
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA monoadduct generating Ru(II) complex induces ribosome biogenesis stress and is a molecular mimic of phenanthriplatin†
Richard J.
Mitchell
a,
Sarah M.
Kriger
d,
Alexander D.
Fenton
a,
Dmytro
Havrylyuk
a,
Ankit
Pandeya
a,
Yang
Sun
a,
Tami
Smith
b,
Jason E.
DeRouchey
 a,
Jason M.
Unrine
b,
Viral
Oza
c,
Jessica S.
Blackburn
c,
Yinan
Wei
a,
David K.
Heidary
*d and
Edith C.
Glazer
*d
a,
Jason M.
Unrine
b,
Viral
Oza
c,
Jessica S.
Blackburn
c,
Yinan
Wei
a,
David K.
Heidary
*d and
Edith C.
Glazer
*d
aDepartment of Chemistry, University of Kentucky, 505 Rose St., Lexington, KY 40506, USA. E-mail: ec.glazer@uky.edu; david.heidary@gmail.com
bDepartment of Plant and Soil Sciences, University of Kentucky, 1100 S. Limestone St, Lexington, KY 40546, USA
cDepartment of Molecular and Cellular Biochemistry, University of Kentucky, 741 S. Limestone St., Lexington, KY 40536, USA
dDepartment of Chemistry, North Carolina State University, 2620 Yarbrough DriveRaleigh, NC, 27695, USA. E-mail: eglazer@ncsu.edu
First published on 27th February 2023
Abstract
Ruthenium complexes are often investigated as potential replacements for platinum-based chemotherapeutics in hopes of identifying systems with improved tolerability in vivo and reduced susceptibility to cellular resistance mechanisms. Inspired by phenanthriplatin, a non-traditional platinum agent that contains only one labile ligand, monofunctional ruthenium polypyridyl agents have been developed, but until now, few demonstrated promising anticancer activity. Here we introduce a potent new scaffold, based on [Ru(tpy)(dip)Cl]Cl (tpy = 2,2′:6′,2′′-terpyridine and dip = 4,7-diphenyl-1,10-phenanthroline) in pursuit of effective Ru(II)-based monofunctional agents. Notably, the extension of the terpyridine at the 4′ position with an aromatic ring resulted in a molecule that was cytotoxic in several cancer cell lines with sub-micromolar IC50 values, induced ribosome biogenesis stress, and exhibited minimal zebrafish embryo toxicity. This study demonstrates the successful design of a Ru(II) agent that mimics many of the biological effects and phenotypes seen with phenanthriplatin, despite numerous differences in both the ligands and metal center structure.
Introduction
Metal-based small molecules that act primarily by damaging DNA are a mainstay of cancer chemotherapy. While traditional platinum agents, such as cisplatin and carboplatin, induce apoptosis through DNA crosslinking mechanisms,1,2 oxaliplatin is now believed to function primarily by induction of ribosome biogenesis stress.3,4 The difference in the mechanisms of action of these drugs is thought to be responsible for the variations in therapeutic efficacy in different cancer types; for example, oxaliplatin is the only platin agent used for colorectal cancer. Very few compounds are known to disrupt ribosome biogenesis, which is an emerging area of interest for targeted chemotherapy, as this mechanism of action should impact cancer cells that are highly dependent on active translation.5In contrast to cisplatin, carboplatin, and oxaliplatin, which can make two bonds to a biomolecule, pyriplatin and phenanthriplatin defied traditional structure–activity relationships (SAR) by exerting their anticancer properties as monoadduct generators.6–9 Recently, a variety of studies have demonstrated that phenanthriplatin acts via multiple biological mechanisms, including topoisomerase inhibition,10 damage to nuclear DNA,9 and, like oxaliplatin, ribosome biogenesis stress.3,4 As with oxaliplatin, the anticancer profile of phenanthriplatin is distinct from traditional platinum agents, suggesting potential application in tumor types that are refractory to the other drugs. It is also possible that monoadduct formers may evade resistance mechanisms, such as DNA repair, that are effective against biadduct formers. However, all platinum agents suffer from other mechanisms of resistance in cancer cells, such as thiol-mediated inactivation and decreased cellular accumulation.1,7 For this reason, other transition metals are intrinsically appealing if they can mimic the biologically efficacious molecular interactions of non-traditional Pt(II) agents, but overcome these limitations.
Polypyridyl ruthenium complexes with one labile ligand are potential replacements for platinum-based monoadduct-generators.11,12 This might seem counterintuitive, due to structural differences, as Ru(II) creates octahedral complexes and Pt(II) is square planar, and the platin agents do not contain polypyridyl ligands. However, both metals are relatively “soft” and exhibit aquation kinetics that typically occur over several hours, a similar time-scale to cell division processes.13 Ru(II) has a lower affinity for free thiols, making its complexes less susceptible to inactivation and efflux.13,14 Despite this fact, currently reported Ru(II) polypyridyl monofunctional agents have poor activity compared to phenanthriplatin.11,12 It is unclear if this is due to a failure to mimic appropriate mechanism of action (MoA), or other chemical or biological factors.
We questioned if it was possible to make a Ru(II) complex that recapitulated phenanthriplatin's activity, which is attributed to multiple different MoAs, but with an emphasis on ribosome biogenesis. This was anticipated to be a challenge, as recently, the DeRose group demonstrated very stringent structural requirements for Pt(II) compounds capable of disrupting ribosome biogenesis. To this date, there are only six molecules that have this effect: oxaliplatin, three oxaliplatin analogues, an oxaliplatin prodrug, and phenanthriplatin.4,15,16 However, we were encouraged by the fact that some Ru(II) complexes are able to localize to the nucleolus,17 as oxaliplatin and phenanthriplatin do, and other ruthenium complexes have been shown to alter translation.18,19 Moreover, specific Ru(II) complexes have also shown topoisomerase inhibition, and can either inhibit the catalytic site of the enzyme20 or generate cleavage complexes.21–23 These studies demonstrate individual Ru(II) compounds can exhibit the various biological activities of phenanthriplatin, and we hypothesized that this may be done in a single molecule if key structural features, such as polyaromatic lipophilic groups are incorporated into the system. Here we report the successful creation of an octahedral Ru(II) “mimic” of phenanthriplatin, which is a new addition to the select group of compounds that induce nucleolar stress.
