
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceArabidopsis thaliana NudiXes have RNA-decapping activity†
Maria-Bianca
Mititelu
 ab,
Oldřich
Hudeček
a,
Agnieszka
Gozdek
c,
Roberto
Benoni
a,
Ondřej
Nešuta
a,
Szymon
Krasnodębski
c,
Joanna
Kufel
c and
Hana
Cahová
*a
ab,
Oldřich
Hudeček
a,
Agnieszka
Gozdek
c,
Roberto
Benoni
a,
Ondřej
Nešuta
a,
Szymon
Krasnodębski
c,
Joanna
Kufel
c and
Hana
Cahová
*a
aInstitute of Organic Chemistry and Biochemistry of the CAS, Flemingovo náměstí 2, Prague 6, Czechia. E-mail: cahova@uochb.cas.cz
bCharles University, Faculty of Science, Department of Cell Biology, Viničná 7, Prague 2, Czechia
cInstitute of Genetics and Biotechnology, Faculty of Biology, University of Warsaw, Pawinskiego 5a, Warsaw 02-106, Poland
First published on 9th January 2023
Abstract
Recent discoveries of various noncanonical RNA caps, such as dinucleoside polyphosphates (NpnN), coenzyme A (CoA), and nicotinamide adenine dinucleotide (NAD) in all domains of life have led to a revision of views on RNA cap function and metabolism. Enzymes from the NudiX family capable of hydrolyzing a polyphosphate backbone attached to a nucleoside are the strongest candidates for degradation of noncanonically capped RNA. The model plant organism Arabidopsis thaliana encodes as many as 28 NudiX enzymes. For most of them, only in vitro substrates in the form of small molecules are known. In our study, we focused on four A. thaliana NudiX enzymes (AtNUDT6, AtNUDT7, AtNUDT19 and AtNUDT27), and we studied whether these enzymes can cleave RNA capped with NpnNs (Ap2–5A, Gp3–4G, Ap3–5G, m7Gp3G, m7Gp3A), CoA, ADP-ribose, or NAD(H). While AtNUDT19 preferred NADH-RNA over other types of capped RNA, AtNUDT6 and AtNUDT7 preferentially cleaved Ap4A-RNA. The most powerful decapping enzyme was AtNUDT27, which cleaved almost all types of capped RNA at a tenfold lower concentration than the other enzymes. We also compared cleavage efficiency of each enzyme on free small molecules with RNA capped with corresponding molecules. We found that AtNUDT6 prefers free Ap4A, while AtNUDT7 preferentially cleaved Ap4A-RNA. These findings show that NudiX enzymes may act as RNA-decapping enzymes in A. thaliana and that other noncanonical RNA caps such as Ap4A and NADH should be searched for in plant RNA.
Introduction
Currently, more than 170 RNA modifications are known.1 Among the least explored RNA modifications are 5′ RNA caps. A typical example of a noncanonical RNA cap is nicotinamide adenine dinucleotide (NAD), which has been detected in all domains of life.2–5 Other caps, such as NADH (a reduced form of NAD),6 coenzyme A (CoA),7 and flavin adenine dinucleotide (FAD),8 have been detected only in some organisms. Recently, we have discovered an entirely new class of RNA caps in bacteria with the structure of dinucleoside polyphosphates (NpnNs).9 The presence of free NpnNs molecules in cells of various organisms has been known more than 50 years.10–12 Their intracellular concentration increases under stress conditions,13 thus they are often called alarmones. For instance, Ap3A and Ap4A are known to act as alarmones in Arabidopsis thaliana (A. thaliana),13 although their mechanism of action is unknown. The discovery of these molecules as RNA caps opened new perspectives on their function and metabolism. Free molecules such as NpnNs, NAD, and CoA are usually cleaved by NudiX enzymes. Plants especially have a high number of NudiX-encoding genes, with a total of 28 in A. thaliana;14 by comparison, there are 24 in human cells and only 13 in Escherichia coli.15 Nevertheless, it is still unclear why so many NudiX enzymes are necessary for cellular metabolism and why many are redundant, i.e. two or more enzymes cleave the same substrates in vitro. Importantly, some NudiX enzymes have also been reported to have RNA-decapping activity.16–20 Identification of the new decapping role of some NudiX enzymes will help us to reveal new potential RNA caps and explore their biological role.In our search for new noncanonical RNA caps, we explored A. thaliana NudiX proteins and their substrate specificity. In particular, we examined four enzymes, cytosolic AtNUDT6, AtNUDT7, and chloroplastic AtNUDT19 and AtNUDT27, since small molecules such as NAD(H), CoA, ADP-ribose, and various NpnNs are known to be their substrates in vitro.14 In addition, these proteins are the closest plant homologs of mammalian Nudt2 and Nudt12, which have been demonstrated to have RNA non-canonical decapping activity in vitro, namely deFADding and deCoAding, respectively.20,21 We therefore tested whether the aforesaid enzymes are capable of cleaving these noncanonical RNA caps directly from RNA. In addition, we compared their ability to cleave small molecules and noncanonically capped RNA. Our results indicate that plant NudiXes may fulfil the role of RNA-decapping enzymes specific for noncanonically capped RNA.
