
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiosynthetic incorporation of fluorinated amino acids into the nonribosomal peptide gramicidin S†
Maximilian
Müll
a,
Farzaneh
Pourmasoumi
a,
Leon
Wehrhan
b,
Olena
Nosovska
c,
Philipp
Stephan
a,
Hannah
Zeihe
a,
Ivan
Vilotijevic
 c,
Bettina G.
Keller
b and
Hajo
Kries
*ad
c,
Bettina G.
Keller
b and
Hajo
Kries
*ad
aJunior Research Group Biosynthetic Design of Natural Products, Leibniz Institute for Natural Product Research and Infection Biology, Hans Knöll Institute (HKI Jena), Jena 07745, Germany. E-mail: hajo.kries@leibniz-hki.de
bFreie Universität Berlin, Department of Biology, Chemistry, and Pharmacy, Institute of Chemistry and Biochemistry, Arnimallee 20, Berlin 14195, Germany
cInstitute of Organic Chemistry and Macromolecular Chemistry, Friedrich Schiller University Jena, Humboldtstr. 10, Jena 07743, Germany
dUniversity of Bayreuth, Organic Chemistry I, Bayreuth 95440, Germany
First published on 25th July 2023
Abstract
Fluorine is a key element in medicinal chemistry, as it can significantly enhance the pharmacological properties of drugs. In this study, we aimed to biosynthetically produce fluorinated analogues of the antimicrobial cyclic decapeptide gramicidin S (GS). However, our results show that the A-domain of the NRPS module GrsA rejects 4-fluorinated analogues of its native substrate Phe due to an interrupted T-shaped aromatic interaction in the binding pocket. We demonstrate that GrsA mutant W239S improves the incorporation of 4-fluorinated Phe into GS both in vitro and in vivo. Our findings provide new insights into the behavior of NRPSs towards fluorinated amino acids and strategies for the engineered biosynthesis of fluorinated peptides.
Introduction
The introduction of fluorine atoms is a widespread strategy in medicinal chemistry to fine-tune drug properties.1,2 Fluorination can improve the fit to a protein binding pocket, or improve pharmacokinetic parameters, as in the case of the “second generation” macrolide antibiotic flurithromycin.3 Throughout the last two decades, many fluorinated drugs transitioned from the clinical stage to the market (Fig. 1), which demonstrates the importance of fluorination. For drugs acting on protein targets, the focus has long been on using fluorine to better fit a binding pocket. This led to an understanding of fluorine–enzyme interactions,1,4 which can be used for rational design of small molecule libraries.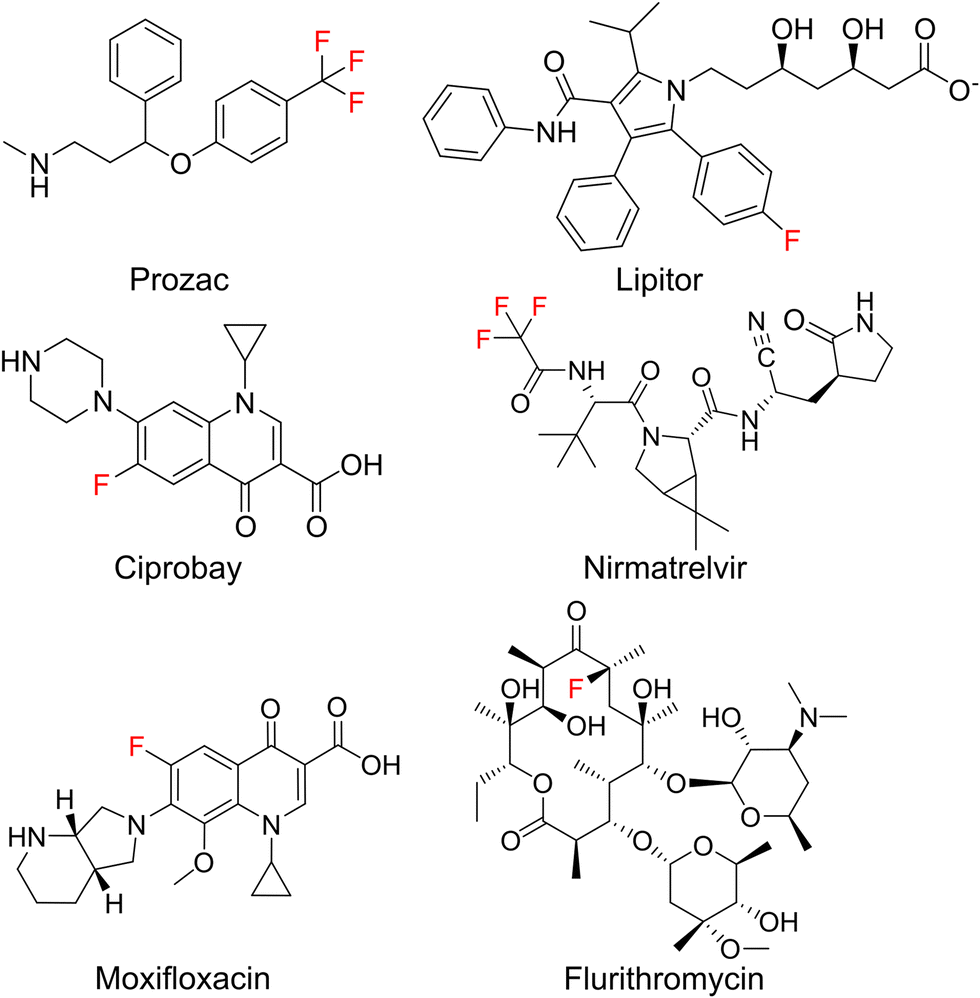 | ||
| Fig. 1 Important fluorinated drug molecules. | ||
