DOI:
10.1039/D2TB00276K
(Review Article)
J. Mater. Chem. B, 2022,
10, 6590-6606
Recent development in the design of artificial enzymes through molecular imprinting technology†
Received
7th February 2022
, Accepted 25th May 2022
First published on 28th May 2022
Abstract
Enzymes, a class of proteins or RNA with high catalytic efficiency and specificity, have inspired generations of scientists to develop enzyme mimics with similar capabilities. Many enzyme mimics have been developed in the past few decades based on small molecules, DNA, and nanomaterials. These artificial enzymes are of great interest because of their low cost and high stability. However, most of these enzyme mimics do not have the desired substrate selectivity. The substrate selectivity of natural enzymes usually stems from a specific binding pocket. A powerful method to create substrate binding cavities is molecular imprinting technology (MIT). Molecularly imprinted polymers (MIPs) have three main characteristics: structural predictability, identification specificity, and application versatility compared with other identification systems. The MIP-based artificial enzymes have the advantages of simple preparation, low cost, and high stability and can realize excellent catalytic activity and selectivity. The development of MIP-based artificial enzymes has been further promoted by optimization methods such as imprinting transition state molecules, post-imprinting modification, opening cross-linked polymers’ internal space, and some special preparation methods. Combining molecular imprinting technology with nanozymes, the synergistic effect of both solved the defect of lack of specificity of nanozymes and improved their catalytic activity. This paper summarizes the recent research progress in preparing high-performance artificial enzymes based on MIPs and molecularly imprinted nanozymes. We hope to provide a reference for the design of artificial enzymes, reduce the gap between artificial enzymes and natural enzymes, and thus broaden the application of artificial enzymes in human life and production.
1. Introduction
Enzymes are potent protein-based biocatalysts. The discovery of ribozymes in the early 1980s expanded the definition of enzymes to RNA.1 Their specific functional sites allow them to be highly selective and significantly active towards target substrates and enable most biochemical processes to occur under physiological conditions.2,3 On the other hand, further applications of enzymes are often limited by poor stability, time-consuming preparation, and high cost.4–6 To overcome these drawbacks, scientists have made considerable efforts to design and synthesize artificial enzymes exploiting organic and inorganic chemistry, extending from semi-synthetic approaches (e.g., genetic modification of natural enzymes) to synthetic systems (e.g., cyclodextrins, metal complexes, porphyrins, dendrimers, and polymers).5,7,8 Artificial enzymes have better stability, ease of preparation, and low cost than natural enzymes. Although these artificial enzymes show promising catalytic activity, they are still not comparable to the activity of natural enzymes.9–11 A significant obstacle is the lack of suitable synthetic strategies to construct multifunctional active sites with precisely localized catalytic groups for complex structures and shapes of substrates.
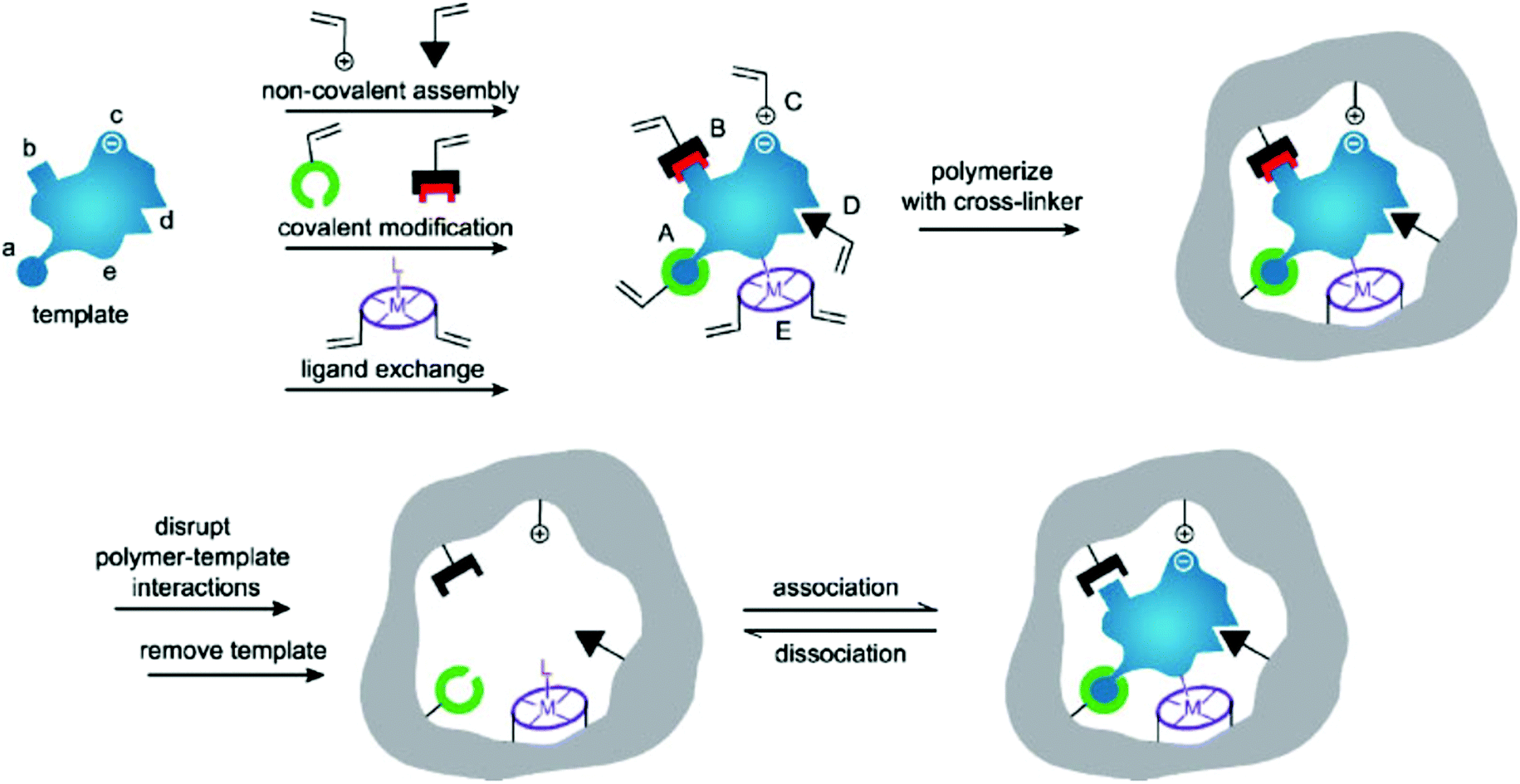
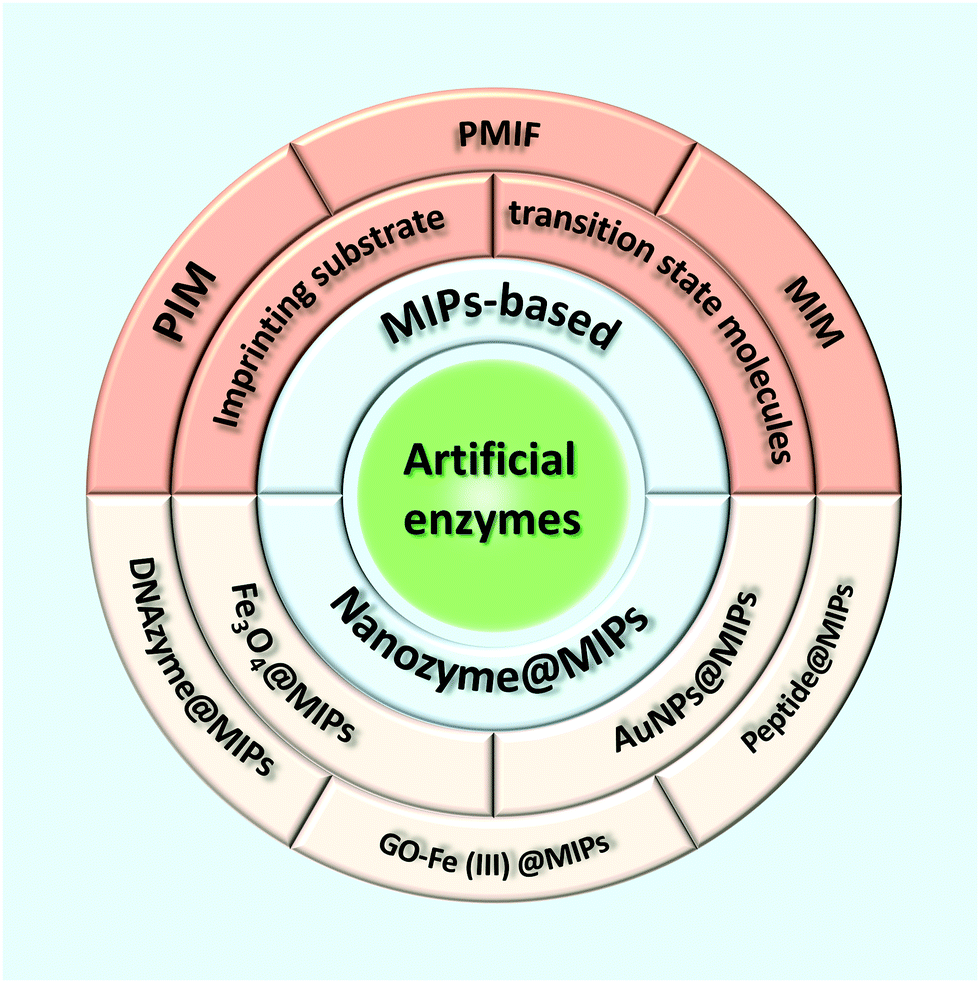
Molecular imprinting is an effective method for creating multifunctional binding sites for molecules of many different sizes.12–15 Moreover, due to the inherent advantages of molecularly imprinting polymers (MIPs) such as low cost, high selectivity, excellent thermochemical stability, and no need for biologically based protocols, molecular imprinting has developed into a feasible method for mimicking natural enzymes.16–21 In its conventional embodiment, the template molecule is pre-assembled with the functional monomer containing functional groups by certain driving forces (e.g., covalent bonding, hydrogen bonding, van der Waals forces, ionic interactions, metal–ligand interactions, or hydrophobic effects) (Fig. 1).22–27 The subsequent monomer–template complexes are copolymerized in a suitable porogenic solvent with the cross-linker. After removing the template from the cross-linked polymer network, the binding sites formed to complement the template in shape, size, and functional group distribution, so that the MIPs acquire selective template-binding capabilities.28,29 Catalytic MIPs at early stages focused on stereo controlled chemical reactions and enantioselective synthesis.30 Subsequently, a variety of catalytically active MIPs were synthesized by taking substrate analogs, transition states, and products or intermediates as templates.31–35 Although the MIP-based artificial enzymes prepared by such a strategy can exhibit high catalytic activity, there are still some obstacles that limit the higher activity, including difficulties in template removal, slow transport of materials inside and outside the polymer, and most importantly, heterogeneity of binding sites leading to extensive binding affinity.36 Therefore, it is desirable to develop suitable methods for the rational preparation of MIP-based artificial enzymes with high catalytic activity and high specific selectivity. Nanozymes are receiving more and more attention for their superior properties, so tailoring MIPs on nanozymes will provide a new way to develop the artificial enzyme field. This article reviews recent advances in preparing high-performance artificial enzymes based on MIPs and molecularly imprinted nanozymes (Scheme 1).
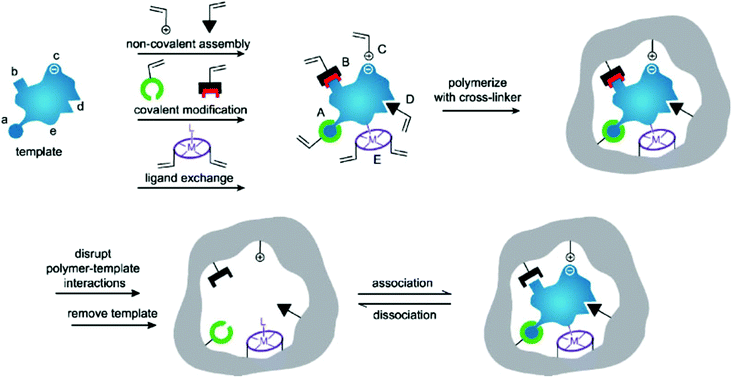 |
| | Fig. 1 Highly schematic representation of the molecular imprinting process: the formation of reversible interactions between the template and polymerizable functionality may involve one or more of the following interactions: [(A) reversible covalent bond(s), (B) covalently attached polymerizable binding groups that are activated for non-covalent interaction by template cleavage, (C) electrostatic interactions, (D) hydrophobic or van der Waals interactions or (E) co-ordination with a metal centre; each formed with complementary functional groups or structural elements of the template, (a–e) respectively]. A subsequent polymerization in the presence of crosslinker(s), a cross-linking reaction or other process, results in the formation of an insoluble matrix (which itself can contribute to recognition through steric, van der Waals and even electrostatic interactions) in which the template sites reside. The template is then removed from the polymer through disruption of polymer template interactions, and extraction from the matrix. The template, or analogues thereof, may then be selectively rebound by the polymer in the sites vacated by template, the imprints. While the representation here is specific to vinyl polymerization, the same basic scheme can equally be applied to sol–gel, polycondensation etc. Reprinted with permission from ref. 22. Copyright 2006 the John Wiley and Sons. | |
 |
| | Scheme 1 Recent advances in the preparation of high-performance artificial enzymes based on MIPs and molecularly imprinted nanozymes. | |
2. The design of molecularly imprinted polymer-based artificial enzymes
In molecular imprinting techniques, template molecules interact with functional monomers to form pre-polymerized complexes. After removing excess cross-linker and template molecules from the polymer, polymerization of such complexes can form imprinted cavities complementary to the template molecules' size, shape, and coordination spheres.37–39 Wulff's seminal report used a template covalently linked to a reactive monomer.40 Interest in molecularly imprinted polymers (MIPs) proliferated when Mosbach reported a more straightforward method using methacrylic acid and a cross-linking agent.41 Molecularly imprinted materials thus hold significant promise as selective adsorbents and specific biological recognition elements in medicine, chemistry, biology, and environmental and food sciences.37,42–46 In addition to using molecularly imprinted polymers called plastic antibodies as biological recognition elements, artificial enzymes called plastic enzymes can be designed. This approach involves imprinting substrates, substrate analogs, or structurally similar transition state molecules into the polymer structure to form cavities, which are artificial active sites for catalytic reactions. Table S1 (ESI†) summarizes the main MIPs-based artificial enzymes in this review.
2.1 Imprinting substrate molecules
Various artificial enzymes with high selectivity have been prepared using target substrates or substrate analogs as templates.47–49 These artificial enzymes can exhibit high catalytic activity because the cavities in the polymer can selectively bind to the corresponding substrate or substrate analogue and the catalytic groups (derived from the functional monomer) are complementary in position to the substrate or substrate analogue. Li's group synthesized a new molecularly imprinted polymer (MIP) by introducing heme as the catalytic center with the substrate (homovanillic acid and HVA) as the template and using several functional monomers to prepare the active site, which successfully simulated the natural peroxidase.50,51 The inherent difficulty of MIPs in recognizing template molecules in polar solutions was overcome by using the unique structure of heme and the multi–sites interactions provided by multiple functional monomers. The newly developed MIPs have about 30-fold higher recognition capacity for homovanillic acid and 8-fold higher catalytic activity than the NIPs, with significant advantages in specificity and stability over natural peroxidases. Nevertheless, the catalytic activity of the imprinted polymers was restricted due to their limited solubility in water. To improve their solubility, they prepared terpolymers of acrylamide, 4-vinyl pyridine, and heme, forming soluble enzyme systems in dimethyl sulfoxide (DMSO)-Tris/HCl (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :95, v/v) buffer.52 They exhibited higher catalytic activity than in pure Tris/HCl buffer. However, it was clear that preparing homogeneous imprinted catalysts with high catalytic activity in pure aqueous solutions was of more interest. Therefore, they constructed a novel horseradish peroxidase mimetic system based on an imprinted tetramer formed by cross-linking of 4-vinylpyridine, hemin, acrylamide, and N-isopropylacrylamide via ethylene glycol dimethacrylate with HVA as a template molecule.53 This novel artificial enzyme has adequate water solubility and dual pH and salt response properties, a further mimic of the natural enzyme.