Results and discussion
First, we screened the in vitro activity of AtNUDT6, AtNUDT7, AtNUDT19, and AtNUDT27 on RNA capped with NAD(H), CoA, ADP-ribose, canonical m7Gp3A(G), or various dinucleoside polyphosphates (Ap2–5A, Gp3–4G, Ap3–5G). We prepared 32P labelled RNA by in vitro transcription with T7 RNA polymerase in the presence of cap precursors using two DNA templates (Table S1, ESI†). Template 35A has at +1 position A and was used in experiments with Ap2–5A, Gp3–4A, NAD(H), CoA, ADP-ribose and m7Gp3A. Template 35G has at position +1 G and was used in experiments with Gp3–4G, Ap3–5G, and m7Gp3G. Asymmetrical NpnNs such as Ap3–5G can be used with both templates in order to obtain RNA with flanking A or G, Ap3–5G-RNA, or Gp3–5A-RNA, respectively.Following in vitro transcription, we purified the capped RNA from uncapped by treatment with 5′ polyphosphatase and Terminator™ exonuclease. This treatment guaranteed that the amount of capped RNA in each reaction was in average 96 ± 4% (only in case of m7Gp3A(G)-RNA the amount of capped RNA was 58 ± 3%). Afterwards, we treated capped RNA with NudiX enzymes (Fig. 1A) and followed the cleavage with PAGE analysis (Fig. 1B and Fig. S1 and S2, ESI†). We observed the highest enzymatic activity on the capped RNA at 500 nM concentration for AtNUDT6, 7, and 19 and 50 nM concentration for AtNUDT27 (Fig. S1 and S2, ESI†). AtNUDT6 and AtNUDT7 had a similar substrate specificity, with the Ap4A-RNA being the best substrate for both of them (Fig. 1B).
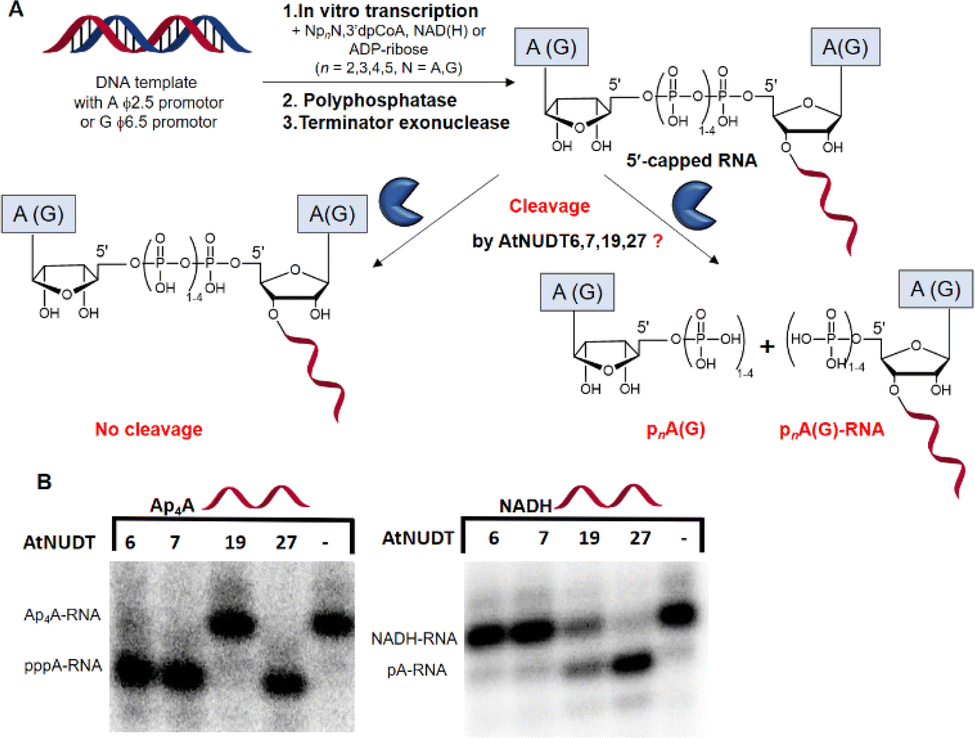 | ||
| Fig. 1 Screening of the RNA-decapping activity of AtNUDT6, 7, 19, and 27. (A) Scheme of the experimental setup. 32P labelled RNA was prepared by in vitro transcription with T7 RNA polymerase in the presence of the small molecules NpnNs, NAD(H), CoA, or ADP-ribose. The side product, uncapped triphosphate RNA, was degraded by treatment with 5′ polyphosphatase and Terminator™ exonuclease. (B) A representative example of PAGE analysis of the RNA-decapping activity of AtNUDT6, 7, 19, and 27 on 5′-capped Ap4A-RNA and NADH-RNA. | ||
In addition, AtNUDT6 also partially cleaved (less than 50%) Ap3A-, Ap5A- and Ap4G-RNA. AtNUDT7 cleaved Ap2,3,5A-RNA, Ap4,5G-RNA, and (to some extent) Gp4A-RNA (Table 1 and Fig. S3, ESI†). The main product of the Ap4A-RNA-decapping reaction by AtNUDT6 and AtNUDT7 is a triphosphate (ppp-)RNA. AtNUDT27 cleaved Ap4A-RNA into a mixture of ppp-RNA and monophosphate (p-)RNA (Fig. S4, ESI†). While AtNUDT6 and AtNUDT7 had quite wide substrate specificities, AtNUDT19 efficiently cleaved only the NADH-RNA, with the NAD-RNA being partially cleaved (Fig. 1B and Table 1 and Fig. S3, ESI†). Surprisingly, at a tenfold lower concentration (50 nM), AtNUDT27 still partially hydrolyzed the majority of capped RNA with a relatively high cleavage efficiency (60–90% after 1 h), except for Ap3A-RNA (less than 30%) (Table 1 and Fig. S3, ESI†).