Although natural products are a major source of new drugs,5 they rarely contain fluorine.6 Therefore, attempts have been made to biosynthetically incorporate fluorine into natural products belonging to the classes of alkaloids, nonribosomal peptides (NRPs), polyketides, and cyclic dinucleotides.7–19 Notable successes have been achieved in engineering polyketide biosynthesis, bringing the biosynthesis of flurithromycin almost within reach.17 While the similarity between a C–H and a C–F bond allows many non-natural, fluorinated analogues to slip through biosynthetic selectivity filters, designing biosynthetic enzymes with binding pockets selective for fluorinated substrates is largely unexplored.18 Hence, directing fluorine incorporation into natural products often remains unpredictable and low yielding.
Incorporation of fluorinated amino acids into NRPs, which are a prolific source of various drugs and antibiotics,20–22 has so far relied on precursor-directed biosynthesis exploiting the natural promiscuity of biosynthetic enzymes.9,11,13–16,19 NRPs are produced by large multimodular enzymes called non-ribosomal peptide synthetases (NRPSs).21,23,24 These NRPSs are built from three core domains (Fig. 2A). Adenylation domains (A-domains) activate a specific amino acid, thiolation domains (T-domains) carry the activated thioester intermediates, and condensation domains (C-domains) catalyze peptide bond formation between substrates bound to adjacent T-domains. A-domains typically exhibit selectivity for specific amino acids and thereby determine the NRP sequence.25–27 This selectivity correlates strongly with the identity of residues in the A-domain binding pocket,25,26 which allows reliable prediction,28 and design to some extent.29 Since fluorinated amino acids have not been described as natural NRPS substrates, A-domain binding pockets specific for them are elusive.
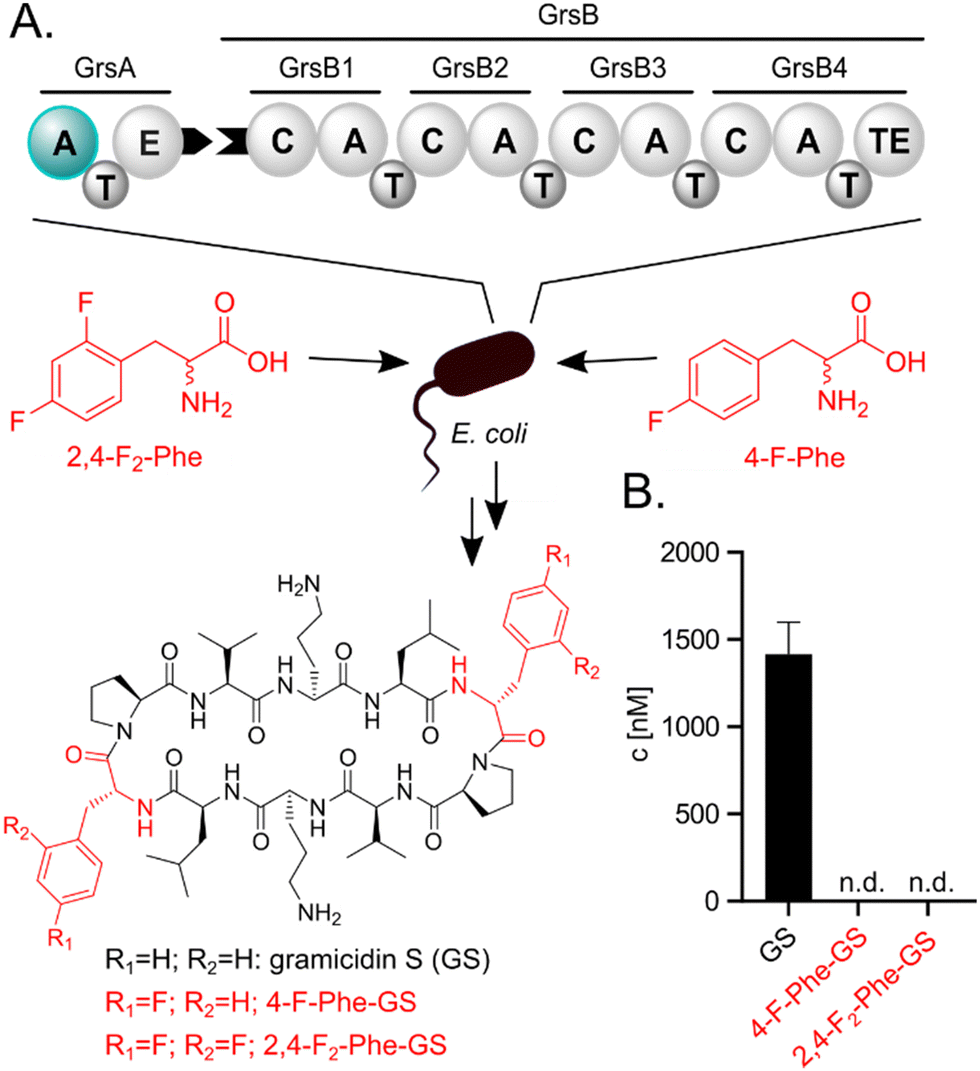 | ||
| Fig. 2 (A) E. coli heterologously expressing the GS NRPS. Either Phe or a fluorinated analog (2,4-F2-Phe or 4-F-Phe) is supplied to the medium. The A-domain of GrsA which selects Phe or analogs is highlighted in teal. (B) Biosynthetic products are quantified via UPLC-MS/MS. N.d.: not detectable. Fluorinated compounds or residues are highlighted in red. | ||
Here, we investigate the specificity for fluorinated Phe analogues of GrsA, the first module of the gramicidin S (GS) NRPS.30 According to adenylation kinetics, GrsA has a large preference for the native Phe substrate over 4-fluorinated analogues, which prevents incorporation of fluorine into GS. However, mutation W239S known to enhance incorporation of para-substituted Phe-derivatives31 yields a preference for the fluorinated substrates and thus allows in vivo production of fluorinated GS. We conclude that A-domain mutagenesis can significantly improve fluorine incorporation into NRPs, which is good news for the biosynthetic engineering of therapeutically valuable peptides.