:95, v/v) buffer.52 They exhibited higher catalytic activity than in pure Tris/HCl buffer. However, it was clear that preparing homogeneous imprinted catalysts with high catalytic activity in pure aqueous solutions was of more interest. Therefore, they constructed a novel horseradish peroxidase mimetic system based on an imprinted tetramer formed by cross-linking of 4-vinylpyridine, hemin, acrylamide, and N-isopropylacrylamide via ethylene glycol dimethacrylate with HVA as a template molecule.53 This novel artificial enzyme has adequate water solubility and dual pH and salt response properties, a further mimic of the natural enzyme.
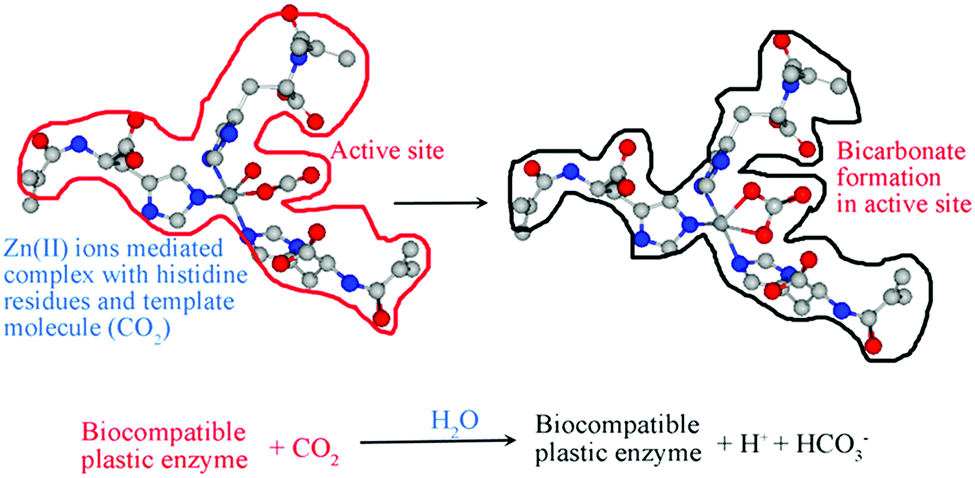
In this direction, Özgür's group has synthesized CO2-imprinted hydroxyethyl methacrylate-based cryogel membranes utilizing polymerizable L-histidine derivatives.54 The active site of carbonic anhydrase was simulated by molecular imprinting, and its CO2 biotransformation activity was examined (Fig. 2). The Wilber–Anderson method was used to calculate the hydratase activity of cryogel membranes. Wilber Anderson units (WAU) were calculated for cryogenic membranes containing MAH Zn2+ complex (1.0 mL), which increased the number of membranes by 1.07, 1.35, 1.40, 1.63, 2.01, and 2.56, respectively. The cryogel membranes also exhibited excellent stability. Repeated measurements were performed 10 times using cryogenic membranes containing MAH-Zn2+ complex (1.0 mL) to calculate the reusability of the membranes, and an activity loss of about 3% was observed in the results obtained.
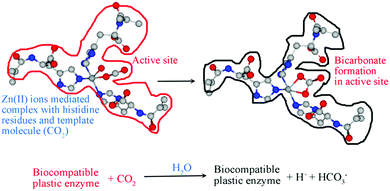 |
| | Fig. 2 CO2-imprinted hydroxyethyl methacrylate-based cryogel membranes (plastic enzymes) for CO2 bioconversion by means of the polymerizable L-histidine derivative (MAH). Reprinted with permission from ref. 54. Copyright 2021 the American Chemical Society. | |
Apart from selecting suitable functional monomers and template molecules, modulating the morphology of MIPs also has a considerable impact on their catalytic performance. Guo's group demonstrated that the hollow morphology of MIPs increased their hydrolysis rate by 88%.55 Therefore, they prepared molecularly imprinted nanocapsules with specially designed functional monomers using the substrate molecule methyl parathion as a template molecule for mimicking phosphotriesterase-catalyzed hydrolysis of organophosphorus pesticides.56 The results indicated that the prepared hollow MIPs not only had a catalytic performance approximately 400 times higher than the self-hydrolysis rate of the substrate but also could effectively adsorb p-nitrophenol, another contaminant to be eliminated, from the hydrolysis of organophosphorus compounds.
2.2 Imprinting transition state molecules
It is generally accepted that enzymes are spatially and electronically complementary to reactants in their rate-determining transition states, accelerating the reaction.57 The preferential binding of the reaction transition state reduces the activation energy of the reaction and thus can effectively enhance the reaction rate. Therefore, molecular imprinting using a stable transition state analog as a template to prepare artificial enzymes with high catalytic activity is a promising strategy. To develop efficient catalytic systems, the binding sites of MIPs need to be designed to stabilize the formation of the reaction transition state, thus reducing the transition energy requirement; suitable functional monomers need to be selected and precisely positioned to increase the transition state binding. After Jencks first proposed this approach and used it to construct so-called “catalytic antibodies”,58 the transition state analog as templates for molecular imprinting gained widespread application.59–63 Liu and Wulff synthesized catalytic MIPs to mimic the activity of Carboxypeptidase A using phenyl-2-pyridyl-phosphate, which mimics the hydrolysis transition state of carboxylic acid esters as a template molecule along with an amidinium functional group and a ligated Zn(II) trialkyl amine group.64 The results showed that the prepared MIP significantly promoted the hydrolysis of the substrate diphenyl carbonate. This is the first example of molecularly imprinted catalysts working more efficiently than catalytic antibodies. After that, they replaced the metal ion Zn2+ with Cu2+ to increase the metal complex's stability and assist the catalytic effect. The reaction rate was increased by a factor of 8000 compared to the background reaction.65 Later, to further improve the performance of the catalytic system, the authors added two amidine units to the trialkyl amine to form a complex that was coordinated to Cu2+ near the two molecules of the substrate interacting with the amidine. The optimized imprinting catalyst increased the hydrolysis rate of the corresponding carbonate by a factor of about 410000.66
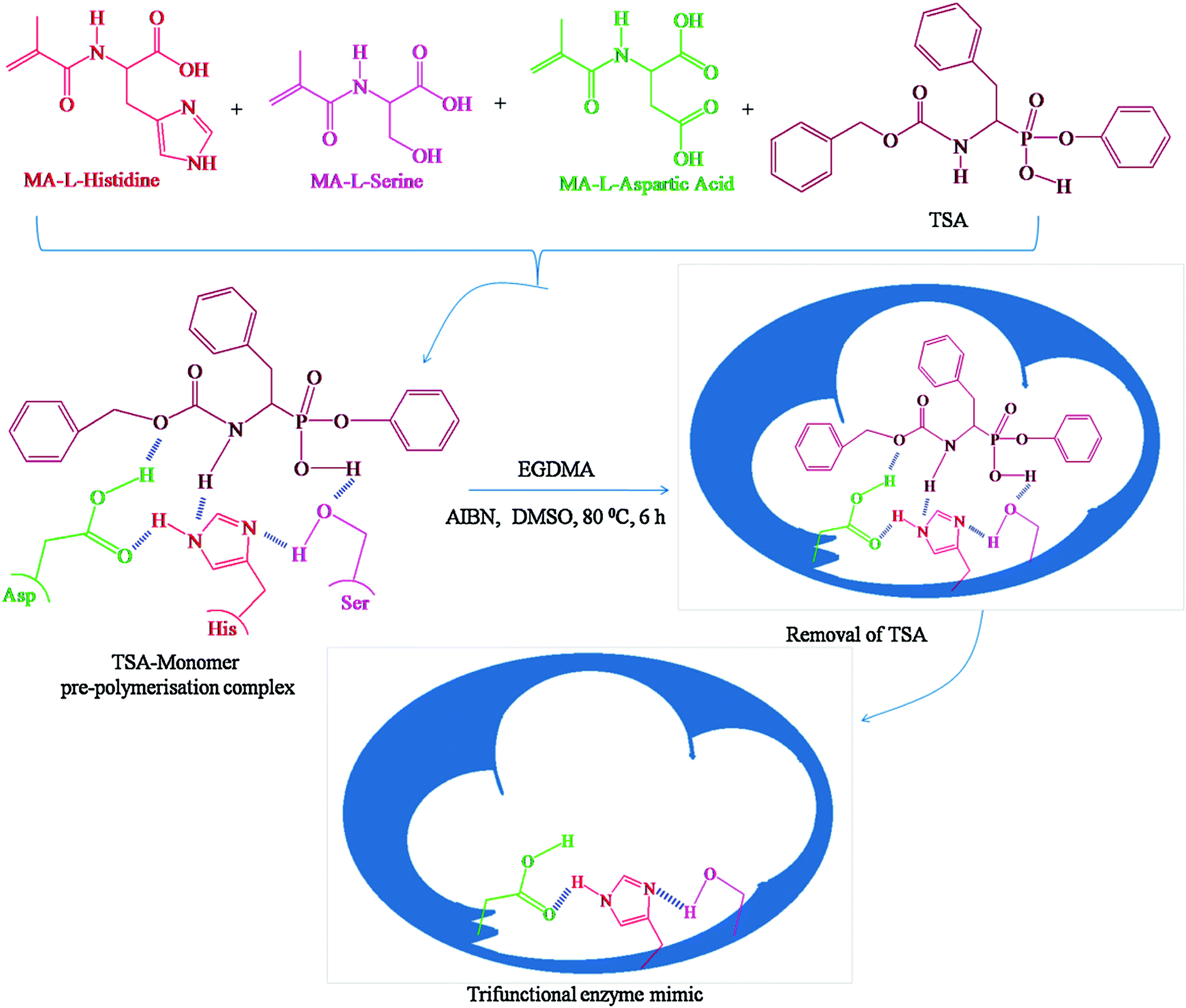
Devaky's group synthesized mimic chymotrypsin for the hydrolysis of anilide using a phosphate template as a stable transition mimic utilizing molecular imprinting techniques.67 Hydrolysis of aniline involves nucleophilic attack of OH− on the carbonyl center to form a tetrahedral carbon atom, which can be mimicked by a tetra-coordinated phosphorus atom (phenyl-1-(N-benzyloxycarbonylamino)-2-(phenyl)ethyl phosphonate) (Fig. 3). The polymer obtained after removal of the template underwent efficient hydrolysis of p-nitroanilide of phenylalanine, and the reaction conformed to pseudo-first-order kinetics while exhibiting stereospecificity and substrate selectivity (Fig. 3). They further synthesized a MIP mimicking chymotrypsin on multiwalled carbon nanotubes (MWCNT) using the transition state analogue phenyl 1-benzyloxycarbonylamino-4-methoxybenzyl phosphonate as a template.68 MWCNTs as a carrier material not only improved the specific surface area of MIPs but also enhanced the removal of the template as well as the binding ability and kinetics of molecular recognition of MIPs. Therefore, MWCNTs-grafted MIPs have better catalytic properties than ungrafted MWCNTs MIPs. They also extended their studies on TSA-imprinted chymotrypsin's hydrolase activity, which mimics targeting dipeptides containing chymotrypsin-specific amino acids as substrates.69 Proper orientation of the reactive functional groups in the hyper-crosslinked macroporous polymer matrix selectively binds the substrate and catalyzes its hydrolysis with high efficiency. Zhu's group prepared molecularly imprinted porous aromatic frameworks (MIPAF) by combining the porous aromatic frameworks with molecularly imprinted sites.70 The catalytic hydrolysis site consists of Zn2+ ligated with 1-vinylimidazole, methacrylic acid, and 1-(diethoxyphosphorylmethyl)-4-nitrobenzene, a transition state like the hydrolysis of paraoxon. The rate of hydrolysis of paraoxon by MIPAF (21 μM min−1) was even higher than that reported for natural organophosphorus hydrolases (1.42 μM min−1). However, this strategy has two serious drawbacks. On the one hand, the transition state structures of many reactions are still unclear and need to be elucidated by further studies. On the other hand, even if the transition state structures are known, the design and synthesis of tsa are still very challenging. Therefore, this strategy is very dependent on the development of organic chemistry.
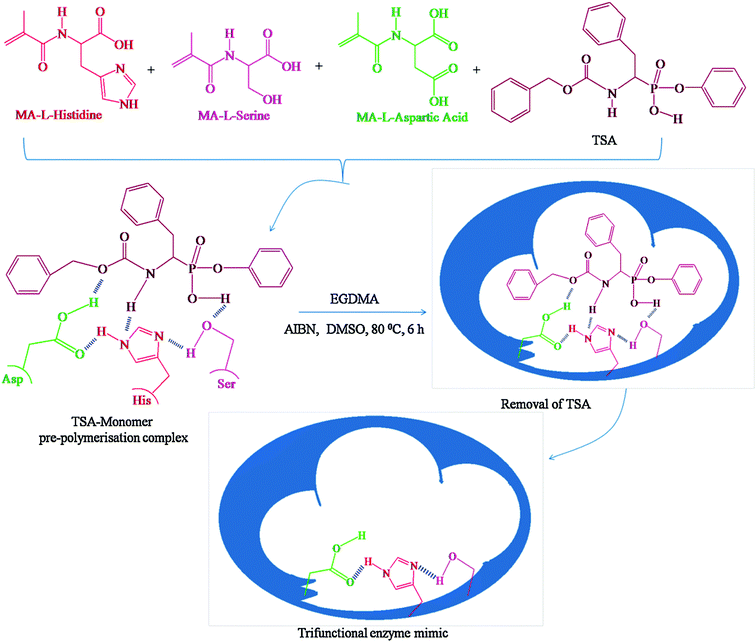 |
| | Fig. 3 Scheme for the synthesis of trifunctional enzyme mimic. Reprinted with permission from ref. 67. Copyright 2016 the Elsevier. | |
2.3 Post-imprinting modification (PIM)
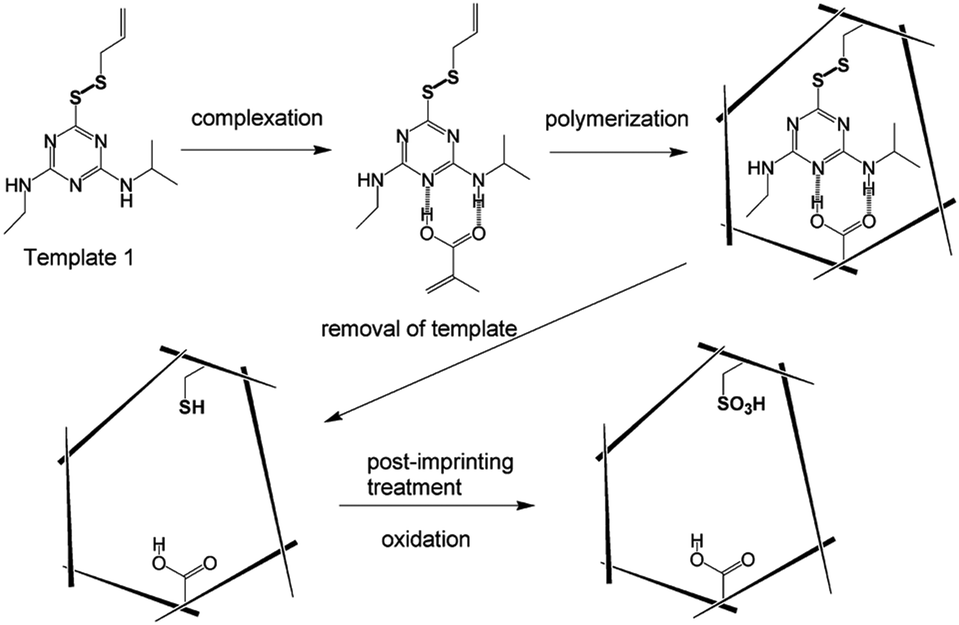
Partial catalytic residues of the functional monomer are present outside the cavity, leading to non-specific catalysis. To achieve higher selectivity, catalytic sites should be present only in the binding pocket. For this purpose, a new post-imprinting modification (PIM) method is proposed.67,71–75 PIM refers to the chemical modification of functional monomer residues in the cavity at a fixed point. The MIP substrate is first prepared using a functional monomer with a modifiable portion, such as a reversible bond (e.g., disulfide or imine bond), an amino group, or a carboxyl group. Then the functional monomer residues are modified by disulfide exchange, hydrolysis of imine bonds or chemical modification of amino, or carboxyl groups within the formation of binding cavities to tailor the functionality of the prepared MIPs.74 An example of using PIM is the construction of an artificial enzyme for the methanolysis of atrazine.76 Carboxylic acid and sulfonic acid are the two functional groups necessary for the cleavage of atrazine since the carboxylate adsorbs the substrate by electrostatic interaction, while the sulfonic acid acts as an acidic catalyst (Fig. 4). In the molecular imprinting process, atrazine analogs are combined with allyl or styryl groups through disulfide bonds at the 6-position to form templates and then copolymerized with methacrylic acid (functional monomer) and styrene/divinylbenzene (cross-linker) to prepare molecularly imprinted polymers with atrazine conversion activity (Fig. 4). Within the imprinting cavity, the atrazine groups covalently bound to the polymer matrix are removed by reducing the disulfide bonds, and the remaining thiol groups are oxidized to generate sulfonic acid (catalytically active group) (Fig. 4). By this post-imprinting treatment, the catalytic residues of the decomposed atrazine are selectively introduced into the catalytic center, efficiently completing the catalytic reaction while reducing the generation of unnecessary by-products.