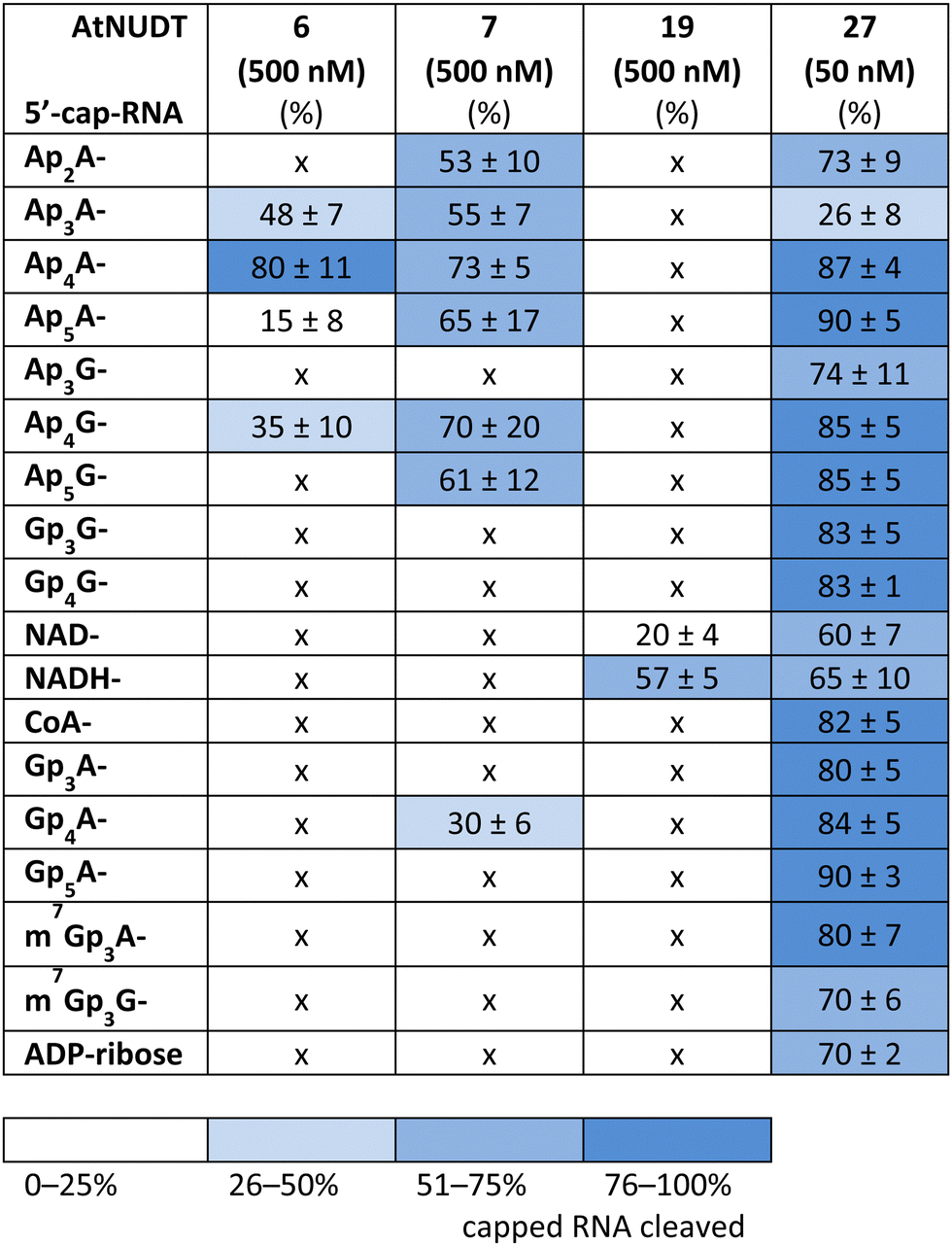
|
While some of the small molecules are known to be in vitro substrates of these NudiX enzymes, the entire set of recently discovered noncanonical caps has not yet been tested as free compounds. Therefore, we screened all the noncanonical RNA caps in their free form as substrates for the four NudiX proteins (Table 2 and Fig. S5–S8, ESI†). AtNUDT6 and AtNUDT7 had a similar substrate specificity on both the small molecules and the capped RNA. Surprisingly, their best substrate was not free Ap4A, as expected based on the results from assays on capped RNA (Table 2 and Fig. S5–S8, ESI†) but rather Ap2A and NADH, both of which have a diphosphate bridge. As in the case of capped RNA, AtNUDT19 hydrolyzed free NADH with the highest efficiency (Table 2 and Fig. S5–S8, ESI†). AtNUDT27 had a very high activity at 500 nM concentration and quantitatively cleaved all the free tetraphosphates (Ap4A, Ap4G, and Gp4G) and Ap5A (Table 2 and Fig. S5–S8, ESI†). To better observe the selectivity of the enzyme towards small free molecules, we decreased its concentration to 50 nM (Fig. S9, ESI†). Under these reaction conditions, AtNUDT27 preferred dinucleoside tetraphosphates (i.e. Ap4A, Ap4G, and Gp4G) over other noncanonical RNA caps.
