
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTemporal resolution of NAIL-MS of tRNA, rRNA and Poly-A RNA is overcome by actinomycin D†
Felix
Hagelskamp
a,
Kayla
Borland
a,
Gregor
Ammann
 b and
Stefanie M.
Kaiser
*ab
b and
Stefanie M.
Kaiser
*ab
aDepartment of Chemistry, Ludwig Maximilians University Munich, Munich, Germany
bInstitute of Pharmaceutical Chemistry, Goethe-University Frankfurt, Max-von-Laue-Str. 9, 60438, Frankfurt, Germany. E-mail: stefanie.kaiser@pharmchem.uni-frankfurt.de
First published on 24th February 2023
Abstract
RNA is dynamically modified and has the potential to respond to environmental changes and tune translation. The objective of this work is to uncover the temporal limitation of our recently developed cell culture NAIL-MS (nucleic acid isotope labelling coupled mass spectrometry) technology and overcome it. Actinomycin D (AcmD), an inhibitor of transcription, was used in the NAIL-MS context to reveal the origin of hybrid nucleoside signals composed of unlabelled nucleosides and labelled methylation marks. We find that the formation of these hybrid species depends exclusively on transcription for Poly-A RNA and rRNA but is partly transcription-independent for tRNA. This finding suggests that tRNA modifications adapt and are dynamically regulated by cells to overcome e.g. stress. Future studies on the tRNA modification mediated stress response are now accessible and the temporal resolution of NAIL-MS is improved by the use of AcmD.
Introduction
Ribonucleic acids (RNA), namely transfer RNA (tRNA), ribosomal RNA (rRNA) and messenger RNA (mRNA), are the core players in translation. All three molecules are post-transcriptionally modified by specific enzymes in a site-specific manner to fulfil and fine-tune the important process of translation. Humans possess over 400 different genes for cytosolic tRNAs, which can be collapsed into 47 tRNA isoacceptors carrying 21 proteinogenic amino acids.1 While we currently only have the complete modification profile of ∼20 of these tRNAs, up to 13 modifications can be expected per tRNA to stabilize the 3D L-shape structure and tune codon-anticodon binding.2–4 Small alterations in the tRNA modification structure and abundance, especially in the anticodon-loop (acl), have been connected to fine-tune translation and in the case of bacteria to overcome e.g. antibiotic stress.5–7 This hypothesis of stress-dependent tRNA modification reprogramming5,6 is supported by the permanent accessibility of the modification sites and writer enzymes by e.g. shuttling of tRNAs between cellular compartments.8,9 Thus, the cells can change the modification profile of the existing tRNAs quickly and independently of transcription. Similar adaptations are difficult to achieve for rRNA, where e.g. 2′-O-methylations are placed immediately after transcription in the nucleolus (from yeast10,11) and modification sites are hidden deep within the ribosome once they are mature. Regulation of rRNA modification abundance is possible by adapting newly transcribed rRNA modification profiles by e.g. tuning the abundance of small nucleolar RNAs (snoRNAs) responsible for rRNA modification.12 mRNA has the ability to get methylated at the N6 of adenine through RNA writers, which attracts or repels effector proteins (readers). Afterwards, m6A marks can be enzymatically removed through erasers without degrading the RNA.13,14With the goal to follow RNA modification processes and observe modification events in existing RNAs, we have developed NAIL-MS (nucleic acid isotope labelling coupled mass spectrometry).15 NAIL-MS studies rely on stable monoisotopic labelling of the nucleoside core structure. In human cell culture, nucleoside labelling is achieved by the addition of 15N5-adenine to the growth medium, which results in the formation of adenosine and guanosine with a mass increase of +5 and +4, respectively. Pyrimidines are labelled using 15N2–13C5-uridine (15 or similar isotopologues16) which results in a +7-mass increase. In addition, (12CD3)-L-methionine (CD3-methionine) can be added to a medium15 to study RNA methylation processes. A combination of nucleoside labelling and CD3-methionine labelling results in mass +8/+7 for monomethylated purines and +10 for monomethylated pyrimidines. Both tRNA and rRNA have a long half-life (3 and 7 days,17 respectively) and unlabelled signals from pre-existing RNAs are detectable by MS for at least 48 h. In our previous work, we observed a hybrid species, which contains the unlabelled nucleoside structure of a pre-existing RNA and a CD3-labelled methylation mark added after nutrient exchange. In our previous work,18 we have named this hybrid species “post-methylated” nucleosides. We hypothesized that these originate from not fully maturated pre-existing RNAs transcribed before the nutrient exchange (Fig. 1A) or pre-existing RNAs adapting after cellular stress.
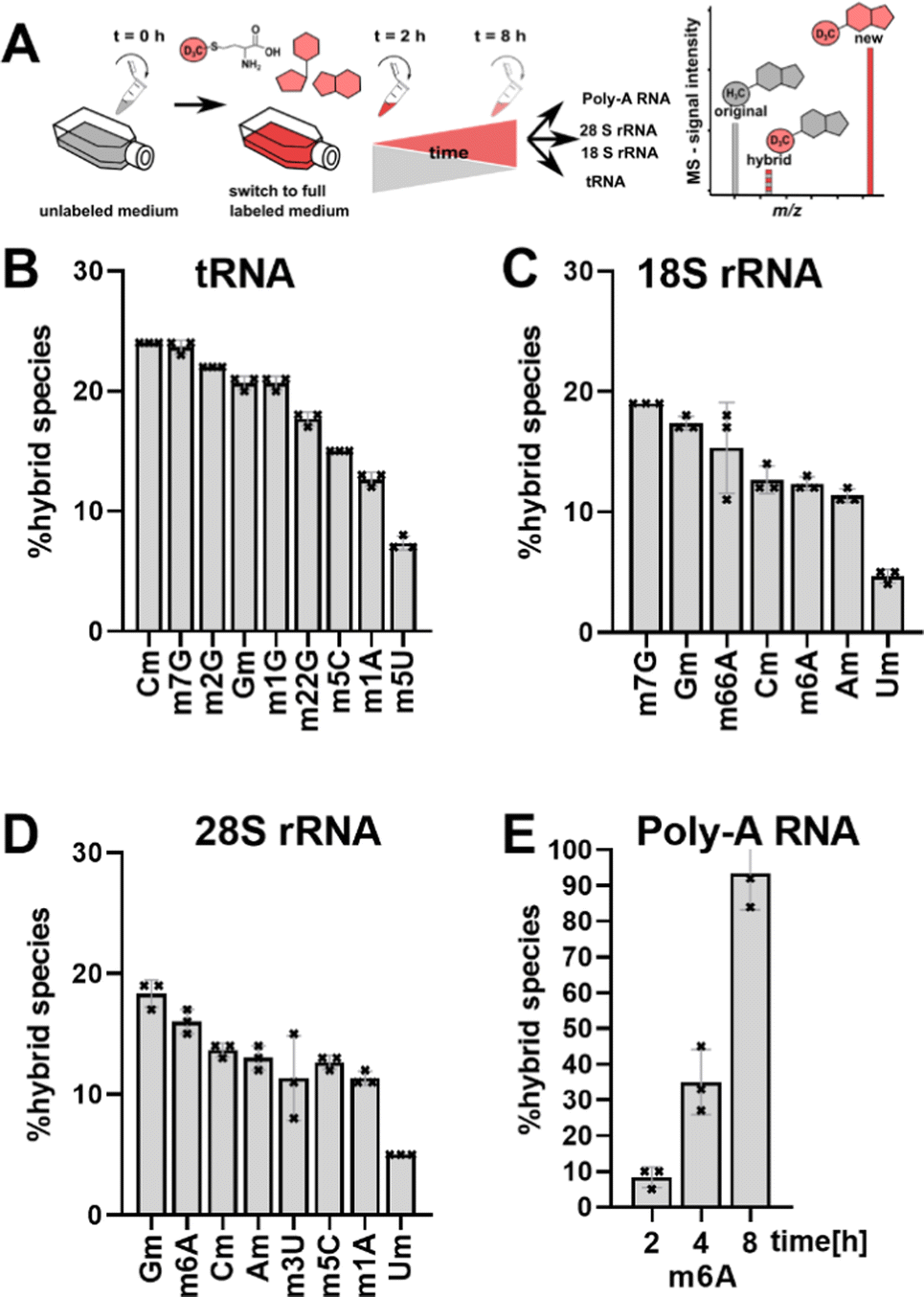 | ||
| Fig. 1 Abundance of hybrid species in various RNAs. HEK 293 cells were harvested after 6 hours (tRNA, and rRNA, data from Heiss et al.) and 2, 4 and 8 hours for Poly-A RNA. Error bars reflect the standard deviation of three biol. replicates. | ||
In this manuscript, we use Actinomycin D (AcmD) to suppress transcription and determine how transcription impacts hybrid species formation. We find that hybrid species in Poly-A RNA and rRNA depend on transcription and thus we refute our hypothesis of post-methylation in these RNAs. In tRNA, at least 50% of the hybrid species depend on transcription and are not caused by post-methylation. Interestingly, some modifications, e.g. Cm, Gm and m5C which can be found in tRNA acl, show substantially high abundances of hybrid species even under AcmD, which supports our previous hypothesis that NAIL-MS captures post-methylation and even adaptation to stress. To exclude a proteome bias in favour of tRNA writers due to the AcmD treatment, we performed shotgun proteomics but found no obvious differences in RNA writer abundances. This new study suggests that tRNA modifications can be adapted to the cell's need and are dynamically regulated to overcome e.g. stress. Future studies on the tRNA modification mediated stress response are now accessible and the temporal resolution of NAIL-MS is improved using AcmD.