Results and discussion
Compound design and synthesis
To test our hypothesis that phenanthriplatin mimics can be created with an octahedral system, two Ru(II) complexes were designed and synthesized that contained a 2,2′:6′,2′′-terpyridine (tpy)-based ligand and 4,7-diphenyl-1,10-phenanthroline (dip). The dip ligand was selected due in part to the improved cellular uptake of dip-containing ruthenium complexes,24 as well as the increased hydrophobicity of its metal complexes. The incorporation of the tridentate tpy ligand with a bidentate ligand ensured only one exchangeable monodentate ligand in the complex. Some metal complexes containing tpy ligands are cytotoxic agents,11,25–27 and this simple ligand allows for facile synthetic modifications that could impact uptake, localization, and cellular targets.11,26,28,29 Notably, aromatic extension at the 4-position in the center pyridine increases the potency of both the free ligands and tpy-coordinated Ru(II) complexes.11,30 Although modification of tpy- and dip-based ligands have independently been shown to improve cellular uptake and cytotoxicity of Ru(II) compounds, they have not yet been combined for anti-cancer compounds. Compound 1, [Ru(tpy)(dip)Cl]Cl (Chart 1) was previously reported,31,32 although biological studies were not performed; [Ru(ph-tpy)(dip)Cl]Cl (2) is a novel compound. Care was taken with both compounds to prevent ligand exchange, since various solvents can coordinate to the metal center.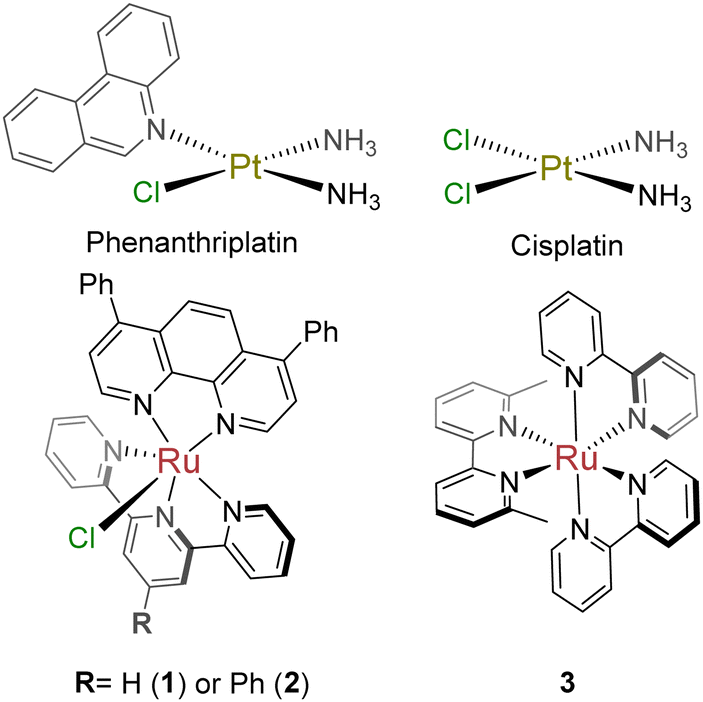 | ||
| Chart 1 Compounds investigated in this study. | ||
Inactivation by free thiols
Inactivation by cellular thiols, such as glutathione (GSH), is a common drawback for platinum-based drugs. When phenanthriplatin was incubated with 5 mM GSH, conversion to the thiol adduct was observed (Fig. S1A, ESI†). The inactivation of phenanthriplatin by GSH could explain why previous in vivo data showed no improvement in mice treated with phenanthriplatin versus cisplatin.33 In contrast, 2 did not react with GSH, as indicated by both HPLC kinetics (Fig. S1B, ESI†) and UV/Vis endpoint experiments (Fig. S1C, ESI†). The only species found was the aqua complex, indicating that 2 is not susceptible to thiol-mediated inactivation. If GSH is the limiting factor for efficacy in murine models, 2 could be a promising replacement.Cell cytotoxicity and zebrafish embryo toxicity
Several cancer cell lines were tested to evaluate compound activity: lung adenocarcinoma (A549), promyelocytic leukemia (HL-60), pancreatic ductal adenocarcinoma (MIA PaCa-2), and prostate carcinoma (DU 145). Compounds 1 and 2 were cytotoxic in all cell lines, with potency in the nM to low μM range (Table 1 and Fig. S2–S6, ESI†). Compound 1 exhibited potencies comparable to cisplatin and similar to 3, a light-activated Ru(II) DNA biadduct former we previously reported.34 Compound 2 was more active, with an 8–11-fold improvement as compared to 1. The DU145 spheroid data highlights the activity of these compounds in a multicellular system, with 2 exhibiting low micromolar activity (Table 1 and Fig. S7, ESI†).| Compound | IC50 (St. dev.) [μM] | Zebrafish viability (%) [20 μM] | ||||||
|---|---|---|---|---|---|---|---|---|
| HL-60 | DU145 | MIA PaCa2 | A549 | HEL299 | SI (HEL299:A549) | DU145 spheroids | ||
| a Average values from triplicate measurements. b The percentage of viable embryos is representative of n = 6. c Reported values are following exposure to indigo (450 nm) light. d Values taken from prior work. | ||||||||
| 1 | 3.33 (0.04) | 1.50 (0.10) | 2.6 (0.2) | 5.73 (0.04) | 28.19 (1.85) | 4.9 | 27.33 (2.14) | 100 |
| 2 | 0.4 (0.1) | 0.17 (0.02) | 0.23 (0.06) | 0.57 (0.02) | 7.56 (0.37) | 13.3 | 4.91 (0.84) | 100 |
| 3 | 1.6 (0.2)d | 7.4 (1.0) | 26.5 (3.5) | 1.1 (0.3)d | 16.72 (4.27) | 15.2 | n.d. | n.d. |
| Cisplatin | 3.1 (0.2)d | 0.62 (0.06) | 5.9 (0.4) | 3.4 (0.6) | 3.05 (0.44) | 0.9 | 5.28 (0.36) | 100 |
| Phenanthriplatin | 1.8 (0.4) | 0.14 (0.01) | 0.15 (0.01) | 0.19 (0.01) | 2.63 (0.14) | 13.8 | 0.84 (0.06) | 86 |
In order to determine the selectivity for cancer cells compared to healthy cells, healthy lung fibroblasts (the HEL299 cell line) were treated with each compound and a selectivity index (SI) was determined for the ratio of IC50 values for HEL299:A549. The SI for cisplatin was 0.9, consistent with previous reports.26 Compound 1 had an SI of 4.9, while phenanthriplatin and 2 had an SI value of 13.8 and 15.2 respectively, displaying the capacity for this scaffold to selectively target cancerous cells over healthy cells. Zebrafish embryos were then used as a screening tool to assess tolerability for small molecules in vivo.35 We found that zebrafish embryos tolerated 20 μM treatment with compounds 1 and 2 (Table 1), demonstrating that these compounds exhibit selective toxic effects.
Different cancer types exhibit varying sensitivities to small molecules. This information can be utilized to understand mechanistic underpinnings of novel molecules.36 To estimate the selectivity of compounds for the various cancer cell lines, the difference in individual pIC50 values versus the average pIC50 was calculated (eqn (S1) and (S2), ESI†). From these data, the sensitivity of each cell line to each of the compounds was visualized, with negative and positive values corresponding to cell line resistance and sensitivity, respectively (Fig. S8, ESI†). The profiles for 1 and 2 were similar to phenanthriplatin, and distinct from cisplatin and 3.
This intriguing result motivated the submission of 1 and 2 to the National Cancer Institute (NCI) for the 60-cell line analysis at a single dose for percent growth inhibition (Fig. S9 and S10, ESI†), and five-dose response (Fig. S11 and S12, ESI†) to correlate the compounds to the Standard Agents GI50 library of small molecules with known mechanisms of action.36 The COMPARE analysis, which correlates specific cell line response profiles between a compound and the Standard Agents GI50 library,36 was used for 2. The resistance-sensitivity data for compounds with high correlations and distinct mechanisms of action were plotted in a heat map (Fig. 1). The COMPARE analysis identified chromomycin A3 as having the highest correlation to 2, with a Pearson correlation coefficient of 0.85 (Table S1, ESI†). Chromomycin A3 binds to DNA with GC-rich regions,37 with a preference for nucleolus organizer regions (NORs) of DNA,38 which have been implicated in nucleolar stress.39 Actinomycin D exhibited a correlation of 0.69; this compound was initially described as a DNA binding agent that interfered with elongating RNA strands,40,41 resulting in inhibition of transcription, but it is now established that actinomycin D is also a ribosome biogenesis stress inducing agent.42 There was also high correlations between 2 and microtubule-targeting molecules paclitaxel (0.71) and vinblastine (0.64). Very few coordination complexes have been reported to interact with microtubules, but hyperstabilization was shown with [Ru(dip)3]2+ in one study,43 and an organometallic osmium compound had a similar effect due to coordination to the sulfhydryl groups of microtubules.44
 | ||
| Fig. 1 NCI 60-Cell Line Panel COMPARE Analysis results. The sensitivity-resistance profile for 2 was compared to the profiles of small molecules in the Standard Agents GI50 library. The difference between individual cell line IC50 and the average IC50 across the 60 cell lines was calculated and plotted. Values greater than +2 were plotted in dark red. No values exceeded −2. Black cells indicate IC50 values not determined. | ||
Notably, the correlation between 2 and DNA alkylating agents such as cisplatin and carboplatin was poor (0.12–(−0.16)), suggesting the primary mechanism of action is not reliant upon this kind of DNA damage (Table S1, ESI†). There was a positive correlation between the topoisomerase inhibitors etoposide and doxorubicin and 2 (0.55–0.39), although it was lower than that of transcription inhibitors. However, doxorubicin also induces hallmarks of nucleolar stress. Oxaliplatin and 1 were also plotted in the heatmap, but only for qualitative comparison, since they are not in the standard agents library. The five-dose data for 1 and oxaliplatin was collected independently for addition to Fig. 1. Oxaliplatin appeared to have a moderate correlation, however, non-small cell lung cancer cell lines exhibited a resistant profile for oxaliplatin but a sensitive profile for 2. Phenanthriplatin was not included in the heatmap because the 60-cell line data is not available on the public NCI DTP database. However, the data available from previous reports allowed for some comparisons.9 Non-small cell lung cancers and central nervous system (CNS) cancers were both sensitive to phenanthriplatin and 2.9 The response across ovarian cancers was comparable, suggesting that phenanthriplatin and 2 exhibit similar behaviors across different cell lines of the same cancer type. The response in renal and breast cancers, in contrast, differed between the two compounds. Renal cancers were sensitive to treatment with phenanthriplatin but breast cancer response was average.9 Renal cancers were resistant to 2 and breast cancers were generally sensitive. Since phenanthriplatin has been identified as having multiple MoAs, and 2 exhibits comparable sensitivity-resistance profiles to phenanthriplatin, we decided to evaluate 2 for each of the plausible mechanisms highlighted by the NCI analysis: DNA damage, topoisomerase inhibition, microtubule dynamics, and ribosome biogenesis stress.
