
New near-infrared absorbing conjugated electron donor–acceptor molecules with a fused tetrathiafulvalene–naphthalene diimide framework†
Qi
Zhou
ab,
Junfang
Yang
b,
Mingxu
Du
a,
Xiaobo
Yu
ab,
Cheng
Li
 ab,
Xi-Sha
Zhang
ab,
Qian
Peng
b,
Guanxin
Zhang
*ab and
Deqing
Zhang
*ab
ab,
Xi-Sha
Zhang
ab,
Qian
Peng
b,
Guanxin
Zhang
*ab and
Deqing
Zhang
*ab
aBeijing National Laboratory for Molecular Sciences, CAS Key Laboratories of Organic Solids, CAS Research/Education Center for Excellence in Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, P. R. China. E-mail: dqzhang@iccas.ac.cn; gxzhang@iccas.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 29th December 2021
Abstract
Molecular materials that can absorb and emit light in the near-infrared (NIR) region are attracting more and more attention in many fields. In this paper, we report the synthesis and studies of the photophysical and semiconducting properties of tetrathiafulvalene (TTF)–naphthalene diimide (NDI) fused molecules (NDITTF2, NDI2TTF and NDI2TTF3). The results reveal that all molecules show strong absorptions in the NIR region, and their absorption maxima can reach 1015 nm with molar extinction coefficients (ε) up to 66![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 M−1 cm−1 in the solution state. Moreover, NIR emissions were found for NDITTF2 and NDI2TTF either in solution or in the film state. The emission maxima in the CHCl3 solution locate at 892 nm for NDI2TTF (ϕ = 1.64%) and 1062 nm for NDITTF2 (ϕ = 0.21%). Furthermore, the thin film of NDITTF2 was found to show p-type semiconducting properties with a hole mobility up to 0.45 cm2 V−1 s−1.
900 M−1 cm−1 in the solution state. Moreover, NIR emissions were found for NDITTF2 and NDI2TTF either in solution or in the film state. The emission maxima in the CHCl3 solution locate at 892 nm for NDI2TTF (ϕ = 1.64%) and 1062 nm for NDITTF2 (ϕ = 0.21%). Furthermore, the thin film of NDITTF2 was found to show p-type semiconducting properties with a hole mobility up to 0.45 cm2 V−1 s−1.
1. Introduction
Organic molecules that can absorb and emit light in the near-infrared (NIR, >750 nm) region are of tremendous interest because of their practical applications in a number of important fields, such as organic photovoltaics, NIR light photodetectors, bioimaging, phototherapy, etc.1–12 Due to the fact that NIR light exhibits low light scattering and deep penetration behaviors and most biological samples absorb weakly in the NIR region which can lower the background absorbance and autofluorescence, these NIR-absorbing and emitting molecules are particularly interesting for photodiagnosis (e.g. NIR fluorescence imaging and photoacoustic imaging) and phototherapy (e.g. photodynamic therapy and photothermal therapy).9–12 To meet the urgent demand for NIR absorbing and emitting materials, various design approaches have been adopted, such as extension of the conjugation length, reduction of bond length alternation, and introduction of appropriate electron donor and acceptor units.1,2 Among them, introduction of a strong electron donor (D) and a strong electron acceptor (A) in small molecules and polymers becomes a common strategy to realize such materials.As a typical strong electron donor, tetrathiafulvalene (TTF) and its derivatives have played a key role in the construction of organic conductors and superconductors in the past several decades.13,14 With the passage of time, abundant properties of TTF have been exploited, which made them intensively studied in many fields including chemical sensors, molecular devices as well as optoelectronic devices in the recent years.15–29 Among these TTF derivatives, TTF based conjugated electron donor–acceptor systems have received extensive attention. This is because the incorporation of TTF units in a conjugated way can raise the highest occupied molecular orbital (HOMO) energy levels of the D–A molecules, redshift the absorption and enhance the intermolecular interaction.23 Such modifications can enrich the physical and chemical properties of TTF derivatives and thus enable them to be applied in organic optoelectronic materials. Up to now, all kinds of TTF based electron donor–acceptor systems had been developed.13–29 However, TTF-based molecules with both NIR-absorbing and emitting properties remain scarcely reported.
Herein, we report three D–A molecules: NDITTF2, NDI2TTF and NDI2TTF3 (see Scheme 1), in which the TTF and naphthalene diimide (NDI) moieties are fused in conjugated ways. The combinations of both strong electron donating and accepting units are likely to redshift their absorptions to the NIR region. In addition, the fusion of TTF with NDI units might endow them with not only NIR absorption but also NIR emission properties.23,27–29 The results manifest that NDITTF2, NDI2TTF and NDI2TTF3 exhibit NIR absorptions and the maximum absorption of NDI2TTF3 in the film state can reach 1072 nm. Moreover, emissions in solution were detected for NDITTF2 and NDI2TTF. The maximum emissions of CHCl3 solutions of NDITTF2 and NDI2TTF appear around 1062 and 892 nm, with quantum yields of 0.21% and 1.64%, respectively. Finally, thin-film OFETs based on NDITTF2 exhibit good a hole mobility up to 0.45 cm2 V−1 s−1.