Results
Methylation of original RNA occurs in all RNA species in the cell
We performed a cell culture NAIL-MS experiment, with the goal to define the extent of hybrid species formation in tRNA, 18S and 28S rRNA (data from ref. 15) and in addition Poly-A RNA. In a forward pulse-chase set-up, cells were first grown in an unlabelled medium and exchanged for a fully labelled medium (15N5-adenine, 15N2-13C5-uridine and (12CD3)-L-methionine) upon experiment initiation as shown in Fig. 1A. Modifications in new transcripts are labelled both at the nucleoside core and the methyl groups resulting in +7/+8/+10 mass signals for guanosine, adenosine and purines, respectively, and are not further analysed at this stage. Pre-existing RNAs with pre-existing methylation marks appear as unlabelled (ul in eqn 1), while methylations added after medium exchange appear as +3 mass signals (CD3–m6A in eqn 1). The relative abundance of such +3 hybrid species is calculated as given for m6A in eqn (1):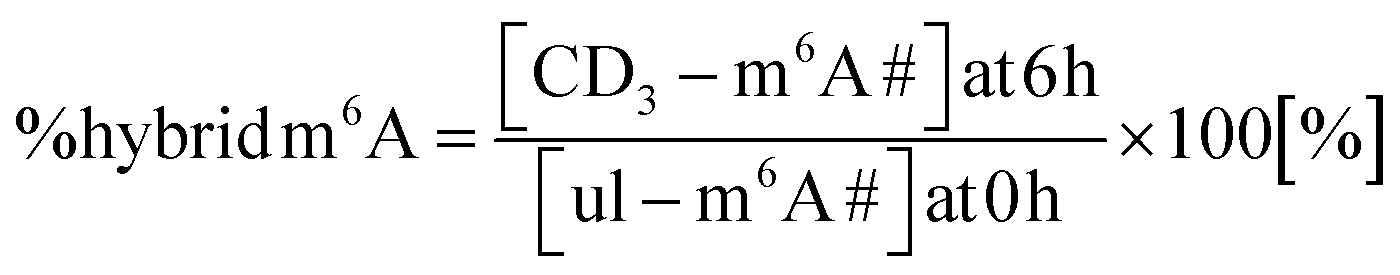 | (1) |
Fig. 1B–E shows the resulting plot of % hybrid species, which we formerly interpreted as post-methylation of pre-mature RNAs. For tRNA (Fig. 1B), Cm is the most abundant hybrid species with ∼ 24% while m5U is the least abundant with ∼7%. For 18S and 28S rRNA, we observed hybrid species for all modifications including all ribose methylated modifications (Fig. 1C and D). From our previous NAIL-MS studies, we know that new 18S rRNA is fully e.g. Cm or m6A methylated 8 hours after medium exchange (Fig. S1, ESI†). Thus, our hypothesis that maturation of pre-existing rRNAs is the cause of hybrid species in rRNA is unlikely. Our doubts were further supported by the abundance of 8%, 35% and 93% hybrid species m6A in Poly-A RNA after, 2 h, 4 h and 8 h, respectively (Fig. 1E). The purity of the analysed Poly-A RNA is documented in Fig. S2 (ESI†). Poly-A RNA is a fully mature mRNA mainly found in the cytosol, which should not be the target of the m6A writer and eraser enzymes, which are located in the nucleus.19 From this data, we conclude that post-methylation cannot be the only explanation of hybrid species formation as the post-methylation hypothesis cannot account for 93% of hybrid species within 8 hours. Another hypothesis for hybrid species formation is the emergence of hybrid RNAs composed of both labelled and unlabelled nucleotides at the early time points of NAIL-MS experiments. To test this hypothesis, transcription can be blocked which should lead to a disappearance of the hybrid species.