Studies in Escherichia coli
Antitumor antibiotics demonstrate utility in cancer chemotherapy as well as functioning as antibiotics, which motivated investigation of 1 and 2 in bacteria. Escherichia coli (E. coli) were used, as this is a simple model organism to probe the DNA-damaging capacity for small molecules. First, the minimum inhibitory concentration (MIC; the lowest concentration of an agent where no bacterial growth is observed) was determined (Fig. S13, ESI†). The MIC values for the Ru(II) complexes were similar to that of phenanthriplatin. Additional positive controls included cisplatin, rifampicin (an RNA polymerase inhibitor), tetracycline (a translation inhibitor) and a negative control, paclitaxel, which is a microtubule disruptor that was implicated by the COMPARE analysis. All controls inhibited bacterial growth except for paclitaxel, as expected. Moreover, while 2 and the other compounds induced filamentation, a sign of cellular stress, no filament formation was seen with paclitaxel, suggesting 2 had a different biological activity. Next, a colony formation assay was performed, as many DNA damaging agents prevent colony formation at their MIC (Fig. S14, ESI†).45,46 In contrast, treatment with cytostatic agents, such as translation inhibitors, does not prevent colony growth.46 No colonies formed with cisplatin- or phenanthriplatin-treated cells (Fig. S14, ESI†), consistent with previous reports.46 Compounds 1 and 2 also prevented colony formation, supporting DNA-damage as a component of the MoA.Cellular uptake of the different metal complexes in E. coli was quantified by Inductively Coupled Plasma Mass Spectrometry (ICP-MS). An uptake of 1.24 × 10−15 and 1.67 × 10−15 moles of Ru per cell was found for 1 and 2, compared to 8.76 × 10−16 moles of Pt (Table S2 and Fig. S15, ESI†). Compound 3 was utilized as a Ru(II) positive control, and approximately 7.7% of the compound was observed in bacteria, similar to earlier reports.46 Next, cellular DNA was isolated and analyzed for metal content. The quantity of metal in the total genomic DNA was determined by ICP-MS, and the nucleotide to metal center ratio was calculated by dividing the number of nucleotides in the total genomic DNA present in each sample by the metal atoms. Phenanthriplatin produced the lowest nucleotide (nt) to metal center (mc) ratio, followed by cisplatin and 1, and then 2 (Table S3, ESI†). These results indicate that both compounds 1 and 2 irreversibly bind DNA. However, despite the fact that compound 2 and phenanthriplatin have the same MIC value in E. coli, the Ru(II) complex formed 10-fold fewer DNA adducts. This is consistent with an interpretation that an indirect mechanism of DNA damage, such as topoisomerase inhibition, or another mechanism, such as ribosome biogenesis stress, plays a greater role in the functional activity of 2. A ratio of 1 DNA-Ru(II) adduct to 106 atoms of Ru taken up into each E. coli cell also suggests a mechanism of cell death independent of DNA damage.
Bacterial cytological profiling
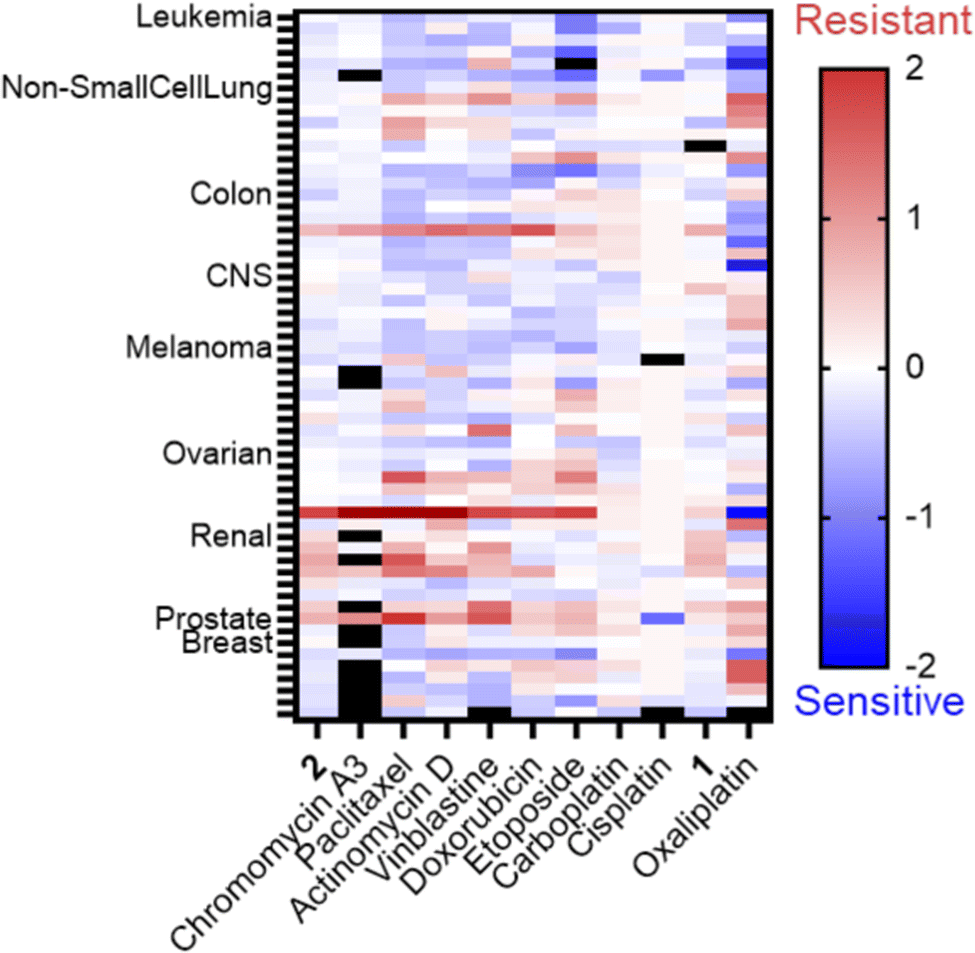
Another approach to identifying the biological impacts of small molecules is bacterial cytological profiling (BCP), which provides a fingerprint of phenotypic characteristics that depend on a compound's MoA. For example, DNA crosslinkers produce a different phenotype than TopoII inhibitors.47 In contrast to healthy E. coli, which are rod-shaped and approximately 2 μm in length (Fig. 2A), cytotoxic stress prompts elongation of the cells into filaments, and the population distribution of filament length can be correlated with the type of damage. E. coli treated with cisplatin at the MIC exhibited an average filament length of 53 μm (Fig. 2B and F). Cells treated with phenanthriplatin (Fig. 2C), compound 1 (Fig. 2D), and 2 (Fig. 2E) also exhibited filaments, with average lengths of 44 μm, 39 μm, and 94 μm, respectively (Fig. 2F). The phenotypic responses are consistent with DNA damage MoA,45 and the distribution of filament lengths for compound 2 and phenanthriplatin were similar (Fig. S16, ESI†). Notably, the shift in distribution to longer filaments is also found for the DNA gyrase inhibitor ciprofloxacin (Fig. 2F and Fig. S16, ESI†). When bacteria were treated with mitoxantrone, a TopIIα inhibitor, the distribution of filament length was comparable to cisplatin (Fig. S16, ESI†). The difference in filamentous length distributions between mitoxantrone and ciprofloxacin could be due to differences in their mechanisms of topoisomerase inhibition and the ability of cells to recover from the stress. Ciprofloxacin binds to the catalytic Mg2+ ion used for re-ligation of DNA via turnover of phosphotyrosine to tyrosine,48 whereas the mitoxantrone interactions are facilitated by non-covalent contacts unique to TopIIα.49 This could result in mitoxantrone acting in the bacteria as an intercalator, which would explain the similarity in filament distribution with DNA damaging agents.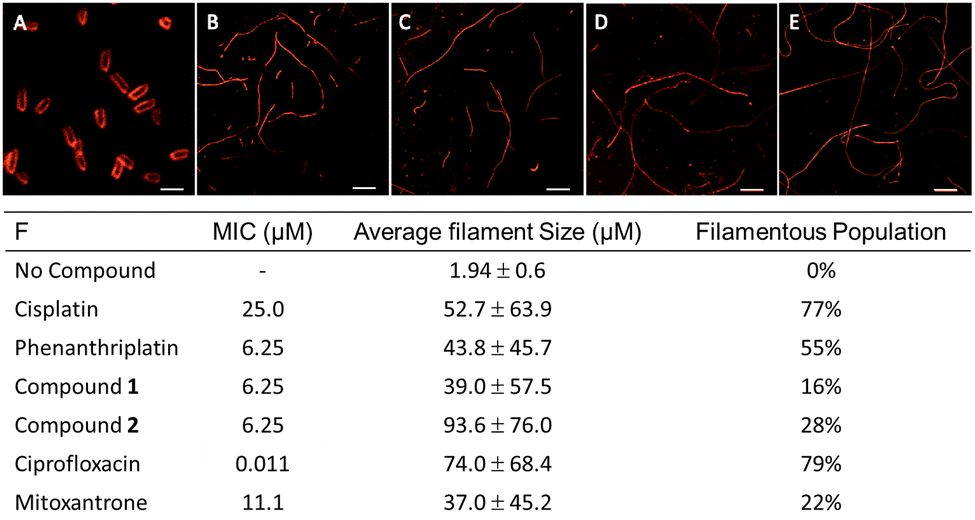 | ||
| Fig. 2 Filamentous bacterial growth observed after treatment. (A) E. coli untreated control; (B) cisplatin (25 μM); (C) phenanthriplatin (6.25 μM); (D) Compound 1 (6.25 μM); (E) Compound 2 (6.25 μM). For images in (B–E), and (G), the cells were treated at the MIC for each compound for 24 hours prior to imaging. The membrane stain FM4-64 is shown in red. The scale bar represents 25 μm in all images except (A) where it represents 2 μm. (F) Quantitative analysis of E. coli filamentous growth and morphology phenotypes. To quantify the population distribution, multiple fields of view were analyzed for a total of 200–800 cells. | ||
