
Carbon dioxide hydrogenation over the carbon-terminated niobium carbide (111) surface: a density functional theory study
Saeedeh
Sarabadani Tafreshi
 *a,
Mahkameh
Ranjbar
a,
Maryam
Jamaati
b,
S. F. K. S.
Panahi
b,
Narges
Taghizade
b,
Mostafa
Torkashvand
a and
Nora H.
de Leeuw
*cd
*a,
Mahkameh
Ranjbar
a,
Maryam
Jamaati
b,
S. F. K. S.
Panahi
b,
Narges
Taghizade
b,
Mostafa
Torkashvand
a and
Nora H.
de Leeuw
*cd
aDepartment of Chemistry, Amirkabir University of Technology (Tehran Polytechnic), No. 350, Hafez Avenue, 1591634311 Tehran, Iran. E-mail: s.s.tafreshi@aut.ac.ir
bDepartment of Physics, Iran University of Science and Technology, Narmak, 16846-13114 Tehran, Iran
cSchool of Chemistry, University of Leeds, LT2 9JT Leeds, UK. E-mail: N.H.deLeeuw@leeds.ac.uk
dDepartment of Earth Sciences, Utrecht University, 3584 CB Utrecht, The Netherlands
First published on 21st December 2022
Abstract
Carbon dioxide (CO2) hydrogenation is an energetic process which could be made more efficient through the use of effective catalysts, for example transition metal carbides. Here, we have employed calculations based on the density functional theory (DFT) to evaluate the reaction processes of CO2 hydrogenation to methane (CH4), carbon monoxide (CO), methanol (CH3OH), formaldehyde (CH2O), and formic acid (HCOOH) over the carbon-terminated niobium carbide (111) surface. First, we have studied the adsorption geometries and energies of 25 different surface-adsorbed species, followed by calculations of all of the elementary steps in the CO2 hydrogenation process. The theoretical findings indicate that the NbC (111) surface has higher catalytic activity towards CO2 methanation, releasing 4.902 eV in energy. CO represents the second-most preferred product, followed by CH3OH, CH2O, and HCOOH, all of which have exothermic reaction energies of 4.107, 2.435, 1.090, and 0.163 eV, respectively. Except for the mechanism that goes through HCOOH to produce CH2O, all favourable hydrogenation reactions lead to desired compounds through the creation of the dihydroxycarbene (HOCOH) intermediate. Along these routes, CH3* hydrogenation to CH4* has the highest endothermic reaction energy of 3.105 eV, while CO production from HCO dehydrogenation causes the highest exothermic reaction energy of −3.049 eV. The surface-adsorbed CO2 hydrogenation intermediates have minimal effect on the electronic structure and interact only weakly with the surface. Our results are consistent with experimental observations.
1. Introduction
Recent years have seen a significant increase in interest in the remarkable physical and chemical features of transition metal carbides (TMCs).1,2 They are carbon-containing close-packed metals exhibiting a remarkable mix of properties, combining the good electric and thermal conductance of transition metals, coupled with the toughness and structural coordination of covalent solids, and the high melting temperatures of ionic solids.3–5 TMCs have been proposed as viable alternatives for valuable metals as a result of their intrinsic advantages of abundance, high reactivity, endurance, and affordability. Owing to their three distinct types of bonding: metallic, ionic, and covalent they are promising materials for a broad spectrum of technical applications of economic value.6,7 The discovery in a seminal study by Levy and Boudart8 of platinum-like behaviour in TMCs equivalent to that of Pt-group metals attracted significant interest in their potential use in a broad variety of catalytic processes, e.g. the water–gas shift,9–11 CO hydrogenation12 and CO2 hydrogenation12–20 reactions.Carbon dioxide is one of the Earth's most significant pollutants and a potent active greenhouse gas, and rising CO2 levels have been linked to global warming and other negative effects on the environment.13,19,21 As a result, substantial research has been conducted into the alleviation of its adverse effects and the transformation of CO2 into useful products, particularly in the context of producing sustainable energy.22–24 However, the stability of CO2 and the high adsorption energy restricts the number of potential viable catalysts.13,20,25 A high activation barrier in C–O bond breaking requires highly efficacious catalysts.
Previous studies have been carried out to gain insight into CO2 adsorbtion and conversion into usable hydrocarbon alternatives at low-index TMC surfaces,13,15,20,22 where niobium carbide has been shown to be a substantial generator of CO and the key product of CH4.15 As such, NbC has been the topic of a number of experiments to investigate and customise its various characteristics.26,27 Two often-researched NbC crystalline surfaces are the (100) plane, containing both metal and carbon atoms, and the (111) surface, which is terminated by either metal or carbon atoms that alternate in each layer.7 The (111) surface of NbC has been shown to be more active towards gas adsorption than the (100) surface, owing to the higher electronic density of states around the Fermi level at the (111) surface.7,28–31
Our previous study has investigated CO2 hydrogenation at the niobium-terminated NbC (111) surface,32 where we found that the most likely products are CH4 and CO. In contrast to the carbon-terminated (111) surfaces of TMCs at the top of the TM group, like the TiC (111) surface which proved to be unstable,33 the carbon-terminated NbC surface was found to be important in a number of studies.5,7,20,34 However, although an experimental study on CO2 hydrogenation at the NbC (111) surface has been carried out by Prosoff and coworkers,15 who found that CO and CH4 are the main products, theoretical investigations of these processes, providing further detail on the reaction mechanism and formed intermediates, are still lacking. In this work, we have therefore carried out a comprehensive computational investigation of the network of reactions involved in CO2 hydrogenation at the carbon-terminated NbC (111) surface.
Identifying the mechanism of CO2 hydrogenation and the energetics of the elementary steps is important if we are to gain a better understanding of the formation and scission of the bonds which can aid in the development of active catalysts for the conversion of CO2 into methane (CH4), carbon monoxide (CO), methanol (CH3OH), formaldehyde (CH2O), and formic acid (HCOOH) during the hydrogenation mechanism. We have used calculations based on the density functional theory (DFT) with periodic surface models to elucidate the CO2 hydrogenation intermediates and elementary steps on the carbon-terminated NbC (111), where we have focused on the geometric and energetic properties of the adsorbed species, as well as an in-depth examination of the electronic characteristics of the CO2 hydrogenation products on the C-terminated NbC (111) surface, which is critical for the development of more effective TMC catalysts. In agreement with experiment,15 we found that CH4 and CO are indeed the predominant products of CO2 hydrogenation at the C-terminated NbC (111) surface.