Impact of actinomycin D (AcmD) on transcription
To assess the impact of transcription on hybrid modification abundances, we used various concentrations of AcmD in HEK 293 cells in a forward NAIL-MS experiment. We find that addition of 1.0 μg mL−1 AcmD to the cell culture medium is sufficient to reduce transcription of all RNAs to below 2% after 8 hours (Fig. S3, ESI†). It is noteworthy that transcription is even further reduced at 5 μg mL−1 AcmD, but cells increasingly died at these high concentrations (Fig. S4, ESI†). We have decided to use 1 μg mL−1 AcmD in the subsequent experiments, and the results for the four RNAs of interest are summarized in Fig. 2. As expected for the untreated controls, Poly-A RNA is transcribed very fast which is in accordance with its short half-life of 9 hours.20 tRNA and rRNA with their 3-to-7-day long half-lives are transcribed less fast. In accordance with the literature, rRNA transcription by RNA-Polymerase I (Pol-I) is inhibited by AcmD the most, followed by inhibition of Poly-A RNA transcription by Pol II and tRNA transcription by Pol III.21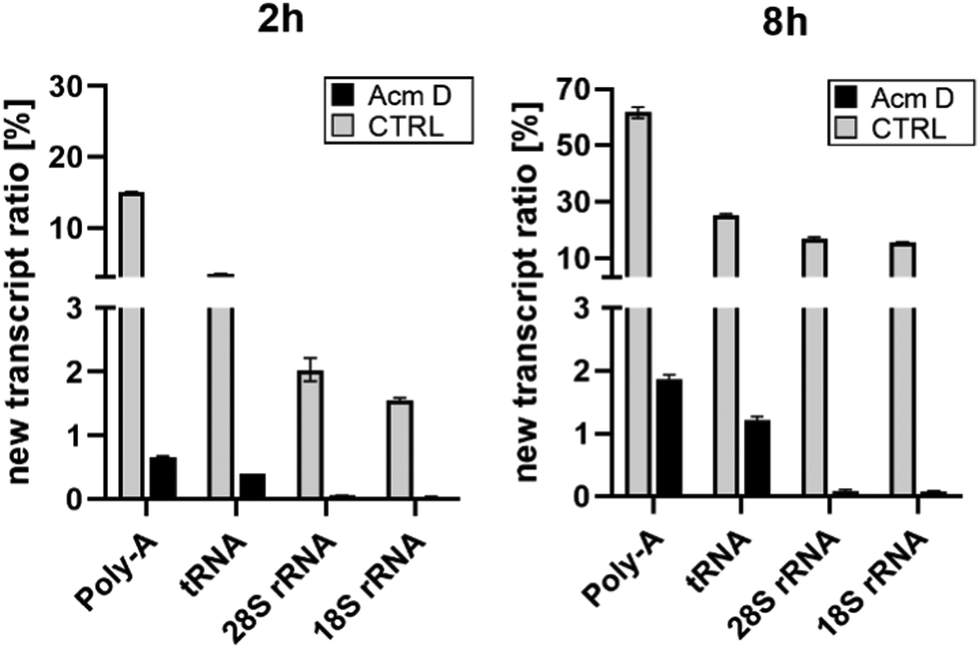 | ||
| Fig. 2 New transcript ratio (TR) of various RNAs after 2 and 8 hours of 1 μg mL−1 AcmD incubation determined by NAIL-MS. Error bars reflect the standard deviation of two biol. replicates. | ||
Hybrid species in rRNA are transcription dependent
We determined the abundance of hybrid species in control and AcmD treated cells and compared these to the starting abundance of the respective modification (see Fig. 1 and eqn (1)). The stacked bar graphs in Fig. 3A and B display the hybrid species detectable in 18S and 28S rRNA after AcmD treatment. For both RNAs, little or no hybrid species are found in the AcmD conditions. Only for m6A and m7G in 18S rRNA, a low abundance of hybrid species is found. However, the abundance of these hybrid species is only 1–2% after 2 hours and remains on the same level even after 8 hours. From our data, we assume that the hybrid species of m6A and m7G reflect post-methylation in the AcmD-treated samples and that both modifications are added later than the ribose methylations as here no hybrid species are detectable. For m7G and m6A, hybrid species formation, potentially post-methylation, is quick and finishes 2 hours after medium exchange. All other 18S and 28S rRNA modifications show no occurrence of hybrid species once transcription is blocked which argues (a) for their co-transcriptional placement and (b) for a stable, non-adaptive rRNA modification profile in human cell culture. From the NAIL-MS perspective, the absence of hybrid species in rRNA and the early high abundance of these modifications in new transcripts (Fig. S1, ESI†), we are confident that rRNAs are composed of unlabelled and labelled nucleotides at early time points of NAIL-MS experiments. The methylation of the unlabelled nucleotides in the hybrid rRNAs results in the occurrence of the hybrid species which we formerly interpreted as post-methylation. In light of our new findings, hybrid species in rRNA should be interpreted as new methylations placed in new, but hybrid RNAs at early time points of NAIL-MS experiments.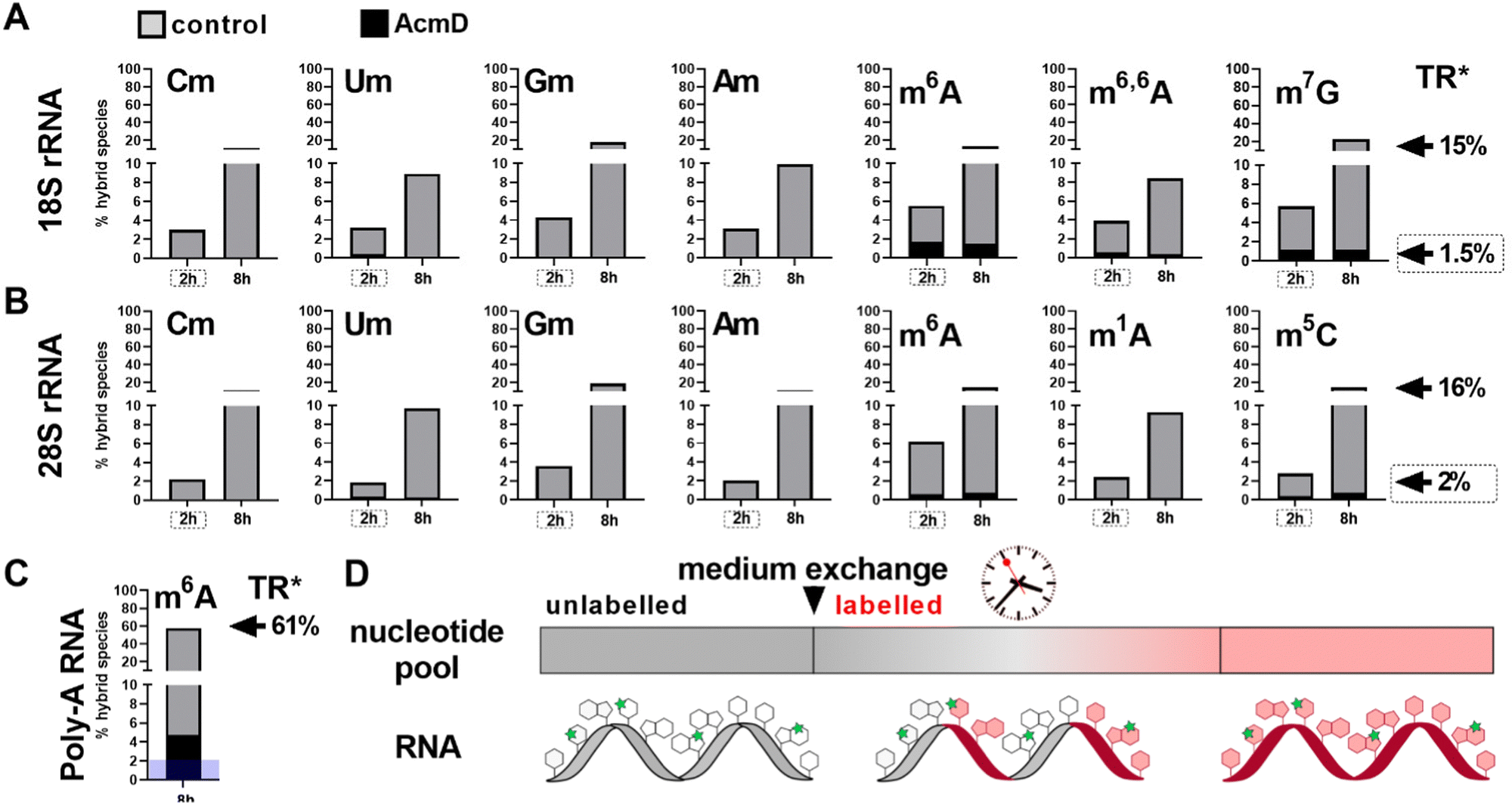 | ||
| Fig. 3 Occurrence of hybrid species in rRNA and Poly-A RNA with (black) and without (grey) AcmD treatment. (A and B) Hybrid species in 18S and 28S rRNA. *The new transcript ratio (TR) of control cells is indicated by arrows on the right for the 2 h (dashed box) and 8 h time points. TR of rRNA under AcmD is <0.1% at 8 h and not indicated. Average of 2 biol. replicates. (C) Hybrid species in Poly-A RNA after 8 h. TR*: new transcript ratio in control, TR in AcmD is highlighted in blue. Average of 2 biol. replicates. (D) Hybrid modification signals hail from hybrid RNAs composed of labelled (red) and unlabelled (grey) nucleotides which can be methylated (green stars) at the early time points of NAIL-MS experiments. | ||
The origin of hybrid species in Poly-A RNA is mainly but not limited to transcription