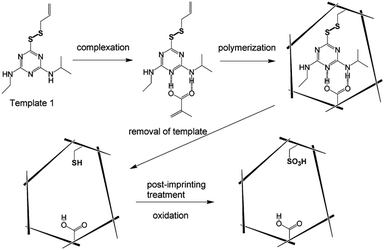 |
| | Fig. 4 Illustration of the preparation of atrazine transforming polymers using the post-imprinting process. Reprinted with permission from ref. 76. Copyright 2006 the Royal Society of Chemistry. | |
Glycosidase is a vital enzyme capable of selective hydrolysis of sugars. Although sugars can be hydrolyzed in acidic water, achieving highly selective hydrolysis using non-enzymatic catalysis is still an unrealized goal. Zhao's group prepared water-soluble polymer nanoparticles by molecular imprinting in cross-linked micelles with a boroxole bound to the sugar in the imprinted site.77 The post-modification installs an acidic group near the oxygen of the target glycosidic bond, with a systematic change in acidity and distance of the acid. The resulting synthetic mimetic glycosidases hydrolyze oligosaccharides and polysaccharides only in hot water in a highly controlled manner. These catalysts not only decompose straight-chain starch with similar selectivity as natural enzymes but can also be designed to have selectivity that biocatalysts do not have. The substrate selectivity depends mainly on the glycosyl residues within the active site, including their spatial orientation. In short, the catalytic groups are precisely installed within the imprinted pocket, which helps to prepare highly selective, high catalytic activity artificial enzymes.
2.4 Porous molecularly imprinted materials (PMIF)
To prepare high-performance MIPs, the first thing to pay attention to is the design of the functional monomer, which is closely related to the affinity between the imprinted sites and the substrate; secondly, it is the morphology control of the polymer, which is directly related to the mass transfer and substrate diffusion. In addition, for the common highly cross-linked MIPs, many functional sites are embedded in the internal region, resulting in the inaccessibility of the substrate to the active site and a decrease in catalytic activity.25 Efforts to improve mass transfer involve a variety of strategies, including making MIPs smaller. Because nano MIPs have some distinct advantages over previously developed MIPs with large size and irregular shape, including their high surface area to volume ratio, easy template removal, fast binding kinetics, and good dispersibility.78 In addition, finding an effective method to open the internal space of highly cross-linked MIPs would be beneficial to improve the accessibility of the active site and the catalytic activity.
2.4.1 Molecularly imprinted porous cross-linked polymers.
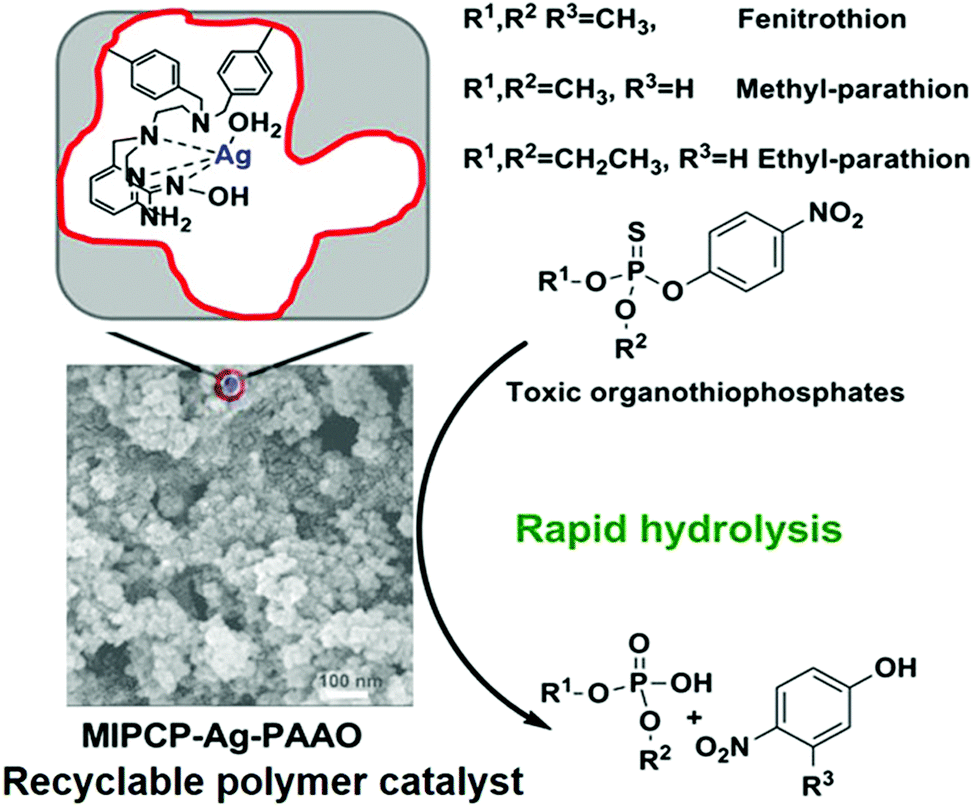
In this regard, porous cross-linked polymers prepared by the solvothermal method can generate porous backbones and provide abundantly accessible sites, which have obvious advantages in catalysis.79 Guo's group designed two novel Ag(I) complexes with synergistic pyridine and amidoxime ligands (Ag-PAAO) as functional monomers to mimic the organophosphorus hydrolase (OPH) catalytic site.80,81Fig. 5 represents the schematic diagram for the preparation of MIPCP-Ag-PAAO. First, the mixed Ag-PAAO and template (EP) were self-assembled to form Ag-PAAO-t; then Ag-PAAO-T was copolymerized with the cross-linker DVB under solvothermal conditions to obtain the polymer catalyst. Finally, the template and metal ions in the resulting polymer were removed and reloaded with metal ions to obtain the recyclable bio-nanocatalyst MIPCP-Ag-PAAO. From the 1H NMR experimental results, the EP- Ag-PAAO pre-assembly spectra are significantly right-shifted in the characteristic peaks compared with those of EP, indicating a strong binding interaction between EP and Ag-PAAO. In addition, density functional theory and experimental spectral analysis suggest that amidoxime has both a critical coordination role with silver ions and a nucleophilic attack to weaken the P-OAr bond at the catalytically active center. The observed pseudo primary rate constants (kobs) of MIPCP-Ag-PAAO-20% p-ethyl for the hydrolysis parathion (EP) were about 1.2 × 104 times higher than those for the self-hydrolysis (30 °C, pH = 9). Such easily prepared and low-cost MIPCP-Ag-PAAO compounds have great potential for practical applications in the field of artificial enzymes. Although MIPCP-Ag-PAAO showed high hydrolytic activity toward the substrate, the imprinting efficiency was not very high, possibly due to the high temperature in the generation of open pores that can affect and damage the imprinting sites. Therefore, this method requires further improvement of the pore-creating technique to improve the imprinting efficiency and catalytic performance.
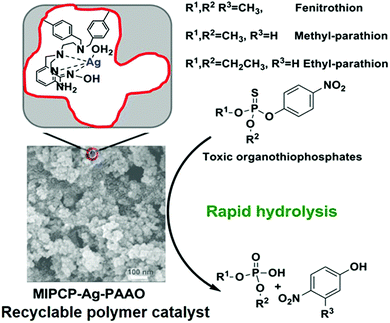 |
| | Fig. 5 The schematic diagram for the preparation of MIPCP-Ag-PAAO. Reprinted with permission from ref. 80. Copyright 2021 the American Chemical Society. | |
2.4.2 Molecularly imprinted MOF.
Metal–organic frameworks (MOFs) are novel porous materials formed by the self-assembly of metal ion clusters and organic linkers in a suitable solvent. Due to their customizable structures, controlled pore properties, and open metal active sites, MOFs have been widely used in catalysis, gas storage, adsorption and separation, and sensing applications.82–84 However, traditional MOFs usually have limited defects and poor binding selectivity, limiting their development in artificial enzymes. Molecular imprinting is a well-established method to generate highly specific cavities. The combination of molecular imprinting and MOF fabrication to produce imprinted MOFs with specific recognition defects will provide a new alternative for designing artificial enzymes. It has been reported that the specific adsorption capacity of MOFs can be enhanced by coating the MIP layer on their surface. AgNPs@ZnMOF coated with a molecularly imprinted layer with catalase-like activity had a low detection limit of 0.06 μmol L−1 for the detection of patulin.85 3D CoFeMOFs/AuNPs coated with polypyrrole (Ppy) based MIPs enabled trace detection of norfloxacin (NOR) electrochemistry.86 The effective surface area of the MIP/CoFe-MOFs/AuNPs/Glassy carbon electrode (GCE) was increased to 0.1058 cm2, indicating that more imprinting sites were generated, resulting in a lower limit of detection of 0.131 pmol L−1 and a wider linear range for MIP/CoFe-MOFs/AuNPs/GCE.
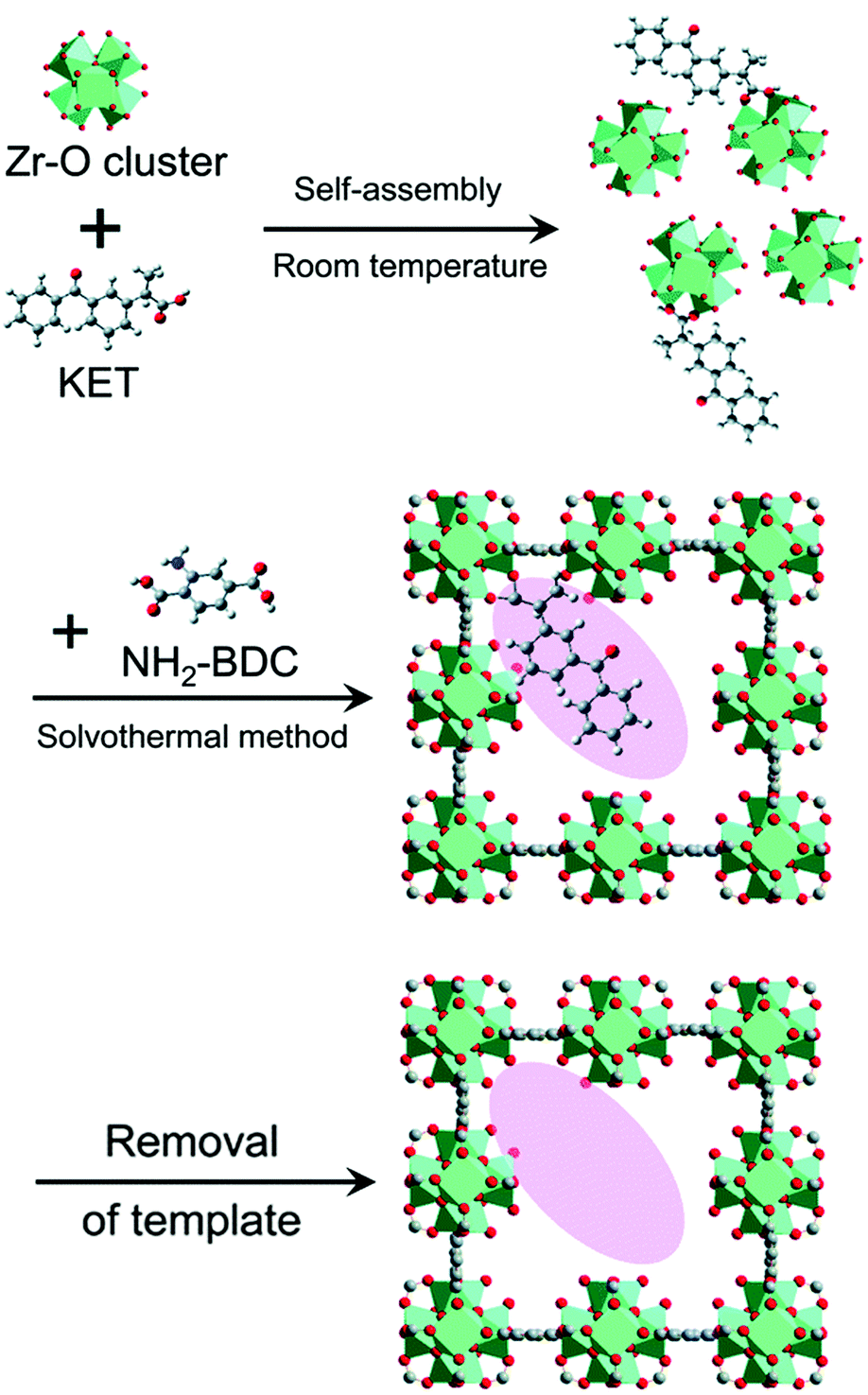
However, because the imprinted cavities are not inside the MOF matrix, the application of such MOF@MIPs in the field of catalysis will be limited. Shen's group added the template to the MOF system and tailored the imprinted cavities within the MOF matrix by exploiting the coordination interactions between the template molecules and the metal sites.87 The resulting active sites complement the template molecule in terms of both coordination interactions and three-dimensional dimensions (Fig. 6). The experimental results showed that different types of imprinted MOFs, including imprinted MIL-101_NH2 and imprinted UiO-66_NH2, had higher adsorption capacity and template selectivity compared with the non-imprinted MOFs, verifying the applicability of this synthetic strategy for the preparation of imprinted MOFs. The binding capacity of the imprinted UiO-66_NH2 to the template KET was as high as 295.2 mg g−1, which was much higher than that of the conventional MIPs. The characterization of the imprinted MOFs showed that the key factor for the generation of imprinting defects in MOFs was the coordination interaction between the template and the metal-centered clusters during the synthesis, which not only provided functional groups and shapes complementary to the target template, but also provided more unsaturated metal sites for catalysis. The presence of imprinted cavities in imprinted MIL-101_NH2 significantly accelerated the H2O2-mediated photocatalytic degradation of SM2, exhibiting higher photocatalytic activity (64%) and higher photocatalytic selectivity (4.74) than the non-imprinted MIL-101_NH2 system. Accordingly, the molecular imprinting strategy provides new insights into the design of artificial enzymes with ordered defects and high binding selectivity of MOFs groups.
