
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Getting drugs to the brain: advances and prospects of organic nanoparticle delivery systems for assisting drugs to cross the blood–brain barrier
Qiuxia
Tan†
a,
Shaojing
Zhao†
a,
Ting
Xu
a,
Qin
Wang
a,
Minhuan
Lan
 *a,
Li
Yan
*b and
Xianfeng
Chen
*c
*a,
Li
Yan
*b and
Xianfeng
Chen
*c
aHunan Provincial Key Laboratory of Micro & Nano Materials Interface Science, College of Chemistry and Chemical Engineering, Central South University, Changsha, 410083, China. E-mail: minhuanlan@csu.edu.cn
bCollege of Health Science and Environmental Engineering, Shenzhen Technology University, Shenzhen, 518118, China. E-mail: yanli@sztu.edu.cn
cSchool of Engineering, Institute for Bioengineering, University of Edinburgh, The King's Buildings, Edinburgh EH9 3JL, UK. E-mail: Michael.Chen@ed.ac.uk
First published on 10th October 2022
Abstract
The blood–brain barrier (BBB) plays an irreplaceable role in protecting the central nervous system (CNS) from bloodborne pathogens. However, the BBB complicates the treatment of CNS diseases because it prevents almost all therapeutic drugs from getting into the CNS. With the growing understanding of the physiological characteristics of the BBB and the development of nanotechnology, nanomaterial-based drug delivery systems have become promising tools for delivering drugs across the BBB to the CNS. Herein, we systematically summarize the recent progress in organic-nanoparticle delivery systems for treating CNS diseases and evaluate their mechanisms in overcoming the BBB with the aim to provide a comprehensive understanding of the advantages, disadvantages, and challenges of organic nanoparticles in delivering drugs across the BBB. This review may inspire new research ideas and directions for applying nanotechnology to treat CNS diseases.
1. Introduction
Central nervous system (CNS) diseases, such as brain tumors, Alzheimer's disease (AD), Parkinson's disease, and Huntington's disease, pose a tremendous threat to human health.1,2 However, it is notoriously difficult to treat CNS diseases due to the presence of the blood–brain barrier (BBB) between the blood plasma and brain cells. The BBB is mainly composed of brain microvascular endothelial cells (BMECs), astrocytes, neuron end-feet, pericytes, resident microglia, and tight junctions;3,4 among which, BMECs are attached closely to one another through tight connection to form the physical barrier of the brain. Astrocytes are responsible for maintaining the homeostasis,5 and pericytes, which are partly wrapped by BMECs, are responsible for the permeability of the BBB.6 These structural properties endow the BBB with selective permeability, allowing the passage of nutrients required by the brain tissue, but blocking that of harmful substances. Although the BBB plays a vital role in maintaining the physiological state of the CNS, it prevents many therapeutic drugs from entering the brain, thereby hindering the treatment of CNS diseases.7,8 Usually, essential molecules and therapeutic agents cross the BBB via various mechanisms such as passive diffusion, receptor-mediated transport (RMT), carrier-mediated transport (CMT), adsorption-mediated transport (AMT) and cell-mediated transport (CET). Currently, the following method approaches are commonly used to increase the efficacy of drug delivery across the BBB; however, each approach has some disadvantages:(1) Improving the lipid solubility of drugs. Changing the physicochemical properties of drugs may reduce their pharmacological activity. In addition, the presence of P-glycoprotein (P-gp) in the cytomembrane can prevent these highly lipid-soluble drugs from entering the brain.
(2) Using P-gp inhibitors for highly lipid-soluble drugs. This approach can avoid the efflux of P-gp and promote the drug delivery into the brain;9 however, inhibiting P-gp may also allow some harmful lipid-soluble compounds to enter the brain.
(3) Loosening of the tight junctions to open the BBB. In general, hypertonic solutions,10 vasoactive substances, and focused ultrasound11 can temporarily and reversibly open the BBB; however, this approach is non-specific and may damage the endothelial cells and tissues of the brain. Moreover, the non-selective opening of the BBB may also allow harmful substances to enter and damage the brain.
(4) Bypassing the BBB. Some invasive approaches (e.g., intrathecal administration) and non-invasive approaches (e.g., intranasal administration) are the commonly used BBB bypassing methods in treating CNS diseases. However, the intrathecal administration is highly invasive and causes patient discomfort,12 whereas the intranasal administration suffers from limited dose, low nasal mucosa membrane permeability, and mucociliary clearance.13
By contrast, advanced strategies used to cross the BBB could realize the brain-targeted administration of drugs with minimal system toxicity, and minimal physical and chemical damage to the BBB. To date, a wide variety of drug delivery systems, including inorganic- and organic-based nanocarriers, have been developed.14–16 However, inorganic-based nanocarriers, such as metal-, carbon-, and semiconductor-based nanomaterials, suffer from non-degradability and inherent cytotoxicity, which is the biggest limitation on the clinical transformation. Additionally, inorganic materials are more frequently used in imaging than carriers. In contrast, organic nanocarriers have the advantages of high biocompatibility, low toxicity, and easy modification and functionalization, thus are potential tools for delivering drugs across the BBB. In 2018, Furtado et al. systematically reviewed the anatomy, physiology, and pathology of the BBB, and summarized the approaches for bypassing or crossing the BBB. However, only a few sections in the article described the use of nanocarriers to cross the BBB.17 In 2019, Xie et al. reviewed different delivery approaches for crossing the BBB, including intranasal delivery, temporary disruption of the BBB, local delivery, and receptor- or peptide-mediated BBB-crossing strategies.18
This field develops rapidly, and many new strategies are reported every year. One attractive approach in reviewing the extensive progress of the field is to categorize the designed strategies and functions based on the nanomaterials used in the applied drug delivery systems. Therefore, in this review, we summarize the recent progress in the development of various types of organic nanocarriers used to overcome the BBB, including lipid-based NPs (i.e., liposomes, solid lipid NPs, and micelles), polymer-based NPs (i.e., amphiphilic copolymers and dendrimers) and biomolecule-based NPs (i.e., protein-, virus-, and nuclear-based NPs) (Scheme 1). The controllable preparation, functional surface modifications, drug loading and release kinetics, working mechanism, and therapeutic effects of these nanocarriers are described in detail. We also compare and discuss the advantages and disadvantages of these organic NPs, and highlight the significance of their physicochemical properties and biological functions (especially surface modifications) in contributing to the BBB-crossing. Lastly, we present our insights into the opportunities and challenges of NP-delivery systems in crossing the BBB for effective treatment of CNS diseases.
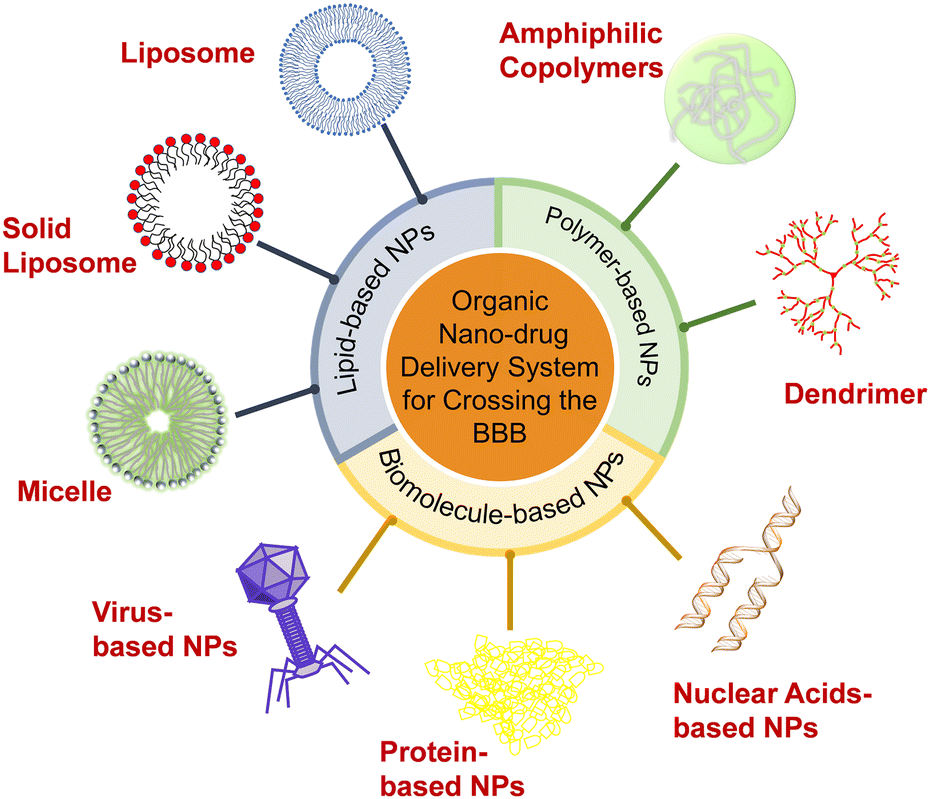 | ||
| Scheme 1 Organic-nanoparticle (NP) delivery systems for crossing the blood–brain barrier. | ||
2. Organic-NP delivery systems
Organic NPs, including lipid-, polymer-, and biomolecule-based NPs which can penetrate the BBB and deliver drugs to the CNS have been developed. After surface modification to improve surface charge, lipophilicity, biocompatibility, and brain targeting ability, these organic NPs can avoid phagocytosis by the reticuloendothelial system (RES) and significantly increase the concentration of drugs in the brain. For example, decorating with polyethylene glycol (PEG) can prolong the retention time of liposomes in blood. Targeting ligands allow NPs to get through the BBB with different transport mechanisms and realize active targeting to promote the carriers to further accumulate in the brain. Furthermore, modification of surfactants can alter the surface charge and improve the BBB-crossing by the enhanced electrostatic interaction.2.1. Lipid-based NPs
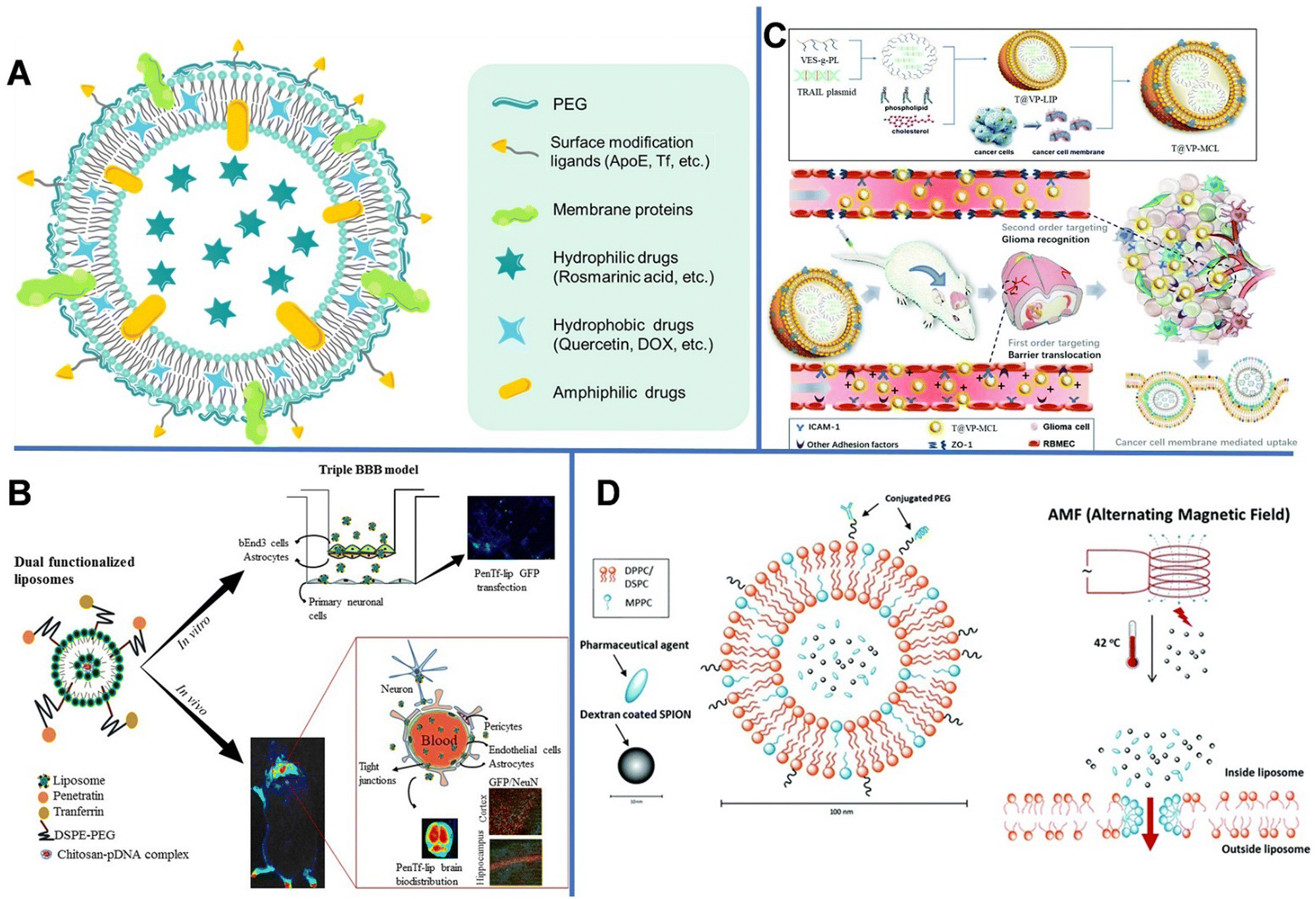 | ||
| Fig. 1 (A) Schematic diagram of the drug-loading liposome. (B) Gene delivery mediated by bifunctional liposomes. Reprinted with permission from ref. 29. Copyright 2018, Elsevier. (C) Tumor cell-membrane modified liposomes were used to specifically target homologous glioma and cross the BBB. Reprinted with permission from ref. 31. Copyright 2020, Royal Society of Chemistry. (D) Magnetic-field mediated drug release from micelles. Reprinted with permission from ref. 35. Copyright 2019, Royal Society of Chemistry. | ||
However, conventional liposomes can be easily removed by the RES, leading to rapid systematic clearance. Liposomes modified with hydrophilic PEG can avoid being captured by the RES, thereby increasing the drug uptake in the brain.22 Xie et al. modified liposomes with glucose (Glu) and PEG with various chain lengths (PEG200, PEG400, PEG1000, and PEG2000) and demonstrated that the PEG length has a great impact on the brain-targeting efficiency of the liposomes,23 and PEG1000-modified liposomes exhibited the most outstanding brain-targeting ability. This is likely because short PEG chains can block the exposure of Glu, which helped the transport of liposomes into the brain by RMT through the multivalent recognition between glucose transporter-1 (GLUT1) in the brain capillary endothelial cells and Glu,24 whereas long PEG chains provided steric hindrance. Zhang et al. prepared a PEGylated liposome modified with a cell penetration peptide (CB5005) containing DOX.25 The prepared liposomes had regular sphere shapes with a diameter of approximately 110 nm. The dual functions of CB5005, namely cell membrane penetration function and nuclear factor-κB (NF-κB) inhibition functions, could effectively penetrate gliomas and inhibit the activation of NF-κB. The modification with CB5005 not only significantly increased the uptake of liposomes by glioma cells, but also greatly improved the killing effect of DOX on U87 tumor cells.