Our analysis further expanded to Poly-A RNA hybrid species. We found low but clear formation of hybrid-m6A even in the presence of AcmD. The abundance of hybrid species m6A is ∼ 4.5% after 8 hours in the AcmD-treated Poly-A RNAs compared to 93% in the control sample (Fig. 3C). These data confirm that most hybrid species in Poly-A RNA are formed by methylation of hybrid Poly-A RNAs which are composed of unlabelled and labelled nucleotides (Fig. 3D). While most hybrid species vanish upon blocking transcription, it is noteworthy that 4.5% hybrid species formation remains during AcmD treatment. For comparison, the remaining transcription under AcmD treatment is ∼ 2% and thus hybrid species abundance is roughly double the amount of remaining transcription. This argues towards some sort of transcription independent mechanism that leads to hybrid species formation, which might well be our previously hypothesized post-methylation process. Future studies inhibiting m6A writers, erasers and potentially the exon junction complex22 might solve this open question.Hybrid species in tRNA are connected to transcription, post-maturation and adaptation
In our next experiment, we calculated the %hybrid species for various modifications of tRNA in the NAIL-MS context in the presence of AcmD and compared the values to untreated cells. Fig. 4A shows in black bars the % of hybrid species detectable in AcmD treated cells for all modifications. For all studied tRNA modifications, the abundance of hybrid species is reduced once transcription is blocked through AcmD. Yet, the abundance of hybrid species is still substantial even if transcription is blocked and exceeds the abundance of new transcripts. Thus, we conclude that hybrid species in tRNA is formed (a) through transcription of hybrid tRNAs at early time points and (b) by post-methylation processes. A careful analysis of the data allows a classification of tRNA modifications mainly forming through transcription or a combination of transcription and post-methylation. In AcmD-treated cells, hybrid species of m1G, m1A, m7G and m22G are only slightly more abundant than the abundance of new transcripts and in addition, their abundance stagnates after 2 hours. This indicates that transcription dominates m1G, m1A, m7G and m22G hybrid species formation and that the remaining post-methylation is likely post-maturation. Other tRNA modifications, namely Cm, Gm and m5C, show high abundances of hybrid species even with blocked transcription, and in addition, the abundance increases over time. We conclude that Cm, Gm and m5C hybrid species hail partly from transcription but also through post-methylation. The continuous increase of Cm, Gm and m5C cannot be explained only by post-maturation but potentially indicates a dynamic adaptation of tRNAs carrying these modifications.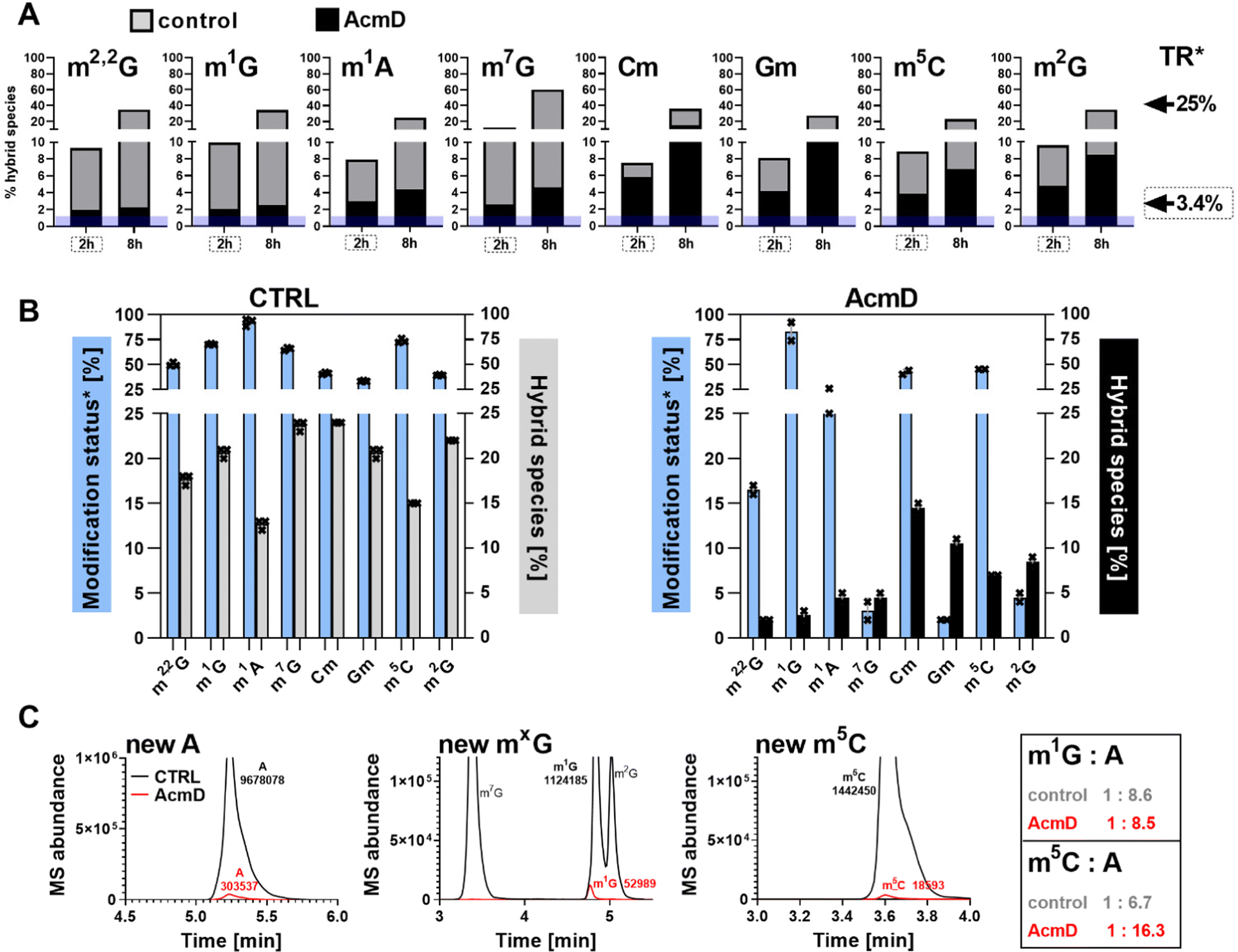 | ||
| Fig. 4 Occurrence of hybrid and new modifications in total tRNA after AcmD treatment. (A) Abundance of hybrid species compared to starting abundance at 2 and 8 hours in control (grey) and AcmD-treated (black) cells. *The new transcript ratio (TR) of control cells is indicated by arrows on the right for the 2 h (dashed box) and 8 h time points. The TR in AcmD is highlighted in blue. Average of n = 2 replicates. (B) Modification status (modification abundance of new tRNAs compared to steady state modification abundance) and hybrid species abundance in control and AcmD treated cells after 8 hours. (C) Extracted ion chromatograms of new A (m/z 273 → 141), new methylated G (mxG m/z 305 → 173) and new m5C (m/z 268 → 131) for control (black) and AcmD treated (red) cells after 8 h. Area under the peak is given in the graph. The ratio of Ctrl-m1G:Ctrl-A and AcmD-m1G:AcmD-A is given in the table to highlight the unchanged modification rate for m1G during AcmD treatment. | ||
With the goal to understand the biological mechanisms of post-methylation in tRNA, we studied the chromatograms for new modifications made in new tRNA transcripts (fully labelled nucleosides with mass +7/+8/+10). We reasoned that slow incorporation of new modifications into new transcripts indicates a slow maturation process and that it is possible to understand which modified nucleoside's hybrid species are caused by post-maturation and which through adaptation. For this purpose, we calculated the modification status in new transcripts (new) compared to the steady state modification abundance (ul) using eqn (2).