2. Computational details
Method
The Vienna Ab Initio Simulation Package (VASP)35,36 has been used to perform the computations. The Perdew–Burke–Ernzerhof (PBE) variant of the extended gradient estimation within the projector augmented wave (PAW) approach has been used for the total energy calculations.37–39 As recommended by Grimme, the DFT-D340 inclusion of the dispersion interactions was also taken into consideration in order to enhance the accuracy of the energy characterization of each system. Plane waves featuring a cut-off energy of 600 eV were used to extend the Kohn–Sham orbitals in the bulk and slab calculations. Monkhorst-Pack grids41 with meshes of 11 × 11 × 11 and 4 × 4 × 1 points were used to sample the Brillouin domain of the bulk and NbC (111) surface, respectively. The remaining forces and energies were adjusted until they fell below 0.01 eV Å−1 and 10−5 eV, respectively. Interference between the simulation system and its images was avoided by creating a vacuum space larger than 20 Å between the surface slabs. DFT-D3 is the primary methodology used for these types of calculations in the field of heterogeneous catalysis, with similar set-ups having been reported widely in the literature, thus enabling direct comparison of our results with previous work.32,42,43 Solvent effects were not included in our calculations. Although these may affect the adsorption characteristics of the various molecules at the surface, they would not alter significantly the relative energies of the adsorbed intermediates and thus would not affect the trends identified in this work.Model
The NbC (111) surface has a hexagonal close-packed (hcp) composition terminated by either Nb or carbon atoms, which comprise the alternating layers. Other than the carbon-terminated NbC (111) surface, previous investigations in the literature have shown that carbon-terminated TMC (111) surfaces were unsustainable.5,7,20,34 Here, we have solely focused on the C-terminated NbC (111) surface, as this termination may play an essential role in the conversion of CO2 (see Fig. 1a). The calculated lattice constant of a0 = 4.53 Å is in excellent agreement with the results of previous numerical (a0 = 4.51, 4.488 Å)3 and empirical (a0 = 4.469 Å)44 studies. To model the surface, a supercell of 112 atoms was used, consisting of seven atomic layers, four of which were kept frozen at their bulk optimised positions to mimic the bulk material. To determine the most stable and favourable adsorption geometry, each adsorbate was initially put in numerous surface sites before full geometry optimisation. The adsorption energy of each adsorbate (Eads) can be determined as follows:| Eads = EA+slab − EA − Eslab | (1) |
| ρ = ρMolecule+NbC(111) − ρNbC(111) − ρMolecule | (2) |
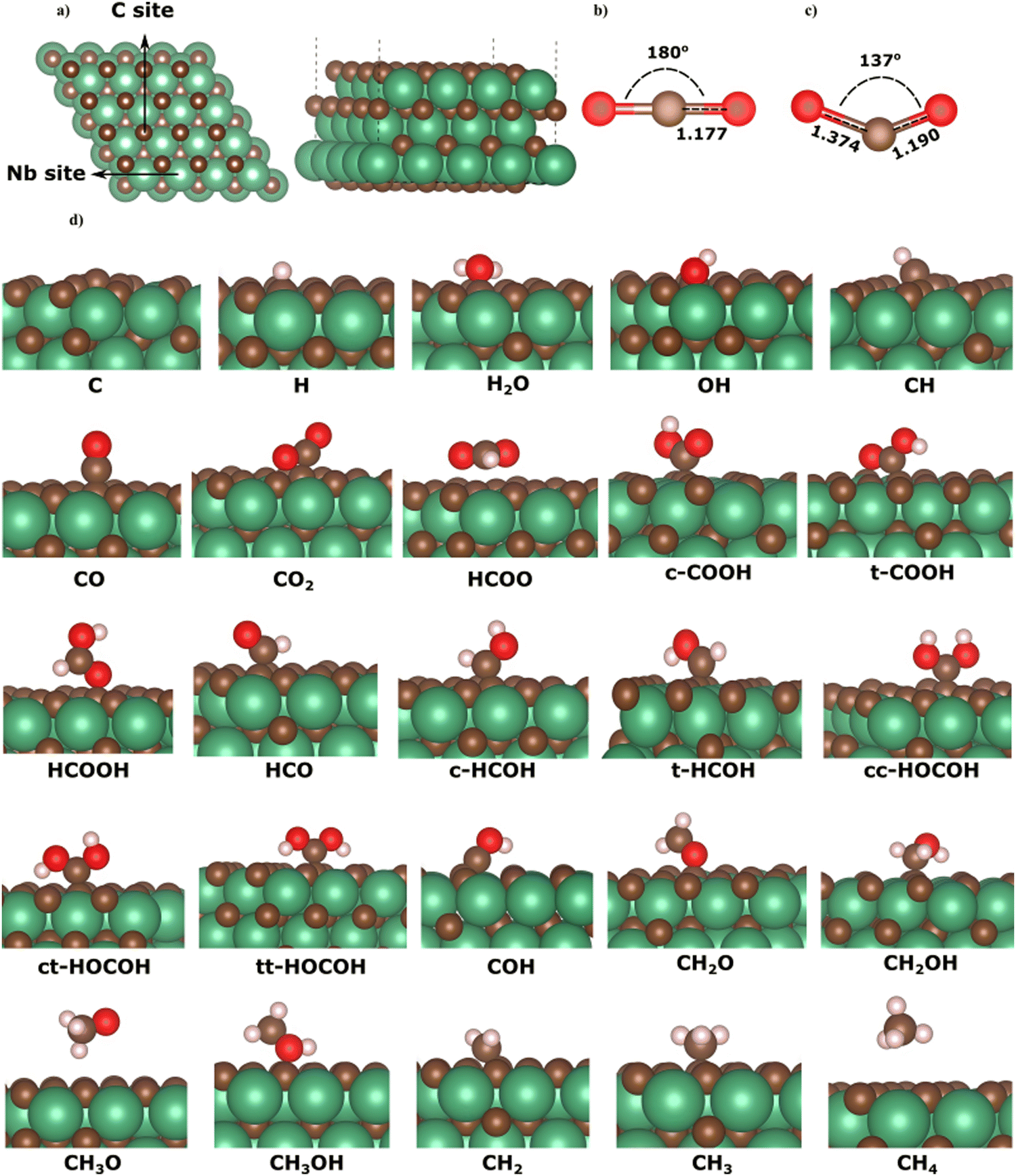 | ||
| Fig. 1 (a) The NbC (111) plane, and the isolated CO2 molecule (b) prior to, and (c) following adsorption, and (d) adsorption structures with minimum energy for all intermediates on the NbC (111) plane. | ||
The methodology employed in this study has already been validated in previous studies reported in the literature.32,45
3. Results
3.1 Adsorption configurations and energies on NbC (111) plane
Here, we describe the adsorption on the C-terminated NbC (111) plane of all potential intermediates during the hydrogenation reactions of CO2. Numerous preliminary configurations of the molecules situated above the NbC (111) plane at different positions were constructed. The most suitable adsorption site was selected following thorough structural optimization of both the NbC (111) plane and the intermediates. Fig. 1 shows the geometries of the lowest energy adsorptions of all intermediates on the NbC (111) plane. Table 1 likewise provides geometry and energy data, where binding between the species and the surface is denoted by the * sign, whereas the chemical bonds in each intermediate following adsorption are depicted without markings.| Species | E ads (eV) | Bond length (Å) | Species | E ads (eV) | Bond length (Å) |
|---|---|---|---|---|---|
| H | −5.130, −3.5245 | d*H–C = 1.10 | t-HCOH | −6.342, −5.3545 | d*C–C = 1.39 |
| d O–C = 1.31 | |||||
| d C–H = 1.10 | |||||
| d O–H = 1.00 | |||||
| C | −11.757 | d*C–C = 1.35 | cc-HOCOH | −4.824 | d*C–C = 1.39 |
| −7.6145 | d O–C = 1.34 | ||||
| d O–H = 0.98 | |||||
| H2O | −1.226, −0.997,32 −1.3310 | d*C–O = 1.48 | ct-HOCOH | −5.302 | d*C–C = 1.40 |
| d H–O = 1.01 | d O–C = 1.32 | ||||
| d H–O = 1.04 | d O–H = 0.98, 1.01 | ||||
| OH | −5.304, −5.52,45 −5.42732 | d*C–O = 1.31 | tt-HOCOH | −5.561 | d*C–C = 1.42 |
| d H–O = 0.98 | d O–C = 1.31 | ||||
| d O–H = 1.00 | |||||
| CH | −9.442, | d*C–C = 1.22 | COH | −7.347, −4.8845 | d*C–C = 1.26 |
| −7.29,45 | d C–H = 1.08 | d O–C = 1.27 | |||
| −7.40432 | d O–H = 1.01 | ||||
| CO | −4.008, −2.4845 | d*C–C = 1.32 | CH2O | −1.908, −2.6345 | d*O–C = 1.36 |
| d O–C = 1.17 | d O–C = 1.28 | ||||
| d C–H = 1.09 | |||||
| CO2 | −0.358, −0.2710 | d*C–C = 1.44 | CH2OH | −4.219, −3.293,32 −3.0945 | d*C–C = 1.50 |
| d*C–O = 1.62 | d O–C = 1.40 | ||||
| d O–C = 1.19, 1.37 | d O–H = 0.99 | ||||
| d C–H = 1.11, 1.12 | |||||
| HCOO | −0.469, −5.22232 | d O–C = 1.25 | CH3O | −0.261 | d O–C = 1.33 |
| d C–H = 1.17 | d C–H = 1.13, 1.12 | ||||
| c-COOH | −4.155, −4.10532 | d*C–C = 1.48 | CH3OH | −1.614, −0.72,45 −0.95932 | d*O–C = 1.46 |
| d O–C = 1.22, 1.36 | d O–H = 1.00 | ||||
| d O–H = 0.98 | d O–C = 1.49 | ||||
| d C–H = 1.09 | |||||
| t-COOH | −4.408, −3.95932 | d*C–C = 1.42 | CH2 | −7.418, | d*C–C = 1.36 |
| d O–C = 1.27, 1.32 | −5.797,32 | d C–H = 1.10 | |||
| d O–H = 0.98 | −5.8145 | ||||
| HCOOH | −1.539, −1.64232 | d*C–O = 1.41 | CH3 | −4.528, −3.58,45 −3.72132 | d*C–C = 1.49 |
| d O–C = 1.28, 1.31 | d H–C = 1.10 | ||||
| d O–H = 0.98 | |||||
| d C–H = 1.10 | |||||
| HCO | −4.214, −3.74,45 −4.24032 | d*C–C = 1.48 | CH4 | −0.161, −0.298,32 −0.1745 | d H–C = 1.10 |
| d O–C = 1.22 | |||||
| d C–H = 1.11 | |||||
| c-HCOH | −6.232 | d*C–C = 1.38 | |||
| d O–C = 1.33 | |||||
| d O–H = 0.98 | |||||
| d C–H = 1.10 |
For each intermediate in the CO2 hydrogenation reaction network, we have reported the geometries and energies of adsorption in the next section.
