. Steinfeld, Nature, 2022, 601, 63–68 CrossRef PubMed.
- E. Prats-Salvado, N. Monnerie and C. Sattler, Energies, 2021, 14, 4818 CrossRef CAS.
- J. Gao, Y. Wang, Y. Ping, D. Hu, G. Xu, F. Gu and F. Su, RSC Adv., 2012, 2, 2358–2368 RSC.
- W. C. Chueh and S. M. Haile, ChemSusChem, 2009, 2, 735–739 CrossRef CAS PubMed.
- F. Lin, A. Wokaun and I. Alxneit, Energy Procedia, 2015, 69, 1790–1799 CrossRef CAS.
- F. Lin, M. Rothensteiner, I. Alxneit, J. A. Van Bokhoven and A. Wokaun, Energy Environ. Sci., 2016, 9, 2400–2409 RSC.
- D. A. Kuznetsov, B. Han, Y. Yu, R. R. Rao, J. Hwang, Y. Román-Leshkov and Y. Shao-Horn, Joule, 2018, 2, 225–244 CrossRef CAS.
- B. Mutz, H. W. P. Carvalho, S. Mangold, W. Kleist and J. D. Grunwaldt, J. Catal., 2015, 327, 48–53 CrossRef CAS.
- A. J. Carrillo, L. E. Chinchilla, A. Iglesias-Juez, S. Gutiérrez-Rubio, D. Sastre, P. Pizarro, A. B. Hungría and J. M. Coronado, Small Methods, 2021, 5, 2100550 CrossRef CAS PubMed.
- C. C. Amoo, C. Xing, N. Tsubaki and J. Sun, ACS Cent. Sci., 2022, 8, 1047–1062 CrossRef CAS PubMed.
- Z. Xie, Y. Xu, M. Xie, X. Chen, J. H. Lee, E. Stavitski, S. Kattel and J. G. Chen, Nat. Commun., 2020, 11, 1–8 CrossRef PubMed.
- A. Lidor, Y. Aschwanden, J. Häseli, P. Reckinger, A. Steinfeld, A. Lidor, Y. Aschwanden, J. Häseli and P. Haueter, Appl. Energy, 2023, 329, 120211 CrossRef CAS.
- U. Ash-Kurlander, O. Martin, L. D. Fontana, V. R. Patil, M. Bernegger, C. Mondelli, J. Pérez-Ramírez and A. Steinfeld, Energy Technol., 2016, 4, 565–572 CrossRef CAS.
- A. de la Calle and A. Bayon, Int. J. Hydrogen Energy, 2019, 44, 1409–1424 CrossRef CAS.
| This journal is © the Owner Societies 2023 |