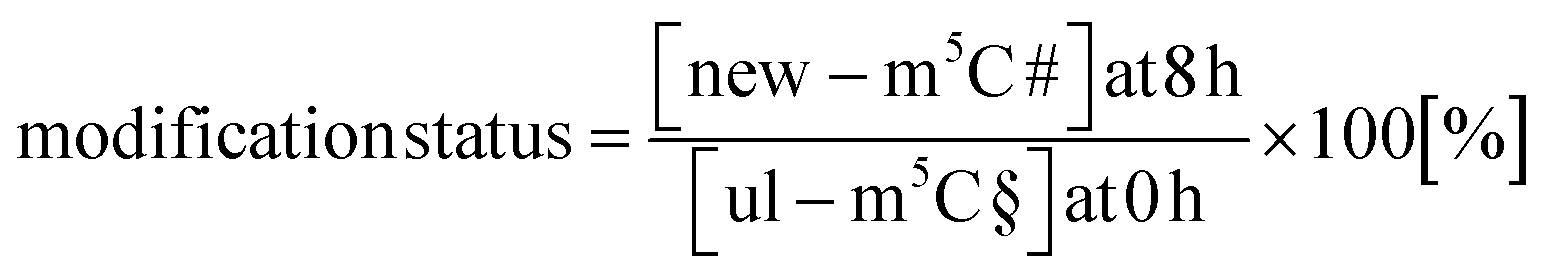 | (2) |
Fig. 4B summarizes the data of modification status next to hybrid species abundance in both control and AcmD-treated cells. The first thing we realized in control cells for e.g. m1A is that the modification status is extremely high in new transcripts after 8 hours and the hybrid species abundance is rather low. This argues for fast incorporation of m1A into tRNA and thus the low and stagnating abundance of post-methylation under AcmD treatment (Fig. 4B left) is explained by little need for post-maturation. Interestingly, m1A is only placed in ∼25% of all newly transcribed tRNAs in AcmD-cells but 90% of all control cell tRNAs. In fact, many modifications are less abundant in new transcripts in the AcmD-background and some such as Gm, m2G and m7G nearly vanish completely. Fig. 4C shows the respective chromatograms, where the complete disappearance of these modifications’ signals becomes even more apparent. The respective writer enzymes appear to be active though as Gm and m2G have a high abundance of hybrid species in AcmD-treated cells. For m5C, Cm and m1G, we noticed a high abundance of new transcripts in both control and AcmD-treated cells. Furthermore, m5C and Cm show a high abundance of hybrid species. For Cm and m5C, both the modification status and hybrid species abundance are high. This argues for the importance of m5C, Cm and m1G to overcome the environmental stress of reduced transcription by quickly modifying all available transcripts. Potentially, Gm and m2G are less critical for AcmD stress survival compared to m5C, Cm and m1G and are not placed in new transcripts. On a mechanistic level, the differences in modification status and hybrid species abundance must be connected to the availability and activity of the respective RNA writers. For E.g. a high abundance of NSUN2 (m5C of most tRNAs) under AcmD conditions would explain the high abundance of m5C and vice versa a low abundance of TARBP1 (Gm) might explain the low modification status for Gm.
AcmD does not impact the abundance of RNA writers
To get a detailed impression on the RNA writer abundance under AcmD conditions, we performed shotgun proteomics experiments. In the first analysis, we quantified the abundance of RNA writers in control and AcmD cells after 8 hours. As shown in Fig. 5, the changes in the global proteome are minimal and limited to enzymes not identified as RNA writers. tRNA writers such as NSUN2, TRMT1 and TRMT2A or rRNA writers such as NSUN5 or NOP56 were detectable in our analyses but no significant changes were observed. Please note that some RNA writers are low abundant and not detectable in our analysis. Although we cannot exclude the AcmD-induced loss of some RNA writers, we are confident that the loss of hybrid modifications in the studied RNAs is not related to a sudden loss of all RNA writers under AcmD. This strengthens our new hypothesis that RNAs with both labelled and unlabelled nucleotides form at the early stages of our NAIL-MS experiments and that hybrid species mainly hail from these early, new transcripts.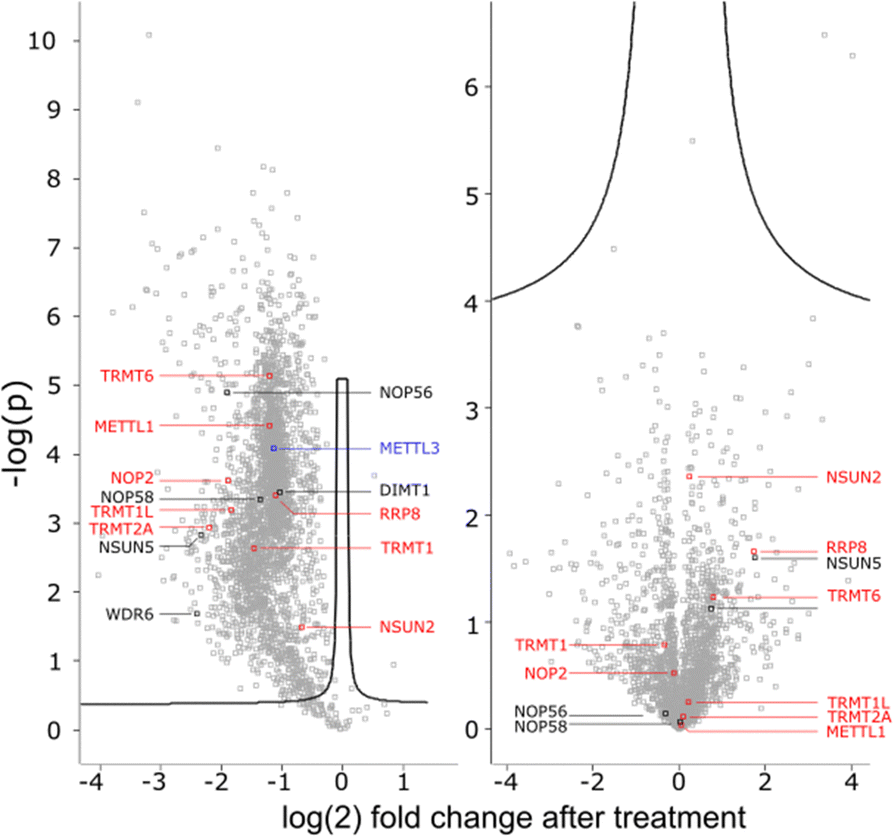 | ||
| Fig. 5 Abundance of tRNA (red) rRNA (black) and mRNA (blue) writers 8 h after AcmD treatment. Left: SILAC pulse-chase displaying new protein abundance compared to control. Right: Shotgun proteomics of remaining proteome. | ||
In addition, we performed a pulse-chase SILAC proteomics experiment to rule out the possibility that translation of certain RNA writers is stopped under AcmD treatment. Overall, the inhibition of mRNA transcription through AcmD leads to a general, significant decrease in new protein abundance. Many RNA writers are found in the average area of reduced abundance. Only NSUN2 is striking as the reduction in abundance is less prominent compared to e.g. TRMT1 or TRMT2A. The pulse chase SILAC data show that translation of some RNA writers is favoured over others. Interestingly, the still highly-translated RNA writers are those with the highest modification abundance in new transcripts under AcmD stress (e.g. NSUN2 – m5C).