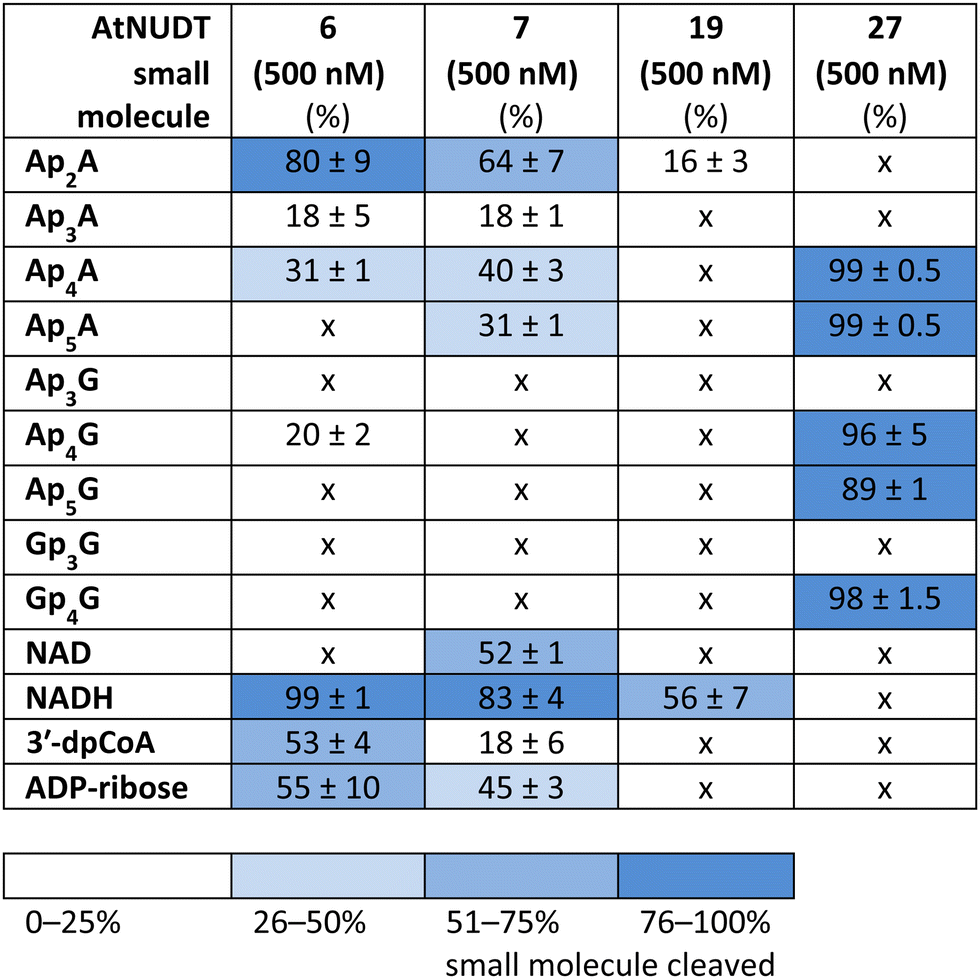
|
In addition, we compared Michaelis–Menten kinetic parameters for AtNUDT6, 7, and 27 and Ap4A as a substrate (Fig. 2A). These parameters were already known for AtNUDT19 and NADH as a substrate.22 Although KM values were comparable for all three enzymes, Vmax and Kcat of AtNUDT27 were both significantly higher (P < 0,05) compared to AtNUDT6 and 7 (Fig. S11a and b, ESI†). This finding may explain observed behaviour of AtNUDT27 to be more efficient in cleaving free Ap4A than AtNUDT6 and 7 (Table 2).