Conjugating liposomes with brain-specific ligands or antibodies can improve the targetability and promote the nanodrugs getting into the brain by RMT. For example, apolipoprotein E (ApoE) can bind to receptors (low-density lipoprotein receptors (LDLRs)) and LDLR-related proteins (LRPs) on the BBB. Transferrin (Tf) is a glycoprotein that can transport iron ions into cells.26 Cheng et al. developed Tf-modified and PEGylated liposomes to encapsulate chemotherapeutic osthole (Ost) (Tf-Ost-Lip) to treat AD.27 Muzykantov et al. constructed vascular cell adhesion molecule 1 (anti-VCAM)-conjugated liposomes, which exhibited improved brain accumulation compared to TfR or intercellular adhesion molecule 1 (ICAM-1)-conjugated liposomes. The uptake efficiency of intravenously injected anti-VCAM-liposomes in an inflamed brain was 27-fold and 8-fold greater than that of TfR-1- and ICAM-1-liposomes, respectively, achieving a brain/blood ratio >300-fold higher than that of immunoglobulin G/liposomes.28
However, RMT is a transport process that can usually become saturated. Dual targeting is one of the fascinating strategies for overcoming the receptor saturation and increasing the delivery efficiency. In 2018, Rodrigues et al. designed liposomes (penetratin [Pen]-Tf liposomes) with dual targeting functions to achieve neuronal transfection, in which Tf could cross the BBB, while the cationic cell-penetrating peptide (Pen) could overcome the receptor saturation and promote the intracellular delivery and endosomal escape.29 The obtained liposomes were spherical with uniform particle sizes of 147–167 nm. Subsequently, the hydrophilic plasmid formed by complexing galactosidase with chitosan was encapsulated in liposomes, in which chitosan could keep DNA from enzymatic degradation and improve its transfection efficiency (Fig. 1B). The encapsulation efficiency of plasmid DNA (pDNA) encoding green fluorescent protein in the liposomes reached up to 93%. In vitro experiments revealed that after 8 h, 15.2% of Pen-Tf liposomes could cross the BBB, which is higher than that of Pen (2.6%) and Tf (3.5%) liposomes.
In the following years, the same research group used liposomes modified with dual targeting molecules (Tf and Pen) to deliver DOX and erlotinib to the brain. Results showed that the accumulation of DOX and erlotinib in the mouse brain increased by 12 fold and 3.3 fold, respectively, by using Tf-Pen liposomes. In addition, mice had a longer survival time and smaller glioblastoma in the brain compared with the control group, demonstrating the excellent anti-tumor effects of Tf-Pen liposomes, as well as its potential in the treatment of aggressive gliomas.30
Unlike the complex ligand modification process, liposomes provide an easy route to use cellular membrane proteins to camouflage the surface of nanocarriers. As shown in Fig. 1C, vitamin E succinate-grafted ε-polylysine (VES-g-PL) polymers could assemble into cationic micelles which were used to condense tumor necrosis factor-related apoptosis-inducing ligand plasmids. The VES-g-PL micelles and the glioma cell membrane were encapsulated in the inner compartment of the liposomes (egg lecithin and cholesterol) and the lipid layer, respectively. This led to the formation of T@VP-MCL with spherical morphology (∼130 nm) and a zeta potential of 14.9 mV.31 The electrostatic adsorption between the liposomes and endothelial cells increases the penetration efficiency of NPs. In addition, the liposomes can target ICAM-1 in gliomas to regulate the expression of the tight junction protein ZO-1. As can be seen in the magnetic resonance images, the tumor volume of rats in the control groups gradually increased over time, whereas that of rats in the experimental group showed relatively slower tumor growth.
A combination of low-intensity pulsed focused ultrasound (LIFU) with systemically administered microbubbles could generate mechanical stress to the brain endothelium to reversibly open the BBB. It has been reported that image-guided LIFU has been used to deliver stable liposomal O6-(4-bromothenyl) guanine (O6BTG) derivative. Liposomes can be used as pseudo-substrates to target O6-methylguanine-DNA methyltransferase (MGMT), an enzyme capable of repairing temozolomide-induced toxic DNA, thus leading to MGMT depletion to overcome MGMT-mediated chemotherapeutic drug temozolomide resistance. Compared with that in the untreated area of the contralateral hemisphere, the accumulation level of dye-labeled liposomes in the LIFU-treated area was significantly higher.32 In addition, Lin et al. prepared a gene delivery system, in which gene-carrying liposomes were combined with microbubbles and ultrasound was used to open the BBB to deliver glia-cell-line-derived neurotrophic factor and brain-derived neurotrophic factor (therapeutic genes commonly tested in Parkinson's disease gene therapy) to the brain.33 The results indicated that the ultrasound helped to open the BBB and increase the penetration of drugs and genes into the brain tissue for the treatment of CNS diseases.
Other than enhanced drug delivery, temperature-sensitive liposomes (TSL) can be used in controlled drug release under thermal stimulation, owing to their phase-change ability at a high temperature. When the temperature reaches the phase-transition temperature (usually >40 °C), liposomes collapse and release encapsulated drugs. The TSLs utilize the intravascular triggered wrapped drug release, in which the drug is released while liposomes are transiting through heated tissue. Some TSL formulations can release drugs within seconds. The higher the temperature or the longer the heating time, the more rapid the drug release. Since the heat stimulation is also helpful to open the BBB, TSL can cross the temporarily disrupted area of the BBB and reach the brain tissue.34 Shi et al. developed a dual-function temperature-sensitive liposome system to encapsulate drugs.35 Therein, glioblastoma multiforme (GBM)-specific cell-penetrating peptide and anti-GBM antibody were coupled to the outer layer of liposomes for targeted delivery. Then, superparamagnetic iron oxide NPs (SPIONs) and DOX were loaded into the liposomes. The alternating magnetic field could induce the SPIONs to generate heat, which subsequently triggered the release of DOX (Fig. 1D). Compared with non-functionalized liposomes, the dual-function liposomes could better enter U-87 human GBM cells and inhibit the proliferation of tumor cells; nonetheless, they had no major effect on the function of healthy brain cells.
The above examples demonstrate that encapsulating drugs into liposomes can prolong the half-life in the blood circulation, reduce side effects, and enhance the therapeutic effects on CNS diseases. However, physical, chemical, and biological factors, such as temperature, phospholipid materials, solvent properties, preparation methods, and stirring speed during the preparation, storage, and application processes can also affect the liposome structure. Thus, these factors directly affect the drug-carrying stability and biological functions of liposomes and need to be further studied before their clinical applications.
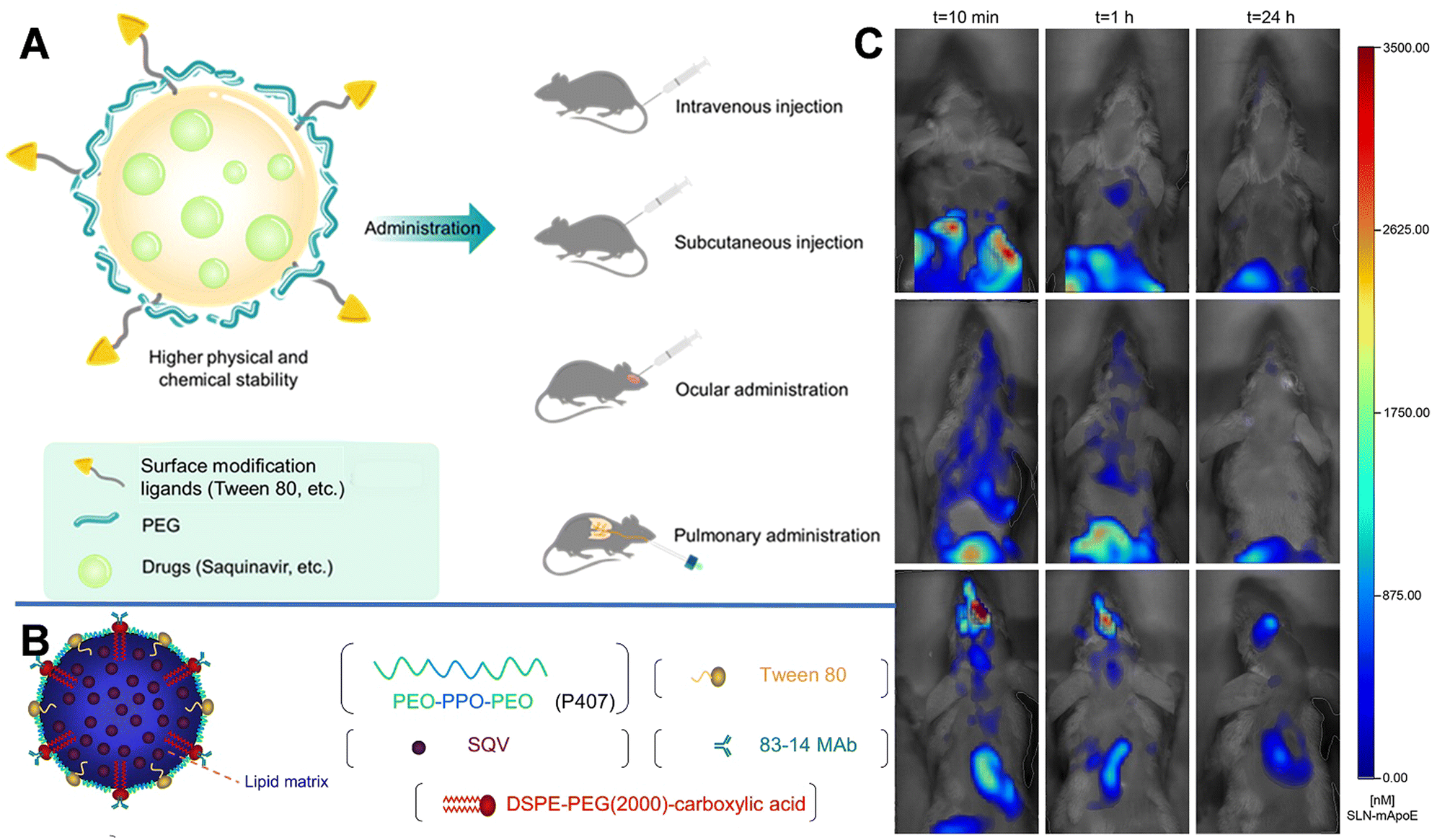 | ||
| Fig. 2 (A) Schematic diagram of SLN and the multiple administrations. (B) Schematic structure of an 83–14 MAb/SQV-SLN. Reprinted with permission from ref. 39. Copyright 2013, Elsevier. (C) Biodistribution of DiR-loaded SLN-mApoE in mice. Reprinted with permission from ref. 42. Copyright 2017, Elsevier. | ||
Furthermore, using Compritol 888 ATO (a mixture of mono-, di-, and tri-glycerides of behenic acid) as the solid lipid and Brij 78 (polyoxyethylene 20 stearyl ether derived from stearic-acid molecules covalently conjugated to PEG 1000) as a surfactant, Graverini et al. prepared SLNs via an emulsification/evaporation/solidification method to encapsulate andrographolide (AG) with an efficiency of 92%.40 The prepared SLNs were spherical with a diameter of 300 nm. And Brij 78 in the SLNs could reduce opsonization, phagocytosis, and clearance by the liver and RES, while protecting NPs from interference of plasma, and prolonging their half-life in the blood circulation. Furthermore, the formulation was highly lipophilic and charged, thus could protect NPs from astrocyte and pericyte attacks and facilitate the permeation of drugs across the neurovascular junction. After incubating the hCMEC/D3 cell monolayer with NaF and 80 μM AG-loaded SLNs for 1 h, the permeability reached 26.8 ± 4.17 × 10−6 cm s−1, which was approximately 3-fold higher than that of free AG.
Decoration with bioactive molecules can also facilitate the BBB-crossing ability. For example, Bozkir et al. used ApoE modification to improve the BBB permeability of donepezil and rhodamine B-loaded liposomes (ApoE-DON-SLNs). After 2 h of incubation, the uptake of ApoE-DON-SLNs by endothelial cells increased by over 4 fold higher than that of non-targeting liposomes.41 SLNs with ApoE-derived peptide (SLN-mApoE) as a specific brain-targeting ligand were also prepared via warm micro-emulsification technology. Magro et al. investigated the effects of various administration methods (including intravenous, intraperitoneal, and pulmonary administrations) on the bioavailability of SLN-mApoE in the brain.42 The biodistribution of SLN-mApoE was subsequently evaluated at different times. As shown in Fig. 2C, after intraperitoneal injection, the abdominal cavity exhibited strong fluorescence originating from SLN-mApoE, whereas the brain was dark. The percentage of the fluorescence signals in the brain were 0.15% and 0.06% of the injected dose at 3 and 24 h after intravenous injection of SLN-mApoE, respectively. In contrast, the pulmonary administration of SLN-mApoE did not cause an acute inflammatory response in the lungs.
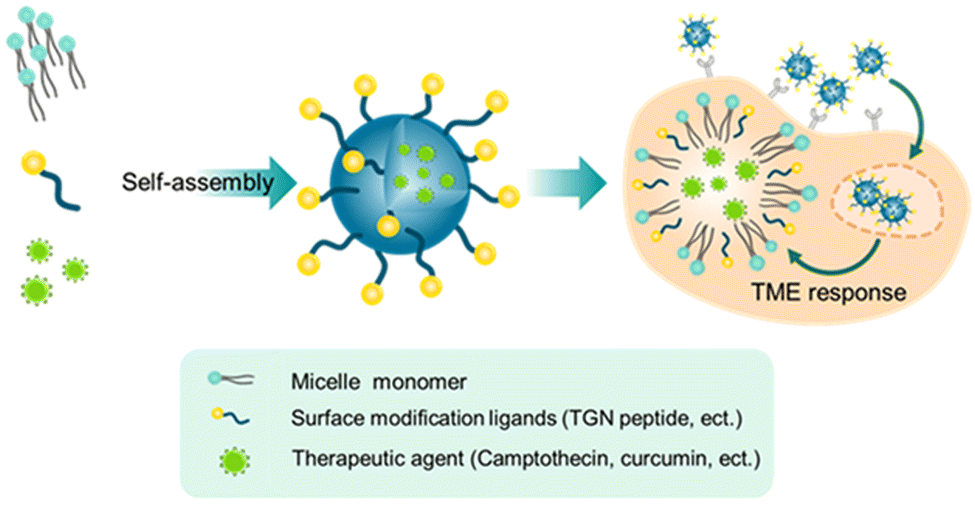 | ||
| Fig. 3 Schematic illustration of drug-loading micelles. | ||
Vasudevan et al. synthesized stearic acid (SA)-modified glycol chitosan (GC) (SAGC) that could spontaneously form spherical core–shell-structured micelles with a diameter of 138 nm in an aqueous solution (Fig. 4A).45 The SA component formed a hydrophobic core to help hold the drug (curcumin, Cur) with a loading efficiency of 76%. The GC component formed the surrounding hydrophilic corona and aids in water solubility. The micelles were further modified with a targeting peptide (TGN peptide-TGNYKALHPHNG) to enhance the BBB-crossing capability. The obtained micelles (TSAGC) could effectively encapsulate Cur (a hydrophobic drug) and deliver it to the brain. In vivo experiments showed that the accumulation of TSAGC in the brain is higher compared with that of SAGC at 1 h post-injection, confirming the TGN peptide-mediated delivery.