Conclusions
RNA and its modifications are not static, but they are temporally orchestrated and regulated with the goal to fine-tune e.g. translation and cellular homeostasis. Cell culture NAIL-MS is a tool for the dynamic resolution of RNA modification processes and depends on metabolic labelling of the RNA. Consequently, at early time points, the nucleotides formed with isotope-labelled nutrients (provided by medium exchange) compete with the remaining unlabelled nucleotides. Using the transcription inhibitor AcmD, we have now proven that there is a period where the nucleotide pool is composed of both labelled and unlabelled nucleotides which results in hybrid RNA formation. From our data, we have learned that especially mRNA, here studied as Poly-A RNA, with its fast transcription, short half-life and thus turnover is most affected. Therefore, hybrid m6A modification is highly abundant in control cell Poly-A-RNA. Under stopped transcription, the abundance of hybrid m6A is drastically reduced. This indicates that AcmD is a valuable tool to overcome the temporal limitation of NAIL-MS in human cell culture and it allows truly studying the dynamics of m6A in mRNA under e.g. stress conditions. As a side note, the Poly-A RNA studied here is mainly composed of mature mRNA used for active translation. Here, the m6A dynamic processes of writing and erasing are mostly finished and thus non-Poly-A-depending separation techniques for true mRNA purification are needed, to catch and resolve the dynamics of early mRNA methylation. For rRNA, hybrid modifications are only observed under conditions of active transcription. For Poly-A RNA and rRNA, we are now confident that hybrid species formation depends strongly on transcription. Our new hypothesis is further supported by the abundance of hybrid species in these RNAs. Slow-turnover RNAs such as rRNA with a half-life of 7 days show a lower abundance of hybrid species and high-turnover RNAs such as Poly-A RNA show a high abundance of hybrid species (Fig. 2). The fast turn-over of Poly-A RNA23,24 leads to the intensive consumption of the nucleotide building block pool which consists at early time points of both labelled and unlabelled nucleotides and thus hybrid RNAs are formed (Fig. 3D). We thus conclude that AcmD is an important control to study m6A turnover in Poly-A RNA such as mRNA.Interestingly, AcmD is a substantial stress to the cells which might result in an adaptation of the translation apparatus towards higher efficiency. For rRNA, the modification density is not adapted in the original transcripts to counter the AcmD stress. Yet, the few remaining new transcripts show interesting modification profiles. In new 18S rRNA only Um, m6A and m7G are incorporated and in new 28S rRNA only Um, Gm, Am, m6A and m5C are incorporated under AcmD stress conditions (Fig. S1, ESI†). This observation indicates that rRNA reprofiling as a mechanism of stress response might exist, but only for new transcripts.
The Dedon lab has previously coined the term “tRNA modification reprogramming” as an active means to overcome cellular stress in yeast and bacteria.5–7 With our first human cell culture NAIL-MS report,15 we have laid the foundation for determining tRNA modification reprogramming in human cells. To this end, we had no opportunity to truly observe the process. With AcmD however, we are now confident that human cells are also capable to adapt their tRNA modification profiles under stress. This conclusion is supported by two observations: (i) Cm, Gm, m5C and m1G are partly located in the anticodon loop, where they have a potential impact on anticodon-codon binding and thus translational speed, efficiency, and fidelity. It is thus likely that these modifications are placed by the cell on a “steady-state” level but in addition on demand. For these modifications, we observe a high abundance of hybrid species even in the presence of AcmD which argues towards their importance in the AcmD stress response. (ii) Furthermore, these modifications are quickly placed into new tRNAs which form despite the presence of AcmD. This argues towards their importance to overcome the AcmD stress. The remaining high abundance of NSUN2 (m5C at tRNA positions 34 and 48–50) according to our pulse chase SILAC proteomics experiment supports the importance of m5C in tRNAs which might explain its role in cellular stress survival and cancer.25
Cell culture NAIL-MS is a valuable tool to study the dynamics of RNA modifications inside living cells and determine the underlying mechanisms. Yet, we have determined a temporal limitation because of the metabolic adaptation to the unlabelled-to-labelled nutrient exchange. In particular, mRNA with its fast turnover requires the use of transcription inhibition to truly follow the fate of original mRNAs and determine modification states in new mRNA. Interestingly, under transcription inhibition, hybrid RNA modifications have the potential to serve as a surrogate to watch mRNA modification processes at early time points where the signal of new mRNAs and their modifications is still too low. We thus conclude that NAIL-MS in human cells has a temporal limitation, which can be reduced by using the transcription inhibitor AcmD.
Materials and Methods
Chemicals and general materials
All salts, reagents and media were obtained from Sigma-Aldrich (Munich, Germany) at molecular biology grade unless stated otherwise. The isotopically labelled compounds 13C5,15N2-Uridine (Ribose-13C5, 98%; 15N2, 96–98%) were obtained from Silantes (Munich, Germany) and 15N5-Adenine (15N5, 98%) was obtained from Cambridge Isotope Laboratories (Tewksbury, MA, USA). All solutions and buffers were made with water from a Sartorious arium® pro ultrapure water system (Goettingen, Germany). 1-Methyladenosine (m1A), N3-methylcytidine (m3C), N6-methyladenosine (m6A), 7-methylguanosine (m7G), 5-methylcytidine (m5C), 5-methyluridine (m5U), 2′-Omethylcytidine (Cm), 2′-O-methylguanosine (Gm), 1-methylguanosine (m1G), N2-methylguanosine (m2G), 2-dimethylguanosine (m22G), 2′-O-methyluridine (Um), and 2′-O-methyladenosine (Am) were obtained from Carbosynth (Newbury, UK).Cell Culture NAIL-MS
All cell culture media and supplements were obtained from Sigma-Aldrich (Munich, Germany) unless stated otherwise. Standard Basal medium for HEK 293 culture was DMEM D6546 high glucose supplemented with 10% FBS and 0.584 g L−1L-glutamine. Cells were split 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 using standard procedures every 2–3 days to counter overgrowth. Cells cultured in the DMEM were kept at 10% CO2 for proper pH adjustment. For all experiments where labelling of nucleosides was involved DMEM D0422 without methionine and cystine were used. DMEM D0422 was supplemented with 10% dialyzed FBS (Biowest, Nuaillé, France), 0.584 g L−1 of L-glutamine, 0.063 g L−1 of cystine (stock concentration 78.75 g L−1 dissolved in 1 M HCl), 0.03 g L−1 of methionine, 0.05 g L−1 of uridine, and 0.015 of g L−1 adenine. Uridine, adenine and methionine were either added as unlabelled or labelled compounds depending on the desired labelling.
:7 using standard procedures every 2–3 days to counter overgrowth. Cells cultured in the DMEM were kept at 10% CO2 for proper pH adjustment. For all experiments where labelling of nucleosides was involved DMEM D0422 without methionine and cystine were used. DMEM D0422 was supplemented with 10% dialyzed FBS (Biowest, Nuaillé, France), 0.584 g L−1 of L-glutamine, 0.063 g L−1 of cystine (stock concentration 78.75 g L−1 dissolved in 1 M HCl), 0.03 g L−1 of methionine, 0.05 g L−1 of uridine, and 0.015 of g L−1 adenine. Uridine, adenine and methionine were either added as unlabelled or labelled compounds depending on the desired labelling.