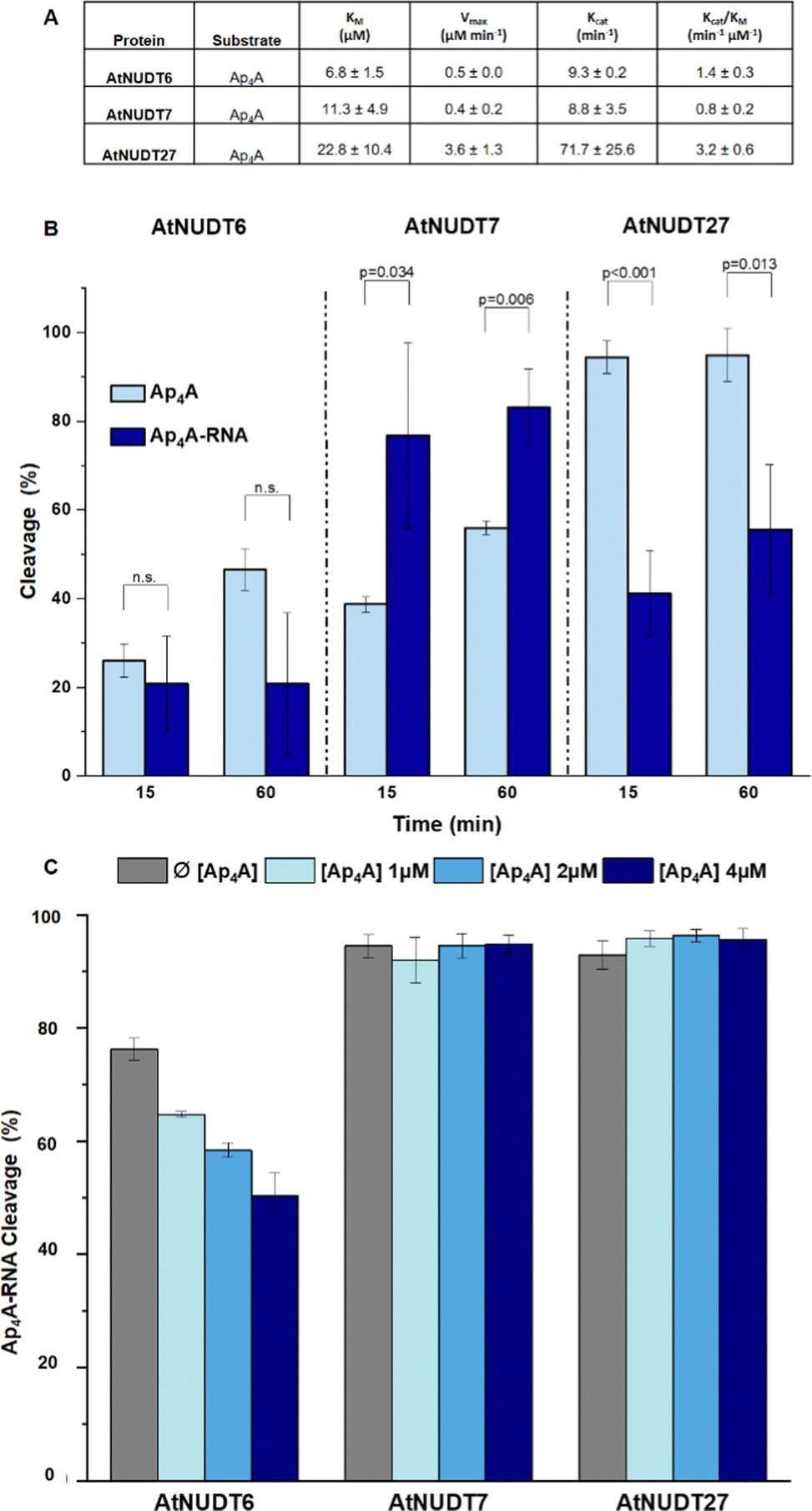 | ||
| Fig. 2 (A) Kinetic parameters of AtNUDT6, AtNUDT7 and AtNUDT27 cleaving Ap4A expressed as mean ± standard deviation. (B) Cleavage efficiency of free Ap4A (2.5 μM) and Ap4A-RNA (2.5 μM) by AtNUDT6, AtNUDT7, and AtNUDT27 (50 nM) at 15- and 60 min time points. (C) Inhibition study of Ap4A-RNA (1 μM) cleavage by AtNUDT6 (500 nM), AtNUDT7 (500 nM) and AtNUDT27 (50 nM) with addition of 1, 2 or 4 μM Ap4A. | ||
Next, we wanted to determine whether NudiX enzymes prefer free small molecules or capped RNA as substrates especially since this could reflect the situation in vivo. Due to the need to use a very large amount of RNA substrate for the entire series of tests, we compared only one set of substrates, free Ap4A and Ap4A-RNA. We observed the cleavage of both substrates at a concentration of 2.5 μM and 15- and 60 minute time points. The cleavage of Ap4A was analysed by HPLC, while decapping of Ap4A-RNA was measured by quantification of corresponding bands of capped RNAs resolved on PAGE (Fig. 2B and Fig. S10, ESI†). Comparison of the substrates showed that AtNUDT27 preferred the small molecule (Ap4A) and cleaved it more efficiently than Ap4A-RNA under the chosen conditions. AtNUDT6 cleaved comparably well both substrates – Ap4A and Ap4A-RNA. AtNUDT7 had higher activity on Ap4A-RNA than on free Ap4A. This finding may explain the redundancy of NudiX enzymes observed in vivo, i.e. some enzymes are used by the cell to hydrolyze free small molecules, while others cleave noncanonically capped RNA.
In order to understand whether free dinucleoside polyphosphates can inhibit the cleavage of capped RNA, we measured cleavage of Ap4A-RNA (1 μM) by AtNUDT6, 7 and 27 in presence of free Ap4A (1,2 and 4 μM). We observed that only AtNUDT6 activity was affected in dose-dependent manner. The activity of other two enzymes AtNUDT7 or AtNUDT27 was not affected by addition of free Ap4A (Fig. 2C and Fig. S12, ESI†). The difference between AtNUDT6 and AtNUDT7, which have comparable kinetic parameters, could be explained by AtNUDT7 preference for Ap4A-RNA instead of free Ap4A as we demonstrated in Fig. 2B. On the other hand, AtNUDT27 showed almost 8 times higher Kcat for Ap4A than AtNUDT6 and 7 suggesting that added free Ap4A would be metabolised at high rate leaving enough enzymatic capacity to hydrolyse Ap4A-RNA as well and thus the inhibition effect was not observed.