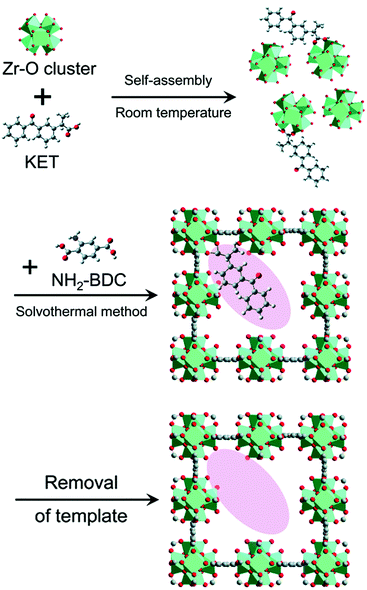 |
| | Fig. 6 Schematic illustration of the preparation of the imprinted UiO-66_NH2. Reprinted with permission from ref. 87. Copyright 2020 the Royal Society of Chemistry. | |
2.4.3 Molecularly imprinted PAF.
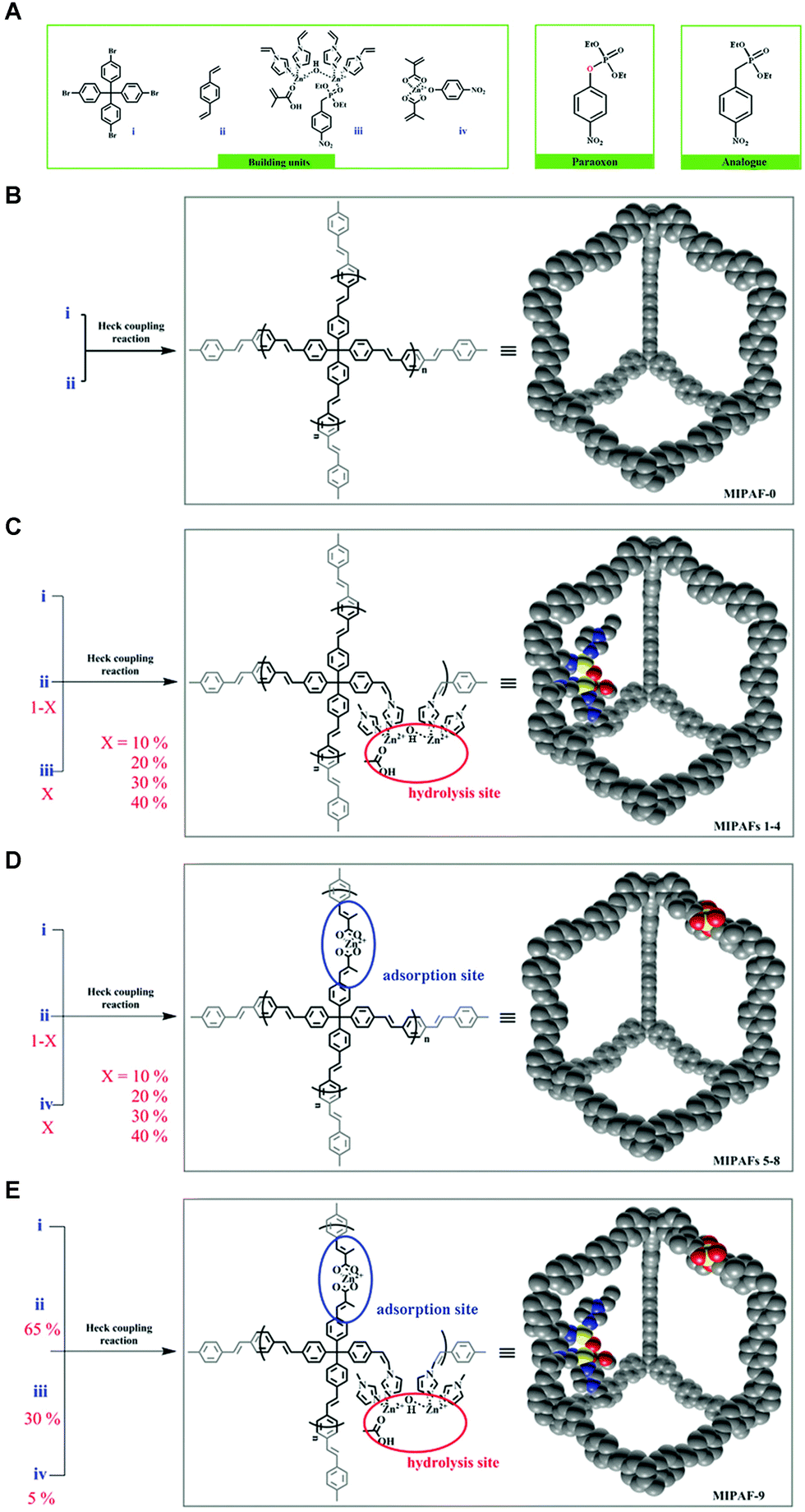
The tailored binding properties of molecularly imprinted polymers (MIPs) have facilitated the rapid development of artificial enzymes.88–90 However, the flexible nature of these polymers results in dense structures where only the active site is exposed on the outer surface of the particle, and most of the functional sites (∼60 to 80%) are embedded in the soft backbone, leading to low catalytic rates and leakage of the imprinted template.25 The PAF is formed by irreversible coupling reactions (generally C–C bonding) and has an ordered local structure.91 In contrast to crystalline materials with dynamic backbones (MOFs and COFs), the internal structure of PAFs, formed by irreversible covalent bonding, guarantees the stable presence of imprinted sites. Their hierarchical pores are suitable for transporting guests to the internal space. Their rigid backbone overcomes the microrheological properties of flexible polymers and facilitates the maintenance of the imprinted site structure.91 So, combining PAF materials with MIT is an effective pathway for constructing artificial enzymes. Zhu's group designed and synthesized molecularly imprinted PAFs for mimicking the activity of natural organophosphorus hydrolases.70 Molecularly imprinted motifs of coordinated zinc complexes containing p-oxa transition state analogs were embedded in PAFs to create hydrolysis sites. MIPAF-1, MIPAF-2, MIPAF-3, and MIPAF-4 (Fig. 7C) with 10%, 20%, 30%, and 40% hydrolysis sites, respectively, were synthesized by Heck coupling reaction. Their BET surface areas were 503, 390, 275, and 160 m2 g−1, respectively. The hydrolysis rate gradually increased from MIPAF-1 to MIPAF-3, and the hydrolysis rate of MIPAF-3 was 9.5 × 10−6 m min−1, which was 26 times higher than that of the conventional non-porous structure MIP. Then it decreased with the increase of MIPAF-4 hydrolysis site content. The hydrolysis product (p-nitrophenol) was used as a template and pre-assembled with zinc dimethacrylate to obtain the product adsorption complexes. Among the product adsorption complexes with 10%, 20%, 30% and 40% of MIPAF-5, MIPAF-6, MIPAF-7, and MIPAF-8 (Fig. 7D), respectively, MIPAF-7 exhibited the strongest uptake for p-nitrophenol with a maximum uptake of 32.06 μg mg−1, which was 4.6 times higher than that of the conventional polymer-based MIP (5.7 μg mg−1). Both substrate hydrolysis and product adsorption experiments reveal that the PAF scaffold can maintain the porous internal space and improve the utilization of the imprinting sites. MIPAF-9 (Fig. 7E) with 30% of substrate hydrolysis sites and 5% of product adsorption sites converted 17% of ethyl paraoxons in 2.5 hours, more than 14 times the rate of natural organophosphorus hydrolase. This indicates that efficient separation of the product from the catalytic site can accelerate the catalytic rate. These results suggest that molecularly imprinted PAFs can effectively mimic the activity of natural enzymes and provide good implications for the design of other artificial enzymes.
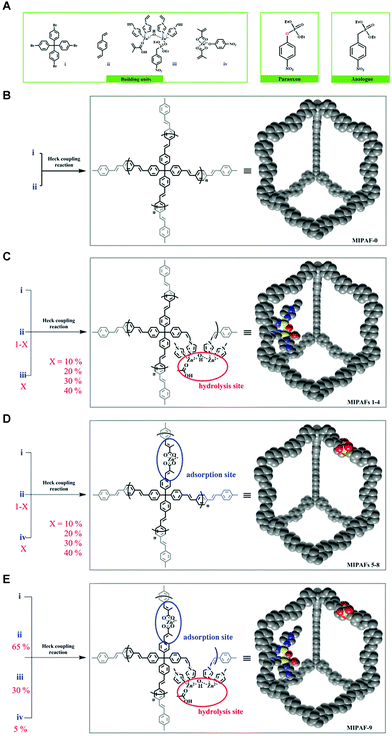 |
| | Fig. 7 (A) Building units of Heck coupling reaction. (B) Synthesis of organic porous material, MIPAF-0, as a scaffold for the design of porous artificial enzymes and the simulated structure to guide the eye. (C) Synthesis of MIPAFs 1 4 with hydrolysis site. (D) Synthesis of MIPAFs 5 6 with product adsorption site. (E) Synthesis of MIPAF-9 with both hydrolysis and product adsorption sites for the cascade process. Reprinted with permission from ref. 70. Copyright 2018 the John Wiley and Sons. | |
2.5 Molecularly imprinted micelle (MIM)
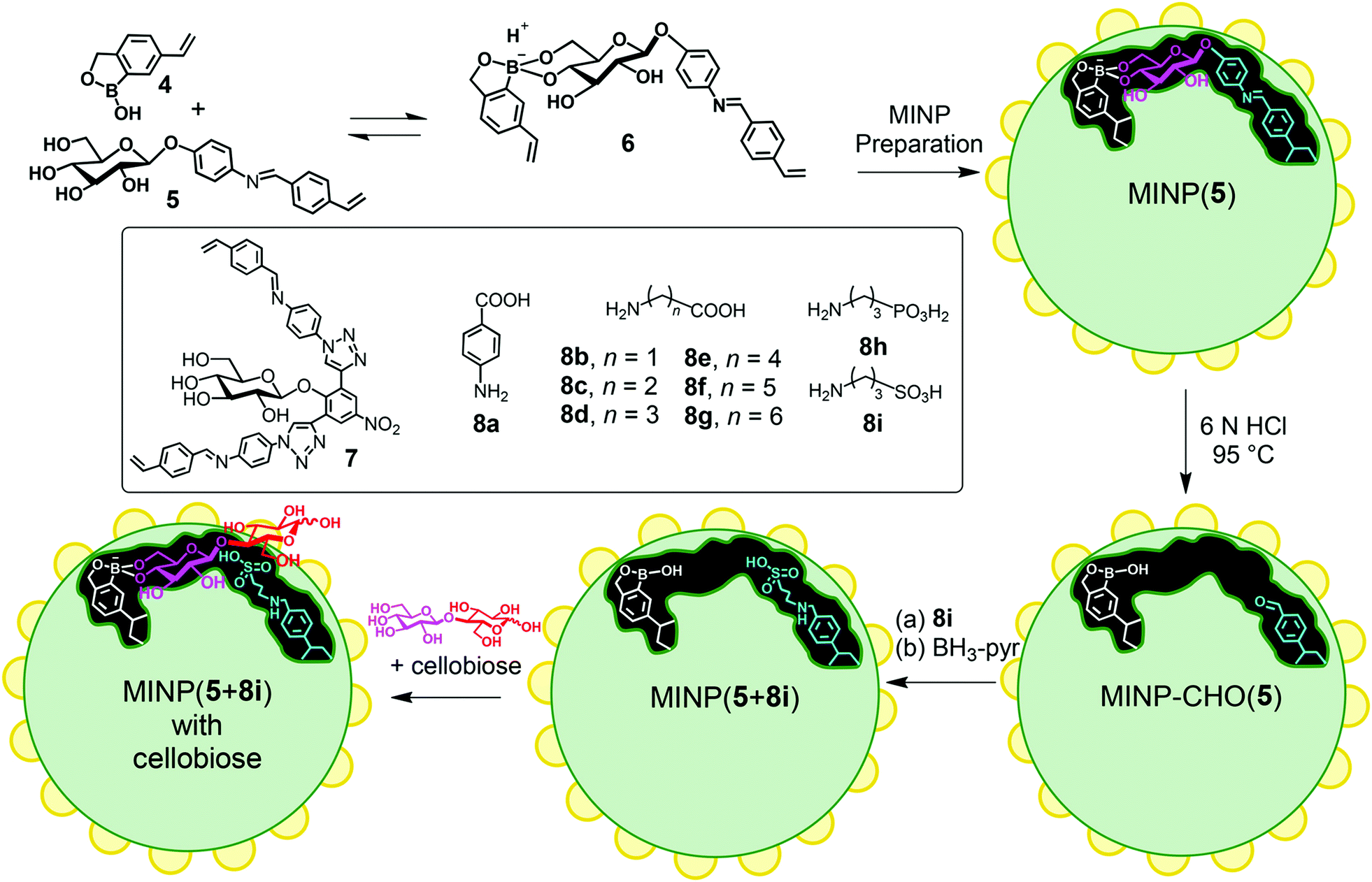
Zhao's group has developed a molecularly imprinted surfactant micelle approach to construct high-efficiency artificial enzymes in recent years.92 The nano-limited domain space generated by surface polymerization and cross-linking of micelles induce extraordinary templating effects, with imprinted/non-imprinted ratios (i.e., imprinting factors) in binding often reaching hundreds or even 10000.93–95 Catalytic groups are further installed in the imprinted pockets to generate mimetic enzymes with high selectivity and catalytic activity. An artificial glucosidase was constructed based on molecular imprinting in cross-linked micelles.96 First, mixed micelles of 1 and 2 were spontaneously formed, which contained template molecules, divinylbenzene (DVB, a free-radical crosslinker), and 2,2-dimethoxy-2-phenylacetophenone (DMPA, photo-initiator) (Fig. 8). The Cu(I) catalyst catalyzes an efficient alkyne azide click reaction to crosslink the micelle surface, and the proximity of reactive groups can facilitate this reaction. Then, UV-induced radical polymerization cross-links the micellar core around the template, and then a layer of hydrophilic ligands is installed on the micelle surface via a second round of click reaction between 3 and the residual alkyne on the micelle. These surface ligands can be cleaned by simple precipitation and organic solvents, while the template molecules are also removed. As shown in Fig. 9, MINP (5 + 8a–i) has a boroxole group at the active site that binds to cellobiose (or cellulose) terminal glucose and has an acid near the glycosidic bond catalyze hydrolysis. The two free hydroxyl groups of boronate 6 facilitate anchoring near the micelle surface. Keeping the active site close to the surface is also essential so that unbound glucose residues in the substrate remain in solution while the terminal glucose forms reversible boronate with boronic acids within the active site. Amino acids 8a–i allows the introduction of different acids at the active site, thus exhibiting different catalytic activities. The ability of MINP (7 + 8f) to hydrolyze cellobiose in aqueous solution approximates that of some natural β-glucosidases and functions at 83% H3PO4 and 90 °C in an ionic liquid/DMSO mixture. The enzyme-like activity of the synthetic glucosidase was 0.719 μmol mg−1 h−1/2:8 [C2 mim] OAc/DMSO with 5% H2O, which is unprecedented. Analogously, Zhao's group developed a site-selective artificial epoxide oxidase by micelle imprinting.97 When the template is removed, the formed imprinted sites generate peroxycarboxylic acid in situ under UV light irradiation, thus precisely fixing the catalytic site within the imprinted pockets. By adjusting the synthesized templates, the size and shape of the imprinted sites can also be regulated, and the position of the catalytic sites can be precisely located, allowing the prepared artificial enzymes to distinguish alkenes whose carbon–carbon double bond positions differ by one carbon. As discussed above, micelle imprinting technology is a method for bottom-up construction of efficient artificial enzymes using carefully designed templates and post-modified catalytic sites.