0.8 million of HEK 293 cells were seeded in 2 mL of “unlabelled NAIL-MS” medium per well on a 6-well plate. Twenty-four hours after seeding (60% confluence), different concentrations of AcmD (part no. A1410-2MG, Sigma Aldrich, Munich, Germany) or DMSO were added to the cell vessels as a control at the same time as the medium was changed to “stable isotope-labelled NAIL-MS” medium. Incubation in the “stable isotope labelled NAIL-MS” medium lasted between 2 and 24 hours and was completed with removal of the medium, a PBS wash step, and inclusion of the cells in 500 μL TRI Reagent®.
tRNA and rRNA isolation
Cells were directly harvested on cell culture dishes using 1 mL of the TRI reagent for T25 flasks or 0.5 mL of TRI reagent for smaller dishes. The total RNA was isolated according to the supplier's manual with chloroform (Roth, Karlsruhe, Germany). tRNA, 28S and 18S rRNA were purified by size exclusion chromatography (AdvanceBio SEC 300 Å, 2.7 μm, 7.8 × 300 mm for tRNA and BioSEC 1000 Å, 2.7 μm, 7.8 × 300 mm for 18S and 28S rRNA, Agilent Technologies) according to published procedures.26 The RNA was suspended in water (35 μL).Poly-A RNA enrichment
Purification of Poly-A RNA was performed from total RNA using oligonucleotide (ON) hybridization. The “Dynabeads mRNA Purification” set (part no. 61006, ThermoFisher Scientific, Waltham, MA, USA) was used to isolate Poly-A RNA following the manufacturer's protocol. Briefly, 75 μg of total RNA dissolved in 100 μL of highly purified water was incubated for 2 min at 65 °C and 500 rpm to break the secondary structure. The sample was stored on ice until further processing. Pre-equilibrated Dynabeads™ (∼200 μL/75 μg total RNA) were used and Poly-A enriched RNA was eluted in 50 μL of water. To remove possible ribosomal RNA contaminants, the sample was purified using the RiboMinus™ Eukaryote Kit v2 (part no. A15020, ThermoFisher Scientific, Waltham, MA, USA) following the manufacturer's instructions. The rRNA depleted Poly-A RNA was concentrated to 100 μL using vacuum concentration and precipitated with 1 μL GlycoBlue™ (Thermo Fisher, Waltham, MA, USA), 10 μL of 5 M NH4OAc, and 250 μL of 100% ice-cold ethanol overnight at −20 °C. Poly-A RNA was dissolved in 20 μL pure water.Sample preparation for LC-MS/MS
Total tRNA (300 ng), 18S or 28S rRNA (500 ng each) or Poly-A RNA (100 ng) were digested in aqueous digestion mix (30 μL) to single nucleosides by using 2 U alkaline phosphatase, 0.2 U phosphodiesterase I (VWR, Radnor, Pennsylvania, USA), and 2 U benzonase in Tris (pH 8, 5 mM) and MgCl2 (1 mM) containing buffer. Furthermore, 0.5 μg tetrahydrouridine (Merck, Darmstadt, Germany), 1 μM butylated hydroxytoluene, and 0.1 μg pentostatin were added. After incubation for 2 h at 37 °C, 20 μL of LC-MS buffer A (QQQ) was added to the mixture and then filtered through 96-well filter plates (AcroPrep Advance 350 10 K Omega, PALL Corporation, New York, USA) at 3000 × g at 4 °C for 30 min. A stable isotope labelled SILIS (gen2,27) was added to each filtrate and calibration solution of synthetic standards before injection into the QQQ MS.LC-MS/MS of nucleosides
For quantitative mass spectrometry, an Agilent 1290 Infinity II equipped with a diode-array detector (DAD) combined with an Agilent Technologies G6470A Triple Quad system and electrospray ionization (ESI-MS, Agilent Jetstream) was used.Nucleosides were separated using a Synergi Fusion-RP column (Synergi® 2.5 μm Fusion-RP 100 Å, 150 × 2.0 mm, Phenomenex®, Torrance, CA, USA). LC buffer consisting of 5 mM NH4OAc pH 5.3 (buffer A) and pure acetonitrile (buffer B) were used as buffers. The gradient starts with 100% buffer A for 1 min, followed by an increase to 10% buffer B over a period of 4 min. Buffer B is then increased to 40% over 2 min and maintained for 1 min before switching back to 100% buffer A over a period of 0.5 min and re-equilibrating the column for 2.5 min. The total time is 11 min and the flow rate is 0.35 mL min−1 at a column temperature of 35 °C.
An ESI source was used for the ionization of the nucleosides (ESI-MS, Agilent Jetstream). The gas temperature (N2) was 230 °C with a flow rate of 6 L min−1. Sheath gas temperature was 400 °C with a flow rate of 12 L min−1. Capillary voltage was 2500 V, skimmer voltage was 15 V, nozzle voltage was 0 V, and nebulizer pressure was 40 Psi. The cell accelerator voltage was 5 V. All methods were performed in the DMRM and positive ion mode.
Data analysis of nucleosides
For calibration, synthetic nucleosides were weighed and dissolved in water to a stock concentration of 1–10 mM. Calibration solutions ranged from 0.0125 pmol to 100 pmol for each canonical nucleoside and from 0.00625 pmol to 5 pmol for each modified nucleoside. The concentrations of Ψ and D ranged from 0.00625 pmol to 20 pmol. Analogous to the samples, 1 μL of SILIS (10×) was co-injected with each calibration. The calibration curve and the corresponding evaluation of the samples were performed using the Excel spreadsheet program and via Agilent's qualitative MassHunter software. All modification abundances were normalized to the amount of RNA injected using the respectively labelled adenosine MS signal. Data were plotted using GraphPad Prism.SILAC
For SILAC experiments, cells were grown under the same conditions as for all other experiments using the SILAC Protein Quantification Kit following manufacturer's instructions (ThermoFisherScientific, Waltham, MA USA). Briefly, cells were grown in a medium (supplemented with 10% dialyzed FBS) containing regular amino acids for 24 h. Then, the medium was aspirated, cells were washed with 5 ml of PBS and a new stable-isotope-labelled medium was added (containing 13C6-15N2-lysine, 13C6-15N4-arginine, as well as 1 μg mL−1 AcmD or DMSO). After 8 h, the cells were harvested using cell culture trypsin, pelleted for 3 min at 2000 rcf and immediately lysed for protein extraction.Proteomics sample preparation
Approximately 6 × 106 cells were lysed in 300 μl of lysis buffer (8 M urea, 10 mM, 40 mM 2-chloroacetamine in 100 mM Tris/HCl pH 8.5), incubated for 5 min at 95 °C, and sonicated for 10 s twice. 30 μl were transferred into 30 kDa cut-off filters (Merck Millipore, Billerica, MA USA), washed using 8 M urea in Tris/HCl pH 8.5 and twice using 50 mM NH4HCO3 before adding 1 μg of trypsin per sample (ThermoFisherScientific, Waltham, MA USA) in 50 mM NH4HCO3. The samples were incubated over night at 37 °C before collecting the peptides in a fresh tube. The samples were acidified using 1 μl of formic acid and desalted using SepPak C18 cartridges (Waters, Milford, MA USA), eluted from the cartridges in 80% acetonitrile supplemented with 0.5% formic acid. Samples were vacuum concentrated and suspended in 200 μl 1% formic acid.LC-Orbitrap measurement
Proteomic mass spectrometry measurements were performed on an Orbitrap Eclipse Tribrid Mass Spectrometer coupled to an UltiMate 3000 Nano-HPLC via an EASY-Spray source and FAIMS interface (ThermoFisherScientific, Waltham, MA USA). 4 μl of the sample were loaded on an Acclaim PepMap 100 μ-precolumn cartridge (5 μm, 100 Å, 300 μm ID × 5 mm ThermoFisherScientific, Waltham, MA USA). Then, peptides were separated at 40 °C on a PicoTip emitter (noncoated, 15 cm, 75 μm ID, 8 μm tip, New Objective, Littleton, MA USA) that was packed in-house with Reprosil-Pur 120 C18-AQ material (1.9 μm, 150 Å, Dr A. Maisch HPLC GmbH, Ammerbuch-Entringen, Germany). The mobile phase consisted of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B). The gradient was as follows: 0–5 min at 4% B, 5–6 min linear increase to 7% B, 7–105 min to 24.8%, 105–126 min to 35.2% B, 126–140 min 80% B, 140–150 min 4% B at a constant flow rate of 300 nl min−1. FAIMS was operated with two alternating CVs (−50 and −70 V). The mass spectrometer was operated in the data dependent MS2 mode with the following parameters: Polarity: positive; MS1 resolution 240 k; MS1 AGC target: standard; MS1 max. inj. time: 50 ms; MS1 scan range: m/z 375–1500, MS 2 ion trap scan rate: rapid; MS 2 AGC target: standard; MS 2 maximum injection time: 35 ms; MS 2 cycle time: 1.7 s; MS 2 isolation window: m/z 1.2; HCD stepped normalized collision energy: 30%; intensity threshold: 1.0 × 104 counts; included charge states: 2–6; dynamic exclusion: 60 s.Proteomics data evaluation
Data obtained by orbitrap measurement was analyzed using MaxQuant version 1.6.5.0 and Perseus version 1.6.15.0 software. MS signals were annotated using an H. sapiens fasta-file (from NCBI) and quantified. Data points originating from known potential contaminants were excluded and missing values were replaced from a normal distribution using Perseus’ default settings (width 0.3, down shift 1.8). The significance line was calculated using Perseus’ default settings (t-test, 250 randomizations, FDR 0.05, S0 0.1). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE28 partner repository with the dataset identifier PXD039549”.Author contributions
Investigations were performed by F. H., K. B. and G. A. G. A. visualized proteomics data and performed statistical analyses. Conceptualization, funding acquisition, supervision and writing of the original draft were performed by S. K.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge advise from Dr. Pavel Kielkovski (LMU Munich) for proteomics support and the Department of Chemistry for infrastructure and financial support. We thank Dr. Klaus Römer for his generous support. This work was funded by the Deutsche Foschungsgemeinschaft (255344185-SPP 1784, 325871075-SFB 1309, KE1943/3-1) and the Horizon 2020 program (ID-952373).Notes and references
- P. P. Chan and T. M. Lowe, Nucleic Acids Res., 2016, 44, D184–189 CrossRef CAS PubMed.