The cytological profile only provides supporting evidence of a MoA, as it reports solely on biological processes in bacteria. Compounds with MoAs that are specific to eukaryotic cells do not produce cytological profiles, as is the case with microtubule disrupting agents and members of the camptothecin family, which are Type I topoisomerase inhibitors that are ineffective in bacteria. The profile of the Ru(II) complex observed in E. coli does not eliminate the possibility of such mechanisms of action in mammalian cells, but it indicates that 2 has other MoA that are effective in bacteria that are also observed with phenanthriplatin and Topo II inhibitors.
DNA damage analysis
In addition to nucleolar stress, phenanthriplatin induces DNA damage by formation of covalent adducts that distort the structure of the nucleic acid.50 As the cytological profiling, colony formation assay, and DNA metalation studies demonstrated the capacity for 1 and 2 to damage DNA in bacteria, this motivated further mechanistic investigations. Docking studies were conducted to determine if 2 was able to bind to DNA in a similar manner to phenanthriplatin. An overlay of phenanthriplatin and 2 showed that both compounds superimpose with one another, with the ligands susceptible to substitution by dG sitting in similar positions (Fig. S17, ESI†). Docking of phenanthriplatin with the Dickerson dodecamer was previously reported,8 and served as the control for this study. An induced-fit model in Schrödinger's Maestro was chosen to allow for greater degrees of freedom in the system.51,52 Overlays of phenanthriplatin and 2 docked into the DNA duplex showed similar interactions with DNA (Fig. 3A). Both the phenanthridine and ph-tpy ligands occupy the major groove, generating distortions along the phosphodiesterase backbone. Moreover, the labile ligand in both molecules (chloride in phenanthriplatin, and water in 2) is situated in close proximity to dG. | ||
| Fig. 3 Ligand exchange and DNA interactions for phenanthriplatin and 2. (A) Docking of phenanthriplatin and 2 into the Dickerson dodecamer. Compound 2 (green, modeled as the aqua species) and phenanthriplatin (purple) were overlayed, with the labile ligand for each positioned at the top. In this orientation, the extended tpy ligand projects over the phenanthridine ligand. Chromatograms of 2 in (B) H2O only and (C) in H2O with excess deoxyguanosine (5 mM). Parental and XPA knockout HeLa cells were treated with (D) cisplatin and (E) compound 2 for 72 h to assess if DNA damage induced cell death. (F) The in vitro complex of enzyme assay (ICE) to identify cleavage complexed DNA (ccDNA) for topoisomerase IIα. The ICEΔ value is the ratio between no drug and positive control; an ICEΔ value of 2–6 indicates a valid result. The ICEΔ value for 5 μg DNA was 3.2. In (A) The condition 2* indicates the complex was dissolved in acetonitrile without water to avoid formation of the aqua complex. | ||
Compounds 1 and 2 were also evaluated as DNA damaging agents via gel electrophoresis. The DNA damage EC50 for phenanthriplatin, 1, and 2 was 8.8 μM, 21.9 μM, and 12.2 μM, respectively (Table S4, Fig. S18 and S19, ESI†). DNA electrophoresis can indicate general DNA damaging capacity in an isolated system, but the shifts in DNA mobility are heavily dependent on distorting adducts being generated, and these studies are limited in their ability to differentiate between non-covalent and covalent adducts. To reconcile the results of the gel electrophoresis experiments with DNA, the reactivity of 1 and 2 for ligand exchange was examined to determine if a covalent or non-covalent MoA was more plausible. When 1 and 2 were dissolved in phosphate buffer, the labile chloride ligands could be exchanged with water. Compound 1 was converted to the aqua, while 2 exhibited some conversion, but some also precipitated out of solution (Fig. S20, ESI†). Both 1 and 2 did not precipitate in the presence of BSA or Opti-MEM, and conversion to the aqua was significantly slowed. The rates of ligand exchange reactions with DNA also differed. Compound 1 had a half-life (t1/2) of 2.5 hours, while 2 exhibited biphasic kinetics, with an average t1/2 of 38.6 hours in the presence of calf thymus (CT) DNA and BSA (Fig. S21, ESI;† compounds 1 and 2 were dosed at 100 μM, and a concentration of 0.66 μg μL−1 for each CT-DNA and BSA was used).
High performance liquid chromatography (HPLC) analysis was conducted to determine if 2 could covalently modify a DNA base over 16 h. First, 2 was evaluated in water alone. A new peak in the chromatogram appeared at 20.8 minutes, and increased in area over time, while the parent peak at 23.3 minutes decreased, indicating the conversion to the aqua complex (Fig. 3A). In the presence of deoxyguanosine (dG), the aqua peak increased initially, and then decreased as a new peak at 20.6 minutes emerged (Fig. 3C). These results indicate that 2 covalently modifies dG within 10 h, either directly or through an aqua intermediate. When phenanthriplatin was incubated at room temperature in water, the complex was stable, with only a small percentage exhibiting exchange of the chloride ligand (Fig. S22A, ESI†). This could be observed from the slight decrease in the peak at 9.5 minutes and a new peak observed at 8 minutes. However, in the presence of 5 mM dG, phenanthriplatin underwent full conversion to the dG-adduct in 16 h (Fig. S22B, ESI†). These data suggest phenanthriplatin does not require an aqua intermediate to react with dG, which is consistent with previous reports.50 When comparing the reaction kinetics of 2 and phenanthriplatin, the dG-conjugates were formed at similar times, but through different processes, as 2 converts to the aqua complex first, and full reaction with dG subsequently takes an additional 8 hours.