Conclusions
In light of recent discoveries of noncanonical RNA caps in various organisms, we focused on potential RNA-decapping NudiX enzymes from A. thaliana. We explored the cleavage properties of four enzymes, cytosolic AtNUDT6, AtNUDT7,23 and chloroplastic AtNUDT19 and AtNUDT27.14,22 First, we tested whether they could cleave canonical and noncanonical caps from RNA using in vitro transcribed RNA capped with NpnNs, CoA, NAD(H), m7Gp3A(G), and ADP-ribose. We found that AtNUDT19 was very specific and cleaved NADH-RNA almost exclusively. Also, AtNUDT6 and AtNUDT7 were relatively specific and cleaved mainly Ap4A-RNA. In contrast, AtNUDT27 effectively hydrolyzed almost all types of capped RNA at a tenfold lower concentration than the other tested enzymes, making it potentially a general RNA-decapping enzyme. As the in vitro substrates were not known for all of these enzymes, we also tested their substrate specificity on free small molecules that can form RNA caps (NpnNs, 3′-dpCoA, NAD(H), m7Gp3A(G), and ADP-ribose). Again, AtNUDT19 was very specific towards NADH, which is in accordance with role of AtNUDT19 in regulating NADH and NADPH levels in chloroplasts, as previously reported.22 Under our experimental conditions AtNUDT19 cleaved only free NADH but not NAD. Interestingly, when capped RNA was used as substrate, AtNUDT19 cleaved NADH-RNA and also partially NAD-RNA. This may indicate its role as an NAD/NADH-RNA-decapping enzyme. Contrary to its promiscuity in the case of capped RNA, AtNUDT27 hydrolyzed dinucleoside tetraphosphates and pentaphosphates almost exclusively. Our observed activity of AtNUDT27 against dinucleoside tetraphosphates is in contrast with previously reported results.22 There, authors observed cleavage of only Ap5A but not of Ap4A. Nevertheless, the presence of reducing agents may positively influence the activity of these enzymes.24 Thus, this discrepancy may be explained by the presence of a higher concentration of reducing agent DTT (2 mM) in our reaction in comparison with their 1 mM DTT concentration. AtNUDT6 and AtNUDT7 mainly cleaved free molecules with the diphosphate bridge. In fact, both AtNUDT6 and AtNUDT7 were demonstrated to be cytosolic NADH pyrophosphohydrolases participating in stress responses through modulation of intracellular NADH levels in A. thaliana.25 Ap4A is also known to be involved in stress signalling in many organisms, and because AtNUDT6, AtNUDT7, and AtNUDT27 efficiently cleaved both the free Ap4A and the Ap4A-RNA, we compared their preferences on cleavage of these substrates. We observed that AtNUDT27 and AtNUDT6 cleaved the free Ap4A more efficiently, while AtNUDT7 favoured the Ap4A-RNA. These differences in substrate specificities suggest that in vivo some NudiX proteins may function in degradation of free small molecules, while others serve as specific decapping enzymes, probably during regulated RNA decay.It has been shown that diadenosine polyphosphates such as Ap3A and Ap4A behave like alarmones in A. thaliana and trigger a cascade of reactions to yield various protective compounds.13 Our experiments show that plant NudiX enzymes have RNA-decapping activity and that they can cleave various noncanonical RNA caps. This finding supports the notion that plant RNA also contains other unknown RNA caps in addition to the canonical m7G structure and NAD cap.3,26 Altogether, the existence of specific enzymes that cleave NADH- and Ap4A-RNA suggest that these RNA caps may play an important role in plant RNA metabolism, signalling, or stress reaction.
Moreover, the specific cleavage properties of the individual NudiX enzymes characterized in this work can be used to develop capturing strategies for noncanonically capped RNA and thus help us to identify the sequence and features of these RNA species.
Materials and methods
Preparation of RNA
Purification of recombinant NudiX enzymes
A. thaliana NUDT6, 7, 19, and 27 genes were cloned into the pET26b vector with the C-terminal His tag (Novagen). Due to the problem with expressing the full-length AtNUDT27, it was cloned without the first 44 aa containing the chloroplast signal sequence. Proteins were overexpressed in Escherichia coli Rosetta DE3 cells grown for 16 hours in auto-inducing medium.27 Cells were lysed by sonication in buffer A (20 mM Tris-HCl, pH 8.0, 300 mM NaCl, 10% glycerol and 5 mM β-mercaptoethanol) and recombinant proteins were purified on TALON Metal Affinity Resin (Clontech), eluted in buffer A supplemented with 250 mM of imidazol and fractionated by gel filtration chromatography on an ENrichTM SEC70 column (BioRad). The purified proteins were concentrated to 50 μM in a buffer containing 20 mM Tris-HCl, pH 8.0, 200 mM NaCl, 2 mM DTT, and 10% glycerol, flash frozen in liquid nitrogen, and stored at −80 °C. Protein concentration was determined at 280 nm using the molar extinction coefficient calculated on the basis of protein composition with the ProtParam tool (https://web.expasy.org/protparam/) (Fig. S13, ESI†).Cleavage of capped RNA by A. thaliana NudiX enzymes