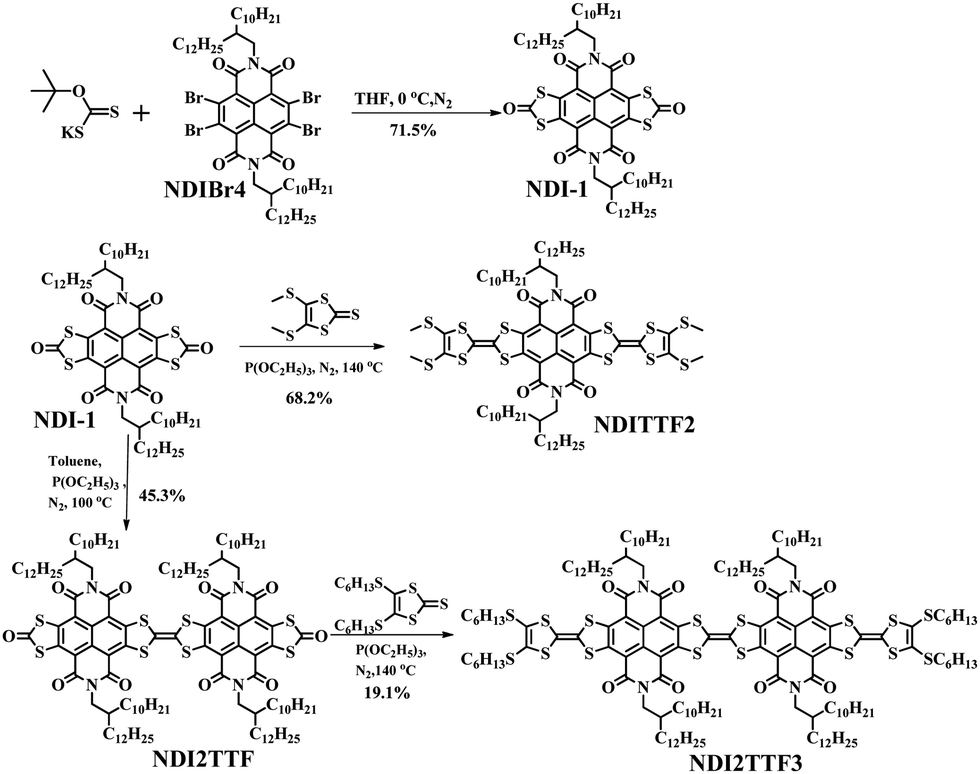 | ||
| Scheme 1 Chemical structures of NDI-1, NDITTF2, NDI2TTF and NDI2TTF3 and the synthetic approach. | ||
2. Results and discussion
Synthesis and characterization
As shown in Scheme 1, the reaction of NDIBr4 with potassium O-t-butyl dithiocarbonate afforded the key intermediate (NDI-1) in 71.5% yield, which can be transformed into NDITTF2 in 68.2% yield after reaction with 5-bis-(methylthio)-1,3-dithiole-2-thione. Meanwhile, self-coupling of NDI-1 in the presence of triethylphosphite formed NDI2TTF in 45.3% yield, which further reacted with 4,5-bis(hexylthio)-1,3-dithiole-2-thione to form NDI2TTF3 in 19.1% yield. All the reactions were carried under a nitrogen atmosphere. NDITTF2, NDI2TTF and NDI2TTF3 were characterized by NMR and high-resolution mass spectrometry (HRMS) as well as elemental analysis. Also, as shown in Fig. S1 (ESI†), the degradation temperatures at 5% weight loss were measured to be 348 °C for NDITTF2, 426 °C for NDI2TTF and 311 °C for NDI2TTF3, indicating that they all show good thermal stability.Spectroscopic studies and theoretical calculations
The UV-vis absorption spectra of NDITTF2, NDI2TTF and NDI2TTF3 in chloroform are shown in Fig. 1 and the respective absorption peaks are collected in Table 1. Intense broad absorptions in the region of 500–1200 nm were detected for NDI2TTF, NDITTF2 and NDI2TTF3. Moreover, these absorptions are red-shifted in the following order: NDI2TTF < NDITTF2 < NDI2TTF3. The absorptions of the thin films of NDITTF2, NDI2TTF and NDI2TTF3 were also measured. As shown in Fig. 1, the absorption spectra of the thin films of NDITTF2, NDI2TTF and NDI2TTF3 were all red-shifted compared to those in solutions owing to the intermolecular interactions within their thin films, which may involve intermolecular electron donor and acceptor interactions. Based on the absorption edges of their thin films, the band gaps of NDITTF2, NDI2TTF and NDI2TTF3 were estimated to be 1.03 eV, 1.09 eV and 0.99 eV, respectively. These are in good agreement with those obtained with cyclic voltammetry data as it will be discussed below.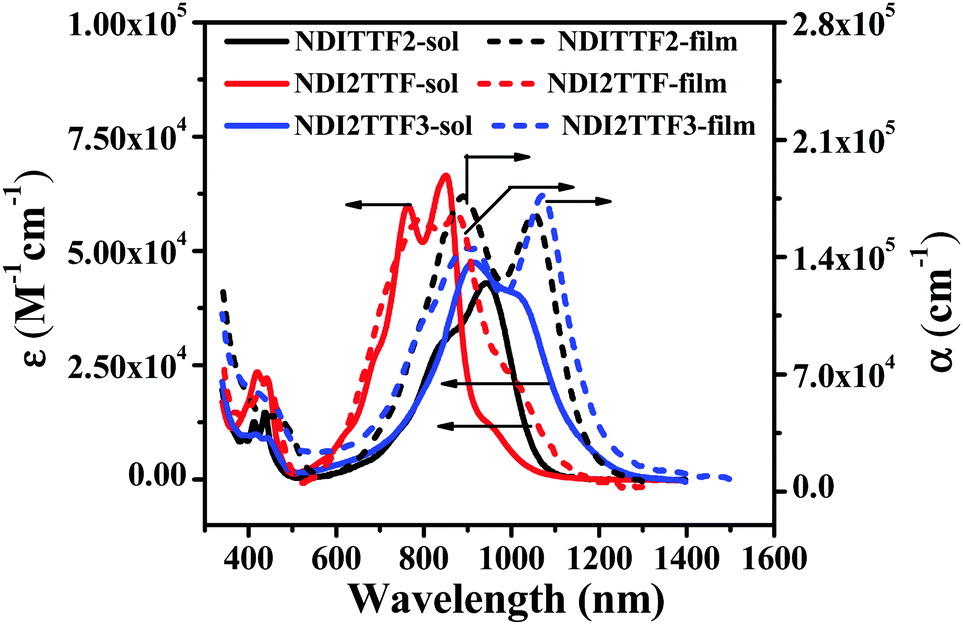 | ||
| Fig. 1 Absorption spectra of chloroform solutions (solid lines) and thin films (dashed lines) of NDITTF2, NDI2TTF and NDI2TTF3. The concentration for each molecule was 10 μM. | ||
| Compd | λ (nm) | λ (nm) | E LUMO (eV) | E HOMO (eV) |
E
cvgg (eV) |
E
optgh (eV) |
|||
|---|---|---|---|---|---|---|---|---|---|
| Solutionb (ε, M−1 cm−1) | Filmc (α, cm−1) | Solution | Film | Φ (%) | |||||
| a Absorption peaks in CHCl3 solution (1.0 × 10−5 M) and spin-coated thin films. b Molar extinction coefficient. c Extinction coefficient for spin-coated thin films. d Emission peaks in CHCl3 solution (1.0 × 10−5 M) and spin-coated thin films. e Quantum yield in CHCl3 solution (1.0 × 10−5 M). f Estimated using the equations: EHOMO = −(E1/2ox1 + 4.8 − EFcFc+ox) eV and ELUMO = − (E1/2red1 + 4.8 − EFcFc+ox) eV, where the E1/2ox1 and E1/2red1 values can be found in Table S4 (ESI), and EFcFc+ox = 0.56 V. g E cvg = ELUMO − EHOMO. h Calculated on the basis of the onset of thin film absorption spectra; ‘—’ means too weak to be detected. | |||||||||
| NDITTF2 | 946 (42900), 858 (31600), 439, 413 |
1052 (165000), 892 (176000) |
1062 | 1188 | 0.21 | −3.60 | −4.96 | 1.36 | 1.03 |
| NDI2TTF | 846 (66900), 765 (60500), 444, 421 |
872 (166000), 790 (161000) |
892, 1005 | 1158 | 1.64 | −4.12 | −5.54 | 1.42 | 1.09 |
| NDI2TTF3 | 1015 (40400), 915 (47800), 448, 413 |
1072 (173000), 915(154000) |
— | — | — | −3.64 | −4.93 | 1.29 | 0.99 |