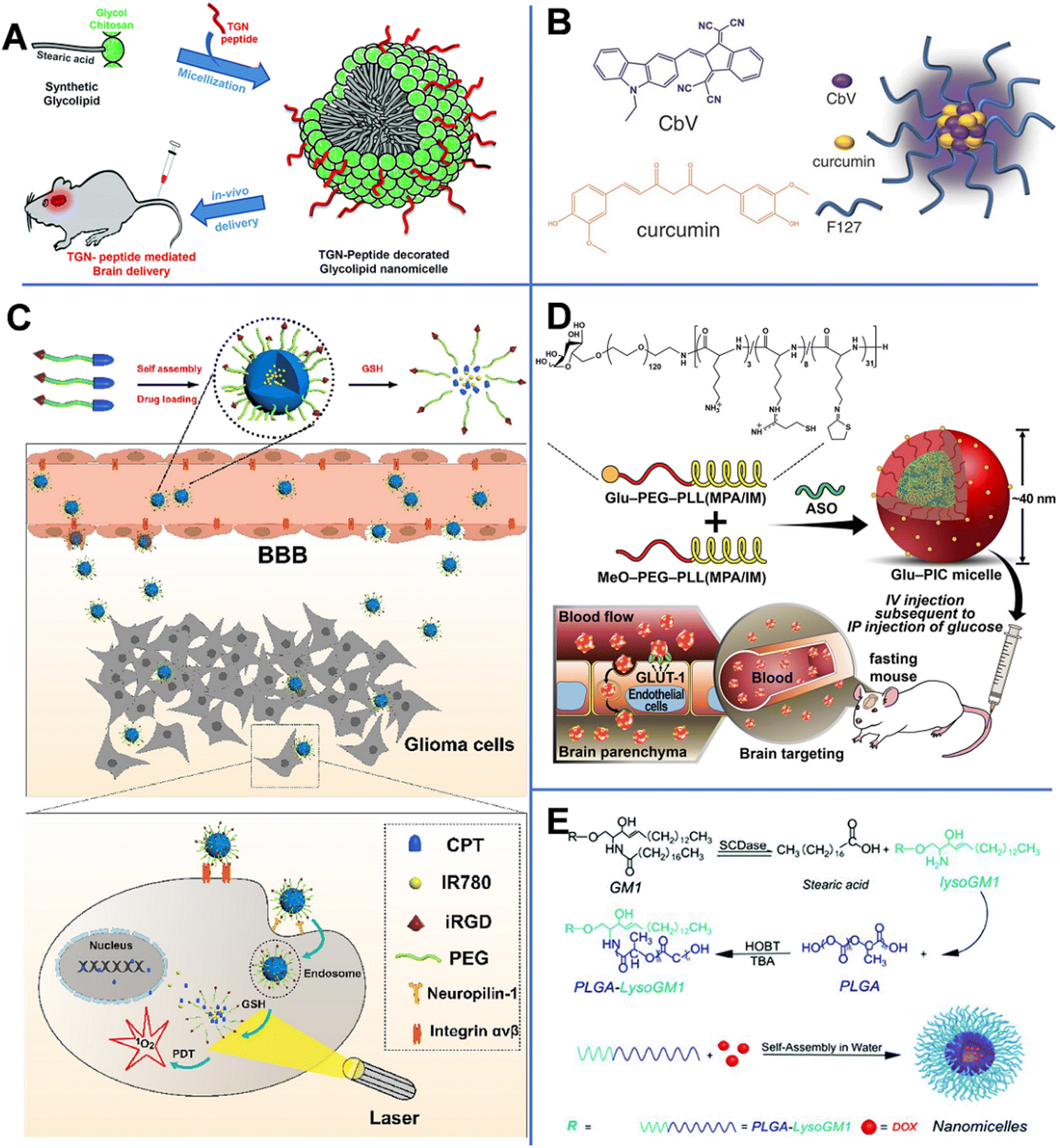 | ||
| Fig. 4 (A) TGN-peptide-modified nano micelles. Reprinted with permission from ref. 45. Copyright 2019, Royal Society of Chemistry. (B) CbV- and Cur-based micelles. Reprinted with permission from ref. 46. Copyright 2016, Wiley-VCH. Schematic illustrations of (C) iRGD-peptide-modified micelles. Reprinted with permission from ref. 47. Copyright 2020, Elsevier. (D) ASO-loaded and Glu-modified micelles. Reprinted with permission from ref. 39. Copyright 2020, Wiley-VCH. (E) PLGA-lysoGM1/DOX micelles. Reprinted with permission from ref. 51. Copyright 2015, Elsevier. | ||
Singh et al. prepared biocompatible multifunctional theranostic and photonic NPs (TPNs) that could cross the BBB and be tracked after systemic administration (Fig. 4B).46 In this work, the TPNs were 20 nm ultra-small nano-micelles (TPN-Cur/encapsulating photonic molecule [CbV]) with a spherical shape, assembled by pluronic (F-127, PEO–PPO–PEO), Cur, and CbV. F-127 could bind to the plasma membrane to enhance the endocytosis-mediated transendothelial migration by inhibiting the activity of P-gp or promoting the vesicular transport by the micellar structure. Besides, CbV exhibited strong solid-state fluorescence in the near-infrared region and could be used to enhance the optical signal of TPNs. In TPN-Cur/CbV micelles, the fluorescence of Cur was quenched by CbV through an energy transfer mechanism. However, after Cur was released from the micelles, its fluorescence was recovered. This mechanism could be used to monitor the in vivo release of Cur. In vivo imaging of temporal signals from TPNs revealed that the obtained TPN-Cur/CbV accumulated in the brain within 30 min, and the intensity reached the maximum at 2–3 h post-injection.
By conjugating camptothecin (CPT) to PEG through a disulfide bond, Lu et al. prepared a pro-drug and then modified it with the tumor-penetrating peptide (iRGD).47 As shown in Fig. 4C, CPT–S–S–PEG–COOH-based spherical micelles with a diameter of about 100 nm could encapsulate photosensitizer IR780. The iRGD peptide enhanced the ability of NPs to target neural GBM cells through the αvβ-integrin- and neuropilin-1-mediated ligand-transport mechanisms. In vivo experiments revealed that iRGD-micelles started to accumulate in the brain at 30 min post-injection. In GBM cells, a high concentration of glutathione (GSH) broke the S–S bond, causing the release of CPT and IR780, recovering their potential of chemical and photodynamic therapies.
Antisense oligonucleotide (ASO), a widely used therapeutic agent to treat CNS diseases, is an artificially synthesized nucleic-acid fragment designed to bind to noncoding and toxic RNAs associated with disease pathogenesis.48 Min et al. prepared ASO-loaded glucosylated-polyion complex micelles (Glu-PIC/M) with a sphere shape and a size of 40 nm, formed by self-assembly of ASO and a mixture of Glu-PEG-PLL (MPA/IM) and MeO-PEG-PLL (MPA/IM) (Fig. 4D).49 PEG-PLL (MPA/IM) is a block copolymer consisting of poly(ethylene glycol)-b-poly(L-lysine), 3-mercaptopropyl amidine, and 2-thiolaneimine. The density of Glu in NPs could be regulated by adjusting the ratio of the two polymers, and ASO could be effectively wrapped inside the core of the micelle. The concentration of ASO-loaded Glu-PIC/Ms in the brain was significantly increased by the overexpressed Glu transporter-1 on brain capillary endothelial cells. Furthermore, disulfide crosslinking could be created in the PIC/M core. The disulfide bond in the micelles was cleaved in the reduced microenvironment of the brain, resulting in the release of the carried ASO. In vivo results showed that ASO was delivered to multiple areas of the brain parenchyma of glycemic-controlled fasting mice at 1 h after tail-vein injection. The optimized nanocarrier efficiently knocked out the long non-coding RNA target gene in different areas of the brain (including the cerebral cortex and hippocampus). These results prove that Glu-modified nanocarriers have great potential in minimally invasive implementation of ASO therapy.
Monosialohexylganglioside (GM1) is the main component of mammalian gangliosides and is mainly composed of hydrophilic sugar chains and lipophilic ceramide.50 LysoGM1 is a hydrolyzed product of GM1. Yin et al. designed anticancer drug DOX-loaded poly(lactic-co-glycolic acid)-LysoGM1 (PLGA-LysoGM1) micelles with sustained and pH-sensitive drug release capacity (Fig. 4E) and proven that the PLGA-LysoGM1/DOX could effectively induce the apoptosis of drug-resistant GBM cells.51 Transmission electron microscopy (TEM) results showed that the spherical PLGA-lysoGM1 and PLGA-lysoGM1/DOX micelles were 33.3 and 27.8 nm, respectively. In vivo studies were also conducted in zebrafish, mice, and rats. In vivo fluorescence imaging indicated that PLGA-lysoGM1/DOX micelles began to enter the brain of zebrafish at 20 min after intracardiac injection and enter the brain of mice at 1 h after intravenous injection. The results confirmed that the micelles could specifically accumulate in the brain to exert a significant anti-neural GBM effect on rats with intracranial nerve GBM.
In attempts to suppress oxidative stress, immune response, and microvascular dysfunction caused by reperfusion injury, Lu et al. designed rapamycin-encapsulated fibrin-binding polymer self-assembled micelles (CPLB/RAPA). Rapamycin can attenuate reperfusion injury. According to an in vitro simulation experiment, only the CPLB/RAPA experimental group could maintain the integrity of the BBB.52 In the same year, the authors prepared a reactive oxygen species (ROS)-responsive polymer micellar system modified with a receptor for advanced glycation end-product (RAGE)-targeting peptide (Ab) derived from Aβ protein (Ab-PEG-LysB/Cur, [APLB/Cur]). These uniform sphere-like micelles with a diameter of 65 nm were found to accumulate at the diseased area via the Aβ-transportation-mimicked pathway and play a synergistic role in ROS clearance and Aβ inhibition under the stimulation by the microenvironment.53
2.2. Polymer-based NPs
Polymer-based NPs are formed by the assembly of polymers. According to the building blocks, polymer-based NPs can be divided into amphiphilic block copolymers and dendrimers. As a drug carrier, polymer-based NPs possess many advantages as follows: (1) the large molecular weight, which can to prolong the retention of drug in the lesion. For example, chitosan with a high molecular weight is an active enhancer.54 (2) High biocompatibility due to reduced enzymatic and hydrolytic degradation.55 (3) Controllable in vivo drug release achieved by tuning the solubility, pH sensitivity, and zeta potential of the components; and (4) flexibility of conjugated functional chemical groups on the polymer surface for targeting delivery.56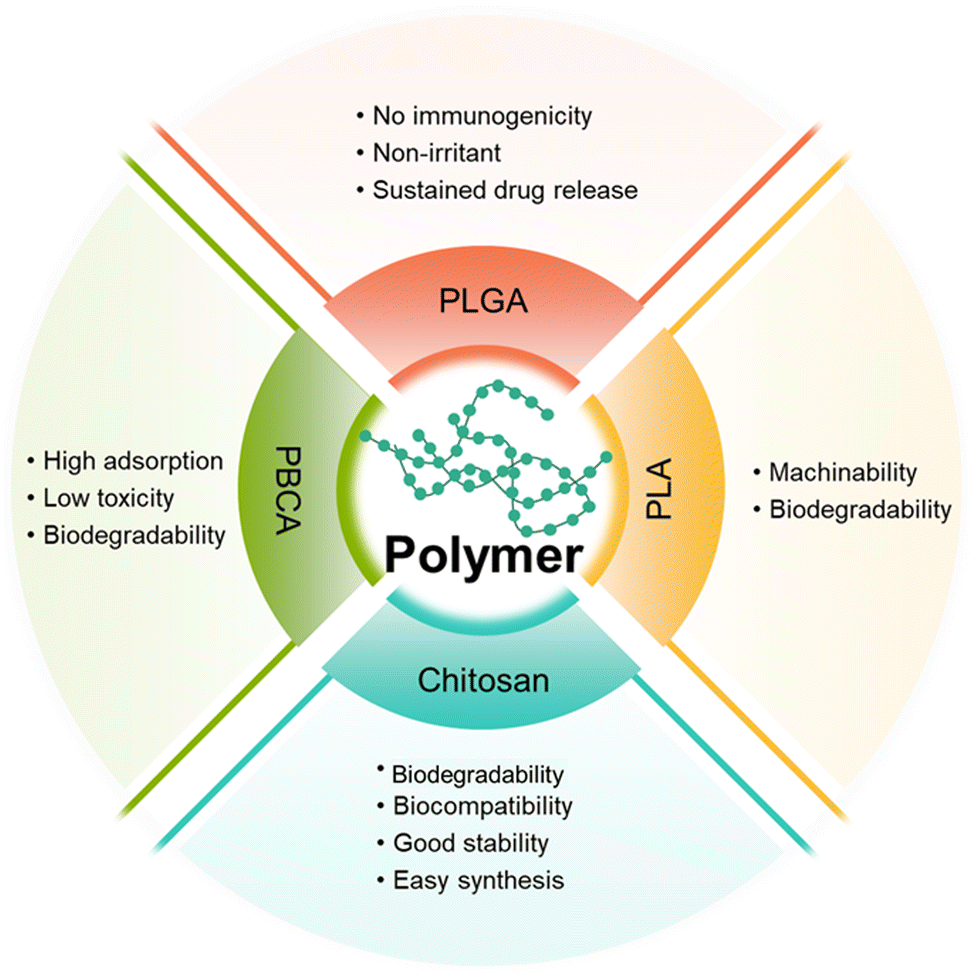 | ||
| Fig. 5 Comparison of different types of amphiphilic copolymers. | ||
2.2.1.1 Poly(butyl cyanoacrylate)-based NPs. Poly(butyl cyanoacrylate) (PBCA) is a promising delivery system with high adsorption, low toxicity, and good biodegradability. Koffie et al. prepared PBCA NPs with spherical and ellipsoid geometry and a mean diameter of 48 nm, and invested their capability for crossing the BBB.58 As shown in Fig. 6A, Texas Red Glucan dissolved in PBS alone could not penetrate the BBB and remained in the blood vessel at 2 h after intravenous injection. In contrast, after covalently incorporating with Texas Red Glucan and modifying with Tween 80, PBCA NPs could pass through the BBB at 30 min post-intravenous injection and stain amyloid plaques, cerebral amyloid angiopathy, and neuron/glial cell bodies. In vivo fluorescence analysis showed that the PBCA NPs crossed the BBB via receptor-mediated endocytosis. Most importantly, they did not induce non-specific BBB disruption. However, the use of PBCA NPs is limited due to the generation of toxic hydrolytic by-products (polyacrylic acid and alcohols). Moreover, the pharmacological effects of PBCA NPs were temporary; thus, daily intravenous injection may be required over the course of treatment.59
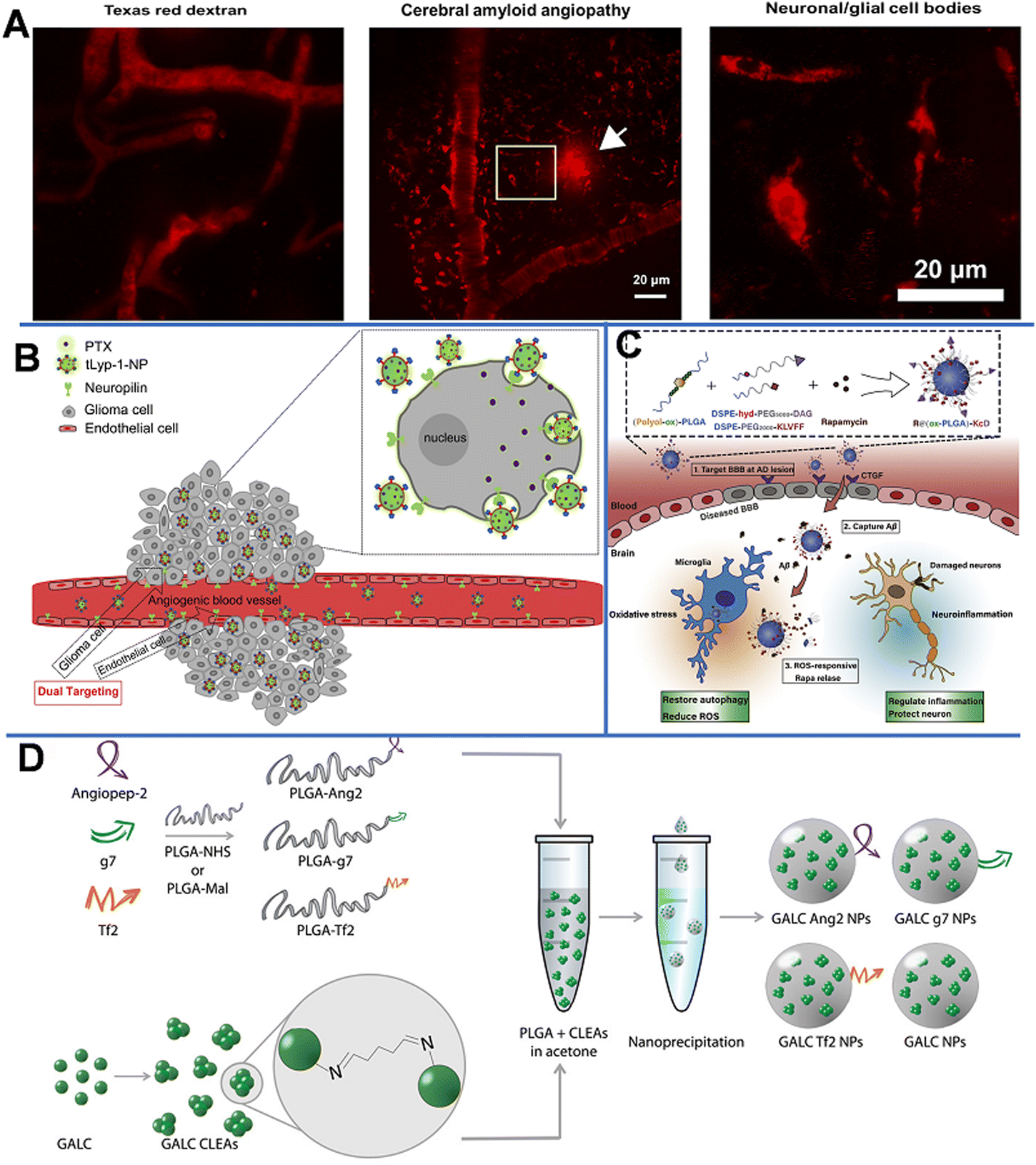 | ||
| Fig. 6 (A) Texas red glucan is covalently linked to PBCA NPs for BBB penetration and marking the neuropathological changes of AD. Reprinted with permission from ref. 58. Copyright 2011, National Academy of Sciences. (B) tLyp-1-conjugated PEG-PLA NP loaded PTX penetrates the BBB through NRP-mediated endocytosis and permeation. Reprinted with permission from ref. 61. Copyright 2013, Elsevier. (C) Scheme of the ROS-responsive specific targeted R@(ox-PLGA)-KcD. Reprinted with permission from ref. 69. (D) Graphical summary of the synthesis of targeted GALC CLEA NPs. Reprinted with permission from ref. 74. Copyright 2019, Elsevier. | ||