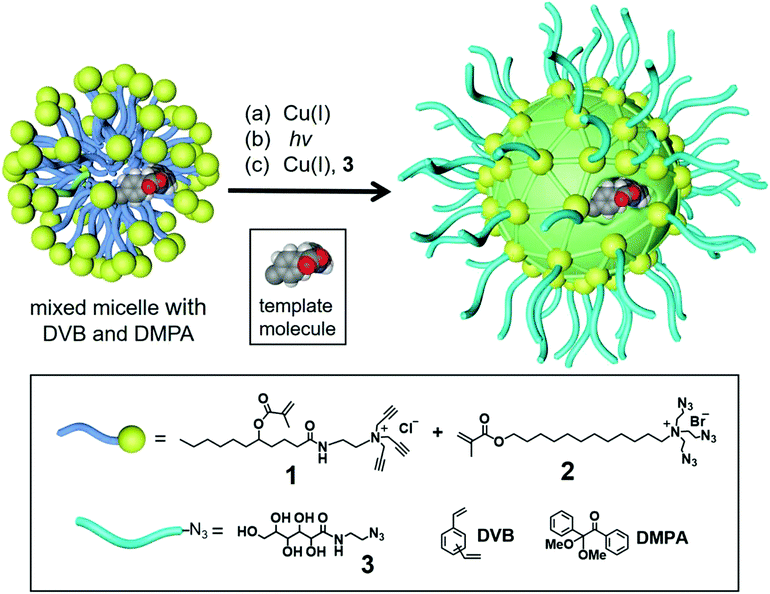 |
| | Fig. 8 General procedure for the MINP preparation from a mixed micelle of 1 and 2 containing DVB and DMPA. Reprinted with permission from ref. 96. Copyright 2021 the American Chemical Society. | |
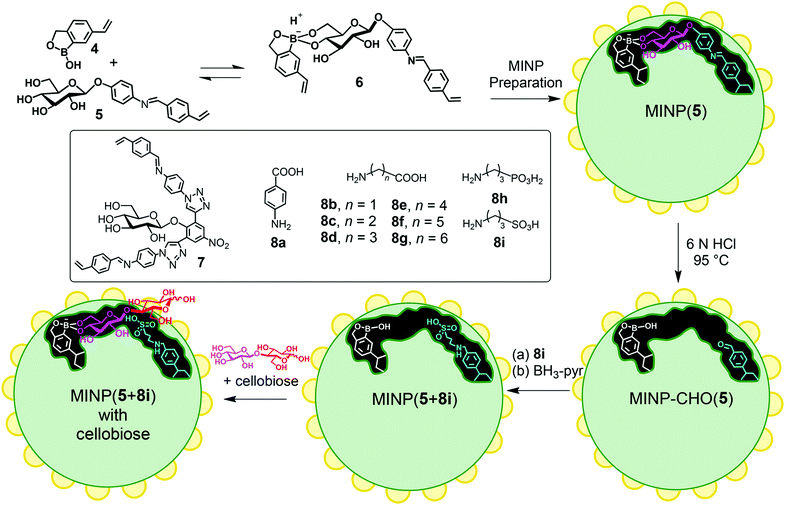 |
| | Fig. 9 Preparation of MINP-based synthetic glucosidase, with a schematic representation of the active site functionalized with 8i and a cellobiose bound by the boroxole group introduced through FM 4. Reprinted with permission from ref. 96. Copyright 2021 the American Chemical Society. | |
3. Molecular imprinting on nanozymes
Nanozymes are a series of nanomaterials that possess enzyme-like activities, such as oxidase-like, peroxidase-like, hydrogen peroxidase-like, superoxide-like dismutase, and laccase like activities, under near-physiological conditions.98–106 Nanozymes have been widely used in biosensor development, environmental remediation, and nanomedicine with higher stability and lower cost than natural enzymes.8,107,108 However, nanozymes lack specific recognition and binding of substrates, one of the major characteristics of most natural enzymes. To solve this problem, compared with biological ligands such as antibodies or aptamers, stable and economic molecularly imprinted polymers are more suitable for constructing substrate binding sites for nanozymes. Molecular imprinting technology can bridge the gap between nanozymes and natural enzymes. Table S2 (ESI†) summarizes the main Nanozyme@MIPs in this review.
3.1 Fe3O4@MIPs
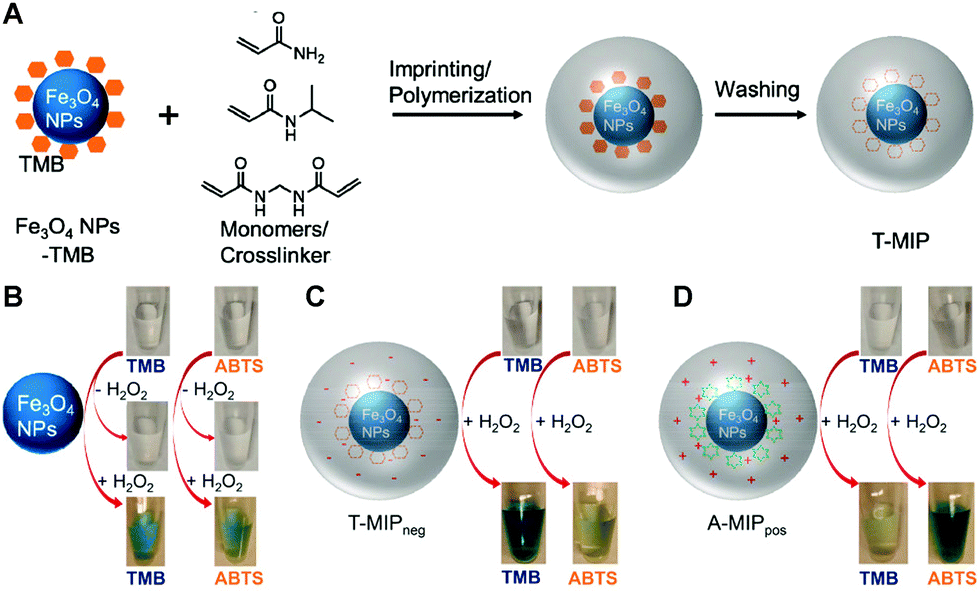
Iron oxide nanoparticles (NPs) are one of the first reported nanozymes with peroxide-like activity.99 Because of their potent activity and good biocompatibility, the group of Liu encapsulated Fe3O4 nanoparticles with peroxidase-like activity in MIP by aqueous precipitation polymerization (Fig. 10A).90 The peroxidase-like activity of Fe3O4 NPs was confirmed by mixing TMB and ABTS substrates with Fe3O4 NPs and H2O2, respectively, and TMB and ABTS turned blue and green, respectively (Fig. 10B). The change in the ζ-potential of Fe3O4 NPs after mixing TMB and ABTS with free Fe3O4 NPs, respectively, indicates that the substrates can adsorb to the surface of Fe3O4 NPs, which is the basis for ensuring that surface imprinting can be achieved. Acrylamide and NIPAAm (monomers), and MBAAm (a crosslinker) are added to the Fe3O4 NPs and TMB (or ABTS) mixture. After adding the initiator, the nanogels are obtained by precipitation polymerization, and then specific recognition pockets are formed when the substrate template is rinsed off. The TMB and ABTS imprinted gels were named T-MIP and A-MIP, respectively. In addition, gels without imprinted TMB or ABTS were named non-imprinted polymers (NIP). The kcat/Km of T-MIP (6.8 × 10−2 s−1 μM−1) is 2.8 times higher than that of bare Fe3O4 (2.4 × 10−2 s−1 μM−1). The same gel oxidized ABTS with kcat/Km about 3 times lower than that of bare Fe3O4. Similarly, the specificity of A-MIP for oxidation of ABTS was 4-fold higher than that of bare Fe3O4 and 1.5-fold lower for oxidation of TMB. Since TMB is positively charged and ABTS is negatively charged, the imprinting can be further improved by adding cationic DMPA or anionic AMPS. The TMB-imprinted nanogel containing anionic AMPS is named T-MIPneg, and the TMB-imprinted nanogel containing cationic DMPA is named T-MIPpos. T-MIPneg has the best catalytic efficiency, 15 times higher than bare Fe3O4 NPs, which is better than T-MIP gels without AMPS monomers. Meanwhile, T-MIPneg showed the lowest oxidation efficiency for ABTS. The same trend was observed in the A-MIPpos gels. In the best case, the selectivity of TMB using T-MIPneg nanogels was 98 times higher than that of ABTS, while the selectivity of ABTS using A-MIPpos was 33 times higher than that of TMB. These results can be reflected in the visual colorimetric tests (Fig. 10C and D) and are the reason for the enhanced activity, and the authors used a surface science approach to decompose the mechanism of the nanozyme-catalyzed reaction into three steps: adsorption of the substrate, reaction, and product release.109 The enrichment of the local substrate concentration is about 8-fold due to the imprinting effect, and the increase in substrate concentration can promote an increase in enzyme-like activity. The MIP gel layer has faster molecular transport kinetics than the NIP sample. The substrate imprinted gel also reduced the reaction energy barrier with the lowest activation energy. The activation energy (Ea) decreases from 29.1 kJ mol−1 (Fe3O4) to 13.8 kJ mol−1 (A-MIPpos). Since the oxidation product has a different geometry, it cannot fit into the imprinting pocket and will be released to regenerate the substrate binding sites. The same approach was successfully applied to the imprinting of gold nanoparticles (peroxidase-like activity) and nanoceria (oxidase-like activity), showing that molecular imprinting is a versatile method to improve activity and specificity. Other examples include the growth of polypyrrole (PPy)-based MIP on Fe3O4 nanoparticles using methylene blue (MB) as a substrate.110 The Fe3O4@PPy composites showed better catalytic performance towards MB in the presence of the sulfate-radical oxidant sodium persulfate compared to bare Fe3O4 nanoparticles. Interestingly, Fe3O4@PPy could still degrade more than 80% of MB after five cycles. The in-depth study of the activity and specificity of the imprinted nanozymes can provide meaningful guidance for the further rational design of molecularly imprinted nanozymes.
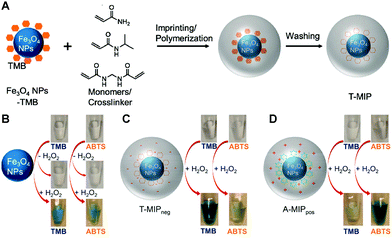 |
| | Fig. 10 (A) A scheme of imprinting TMB on Fe3O4 NPs. Photographs showing the activity and specificity of (B) Fe3O4 NPs, (C) T-MIPneg and (D) A-MIPpos nanogels for the oxidation of TMB and ABTS with or without H2O2. Fe3O4 NPs (50 μg mL−1, free or in nanogels) were used in all experiments oxidizing 0.5 mM substrates with 10 mM H2O2 in buffer A (20 mM acetate, pH 4.0) for 30 min at room temperature. Reprinted with permission from ref. 90. Copyright 2017 the American Chemical Society. | |
3.2 AuNPs@MIPs
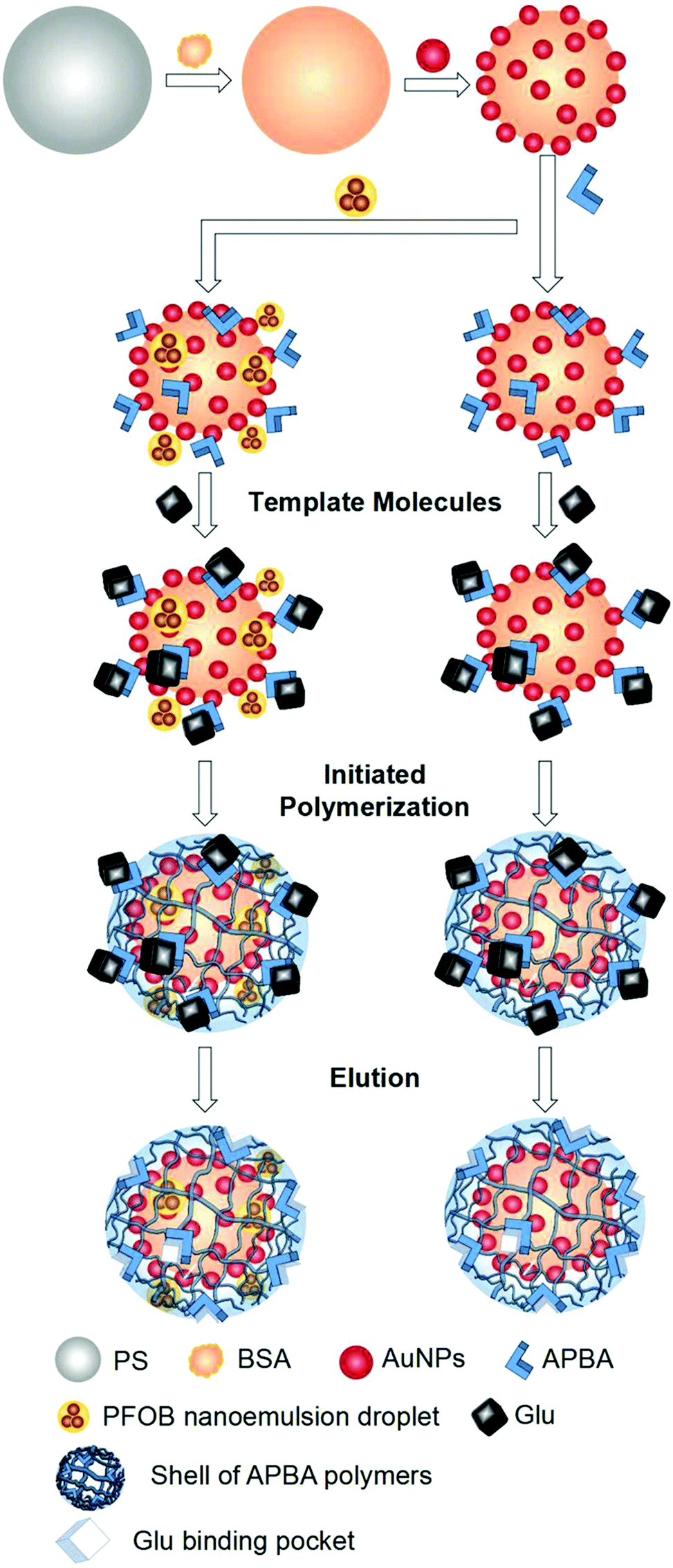
To improve the selectivity of AuNP-based glucose oxidase (GOD) mimics, Zhang's group introduced molecular imprinting technology to construct mimics with higher catalytic activity and specific recognition of glucose (Glu).111 Polystyrene microspheres (PS) were used as carriers to which AuNPs were adsorbed. Aminophenylboronic acid (APBA) acts as a recognition molecule that can bind to the adjacent hydroxyl groups of the glycans under alkaline or neutral conditions (Fig. 11). APBA was adsorbed to AuNPs by electrostatic adsorption and N-Au bonding between AuNPs and APBA amine groups and then bound to the template molecule of Glu under anaerobic conditions. APBA can be used to construct molecularly imprinted polymer (MIP) shell layers by forming brown polymers through ammonium persulphate (APS)-initiated reactive amino groups in addition to binding to neighboring hydroxyl groups (Fig. 11). In the presence of N,N-methylene bisacrylamide (MBA) as a cross-linking agent, the free APBA polymerizes with the APBA adhered to the Au–PS surface to form a net-like structure. Then, the polymer was deposited on the surface of microspheres to form an APBA shell layer around Au–PS. After removal of the template molecules, a shell layer with Glu-binding pockets was formed on the Au–PS surface capable of specifically recognizing, trapping, and enriching Glu with significant selectivity for the substrate (Fig. 11). On this basis, a heptadecafluoro-n-octyl bromide nanoemulsion with oxygen supply function was also introduced to further improve the catalytic activity, successfully increasing the catalytic efficiency (kcat/Km) by about 270-fold. This improved catalytic efficiency could also be attributed to the spatial confinement effect of molecularly imprinted porous shells. These AuNPs-based GOD mimics have been successfully employed for actual Glu detection in drinks and blood glucose.