Density functional theory (DFT) and time-dependent DFT (TDDFT) calculations were performed to figure out the origin of the absorption spectra of the three compounds by using the B3LYP functional and the 6-31G(d) basis set. Briefly, the calculated results are given in Fig. S2, S3 and Tables S1–S3 (ESI†), including the absorption peak positions, oscillator strengths, transition properties and the charge density of the frontier molecular orbitals. Comparing the calculated and experimental results, it can be inferred that (i) the absorptions are red-shifted gradually from NDI2TTF through NDITTF2 to NDI2TTF3, in good agreement with the respective absorption spectra measured experimentally (see Fig. S3, ESI†); (ii) the absorptions in the long-wavelength regions mainly stem from the transition from S0 to S1 with large oscillator strength for the three compounds, which agrees with the strong absorption intensity observed in the experimental spectra. The fine structures may be caused by the contribution from vibration structures, which are very common in rigid organic compounds.30 The maximum absorption for NDI2TTF3 deviates from the 0–0 transition and blue-shifted to 915 nm owing to the vibration-caused satellite band, which results in the difference between the experimental value and the calculated one without considering the vibration effect. The electronic transition and orbital properties indicate that NDITTF2 and NDI2TTF3 have similar features: the transition is mainly from the HOMO to the LUMO, and the HOMO is delocalized over TTF and the naphthalene ring of NDI and the LUMO is largely concentrated on the NDI moiety, which leads to a very similar absorption spectrum. At the same time, it suggests a bit stronger conjugation degree in NDI2TTF3 than in NDITTF2, which results in a slight enhancement of the HOMO and the decrease of the LUMO in energy for NDI2TTF3 by comparing NDITTF2; thus, it rationally explains the red-shift of the absorption bands. Differently, the absorption of NDI2TTF is caused by the transition between the HOMO and LUMO which both spread over the whole molecule, resulting in a blue-shifted absorption spectrum relative to NDITTF2 and NDI2TTF3. (iii) The short-wavelength absorptions mainly come from S3 (HOMO−2 → LUMO) and S5 (HOMO−6 → LUMO and HOMO−3 → LUMO) for NDITTF2, S4 (HOMO−2 → LUMO) and S7 (HOMO−1 → LUMO+1) for NDI2TTF and S9 (HOMO−4 → LUMO and HOMO−3 → LUMO) and S10 (HOMO−3 → LUMO and HOMO−4 → LUMO) for NDI2TTF3 (Table S1–S3, ESI†).
NIR fluorescence was detected for NDITTF2 and NDI2TTF both in the solution state and in the film state (see Fig. 2). For example, the emission maxima locate at 892 nm for NDI2TTF and 1062 nm for NDITTF2 after being excited with an 808 nm light source, and their quantum yields were 1.64% and 0.21%, respectively (see Table 1). Compared with their solution fluorescence spectra, large red-shifted emissions up to 1188 and 1158 nm were observed for thin films of NDITTF2 and NDI2TTF (see Fig. 2), respectively, though their quantum yields were too low to be detected. But almost no fluorescence was detected for NDI2TTF3 in both solution and thin film states.
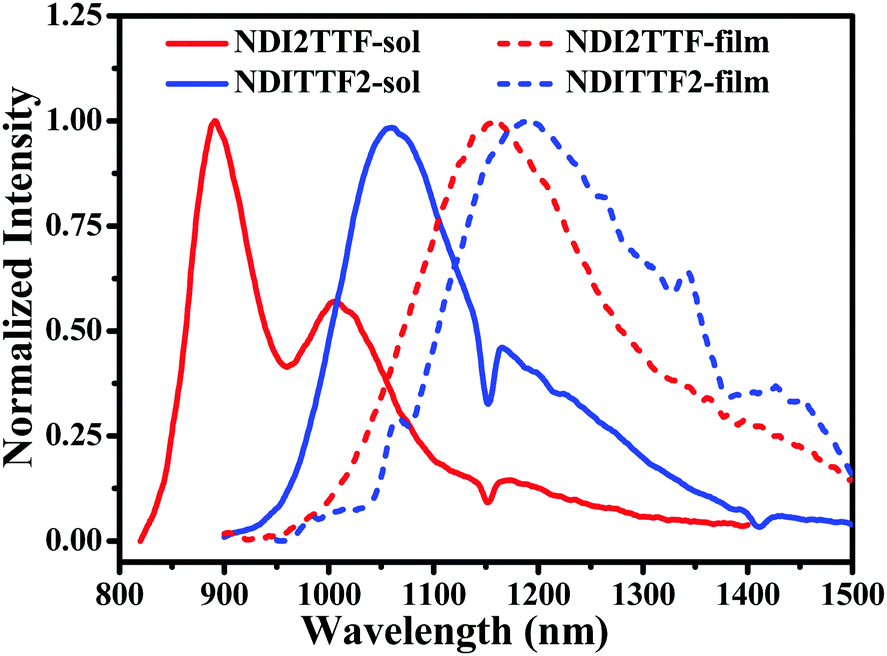 | ||
| Fig. 2 Normalized emission spectra of the CHCl3 solutions (solid lines) and thin films (dashed lines) of NDITTF2 and NDI2TTF. The concentration for each molecule was 10 μM and the excitation wavelength was 808 nm. | ||
It is well known that the absorption and emission spectra of D–A molecules show solvatochromic behavior. Fig. S4 and S5 (ESI†) show the absorption of NDITTF2, NDI2TTF and NDI2TTF3 and emission spectra of NDITTF2 and NDI2TTF in chloroform, chlorobenzene and toluene with different polarities. Redshifts (albeit slight) of both absorption and emission spectra of these molecules were observed by increasing the solvent polarities. This indicates the existence of charge transfer character in these molecules.25–27