The decapping assay was performed at 37 °C in 10 μL reaction. To test the cleavage of the 5′-caps, the RNA samples were divided into two parts. The positive control contained 100 ng of the pure RNA (in vitro transcription, DNAse I treatment, purified on RNA mini Quick Spin Columns), 1 μL of 10× buffer (100 mM Tris-HCl, pH 7.5, 1 M KCl, 20 mM MgCl2, 20 mM MnCl2, 20 mM DTT), and 500 nM of the recombinant AtNUDT6, 7, 19, and 50 nM AtNUDT27. Water was used as the negative control. The mixtures were incubated at 37 °C for 1 h and purified on RNA mini Quick Spin Columns. Then 3 μL of the purified samples were mixed with 2× RNA loading dye (NEB) for analysis by 12% PAGE (600 V, 3.5 h). Typhoon FLA 9500 was used as a visualization imaging system.Calculations of RNA decapping
The experiments were performed in triplicates, and the amount of capped RNA was calculated from PAGE analysis using the software ImageJ.28 Areas under the peaks (AUC) corresponding to the 5′-capped RNA (Arp) in the control without enzyme and 5′-capped RNA (Arcap) in samples with enzyme were calculated. The percentage of 5′-capped RNA species was calculated according to: (Arcap/Arp) × 100.Enzymatic assay and kinetics assay on small free molecules
The hydrolytic activities of AtNUDT proteins towards different NpnNs (Ap2–5A, Gp3–4G, Ap3–5G, m7Gp3G, m7Gp3A), 3′-dpCoA, ADP-ribose, and NAD(H) were performed using 500 nM of the AtNUDT6, 7, 19, and 27 and 400 μM of substrate in 10 mM Tris-HCl (pH 7.5), 100 mM KCl, 2 mM magnesium acetate, 2 mM DTT, at 37 °C. The enzyme was heat deactivated at 75 °C for 10 min followed by cooling on ice. The mixture was then analysed by monitoring absorbance at 259 nm and retention time compared to standards on high-performance liquid chromatography using a Kinetex C18 column (4.6 × 150 mm, 5 μm, Phenomenex) at a flow rate of 1 ml min−1. The mobile phase consisted of 100 mM triethylammonium acetate pH 7 (buffer A) and Acetonitrile (buffer B). Separation was achieved using a 25 minute gradient: 100% A for 2 min; linear decrease to 95% A over 4 min; linear decrease to 70% A over 9 min; maintaining 70% A for 3 min; returning linearly to 100% A over 1 min. Chromatograms were integrated, and the percentage of substrate cleavage was calculated from corresponding AUC compared to the control without enzyme.Kinetic parameter calculation
Kinetic parameters of AtNUDT enzymes were calculated from experiments using increasing concentrations of Ap4A (0, 5, 10, 20, 30, 50, 70, and 100 μM) and 50 nM of AtNUDT6, 7, and 27. The 500 μL mixture was incubated at 37 °C, and aliquots of 100 μL were collected at 0, 2.5, 5, 10, and 15 min and immediately heated to 75 °C for 10 min to stop the reaction. All samples were analysed using the HPLC method described above. Calculated initial reaction velocities were plotted against Ap4A concentration and fitted to the Michaelis–Menten model to obtain Km and Vmax values using Origin software (Northampton, MA). Student t-test and analysis of variance (ANOVA) with post hoc Bonferroni analysis were performed in Microsoft Excel to assess significant differences between measured parameters where needed.Inhibition test with Ap4A for AtNUDT enzymes
To test the inhibition of AtNUDT6, AtNUD7 and AtNUDT27 enzymes in the presence of Ap4A we measured the cleavage of Ap4A-RNA at 37 °C in 10 μL reaction using 1 μM of purified Ap4A-RNA and increased concentrations of Ap4A (1, 2 and 4 μM). The concentration used for AtNUDT6 and AtNUD7 was 500 nM while for AtNUDT27 was 50 nM. The reaction was stopped using 2× RNA Loading Dye (NEB).Author contributions
M.-B.M., O.H., A.G., S.K. performed experiments. M.-B.M. and O.H. studied RNA and small molecule behaviour and designed experiments, A.G. and S.K. purified plant proteins. R.B. and O.N. designed kinetic assays. J.K. and H.C. designed experiments and coordinated study. M.-B.M., O.N., and H.C. wrote the manuscript.Conflicts of interest
Authors declare no conflicts of interests.Acknowledgements
We are grateful to all members of the Cahova Group for their help and advice. We acknowledge Martin Lepšík for figure of AtNudt7 for TOC. We acknowledge funding from the Ministry of Education, Youth and Sports (Czech Republic), programme ERC CZ (LL1603) and European Research Council Executive Agency (ERCEA) under the European Union’s Horizon Europe Framework Programme for Research and Innovation (grant agreement No 101041374 - StressRNaction). J. K. acknowledges funding from National Science Centre UMO-2018/29/B/NZ3/01980.Notes and references
- P. Boccaletto, M. A. Machnicka, E. Purta, P. Piątkowski, B. Bagiński, T. K. Wirecki, V. de Crécy-Lagard, R. Ross, P. A. Limbach, A. Kotter, M. Helm and J. M. Bujnicki, Nucleic Acids Res., 2017, 46, D303–D307 CrossRef PubMed
.