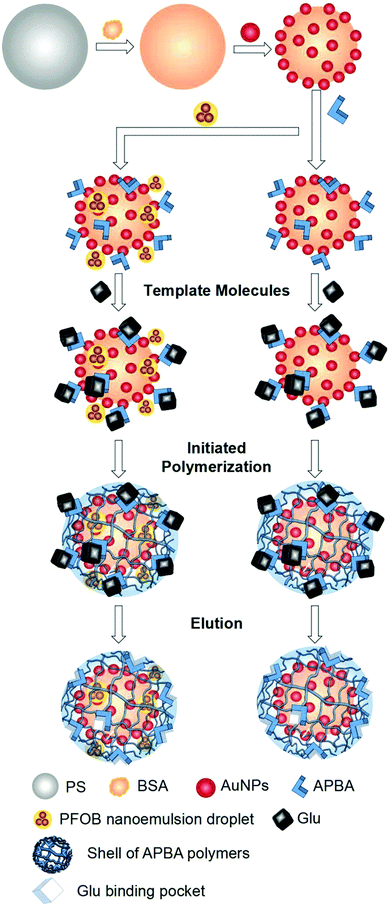 |
| | Fig. 11 Principle of the AuNP-based GOD mimic with enhanced activity and selectivity constructed by molecular imprinting. Reprinted with permission from ref. 111. Copyright 2018 the John Wiley and Sons. | |
3.3 DNAzyme@MIPs
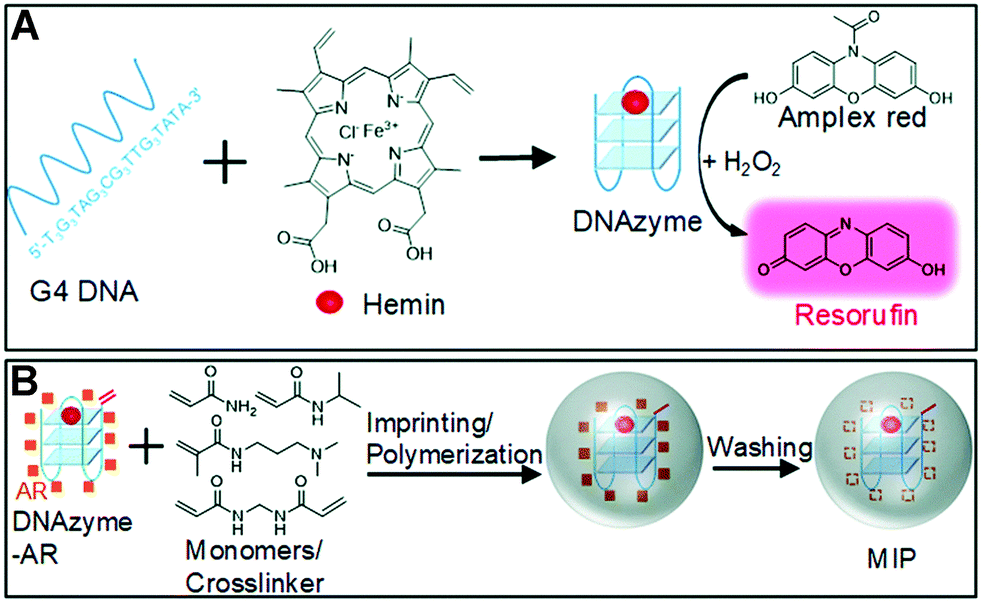
This peroxidase DNAzyme is based on guanine tetraploid (G4) DNA, with heme as a cofactor (Fig. 12A). It catalyzes the oxidation of many substrates in the presence of H2O2, such as 3,3,5,5-tetramethylbenzidine (TMB), 2,2-azidobis (3-ethylbenzothiazoline-6-sulfonic acid) (ABTS), dopamine, and 10-acetyl-3,7-dihydroxyphenoxazine (Amplex Red, AR).112 Liu's group integrated the DNAzyme into the nanogel by molecular imprinting and polymerization to achieve specific oxidation (Fig. 12B).112 An acrylate group was added to the 5′-end of the G4 DNA to make it copolymerized. The acrydite-modified DNAzyme complex was first mixed with AR (template) in the absence of H2O2, thus avoiding being oxidized. At pH 7.4 (imprinting conditions), AR is close to charge-neutral and therefore not electrostatically repelled by DNAzyme. Its aromatic ring and polar groups can interact with DNA bases and facilitate adsorption on the DNAzyme surface. In addition, acrylamide (AAm) and N-isopropyl acrylamide (NIPAAm) and partially (15 mol%) positively charged N-[3-(dimethylamino)propyl] methacrylamide (DMPA) (functional monomers) and N,N-methylenebisacrylamide (MBAAm) (cross-linker) were added. After reaction initiation, the system underwent precipitation polymerization to obtain gel nanoparticles. After removal of the AR template by extensive washing, the imprinted DNAzyme nanogels were collected. MIP gels (Km = 1.8 ± 0.6 μM) exhibited the highest affinity for the substrate, compared to free DNAzyme (Km = 5.5 ± 1.5 μM) and NIP gels (Km = 8.5 ± 1.5 μM). The oxidation rate of MIP nanogels (at ∼250 ΔF min−1) was over 2.7 times faster than that of NIP gels and over 3.5 times faster than that of free DNAzyme, confirming that MIP favors substrate binding and thus enhances catalytic activity. The colorimetric test showed that MIP nanogels could promote the catalysis of AR compared with free DNAzyme, while the other three substrates (TMB, ABTS and DOPA) were even inhibited, proving that MIP nanogels were indeed capable of selective oxidation of AR.
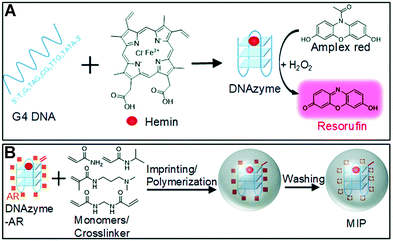 |
| | Fig. 12 (A) A schematic representation of the peroxidase mimicking DNAzyme and its oxidation of AR to fluorescent resorufin in the presence of H2O2. (B) A scheme of preparing DNAzyme containing molecularly imprinted nanogels (MIP) using AR as a template. Reprinted with permission from ref. 112. Copyright 2018 the Royal Society of Chemistry. | |
3.4 Graphene oxide (GO) iron(III) complexes@MIPs
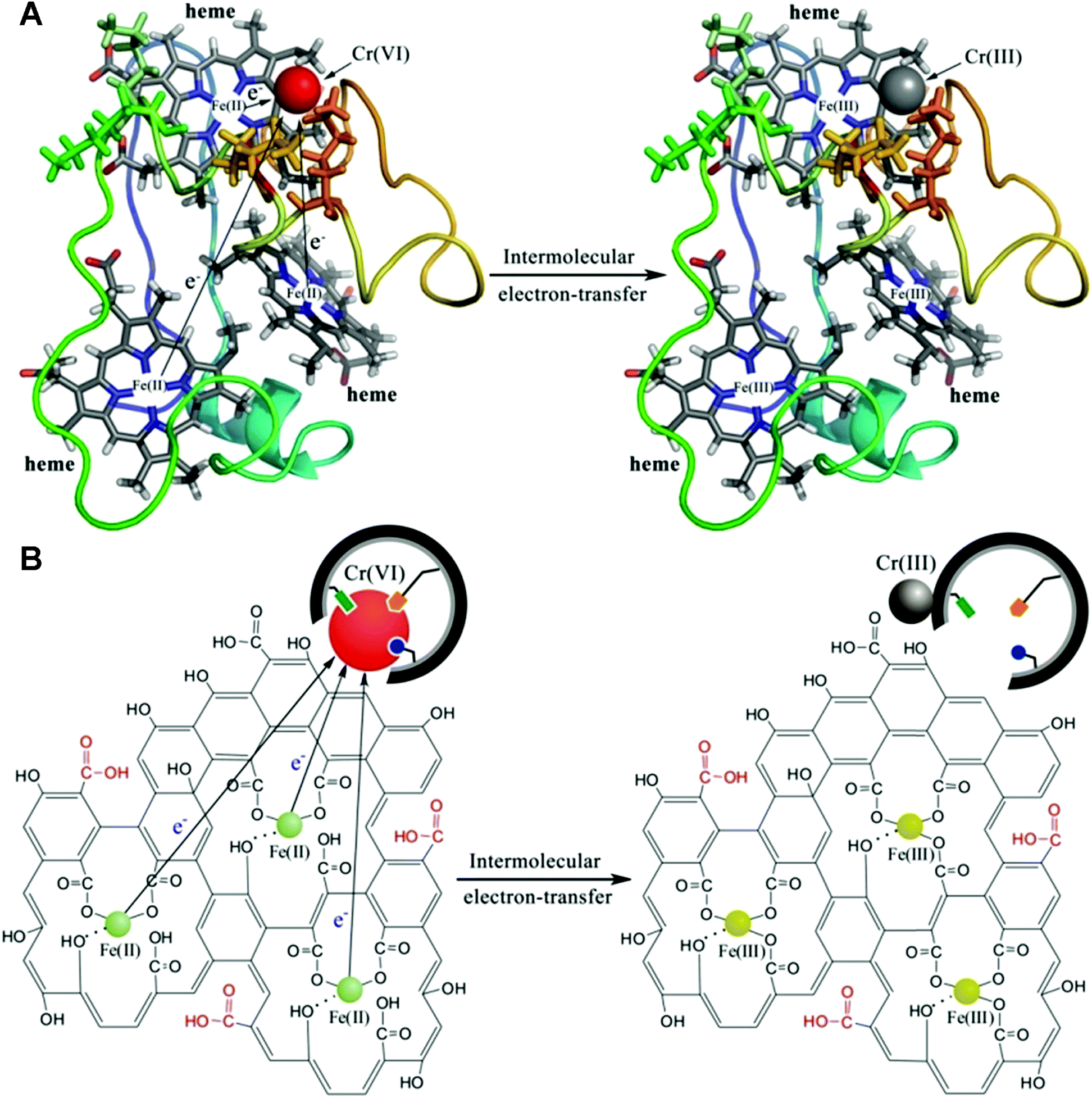
Combining molecular imprinting technology with nanomaterials with enzyme-mimetic catalytic activity is a proven method for constructing efficient artificial enzymes. It is challenging to integrate MIPs as identification materials and nanomaterials as active centers to play the ‘1 + 1 > 2’ effect. Shen's prepared artificial cyt c mimic (ACM) using molecularly imprinted colloidosomes (CSs) encapsulated by graphene oxide (GO) iron(III) complexes via Pickering emulsion polymerization.113 Briefly, Cr(VI) ion-imprinted polymers were first synthesized using GO nanosheet stabilized emulsions. The ACM was then successfully constructed by loading Fe(III) centers into GO nanosheets located on the surface of the imprinted CSs. Fig. 13 represents Cr(VI) reduction mechanism by cyt c7 and ACMs, showing the specific affinity of imprinted CSs for Cr(VI) ions and the electron transfer pathway during catalytic reduction. The maximum binding of Cr on MIP CSs (0.68 mg g−1) was about 1.6 times higher than that of NIP CSs (0.43 mg g−1), which indicates that the prepared MIP CSs have specific binding cavities. However, the adsorption of Cr(VI) by MIP CSs was lower than that of the imprinted nanoparticles (0.3 μm), which may be due to the dense structure of the highly cross-linked MIP CSs (72.3 μm) resulting in the inability of the internal cavities to adsorb Cr(VI). The binding ability of MIP CSs can be further improved by modulating the proportion of porogenic agents in the oil phase to improve the porosity and accessibility of the imprinting sites. In this work, the Michaelis Menten model was also used to characterize the activity of ACMs. The Vmax (the max reduction velocity) value of ACMs (1.53 μM min−1) were approximately triple that of non-ACMs (0.48 μM min−1), and the Km (the affinity of the enzyme to the substrate) value of ACMs (4.26 mM) was much lower than that of non-ACMs (9.20 mM), indicating that the affinity of Cr(VI) on ACMs is much higher than that of non-ACMs. The Kcat/kM (a measurement of catalytic efficiency) values of ACMs (0.035 mM−1 min−1) were much larger than those of non-ACMs (0.004 mM−1 min−1), indicating that the catalytic efficiency of ACMs is much higher than that of non-ACMs.