Results
We initially hypothesized that one or two fluorine substituents would be well tolerated by the biosynthetic machinery and aimed to use precursor feeding to make fluorinated analogues of the cyclic decapeptide GS (Fig. 2). For this purpose, we used E. coli HM007932 carrying the gene cluster for GS biosynthesis on plasmid pSU18-GrsTAB as a heterologous production platform.33 The pentamodular GS NRPS consisting of GrsA (one module) and GrsB (four modules) produces a DPhe-Pro-Val-Orn-Leu pentapeptide, which is dimerized and cyclized. Addition of racemic 4-F-Phe or 2,4-F2-Phe did not impair the growth of the GS producing E. coli cultures. However, the expected products 2,4-F2-Phe-GS and 4-F-Phe-GS (Fig. 2B), in which Phe residues should have been substituted with the fluorinated analogues, were not detectable by LC-MS/MS. We hypothesized that the A-domain of module GrsA, which incorporates Phe, rejects the fluorinated amino acids despite the seemingly small structural perturbation caused by fluorination.To explain the rejection of fluorinated amino acids by GrsA, we investigated the adenylation specificity of this NRPS module using the MESG/hydroxylamine assay34 and recorded saturation kinetics. For this purpose, GrsA was expressed in His-tagged form and purified (Fig. S1, ESI†).31 A comparison of the specificity constants (kcat/KM's; Table 1 and Fig. S2, ESI†) revealed 8- and 31-fold preferences for Phe over 2,4-F2-Phe and 4-F-Phe, respectively. The turnover rates at substrate saturation (kcat) are in a similar range for all substrates and the difference is mostly caused by an increase in the Michaelis constant (KM) for the non-native substrates. Interestingly, the detrimental effect of fluorination in the 2- and 4-position of the phenyl ring is not additive, as 2,4-F2-Phe (kcat/KM = 44 mM−1 min−1) is slightly superior to 4-F-Phe (kcat/KM = 11 mM−1 min−1) as a substrate. These results indicate that GrsA-A is catalytically capable of adenylating fluorinated analogs of Phe, but Phe outcompetes these analogs when both substrates are present. To compare the poor fluorine selectivity of GrsA with other NRPS modules, we additionally tested the Val-specific A-domain SrfA-B1 from surfactin A biosynthesis.35 While adenylation activity for hexafluorinated F6-Val was not even detectable, rac-3-F-Val showed a similar trend as GrsA with a 39-fold selectivity for the native over the monofluorinated substrate.