2.2.1.2 Polylactic-acid-based NPs. Polyester polymers, such as polylactic acid (PLA) can produce non-toxic lactic acid and glycolic acid oligomers that can be further degraded into CO2 and H2O. Wang et al. prepared Cur-encapsulated mPEG-b-PLA (NP Cur), which had a smooth surface, a spherical morphology, and a narrow size distribution (147.8 ± 5.7 nm), to effectively deliver Cur to the brain.60 The Cur-loading efficiency reached as high as 51.7 ± 3.1%. NP Cur can reduce oxidative stress and inflammation by protecting the BBB and inhibiting the activity of M1 microglia. Triphenyltetrazole (TTC) staining was used to evaluate the area and volume of infarct tissue due to inadequate blood supply. In this study, Cur could accumulate to ischemic penumbra at 3 h post-injection. The result showed that the infarct volumes of PBS, Cur, and NP Cur groups were 40.1%, 32.4%, and 18.3%, respectively. Compared with that of the PBS- and Cur-treated groups, the infarct (dead tissue due to inadequate blood supply) volume of the NP Cur-treated group was significantly lower at 3 days after ischemia-reperfusion injury. The data indicated the successful protection of neurons against ischemia-reperfusion injury by NP Cur.
Neuroprotein is a modular transmembrane protein expressed on both angiogenic glioma and endothelial cells; it is a promising receptor for targeted anti-glioma drug delivery. As shown in Fig. 6B, paclitaxel (PTX)-loaded PEG-PLA NPs modified with tLyp-1 peptide (cell penetrating peptides tLyp-1, the ligand of neuroprotein) had a strong affinity to neuroprotein-overexpressed glioma cells; the presence of neuroproteins could significantly enhance the cellular uptake, thus increasing the PTX uptake of the target cells, leading to the inhibition of tumor development.61 A strong fluorescence signal at the glioma foci indicates that tLyp-1-NP had excellent glioma permeability and targeting ability. In addition, the penetration depth of tLyp-1-NP (121.69 mm) was 1.32-fold higher than that of blank NP (92.02 mm). Compared with NP-PTX (28 days), PTX (23 days), and saline (18 days), tLyp-1-NP-PTX (37 days) could significantly increase the survival time in vivo.
2.2.1.3 Poly(lactic-co-glycolic acid)-based NPs. Poly(lactic-co-glycolic acid) (PLGA) is formed by random polymerization of lactic and glycolic acids. PLGA has been approved as a pharmaceutical excipient for clinical use by the U.S. Food and Drug Administration.62
The formation of extracellular aggregates of Aβ1–42 is a sign of AD.63,64 It has been reported that Cur not only inhibits the formation of new Aβ aggregates, but also shows anti-amyloid activity by breaking down existing Aβ aggregates.65 Therefore, Barbara et al. prepared Cur-encapsulated PLGA NPs with a loading efficiency of 3% (w/w) and modified them with the g7 ligand to enhance their BBB penetration capability.66 It was found that the obtained g7-NPs-Cur could significantly decrease the amount of Aβ aggregates. In another work, by using a high-pressure emulsification-solvent evaporation method, Tsai et al. prepared Cur-loaded 163 nm PLGA NPs (C-NPs) with a drug-loading efficiency of 46.9%.67 With this approach, the retention time of Cur in specific brain areas (the cerebral cortex and hippocampus) was significantly prolonged. Similarly, Bhatt et al. synthesized Tet1-peptide-conjugated PLGA-encapsulated nattokinase NPs and found that these NPs can inhibit the formation of Aβ40 plaques in AD and exhibit antifibrinolytic activity.68 However, due to low drug accumulation in the brain and difficulty in inhibiting the Aβ aggregates, Gao et al. designed a nano-cleaner with a rapamycin-loaded ROS-responsive PLGA core and surface modified with KLVFF peptide and acid-cleavable DAG peptide [R@(ox-PLGA)-KcD]. The DGA peptide was an acid-responsive targeting ligand that could be separated from the nano-cleaner to promote transcytosis from endothelial cells into brain parenchyma. The exposed KLVFF could capture Aβ, and in response to the presence of ROS, rapamycin was released to improve Aβ degradation and normalize inflammatory conditions. The physiological evaluation and behavioral experiments in an AD mice model demonstrated the effectiveness and biocompatibility of the nano-cleaner in AD therapy (Fig. 6C).69
Cui et al. reported magnetic PLGA NPs with transferrin receptor-binding peptide T7 that could co-deliver PTX and Cur.70 The NPs could increase the drug delivery to the brain by >5 times compared to non-targeting NPs. Furthermore, these NPs could enhance the survival rate compared with free drugs (100% vs. 62.5%). In another study by Bi et al., the T7 peptide was used to modify PLGA-PEG micelles.71 The study revealed that these micelles achieved more efficient accumulation of the micelles in the brain tumor, providing better tumor inhibition, and better prolonging the survival time compared with the unconjugated counterparts. In addition to the T7 peptide, the iNGR peptide has also been used to functionalize PEG-PLGA NPs and to penetrate gliomas. In this design, the iNGR peptide initially binds to the overexpressed aminopeptidase N in tumors and is then cleaved to CRNGR that could subsequently bind to neuropilin-1 for deep penetration into the tumor parenchyma.72 Importantly, the size of PLGA NPs can impact the efficiency of drug delivery to the brain. One study has suggested that PLGA NPs with a size of approximately 100 nm could be transported to the brain more efficiently than those with a size of 200 or 800 nm.73
Grosso et al. developed a new enzyme delivery nanoplatform by encapsulating the cross-linked enzyme aggregate (CLEA) into PLGA NPs, which could encapsulate galactosylceramidase (GALC) and retain good enzyme activity.74 As shown in Fig. 6D, the obtained NPs were further modified with functional peptides, such as angiopep-2 (ANG-2), g7, or Tf-binding (Tf2) peptides, to enhance the BBB penetration. The in vitro study on the transport of NPs in cells and their ability to restore enzyme activity demonstrated that these NPs could effectively deliver GALC into human cells and maintain the enzyme activity up to 96 h. This effective approach provides a promising therapeutic opportunity for lysosomal-storage disorders involving the CNS.
2.2.1.4 Chitosan. Chitosan is obtained by deacetylation of chitin and is broadly utilized in biomedical applications due to the merits of degradability, biocompatibility, good stability, easy synthesis and functions,75,76 and mucoadhesivity. In 2006, chitosan was approved as a “generally recognized as safe” (GRAS) class of natural products by the FDA.
Trapani et al. prepared dopamine-encapsulated chitosan NPs to cross the BBB for Parkinson's disease.77 Gu et al. designed double antibody-modified (Tf antibody and bradykinin B2 antibody) chitosan/siRNA NPs (chitosan-NPs) with a spherical shape and an average particle size of 235.7 ± 10.2 nm, which could target HIV-infected brain astrocytes and deliver siRNAs to inhibit HIV replication.78 Double-ligand modification of NPs is a potential strategy for improving drug-delivery efficiency. Herein, transferrin antibody and bradykinin B2 antibody could specifically bind to transferrin and bradykinin B2 receptors, respectively, and then deliver the siRNA across the BBB into the target cells astrocytes. In vitro experimental results demonstrated that this siRNA carrier could effectively silence the expression of the proteins SART3 and hCycT1 proteins related to HIV replication. The knockout rates of SART3 and hCycT1 by chitosan-NPs were 80.9% and 66.6%, respectively.
The deposition of Aβ1–42 peptide aggregates in the brain is a sign of AD. Therefore, amyloid aggregates are widely used as the therapeutic targets in AD. For example, Jha et al. studied the anti-amyloidogenic activity of naked chitosan and chitosan-based NPs copolymerized from chitosan and PLGA.79 The chitosan-based NPs could also break down preformed amyloid fibres derived from the Aβ1–42 peptide. The anti-amyloid activity of chitosan-based NPs was 200-fold stronger than that of PLGA-chitosan-based NPs, and the enhancement was thought to be the surface charge. To improve the efficiency of the drug delivery to the brain, Yang et al. synthesized Cur-loaded chitosan-BSA NPs.80 The encapsulation rate by NPs was measured to be 95.4%. Experimental results showed that chitosan-BSA NPs could improve the BBB-crossing ability of the drug, activate microglia, and stimulate phagocytosis of the Aβ peptide. Recently, Zheng et al. applied dihydromyricetin (DMY)-coated Se NPs that were further wrapped by chitosan (which served as a stabilizer) and decorated with the BBB targeting peptide Tg (TGNYKALHPHNG) to yield Tg-CS/DMY@SeNPs for AD treatment.81 The prepared NPs were sphere with a diameter of less than 50 nm. The results showed that Tg-CS/DMY@SeNPs could successfully cross the BBB to inhibit the aggregation of Aβ and the secretion of inflammatory cytokines involved in the progression of AD. Thus, the Tg-CS/DMY@SeNPs are ideal candidates for AD therapy.
Overall, highly biocompatible polymer-based NPs can enhance brain permeation and tumor accumulation of drugs, and realize the controlled release of drugs under the tumor microenvironment (TME), and thus have been widely used in biomedicine. However, traditional linear polymers face the limitation of a low number of interaction sites and low drug-loading areas. Multi-dimensional polymer-based nanocarriers with abundant active groups and high drug-loading capacity are therefore needed.