The calculated adsorption energies of the intermediates on the C-terminated NbC (111) surface show highly localized, strong interaction between the adsorbates and the surface. All adsorption geometries where the intermediates adsorb on the surface via their carbon atoms are energetically preferred over those structures that attach to the surface via their oxygen atoms. These results indicate the formation of C–C bonds between the adsorbates and surface, in accord with the large energies released, when these species adsorb on the molybdenum carbide surface via attachment to a surface carbon atom.45
3.2. Reaction networks in the CO2 hydrogenation process
Fig. 2 depicts the reaction pathways that result in the production of the products, e.g. CO, HCOOH, CH2O, CH3OH, and CH4, that were considered for this study. Mechanisms via HCOO or (trans,cis)-COOH intermediates act as the foundation for the overall reaction network. In the next section, the reaction pathways are discussed, using the optimum adsorption geometries for all intermediates.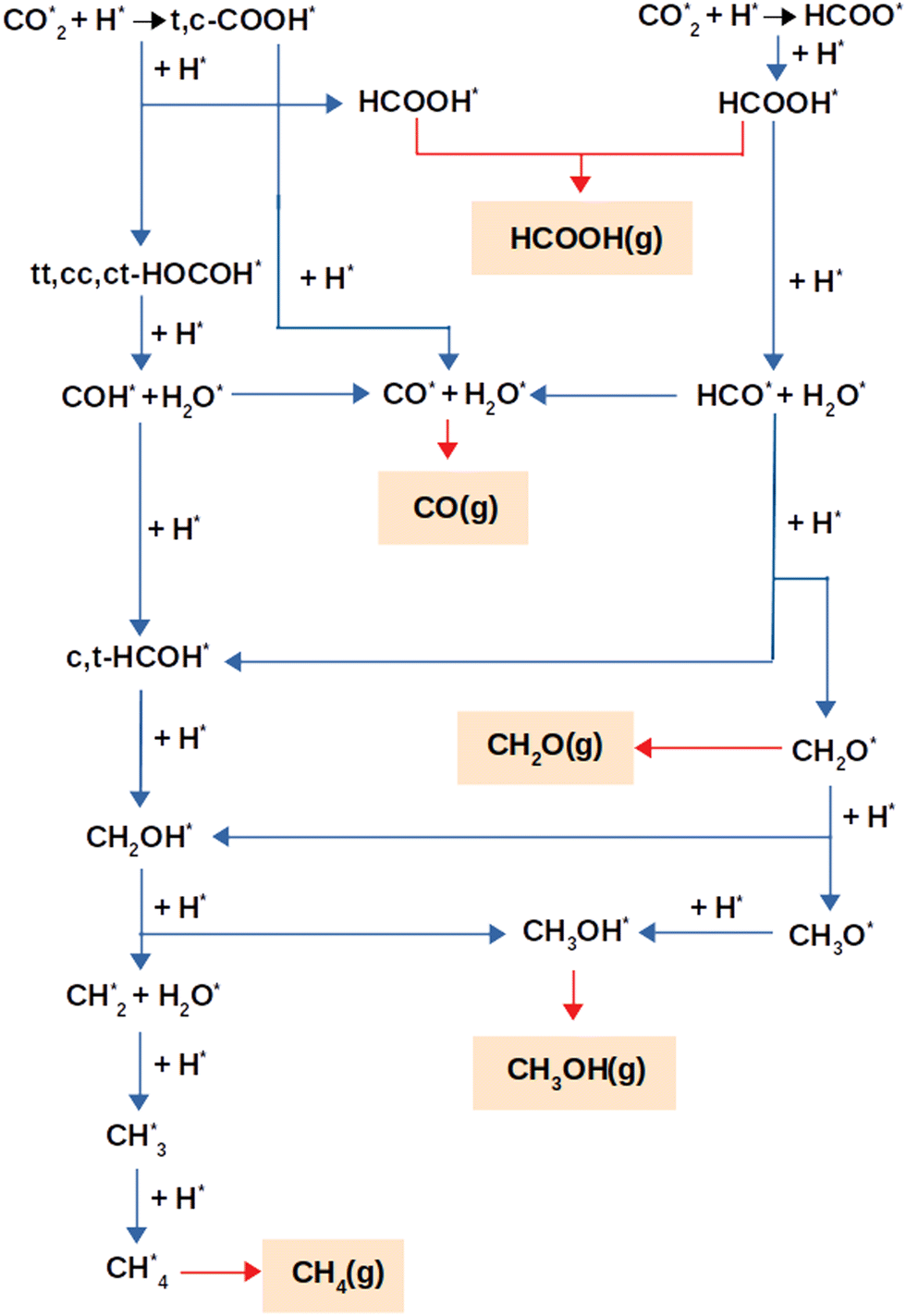 | ||
| Fig. 2 Possible CO2 hydrogenation reaction mechanisms on the NbC (111) surface. | ||
3.3. Chemical reactions
We have studied the possible processes of CO2 hydrogenation over the NbC (111) plane, which are discussed in detail in this section. Table 2 shows the predicted reaction energies for all elementary steps in the hydrogenation at the NbC (111) surface.| Elementary reactions | ΔE (eV) | Elementary reactions | ΔE (eV) |
|---|---|---|---|
| CO2 (g) → CO2* | −0.358 | c-COOH* + H* → cc-HOCOH* | 0.676 |
| CO2* + H* → HCOO* | 4.334 | t-COOH* + H* → tt-HOCOH* | 0.082 |
| CO2* + H* → c-COOH* | 0.369 | c-COOH* + H* → ct-HOCOH* | −0.038 |
| CO2* + H* → t-COOH* | −0.005 | t-COOH* + H* → ct-HOCOH* | 0.336 |
| HCOO* + H* → HCOOH* | −2.080 | cc-HOCOH* → tt-HOCOH* | −0.968 |
| c-COOH* + H* → CO* + H2O* | −0.930 | ct-HOCOH* → tt-HOCOH* | −0.254 |
| t-COOH* + H* → CO* + H2O* | -0.556 | tt-HOCOH* + H* → COH* + H2O* | 1.205 |
| c-COOH* + H* → HCOOH* | 1.886 | ct-HOCOH* + H* → COH* + H2O* | 0.951 |
| t-COOH* + H* → HCOOH* | 2.259 | cc-HOCOH* + H* → COH* + H2O* | 0.237 |
| HCOOH* + H* → HCO* + H2O* | 0.234 | COH* → CO* + H* | −1.843 |
| HCOOH* → HCOOH (g) | 1.539 | COH* + H* → t-HCOH* | 1.116 |
| HCO* + H* → t-HCOH* | −0.090 | COH* + H* → c-HCOH* | 1.429 |
| HCO* + H* → c-HCOH* | 0.223 | t-HCOH* + H* → CH2OH* | 2.711 |
| HCO* → CO* + H* | −3.049 | c-HCOH* + H* → CH2OH* | 2.398 |
| CO* → CO (g) | 4.008 | CH2OH* + H* → CH2* + H2O* | −0.357 |
| HCO* + H* → CH2O* | 2.068 | CH2* + H* → CH3* | 1.830 |
| CH2O* → CH2O (g) | 1.908 | CH3* + H* → CH4* | 3.105 |
| CH2O* + H* → CH3O* | 4.857 | CH4* → CH4(g) | 0.161 |
| CH2O* + H* → CH2OH* | 0.553 | CH3OH* → CH3OH (g) | 1.641 |
| CH3O* + H* → CH3OH* | −2.338 | H2O* → H2O (g) | 1.226 |
| CH2OH* + H* → CH3OH* | 1.967 |
Formate, HCOO*, or carboxyl species, trans-COOH* (t-COOH*) and/or cis-COOH* (c-COOH*) were formed as a consequence of the initial step of CO2 hydrogenation via the reactions:
| CO2* + H* → HCOO* | (3) |
| CO2* + H* → (t,c)-COOH* | (4) |
This network of reactions results in additional products using two different hydrogenation mechanisms, one through HCOOH* and the other through a HOCOH*. Detailed descriptions of these mechanisms are shown below.