- P. F. Agris, A. Narendran, K. Sarachan, V. Y. P. Vare and E. Eruysal, Enzymes, 2017, 41, 1–50 CAS.
- P. Boccaletto, M. A. Machnicka, E. Purta, P. Piatkowski, B. Baginski, T. K. Wirecki, V. de Crecy-Lagard, R. Ross, P. A. Limbach, A. Kotter, M. Helm and J. M. Bujnicki, Nucleic Acids Res., 2018, 46, D303–D307 CrossRef CAS PubMed.
- N. Manickam, K. Joshi, M. J. Bhatt and P. J. Farabaugh, Nucleic Acids Res., 2016, 44, 1871–1881 CrossRef PubMed.
- C. T. Chan, M. Dyavaiah, M. S. DeMott, K. Taghizadeh, P. C. Dedon and T. J. Begley, PLoS Genet., 2010, 6, e1001247 CrossRef CAS PubMed.
- C. T. Chan, Y. L. Pang, W. Deng, I. R. Babu, M. Dyavaiah, T. J. Begley and P. C. Dedon, Nat. Commun., 2012, 3, 937 CrossRef PubMed.
- Y. H. Chionh, M. McBee, I. R. Babu, F. Hia, W. Lin, W. Zhao, J. Cao, A. Dziergowska, A. Malkiewicz, T. J. Begley, S. Alonso and P. C. Dedon, Nat. Commun., 2016, 7, 13302 CrossRef CAS PubMed.
- E. M. Phizicky and A. K. Hopper, Genes Dev., 2010, 24, 1832–1860 CrossRef PubMed.
- A. K. Hopper and H. Y. Huang, Mol. Cell. Biol., 2015, 35, 2052–2058 CrossRef CAS PubMed.
- M. Kos and D. Tollervey, Mol. Cell, 2010, 37, 809–820 CrossRef CAS PubMed.
- U. Birkedal, M. Christensen-Dalsgaard, N. Krogh, R. Sabarinathan, J. Gorodkin and H. Nielsen, Angew. Chem., Int. Ed., 2015, 54, 451–455 CAS.
- M. F. Burke, D. M. McLaurin, M. K. Logan and M. D. Hebert, Biol Open, 2019, 8, bio041848 CrossRef CAS PubMed.
- S. Zaccara, R. J. Ries and S. R. Jaffrey, Nat. Rev. Mol. Cell Biol., 2019, 20, 608–624 CrossRef CAS PubMed.
- H. Shi, J. Wei and C. He, Mol. Cell, 2019, 74, 640–650 CrossRef CAS PubMed.
- M. Heiss, F. Hagelskamp, V. Marchand, Y. Motorin and S. Kellner, Nat. Commun., 2021, 12, 389 CrossRef CAS PubMed.
- M. Taoka, Y. Nobe, Y. Yamaki, K. Sato, H. Ishikawa, K. Izumikawa, Y. Yamauchi, K. Hirota, H. Nakayama, N. Takahashi and T. Isobe, Nucleic Acids Res., 2018, 46, 9289–9298 CrossRef CAS PubMed.
- B. G. Miller, J. Endocrinol., 1973, 59, 81–85 CAS.
- M. Heiss, V. F. Reichle and S. Kellner, RNA Biol., 2017, 14, 1260–1268 CrossRef PubMed.
- G. Zheng, J. A. Dahl, Y. Niu, P. Fedorcsak, C. M. Huang, C. J. Li, C. B. Vagbo, Y. Shi, W. L. Wang, S. H. Song, Z. Lu, R. P. Bosmans, Q. Dai, Y. J. Hao, X. Yang, W. M. Zhao, W. M. Tong, X. J. Wang, F. Bogdan, K. Furu, Y. Fu, G. Jia, X. Zhao, J. Liu, H. E. Krokan, A. Klungland, Y. G. Yang and C. He, Mol. Cell, 2013, 49, 18–29 CrossRef CAS PubMed.
- B. Schwanhausser, D. Busse, N. Li, G. Dittmar, J. Schuchhardt, J. Wolf, W. Chen and M. Selbach, Nature, 2011, 473, 337–342 CrossRef PubMed.
- R. P. Perry and D. E. Kelley, J. Cell. Physiol., 1970, 76, 127–139 CrossRef CAS PubMed.
- A. Uzonyi, D. Dierks, R. Nir, O. S. Kwon, U. Toth, I. Barbosa, C. Burel, A. Brandis, W. Rossmanith, H. Le Hir, B. Slobodin and S. Schwartz, Mol. Cell, 2023, 83(237–251), e237 CrossRef PubMed.
- Q. Wu, S. G. Medina, G. Kushawah, M. L. DeVore, L. A. Castellano, J. M. Hand, M. Wright and A. A. Bazzini, eLife, 2019, 8, e45396 CrossRef PubMed.
- V. Agarwal and D. R. Kelley, Genome Biol., 2022, 23, 245 CrossRef CAS PubMed.
- A. Chellamuthu and S. G. Gray, Cells, 2020, 9, 1758 CrossRef CAS PubMed.
- F. Hagelskamp, K. Borland, J. Ramos, A. G. Hendrick, D. Fu and S. Kellner, Nucleic Acids Res., 2020, 48, e41 CrossRef CAS PubMed.
- M. Heiss, K. Borland, Y. Yoluc and S. Kellner, Methods Mol. Biol., 2021, 2298, 279–306 CrossRef CAS PubMed.
- E. W. Deutsch, N. Bandeira, V. Sharma, Y. Perez-Riverol, J. J. Carver, D. J. Kundu, D. Garcia-Seisdedos, A. F. Jarnuczak, S. Hewapathirana, B. S. Pullman, J. Wertz, Z. Sun, S. Kawano, S. Okuda, Y. Watanabe, H. Hermjakob, B. MacLean, M. J. MacCoss, Y. Zhu, Y. Ishihama and J. A. Vizcaino, Nucleic Acids Res., 2020, 48, D1145–D1152 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cb00243d |
| This journal is © The Royal Society of Chemistry 2023 |