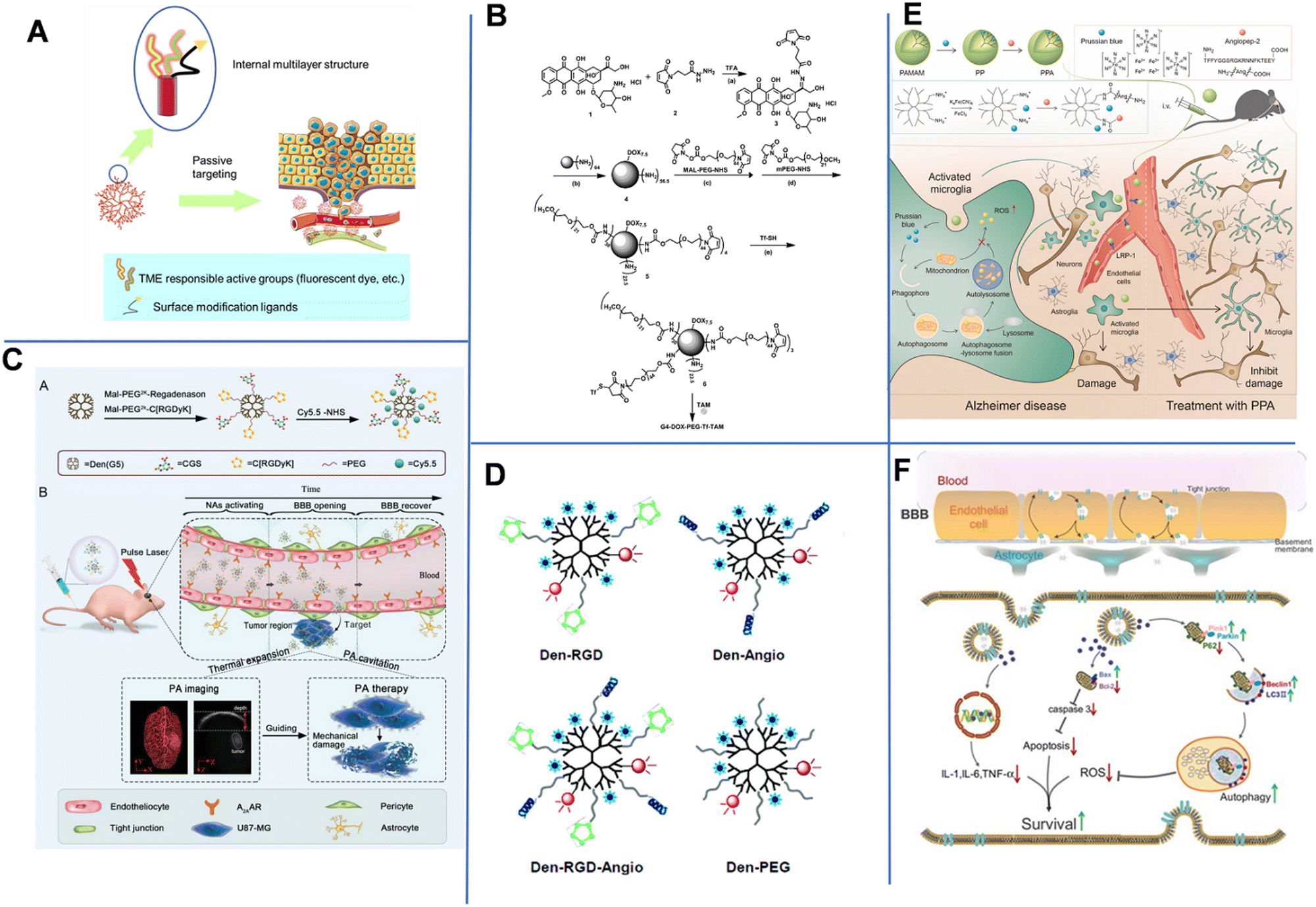 | ||
| Fig. 7 (A) Schematic diagram of the structure and administration of dendrimers. (B) Synthetic route of the dual-targeting drug carrier. Reprinted with permission from ref. 87. Copyright 2012, Elsevier. (C) Schematic summary of the synthesis and photoacoustic precision glioblastoma therapy of the Dendrimers-RGD/CGS/Cy5.5 (Dendrimers-Den). Reprinted with permission from ref. 90. Copyright 2019, Wiley-VCH. (D) Schematics of the targeted and control nanoprobes. Reprinted with permission from ref. 91. Copyright 2012, American Chemical Society. (E) Illustration of the microglia induced AD microenvironment and mechanisms of regulating the metabolism of microglia.92 (F) Scheme of the construction of the T-SA-NPs NPs and the potential mechanism of action.93 | ||
Poly(amidoamine) (PAMAM) contains many cavities that can encapsulate drug molecules, and terminal –NH2 groups that can bind to antibodies and other bioactive substances to form a stable hybrid system.85 Teow et al. prepared a Den-drug complex by conjugating PTX to the surface of PAMAM via a glutamate linker.86In vitro experiments demonstrated that these Den-drug conjugates could cross the endothelial cell monolayers of a porcine brain within 3 h and could be one-way transported (basolateral to apical direction) across the cell monolayers. The prepared G3 PAMAM dendrimers could enhance paracellular transport by opening the tight junctions, thereby successfully delivering the drug into the brain. Conversely, the free drugs could not effectively and unidirectionally permeate the cell monolayers due to the presence of P-gp.
To enhance the drug delivery across the BBB and improve the tumor-targeting delivery efficiency, Li et al. constructed a dual brain-targeted drug delivery system (G4-DOX-PEG-Tf) with a hydrodynamic diameter of approximately 19 nm using Tf and tamoxifen (TAM)-modified fourth-generation PAMAM Den that can be used for the treatment of GBM.87 As shown in Fig. 7B, Den was a pH-sensitive drug vector that could co-load TAM and DOX through pH-sensitive chemical bonds. Additionally, there were one Tf group, seven DOX molecules and over 30 PEG1000 and PEG2000 chains on the surface of one Den molecule. The Tf group improved the BBB-crossing capability, while TAM promoted the accumulation of drug in tumor by inhibiting the efflux transports. In an acidic tumor environment, the pH-sensitive chemical bond was broken, allowing DOX to be released from the system into the tumor tissue. Later, Wang et al. demonstrated that conjugating Tf to an intrinsically fluorescent PAMAM could enhance the BBB-crossing ability of nanosystems.88 Unlike Tf, ANG-2 could effectively enter the brain parenchymal cells through a mechanism mediated by low-density lipoprotein-receptor-related protein-1 (LRP1). Therefore, Huang et al. conjugated PAMAM and ANG-2 to a bifunctional PEG, and then hybridized it with pDNA to produce gene-carrying NPs (PAMAM-PEG-ANG/DNA NPs) with a diameter of 110 nm. The NPs could cross the BBB and target glial cells.89
In 2019, Liu et al. constructed NP-containing PAMAM dendrimers with a diameter of 10 nm. The NPs could cross the BBB through the reorganization of the cytoskeleton of endothelial cells. The NPs were labeled with red-absorbing dye Cy5.5, cyclic [RGDyK] peptide (which targets tumor vasculature and U87-MG glioblastoma cells), and A2AAR agonist agent (CGS) (Fig. 7C).90 The dendrimers-RGD/CGS/Cy5.5 NPs first target the αvβ3 integrin overexpressed in the blood vessels of brain glioblastoma. The CGS agonist activated A2AAR on vascular endothelial cells to open the BBB, allowing the NPs to enter the glioblastoma. Under pulsed laser irradiation, NPs absorbed the photon energy and generated photoacoustic (PA) shock waves that kill tumor cells. In addition, PA imaging was utilized to clearly locate tumors after Den-RGD/CGS/Cy5.5 administration. A moderate PA signal was detected at the tumor site after 1 h of injection, and the signal gradually increased over time and reached its maximum value at 12 h post-injection.
Magnetic resonance imaging (MRI) is the main approach used in diagnosing brain tumors. Unfortunately, the accuracy of MRI in brain imaging is limited by the short cycle life, non-specificity, and poor BBB permeability of commonly used MRI agents. Yan et al. developed a dual-targeting nanoprobe for MRI and optical imaging of brain tumors.91 As shown in Fig. 7D, PAMAM-G5 was simultaneously labeled with the tumor-vessel-targeting cyclic (RGDyK) peptide and ANG-2 peptide to produce Den-RGD-Angio with a hydrodynamic diameter of 15.6 nm. The near-infrared (NIR) fluorophore Cy5.5, rhodamine, and the MRI contrast agent Gd3+-DOTA were conjugated to PAMAM-G5. This dual-targeting strategy could achieve targeted delivery of imaging agents to tumors without disturbing or damaging the BBB. With a strong tissue penetration ability of NIR fluorescent agent, the probe could be detected in the brain in real-time within 2 h of injection. In vivo imaging results revealed that the Den-RGD-Angio-modified nanoprobe could effectively penetrate the BBB in normal mice, and the tumor boundary could be accurately determined. Similarly, Shi et al. constructed Prussian blue/PAMAM dendrimer/Angiopep-2 (PPA) NPs for effective AD treatment. The component PB held ROS scavenging capacity to facilitate mitochondrial autophagy and inhibit amyloid plaques, thus compromise AD. Angiopep-2 had increased the brain-targeted accumulation in the brain. MRI images showed that PPA NPs had a high positive signal after 4 h of injection (Fig. 7E).92
Besides, environment-responsive dendrimers are also attractive due to their highly targeted drug delivery, which can avoid systematic toxicity more probably compared to non-responsive dendrimers. In 2020, Du et al. constructed O2˙−-responsive dendrimers modified with an antioxidant salvianic acid A (SA) and a targeting peptide COG1410 (T-SA-NPs) to aid in BBB crossing when treating ischemic stroke. Fluorescence imaging showed that COG1410-modified NPs conjugated with Texas Red was specifically accumulated in the brain after 6 h of injection (Fig. 7f). Laser confocal microscopy analysis of immunostaining demonstrated the NPs could permeate into the penumbra area of cerebral ischemia. NP-treated mice also showed a better behavioral ability than the control groups.93 Jin et al. prepared a pH-sensitive siLSINCT5-loaded dendrimer tLyp-1 and anti-NKG2A monoclonal antibody (aNKG2A, a checkpoint inhibitor) modified tLyp/aNKNP-siRNA. Once the tLyp/aNKNP-siRNA was targeted and accumulated into the brain, the hydrazone bond between aNKNP and the NPs would be pH-responsively broken and aNKNP released to induce anti-tumor immunity. Residual NPs were further captured by tumor cells to inhibit the LSINCT5-activated signaling pathways. In vitro experiments showed the tLyp/aNKNP-siRNA had a strong anti-proliferation effect. The modification with tLyp-1 endowed the NPs with an excellent BBB-crossing ability. After 24 h of injection, the accumulation of tLyp/aNKNP-siRNA in the brain tumor reached its maximum value.94
Dendrimers are a type of nanoscale drug carrier with broad space for development in the field of pharmacy due to their inherent unique advantages. However, the current research on dendrimers is still in the early stages, and the release behavior, targeting ability, and metabolic pathways of drugs need to be further studied both in vitro and in vivo.
2.3. Biomolecule-based NPs
Biomolecule-based NPs are another type of BBB-crossing nanocarrier that have been widely used. These include engineering and modification of several common biomolecules including proteins, nucleic acids, and virus.For example, a simple small 12 amino-acid peptide originating from melanotransferrin protein (MTf, belongs to the Tf family) was found to possess improved brain delivery properties.97 Jefferies et al. conjugated MTf with siRNA to construct a peptide-oligonucleotide conjugate (POC) for the therapy of ischemic stroke.98 Besides, Zhang et al. used a high-density lipoprotein-mimicking peptide-phospholipid scaffold (HPPS) to load Cur for the targeted immunomodulation of inflammatory monocytes for further treatment of encephalomyelitis (EVE). The constructed Cur-loaded HPPS (Cur-HPPS) could be internalized specifically by monocytes through the scavenger receptor class B type I (SR-B1) receptor. After incubating for 3 h, over 80% of Cur-HPPS was found to be taken up by monocytes. To make NPs trackable, fluorescent dye DiR-BOA (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide bisoleate) was also loaded into the NPs. At 6 and 24 h after injection, a strong fluorescence signal of DiR-BOA was observed in the brain, which was indicative of effective BBB-crossing and brain accumulation (Fig. 8A).99 To further improve the drug delivery efficiency, Giralt designed the third generation of BBB-shuttles, branched BBB-shuttle peptides. Compared with a single copy of the BBB-shuttle, the branched BBB-shuttle showed higher uptake and improved permeability of the model protein in endothelial cells.100
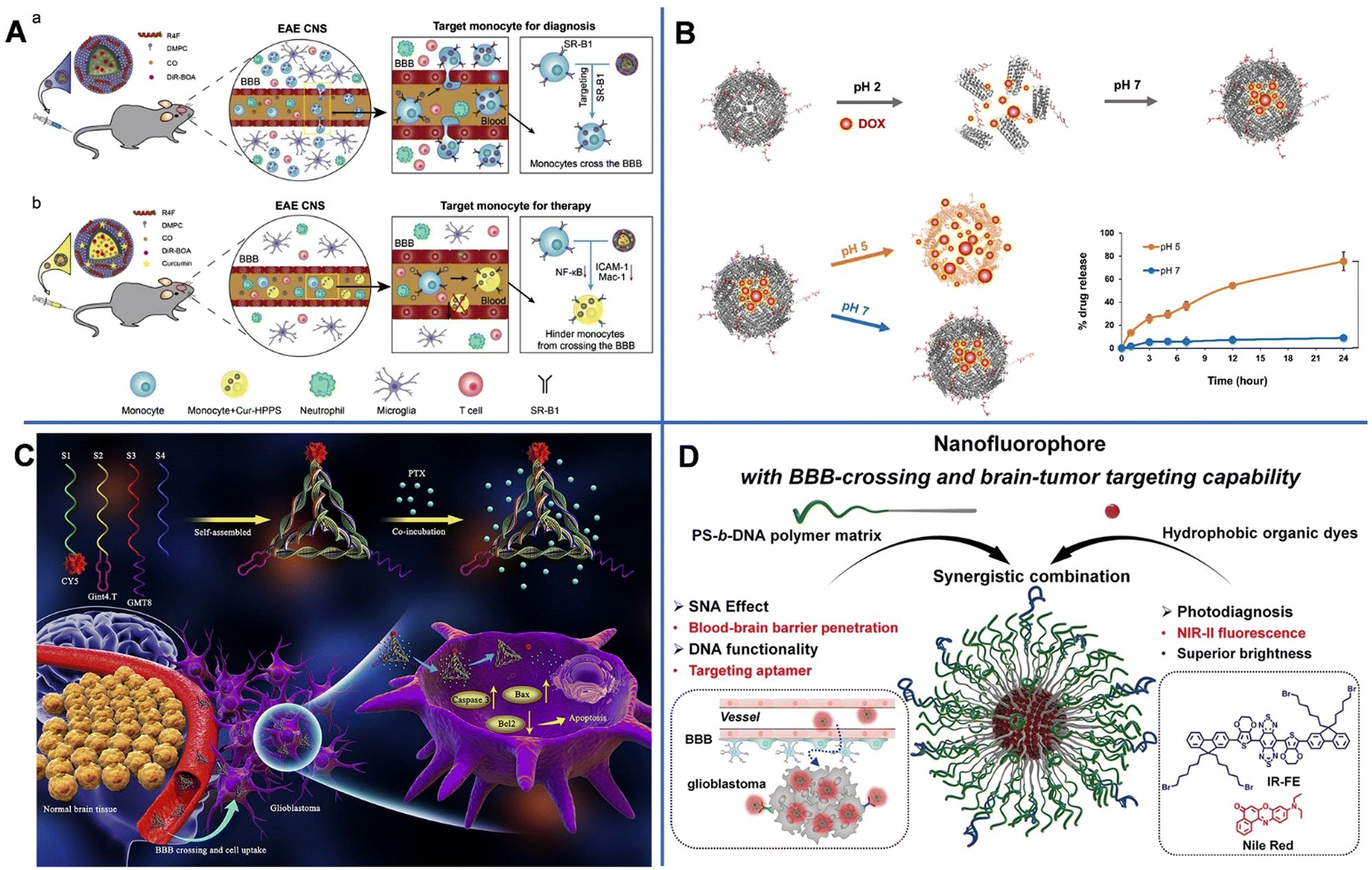 | ||
| Fig. 8 (A) The design of the HPPS for targeted immunomodulation of inflammatory monocytes.99 (B) Integrin α2β1-targeting ferritin nanocarrier traverses the blood–brain barrier for effective glioma chemotherapy.102 (C) The preparation of GPC and the mechanism of GPC inducing apoptosis of U87MG cells via crossing the blood–brain barrier and targeting U87MG cells.120 (D) Illustration of organic fluorescence-emitting spherical nucleic acid self-assembled from PS-b-DNA and organic dyes.121 | ||
Above all, TME-responsive delivery systems are more attractive approaches. Zuhorn reported dodecamer peptide (G23)-functionalized polydopamine (pD)-coated Cur-loaded zein NPs (CUR-ZpD-G23 NPs) to traverse the BBB in the treatment of glioblastoma. Zein originating from corn could be easily self-assembled into NPs and had a sustained drug release capability. The polydopamine (pD) coating provided NPs with hydrophilicity, colloidal stability, and biocompatibility, and endowed NPs with pH-responsive drug release capability. At pH 7.4, about 45% of Cur was released from CUR-ZpD-G23 NPs, and the released Cur reached up to 80% in 48 h when pH decreased to 5. After incubation for another 8 h, approximately 17% of NPs passed by the in vitro BBB model. And the viability of C6 glioma cells incubated with CUR-ZpD-G23 NPs for 48 h dramatically decreased to 25%. All the above results demonstrated the BBB-crossing ability and glioma-treating capacity of NPs.101 Huang et al. constructed an integrin α2β1 targeting ligand (DGEAGGDGEA) modified with human H-ferritin (HFn) and loaded with DOX (DOX-2D-HFn NPs) for orthotopic glioma therapy. HFn could self-assemble into a cage-like structure that was highly tolerant to extreme acidic conditions (pH = 1–2) or extreme basic conditions (pH = 11–13) and high temperatures of up to 80 °C, thus realizing pH-responsive DOX release. At pH 7, only 9% of DOX was released from the NPs after 24 h of incubation, whereas at pH 5, 75% of DOX was released. Besides, the interaction between HFn and TfR1, integrin α2β1 and peptide-DGEAGGDGEA were greatly beneficial to the BBB-crossing capacity of NPs. The amount of integrin α2β1-targeted HFn passed through the BBB and bound to U-87MG cells was almost 2 times higher than that of HFn after four hours of incubation. And the cell toxicity of DOX-2D-HFn (69%) was also 2 times higher than that of free DOX (29%). In an intracranial glioblastoma mouse model, the median survival time of the DOX-2D-HFn NP-treated group was dramatically higher than that of the untreated groups (Fig. 8B).102
However, not all proteins are suitable for BBB crossing. Protein-based NPs also face challenges of enzymatic degradation and RES clearance; thus, further surface modification is still required. Gao used erythrocytes (ETm) coating to prevent the loss of nano-drugs after systematic administration.103
Twelve AAV serotypes from AAV-1 to AAV-12 have been reported,106 and AAV-9 had the potential to penetrate the BBB and transduce the CNS cells.107 In 2017, Gradinaru et al. reported that AAV-PHP.eB (a variant of the neurotropic vector AAV) could effectively pass through the BBB and could greatly improve the treatment of neurological diseases.108 Lin et al. reported that the ApoE-LDLR pathway was required for intravenous AAV-PHP.eB to penetrate the BBB. Additionally, blood B cells (not T cells) could reduce the transduction of AAV-PHP.eB to the CNS.109 Wilson et al. demonstrated that glycosylphosphatidylinositol (GPI)-linked protein LY6A was involved in BBB transport. There appears to be a direct link between LY6A and AAV-PHP, which held the excellent ability of AAV-PHP to deliver transgenes across the BBB in C57BL/6J mice. Moreover, murine GPI-anchored protein may influence the viral interaction and transcytosis at the BBB.110