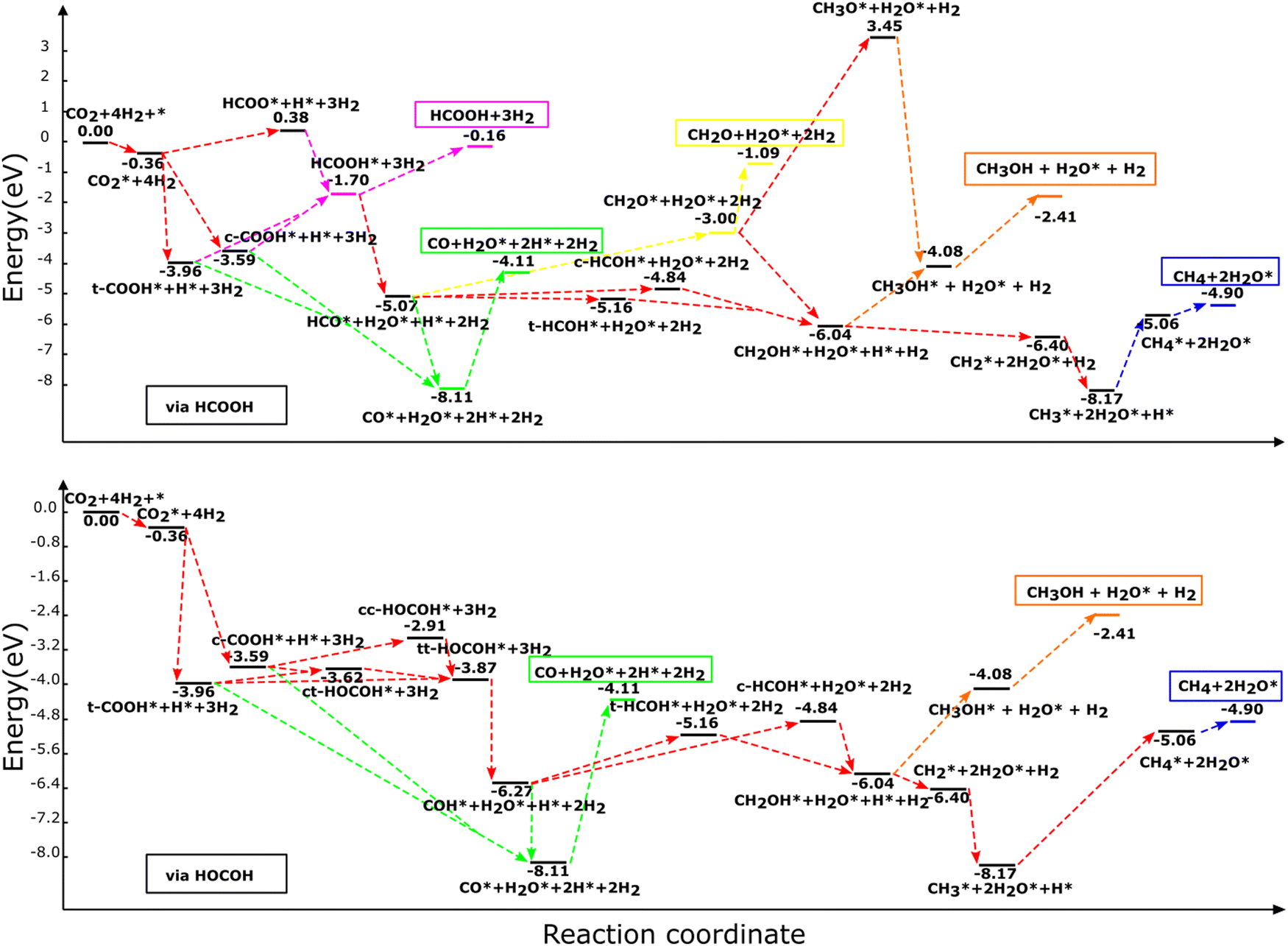 | ||
| Fig. 3 Reaction profile for the hydrogenation of CO2 and all intermediates to HCOOH, CO, CH2O, CH3OH, and CH4 molecules via (a) HCOOH and (b) HOCOH on the NbC (111) plane. The illustrated profiles are associated with the total free energy of the catalyst, CO2, and four H2 molecules in the gas phase.46 | ||
3.3.1.1. Formic acid formation. HCOOH may be formed in any one of three ways, as was stated already. Initially, CO2 is hydrogenated to generate HCOO* which subsequently receives another H to produce HCOOH. The other process goes through hydrogenation of CO2 to t-COOH* or c-COOH* with the production of HCOOH* as an end product of further hydrogenation of (t,c)-COOH*. The hydrogenation of CO2 to generate HCOO* requires 4.33 eV, whereas the synthesis of HCOOH from the HCOO* product is an exothermic reaction. Thus, the generation of HCOOH through the (t,c)-COOH* intermediate is thermodynamically more advantageous than the HCOO* pathway (see Table 2).
3.3.1.2 Carbon monoxide formation via formic acid. The (t,c)-COOH* and the HCOOH* pathways are the two main routes by which carbon monoxide is produced. In the first, CO is formed through the degradation of (t,c)-COOH* using the chemical mechanism of (t,c)-COOH* + H* → CO* + H2O*, which are exothermic processes with reaction energies of −0.556 and −0.930 eV, for the trans and cis conformers, respectively.
Breakdown of HCOOH* to HCO* (HCOOH* + H* → HCO* + H2O*) leads to an endothermic reaction energy of 0.234 eV, which is followed by HCO* dehydrogenation (HCO* → CO* + H*) to produce CO with a reaction energy of −3.049 eV. In consequence, the general reactions for CO production using HCOOH may be written as:
| CO2* → (t,c)-COOH* → CO* | (5) |
| CO2* → t-COOH* → HCOOH* → HCO* → CO* | (6) |
3.3.1.3 Formaldehyde formation via formic acid. The sole pathway to obtain CH2O from CO2 is via HCO*, where HCO* accepts hydrogen to generate CH2O. Formaldehyde (CH2O*) or (c,t)-HCOH* may be formed via the hydrogenation of HCO*, with an endothermic energy of 2.068 eV for HCO* + H* → CH2O*. As a result, the following are the main reactions involved in producing CH2O:
| CO2* → HCOO*, (t,c)-COOH* → HCOOH* → HCO* → CH2O* | (7) |
3.3.1.4. Methanol formation via formic acid. It has been suggested previously that with reaction energies of 0.223 eV and −0.090 eV, respectively, for the cis and trans conformers, hydrogenation of HCO* may result in the creation of (c,t)-HCOH* through the reaction HCO* + H* → (c,t)-HCOH*. HCO* hydrogenation should therefore generate the t-HCOH* conformer more often, since it is an exothermic reaction.