| Enzyme | Substrateb | k cat [min−1] | K M [mM] | k cat/KM [min−1 mM−1] | Selectivityc |
|---|---|---|---|---|---|
| a Adenylation kinetics were measured using the MESG/hydroxylamine assay (Fig. S2, ESI).34 N.a.: not applicable; n.d.: not detectable. b All amino acids are used in racemic form. c The selectivity is calculated as (kcat/KM[native substrate])/(kcat/KM[fluorinated analog]). | |||||
| GrsA | Phe | 16 ± 2 | 0.05 ± 0.01 | 340 | N.a. |
| 4-F-Phe | 26 ± 3 | 2.3 ± 0.7 | 11 | 31 | |
| 2,4-F2-Phe | 42 ± 3 | 0.9 ± 0.2 | 44 | 7.6 | |
| 2-F-Phe | 27.5 ± 0.8 | 0.020 ± 0.002 | 1400 | 0.24 | |
| 3-F-Phe | 21 ± 2 | 0.014 ± 0.005 | 1500 | 0.23 | |
| 3,5-F2-Phe | 31 ± 2 | 0.014 ± 0.003 | 2200 | 0.15 | |
| GrsA-W239S | Phe | 21 ± 3 | 5 ± 1 | 4.1 | N.a. |
| 4-F-Phe | 20 ± 10 | 3 ± 5 | 5.7 | 0.7 | |
| 2,4-F2-Phe | 15 ± 4 | 2 ± 2 | 7.2 | 0.57 | |
| 2-F-Phe | 40 ± 10 | 3 ± 1 | 16 | 0.25 | |
| 3-F-Phe | 14 ± 1 | 0.9 ± 0.1 | 15 | 0.27 | |
| 3,5-F2-Phe | 10 ± 2 | 2.2 ± 0.8 | 4.3 | 0.9 | |
| SrfA-B1 | Val | 8 ± 2 | 0.06 ± 0.02 | 140 | N.a. |
| 3-F-Val | 28 ± 3 | 8 ± 1 | 3.5 | 39 | |
| F6-Val | N.d. | N.d. | N.a. | N.a. | |
To directly test the selectivity of GrsA under competition conditions, which are similarly found inside cells fed with fluorinated amino acids, we used a multiplexed hydroxamate assay (HAMA, Fig. 3a and b). HAMA is based on direct detection of amino acid hydroxamates formed from amino acyl adenylates in the presence of 150 mM hydroxylamine.36 HAMA confirmed strong preference of wildtype GrsA for Phe over fluorinated Phe (Fig. 3a).
 | ||
| Fig. 3 Impact of mutation W239S on substrate specificity. (A) HAMA profiles of GrsA and (B) GrsA-W239S. All proteinogenic amino acids were present as substrates, but only detectable hydroxamates are shown. Deuterated Phe was used to distinguish D- and L-Phe.36 Fluorinated substrates were added in racemic form. (C) Crystal structure of GrsA with Phe and AMP bound as ligands (PDB ID 1 amu)37 and (D) a computational model of 4-F-Phe bound to the same active site. (E) Potential energy of a model system, calculated from classical force field parameters, as a function of the distance between Phe (carbon atom in 4-position) and the aromatic system of Trp. | ||
To understand the origins of the high KM-values for fluorinated substrates, we performed binding studies using isothermal titration calorimetry (ITC, Fig. S3, ESI†). For ITC experiments, we expressed only the N-terminal fragment (Acore) of GrsA-A, which contains the amino acid binding pocket but not the catalytic lid of the A-domain. The KD values determined by ITC were 600 ± 300 μM for 4-F-Phe, 420 ± 40 μM for 2,4-F2-Phe, and 60 ± 10 μM for Phe binding to GrsA-Acore. Apparently, the increase in KM caused by fluorination of the substrate is due to a higher KD (weaker binding) of the fluorinated substrates. Previously, fluorination of drug molecules has been observed to cause entropy-enthalpy trade-offs upon binding38 resulting only in minor changes in KD. We did not observe this effect with GrsA-Acore. If there are differences, these were obscured by the errors on the measured ΔH and ΔS values (Table S2, ESI†).