Although compound 2 can covalently modify DNA, it was unknown if this was a cause of cell death. Nucleotide excision repair (NER) is the DNA damage response mechanism utilized to repair distorting adducts, such as those seen with cisplatin. Xeroderma Pigmentosum Group A (XPA) is an initiator protein involved in global genome and transcription-coupled nucleotide excision repair mechanisms. When XPA is knocked out, distorting adducts are not repaired, resulting in cellular sensitization to DNA damaging agents.53,54 Thus, XPA knock out HeLa cells were treated with phenanthriplatin, cisplatin, 1, and 2. The XPA knockout was sensitized 4-fold versus the parental cell line to cisplatin (Fig. 3D). A 2-fold sensitization occurred for 1 (Fig. S24, ESI†), and no sensitization was observed for phenanthriplatin (Fig. S24, ESI†) or 2 (Fig. 3E), indicating that DNA adducts resolved by NER were not the cause of cell death for either compound. This result was consistent with previous reports for phenanthriplatin.9
Another commonly utilized reporter for DNA damage is phosphorylation of serine 139 on Histone 2A; phospho-H2AX (p-γH2AX) initiates DNA damage repair (DDR). Immunoblotting for p-γH2AX thus is a common approach to probe small molecules that covalently modify DNA and initiate DDR. To further investigate DNA damage, the lysates from A549 cells treated with cisplatin (5 μM), phenanthriplatin (0.5 μM), or 2 (0.5 μM) were probed for p-γH2AX at 4 and 24 h (Fig S25, ESI†). The 24 h treatment with phenanthriplatin yielded a significant p-γH2AX response. Compound 2-treated samples, however, did not exhibit p-γH2AX, indicating that another mechanism of action is involved.
Topoisomerase inhibition
Topoisomerases play an essential role in cell replication by relaxing DNA supercoils at the replication fork,55 and are upregulated in proliferating cells.56–58 Several FDA approved drugs act by either direct inhibition of topoisomerases or as topoisomerase poisons.56,59 DNA gyrase, a bacterial Type IIA topoisomerase, was utilized to compare the topoisomerase inhibiting activity of phenanthriplatin, 1, and 2. In this assay, plasmid DNA that is a mixture of relaxed topoisomers is supercoiled by DNA gyrase; inhibition, either by binding to the enzyme, the enzyme DNA cleavage-complex, or DNA itself, is observed by a reduction in the fraction of the supercoiled form. Ciprofloxacin was used as a positive control, and the supercoiled form was lost at a concentration of 0.75 μM. Notably, distinct bands for topoisomers were observed (Fig. S26D, ESI†). In contrast, treatment of DNA with phenanthriplatin caused a decrease in DNA electrophoretic mobility, with significant smearing of the bands (Fig. S26A, ESI†). Phenanthriplatin inhibits gyrase, but also induces covalent modification of the DNA, as is observed in Fig. S26A (ESI†). Compounds 1 and 2 inhibited supercoiling of the plasmid at concentrations 3-fold lower than phenanthriplatin (Table S4 and Fig S26B, C, and S27, ESI†). Moreover, distinct topoisomers were observed, as with ciprofloxacin (Fig. S26D, ESI†), indicating that both 1 and 2 act as topoisomerase inhibitors. Notably, while the Ru(II) complexes are able to directly damage DNA (Table S4, ESI†), this occurs more slowly than in the 1 h time frame of the experiment.In order to determine the topoisomerase inhibiting capacity of 2in vitro, the in vitro complex of enzyme (ICE) assay was conducted. If topoisomerase is unable to re-ligate the DNA, it stalls on the DNA as a cleavage complex (ccDNA). In the ICE assay, cells are treated acutely with high concentrations of the compound of interest, lysed, the DNA is extracted, and stalled topoisomerase is detected by immunoblotting. In contrast to phenanthriplatin, compound 2 did not generate ccDNA at 50 μM or 100 μM, indicating that 2 is not inhibiting topoisomerase function via cleavage complex generation.
To reconcile the results for the two topoisomerase assays, the capacity for 2 to directly inhibit topoisomerase IIα (TopIIα) was evaluated using a similar computational approach as described above, with Mitoxantrone docked into TopIIα as a control.60 Phenanthriplatin and 2 were both predicted to insert into the DNA-binding channel of TopIIα. A single binding mode was observed for both metal complexes, despite the entirety of the channel being included in the active site definition (Fig. S28 and S29, ESI†). However, compound 2 had fewer interactions with the binding channel than phenanthriplatin, suggesting that 2 has a lower affinity for the enzyme or is unable to compete with biological substrates. This data, in combination with the experimental results for phenanthriplatin and 2 indicates that the Ru(II) complex is unlikely to bind to and inhibit TopoIIα. It also highlights the limitations of an isolated system, as shown by the differences in the ICE assay and gyrase data.
Influence on microtubule dynamics
Recently, [Ru(4,7-diphenyl-1,10-pehanthroline)3]2+ was evaluated as a microtubule stabilizer,43 preventing microtubule depolymerization and inducing apoptosis. This, in addition to the relatively high correlation between 2, paclitaxel, and vinblastine, which influence microtubule dynamics, motivated immunohistochemistry studies in A549 cells following treatment with these compounds. Compound 2 did not impact microtubule dynamics (Fig. 4A–D). Cell membrane morphology changed following treatment with 2, but this was attributed to a general cell stress response. The absence of α-tubulin from around the nucleus for paclitaxel as well as the increased abundance of free α-tubulin for vinblastine were consistent with previous reports.61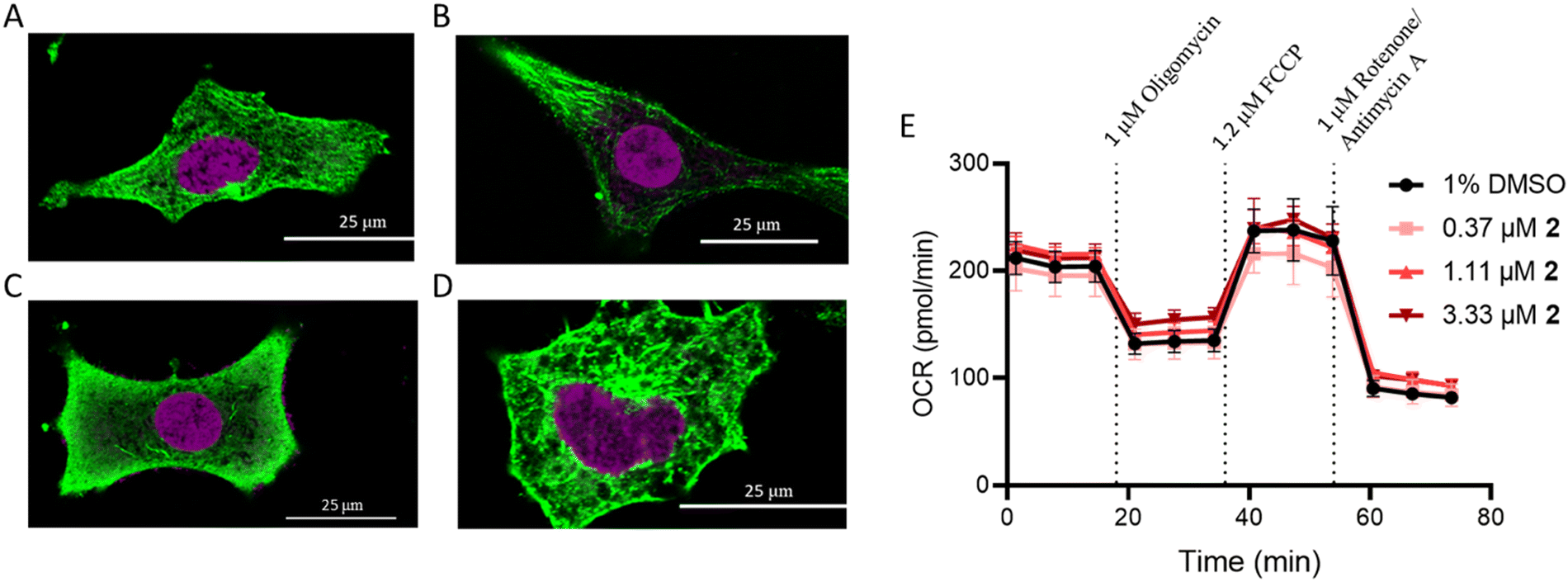 | ||