Electrochemical and spectroelectrochemical studies
Cyclic voltammograms (CVs) and differential pulse voltammograms (DPVs) of NDITTF2, NDI2TTF and NDI2TTF3 were measured (see Fig. 3) by using a ferrocene/ferrocenium (Fc/Fc+) redox couple as the reference standard (see Fig. S6, ESI†), and the respective oxidation and reduction potentials are presented in Table S4 (ESI†). As shown in Fig. 3, all the three molecules show multistate redox properties. For example, NDITTF2 undergoes six reversible redox processes (E1/2ox1 = 0.72 V, E1/2ox2 = 0.96 V, E1/2ox3 = 1.30 V, E1/2ox4 = 1.43 V, E1/2red1 = −0.64 V, E1/2red2 = −1.01 V), which can be assigned to the oxidation of the TTF units and the reduction of the NDI moiety. NDI2TTF and NDI2TTF3 also exhibit quasi-reversible redox waves. As compared to those of NDI2TTF3 (E1/2ox1 = 0.69 V, E1/2red1 = −0.60 V), the redox potentials (E1/2ox1 =1.3 V and E1/2red1 = −0.11 V) of NDI2TTF are positively shifted. This may be caused by the two electron withdrawing carbonyl groups in NDI2TTF.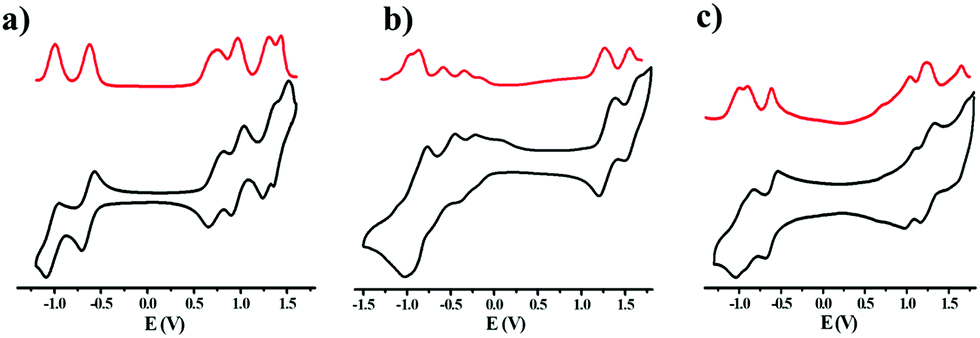 | ||
| Fig. 3 DPVs (red line) and CVs (black line) of NDITTF2 (a), NDI2TTF (b) and NDI2TTF3 (c) in a mixture of o-dichlorobenzene and CH2Cl2 (1:1, v/v) at a scan rate of 100 mV s−1 with a Pt disc (2 mm diameter) as the working electrode, a Pt wire as the counter electrode, a Ag wire as the reference electrode, and n-Bu4NPF6 (0.1 M) as the supporting electrolyte. The concentration of each sample was 1 mM. | ||
The HOMO/LUMO energies were estimated to be −4.96/−3.6, −5.54/−4.12 and −4.93/−3.64 eV for NDITTF2, NDI2TTF and NDI2TTF3, respectively (see Table 1). Obviously, the HOMO energy increases in the following order: NDI2TTF < NDITTF2 < NDI2TTF3, which agrees well with the order obtained from theoretical calculations (see Table S4, ESI†). The band gaps were estimated to be 1.36 eV, 1.42 eV and 1.29 eV for NDITTF2, NDI2TTF and NDI2TTF3, respectively, based on their HOMO/LUMO energies.
We also investigated the variation of the absorption spectrum upon electrochemical oxidation/reduction for NDITTF2. Oxidation of the dichlorobenzene/CH2Cl2 (1:1, v/v) solution of NDITTF2 containing n-Bu4NPF6 (0.15 M) was performed by applying an oxidation potential of 0.8 V (vs Ag wire). Obviously, as shown in Fig. S7A (ESI†), a steady decrease in the absorption band around 918 nm and emergence of an absorption band around 1083 nm as well as a broad absorption from 2044 nm to 3075 nm were observed. This variation of the absorption spectra should be attributed to the transformation of neutral NDITTF2 into its radical cation form. This was proved by chemical oxidation of NDITTF2. For example, as shown in Fig. S8 (ESI†), the absorption band around 1083 nm and the broad absorption from 2044 nm to 3075 nm appeared and reached their maxima after the addition of 2.66 eq. of Fe(ClO4)3. Interestingly, application of a reduction potential of 0.4 V (vs. Ag wire) to the solution to which an oxidation potential was applied for 2000 s resulted in a gradual decrease of the absorption band to around 1083 nm and the broad absorption from 2044 nm to 3075 nm (Fig. S7B, ESI†). After the electrochemical reduction was carried out for 500 s, the initial absorption spectrum of the solution was almost recovered. Briefly, the NIR absorption of NDITTF2 can be reversibly modulated by the sequential electrochemical oxidation and reduction.