GrsA's surprisingly strong discrimination against 4-fluorinated Phe derivatives also suggested that this preference could perhaps be inverted through mutagenesis to allow efficient and selective biosynthesis of fluorinated peptides. According to the three-dimensional structure of GrsA-A (Fig. 3C),37 the para-position of the substrate's phenyl side chain points towards the indole side chain of protein residue Trp239. Since the substrate side chain is accommodated in a tightly packed, hydrophobic space, the fluorinated substrates might be rejected due to the slightly larger size and higher polarity of fluorine compared to hydrogen (Fig. 3D). To better accommodate the fluorine substituent, we mutated residue Trp239 to Ser which is smaller and more polar. This mutation has previously been shown to promote activation of para-substituted Phe in GrsA and related A-domains.31,39 Gratifyingly, adenylation kinetics show that the kcat is barely influenced by mutation W239S (Table 1 and Fig. S2, ESI†). At the same time, the KM value for Phe increases 100-fold, while it remains nearly unchanged for the 4-fluorinated analogs. As a result, the specificity constant (kcat/KM) of GrsA-W239S is higher for the fluorinated Phe analogs than for Phe. For 4-F-Phe, mutation W239S causes a 43-fold switch in specificity. To test the performance of the mutant under cell-like substrate competition, we again employed HAMA (Fig. 3B). The specificity switch in favor of the fluorinated substrates was confirmed, although Tyr turned out to be the overall preferred substrate. Screening of other side-chains in position 239 using HAMA in 96-well plate format29 showed that mutants W239A and W239L also have increased preference for the 4-fluorinated substrates but slightly less than W239S (Fig. S5, ESI†).
The effect of a fluorine substituent on substrate binding to GrsA and GrsA-W239S was further investigated by computational modelling (Fig. 3C–E). We hypothesized that the fluorine substituent might disturb the edge-to-face (T-shaped) aromatic interaction40 between the substrate side-chain and Trp239 (Fig. 3C). To gauge the strength of this interaction, the potential energy was calculated for a model system consisting only of capped Trp and Phe in vacuum using the Amber14SB/GAFF2 force field (Fig. 3E). With one or two fluorine substituents, the energy well for the interaction is about 10 kJ mol−1 shallower than with an unsubstituted phenyl ring, explaining the lower binding of the fluorinated substrates. To understand why F-Phe binds GrsA-W239S better than Phe, we considered the cavity generated by the large-to-small mutation in a 10 ns molecular dynamics simulation. While GrsA with Phe as a ligand has zero water molecules near the tip of the phenyl side-chain, two or three waters are found in GrsA-W239S (Table S3, ESI†). With F-Phe as a ligand, that number is reduced to zero to two waters, indicating a better fit into the cavity. No binding interaction between Ser239 and fluorine was detected, but the aqueous environment may accommodate the electronegative fluoro-substituent better than the hydrophobic pocket in GrsA. However, the fluoro-substituent fills the enlarged GrsA-W239S binding pocket less efficiently than the previously tested O-propargyl-Tyr, that was also docked into the pocket (Table S4 and Fig. S4, ESI†) and for which the mutation causes a more dramatic 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold specificity switch.31
000-fold specificity switch.31
After identifying the T-shaped aromatic interaction as crucial for substrate binding in GrsA, we were eager to investigate the behavior of fluorinated Phe analogues that lacked substitution at the 4-position. We expected strongly electron-withdrawing fluoro-substituents to strengthen the electrostatic interaction with the electron-rich indole side-chain when they were not interrupting the interaction at the 4-position. These expectations were substantiated by additional simulations also indicating a slightly lowered energy minimum for the T-shaped aromatic interaction (Fig. 3E). Consequently, we recorded saturation kinetics with 2-F-Phe, 3-F-Phe, and 3,5-F2-Phe (Table 1). Indeed, GrsA prefers 2-F-Phe, 3-F-Phe, and 3,5-F2-Phe over Phe. The best substrate was 3,5-F2-Phe, which was adenylated with a 6.5-fold higher catalytic efficiency than the wild-type substrate Phe. In agreement with our binding model, this substantial improvement is absent in the W239S mutant where the crucial aromatic interaction is absent.