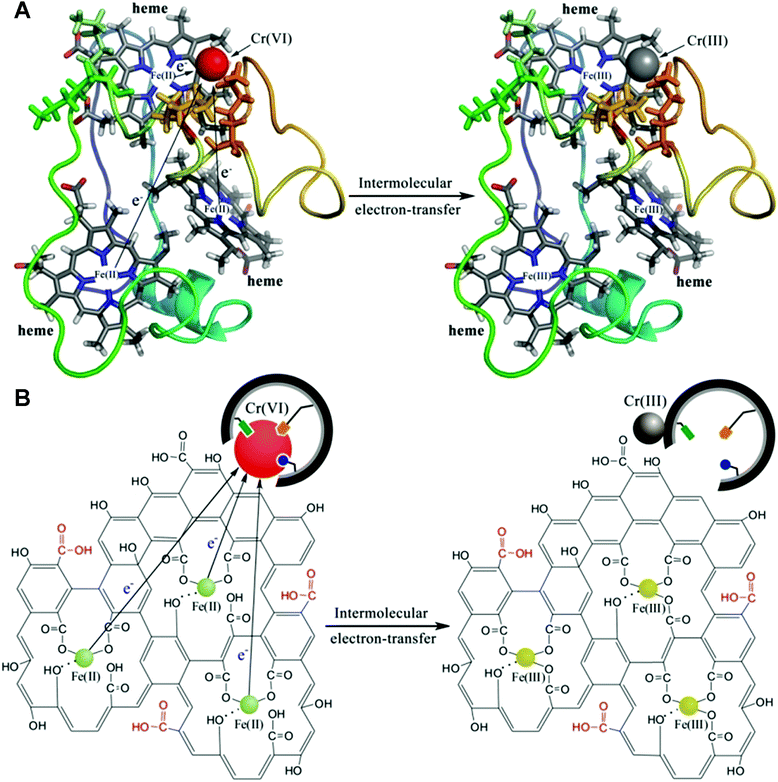 |
| | Fig. 13 Schematic representation of the reduction of Cr(VI) via (a) cyt c7 and (b) ACMs (GO-Fe(III) complex-coated MIP CSs). Reprinted with permission from ref. 113. Copyright 2019 the American Chemical Society. | |
3.5 Peptide-based materials@MIPs
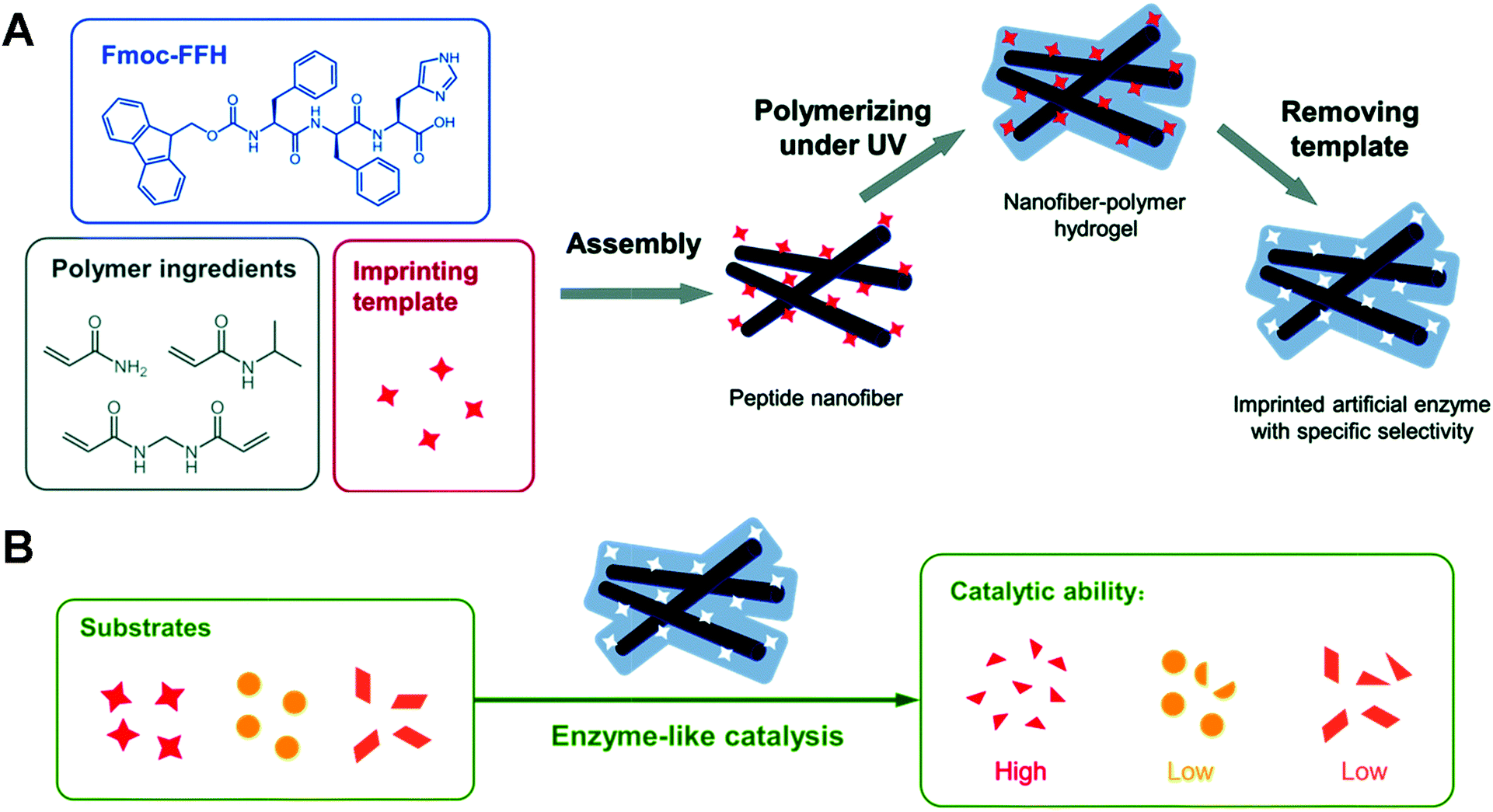
In recent years, peptide-based materials have received increasing attention in the biomedical field because of their structural versatility and good biocompatibility.114–116 Functionalizable peptide materials can be the basis for constructing artificial enzymes. Fry et al. prepared an artificial peroxidase with tunable catalytic activity by modulating the peptide sequence and the structure of the peptide-based assemblies.117 Woolfson et al. modified the peptide sequence with a catalytically active Cys–His–Glu triplet, resulting in the formation of a heptameric α-helical cylinder assembly with hydrolytic activity.118 In contrast to natural enzymes, these peptide-based catalysts with a simple structure and chemical composition do not provide a complex binding pocket for the substrate and lack substrate selectivity. He's group co-assembled Fmoc–Phe–Phe–His–COOH (Fmoc-FFH), Fmoc–Phe–Phe–Ser–COOH (Fmoc-FFS) and fmoc–phe–phe–ap–cooh (Fmoc-FFD) with p-nitrophenyl acetate (p-NPA) as a template. The resulting peptide nanofibers exhibited vigorous hydrolase-like activity and selectivity for p-NPA.119
However, the instability of supramolecular co-assembly makes this artificial enzyme unsatisfactory in terms of reusability. They next combined molecularly imprinted polymers with the peptide assemblies to develop a stable peptide-based artificial hydrolase.120 After mixing the Fmoc-FFH-containing catalytic histidine peptide with the polymer component and the imprinted template, the peptide molecules can self-assemble into supramolecular nanofibers driven by non-covalent forces, exhibiting apparent hydrolase activity. At the same time, the template and functional monomers form a pre-organization around the peptide assemblies. After polymerization under UV light irradiation followed by removal of the template, nanofiber polymer hydrogels containing specific cavities that bind more readily to the template or analogues are formed (Fig. 14). The relative activity of AMIP-H remained more than 80% after four cycles. In contrast, SA-H is difficult to reuse after a one-time use owing to the intrinsic instability within the supramolecular assembly. The activity of AMIP-Hmax (7.68 mM min−1) was 2.5 times higher than the activity of SA-H (3.03 mM min−1). Similarly, Ge's group found that imprinted polyacrylamide porous shells around natural lipase could promote substrate adsorption and enhance the catalytic activity of lipase nanogels.121 The results suggest that using peptide-based materials as a platform combined with molecular imprinting techniques to construct artificial enzymes with tailored substrate selectivity and stability is practical.
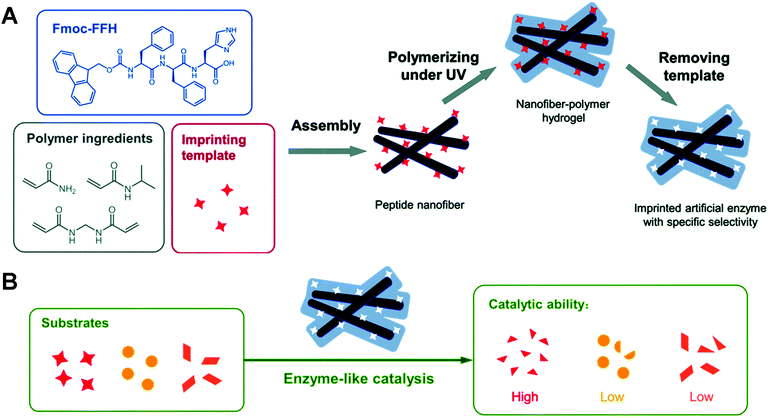 |
| | Fig. 14 The preparation (A) and catalytic reaction (B) of a peptide-based artificial hydrolase with customized selectivity. Reprinted with permission from ref. 120. Copyright 2019 the Royal Society of Chemistry. | |
4. Conclusions and outlook
In this article, we describe the recent progress in preparing high-efficiency artificial enzymes by MIT. MIP-based artificial enzymes exhibit excellent enzymatic catalytic activity and selectivity and possess the advantages of easy preparation, low cost, and high stability. The development of MIP-based artificial enzymes has been further promoted by some optimized methods such as imprinting transition state molecules, post-imprinting modifications, opening the internal space of cross-linked polymers, and some special preparation methods. Establishing selective substrate binding sites on the nanozyme by utilizing molecular imprinting technology to obtain synergistic effects solved the problem of the lack of specificity of the nanozyme and further improved its catalytic activity.
Although molecular imprinting technology has made tremendous development in the field of preparing artificial enzymes, many types of research are still at the laboratory level, and there are still problems such as low binding capacity, template leakage, and a low mass transfer rate, which hinder the practical and industrial applications of artificial enzymes. Due to the lack of sound theory to guide rational design and controlled preparation, the development of enzyme-mimetic MIPs still lags significantly behind their antibody-mimetic counterparts, which have found applications in many essential areas such as disease diagnosis, drug delivery, targeted bioimaging, and cancer therapy. Moreover, the variety of artificial enzymes synthesized so far has been limited, and an understanding of the catalytic mechanism of more natural enzymes is lacking. Even if the same kinds of artificial enzymes are prepared, comparison of their activities lacks a standardized criterion. Accordingly, we propose several recommendations on MIT.
First, the integration of MIT with a variety of technologies needs to be continuously enhanced to drive MIT forward. Various polymer synthesis techniques have been introduced into MIT, such as free radical polymerization and sol–gel methods, as well as grafting. Polymer technology, nanotechnology, click chemistry, microfluidics, stimulus-response technology, biotechnology, and optical technology have advanced in recent years. The rational integration of MIT and other technologies (e.g., nanoimprinting, immobilized enzyme techniques, solid-phase synthesis and computer-assisted techniques, etc.) will accelerate the development of molecular imprinting technology, which will achieve various MIPs with better performance. For example, nanoimprinting can significantly increase the number of effective recognition sites and thus significantly improve the imprinting ability. MIT can also be combined with natural or immobilized enzymes to further enhance their activity and applicability. Some scientists have already made some attempts in this regard,122,123 with molecularly imprinted polymeric layers of immobilized elastase nanomaterials that can effectively recognize and enrich substrates, hydrolyze interleukin 6 more efficiently, and be applied to living organisms.123 Advanced computer-assisted techniques (process simulation, chemometric techniques, molecular modeling, etc.) facilitate the selection of templates, functional monomers, solvents, and the estimation of the type of interaction between templates and functional monomers, which will significantly improve the probability of success in the preparation of molecularly imprinted artificial enzymes.
Second, the design and synthesis of new functional monomers is also an effective way to improve the selectivity of molecularly imprinted polymers. The combination of theoretical calculations (computer simulations) and organic synthetic chemistry has become an essential tool to obtain desirable monomers. The development of monomers will significantly broaden the scope of MIT applications. In addition, it is generally believed that the imprinted cavities determine the recognition properties of the prepared MIPs. In fact, MIPs are composed of imprinted cavities and non-imprinted regions, so the overall binding performance is not only determined by the imprinted cavities but also by the non-imprinted regions. The non-imprinted surfaces are functionally rich and inevitably lead to significant non-specific adsorption, which in turn has an impact on the affinity and specificity of the MIPs. Therefore increasing the attention to the non-imprinted regions and reducing their non-specific adsorption will have a positive effect on the catalytic activity of molecularly imprinted artificial enzymes.
Thirdly, the relatively large size of conventional MIPs makes it challenging to remove the internal template molecules, affecting the formation of the imprinting cavity. The irregular shape increases the resistance to mass transfer. In some cases, some large analytes may easily be trapped permanently inside the MIPs due to the long diffusion distance and very slow binding rate.78,124 This dramatically limits their performance and applications of MIPs. Scientists have therefore developed a series of nanoMIPs. The template molecules inside are easier to remove. The inprinting sites are easier to retain while reducing the diffusion distance of the target molecule and enhancing the imprinting efficiency for nano-sized MIPs. They also have a high specific surface area to volume ratio and regular geometry, providing better recognition sites for analytes and lower mass transfer resistance for fast binding kinetics.78,125,126 Their good dispersion ability and high compatibility with various nanodevices make nanoMIPs widely used in chemical sensing and biological fields,127–129 providing a possible direction for developing molecularly imprinted artificial enzymes.
The final point is to better understand and describe the mechanisms of enzyme-catalyzed reactions so that they can be simulated more effectively. More efforts must be focused on understanding the factors that affect the catalytic activity and preparing polymeric matrices that change their properties in response to external changes. Furthermore, a unified standard for enzyme activity comparison and a library of molecularly imprinted artificial enzymes should be established to facilitate people's reference, screening, and lateral comparison, reduce repetitive studies and simplify the tedious process of optimizing MIP formulations. It is reasonable to believe that with the continuous progress of molecular imprinting technology and a deeper understanding of natural enzymes, more productive artificial enzymes will be developed. We believe that shortly, more molecularly imprinted artificial enzymes with different mimetic enzymatic activities will be created and can be applied in biosensing, molecular and ion detection, imaging, cancer therapy, inflammation therapy, batteries, and beyond.
Author contributions
Ruizhen Tian: writing – original draft; Yijia Li, Jiayun Xu, Chunxi Hou and Quan Luo – review & editing, supervision; Junqiu Liu: writing – review & editing, funding acquisition, supervision.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Key R&D Program of China (Grant no. 2020YFA0908500 and No. 2018YFA0901600), and the Natural Science Foundation of China (No. 22161142015).
Notes and references
- A. J. Zaug and T. R. Cech, Cell, 1980, 19, 331–338 CrossRef CAS PubMed.
- J. Mates and C. Pérez-Gómez, Clin. Biochem., 1999, 32, 595–603 CrossRef CAS PubMed.
- T. Saleh and C. G. Kalodimos, Science, 2017, 355, 247–248 CrossRef CAS PubMed.
- X. Tao, X. Wang, B. Liu and J. Liu, Biosens. Bioelectron., 2020, 168, 112537 CrossRef CAS PubMed.
- M. Resmini, Anal. Bioanal. Chem., 2012, 402, 3021–3026 CrossRef CAS PubMed.
- Y. Lin, J. Ren and X. Qu, Acc. Chem. Res., 2014, 47, 1097–1105 CrossRef CAS PubMed.
- H. Wei and E. Wang, Chem. Soc. Rev., 2013, 42, 6060–6093 RSC.
- J. Wu, X. Wang, Q. Wang, Z. Lou, S. Li, Y. Zhu, L. Qin and H. Wei, Chem. Soc. Rev., 2019, 48, 1004–1076 RSC.
-
R. Breslow, Artificial Enzymes, Wiley-VCH, Weinheim, Germany, 2005, pp. 1–35 Search PubMed.
-
A. J. Kirby and F. Hollfelder, From enzyme models to model enzymes, Royal Society of Chemistry, 2009 Search PubMed.
- M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. Van Leeuwen, Chem. Soc. Rev., 2014, 43, 1734–1787 RSC.
- H. Chen, F. Bian, L. Sun, D. Zhang, L. Shang and Y. Zhao, Adv. Mater., 2020, 32, 2005394 CrossRef PubMed.
- K. K. Dar, S. Shao, T. Tan and Y. Lv, Biotechnol. Adv., 2020, 45, 107640 CrossRef CAS PubMed.
- D. Wu, W. Baaziz, B. Gu, M. Marinova, W. Y. Hernández, W. Zhou, E. I. Vovk, O. Ersen, O. V. Safonova and A. Addad, Nat. Catal., 2021, 4, 595–606 CrossRef CAS.
- Y. Zhao, H. Wang and T. Wang, Chem. Eng. J., 2016, 306, 832–839 CrossRef CAS.
- D. Sharabi and Y. Paz, Appl. Catal., B, 2010, 95, 169–178 CrossRef CAS.
- S. Striegler, J. D. Barnett and N. A. Dunaway, ACS Catal., 2012, 2, 50–55 CrossRef CAS.
- J. Q. Liu and G. Wulff, J. Am. Chem. Soc., 2004, 126, 7452–7453 CrossRef CAS PubMed.
- A. Servant, K. Haupt and M. Resmini, Chem. – Eur. J., 2011, 17, 11052–11059 CrossRef CAS PubMed.
- H. Henschel, N. Kirsch, J. Hedin-Dahlström, M. J. Whitcombe, S. Wikman and I. A. Nicholls, J. Mol. Catal. B: Enzym., 2011, 72, 199–205 CrossRef CAS.
- A. R. Cardoso, M. F. Frasco, V. Serrano, E. Fortunato and M. G. F. Sales, Biosensors, 2021, 11, 152 CrossRef CAS PubMed.
- C. Alexander, H. S. Andersson, L. I. Andersson, R. J. Ansell, N. Kirsch, I. A. Nicholls, J. O'Mahony and M. J. Whitcombe, J. Mol. Recognit., 2006, 19, 106–180 CrossRef CAS PubMed.
- J. K. Awino, R. W. Gunasekara and Y. Zhao, J. Am. Chem. Soc., 2016, 138, 9759–9762 CrossRef CAS PubMed.
- M. Gaeta, I. P. Oliveri, M. E. Fragalà, S. Failla, A. D’Urso and R. Purrello, Chem. Commun., 2016, 52, 8518–8521 RSC.
- L. Chen, S. Xu and J. Li, Chem. Soc. Rev., 2011, 40, 2922–2942 RSC.