Thin film semiconducting properties
The semiconducting properties of NDITTF2, NDI2TTF and NDI2TTF3 were explored by fabrication of top-gate/bottom-contact (TGBC) OFETs with their thin films on OTS (octadecyltrichlorosilane) modified SiO2/Si substrates (see the ESI†). Fig. 4 and Fig. S9 (ESI†) show the transfer and output curves for the FET with a thin film of NDITTF2 before and after thermal annealing. Clearly, IDS increases when applying a negative VGs, which means that NDITTF2 exhibits p-type semiconducting behavior, agreeing well with its HOMO and LUMO energy levels as discussed above.31 The average hole mobility of the as-prepared spin-coated thin film of NDITTF2 was extracted to be 0.31 cm2 V−1 s−1. Further thermal annealing of the thin film of NDITTF2 has no remarkable effect on the enhancement of mobility. For example, the maximum mobility can be boosted to only 0.45 cm2 V−1 s−1 even after thermal annealing at 160 °C for 15 min (see Table S5, ESI†). However, the semiconducting properties of thin films of NDI2TTF and NDI2TTF3 are too poor to be detected.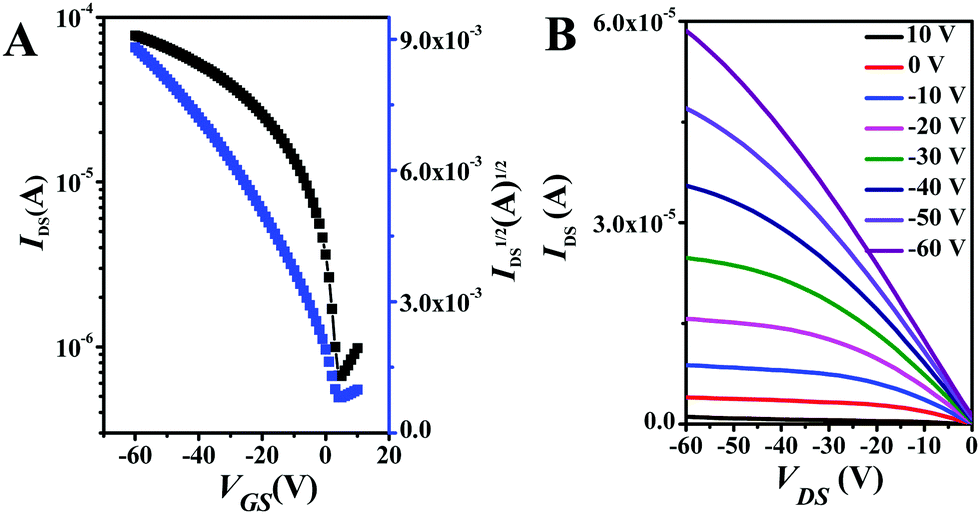 | ||
| Fig. 4 The transfer and output characteristics of the FET device based on NDITTF2 after being annealed at 160 °C for 15 min. The transistor channel width and channel length were 1440 μm and 40 μm, respectively. | ||
Thin films of NDITTF2 before and after annealing were investigated with X-ray diffraction (XRD) and atomic force microscopy (AFM). As depicted in Fig. 5, two very weak diffraction peaks were detected for the thin-film of NDITTF2 before annealing. After annealing at 160 °C for 15 min, the diffraction intensities of both peaks (3.37° and 6.75°) were slightly enhanced. These results reveal that the crystallinity of the thin film of NDITTF2 is slightly enhanced after annealing. The AFM images of thin-films of NDITTF2 before and after annealing were further recorded as depicted in Fig. S10 (ESI†). After annealing, the morphology of the thin film of NDITTF2 was smoother and more uniform with a better continuity and the root-mean square roughness (RRMS) values varied from 6.01 nm to 4.64 nm. Thus, the results of both XRD and AFM studies are in agreement with the fact that the hole mobility of the thin-film of NDITTF2 can be enhanced slightly after annealing.
 | ||
| Fig. 5 XRD pattern of the as-cast thin-film of NDITTF2 and that after annealing at 160 °C for 15 min. | ||
3. Conclusion
In summary, three TTF–NDI fused and conjugated D–A systems (NDITTF2, NDI2TTF and NDI2TTF3) were synthesized and studied. They all exhibit rigid and planar structures, and their HOMO orbitals are delocalized over their entire π system, while their LUMOs were mainly localized on the NDI fragments, based on the DFT calculations. They all show strong absorptions in the NIR region, and the absorption maximum for the thin film of NDI2TTF3 can be extended to 1072 nm. Moreover, the CHCl3 solutions of NDITTF2 and NDI2TTF are emissive in the NIR region with emission maxima at 1062 and 892 nm, with quantum yields of 0.21% and 1.64%, respectively. Furthermore, the thin film of NDITTF2 was found to show p-type semiconducting properties with a hole mobility up to 0.45 cm2 V−1 s−1. Further structural modifications of these molecules are underway with the end to improve their quantum yields in the NIR region as well as explore their applications in photodiagnosis and phototherapy.4. Experimental section
Synthesis of NDI-1
Under a nitrogen atmosphere, a mixture of NDIBr4 (2.5 g, 2 mmol) and potassium O-t-butyl dithiocarbonate (1.12 g, 6 mmol) in THF (100 mL) was stirred at 0 °C for 1 hour. After the reaction, the solvent was removed by rotary evaporation and the crude product was purified by silica gel chromatography with petroleum ether (60–90 °C) and CH2Cl2 (4:1, v/v) as the eluent to give NDI-1 (1.59 g) as a yellow solid in 71.5% yield. 1H NMR (300 MHz, CDCl3) δ 4.19 (d, J = 7.2 Hz, 4H), 2.01 (br, 2H), 1.68–0.98 (m, 80H), 0.87 (t, J = 6.0 Hz, 12H). 13C NMR (75 MHz, CDCl3) δ 195.23, 162.16, 143.21, 123.75, 117.50, 46.04, 36.40, 31.94, 31.45, 30.01, 29.70, 29.66, 29.60, 29.38, 26.22, 22.71, 14.13. HRMS (MALDI-TOF): calcd for C64H98N2O6S4, 1118.6313; found, 1118.6308. Elemental analysis: calcd for C64H98N2O6S4: C 68.65, H 8.82, N 2.50; found: C 68.73, H 8.84, N 2.50.