Having identified the selectivity of GrsA as critical for the biosynthesis of fluorinated GS analogs, we eliminated competition with Phe by using an in vitro system to produce fluorinated GS (Fig. 4A). Therefore, we expressed and purified GrsA and GrsB in His-tagged form from E. coli HM0079.33,41 To a reaction with GrsA and GrsB, we supplied either L-Phe, 2,4-F2-Phe, or 4-F-Phe in addition to ATP and all other required amino acids. In vitro, 4-F-Phe-GS reaches a concentration of 1100 ± 100 nM, which is 61% of the GS concentration obtained under the same conditions (Fig. 4A). Apparently, the fluorinated substrates are tolerated by all downstream domains once they have passed the selectivity filter of GrsA-A. Under competitive conditions with both 4-F-Phe and Phe added, wild-type GrsA allows less than 2% of fluorine incorporation into GS, which is in good agreement with the adenylation preference of GrsA-A. With mutant GrsA-W239S, the fraction of 4-F-Phe-GS increases to 50% (Fig. 4B). A similar biosynthetic preference is observed in vitro for incorporation of 2,4-F2-Phe (Fig. 4C).
 | ||
| Fig. 4 Enhancement of GS formation with mutation W239S in vitro and in vivo. (A) In vitro production of GS and fluorinated GS analogs without competition using purified GrsA and GrsB. (B) In vitro production of GS and 4-F-GS under competitive conditions, using GrsA or GrsA-W239S. (C) In vitro production of GS and 2,4-F2-GS under competitive conditions, using GrsA or GrsA-W239S. (D) In vivo production of GS and the fluorinated variants using a producer strain harboring the W239S mutation. The indicated fluorinated amino acids are supplemented to the growth medium. The error bars represent two biological replicates. | ||
Encouraged by the successful production of fluorinated GS in vitro, we revisited the in vivo conditions, which initially yielded no detectable fluorine incorporation. In vivo conditions are generally preferable because they do not rely on the tedious and low-yielding expression of the fragile NRPS proteins and are thus more easily scalable. To test whether mutation W239S would convey sufficient specificity for fluorinated Phe analogs in vivo, we generated the mutated plasmid pSU18-GrsTAB-W239S (Table S1, ESI†). Indeed, when 4 mM of 4-F-Phe or 2.5 mM 2,4-F2-Phe were added to cultures of E. coli HM0079 carrying this plasmid, LC-MS/MS analysis revealed production of 68 nM 4-F-GS and 67 nM 2,4-F2-GS, respectively. These concentrations correspond to volumetric yields of 0.079 mg L−1 for 4-F-GS and 0.081 mg L−1 for 2,4-F2-GS from 3 mL cultures (Fig. 4D).
Conclusion
Hydrogen-fluorine exchange is one of the most subtle, yet powerful changes that can be introduced into a molecule. Our results show that nonribosomal A-domains can be unexpectedly sensitive to this change, which may prevent the incorporation of fluorinated amino acids into nonribosomal peptides. While the observed difference in kcat/KM for Phe and 4-F-Phe in GrsA is 31-fold, the discrimination under in vivo conditions seems to be even stronger (Fig. 2B). Computational modelling indicates that fluorine-substitution in the 4-position of the Phe side-chain interrupts a crucial T-shaped aromatic interaction between Trp239 and the substrate. Perhaps, the poor acceptance of 4-F-Phe is compounded by poor uptake into the cell. With the wild-type GS NRPS, biosynthesis of 4-F-GS was only possible in vitro, where competition with the native substrate Phe is eliminated. However, for synthesis of nonribosomal peptides at scale, in vitro conditions are hardly viable since the NRPS proteins are not sufficiently high-yielding and robust.For efficient in vivo biosynthesis of a fluorinated peptide analog, the NRPS must be able to fend off competition from the unfluorinated building block. Here we show how a single mutation within the binding pocket of GrsA-A, W239S, shifts the selectivity 43-fold in favor of 4-F-Phe. With this