Besides, Gradinaru et al. employed a Cre-transgenic-based screening platform and sequential engineering to engineer AAV-PHP.eB between the surface-exposed AA452 and AA460 in VP3 to develop a variant AAV named AAV.CAP-B10. After intravenous administration, most AAV.CAP-B10 accumulated into the brain, and a small portion accumulates in the liver. The differential accumulation in an adult marmoset was also more distinct compared to that in AAV9 and AAV-PHP.eB.111
Although engineered AAV variants have shown success in neuron transduction in preclinical trials, the related animal studies are usually undesirable.112 Furthermore, small biomolecule modification seems to be a shortcut in the background of the widely used various BBB shuttle peptides in drug delivery.113,114 Li et al. specifically linked AAV9 with PB5-3 peptide (BBB shuttle peptide) to increase its brain accumulation and transduction. Mechanistic studies revealed that the PB5-3 peptide could effectively improve AAV9 trafficking and transcytosis in hCMEC/D3 cells, as well as decrease the clearance of AAV9 from blood.115
For example, Shi et al. utilized two aptamers, GMT8 and Gint4.T conjugated with tetrahedral framework nucleic acid (tFNA) to carry chemotherapeutics PTX (GTG) for treating glioblastomas (Fig. 8C). Both the aptamers had a strong affinity towards platelet-derived growth factor receptor β (PDGFRβ), which is expressed on endothelial cells and is related to tumor progression. Gint4.T was used for targeted delivery. The binding of GMT8 and PDGFRβ would suppress the growth of tumors. The results showed that GTG could effectively permeate the BBB in the in vitro BBB model. The migration and invasion inhibition were also significantly observed.120 Tian et al. prepared a block copolymer of DNA and polystyrene-b-polyethylene glycol (PS-b-PEG) (PS-b-DNA) (Fig. 8D) that can self-assemble into 13 nm spherical nucleic acids (SNAs) with the ability to deliver NIR-II dye into the brain (NR@PS-b-DNA) and the highest dye-loading amounts of 30 wt%. The NR@PS-b-DNA showed 4.5-fold higher traversing efficiency and 3-fold higher accumulation in U87MG cells compared to NR@PS-b-PEG. CLSM images of NR@PS-b-DNA in coronal slices taken at 3 h post-injection showed prominent fluorescence signals.121
More interestingly, Tam et al. reported adapt-free and DOX-loaded DNA nanocages produced by supramolecular self-assembly. The nanocages could effectively pass through the BBB via endocytosis and be internalized by endothelial cells and U-87 MG cells without peptide modification. The drug-loading efficiency was calculated to be 15.1 ± 2.7%. And the spherical structures held better BBB-crossing than the tubular structures. In vitro and in vivo experiments demonstrated that the adaptor-free nanocages exhibited less BBB crossing and glioma. Thus, a DNA nanocage-based platform is a safe and cost-effective tool for targeted drug delivery to the brain.122
In summary, virus-based NPs are widespread in gene therapy with extensive host cells and high transfection efficiency. However, inherent immunogenicity can cause the NPs to be easily captured by RES, thus need to be further investigated. The natural receptor-ligand interaction can help in targeting the delivery of protein-based NPs, and components of amino acids can resolve the problem of biosafety. Nucleic acid-functionalized nanomaterials have been widely studied in the field of drug delivery due to their molecular recognition characteristics. Finally, the design of sequences and the use of programmable material can endow NPs with adjustable structures and controlled drug release ability.
In Table 1, we summarize some important examples of organic-NP delivery systems for crossing the BBB.
| Types of nanocarrier | Delivery system | Surface modification | Size (nm) | Drug/gene | Drug loading efficiency | Targets | Diseases | Ref. | Advantages | Disadvantages |
|---|---|---|---|---|---|---|---|---|---|---|
| Liposome | Phospholipid\CHL | Polyethylene\glucose | 100 | Coumarin 6 | NA | Glucose | NA | 23 | Biocompatibility; low toxicity; aptitude to trap hydrophilic and lipophilic drugs; prolong drugs’ blood circulation time | Limited drug-carrying stability and biological functions; small scale production |
| DPPC\DSPC\SPIONs | Cell-penetrating peptide\Anti-GBM antibody | 114 | DOX | 75% | Glioblastoma | Glioma | 35 | |||
| SLN | Dynasan@114\palmitic acid | 83–14 MAb\Tween 80\poloxamer 407\DSPE-PEG(2000)-carboxylic acid | 120–450 | Saquinavir | 79% | α-Subunit;LDLRs | AIDS | 39 | High monodispersity; long temporal stability; low toxicity; sustained drug release; multi-administration | Limited drug loading capacity; the drug leakage during storage; the storage of solid lipids may form crystals resulting in performance degradation |
| Dynasan 116\epikuron 200\short chain alcohols | DPM\ApoE | 119 | NA | NA | LDLRs | NA | 42 | |||
| Micelle | CPT–S–S–PEG–COOH | PEG\iRGD peptide | 100 | CPT\IR780 | CPT-11.07 | αvβ-Integrin- and neuropilin-1 | Glioma | 47 | Small particle size; flexibility of modification with targeting ligands; prevent or minimize drug degradation; lower adverse side effects; ease of preparation | The degradability of micelles needs to be improved; the probable drug leakage |
| Glycol chitosan\stearic acid | TGN peptide | 146.2 | Cur | 76.2% | NA | Glioma | 45 | |||
| 3-Mercaptopropyl amidine\2-thiolaneimine\poly(L-lysine) | Glucose\poly(ethylene glycol)-b-poly(lysine) | 45 | ASO | NA | Glut 1 | NA | 49 | |||
| F-127 | F-127 | 14 | Cur | NA | NA | Glioma | 46 | |||
| PLGA-lysoGM1 | LysoGM1 | 27.8 | DOX | 61.6% | NA | Glioma | 51 | |||
| Amphiphilic copolymer-based NP | PBCA | Tween 80 | 200 | Fluorophores (dextran Trypan blue)\Gadobutrol | NA | LDLRs | AD | 58 | Controlled drug release; structural controllability; high stability; ease of synthesis; strong drug loading efficiency | Complicated design; poor repeatability |
| mPEG-b-PLA | mPEG | 147.8 | Cur | 4.92% | NA | Ischemia/reperfusion | 60 | |||
| MePEG-PLA\male–PEG–PLA | tLyp-1 peptide | 111 | PTX | NA | Neuroproteins | Glioma | 61 | |||
| PLGA | g7 | 200–250 | Cur | 60% | NA | AD | 66 | |||
| PLGA | Angiopep-2\g7\Tf | 200 | GALC CLEAs | Angiopep-2(60%)\g7(74%)\Tf (39%) | TfR;LPR-1 receptor | Krabbe disease | 74 | |||
| Chitosan | NA | 110 | Dopamine | 81% | NA | Parkinson | 77 | |||
| Dendrimer | G3-PAMAM | Lauryl chains | 13.7 | PTX | NA | NA | NA | 86 | High drug loading efficiency due to the branched structure and internal cavity; adjustable molecular weight and shape; abundant terminal groups for functionalization | Chronic toxicity; poor accumulation in vivo |
| G4-PAMAM | Tf\TAM | 19 | DOX | NA | TfR | Glioma | 87 | |||
| G5-PAMAM | Cyclic [RGDyK] peptides | 10 | A2AAR agonis | NA | Integrin | Glioma | 90 | |||
| G5-PAMAM | Cyclic [RGDyK] peptide\Angiopep-2 peptides | 15.6 | NA | NA | Integrin\LPR-1 receptor | Glioma | 91 | |||
| Virus-based NPs | AAV9 | PB5-3 peptide | NA | NA | NA | NA | NA | 115 | Extensive host cells and high transfection efficiency | Inherent immunogenicity |
| Protein-based NPs | HPPS | NA | 15 ± 2.3 | Cur | 80 μg mg−1 | SR-B1 | Encephalomyelitis | 99 | Biocompatibility, good biodegradability; targeting; easy modification | Enzymic degradation, RES clearance; low drug loading efficiency |
| Zein | G23-peptide | <100 | Cur | 94.4% | NA | Glioma | 101 | |||
| HFn | HFn, DGEAGGDGEA peptide | 12 | DOX | NA | Integrin α2β1, TfR1 | Glioma | 102 | |||
| Nucleic acids-based NPs | tFNA | GMT8, Gint4.T | 20 | PTX | 250 nM | U87MG glioma cells, PDGFRβ | Glioma | 120 | Excellent biodegradability; adjustable structure and responsible drug-release | |
| DNA nanocage | NA | 19.7 | DOX | 15.7 | NA | Glioma | 122 |
3. Conclusion and outlook
The use of NP delivery systems in treatment of CNS diseases is particularly important because most free drugs or therapeutic molecules cannot pass through the BBB and be delivered to the brain. Many studies, including many clinical trials, have shown that the drug delivery strategies relying on BBB avoidance rather than facilitating BBB delivery are generally ineffective.123 Therefore, it is essential to develop effective BBB drug delivery strategies. The development of a NP-delivery system is an innovative and flexible way for delivering drugs to the brain for treating various major diseases. A wide variety of drug delivery systems based on organic NPs (e.g., lipid-, polymer-, and protein-based NPs) have been constructed using suitable carrier materials to achieve effective delivery of anti-tumor drugs.Generally, all the above-mentioned organic delivery systems exhibit excellent biocompatibility and hold clinical potential. Size, shape, ligand density, lipophilicity and surface chemistry are the main factors for the design of nanocarriers and the selection criteria. For example, smaller NPs can penetrate the BBB more easily than larger NPs, but they are restricted by a smaller surface area and relatively low drug loading amount. Liposomes and SLNs can be used to deliver both hydrophilic and hydrophobic drugs. Micelles are easily prepared, but can only be applied in the delivery of hydrophobic drugs. Polymer-based NPs could avoid the enzymatic and hydrolytic degradation of drugs with a high drug loading efficiency. Amphiphilic copolymers are suitable for hydrophobic drugs. Virus-based nanomedicines are widely used in gene therapy (such as DNA, RNA, and so on), because of their high transfection efficacy. Protein-based NPs, nucleic-based NPs and dendrimers have a number of active groups that can be easily functionalized.
The development of effective nanocarriers requires synergistic improvement of their physicochemical properties and biological functions, in order to broaden their applications in the field of biomedicine. In the optimization of nanomedicines for CNS treatment, it is essential to understand the impact of structures and surface chemical compositions of nanocarriers (e.g., morphology, size, surface functional group, and surface charge) on the BBB-crossing efficiency and safety. Generally, NPs with an average size of approximately 100 nm and neutral and hydrophilic surfaces can be maintained in the blood circulation for a long time and exhibit an improved delivery to tumors.124 Small-sized NPs may pass through the BBB by endocytosis which is related to the reorganization of the cytoskeleton of endothelial cells.87 For surface modification, targeting ligands can promote the accumulation of carriers in the brain and enable NPs to cross the BBB via different transport mechanisms. Decoration with surfactants is also another plausible approach, as this not only improves the stability of NPs, but can also alter their targeting capacity and increase their cellular uptake. As an example, Tween 80 and F-127 promote the cellular uptake of NPs, while inhibiting P-gp. Other surfactants with similar effects have also been discovered. Although the biosafety of nanomaterials is generally good, their clinical use still has a long way to go.
Although great progress in effective drug delivery to the brain has been achieved by using nano-carriers, further effort is still required to improve their efficacy and safety in clinical use. It is desirable that nanocarriers should have the following characteristics: (1) tumor-targeting properties to promote the targeted delivery of drugs to diseased sites; (2) clinical imaging ability to monitor the delivery, transport, distribution, and clearance of drugs; (3) ability to carry and controllably release different drugs, which is a useful characteristic for combination therapy or therapy in which the drug concentration needs to be in the effective therapeutic window; (4) responsive release mechanism to improve the targeting efficiency and achieve the corresponding results based on the change in the disease sites; (5) ability to effectively cross the BBB without causing irreversible damage; (6) quick degradability and rapid clearance; (7) high safety profiles. While effectiveness in delivering drugs to the CNS and high therapeutic efficacy are important characteristics of nanocarriers, the safety profiles are equally important. NPs have been found to induce brain dysfunction in normal animals and cause oxidative stress and injuries in the brain. To alleviate these problems and facilitate the clinical translation of NPs in the treatment of CNS diseases, various strategies for alleviating the toxicity of NPs have been explored. The neurotoxicity of NPs and the corresponding potential solutions have been described in detail in the literature.125–129
It is important to recognize that nanomedicines are being developed to be more effective and safer than ever. With the development of nanotechnology, brain-targeting nanomedicines can be developed for more effective delivery of drugs to the brain. The techniques presented may also have broader applications and be used as a promising platform for the treatment of CNS diseases in the future.