Synthesis of NDITTF2
Under a nitrogen atmosphere, to a mixture of 4, 5-bis(methylthio)-1,3-dithiole-2-thione (204 mg, 0.9 mmol) and NDI-1 (100 mg, 0.09 mmol) in a Schlenk tube, triethylphosphite (10 mL) was added. Then the mixture was stirred at 140 °C for 4 h. After being cooled to room temperature, the resulting mixture was poured into methanol (30 mL) and filtered. The residue was purified by silica gel chromatography with gradient elution: firstly, the mixture of petroleum ether (60–90 °C) and CH2Cl2 (4:1, v/v) was used to remove the impurities, then CHCl3 was used to wash the product out of the silica gel column. Finally, NDITTF2 (0.091 g) was obtained in 68.2% yield. 1H NMR (500 MHz, CDCl2) δ 4.18 (d, J = 6.9 Hz, 4H), 2.52 (s, 12H), 2.12 (br, 2H), 1.61–1.01 (m, 80H), 0.89–0.87 (br, 12H).13C NMR (125 MHz, o-dichlorobenzene-d4) δ 162.39, 147.88, 128.09, 125.71, 116.81, 115.57, 114.02, 46.18, 37.15, 32.39, 32.01, 30.48, 29.88, 29.43, 26.90, 22.70, 18.71, 13.92. HRMS (MALDI-TOF): calcd for C74H110N2O4S12, 1474.5109; found, 1474.5101. Elemental analysis: calcd for C74H110N2O4S12: C 60.20, H 7.51, N 1.90; found: C 60.16, H 7.53, N 1.92.
Synthesis of NDI2TTF
Under a nitrogen atmosphere, a toluene solution of NDI-1 (800 mg, 0.7 mmol) and triethylphosphite (1.0 mL) was stirred at 100 °C for one hour. After being cooled to room temperature, the resulting mixture was poured into methanol (30 mL) and filtered, and the residue was purified by silica gel chromatography with petroleum ether (60–90 °C) and CH2Cl2 (4:1, v/v) to give NDI2TTF (0.35 g, 45.3%). 1H NMR (500 MHz, CDCl2) δ 4.30 (d, J = 6.5 Hz, 8H), 2.18 (br, 4H), 1.94–1.01 (m, 160H), 0.89–0.87 (br, 24H); 13C NMR (126 MHz, toluene-d8) δ 193.38, 162.11, 162.02, 148.15, 140.95, 124.85, 122.99, 117.27, 115.14, 46.67, 36.93, 32.35, 31.87, 30.43, 29.84, 29.70, 29.65, 29.33, 29.27, 26.91, 22.50, 13.54; HRMS (MALDI-TOF): calcd for C128H196N4O10S8, 2205.2723; found, 2205.2747. Elemental analysis for C128H196N4O10S8: C 69.65, H 8.95, N 2.54; found: C 69.62, H 8.96, N 2.39.
Synthesis of NDI2TTF3
Under a nitrogen atmosphere, a toluene solution of NDI2TTF (100 mg, 0.046 mmol), 4,5-bis(hexylthio)-1,3-dithiole-2-thione (366 mg, 1 mmol) and triethylphosphite (1.0 mL) was stirred at 120 °C for 12 hours. After being cooled to room temperature, the resulting mixture was poured into methanol (30 mL) and filtered. The residue was purified by silica gel chromatography with gradient elution: firstly, the mixture of petroleum ether (60–90 °C) and CH2Cl2 (4:1, v/v) and CH2Cl2 were used to remove the impurities sequentially; then CHCl3 was used to wash the product out of the silica gel column. Finally, NDI2TTF3 (0.025 g) was obtained in 19.1% yield. 1H NMR (500 MHz, C6D4Cl2) δ 4.54 (br, 8H), 3.08 (br, 8H), 2.60 (br, 4H), 2.28–1.13 (m, 192H), 1.03 (br, 36H). 13C NMR (100 MHz, solid) δ 161.44, 146.61, 124.91, 113.64, 46.54, 36.20, 30.65, 23.53, 14.77. HRMS (MALDI-TOF): calcd for C158H248N4O8S16, 2841.4648; found, 2841.4667. Elemental analysis: calcd for C142H216N4O10S16: C 66.71; H 8.79, N 1.97, found: C 67.01, H 8.76, N 1.91.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Ministry of Science and Technology of China (2018YFA0703200), the National Natural Science Foundation of China (22090021, 21871271, 21833005 and 22021002), and Chinese Academy of Sciences (GJTD-2020-02) is gratefully acknowledged. This work was also supported by the CAS-Croucher Funding Scheme for Joint Laboratories.Notes and references
- Z. Wang, Near-Infrared Organic Materials and Emerging Application, CRC Press,New York, 2013 Search PubMed.
- (a) J. Fabian, H. Nakazumi and M. Matsuoka, Chem. Rev., 1992, 92, 1197 CrossRef CAS; (b) J. Qi, W. Qiao and Z. Wang, Chem. Rec., 2016, 16, 1531 CrossRef CAS PubMed; (c) G. Qian and Z. Y. Wang, Chem. – Asian J., 2010, 5, 1006 CrossRef CAS PubMed.
- (a) C. Liu, K. Wang, X. Gong and A. J. Heeger, Chem. Soc. Rev., 2016, 45, 4825 RSC; (b) Z. Hu, J. Wang, X. Ma, J. Gao, C. Xu, K. Yang, Z. Wang, J. Zhang and F. Zhang, Nano Energy, 2020, 78, 105376 CrossRef CAS; (c) Z. Zhang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 4422 CrossRef CAS PubMed.
- (a) D. Lim, J. Ha, H. Choi, S. C. Yoon, B. R. Lee and S. Ko, Nanoscale Adv., 2021, 3, 4306 RSC; (b) B. Xie, Z. Chen, L. Ying, F. Huang and Y. Cao, InfoMat, 2020, 2, 57–91 CrossRef CAS.
- (a) S. Dai, T. Li, W. Wang, Y. Xiao, T. Lau, Z. Li, K. Liu and X. Zhan, Adv. Mater., 2018, 30, 1706571 CrossRef PubMed; (b) Y. Sun, Y. Zhang, Y. Ran, L. Shi, Q. Zhang, J. Chen, Q. Li, Y. Guo and Y. Liu, J. Mater. Chem. C, 2020, 8, 15168 RSC; (c) Z. Xie, D. Liu, Y. Zhang, Q. Liu, H. Dong and W. Hu, Chem. J. Chin. Univ., 2020, 41, 1179 CAS.
- (a) Z. Wu, Y. Zhai, H. Kim, J. D. Azoulay and T. N. Ng, Acc. Chem. Res., 2018, 51, 3144 CrossRef CAS PubMed; (b) X. Liu, Y. Lin, Y. Liao, J. Wu and Y. Zheng, J. Mater. Chem. C, 2018, 6, 3499 RSC; (c) N. Li, Z. Lan, L. Cai and F. Zhu, J. Mater. Chem. C, 2019, 7, 3711 RSC.