Additional hydrogenation of (c,t)-HCOH* or CH2O* may lead to the formation of CH3OH. t-HCOH* and CH2O* are hydrogenated, but the former occurs through the t-HCOH* + H* → CH2OH* mechanism, whereas the latter is achieved through CH2O* via CH2O* + H* → CH2OH*. In order to produce CH2OH*, a total of 2.711 eV and 0.553 eV of energy is required for the two separate processes. CH2OH* is more likely to be generated via CH2O* hydrogenation than through t-HCOH*, because the hydrogenation of CH2O* to CH2OH* is more efficient in terms of energy than the hydrogenation of t-HCOH*. Continuing the hydrogenation of CH2OH* results in the formation of CH3OH* (CH2OH* + H* → CH3OH*), distinguished by its endothermic nature with an energy of 1.967 eV. Hydrogenation of CH2O* may also produce methoxy (CH3O*), in addition to CH2OH*. However, whereas this mechanism happens via CH2O* + H* → CH3O* and consumes a significant amount of energy (4.857 eV), the following hydrogenation stage leads to CH3OH* via CH3O* + H* → CH3OH* which releases just 2.338 eV of reaction energy. Although it is energetically possible to produce CH3OH using CH3O, the recommended method is to go through CH2OH* hydrogenation to produce CH3OH* owing to the less endothermic reaction involved. For CH3OH* synthesis, the suggested sequence of steps is as follows:
| CO2*→ HCOO*, (t,c)-COOH* → HCOOH* → HCO* → CH2O*, (c,t)-HCOH* → CH2OH* → CH3OH* | (8) |
3.3.1.5. Methane formation via formic acid. Only dissociation of CH2OH* (CH2OH* + H* → CH2* + H2O*) results in the synthesis of CH2*, and the end products CH3* and CH4*. Reactions CH2* + H* → CH3 and CH3* + H* → CH4* may generate CH4* by hydrogenating CH2* and CH3* with reaction energies of 1.830 eV and 3.105 eV for the production of CH3* and CH4*, respectively. The following is an ordinary sequence, indicating some reactions that contribute to the synthesis of CH4* from HCOOH*:
| CO2* → HCOO*, (t,c)-COOH* → HCOOH* → HCO* → CH2O*, (c,t)-HCOH* → CH2OH* → CH2* → CH3* → CH4* | (9) |
3.3.2.1. Carbon monoxide formation via dihydroxycarbene. From the direct breakdown of (t,c)-COOH*, CO* may be produced, although it can also be produced via COH*. The first route was described in detail in Section 3.3.1.2, where the processes were found to be exothermic with energies of −0.556 eV via the cis and −0.930 eV via the trans conformer. In the second route, COH* and H2O* are formed via the breakdown of the most suitable HOCOH* conformers (tt-HOCOH*) via the reaction tt-HOCOH* + H* → COH* + H2O* with a reaction energy of 1.205 eV. Further breakdown of COH* (COH* → CO* + H*) leads to the generation of CO*. CO* tends to be created through the COH* route with the exothermic mechanism of creating CO* from COH* with a reaction energy of −1.843 eV. As a consequence, the reactions for CO production using HOCOH* could be written as:
| CO2* → (t,c)-COOH* → CO* | (10) |
| CO2* → (t,c)-COOH* → (cc,ct,tt)-HOCOH* → COH* → CO* | (11) |
3.3.2.2. Methanol production from dihydroxycarbene. Trans-HCOH* or cis-HCOH* is formed after addition of a hydrogen atom via COH* + H* → t-HCOH* and COH* + H* → c-HCOH* with endothermic energies of 1.116 eV and 1.429 eV, respectively. Further hydrogenation of (t,c)-HCOH* results in the formation of CH2OH* via the process (t,c)-HCOH* + H* → CH2OH*. CH3OH* is then generated through additional hydrogenation of CH2OH*. The creation of CH2OH* from (t,c)-HCOH* and the generation of CH3OH* from CH2OH* are endothermic processes with reaction energies of 2.711 eV, 2.398 eV and 1.967 eV. Based on the relative energies, we suggest that the following route is the most likely to lead to the generation of CH3OH*:
| CO2* → (t,c)-COOH* → (cc,ct,tt)-HOCOH* → COH* → (t,c)-HCOH* → CH2OH* → CH3OH* | (12) |
3.3.2.3. Methane formation via dihydroxycarbene. Decomposition of CH2OH* into either HOCOH or HCOOH results in the production of CH2* and its further hydrogenation. As a result, the following route is suggested to generate CH4* via HOCOH*:
| CO2* → (t,c)-COOH* → (cc,ct,tt)-HOCOH* → COH* → (t,c)-HCOH* → CH2OH* → CH2* → CH3* → CH4* | (13) |
3.4. Electronic properties from CO2 adsorption