- H. Cahová, M.-L. Winz, K. Höfer, G. Nübel and A. Jäschke, Nature, 2014, 519, 374 CrossRef PubMed
- Y. Wang, S. Li, Y. Zhao, C. You, B. Le, Z. Gong, B. Mo, Y. Xia and X. Chen, Proc. Natl. Acad. Sci., 2019, 116, 12094–12102 CrossRef CAS PubMed
- X. Jiao, S. K. Doamekpor, J. G. Bird, B. E. Nickels, L. Tong, R. P. Hart and M. Kiledjian, Cell, 2017, 168, 1015–1027.e1010 CrossRef CAS PubMed
- O. Ruiz-Larrabeiti, R. Benoni, V. Zemlianski, N. Hanišáková, M. Schwarz, B. Brezovská, B. Benoni, J. Hnilicová, V. R. Kaberdin, H. Cahová, M. Vítězová, M. Převorovský and L. Krásný, bioRxiv, 2021, Preprint, 2021.2012.2014.472595 DOI:10.1101/2021.12.14.472595.
- J. G. Bird, U. Basu, D. Kuster, A. Ramachandran, E. Grudzien-Nogalska, A. Towheed, D. C. Wallace, M. Kiledjian, D. Temiakov, S. S. Patel, R. H. Ebright and B. E. Nickels, eLife, 2018, 7, e42179 CrossRef PubMed
- W. E. Kowtoniuk, Y. Shen, J. M. Heemstra, I. Agarwal and D. R. Liu, Proc. Natl. Acad. Sci., 2009, 106, 7768–7773 CrossRef CAS PubMed
- J. Wang, B. L. Alvin Chew, Y. Lai, H. Dong, L. Xu, S. Balamkundu, W. M. Cai, L. Cui, C. F. Liu, X.-Y. Fu, Z. Lin, P.-Y. Shi, T. K. Lu, D. Luo, S. R. Jaffrey and P. C. Dedon, Nucleic Acids Res., 2019, 47, e130–e130 CrossRef CAS PubMed
- O. Hudeček, R. Benoni, P. E. Reyes-Gutierrez, M. Culka, H. Šanderová, M. Hubálek, L. Rulíšek, J. Cvačka, L. Krásný and H. Cahová, Nat. Commun., 2020, 11, 1052 CrossRef PubMed
- E. Rapaport and P. C. Zamecnik, Proc. Natl. Acad. Sci., 1976, 73, 3984–3988 CrossRef CAS PubMed
- P. Zamecnik, Anal. Biochem., 1983, 134, 1–10 CrossRef CAS PubMed
- P. G. Zamecnik, M. L. Stephenson, C. M. Janeway and K. Randerath, Biochem. Biophys. Res. Commun., 1966, 24, 91–97 CrossRef CAS PubMed
- M. Pietrowska-Borek, K. Nuc, M. Zielezińska and A. Guranowski, FEBS Open Bio, 2011, 1, 1–6 CrossRef CAS PubMed
- K. Yoshimura and S. Shigeoka, Biosci., Biotechnol., Biochem., 2015, 79, 354–366 CrossRef CAS PubMed
- A. G. McLennan, Cell. Mol. Life Sci. CMLS, 2006, 63, 123–143 CrossRef CAS PubMed
- M. G. Song, S. Bail and M. Kiledjian, RNA, 2013, 19, 390–399 CrossRef CAS PubMed
- S. Kramer and A. G. McLennan, Wiley Interdiscip. Rev.: RNA, 2019, 10, e1511 Search PubMed
- E. Grudzien-Nogalska, X. Jiao, M. G. Song, R. P. Hart and M. Kiledjian, RNA, 2016, 22, 773–781 CrossRef CAS PubMed
- M.-G. Song, Y. Li and M. Kiledjian, Mol. Cell, 2010, 40, 423–432 CrossRef CAS PubMed
- E. Grudzien-Nogalska, Y. Wu, X. Jiao, H. Cui, M. K. Mateyak, R. P. Hart, L. Tong and M. Kiledjian, Nat. Chem. Biol., 2019, 15, 575–582 CrossRef CAS PubMed
- S. Sharma, E. Grudzien-Nogalska, K. Hamilton, X. Jiao, J. Yang, L. Tong and M. Kiledjian, Nucleic Acids Res., 2020, 48, 6788–6798 CrossRef CAS PubMed
- T. Ogawa, K. Yoshimura, H. Miyake, K. Ishikawa, D. Ito, N. Tanabe and S. Shigeoka, Plant Physiol., 2008, 148, 1412–1424 CrossRef CAS PubMed
- T. Ogawa, Y. Ueda, K. Yoshimura and S. Shigeoka, J. Biol. Chem., 2005, 280, 25277–25283 CrossRef CAS PubMed
- K. Olejnik, M. W. Murcha, J. Whelan and E. Kraszewska, FEBS J., 2007, 274, 4877–4885 CrossRef CAS PubMed
- T. Ogawa, K. Muramoto, R. Takada, S. Nakagawa, S. Shigeoka and K. Yoshimura, Plant Cell Physiol., 2016, 57, 1295–1308 CrossRef CAS PubMed
- H. Zhang, H. Zhong, S. Zhang, X. Shao, M. Ni, Z. Cai, X. Chen and Y. Xia, Proc. Natl. Acad. Sci., 2019, 116, 12072–12077 CrossRef CAS PubMed
- F. W. Studier, Protein Expression Purif., 2005, 41, 207–234 CrossRef CAS PubMed
- W. S. Rasband, ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, 1997–2018 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cb00213b |
| This journal is © The Royal Society of Chemistry 2023 |