- X. Gong, M. Tong, Y. Xia, W. Cai, J. Moon, Y. Cao, G. Yu, C. Shieh, B. Nilsson and A. Heeger, Science, 2009, 325, 1665 CrossRef CAS PubMed.
- (a) J. Kim, A. Liess, M. Stolte, A. Krause, V. Stepanenko, C. Zhong, D. Bialas, F. Spano and F. Würthner, Adv. Mater., 2021, 33, 2100582 CrossRef CAS PubMed; (b) J. Huang, J. Lee, J. Vollbrecht, V. V. Brus, A. L. Dixon, D. Cao, Z. Zhu, Z. Du, H. Wang, K. Cho, G. C. Bazan and T. Nguyen, Adv. Mater., 2020, 32, 1906027 CrossRef CAS PubMed; (c) S. Anantharaman, K. Strassel, M. Diethelm, A. Gubicza, E. Hack, R. Hany, F. Nüesch and J. Heier, J. Mater. Chem., 2019, 7, 14639 CAS.
- (a) S. He, J. Song, J. Qu and Z. Cheng, Chem. Soc. Rev., 2018, 47, 4258 RSC; (b) Z. Guo, S. Park, J. Yoon and I. Shin, Chem. Soc. Rev., 2014, 43, 16 RSC; (c) C. Yan, Y. Zhang and Z. Guo, Coord. Chem. Rev., 2021, 427, 213556 CrossRef CAS; (d) J. Li and K. Pu, Chem. Soc. Rev., 2019, 48, 38 RSC; (e) H. Kobayashi and P. L. Choyke, Acc. Chem. Res., 2019, 52, 2332 CrossRef CAS PubMed.
- (a) Y. Su, B. Yu, S. Wang, H. Cong and Y. Shen, Biomaterials, 2021, 271, 120717 CrossRef CAS PubMed; (b) P. Liu, X. Mu, X. Zhang and D. Ming, Bioconjugate Chem., 2020, 31, 260 CrossRef CAS PubMed; (c) P. Reineck and B. C. Gibson, Adv. Opt. Mater., 2017, 5, 1600446 CrossRef.
- (a) K. Pu, J. Mei, J. Jokerst, G. Hong, A. Antaris, N. Chattopadhyay, A. Shuhendler, T. Kurosawa, Y. Zhou, S. Gambhir, Z. Bao and J. Rao, Adv. Mater., 2015, 27, 5184 CrossRef CAS PubMed; (b) Q. Zhao and J. Sun, J. Mater. Chem. C, 2016, 4, 10588 RSC; (c) X. Liu, Z. Yang, W. Xu, Y. Chu, J. Yang, Y. Yan, Y. Hu, Y. Wang and J. Hua, J. Mater. Chem. C, 2019, 7, 12509 RSC.
- (a) H. Zhu, Y. Fang, Q. Miao, X. Qi, D. Ding, P. Chen and K. Pu, ACS Nano, 2017, 11, 8998 CrossRef CAS PubMed; (b) W. Wu, D. Mao, F. Hu, S. Xu, C. Chen, C. Zhang, X. Cheng, Y. Yuan, D. Ding, D. Kong and B. Liu, Adv. Mater., 2017, 29, 1700548 CrossRef PubMed; (c) L. Li, C. Shao, T. Liu, Z. Chao, H. Chen, F. Xiao, H. He, Z. Wei, Y. Zhu, H. Wang, X. Zhang, Y. Wen, B. Yang, F. He and L. Tian, Adv. Mater., 2020, 32, 2003471 CrossRef CAS PubMed.
- (a) J. Yamada and T. Sugimoto, TTF Chemistry: Fundamentals & applications of Tetrathiafulvalene, Springer Verlag, Berlin, 2004 Search PubMed; (b) M. Bendikov, F. Wudl and D. Perepichka, Chem. Rev., 2004, 104, 4891 CrossRef CAS PubMed; (c) T. Mori, Chem. Rev., 2004, 104, 4947 CrossRef CAS PubMed.
- (a) F. Wudl, D. Wobschall and E. J. Hufnagel, J. Am. Chem. Soc., 1972, 94, 670 CrossRef CAS; (b) L. B. Coleman, M. J. Cohen, D. J. Sandman, F. G. Yamagishi, A. F. Garito and A. J. Heeger, Solid State Commun., 1973, 12, 1125 CrossRef CAS.
- (a) M. Bryce, J. Mater. Chem., 2000, 10, 589 RSC; (b) N. Martín and J. Segura, Angew. Chem., Int. Ed., 2001, 40, 1372 CrossRef; (c) M. Nielsen, C. Lomholt and J. Becher, Chem. Soc. Rev., 2000, 29, 153 RSC; (d) T. Jørgensen, T. Hansen and J. Becher, Chem. Soc. Rev., 1994, 23, 41–51 RSC.
- (a) D. Canevet, M. Sallé, G. Zhang, D. Zhang and D. Zhu, Chem. Commun., 2009, 2245 RSC; (b) A. Jana, S. Bähring, M. Ishida, S. Goeb, D. Canevet, M. Sallé, J. Jeppesen and J. Sessler, Chem. Soc. Rev., 2018, 47, 5614 RSC; (c) A. Jana, M. Ishida, J. Park, S. Bahring, J. Jeppesen and J. Sessler, Chem. Rev., 2017, 117, 2641 CrossRef CAS PubMed.
- C. Rovira, Chem. Rev., 2004, 104, 5289 CrossRef CAS PubMed.
- N. Duvva, U. Chilakamarthi and L. Giribabu, Sustainable Energy Fuels, 2017, 1, 678 RSC.
- (a) J. Spruell, A. Coskun, D. Friedman, R. Forgan, A. Sarjeant, A. Trabolsi, A. Fahrenbach, G. Barin, W. Paxton, S. Dey, M. Olson, D. Benítez, E. Tkatchouk, M. Colvin, R. Carmielli, S. Caldwell, G. Rosair, S. Hewage, F. Duclairoir, J. Seymour, A. Slawin, W. Goddard III, M. Wasielewski, G. Cooke and J. F. Stoddart, Nat. Chem., 2010, 2, 870 CrossRef CAS PubMed; (b) J. Xiao, Z. Yin, H. Li, Q. Zhang, F. Boey, H. Zhang and Q. Zhang, J. Am. Chem. Soc., 2010, 132, 6926 CrossRef CAS PubMed.