Charge densities and total densities of state (TDOS) are calculated to gain more detailed insight into the CO2 adsorption at the NbC (111) surface. The density of states is large around the Fermi level, as seen in Fig. 4a, and there is no energy gap at this level. A partly filled d band and potentially occupied s orbital in NbC may contribute to the metal characteristics of the compound, which is consistent with previous observations.3,4 The TDOS behaves in a manner comparable to the behavior of the DOS of the Nb atoms, and both above and below the Fermi level, the participation of Nb atoms in the TDOS is much greater than that of the C atoms, because the electronic structure of NbC is defined by surface states formed of Nb 4d orbitals at about EF.47,48 However, Fig. 4b demonstrates that whenever a CO2 molecule is adsorbed on the NbC (111) surface, there is no substantial variation in the TDOS, apart from a slight increase at the Fermi level.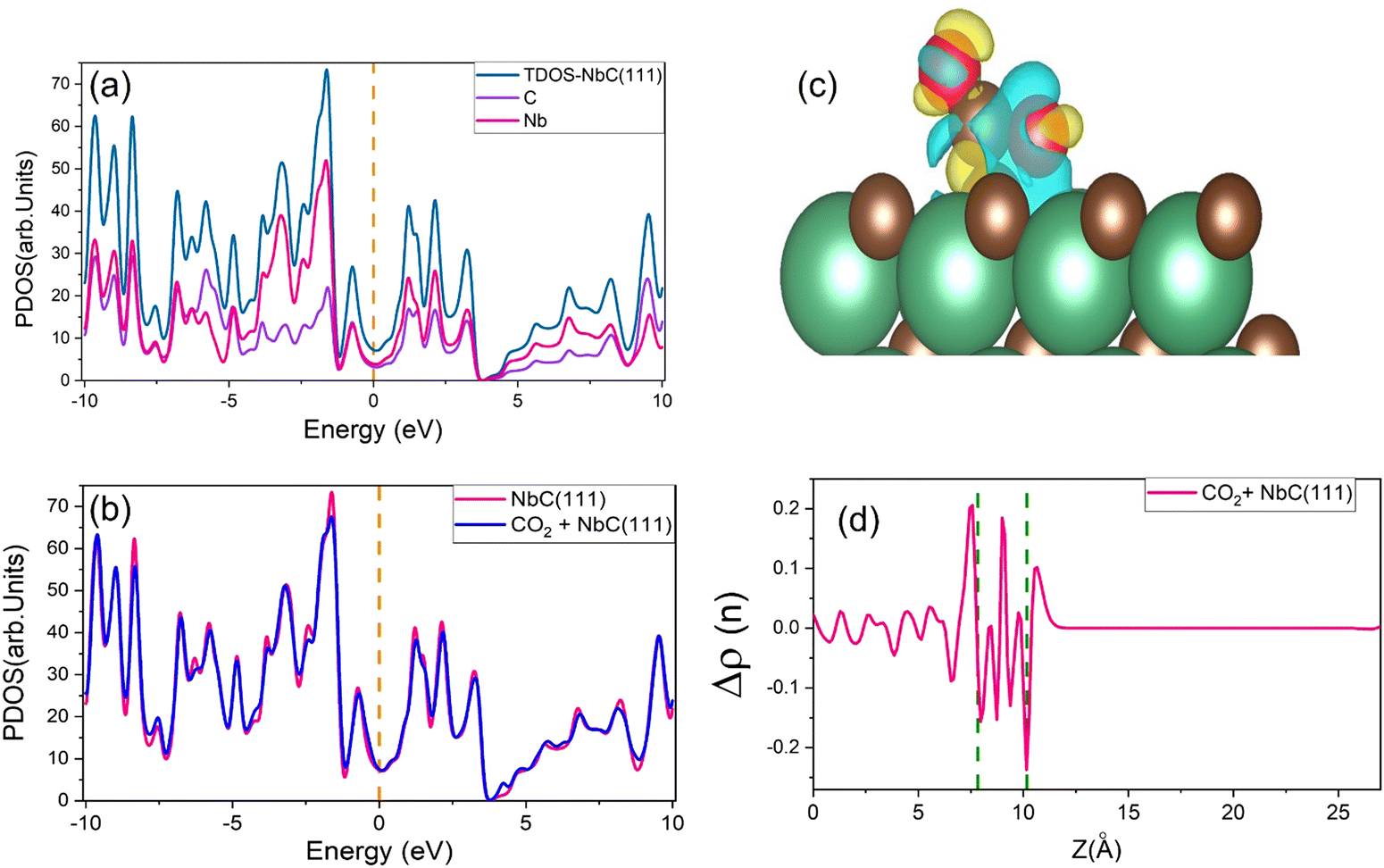 | ||
| Fig. 4 (a) The PDOS for the NbC (111) surface, (b) the TDOS of NbC (111) prior to and after adsorption of CO2, (c) view of the charge density differences, with the C and O atoms illustrated in brown and red (isovalue = ±0.002) and, (d) the planar average charge density. The Fermi level is set at zero. | ||
Next, we have used equation (2) to determine the charge density difference caused by CO2 adsorption by considering separately the charge density of CO2 + NbC (111), the bare slab, and the isolated CO2. Fig. 4c depicts the rearrangement of charge density in the system following CO2 adsorption, where yellow indicates a positive charge difference and blue indicates a negative charge difference. CO2 is seen to have accumulated negative charge upon adsorption at the surface. We have also used the planar mean charge density differential Δρ(z)23 to investigate the charge rearrangement of CO2 following adsorption on NbC (111), as shown below:
| Δρ(z) = Δρ(z)Molecule+NbC(111) − Δρ(z)NbC(111) − Δρ(z)Molecule | (14) |
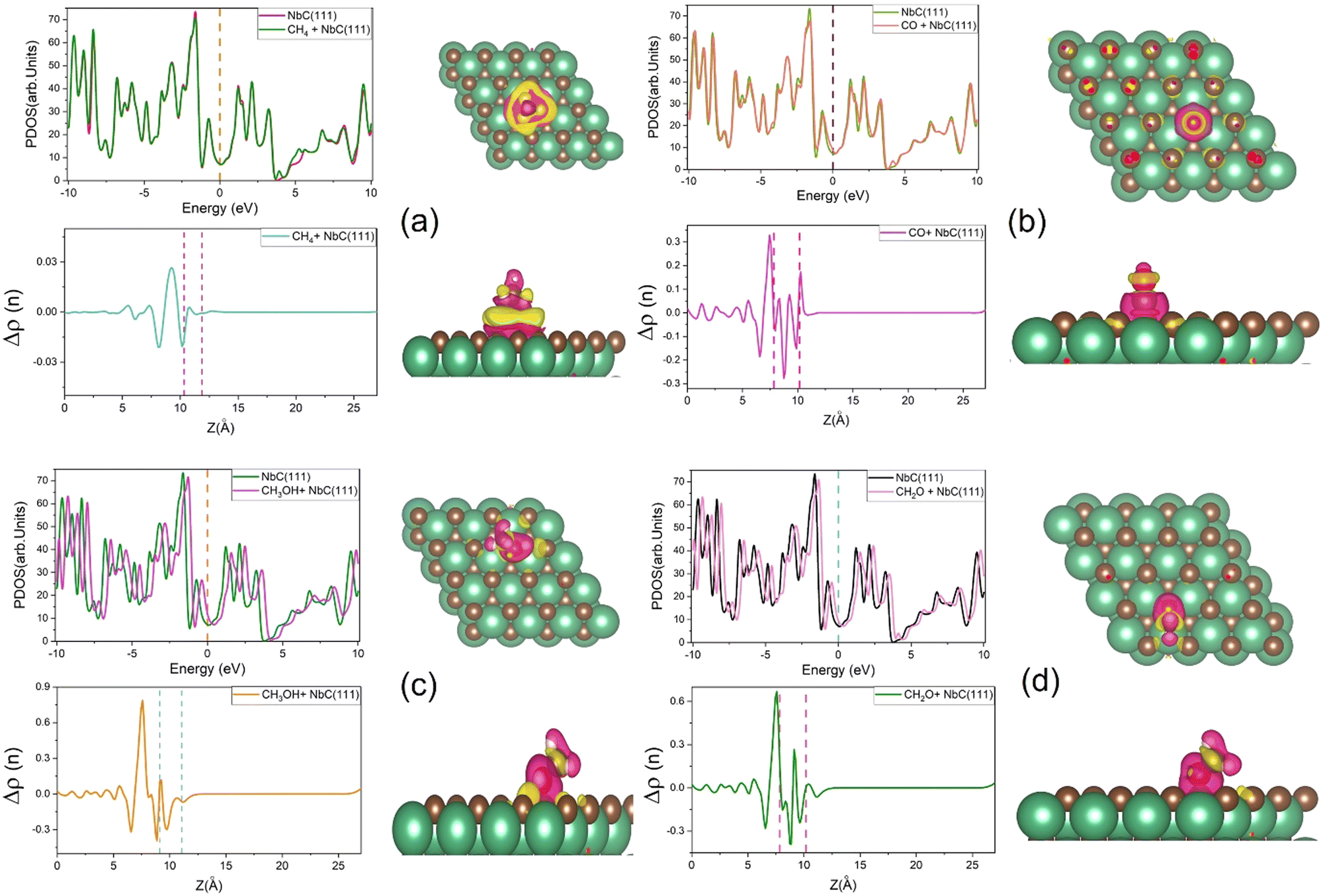 | ||
| Fig. 5 The TDOS, the planar average charge densities, and charge density differences for (a) CH4 (isovalue = ±0.00005), (b) CO (isovalue = ±0.001), (c) CH3OH (isovalue = ±0.002) and, (d) CH2O (isovalue = ±0.002). The Fermi level is set at zero. | ||
4. Conclusion
In this investigation, we have calculated the energetics of the interaction of 25 reactant, intermediate and product species at the carbon-terminated NbC (111) surface and we have evaluated their reactions in the CO2 hydrogenation process, where it has been shown that methane has the maximum capacity for hydrogen of all the investigated products.CH2O generation is expected to be produced predominantly from HCOOH rather than other intermediates, because the endothermic reaction energies for the production of CH4, CH3OH and CO via HCOOH are rather high, and the route through HCOOH is thus not practical for the synthesis of CH3OH, CH4 or CO. The suggested pathways for each product have been collected in Fig. 6 to allow direct comparison. According to our findings, 4.902 eV in energy is generated during CO2 hydrogenation to methane over the C-terminated NbC (111) surface through the route:
| CO2* → t-COOH* → tt-HOCOH* → COH* → t-HCOH* → CH2OH* → CH2* → CH3* → CH4* → CH4 (g) | (15) |
| CO2* → t-COOH* → tt-HOCOH* → COH* → CO* → CO (g) | (16) |
| CO2* → t-COOH* → CO* → CO (g) | (17) |
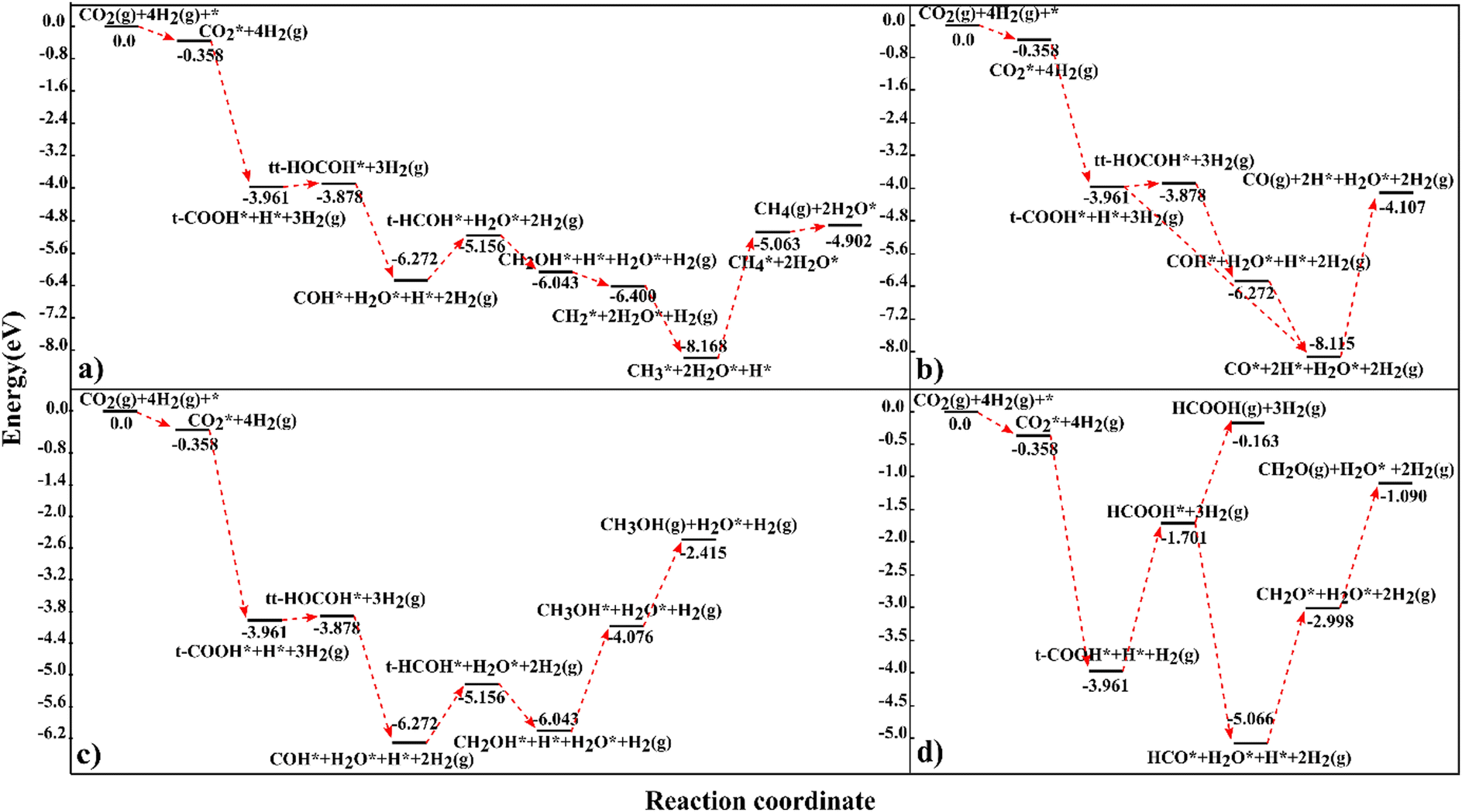 | ||
| Fig. 6 The preferred reaction profiles for CO2 hydrogenation to (a) CH4, (b) CO, (c) CH3OH, and (d) CH2O and HCOOH on the NbC (111) plane. The illustrated profiles are compared to the summation of the total free energies of the catalyst, CO2, and four H2 in the gas phase.46 | ||
CH3OH is the third possible product with an exothermic energy of 2.415 eV, through the preferred route (Fig. 6c):
| CO2* → t-COOH* → tt-HOCOH* → COH* → t-HCOH* → CH2OH* → CH3OH* → CH3OH(g) | (18) |
| CO2* → t-COOH* → HCOOH* → HCO* → CH2O* → CH2O(g) | (19) |
| CO2* → t-COOH* → HCOOH* → HCOOH (g) | (20) |