mutation, production of 4-fluorinated GS analogs becomes possible both under in vitro and in vivo conditions, although native GS remains the major product in vivo. A similar mutational effect was observed by Sirirungruang and coworkers, when they introduced mutation F190V into the trans-AT DszAT that they used for activation of fluoromalonyl-CoA.18 Based on computational modelling, we speculate that the mutational effect is explained by the entropic contribution of ordered water molecules rather than specific binding interactions to the fluorine substituent, which were not found here or in a previous study investigating another protein–ligand interaction.42 Importantly, the observed enhancement of the fluorine specificity in GrsA upon mutation augurs well for future projects aiming to further increase this specificity.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Donald Hilvert (ETH Zurich) for kindly providing pSU18-GrsA and Vanessa Rongen for technical assistance with kinetic measurements. We are thankful for the financial support of the Jena School for Microbial Communication (F. P.), Deutscher Akademischer Austauschdienst (F. P.), Konrad Adenauer Stiftung (O. N.), International Leibniz Research School (P. S.), the Daimler und Benz Stiftung (H. K.), the Fonds der Chemischen Industrie, and the Bundesministerium für Bildung und Forschung (ZIK Septomics, FKZ 03Z22JI1). This project has been supported by a Max-Buchner-Fellowship of the DECHEMA. Partially funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – project ID 387284271 – SFB 1349 Fluorine-Specific Interactions and project ID 441781663.References
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- T. Kaneko, T. J. Dougherty and T. V. Magee, in Comprehensive Medicinal Chemistry II, ed. J. B. Taylor and D. J. Triggle, Elsevier, Oxford, 2007, pp. 519–566 Search PubMed.
- A. A. Berger, J.-S. Völler, N. Budisa and B. Koksch, Acc. Chem. Res., 2017, 50, 2093–2103 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- M. C. Walker and M. C. Y. Chang, Chem. Soc. Rev., 2014, 43, 6527–6536 RSC.
- K. Rosenthal, M. Becker, J. Rolf, R. Siedentop, M. Hillen, M. Nett and S. Lütz, ChemBioChem, 2020, 21, 3225–3228 CrossRef CAS PubMed.
- J. Rivera-Chávez, H. A. Raja, T. N. Graf, J. E. Burdette, C. J. Pearce and N. H. Oberlies, J. Nat. Prod., 2017, 80, 1883–1892 CrossRef PubMed.
- S. Weist, B. Bister, O. Puk, D. Bischoff, S. Pelzer, G. J. Nicholson, W. Wohlleben, G. Jung and R. D. Süssmuth, Angew. Chem., Int. Ed., 2002, 41, 3383–3385 CrossRef CAS PubMed.
- W. Runguphan, J. J. Maresh and S. E. O’Connor, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13673–13678 CrossRef CAS PubMed.
- N. K. O’Connor, D. K. Rai, B. R. Clark and C. D. Murphy, J. Fluorine Chem., 2012, 143, 210–215 CrossRef.
- M. C. Walker, B. W. Thuronyi, L. K. Charkoudian, B. Lowry, C. Khosla and M. C. Y. Chang, Science, 2013, 341, 1089–1094 CrossRef CAS PubMed.
- N. K. O’Connor, A. S. Hudson, S. L. Cobb, D. O’Neil, J. Robertson, V. Duncan and C. D. Murphy, Amino Acids, 2014, 46, 2745–2752 CrossRef PubMed.
- K. M. J. De Mattos-Shipley, C. Greco, D. M. Heard, G. Hough, N. P. Mulholland, J. L. Vincent, J. Micklefield, T. J. Simpson, C. L. Willis, R. J. Cox and A. M. Bailey, Chem. Sci., 2018, 9, 4109–4117 RSC.
- C. S. M. Amrine, J. L. Long, H. A. Raja, S. J. Kurina, J. E. Burdette, C. J. Pearce and N. H. Oberlies, J. Nat. Prod., 2019, 82, 3104–3110 CrossRef CAS PubMed.
- A. Sester, K. Stüer-Patowsky, W. Hiller, F. Kloss, S. Lütz and M. Nett, ChemBioChem, 2020, 21, 2268–2273 CrossRef CAS PubMed.
- A. Rittner, M. Joppe, J. J. Schmidt, L. M. Mayer, S. Reiners, E. Heid, D. Herzberg, D. H. Sherman and M. Grininger, Nat. Chem., 2022, 14(9), 1000–1006 CrossRef CAS PubMed.
- S. Sirirungruang, O. Ad, T. M. Privalsky, S. Ramesh, J. L. Sax, H. Dong, E. E. K. Baidoo, B. Amer, C. Khosla and M. C. Y. Chang, Nat. Chem. Biol., 2022, 18(8), 886–893 CrossRef CAS PubMed.