Abbreviations
| NA | Not available |
| Papp | Apparent permeability coefficient |
| DSPE | 1,2-distearoyl-sn-glycero-3-phosphotidylethanolamine |
| SLE | Soya-a-lecithin |
| CHL | Cholesterol |
| DPPC | 1,2-Dipalmitoyl-sn-glycero-3-phosphocholine |
| PEG | Poly ethylene glycol |
| DHDP | Dihexadecyl phosphate |
| ApoE | Apolipoprotein E |
| QU | Quercetin |
| RA | Rosmarinic acid |
| AD | Alzheimer's disease |
| Tf | Transferrin |
| DSPC | 1,2-Distearoyl-sn-glycero-3-phosphocholine |
| SPIONs | Superparamagnetic iron oxide NPs |
| DOX | Doxorubicin |
| MAb | Monoclonal antibody |
| AIDS | Acquired immunodeficiency syndrome |
| MA | Melanotransferrin antibody |
| TX/TAM | Tamoxifen |
| DPM | 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000] |
| CPT | Camptothecin |
| iRGD, CRGDRGPDC | Internalizing RGD peptide |
| ASO | Antisense oligonucleotide |
| PLGA | Poly(lactic-co-glycolic acid) |
| GM1 | Monosialohexylganglioside |
| PBCA | Poly(butyl cyanoacrylate) |
| mPEG-b-PLA | Poly(ethylene glycol)-b-poly(D,L-lactide) |
| PLA-TPGS | Poly(lactide)-D-α-tocopheryl polyethylene glycol succinate |
| MePEG-PLA | Methoxypoly(ethylene glycol)3000-poly(lactic acid)34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
| Male–PEG–PLA | Maleimide–poly(ethylene glycol)3400–poly(lactic acid)34000 |
| PTX | Paclitaxel |
| SSTR2 | Somatostatin receptor 2 |
| g7 | Glycopeptide |
| HAND | HIV-1-associated neurocognitive disorders |
| GALC CLEAs | Galactosylceramidase cross-linked enzyme aggregate |
| Cur | Curcumin |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 62175262, 81803480), The Science and Technology Innovation Program of Hunan Province (No. 2022RC1201), the Fundamental Research Funds for Central Universities of the Central South University (No. 2020CX021), and the Key R&D Plan of Hunan Province (No. 2022SK2101).Notes and references
- J. Hardy and D. J. Selkoe, Science, 2002, 297, 353 CrossRef CAS PubMed.
- J. M. Long and D. M. Holtzman, Cell, 2019, 179, 312 CrossRef CAS PubMed.
- C. Zihni, C. Mills, K. Matter and M. S. Balda, Nat. Rev. Mol. Cell Biol., 2017, 17, 564–580 CrossRef PubMed.
- S. Ding, A. I. Khan, X. Cai, Y. Song, Z. Lyu, D. Du, P. Dutta and Y. Lin, Mater. Today, 2020, 37, 112–125 CrossRef CAS PubMed.
- M. R. Freeman and D. H. Rowitch, Neuron, 2013, 80, 613–623 CrossRef CAS PubMed.
- R. Daneman, L. Zhou, A. A. Kebede and B. A. Barres, Nature, 2010, 468, 562–566 CrossRef CAS PubMed.
- M. D. Sweeney, K. Kisler, A. Montagne, A. W. Toga and B. V. Zlokovic, Nat. Neurosci., 2018, 21, 1318 CrossRef CAS PubMed.
- Y. H. Tsou, X. Q. Zhang, H. Zhu, S. Syed and X. Y. Xu, Small, 2018, 14, 1801588 CrossRef PubMed.
- T. P. Heffron, J. Med. Chem., 2016, 59, 10030 CrossRef CAS PubMed.
- T. Kheirbek and J. L. Pascual, Curr. Neurol. Neurosci. Rep., 2014, 14, 482 CrossRef PubMed.
- Y. C. Chen, C. F. Chiang, S. K. Wu, L. F. Chen, W. Y. Hsieh and W. L. Lin, J. Controlled Release, 2015, 211, 53 CrossRef CAS PubMed.
- N. U. Barua, S. S. Gill and S. Love, Brain Pathol., 2014, 2, 117–127 CrossRef PubMed.
- R. Awad, A. Avital and A. Sosnik, Acta Pharm. Sin. B, 2022 DOI:10.1016/j.apsb.2022.07.003.
- C. Ferraris, R. Cavalli, P. P. Panciani and L. Battaglia, Int. J. Nanomed., 2020, 15, 2999 CrossRef CAS PubMed.
- M. Nowak, M. E. Helgeson and S. Mitragotri, Adv. Ther., 2020, 3, 1900073 CrossRef.
- Y. H. Tsou, X. Q. Zhang, H. Zhu, S. Syed and X. Y. Xu, Small, 2017, 13, 1701921 CrossRef PubMed.
- D. Furtado, M. Bjornmalm, S. Ayton, A. I. Bush, K. Kempe and F. Caruso, Adv. Mater., 2018, 30, 1801362 CrossRef PubMed.
- J. B. Xie, Z. Y. Shen, Y. Anraku, K. Kataoka and X. Y. Chen, Biomaterials, 2019, 224, 119491 CrossRef CAS PubMed.
- E. Rideau, R. Dimova, P. Schwille, F. R. Wurm and K. Landfester, Chem. Soc. Rev., 2018, 47, 8572 RSC.
- M. Agrawal, Ajazuddin, D. K. Tripathi, S. Saraf, S. Saraf, S. G. Antimisiaris, S. Mourtas, M. Hammarlund-Udenaes and A. Alexander, J. Controlled Release, 2017, 260, 61 CrossRef CAS PubMed.
- C. Ross, M. Taylor, N. Fullwood and D. Allsop, Int. J. Nanomed., 2018, 13, 8507 CrossRef CAS PubMed.
- R. Alyautdin, I. Khalin, M. I. Nafeeza, M. H. Haron and D. Kuznetsov, Int. J. Nanomed., 2014, 9, 795 CAS.
- F. L. Xie, N. Yao, Y. Qin, Q. Y. Zhang, H. L. Chen, M. Q. Yuan, J. Tang, X. K. Li, W. Fan, Q. Zhang, Y. Wu, L. Hai and Q. He, Int. J. Nanomed., 2012, 7, 163 CrossRef CAS PubMed.
- Y. Zhang, H. Qu and X. Xue, Biomater. Sci., 2022, 10, 423 RSC.
- Y. Y. Zhang, L. Zhang, Y. Hu, K. Jiang, Z. Q. Li, Y.-Z. Lin, G. Wei and W. Y. Lu, J. Controlled Release, 2018, 289, 102 CrossRef CAS PubMed.
- G. Sharma, A. Modgil, B. Layek, K. Arora, C. W. Sun, B. Law and J. Singh, J. Controlled Release, 2013, 167, 1 CrossRef CAS PubMed.
- L. Kong, X. Li, Y. Ni, H. Xiao, Y. Yao, Y. Wang, R. Ju, H. Li, J. Liu, M. Fu, Y. Wu, J. Yang and L. Cheng, Int. J. Nanomed., 2020, 15, 2841–2858 CrossRef CAS PubMed.
- O. A. Marcos-Contreras, C. F. Greineder, R. Y. Kiseleva, H. Parhiza, L. R. Walsh, V. Z. Ramirez, J. W. Myerson, E. D. Hood, C. H. Villa, I. Tombacz, N. Pardi, A. Seliga, B. L. Mui, Y. K. Tam, P. M. Glassman, V. V. Shuvaev, J. Nong, J. S. Brenner, M. Khoshnejad, T. Madden, D. Weissmann, Y. Persidsky and V. R. Muzykantov, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 3405–3414 CrossRef CAS PubMed.
- B. D. S. Rodrigues, H. Oue, A. Banerjee, T. Kanekiyo and J. Singh, J. Controlled Release, 2018, 286, 264 CrossRef PubMed.
- S. Lakkadwala, B. D. S. Rodrigues, C. W. Sun and J. Singh, J. Controlled Release, 2019, 307, 247 CrossRef CAS PubMed.
- Y. Z. Zhao, B. X. Shen, X. Z. Li, M. Q. Tong, P. P. Xue, R. Chen, Q. Yao, B. Chen, J. Xiao and H. L. Xu, Nanoscale, 2020, 12, 15473 RSC.
- A. Papachristodoulou, R. D. Signorell, B. Werner, D. Brambilla, P. Luciani, M. Cavusoglu, J. Grandjean, M. Silginer, M. Rudin, E. Martin, M. Weller, P. Roth and J.-C. Leroux, J. Controlled Release, 2019, 295, 130 CrossRef CAS PubMed.
- C. Y. Lin, Y. C. Lin, C. Y. Huang, S. R. Wu, C. M. Chen and H. L. Liu, J. Controlled Release, 2020, 321, 519 CrossRef CAS PubMed.
- A. L. Bredlau, A. Motamarry, C. Chen, M. A. McCrackin, K. Helke, K. E. Armeson, K. Bynum, A. M. Broome and D. Haemmerich, Drug Delivery, 2018, 25, 973 CrossRef CAS PubMed.
- D. Shi, G. J. Mi, Y. Shen and T. J. Webster, Nanoscale, 2019, 11, 15057 RSC.
- C. Tapeinos, M. Battaglini and G. Ciofani, J. Controlled Release, 2017, 264, 306 CrossRef CAS PubMed.
- P. F. Zhao, Z. C. Le, L. X. Liu and Y. M. Chen, Nano Lett., 2020, 20, 5415 CrossRef CAS PubMed.
- Y. Zhao, Y. X. Chang, X. Hu, C. Y. Liu, L. H. Quan and Y. H. Liao, Int. J. Pharm., 2017, 516, 364 CrossRef CAS PubMed.
- Y. C. Kuo and H. F. Ko, Biomaterials, 2013, 34, 4818 CrossRef CAS PubMed.
- G. Graverini, V. Piazzini, E. Landucci, D. Pantano, P. Nardiello, F. Casamenti, D. E. Pellegrini-Giampietro, A. R. Bilia and M. C. Bergonzi, Colloids Surf., B, 2018, 161, 302–313 CrossRef CAS PubMed.
- G. R. Topal, M. Mészáros, G. Porkoláb, A. Szecskó, T. F. Polgár, L. Siklós, M. A. Deli, S. Veszelka and A. Bozkir, Pharmaceutics, 2022, 13, 38 CrossRef PubMed.
- R. D. Magro, F. Ornaghi, I. Cambianica, S. Beretta, F. Re, C. Musicanti, R. Rigolio, E. Donzelli, A. Canta, E. Ballarini, G. Cavaletti, P. Gasco and G. Sancini, J. Controlled Release, 2017, 249, 103 CrossRef PubMed.
- H. Cabralnull, K. Miyatanull, K. Osadanull and K. Kataoka, Chem. Rev., 2018, 118, 6844 CrossRef PubMed.
- Y. P. Li, K. Xiao, W. Zhu, W. B. Deng and K. S. Lam, Adv. Drug Delivery Rev., 2014, 66, 58 CrossRef CAS PubMed.
- S. M. Vasudevan, N. Ashwanikumar and G. S. V. Kumar, Biomater. Sci., 2019, 7, 4017 RSC.
- A. Singh, W. Kim, Y. Kim, K. Jeong, C. S. Kang, Y. Kim, J. Koh, S. D. Mahajan, P. N. Prasad and S. Kim, Adv. Funct. Mater., 2016, 26, 7057 CrossRef CAS PubMed.
- L. Lu, X. J. Zhao, T. W. Fu, K. Li, Y. He, Z. Luo, L. L. Dai, R. Zeng and K. Y. Cai, Biomaterials, 2020, 230, 119666 CrossRef CAS PubMed.
- C. Rinaldi and M. J. A. Wood, Nat. Rev. Neurol., 2018, 14, 9 CrossRef CAS PubMed.
- H. S. Min, H. J. Kim, M. Naito, S. Ogura, K. Toh, K. Hayashi, B. S. Kim, S. Fukushima, Y. Anraku, K. Miyata and K. Kataoka, Angew. Chem., Int. Ed., 2020, 59, 8173 CrossRef CAS PubMed.
- R. W. Ledeen and G. Wu, Trends Biochem. Sci., 2015, 40, 407 CrossRef CAS PubMed.
- Y. Yin, J. Wang, M. Yang, R. L. Du, G. Pontrelli, S. McGinty, G. X. Wang, T. Y. Yin and Y. Z. Wang, Nanoscale, 2020, 12, 2946–2960 RSC.
- Y. F. Lu, C. Li, Q. J. Chen, P. X. Liu, Q. Guo, Y. Zhang, X. L. Chen, Y. J. Zhang, W. X. Zhou, D. H. Liang, Y. W. Zhang, T. Sun, W. G. Lu and C. Jiang, Adv. Mater., 2019, 31, 1808361 CrossRef PubMed.
- Y. F. Lu, Z. Y. Guo, Y. J. Zhang, C. Li, Y. Zhang, Q. Guo, Q. J. Chen, X. L. Chen, X. He, L. S. Liu, C. H. Ruan, T. Sun, B. Ji, W. G. Lu and C. Jiang, Adv. Sci., 2018, 6, 1801586 CrossRef PubMed.
- N. G. Schipper, K. M. Vårum and P. Artursson, Pharm. Res., 1996, 13, 16861692 Search PubMed.
- Y. Chen and L. Liu, Adv. Drug Delivery Rev., 2012, 64, 640–665 CrossRef CAS PubMed.
- T. Patel, J. B. Zhou, J. M. Piepmeier and W. M. Saltzman, Adv. Drug Delivery Rev., 2012, 64, 701 CrossRef CAS PubMed.
- X. Guo, L. Wang, X. Wei and S. Zhou, J. Polym. Sci. Pol. Chem., 2016, 54, 3525–3550 CrossRef CAS.
- R. M. Koffie, C. T. Farrar, L. J. Saidi, C. M. William, B. T. Hyman and T. L. Spires-Jones, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 18837 CrossRef CAS PubMed.
- A. Friese, E. Seiller, G. Quack, B. Lorenz and J. Kreuter, Eur. J. Pharm. Biopharm., 2000, 49, 103 CrossRef CAS.
- Y. Wang, J. Luo and S. Y. Li, ACS Appl. Mater. Interfaces, 2019, 11, 3763 CrossRef CAS PubMed.
- Q. Y. Hu, X. L. Gao, G. Z. Gu, T. Kang, Y. F. Tu, Z. Y. Liu, Q. X. Song, L. Yao, Z. Q. Pang, X. G. Jiang, H. Z. Chen and J. Chen, Biomaterials, 2013, 34, 5640 CrossRef CAS PubMed.
- Q. Chen, J. W. Chen, Z. J. Yang, J. Xu, L. G. Xu, C. Liang, X. Han and Z. Liu, Adv. Mater., 2019, 31, 1802228 CrossRef PubMed.