- Y. Yuan, Y. Yang, X. Ma, Q. Meng, L. Wang, S. Zhao and G. Zhu, Adv. Mater., 2018, 30, 1706507 CrossRef PubMed.
- C. Boitard, A. Bée, C. Ménager and N. Griffete, J. Mater. Chem. B, 2018, 6, 1563–1580 RSC.
- L. Chen, X. Wang, W. Lu, X. Wu and J. Li, Chem. Soc. Rev., 2016, 2016, 2137–2211 RSC.
- J. J. BelBruno, Chem. Rev., 2019, 119, 94–119 CrossRef CAS PubMed.
- Z. Chen, S. Huang and M. Zhao, Mol. Imprinted Catal., 2016, 229–239 CAS.
- D. Mathew, B. Thomas and K. Devaky, Polym. Bull., 2018, 75, 3883–3896 CrossRef CAS.
- G. n Wulff and J. Liu, Acc. Chem. Res., 2012, 45, 239–247 CrossRef CAS PubMed.
- D. K. Robinson and K. Mosbach, J. Chem. Soc., Chem. Commun., 1989, 969–970 RSC.
- A. Leonhardt and K. Mosbach, React. Polym., Ion Exch., Sorbents, 1987, 6, 285–290 CrossRef CAS.
- J. Matsui, I. A. Nicholls, I. Karube and K. Mosbach, J. Org. Chem., 1996, 61, 5414–5417 CrossRef CAS.
- J. Chen, E. S. Garcia and S. C. Zimmerman, Acc. Chem. Res., 2020, 53, 1244–1256 CrossRef CAS PubMed.
- H. R. Culver and N. A. Peppas, Chem. Mater., 2017, 29, 5753–5761 CrossRef CAS PubMed.
- J. E. Lofgreen and G. A. Ozin, Chem. Soc. Rev., 2014, 43, 911–933 RSC.
- A.-M. Poller, E. Spieker, P. A. Lieberzeit and C. Preininger, ACS Appl. Mater. Interfaces, 2017, 9, 1129–1135 CrossRef CAS PubMed.
- G. Wulff, Angew. Chem., Int. Ed. Engl., 1972, 11, 341 CAS.
- L. Andersson, B. Sellergren and K. Mosbach, Tetrahedron Lett., 1984, 25, 5211–5214 CrossRef CAS.
- A. Martín-Esteban, Trends Analyt. Chem., 2013, 45, 169–181 CrossRef.
- J. Pan, W. Chen, Y. Ma and G. Pan, Chem. Soc. Rev., 2018, 47, 5574–5587 RSC.
- X. Shen, L. Zhu, N. Wang, L. Ye and H. Tang, Chem. Commun., 2012, 48, 788–798 RSC.
- G. Wulff, Microchim. Acta, 2013, 180, 1359–1370 CrossRef CAS.
- Y. Zhang, P. Huang, J. Guo, R. Shi, W. Huang, Z. Shi, L. Wu, F. Zhang, L. Gao, C. Li, X. Zhang, J. Xu and H. Zhang, Adv. Mater., 2020, 32, 2070175 CrossRef.
- M. Komiyama, T. Mori and K. Ariga, Bull. Chem. Soc. Jpn., 2018, 91, 1075–1111 CrossRef CAS.
- G. Wulff, T. Gross and R. Schönfeld, Angew. Chem., Int. Ed. Engl., 1997, 36, 1962–1964 CrossRef CAS.
- O. Ramström and K. Mosbach, Curr. Opin. Chem. Biol., 1999, 3, 759–764 CrossRef.
- Z. Cheng, L. Zhang and Y. Li, Chem. – Eur. J., 2004, 10, 3555–3561 CrossRef CAS PubMed.
- Z. Cheng and Y. Li, J. Mol. Catal. A: Chem., 2006, 256, 9–15 CrossRef CAS.
- Z. Chen, Z. Hua, J. Wang, Y. Guan, M. Zhao and Y. Li, Appl. Catal., A, 2007, 328, 252–258 CrossRef CAS.
- Z. Chen, L. Xu, Y. Liang and M. Zhao, Adv. Mater., 2010, 22, 1488–1492 CrossRef CAS PubMed.
- E. A. Özgür, Ind. Eng. Chem. Res., 2021, 60, 8714–8719 CrossRef.
- Y. Guo and T. Guo, Chem. Commun., 2013, 49, 1073–1075 RSC.
- R. Wang, J. Pan, M. Qin and T. Guo, Eur. Polym. J., 2019, 110, 1–8 CrossRef CAS.
- L. Pauling, Nature, 1948, 161, 707–709 CrossRef CAS PubMed.
-
W. P. Jencks, Catalysis in chemistry and enzymology, Courier Corporation, 1987 Search PubMed.
- M. A. Markowitz, P. R. Kust, G. Deng, P. E. Schoen, J. S. Dordick, D. S. Clark and B. P. Gaber, Langmuir, 2000, 16, 1759–1765 CrossRef CAS.
- K. Polborn and K. Severin, Chem. Commun., 1999, 2481–2482 RSC.
- K. Polborn and K. Severin, Eur. J. Inorg. Chem., 2000, 1687–1692 CrossRef CAS.
- B. Sellergren, R. N. Karmalkar and K. J. Shea, J. Org. Chem., 2000, 65, 4009–4027 CrossRef CAS PubMed.
- A. G. Strikovsky, D. Kasper, M. Grün, B. S. Green, J. Hradil and G. Wulff, J. Am. Chem. Soc., 2000, 122, 6295–6296 CrossRef CAS.
- J. q Liu and G. Wulff, Angew. Chem., Int. Ed., 2004, 43, 1287–1290 CrossRef CAS PubMed.
- J.-q Liu and G. Wulff, J. Am. Chem. Soc., 2004, 126, 7452–7453 CrossRef CAS PubMed.
- J.-q Liu and G. Wulff, J. Am. Chem. Soc., 2008, 130, 8044–8054 CrossRef CAS PubMed.
- D. Mathew, B. Thomas and K. Devaky, J. Mol. Catal. A: Chem., 2016, 415, 65–73 CrossRef CAS.
- C. Philip and K. S. Devaky, Mol. Catal., 2017, 436, 276–284 CrossRef CAS.
- D. Mathew, B. Thomas and K. S. Devaky, React. Funct. Polym., 2018, 124, 121–128 CrossRef CAS.
- Y. Yuan, Y. Yang, M. Faheem, X. Zou, X. Ma, Z. Wang, Q. Meng, L. Wang, S. Zhao and G. Zhu, Adv. Mater., 2018, 30, 1800069 CrossRef PubMed.
- G. Gasparini, M. Dal? Molin, S. Corrà, P. Galzerano, P. Scrimin and L. J. Prins, Isr. J. Chem., 2013, 53, 122–126 CrossRef CAS.
- J. Bos, W. R. Browne, A. Driessen and G. Roelfes, J. Am. Chem. Soc., 2015, 137, 9796–9799 CrossRef CAS PubMed.
- C. Zhang, X. Xue, Q. Luo, Y. Li, K. Yang, X. Zhuang, Y. Jiang, J. Zhang, J. Liu and G. Zou, ACS Nano, 2014, 8, 11715 CrossRef CAS PubMed.
- T. Takeuchi and H. Sunayama, Chem. Commun., 2018, 54, 6243–6251 RSC.
- J. Bos, F. Fusetti, A. J. M. Driessen and G. Roelfes, Angew. Chem., Int. Ed., 2012, 51, 7472–7475 CrossRef CAS PubMed.
- T. Yane, H. Shinmori and T. Takeuchi, Org. Biomol. Chem., 2006, 4, 4469–4473 RSC.
- X. Li and Y. Zhao, Chem. Sci., 2021, 12, 374–383 RSC.
- H. Zhang, Adv. Mater., 2020, 32, 1806328 CrossRef CAS PubMed.
- Q. Sun, X. Meng, X. Liu, X. Zhang, Y. Yang, Q. Yang and F.-S. Xiao, Chem. Commun., 2012, 48, 10505–10507 RSC.
- S. Zheng, J. Pan, J. Wang, S. Liu, T. Zhou, L. Wang, H. Jia, Z. Chen, Q. Peng and T. Guo, ACS Appl. Mater. Interfaces, 2021, 13, 34428–34437 CrossRef CAS PubMed.
- K. Dong, Q. Sun, X. Meng and F.-S. Xiao, Catal. Sci. Technol., 2017, 7, 1028–1039 RSC.
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- X. Lv, L. Li, S. Tang, C. Wang and X. Zhao, Chem. Commun., 2014, 50, 6886–6889 RSC.
- S. Khatua, S. Goswami, S. Biswas, K. Tomar, H. S. Jena and S. Konar, Chem. Mater., 2015, 27, 5349–5360 CrossRef CAS.
- N. Bagheri, A. Khataee, B. Habibi and J. Hassanzadeh, Talanta, 2018, 179, 710–718 CrossRef CAS PubMed.
- C. Ye, X. Chen, D. Zhang, J. Xu and G. Huang, Electrochim. Acta, 2021, 379, 138174 CrossRef CAS.
- L. Wan, H. Liu, C. Huang and X. Shen, J. Mater. Chem. A, 2020, 8, 25931–25940 RSC.
- L. Chen, X. Wang, W. Lu, X. Wu and J. Li, Chem. Soc. Rev., 2016, 45, 2137–2211 RSC.
- Z. Zhang, Y. Liu, X. Zhang and J. Liu, Nano Lett., 2017, 17, 7926–7931 CrossRef CAS PubMed.
- Z. Zhang, X. Zhang, B. Liu and J. Liu, J. Am. Chem. Soc., 2017, 139, 5412–5419 CrossRef CAS PubMed.
- Y. Yuan, Y. Yang and G. Zhu, ACS Cent. Sci., 2020, 6, 1082–1094 CrossRef CAS PubMed.
- J. K. Awino and Y. Zhao, J. Am. Chem. Soc., 2013, 135, 12552–12555 CrossRef CAS PubMed.
- K. Chen and Y. Zhao, Org. Biomol. Chem., 2019, 17, 8611–8617 RSC.
- L. Duan, M. Zangiabadi and Y. Zhao, Chem. Commun., 2020, 56, 10199–10202 RSC.
- M. Zangiabadi and Y. Zhao, ACS Appl. Polym. Mater., 2020, 2, 3171–3180 CrossRef CAS.
- X. Li, M. Zangiabadi and Y. Zhao, J. Am. Chem. Soc., 2021, 143, 5172–5181 CrossRef CAS PubMed.
- I. Bose and Y. Zhao, ACS Catal., 2022, 12, 3444–3451 CrossRef CAS PubMed.
- F. Manea, F. B. Houillon, L. Pasquato and P. Scrimin, Angew. Chem., Int. Ed., 2004, 116, 6291–6295 CrossRef.
- L. Gao, J. Zhuang, L. Nie, J. Zhang, Y. Zhang, N. Gu, T. Wang, J. Feng, D. Yang and S. Perrett, Nat. Nanotechnol., 2007, 2, 577–583 CrossRef CAS PubMed.
- H. Wang, K. Wan and X. Shi, Adv. Mater., 2019, 31, 1805368 CrossRef CAS PubMed.
- B. Yang, Y. Chen and J. Shi, Adv. Mater., 2019, 31, 1901778 CrossRef PubMed.
- M. Comotti, C. Della Pina, R. Matarrese and M. Rossi, Angew. Chem., Int. Ed., 2004, 43, 5812–5815 CrossRef CAS PubMed.
- A. Asati, S. Santra, C. Kaittanis, S. Nath and J. M. Perez, Angew. Chem., Int. Ed., 2009, 121, 2344–2348 CrossRef.
- M. Jiao, Z. Li, X. Li, Z. Zhang, Q. Yuan, F. Vriesekoop, H. Liang and J. Liu, Chem. Eng. J., 2020, 388, 124249 CrossRef CAS.
- Y. Tao, E. Ju, J. Ren and X. Qu, Adv. Mater., 2015, 27, 1097–1104 CrossRef CAS PubMed.
- C. Xu, Z. Liu, L. Wu, J. Ren and X. Qu, Adv. Funct. Mater., 2014, 24, 1624–1630 CrossRef CAS.
- M. Liang and X. Yan, Acc. Chem. Res., 2019, 52, 2190–2200 CrossRef CAS PubMed.
- L. Cheng, X. Wang, F. Gong, T. Liu and Z. Liu, Adv. Mater., 2020, 32, 1902333 CrossRef CAS PubMed.
- Z. Zhang, Y. Li, X. Zhang and J. Liu, Nanoscale, 2019, 11, 4854–4863 RSC.
- Y. Hu, J. Liu, H. Xing, H. Zhou and M. Wu, ChemistrySelect, 2020, 5, 8284–8288 CrossRef CAS.
- L. Fan, D. Lou, H. Wu, X. Zhang, Y. Zhu, N. Gu and Y. Zhang, Adv. Mater. Interfaces, 2018, 5, 1801070 CrossRef.
- Z. Zhang and J. Liu, Mater. Horiz., 2018, 5, 738–744 RSC.
- Z. Chen, X. Liu, C. Huang, J. Li and X. Shen, ACS Appl. Mater. Interfaces, 2020, 12, 6615–6626 CrossRef CAS PubMed.
- E. Gazit, Chem. Soc. Rev., 2007, 36, 1263–1269 RSC.
- J. Kong, Y. Wang, J. Zhang, W. Qi, R. Su and Z. He, Angew. Chem., Int. Ed., 2018, 130, 14228–14232 CrossRef.
- A. Lakshmanan, S. Zhang and C. A. Hauser, Trends Biotechnol., 2012, 30, 155–165 CrossRef CAS PubMed.
- L. A. Solomon, J. B. Kronenberg and H. C. Fry, J. Am. Chem. Soc., 2017, 139, 8497–8507 CrossRef CAS PubMed.
- A. J. Burton, A. R. Thomson, W. M. Dawson, R. L. Brady and D. N. Woolfson, Nat. Chem., 2016, 8, 837–844 CrossRef CAS PubMed.
- M. Wang, Y. Lv, X. Liu, W. Qi, R. Su and Z. He, ACS Appl. Mater. Interfaces, 2016, 8, 14133–14141 CrossRef CAS PubMed.
- M. Zhu, M. Wang, W. Qi, R. Su and Z. He, J. Mater. Chem. B, 2019, 7, 3804–3810 RSC.
- R. Wang, Y. Zhang, J. Huang, D. Lu, J. Ge and Z. Liu, Green Chem., 2013, 15, 1155–1158 RSC.
- T. Chen, A. Zhang, Y. Cheng, Y. Zhang, D. Fu, M. Liu, A. Li and J. Liu, Biosens. Bioelectron., 2021, 188, 113355 CrossRef CAS PubMed.
- L. Zhong, J. Zhai, Y. Ma, Y. Huang, Y. Peng, Y.-E. Wang, Z. Peng, H. Gan, Z. Yuan, P. Yan, Q. Li and S. Guan, ACS Nano, 2022, 16, 3797–3807 CrossRef CAS PubMed.
- J. Wackerlig and R. Schirhagl, Anal. Chem., 2016, 88, 250–261 CrossRef CAS PubMed.
- A. Poma, A. P. F. Turner and S. A. Piletsky, Trends Biotechnol., 2010, 28, 629–637 CrossRef CAS PubMed.
- J. Wackerlig and P. A. Lieberzeit, Sens. Actuators, B, 2015, 207, 144–157 CrossRef CAS.
- M. Dabrowski, P. Lach, M. Cieplak and W. Kutner, Biosens. Bioelectron., 2018, 102, 17–26 CrossRef CAS PubMed.
- A. F. Nahhas and T. J. Webster, J. Nanobiotechnol., 2021, 19, 305 CrossRef CAS PubMed.
- S. Xu, L. Wang and Z. Liu, Angew. Chem., Int. Ed., 2021, 60, 3858–3869 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a and
Junqiu
Liu
*a and
Junqiu
Liu