In conclusion, we have used DFT calculations to investigate a comprehensive network of reaction pathways for CO2 hydrogenation to CH4, CO, CH3OH, CH2O and HCOOH over the carbon-terminated NbC (111) surface. By evaluating the predicted reaction energies, we can conclude that the most abundant products should be CH4 and CO, followed to a lesser extent by CH3OH, CH2O, and HCOOH, which findings are in excellent agreement with experimental findings.
The calculated adsorption energies of the intermediates on the surface show highly localized strong interactions between the adsorbates and the surface, which lead to an overall exothermic reaction pathway that provides sufficient energy to overcome the desorption energy required to release the products from the surface. Furthermore, where there is co-adsorption of the hydrogen and the intermediates at the surface, the hydrogenation step energies indicate that the presence of hydrogen on the surface makes the intermediates more reactive, which as a result readily undergo further surface-mediated reactions to form CH4, CO and the other products.
The carbon-terminated (111) surfaces of TMCs are generally less stable than the metal-terminated planes. However, Quesne et al.5 calculated the energies of both carbon- and metal-terminated surfaces across the TMC series, where the carbon-terminated (111) surfaces of NbC, TaC, MoC and WC were found to be preferred over the metal-terminated surfaces. The carbon-terminated NbC (111) was also found to be important in a number of other studies,5,7,20,34 whereas our results also indicate that the carbon-terminated NbC (111) surface remains stable during the adsorption of CO2. Furthermore, the results from this study indicate that CO2 adsorption on the carbon-terminated NbC (111) surface at Eads = −0.358 eV is weaker than at the metal-terminated NbC (111) with Eads = −1.863 eV (calculated in our previous study32). We have also found a larger exothermic reaction energy for CO2 methanation on the carbon-terminated surface, releasing 4.902 eV, than the same process at the metal-terminated surface, releasing 2.747 eV, as calculated in our previous work.32 Overall, the results suggest that the carbon-terminated surface should be a promising catalyst for CO2 hydrogenation, allowing an energetically favourable hydrogenation process.
From our calculations, we can make a number of secondary observations. For example, it is much more advantageous to produce CO immediately from either t-COOH or COH* degradation through the tt-HOCOH route. Furthermore, the majority of CH4, CO, and CH3OH is generated by t-COOH* via the tt-HOCOH route, whereas the majority of CH2O originates from HCO* via the HCOOH reaction. Along these routes, HCO* fragmentation has the highest exothermic reaction energy of −3.049 eV, whereas CH3* hydrogenation to CH4* requires the highest endothermic reaction energy of 3.105 eV.
It is worth mentioning that in this study we have only investigated the thermodynamics of the reactions, without considering the kinetics and activation energies. The proposed mechanisms were suggested based on the comparative reaction energies of the different pathways, which identified the energetically favoured reaction mechanism. The inclusion of transition states in the catalytic pathways would provide further insight, for example allowing us to identify the rate-determining step, but it would not invalidate the thermodynamically favourable reaction pathway.
We have also assessed the electronic features of the system of CO2 and the intermediates formed as a result of its hydrogenation over the C-terminated NbC (111) surface, which have indicated that the adsorbates have little effect on the surface.
This work has led to detailed understanding of some of the fundamental steps in the CO2 catalytic hydrogenation mechanism at the C-terminated NbC (111) surface which results in the production of C1 type surface species. Since the field of CO2 hydrogenation still requires significant advances in the understanding of reaction mechanisms for C–C coupling, for synthesis of higher alcohols and higher hydrocarbons, further potential pathways leading to the production of other useful compounds have yet to be explored. However, we consider that the results of this work may assist experiment in the development of effective catalysts based on transition metal carbides.