- (a) X. Li, G. Zhang, H. Ma, D. Zhang, J. Li and D. Zhu, J. Am. Chem. Soc., 2004, 126, 11543 CrossRef CAS PubMed; (b) G. Zhang, D. Zhang, X. Guo and D. Zhu, Org. Lett., 2004, 6, 1209 CrossRef CAS PubMed; (c) H. Wu, D. Zhang, L. Su, K. Ohkubo, C. Zhang, S. Yin, L. Mao, Z. Shuai, S. Fukuzumi and D. Zhu, J. Am. Chem. Soc., 2007, 129, 6839 CrossRef CAS PubMed.
- (a) S. Bivaud, S. Goeb, V. Croué, P. I. Dron, M. Allain and M. Sallé, J. Am. Chem. Soc., 2013, 135, 10018 CrossRef CAS PubMed; (b) S. Bivaud, J. Y. Balandier, M. Chas, M. Allain, S. Goeb and M. Sallé, J. Am. Chem. Soc., 2012, 134, 11968 CrossRef CAS PubMed; (c) S. Krykun, M. Dekhtiarenko, D. Canevet, V. Carré, F. Aubriet, E. Levillain, M. Allain, Z. Voitenko, M. Sallé and S. Goeb, Angew. Chem., Int. Ed., 2020, 59, 716 CrossRef CAS PubMed.
- (a) C. Wang, D. Zhang and D. Zhu, J. Am. Chem. Soc., 2005, 127, 16372 CrossRef CAS PubMed; (b) T. Kitamura, S. Nakaso, N. Mizoshita, Y. Tochigi, T. Shimomura, M. Moriyama, K. Ito and T. Kato, J. Am. Chem. Soc., 2005, 127, 14769 CrossRef CAS PubMed; (c) T. Kitahara, M. Shirakawa, S. I. Kawano, U. Beginn, N. Fujita and S. Shinkai, J. Am. Chem. Soc., 2005, 127, 14980 CrossRef CAS PubMed; (d) C. Wang, Q. Chen, F. Sun, D. Zhang, G. Zhang, Y. Huang, R. Zhao and D. Zhu, J. Am. Chem. Soc., 2010, 132, 3092 CrossRef CAS PubMed.
- J. Bergkamp, S. Decurtins and S. Liu, Chem. Soc. Rev., 2015, 44, 863 RSC.
- (a) X. Gao, Y. Wang, X. Yang, Y. Liu, W. Qiu, W. Wu, H. Zhang, T. Qi, Y. Liu, K. Lu, C. Du, Z. Shuai, G. Yu and D. Zhu, Adv. Mater., 2007, 19, 3037 CrossRef CAS; (b) Y. Hu, Z. Wang, X. Zhang, X. Yang, C. Ge, L. Fu and X. Gao, Org. Lett., 2017, 19, 468 CrossRef CAS PubMed.
- (a) G. Yang, C. Di, G. Zhang, J. Zhang, J. Xiang, D. Zhang and D. Zhu, Adv. Funct. Mater., 2013, 23, 1671 CrossRef CAS; (b) L. Tan, Y. Guo, Y. Yang, G. Zhang, D. Zhang, G. Yu, W. Xu and Y. Liu, Chem. Sci., 2012, 3, 2530 RSC.
- (a) H. Yamada, M. Yamashita, H. Hayashi, M. Suzuki and N. Aratani, Chem. – Eur. J., 2018, 24, 18601 CrossRef CAS PubMed; (b) N. Tucker, A. Briseno, O. Acton, H. Yip, H. Ma, S. Jenekhe, Y. Xia and A. Jen, ACS Appl. Mater. Interfaces, 2013, 5, 2320 CrossRef CAS PubMed; (c) M. Yamashita, K. Kawano, A. Matsumoto, N. Aratani, H. Hayashi, M. Suzuki, L. Zhang, A. Briseno and H. Yamada, Chem. – Eur. J., 2017, 23, 15002 CrossRef CAS PubMed.
- (a) C. Jia, S. Liu, C. Tanner, C. Leiggener, A. Neels, L. Sanguinet, E. Levillain, S. Leutwyler, A. Hauser and S. Decurtins, Chem. – Eur. J., 2007, 13, 3804 CrossRef CAS PubMed; (b) Y. Geng, R. Pfattner, A. Campos, J. Hauser, V. Laukhin, J. Puigdollers, J. Veciana, M. Mas-Torrent, C. Rovira, S. Decurtins and S. Liu, Chem. – Eur. J., 2014, 20, 7136 CrossRef CAS PubMed; (c) H. Jia, J. Ding, Y. Ran, S. Liu, C. Blum, I. Petkova, A. Hauser and S. Decurtins, Chem. – Asian J., 2011, 6, 3312 CrossRef CAS PubMed.
- (a) X. Lu, J. Sun, Y. Liu, J. Shao, L. Ma, S. Zhang, J. Zhao, Y. Shao, H. Zhang, Z. Wang and X. Shao, Chem. – Eur. J., 2014, 20, 9650 CrossRef CAS PubMed; (b) L. Liu, C. Yan, Y. Li, Z. Liu, C. Yuan, H. Zhang and X. Shao, Chem. – Eur. J., 2019, 26, 7083 CrossRef PubMed; (c) C. Jia, J. Zhang, J. Bai, L. Zhang, Z. Wan and X. Yao, Dyes Pigm., 2012, 94, 403 CrossRef CAS.
- S. Keshri, D. Asthana, S. Chorol, Y. Kumar and P. Mukhopadhyay, Chem. – Eur. J., 2018, 24, 1821 CrossRef CAS PubMed.
- Y. Niu, Q. Peng, C. Deng, X. Gao and Z. Shuai, J. Phys. Chem. A, 2010, 114, 7817 CrossRef CAS PubMed.
- Electron transport for the OFET devices with thin films of NDITTF2 was not observed even under a N2 atmosphere or vacuum. This may be due to the fact that the LUMO of NDITTF2 is almost concentrated on the NDI moiety, while the HOMO is delocalized over TTF and the naphthalene ring of NDI. Thus, the conduction channel for electron transport cannot be formed efficiently.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc04291b |
| This journal is © The Royal Society of Chemistry 2022 |