| Fig. 4 Excluded mechanisms of action. Tubulin polymerization was measured in (A) untreated A549 cells and cells treated with 1 μM compounds: (B) paclitaxel, (C) vinblastine, and (D) 2. The cells were incubated with each compound for 16 h. (E) Mitochondrial stress was evaluated following treatment with increasing concentrations 2 at 1 h. | ||
Mitochondrial dysfunction
Ru(II) molecules containing dip ligands have been shown to induce mitochondrial dysfunction,62 due to their lipophilicity and cationic charge.63 As compound 2 contains the dip ligand, its impact on mitochondrial function was assessed. The Seahorse MitoStress profile for cells pre-treated with 2 at 6× the IC50 value did not show any significant difference in response compared to the vehicle control (Fig. 4E). This result reveals that 2 does not interact directly with mitochondria or cause depolarization.Ribosome biogenesis stress
Unfortunately, oxaliplatin and phenanthriplatin are not part of the Standard Agents GI50 library, so it was not possible to use COMPARE to perform a direct correlation with the Ru(II) complexes. However, both oxaliplatin and phenanthriplatin have been shown to induce ribosome biogenesis stress, as indicated by several techniques,3 including work from DeRose demonstrating that nucleophosmin (NPM1) redistribution is a phenotypic marker of nucleolar stress.4,15,16 Nucleophosmin is a ribosomal protein chaperone that dissociates from the nucleolus to the nuclear matrix when ribosome biogenesis stress occurs.64,65 Cells treated with oxaliplatin and phenanthriplatin exhibited this response, whereas cisplatin-treated cells exhibited NPM redistribution only at later time points, after initiation of DNA damage response machinery.4,15,16Inspired by this, we used immunofluorescent microscopy to evaluate NPM localization. Concentrated NPM1 foci were observed in the localized phenotype of untreated cells (Fig. 5A and H), while the delocalized NPM1 phenotype was observed with treatment with actinomycin D (Fig. 5B), a positive control,42 consistent with prior reports.4,15,16 Analysis of the images revealed sharp and abundant spikes in fluorescent intensity across the diameter of the nucleus in the untreated population (Fig. 5J). By contrast, the translocated, diffuse NPM1 phenotype displayed overall reduced emission intensity and fewer spikes. Furthermore, the appearance of rounded nucleolar morphology, with NPM1 at the periphery in “cap-like” structures3,4 was reflected with a concave pattern for the pixel intensity across the nucleolus (Fig. 5I and J). Treatment with either phenanthriplatin or 2 induced the translocated NPM1 phenotype in 49% and 36% of the cell population respectively (Fig. 5D, F, and G). Moreover, compound 2 induced the translocated NPM1 distribution at the same concentration as phenanthriplatin (0.5 μM), very close to the IC50 values for cytotoxicity, supporting the implication that induction of ribosome biogenesis stress is correlated to cell death. Smaller changes in NPM1 distribution were observed in cells treated with cisplatin and 1 (Fig. 5C and E). The difference in phenotype between 1 and 2 suggest NPM1 translocation is not a general property of monofunctional Ru(II) compounds. The biological effect is likely a result of a specific combination of molecular features, including sterics and hydrophobicity, as is the case with monofunctional platinum compounds. Indeed, out of five monofunctional compounds, DeRose showed the NPM1 redistribution phenotype was unique to phenanthriplatin,4 highlighting the sensitivity of the cellular response to compound structure.
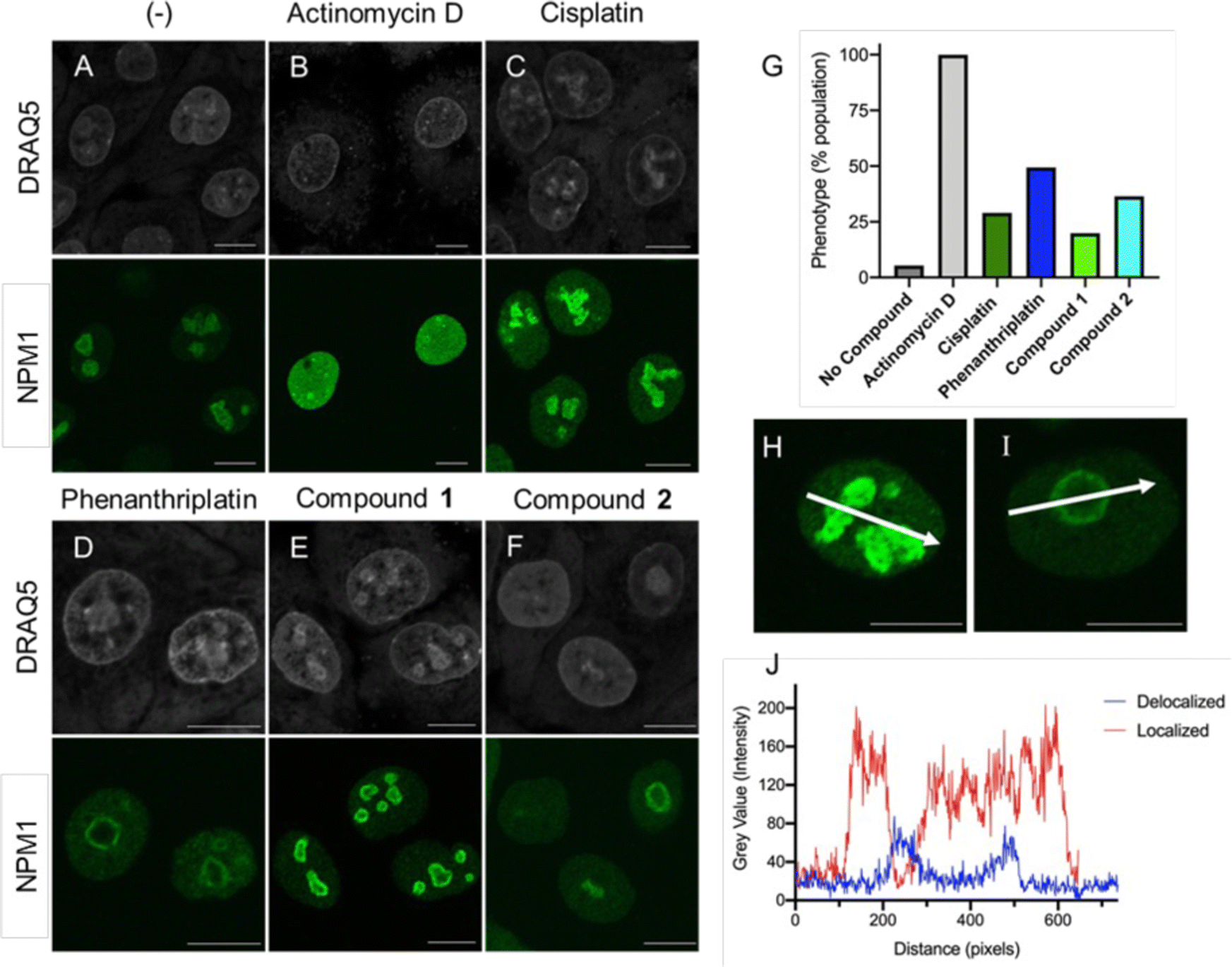 | ||
| Fig. 5 Redistribution of NPM1 indicates nucleolar stress. Immunostaining of NPM1 (green) in A549 cells at 24 h after treatment with (A) no compound, (B) 5 nM actinomycin D, (C) 5 μM cisplatin, (D) 0.5 μM phenanthriplatin, (E) 5 μM compound 1, and (F) 0.5 μM compound 2. Concentrations close to the compound IC50 were used to ensure a correlation with biological activity. DRAQ5 staining (gray) is nuclear DNA. All 488 nm channel images were captured using 3% laser power and 55% gain to facilitate comparison. (G) Quantification of the redistributed NPM1 phenotype as a percentage of cell population. (J) Representative intensity profiles taken from (H) localized and (I) delocalized phenotypes in individual cells. Scale bars represent 10 μm. | ||
Conclusions
The finding that oxaliplatin, a vital drug for the treatment of several cancers, functions primarily by induction of ribosome biogenesis stress,3,4 is strong motivation for the design and validation of other compounds that cause this biological effect. Cancer cells are known to be highly reliant on protein production, providing a potential therapeutic window for compounds that act via mechanisms that ultimately result in the inhibition of transcription and/or translation. Indeed, several established chemotherapeutics exploit this weakness in cancer cells, including cyclin dependent kinase inhibitors (CDKs), which are also acknowledged to act through transcription inhibition,66 and the transcription inhibitor and ribosome biogenesis stress inducer, actinomycin D, which is used to treat Wilms tumor and Ewing's sarcoma. However, actinomycin D suffers from poor selectivity,66 so the development of other molecules with similar mechanisms of action but radically different structures are needed to avoid such drawbacks.Using a semi-rational approach, we designed and characterized a potent Ru(II) compound that acts as a molecular mimic of oxaliplatin and phenanthriplatin. The ability of this Ru(II) complex to induce the hallmarks of ribosome biogenesis stress is noteworthy. To the best of our knowledge, there are only six inorganic compounds that have this effect, five of which are related to oxaliplatin, and phenanthriplatin. There are a few other Ru compounds that have been reported to interact with the nucleolus,19 but none have been reported to cause nucleolar stress. The incorporation of a phenyl-modified terpyridine and a dip ligand were key features in the design of the Ru(II) complex, and given the simplicity of this octahedral scaffold, there are many opportunities for additional synthetic modulation. Indeed, compound 2 opens the door to rational design of improved derivatives, and the use of phenotypic assessment streamlines planning of further biological evaluation. In sum, we believe that Ru(II) complex 2 provides a readily modifiable chemical scaffold for the development of improved ribosome biogenesis stress inducers.
What remains to be determined is if there is any particular significance to the ability of compound 2 to form monoadducts, or if the chemical structure, rather than the chemical reactivity at a single site, is the key to the biological effects observed. We also note the chemical or biological features of phenanthriplatin that prevented its advance into the clinic also unpublished, and are possibly unknown. This information is needed to determine whether Ru(II) analogues have a chance at a more successful ultimate outcome, and to allow for logically directed optimization of properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the National Institutes of Health (Grant GM107586) and the National Science Foundation (Grant MCB-1453168) for the support of this research. We also acknowledge the University of Kentucky Light Microscopy Core, which is supported in part by the Office of the Vice President for Research. We are profoundly thankful to Professor Richard Wood's lab for allowing us to use their HeLa XPA knockout cell line. This research was also supported by the Redox Metabolism Shared Resource Facility of the University of Kentucky Markey Cancer Center (P30CA177558).Notes and references
- V. Cepeda, M. A. Fuertes, J. Castilla, C. Alonso, C. Quevedo and J. M. Pérez, Anti-Cancer Agents Med. Chem., 2007, 7, 3–18 CrossRef CAS PubMed.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- P. M. Bruno, Y. Liu, G. Y. Park, J. Murai, C. E. Koch, T. J. Eisen, J. R. Pritchard, Y. Pommier, S. J. Lippard and M. T. Hemann, Nat. Med., 2017, 23, 461–471 CrossRef CAS PubMed.
- C. E. McDevitt, M. V. Yglesias, A. M. Mroz, E. C. Sutton, M. C. Yang, C. H. Hendon and V. J. DeRose, J. Biol. Inorg. Chem., 2019, 24, 899–908 CrossRef CAS PubMed.
- K. Yang, J. Yang and J. Yi, Cell Stress, 2018, 2, 125–140 CrossRef PubMed.
- L. S. Hollis, A. R. Amundsen and E. W. Stern, J. Med. Chem., 1989, 32, 128–136 CrossRef CAS PubMed.
- K. S. Lovejoy, M. Serova, I. Bieche, S. Emami, M. D'Incalci, M. Broggini, E. Erba, C. Gespach, E. Cvitkovic, S. Faivre, E. Raymond and S. J. Lippard, Mol. Cancer Ther., 2011, 10, 1709–1719 CrossRef CAS PubMed.
- A. A. Almaqwashi, W. Zhou, N. M. Naufer, I. A. Riddell, O. H. Yilmaz, S. J. Lippard and M. C. Williams, J. Am. Chem. Soc., 2019, 141, 1537–1545 CrossRef CAS PubMed.
- G. Y. Park, J. J. Wilson, Y. Song and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11987–11992 CrossRef CAS PubMed.
- I. A. Riddell, K. Agama, G. Y. Park, Y. Pommier and S. J. Lippard, ACS Chem. Biol., 2016, 11, 2996–3001 CrossRef CAS PubMed.
- H. Huang, P. Zhang, Y. Chen, L. Ji and H. Chao, Dalton Trans., 2015, 44, 15602–15610 RSC.
- P. Čanović, A. R. Simović, S. Radisavljević, I. Bratsos, N. Demitri, M. Mitrović, I. Zelen and Ž. D. Bugarčić, J. Biol. Inorg. Chem., 2017, 22, 1007–1028 CrossRef PubMed.
- E. S. Antonarakis and A. Emadi, Cancer Chemother. Pharmacol., 2010, 66, 1–9 CrossRef CAS PubMed.
- D. K. Heidary and E. C. Glazer, ChemBioChem, 2014, 15, 507–511 CrossRef CAS PubMed.
- E. C. Sutton, C. E. McDevitt, J. Y. Prochnau, M. V. Yglesias, A. M. Mroz, M. C. Yang, R. M. Cunningham, C. H. Hendon and V. J. DeRose, J. Am. Chem. Soc., 2019, 141, 18411–18415 CrossRef CAS PubMed.
- E. C. Sutton and V. J. DeRose, J. Biol. Chem., 2021, 296, 100633 CrossRef CAS PubMed.
- X. Li, A. K. Gorle, T. D. Ainsworth, K. Heimann, C. E. Woodward, J. Grant Collins and F. Richard Keene, Dalton Trans., 2015, 44, 3594–3603 RSC.
- E. Ortega, G. Vigueras, F. J. Ballester and J. Ruiz, Coord. Chem. Rev., 2021, 446, 214129 CrossRef CAS.
- B. Neuditschko, A. A. Legin, D. Baier, A. Schintlmeister, S. Reipert, M. Wagner, B. K. Keppler, W. Berger, S. M. Meier-Menches and C. Gerner, Angew. Chem., Int. Ed., 2021, 60, 5063–5068 CrossRef CAS PubMed.
- K. Xiong, C. Qian, Y. Yuan, L. Wei, X. Liao, L. He, T. W. Rees, Y. Chen, J. Wan, L. Ji and H. Chao, Angew. Chem., Int. Ed., 2020, 59, 16631–16637 CrossRef CAS PubMed.
- C. Qian, J. Wu, L. Ji and H. Chao, Dalton Trans., 2016, 45, 10546–10555 RSC.
- K. Du, J. Liang, Y. Wang, J. Kou, C. Qian, L. Ji and H. Chao, Dalton Trans., 2014, 43, 17303–17316 RSC.
- M. S. Legina, J. J. Nogueira, W. Kandioller, M. A. Jakupec, L. González and B. K. Keppler, J. Biol. Inorg. Chem., 2020, 25, 451–465 CrossRef CAS PubMed.
- C. A. Puckett, R. J. Ernst and J. K. Barton, Dalton Trans., 2010, 39, 1159–1170 RSC.
- J. Karges, O. Blacque, M. Jakubaszek, B. Goud, P. Goldner and G. Gasser, J. Inorg. Biochem., 2019, 198, 110752 CrossRef CAS PubMed.
- Z. Deng, P. Gao, L. Yu, B. Ma, Y. You, L. Chan, C. Mei and T. Chen, Biomaterials, 2017, 129, 111–126 CrossRef CAS PubMed.
- M. Adams, M. P. Sullivan, K. K. H. Tong, D. C. Goldstone, M. Hanif, S. M. F. Jamieson and C. G. Hartinger, Inorg. Chem., 2021, 2414–2424 CrossRef CAS PubMed.
- P. Shi, Q. Jiang, Y. Zhao, Y. Zhang, J. Lin, L. Lin, J. Ding and Z. Guo, J. Biol. Inorg. Chem., 2006, 11, 745–752 CrossRef CAS PubMed.
- V. Uma, M. Elango and B. U. Nair, Eur. J. Inorg. Chem., 2007, 3484–3490 CrossRef CAS.
- K. Malarz, D. Zych, R. Gawecki, M. Kuczak, R. Musioł and A. Mrozek-Wilczkiewicz, Eur. J. Med. Chem., 2020, 1–14 Search PubMed.
- N. Yoshikawa, S. Yamabe, N. Kanehisa, Y. Kai, H. Takashima and K. Tsukahara, Inorg. Chim. Acta, 2006, 359, 4585–4593 CrossRef CAS.
- N. Yoshikawa, T. Matsumura-Inoue, N. Kanehisa, Y. Kai, H. Takashima and K. Tsukahara, Anal. Sci., 2004, 20, 1639–1644 CrossRef CAS PubMed.
- A. E. Czapar, Y.-R. Zheng, I. A. Riddell, S. Shukla, S. G. Awuah, S. J. Lippard and N. F. Steinmetz, ACS Nano, 2016, 10, 4119–4126 CrossRef CAS PubMed.
- B. S. Howerton, D. K. Heidary and E. C. Glazer, J. Am. Chem. Soc., 2012, 134, 8324–8327 CrossRef CAS PubMed.
- Q. Chen, J.-A. Cuello-Garibo, L. Bretin, L. Zhang, V. Ramu, Y. Aydar, Y. Batsiun, S. Bronkhorst, Y. Husiev, N. Beztsinna, L. Chen, X.-Q. Zhou, C. Schmidt, I. Ott, M. J. Jager, A. M. Brouwer, B. E. Snaar-Jagalska and S. Bonnet, Chem. Sci., 2022, 13, 6899–6919 RSC.
- M. Monga and E. A. Sausville, Leukemia, 2002, 16, 520–526 CrossRef CAS PubMed.
- D. C. Ward, E. Reich and I. H. Goldberg, Science, 1965, 149, 1259–1263 CrossRef CAS PubMed.
- C. T. Amemiya and J. R. Gold, Copeia, 1986, 1986, 226–231 CrossRef.
- A. Németh and I. Grummt, Curr. Opin. Cell Biol., 2018, 52, 105–111 CrossRef PubMed.
- H. L. Cooper and R. Braverman, Nature, 1977, 269, 527–529 CrossRef CAS PubMed.
- H. M. Sobell, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 5328–5331 CrossRef CAS PubMed.
- B. Y.-M. Yung, A. M.-S. Bor and P.-K. Chan, Cancer Res., 1990, 50, 5987 CAS.
- N. Alatrash, F. H. Issa, N. S. Bawazir, S. J. West, K. E. Van Manen-Brush, C. P. Shelor, A. S. Dayoub, K. A. Myers, C. Janetopoulos, E. A. Lewis and F. M. MacDonnell, Chem. Sci., 2020, 11, 264–275 RSC.
- K. V. Kong, W. K. Leong and L. H. Lim, Chem. Res. Toxicol., 2009, 22, 1116–1122 Search PubMed.
- T. C. Johnstone, S. M. Alexander, W. Lin and S. J. Lippard, J. Am. Chem. Soc., 2014, 136, 116–118 CrossRef CAS PubMed.
- Y. Sun, D. K. Heidary, Z. Zhang, C. I. Richards and E. C. Glazer, Mol. Pharm., 2018, 15, 3404–3416 CrossRef CAS PubMed.
- P. Nonejuie, M. Burkart, K. Pogliano and J. Pogliano, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16169 CrossRef CAS PubMed.
- A. Wohlkonig, P. F. Chan, A. P. Fosberry, P. Homes, J. Huang, M. Kranz, V. R. Leydon, T. J. Miles, N. D. Pearson, R. L. Perera, A. J. Shillings, M. N. Gwynn and B. D. Bax, Nat. Struct. Mol. Biol., 2010, 17, 1152–1153 CrossRef CAS PubMed.
- F. Mohamadi Farsani, M. R. Ganjalikhany, M. Dehbashi, M. M. Naeini and S. Vallian, Med. Chem. Res., 2016, 25, 1250–1259 CrossRef CAS.
- G. Y. Park, J. J. Wilson, Y. Song and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11987–11992 CrossRef CAS PubMed.
- Schrödinger, Maestro 2020-2, Schrödinger, LLC, New York, NY, 2018 Search PubMed.
- This facilitated a more robust analysis of how phenanthriplatin and 2 not only interact with DNA, but how the interaction may change its secondary structure.
- B. Köberle, V. Roginskaya and R. D. Wood, DNA Repair, 2006, 5, 641–648 CrossRef PubMed.
- Z. Cierna, V. Miskovska, J. Roska, D. Jurkovicova, L. B. Pulzova, Z. Sestakova, L. Hurbanova, K. Machalekova, M. Chovanec, K. Rejlekova, D. Svetlovska, K. Kalavska, K. Kajo, P. Babal, J. Mardiak, T. A. Ward, M. Mego and M. Chovanec, BMC Cancer, 2020, 20, 17 CrossRef CAS PubMed.
- W. Goedecke, in xPharm: The Comprehensive Pharmacology Reference, ed. S. J. Enna and D. B. Bylund, Elsevier, New York, 2007, pp. 1–2 DOI:10.1016/B978-008055232-3.60609-9.
- J. L. Nitiss, Nat. Rev. Cancer, 2009, 9, 338–350 CrossRef CAS PubMed.
- B. J. Lynch, D. G. Guinee and J. A. Holden, Human Pathol., 1997, 28, 1180–1188 CrossRef CAS PubMed.
- T. Chen, Y. Sun, P. Ji, S. Kopetz and W. Zhang, Oncogene, 2015, 34, 4019–4031 CrossRef CAS PubMed.
- Y. Pommier, Nat. Rev. Cancer, 2006, 6, 789–802 CrossRef CAS PubMed.
- M. A. Saleh, M. Solayman, M. M. Hoque, M. A. K. Khan, M. G. Sarwar and M. A. Halim, BioMed Res. Int., 2016, 2016, 6817502 Search PubMed.
- C. Sum, D. Nickischer, M. Lei, A. Weston, L. Zhang and L. Schweizer, Curr. Chem. Genomics Transl. Med., 2014, 8, 16–26 CrossRef CAS PubMed.
- M. Dickerson, Y. Sun, B. Howerton and E. C. Glazer, Inorg. Chem., 2014, 53, 10370–10377 CrossRef CAS PubMed.
- A. Gandioso, A. Vidal, P. Burckel, G. Gasser and E. Alessio, ChemBioChem, 2022, 1–12 Search PubMed.
- J. K. Box, N. Paquet, M. N. Adams, D. Boucher, E. Bolderson, K. J. O’Byrne and D. J. Richard, BMC Mol. Biol., 2016, 17, 19 CrossRef PubMed.
- S. Grisendi, C. Mecucci, B. Falini and P. P. Pandolfi, Nat. Rev. Cancer, 2006, 6, 493–505 CrossRef CAS PubMed.
- O. Bensaude, Transcription, 2011, 2, 103–108 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental approaches, additional figures and tables. See DOI: https://doi.org/10.1039/d2cb00247g |
| This journal is © The Royal Society of Chemistry 2023 |