Data availability statement
The data that support the findings of this study are available on request from the corresponding author.Conflicts of interest
There are no competing interest to declare.Acknowledgements
S. S. T thanks the Iran National Science Foundation (INSF) Grant No. 97020912 for the financial support of this investigation. The authors are also grateful to the Research Affairs Division of the Amirkabir University of Technology (AUT), Tehran, Iran, for their financial support. This work has used the computational facilities of the Advanced Research Computing at Cardiff (ARCCA) Division, Cardiff University, and HPC Wales. Via our membership of the UK's HEC Materials Chemistry Consortium, which is funded by EPSRC (EP/R029431), this work has also used the ARCHER2 UK National Supercomputing Service (http://archer2.ac.uk).References
- B. I. Sharma, J. Maibam, R. Paul, R. Thapa and R. B. Singh, Indian J. Phys., 2010, 84, 671–674 CrossRef.
- J. A. Rodriguez, J. Evans, L. Feria, A. B. Vidal, P. Liu, K. Nakamura and F. Illas, J. Catal., 2013, 307, 162–169 CrossRef.
- F. Viñes, C. Sousa, P. Liu, J. Rodriguez and F. Illas, J. Chem. Phys., 2005, 122, 174709 CrossRef PubMed.
- K. Kobayashi, Jpn. J. Appl. Phys., 2000, 39, 4311 CrossRef.
- M. G. Quesne, A. Roldan, N. H. de Leeuw and C. R. A. Catlow, Phys. Chem. Chem. Phys., 2018, 20, 6905–6916 RSC.
- H. W. Hugosson, O. Eriksson, U. Jansson and B. Johansson, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 134108 CrossRef.
- H. H. Hwu and J. G. Chen, Chem. Rev., 2005, 105, 185–212 CrossRef PubMed.
- A. Vojvodic, C. Ruberto and B. I. Lundqvist, J. Phys.: Condens. Matter, 2010, 22, 375504 CrossRef PubMed.
- F. Viñes, J. A. Rodriguez, P. Liu and F. Illas, J. Catal., 2008, 260, 103–112 CrossRef.
- P. Liu and J. A. Rodriguez, J. Phys. Chem. B, 2006, 110, 19418–19425 CrossRef CAS PubMed.
- N. M. Schweitzer, J. A. Schaidle, O. K. Ezekoye, X. Pan, S. Linic and L. T. Thompson, J. Am. Chem. Soc., 2011, 133, 2378–2381 CrossRef CAS PubMed.
- P. M. Patterson, T. K. Das and B. H. Davis, Appl. Catal., A, 2003, 251, 449–455 CrossRef CAS.
- C. Kunkel, F. Vines and F. Illas, Energy Environ. Sci., 2016, 9, 141–144 RSC.
- S. Posada-Pérez, F. Vines, P. J. Ramirez, A. B. Vidal, J. A. Rodriguez and F. Illas, Phys. Chem. Chem. Phys., 2014, 16, 14912–14921 RSC.
- M. D. Porosoff, S. Kattel, W. Li, P. Liu and J. G. Chen, Chem. Commun., 2015, 51, 6988–6991 RSC.
- S. Posada-Pérez, F. Viñes, P. J. Ramirez, A. B. Vidal, J. A. Rodriguez and F. Illas, Phys. Chem. Chem. Phys., 2014, 16, 14912–14921 RSC.
- M. D. Porosoff, X. Yang, J. A. Boscoboinik and J. G. Chen, Angew. Chem., 2014, 126, 6823–6827 CrossRef.
- Q. Zhang, L. Pastor-Pérez, W. Jin, S. Gu and T. R. Reina, Appl. Catal., B, 2019, 244, 889–898 CrossRef CAS.
- S. Posada-Pérez, P. J. Ramírez, R. A. Gutiérrez, D. J. Stacchiola, F. Viñes, P. Liu, F. Illas and J. A. Rodriguez, Catal. Sci. Technol., 2016, 6, 6766–6777 RSC.
- M. G. Quesne, A. Roldan, N. H. de Leeuw and C. R. A. Catlow, Phys. Chem. Chem. Phys., 2019, 21, 10750–10760 RSC.
- Z. Ou, C. Qin, J. Niu, L. Zhang and J. Ran, Int. J. Hydrogen Energy, 2019, 44, 819–834 CrossRef CAS.
- S. Posada-Pérez, P. J. Ramírez, J. Evans, F. Viñes, P. Liu, F. Illas and J. A. Rodriguez, J. Am. Chem. Soc., 2016, 138, 8269–8278 CrossRef.
- N. Y. Dzade and N. H. d Leeuw, Catalysts, 2021, 11, 127 CrossRef.
- S. S. Tafreshi, A. Z. Moshfegh and N. H. de Leeuw, J. Phys. Chem. C, 2019, 123, 22191–22201 CrossRef.
- X. Liu, C. Kunkel, P. Ramírez de la Piscina, N. Homs, F. Viñes and F. Illas, ACS Catal., 2017, 7, 4323–4335 CrossRef.
- K. Edamoto, E. Shiobara, T. Anazawa, M. Hatta, E. Miyazaki, H. Kato and S. Otani, J. Chem. Phys., 1992, 96, 842–847 CrossRef.
- K. Edamoto, E. Miyazaki and H. Kato, Vacuum, 1990, 41, 547–549 CrossRef.
- S. Tokumitsu, T. Anazawa, K. Ozawa, R. Sekine, E. Miyazaki, K. Edamoto, H. Kato and S. Otani, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 4516 CrossRef PubMed.
- K. Edamoto, Y. Abe, T. Ikeda, N. Ito, E. Miyazaki, H. Kato and S. Otani, Surf. Sci., 1990, 237, 241–247 CrossRef.
- K. Edamoto, S. Maehama, E. Miyazaki and H. Kato, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 39, 7461–7465 CrossRef.
- K. Ozawa, S. Ishikawa, E. Miyazaki, K. Edamoto, H. Kato and S. Otani, Surf. Sci., 1997, 375, 250–256 CrossRef.
- S. Sarabadani Tafreshi, M. Ranjbar, N. Taghizade, S. F. K. S. Panahi, M. Jamaati and N. H. de Leeuw, ChemPhysChem, 2022, 23, e202100781 CrossRef.
- K. E. Tan, M. W. Finnis, A. P. Horsfield and A. P. Sutton, Surf. Sci., 1996, 348, 49–54 CrossRef.
- F. Silveri, M. G. Quesne, A. Roldan, N. H. De Leeuw and C. R. A. Catlow, Phys. Chem. Chem. Phys., 2019, 21, 5335–5343 RSC.
- A. S. Botana and M. R. Norman, Phys. Rev. Mater., 2019, 3, 044001 CrossRef.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef.
- J. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1998, 80, 891 CrossRef.
- G. Kresse and J. Hafner, J. Phys.: Condens. Matter, 1994, 6, 8245 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- S. S. Tafreshi, A. Roldan and N. H. de Leeuw, Phys. Chem. Chem. Phys., 2015, 17, 21533–21546 RSC.
- S. Sarabadani Tafreshi, S. F. K. S. Panahi, N. Taghizade, M. Jamaati, M. Ranjbar and N. H. de Leeuw, Catalysts, 2022, 12, 1275 CrossRef.
- https://icsd.products.fiz-karlsruhe.de .
- K.-Z. Qi, G.-C. Wang and W.-J. Zheng, Surf. Sci., 2013, 614, 53–63 CrossRef.
- R. A. Angnes, mechaSVG, GitHub repository, 2020 DOI:10.5281/zenodo.3970267.
- K. Ozawa, S. Ishikawa, S. Tokumitsu, R. Sekine, E. Miyazaki, K. Edamoto, H. Kato and S. Otani, Surf. Sci., 1996, 364, L612–L616 CrossRef.
- K. Ozawa, S. Ishikawa, K. Edamoto, H. Kato and S. Otani, Surf. Sci., 1999, 419, 226–235 CrossRef.
| This journal is © the Owner Societies 2023 |