- M. S. Lichstrahl, L. Kahlert, R. Li, T. A. Zandi, J. Yang and C. A. Townsend, Chem. Sci., 2023, 14(14), 3923–3931 RSC.
- L. J. Stevenson, J. G. Owen and D. F. Ackerley, ACS Chem. Biol., 2019, 14, 2115–2126 CAS.
- R. D. Süssmuth and A. Mainz, Angew. Chem., Int. Ed., 2017, 56, 3770–3821 CrossRef PubMed.
- Y. Liu, S. Ding, J. Shen and K. Zhu, Nat. Prod. Rep., 2019, 36, 573–592 RSC.
- K. A. Bozhüyük, J. Micklefield and B. Wilkinson, Curr. Opin. Microbiol., 2019, 51, 88–96 CrossRef PubMed.
- J. M. Reimer, A. S. Haque, M. J. Tarry and T. M. Schmeing, Curr. Opin. Struct. Biol., 2018, 49, 104–113 CrossRef CAS PubMed.
- G. L. Challis, J. Ravel and C. A. Townsend, Chem. Biol., 2000, 7, 211–224 CrossRef CAS PubMed.
- T. Stachelhaus, H. D. Mootz and M. A. Marahiel, Chem. Biol., 1999, 6, 493–505 CrossRef CAS PubMed.
- A. Stanišić and H. Kries, ChemBioChem, 2019, 20, 1347–1356 CrossRef PubMed.
- M. H. Medema, K. Blin, P. Cimermancic, V. De Jager, P. Zakrzewski, M. A. Fischbach, T. Weber, E. Takano and R. Breitling, Nucleic Acids Res., 2011, 39(suppl_2), W339–W346 CrossRef CAS PubMed.
- A. Stanišić, C.-M. Svensson, U. Ettelt and H. Kries, bioRxiv, 2022, preprint DOI:10.1101/2022.08.30.505883.
- J. Krätzschmar, M. Krause and M. A. Marahiel, J. Bacteriol., 1989, 171, 5422–5429 CrossRef PubMed.
- H. Kries, R. Wachtel, A. Pabst, B. Wanner, D. Niquille and D. Hilvert, Angew. Chem., Int. Ed., 2014, 53, 10105–10108 CrossRef CAS PubMed.
- S. Gruenewald, H. D. Mootz, P. Stehmeier and T. Stachelhaus, Appl. Environ. Microbiol., 2004, 70, 3282–3291 CrossRef CAS PubMed.
- F. Pourmasoumi, S. De, H. Peng, F. Trottmann, C. Hertweck and H. Kries, ACS Chem. Biol., 2022, 17, 2382–2388 CrossRef CAS PubMed.
- D. J. Wilson and C. C. Aldrich, Anal. Biochem., 2010, 404, 56–63 CrossRef CAS PubMed.
- A. Koglin, F. Löhr, F. Bernhard, V. V. Rogov, D. P. Frueh, E. R. Strieter, M. R. Mofid, P. Güntert, G. Wagner, C. T. Walsh, M. A. Marahiel and V. Dötsch, Nature, 2008, 454, 907–911 CrossRef CAS PubMed.
- A. Stanišić, A. Hüsken and H. Kries, Chem. Sci., 2019, 10, 10395–10399 RSC.
- E. Conti, T. Stachelhaus, M. A. Marahiel and P. Brick, EMBO J., 1997, 16, 4174–4183 CrossRef CAS PubMed.
- B. Breiten, M. R. Lockett, W. Sherman, S. Fujita, M. Al-Sayah, H. Lange, C. M. Bowers, A. Heroux, G. Krilov and G. M. Whitesides, J. Am. Chem. Soc., 2013, 135, 15579–15584 CrossRef CAS PubMed.
- D. L. Niquille, I. B. Folger, S. Basler and D. Hilvert, J. Am. Chem. Soc., 2021, 143, 2736–2740 CrossRef CAS PubMed.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191 RSC.
- D. L. Niquille, D. A. Hansen, T. Mori, D. Fercher, H. Kries and D. Hilvert, Nat. Chem., 2017, 10, 282–287 CrossRef PubMed.
- L. Wehrhan, J. Leppkes, N. Dimos, B. Loll, B. Koksch and B. G. Keller, J. Phys. Chem. B, 2022, 126, 9985–9999 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cb00061c |
| This journal is © The Royal Society of Chemistry 2023 |