- A. W. P. Fitzpatrick, B. Falcon, S. He, A. G. Murzin, G. Murshudov, H. J. Garringer, R. A. Crowther, B. Ghetti, M. Goedert and S. H. W. Scheres, Nature, 2017, 547, 185 CrossRef CAS PubMed.
- W. Qiang, W. M. Yau, J.-X. Lu, J. Collinge and R. Tycko, Nature, 2017, 541, 217 CrossRef CAS PubMed.
- S. N. Fan, Y. Q. Zheng, X. Liu, W. L. Fang, X. Y. Chen, W. Liao, X. N. Jing, M. Lei, E. X. Tao, Q. L. Ma, X. M. Zhang, R. Guo and J. Liu, Drug Delivery, 2018, 25, 1091 CrossRef CAS PubMed.
- R. Barbara, D. Belletti, F. Pederzoli, M. Masoni, J. Keller, A. Ballestrazzi, M. A. Vandelli, G. Tosi and A. M. Grabrucker, Int. J. Pharm., 2017, 526, 413 CrossRef CAS PubMed.
- Y. M. Tsai, C. F. Chiena, L. C. Lina and T.-H. Tsai, Int. J. Pharm., 2011, 416, 331 CrossRef CAS PubMed.
- P. C. Bhatt, A. Verma, F. A. Al-Abbasi, F. Anwar, V. Kumar and B. P. Panda, Int. J. Nanomed., 2017, 12, 8749 CrossRef CAS PubMed.
- T. Lei, Z. Yang, X. Xia, Y. Chen, X. Yang, R. Xie, F. Tong, X. Wan and H. Gao, Acta Pharm. Sin. B, 2021, 11, 4032–4044 CrossRef CAS PubMed.
- Y. N. Cui, M. Zhang, F. Zeng, H. Y. Jin, Q. Xu and Y. Z. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 32159 CrossRef CAS PubMed.
- Y. K. Bi, L. S. Liu, Y. F. Lu, T. Sun, C. Shen, X. L. Chen, Q. J. Chen, S. An, X. He, C. H. Ruan, Y. H. Wu, Y. J. Zhang, Q. Guo, Z. X. Zheng, Y. H. Liu, M. Q. Lou, S. G. Zhao and C. Jiang, ACS Appl. Mater. Interfaces, 2016, 8, 27465 CrossRef CAS PubMed.
- T. Kang, X. L. Gao, Q. Y. Hu, D. Jiang, X. Y. Feng, X. Zhang, Q. X. Song, L. Yao, M. Huang, X. G. Jiang, Z. Q. Pang, H. Z. Chen and J. Chen, Biomaterials, 2014, 35, 4319 CrossRef CAS PubMed.
- L. J. Cruz, M. A. Stammes, I. Que, E. Rv Beek, V. T. Knol-Blankevoort, T. J. A. Snoeks, A. Chan, E. L. Kaijzel and C. W. G. M. Lowik, J. Controlled Release, 2016, 223, 31 CrossRef CAS PubMed.
- A. D. Grosso, M. Gallianil, L. Angella, M. Santi, I. Tonazzini, G. Parlanti, G. Signore and M. Cecchini, Sci. Adv., 2019, 5, eaax7462 CrossRef PubMed.
- M. N. V. R. Kumar, R. A. A. Muzzarelli, C. Muzzarelli, H. Sashiwa and A. J. Domb, Chem. Rev., 2004, 104, 6017 CrossRef.
- S. W. Yu, X. L. Xu, J. F. Feng, M. Liu and K. L. Hu, Int. J. Pharm., 2019, 560, 282 CrossRef CAS PubMed.
- A. Trapani, E. D. Giglio, D. Cafagna, N. Denora, G. Agrimi, T. Cassano, S. Gaetani, V. Cuomo and G. Trapani, Int. J. Pharm., 2011, 419, 296 CrossRef CAS.
- J. J. Gu, K. Al-Bayati and E. A. Ho, Drug Delivery Transl. Res., 2017, 7, 497 CrossRef CAS.
- A. Jha, V. Ghormade, H. Kolge and K. M. Paknikar, J. Mater. Chem. B, 2019, 7, 3362 RSC.
- R. Yang, Y. Zheng, Q. J. Wang and L. Zhao, Nanoscale Res. Lett., 2018, 13, 330 CrossRef PubMed.
- L. Yang, Y. Cui, H. Liang, Z. Li, N. Wang, Y. Wang and G. Zheng, ACS Appl. Mater. Interfaces, 2022, 14(27), 30557–30570 CrossRef CAS PubMed.
- S. Mignani, J. Rodrigues, H. Tomas, M. Zablocka, X. Y. Shi, A. M. Caminade and J.-P. Majoral, Chem. Soc. Rev., 2018, 47, 514 RSC.
- H. He, Y. Li, X.-R. Jia, J. Du, X. Ying, W.-L. Lu, J.-N. Lou and Y. Wei, Biomaterials, 2011, 32, 478 CrossRef CAS PubMed.
- V. Leiro, S. D. Santos, C. D. F. Lopes and A. P. Pego, Adv. Funct. Mater., 2018, 28, 1700313 CrossRef.
- J. Li, H. M. Liang, J. Liu and Z. Y. Wang, Int. J. Pharm., 2018, 546, 215 CrossRef CAS.
- H. M. Teow, Z. Y. Zhou, M. Najlah, S. R. Yusof, N. J. Abbott and A. D'Emanuele, Int. J. Pharm., 2013, 441, 701 CrossRef CAS PubMed.
- Y. Li, H. He, X. R. Jia, W.-L. Lu, J. N. Lou and Y. Wei, Biomaterials, 2012, 33, 3899 CrossRef CAS PubMed.
- G. Y. Wang, X. W. Zhao, H. G. Wu, D. B. Lovejoy, M. Zheng, A. Lee, L. B. Fu, K. T. Miao, Y. An, N. Sayyadi, K. J. Ding, R. S. Chung, Y. Q. Lu, J. Li, M. Morsch and B. Y. Shi, Small, 2020, 16, 2003654 CrossRef CAS PubMed.
- S. X. Huang, J. F. Li, L. Han, S. H. Liu, H. J. Ma, R. Q. Huang and C. Jiang, Biomaterials, 2011, 32, 6832 CrossRef CAS PubMed.
- L. M. Liu, Q. Chen, L. W. Wen, C. Li, H. Qin and D. Xing, Adv. Funct. Mater., 2019, 29, 1808601 CrossRef.
- H. H. Yan, L. Wang, J. Y. Wang, X. F. Weng, H. Lei, X. X. Wang, L. Jiang, J. H. Zhu, W. Y. Lu, X. B. Wei and C. Li, ACS Nano, 2012, 6, 410 CrossRef CAS PubMed.
- G. Zhong, H. Long, T. Zhou, Y. Liu, J. Zhao, J. Han, X. Yang, Y. Yu, F. Chen and S. Shi, Biomaterials, 2022, 288, 121690 CrossRef CAS PubMed.
- Y. Li, X. Zhang, Z. Qi, X. Guo, X. Liu, W. Shi, Y. Liu and L. Du, Nano Res., 2020, 13, 2791–2802 CrossRef CAS.
- Z. Jin, L. Piao, G. Sun, C. Lv, Y. Jing and R. Jin, J. Drug Targeting, 2021, 29, 323–335 CrossRef CAS PubMed.
- Z. Haidar, R. Hamdy and M. Tabrizian, Biomaterials, 2008, 29, 1207–1215 CrossRef CAS PubMed.
- W. A. Jefferies, M. R. Brandon, S. V. Hunt, A. F. Williams, K. C. Gatter and D. Y. Mason, Nature, 1984, 312, 162–163 CrossRef CAS PubMed.
- G. Thom, M. Tian, J. P. Hatcher, N. Rodrigo, M. Burrell, I. Gurrell, T. Z. Vitalis, T. Abraham, W. A. Jefferies, C. I. Webster and R. Gabathuler, J. Cereb. Blood Flow Metab., 2019, 39, 2074–2088 CrossRef CAS PubMed.
- B. A. Eyford, C. S. B. Singh, T. Abraham, L. Munro, K. B. Choi, T. Hill, R. Hildebrandt, I. Welch, T. Z. Vitalis, R. Gabathuler, J. A. Gordon, H. Adomat, E. S. T. Guns, C. Lu, C. G. Pfeifer, M. Tian and W. A. Jefferies, Front. Mol. Biosci., 2021, 8, 611367 CrossRef CAS PubMed.
- L. Lu, S. Qi, Y. Chen, H. Luoa, S. Huang, X. Yu, Q. Lu and Z. Zhang, Biomaterials, 2020, 245, 119987 CrossRef CAS PubMed.
- D. P. Cristina, O. S. Benjam, S. N. Macarena, T. Meritxell and G. Ernest, Chem. Sci., 2018, 9, 8409–8415 RSC.
- H. Zhang, W. L. van Os, X. Tian, G. Zu, L. Ribovski, R. Bron, J. Bussmann, A. Kros, Y. Liu and I. S. Zuhorn, Biomater. Sci., 2021, 9, 7092–7103 RSC.
- C. Huang, C. Chuang, Y. Chen, H. Wang, J. Lin, C. Huang, K. Wei and F. Huang, J. Nanobiotechnol., 2021, 19, 180 CrossRef CAS PubMed.
- C. Gao, W. Gong, M. Yang, X. Chu, Y. Wang, Z. Li, Y. Yang and C. Gao, J. Drug Targeting, 2020, 28, 1085–1095 CrossRef CAS PubMed.
- G. Massaro, C. N. Z. Mattar, A. M. S. Wong, E. Sirka, S. M. K. Buckley, B. R. Herbert, S. Karlsso, D. P. Perocheau, D. Burke, S. Heales, A. Richard-Londt, S. Brandner, M. Huebecker, D. A. Priestman, F. M. Platt, K. Mills, A. Biswas, J. D. Cooper, J. K. Y. Chan, S. H. Cheng, S. N. Waddington and A. A. Rahim, Nat. Med., 2018, 24, 1317–1323 CrossRef CAS PubMed.
- D. Blessing and N. Déglon, Curr. Opin. Virol., 2016, 21, 61–66 CrossRef CAS PubMed.
- P. Colella, G. Ronzitti and F. Mingozzi, Mol. Ther.–Methods Clin. Dev., 2017, 8, 87–104 CrossRef PubMed.
- H. Fu, J. Dirosario, S. Killedar, K. Zaraspe and D. M. McCarty, Mol. Ther., 2011, 19, 1025–1033 CrossRef CAS PubMed.
- K. Y. Chan, M. J. Jang, B. B. Yoo, A. Greenbaum, N. Ravi, W. Wu, L. S. Guardado, C. Lois, S. K. Mazmanian, B. E. Deverman and V. Gradinaru, Nat. Neurosci., 2017, 20, 1172–1179 CrossRef CAS PubMed.
- B. Xie, X. Wang, Y. Pan, G. Jiang, J. Feng and Y. Lin, Theranostics, 2021, 11, 1177–1191 CrossRef CAS PubMed.
- J. Hordeaux, Y. Yuan, P. M. Clark, Q. Wang, R. A. Martino, J. J. Sims, P. Bell, A. Raymond, W. L. Stanford and J. M. Wilson, Mol. Ther., 2019, 27, 912–921 CrossRef CAS PubMed.
- D. Goertsen, N. C. Flytzanis, N. Goeden, M. R. Chuapoco, A. Cummins, Y. Chen, Y. Fan, Q. Zhang, J. Sharma, Y. Duan, L. Wang, G. Feng, Y. Chen, N. Y. Ip, J. Pickel and V. Gradinaru, Nat. Neurosci., 2022, 25, 106 CrossRef CAS PubMed.
- J. Hordeaux, Q. Wang, N. Katz, E. L. Buza, P. Bell and J. M. Wilson, Mol. Ther., 2018, 26, 664–668 CrossRef CAS PubMed.
- C. Chen, Z. Duan, Y. Yuan, R. Li, L. Pang, J. Liang, X. Xu and J. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 5864–5873 CrossRef CAS PubMed.
- B. Zhang, X. Sun, H. Mei, Y. Wang, Z. Liao, J. Chen, Q. Zhang, Y. Hu, Z. Pang and X. Jiang, Biomaterials, 2013, 34, 9171–9182 CrossRef CAS PubMed.
- X. Zhang, Z. Chai, A. L. Dobbins, M. S. Itano, C. Askew, Z. Miao, H. Niu, R. J. Samulski and C. Li, Biomaterials, 2022, 281, 121340 CrossRef CAS PubMed.
- P. K. Lo, P. Karam, F. A. Aldaye, C. K. McLaughlin, G. D. Hamblin, G. Cosa and H. F. Sleiman, Nat. Chem., 2010, 2, 319–328 CrossRef CAS PubMed.
- D. Jiang, Y. Sun, J. Li, Q. Li, M. Lv, B. Zhu, T. Tian, D. Cheng, J. Xia, L. Zhang, L. Wang, Q. Huang, J. Shi and C. Fan, ACS Appl. Mater. Interfaces, 2016, 8, 4378–4384 CrossRef CAS PubMed.
- K. R. Kim, D. Bang and D. R. Ahn, Biomater. Sci., 2016, 4, 605–609 RSC.
- N. Li, M. Wang, X. Gao, Z. Yu, W. Pan, H. Wang and B. Tang, Anal. Chem., 2017, 89, 6670–6677 CrossRef CAS PubMed.
- S. Shi, W. Fu, S. Lin, T. Tian, S. Li, X. Shao, Y. Zhang, T. Zhang, Z. Tang, Y. Zhou, Y. Lin and X. Cai, Nanomedicine, 2019, 21, 102061 CrossRef CAS PubMed.
- F. Xiao, L. Lin, Z. Chao, C. Shao, Z. Chen, Z. Wei, J. Lu, Y. Huang, L. Li, Q. Liu, Y. Liang and L. Tian, Angew. Chem., Int. Ed., 2020, 5, 9702–9710 CrossRef.
- D. Y. Tam, J. W. Ho, M. S. Chan, C. H. Lau, T. J. H. Chang, H. M. Leung, L. S. Liu, F. Wang, L. L. H. Chan, C. Tin and P. K. Lo, ACS Appl. Mater. Interfaces, 2020, 12, 28928–28940 CrossRef CAS PubMed.
- W. M. Pardridge, Front. Aging Neurosci., 2020, 11, 373 CrossRef PubMed.
- S. D. Li and L. Huang, Mol. Pharmaceutics, 2008, 5, 496 CrossRef CAS.
- Z. Yang, Z. W. Liu, R. P. Allaker, P. Reip, J. Oxford, Z. Ahmad and G. Ren, J. R. Soc., Interface, 2010, 7, S411 CrossRef CAS PubMed.
- C. Vinod and S. Jena, Front. Pharmacol., 2021, 12, 612692 CrossRef CAS PubMed.
- Y. Hu and J. Gao, Int. J. Pharm., 2010, 394, 115 CrossRef CAS PubMed.
- A. B. Engin and A. Engin, Prog. Brain Res., 2019, 245, 281 Search PubMed.
- Q. Du, D. Ge, V. Mirshafiee, C. Chen, M. Li, C. Xue, X. Maab and B. Sun, Nanoscale, 2019, 11, 12965 RSC.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |