
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA sequential cyclization/π-extension strategy for modular construction of nanographenes enabled by stannole cycloadditions†
Harrison M.
Bergman
 a,
D. Dawson
Beattie
a,
Gavin R.
Kiel
a,
Rex C.
Handford
a,
Yi
Liu
*b and
T. Don
Tilley
*a
a,
D. Dawson
Beattie
a,
Gavin R.
Kiel
a,
Rex C.
Handford
a,
Yi
Liu
*b and
T. Don
Tilley
*a
aDepartment of Chemistry, University of California, Berkeley, California 94720, USA. E-mail: tdtilley@berkeley.edu
bMolecular Foundry, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA. E-mail: yliu@lbl.gov
First published on 1st April 2022
Abstract
The synthesis of polycyclic aromatic hydrocarbons (PAHs) and related nanographenes requires the selective and efficient fusion of multiple aromatic rings. For this purpose, the Diels–Alder cycloaddition has proven especially useful; however, this approach currently faces significant limitations, including the lack of versatile strategies to access annulated dienes, the instability of the most commonly used dienes, and difficulties with aromatization of the [4 + 2] adduct. In this report we address these limitations via the marriage of two powerful cycloaddition strategies. First, a formal Cp2Zr-mediated [2 + 2 + 1] cycloaddition is used to generate a stannole-annulated PAH. Secondly, the stannoles are employed as diene components in a [4 + 2] cycloaddition/aromatization cascade with an aryne, enabling π-extension to afford a larger PAH. This discovery of stannoles as highly reactive – yet stable for handling – diene equivalents, and the development of a modular strategy for their synthesis, should significantly extend the structural scope of PAHs accessible by a [4 + 2] cycloaddition approach.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are a versatile class of organic compounds with diverse applications as components of electronic devices such as OLEDs, OFETs, and OPVs,1–3 as well as in biology and medicine.4 In general, the utility of these compounds stems from the diverse array of properties that can be synthetically controlled by tuning (for example) molecular shape, edge structure, and functionality.5,6 Despite established advances in PAH chemistry, there remain substantial synthetic limitations that restrict possible architectures and new applications.The main challenge associated with PAH synthesis is the lack of general, reliable methods for the selective fusion of aromatic rings, which generally rely on harsh conditions or specific functional handles to facilitate fusion. Significant effort has gone into developing such strategies,7 and several reaction classes have emerged to greatly expand the diversity of accessible PAHs. These include oxidative couplings such as the Scholl reaction, electrophilic cyclization of alkynes,8 radical cascades, annulative π-extension,9 transition metal mediated [2 + 2 + n] cycloadditions,10–13 and the Diels Alder reaction.14
The Diels Alder reaction, or [4 + 2] cycloaddition, has attracted particular interest in the synthesis of PAHs due to its versatility and operational simplicity.14,15 For this application, dienes capable of undergoing facile aromatization after cycloaddition are necessary, and several systems are commonly employed. Perhaps the most ubiquitous are furans which generate epoxy-bridged intermediates, with aromatization occurring in a second, deoxygenation step (Scheme 1a).
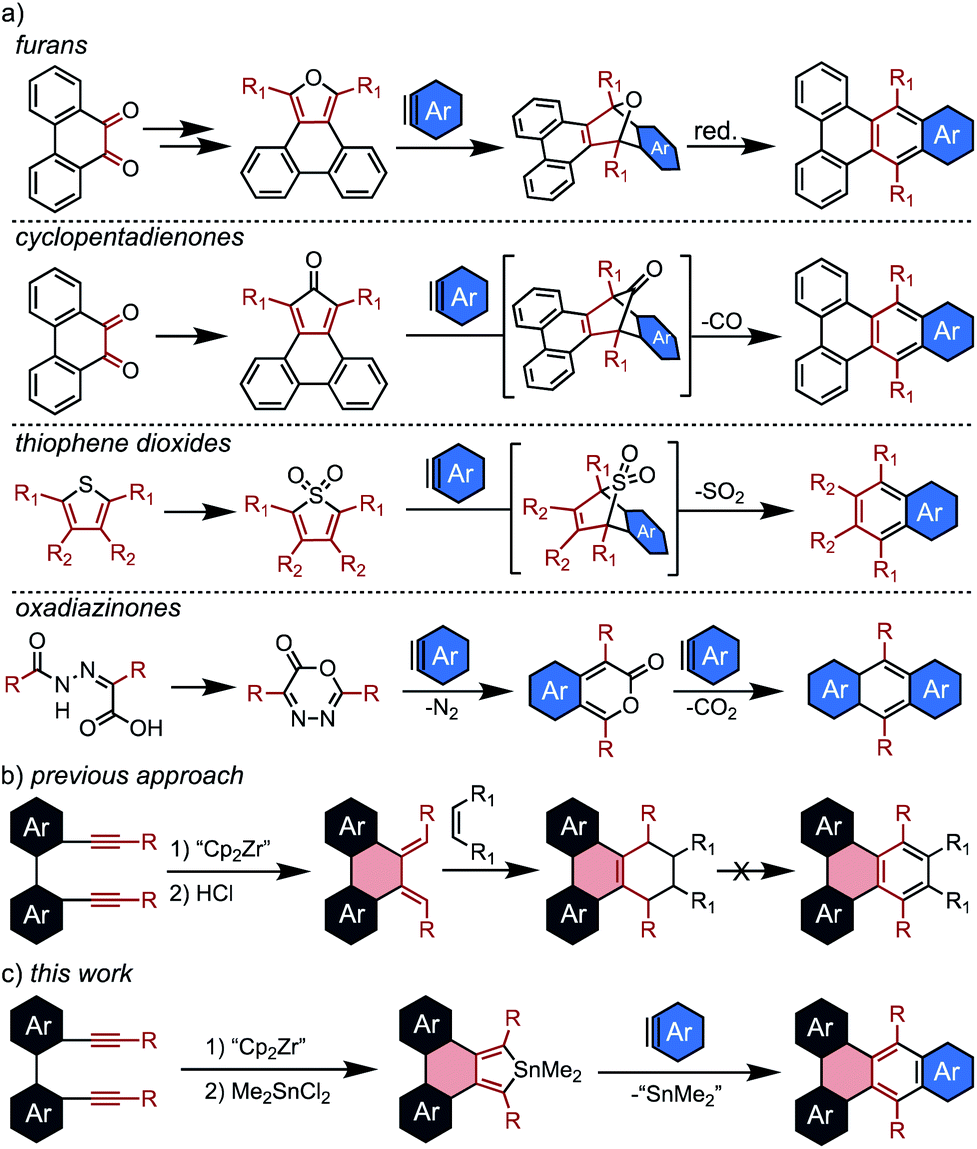 | ||
| Scheme 1 (a) Commonly employed dienes for the synthesis of nanographenes via [4 + 2] cycloadditions; (b) prior development of a zirconocene coupling approach to the synthesis of PAHs from tethered diynes; (c) this work, combining the zirconocene coupling approach to PAH synthesis with π-extension via the [4 + 2] cycloaddition of stannoles. | ||
While this method has proven valuable, particularly in the synthesis of sterically crowded acenes known as twistacenes,16 it suffers from low overall yields and poor functional group tolerance due to the harsh aromatization step. Thus, the development of diene sources that undergo spontaneous aromatization is of high interest. There are three commonly employed dienes of this type: cyclopentadienones,17–19 thiophene dioxides,20,21 and oxadiazinones.22–24 In each case, the cycloaddition intermediate aromatizes with liberation of a gas. Although these examples generate aromatic products in a single step, the electron-withdrawing character of the eliminated substituent (CO, SO2, and CO2) causes the dienes to be sluggish partners for standard Diels Alder reactions. This generally corresponds to limited yields for cycloadditions with benzynes, despite their promise as crucial building blocks in syntheses of nanocarbons.
Another significant roadblock in the application of these dienes to nanographene synthesis is the challenge of incorporating them into complex fused ring systems. Annulated furans and cyclopentadienones are most commonly generated from o-diones, and thus can only be appended to PAHs for which the diones are readily accessible. There are currently no straightforward ways to generate PAH-thiophene dioxides, and oxadiazinones are fundamentally incapable of being annulated to larger fused systems. Thus, the development of new strategies for appending reactive dienes to nanographene cores is critical for expanding the utility of Diels Alder cycloadditions in PAH synthesis.
Previously, this group attempted to resolve this issue via the use of a [2 + 2 + 1] zirconocene-mediated coupling of tethered diynes (Scheme 1b).25 This strategy enabled the synthesis of a wide range of nanographenes from unfused precursors, and facile protodemetallation of the resulting zirconacycles generated PAH-appended dienes. Although the dienes were reactive towards dienophiles, aromatization of the resulting cycloaddition products was not possible.
To address this limitation, we sought to develop a new Diels Alder diene source that is broadly reactive with arynes and readily installed into a wide range of complex PAHs via the zirconocene coupling approach. To this end, several papers from Hitomi26 and Neumann27 that describe stannoles as sources of stannylene (:SnR2) upon reaction with dienophiles served as inspiration. Although these studies focused primarily on the liberation of stannylene, they described high yields of the aromatic byproducts, indicating that stannoles not only undergo efficient [4 + 2] cycloadditions but also spontaneously aromatize upon liberation of divalent tin. Despite a few early observations, stannole cycloaddition chemistry has not been developed into a generally applicable methodology.28,29 Although stannoles have been shown to be effective cross-coupling partners for direct annulation via Pd catalysis,30,31 this is the first report of their general use as efficient dienes for [4 + 2] cycloadditions to generate π-extended polycyclic aromatic hydrocarbons.
In this contribution, stannoles are shown to be readily incorporated into a wide range of PAH structures using the zirconocene coupling of tethered diynes. These stannoles then serve as highly efficient dienes in Diels Alder cycloadditions with arynes (Scheme 1c), most strikingly from ortho-dihalo substrates which are generally incompatible with other spontaneously aromatizing dienes. Furthermore, in a demonstration of the potential for stannoles to engage in a more diverse array of cycloaddition reactions, one example is shown to undergo an unusual dimerization at high temperatures to generate a helically chiral, 1,2,4,5-cyclooctatetraene in exceptional yield (≥95%).
Results and discussion
It seemed that stannoles could be appended to PAHs via zirconocene coupling of tethered alkynes, followed by transmetalation to the stannole via the CuCl-mediated conditions developed by Takahashi.32 This strategy was initially tested on 1a to confirm that the reaction conditions were effective for previously unexplored tethered diynes (Scheme 2). It was determined that the sequence can either be performed in a stepwise manner through isolation of the intermediate zirconacycle, or in a convenient one-pot procedure using the Negishi protocol in essentially the same yield (see page S7† for all reported preparations).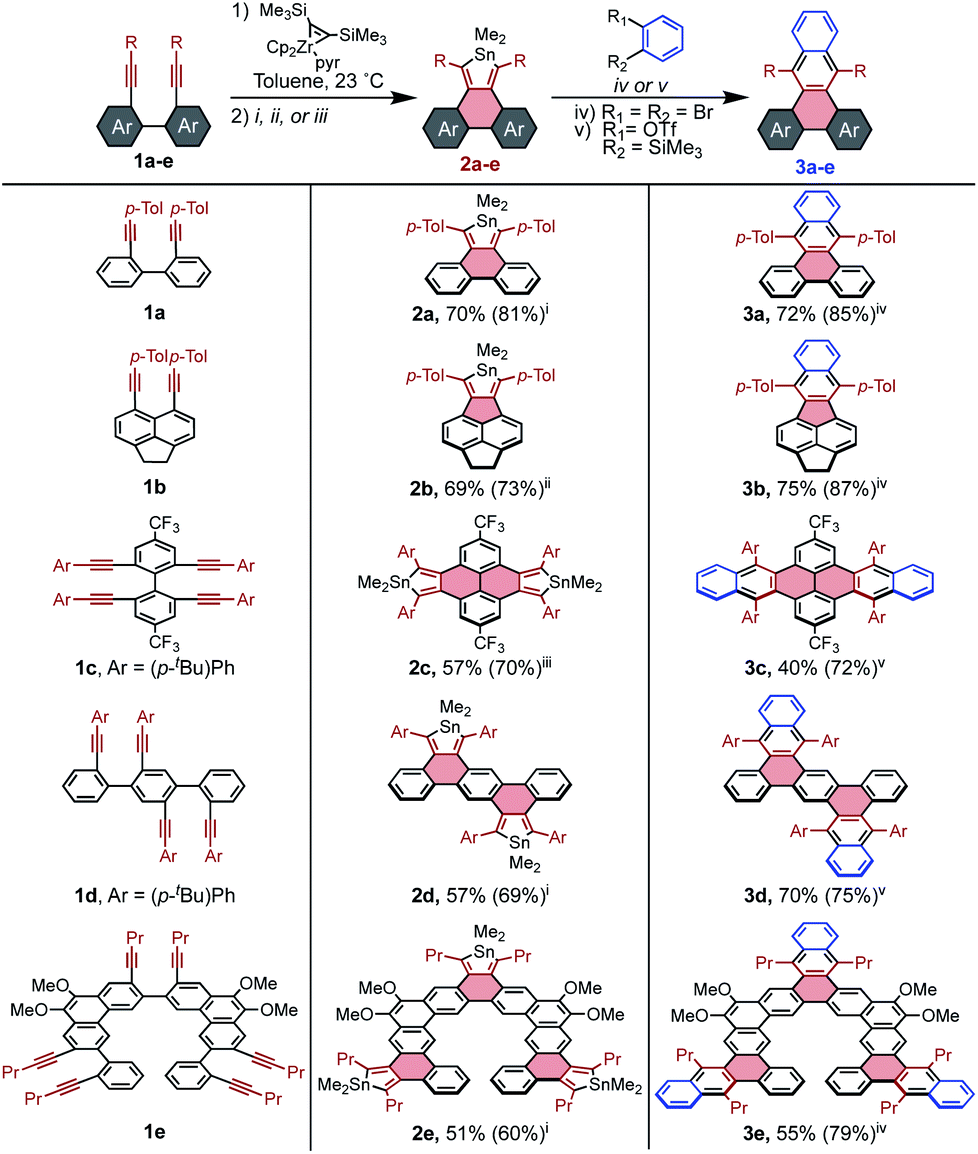 | ||
| Scheme 2 Substrate scope of stannole synthesis and subsequent π-extension via [4 + 2] cycloaddition with benzyne. For 2a–2e, yields represent total isolated yield across two steps from the diyne, while yields from isolated zirconacycles are given in parenthesis. For 3a–3e, in situ yields as determined by 1H NMR spectroscopy are given in parenthesis. Reagents and conditions: (i) Me2SnCl2, CuCl, LiCl, toluene, 23 °C; (ii) Me2SnCl2, CuCl, LiCl, toluene, 60 °C; (iii) Me2SnCl2, CuCl, ZnCl2, toluene, 23 °C; (iv) 1,2-dibromobenzene (1.2 equiv.), nBuLi (1.1 equiv.), toluene, 23 °C; (v) 2-(Me3Si)C6H4(OTf) (1.1 equiv.), [NBu4]Ph3SiF2 (3.3 equiv.), dimethylacetylenedicarboxylate (1.5 equiv.), toluene, 60 °C. | ||
Beyond the model stannole 2a, it is possible to construct more complex stannole-containing PAHs using this methodology (Scheme 2). Zirconocene coupling of alkynes may also be used to generate PAHs containing 5-member rings, which can similarly undergo transmetalation to stannoles as evidenced by the synthesis of stannole 2b in 73% yield. The efficiency of stannole formation makes it possible to synthesize bis- and tris-stannoles in good yield, which enables the construction of more complex diene-containing PAH architectures such as 2c–2e, which would be difficult to access by established methods.
Compound 2a was utilized as a model system to explore the reactivity of stannoles for [4 + 2] cycloadditions with benzyne, beginning with the widely used and mild benzyne precursor 2-(Me3Si)C6H4(OTf). Initially, simple mixing of 2a, the benzyne precursor, and the soluble fluoride source [nBu4N][Ph3SiF2] in toluene furnished 3a in 78% yield by 1H NMR spectroscopy after some optimization (page S18†). A significant improvement to 88% was seen upon addition of dimethylacetylenedicarboxylate (DMAD) as a stannylene trap,25 but no other commonly employed traps were effective (diphenyl disulfide, diacetyl, butylbromide).
Although ortho-dihalo precursors to arynes generally require harsher reaction conditions, they are highly valuable building blocks due to their availability. Significantly, stannoles are robust in the presence of nBuLi, and treatment of o-dibromobenzene with nBuLi at 23 °C in the presence of stannole 2a resulted in the formation of 3a in 85% yield. This efficient transformation contrasts with the absence of analogous reactions involving cyclopentadienones, thiophene dioxides, and pyrones, which are ostensibly reactive toward organolithium species.
Generalization of these cycloaddition conditions to stannoles 2b–2e enabled facile access to π-extended PAHs 3b–3e. In each case except 2d, stannoles were compatible with both types of benzyne precursors, and the method that resulted in the higher isolated yield is reported in Scheme 2. It is believed that the reaction of 2d with o-dibromobenzene failed due to its very limited solubility in aromatic solvents, making its reaction with the generated benzyne too sluggish to compete with benzyne decomposition.
The range of additional benzyne sources that are compatible with [4 + 2] cycloadditions to stannoles was further explored using 2a as a model system. For the ortho-Me3Si-OTf derivative of naphthalene, the cycloaddition product 4 was accessed in 85% yield, suggesting the generality of these conditions to other ortho-Me3Si-OTf PAHs. Other dihalo reactant partners may be employed, as observed for 2-chloro and 2-fluorobromobenzene which both gave 3a in greater than 80% yield (Scheme 3).
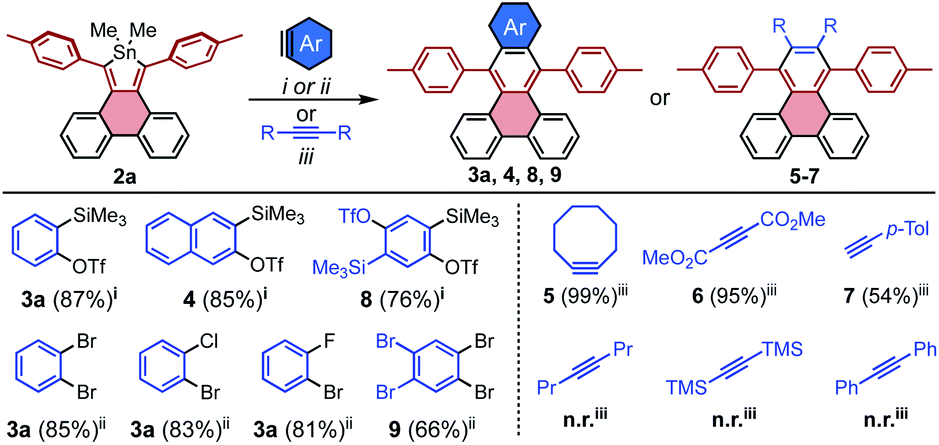 | ||
| Scheme 3 Benzyne and alkyne substrate scope for [4 + 2] cycloadditions with model stannole system 2a. Given yields were determined in situ via1H NMR spectroscopy. Reagents and conditions: (i) aryne (1.1 equiv.), [nBu4N][Ph3SiF2] (3 equiv.), dimethylacetylenedicarboxylate (1.5 equiv.), toluene, 60 °C (ii) aryne (1.2 equiv.), nBuLi (1.1 equiv.), toluene, 23 °C; (iii) alkyne (2.5 equiv.), toluene, 100–160 °C. | ||
Reactions of stannoles with common alkynes were also explored. Generally, 2a reacted rapidly and efficiently at moderate temperatures with activated internal alkynes such as strained cyclooctyne and electron-deficient dimethyacetylene dicarboxylate, to furnish 5 and 6 in 99% and 95% yields, respectively. The unactivated terminal alkyne 4-ethynyl toluene required elevated temperature but gave reasonable conversion to the unsymmetrical PAH 7 in 54% yield (Scheme 3, R,R = H, p-Tol). The unactivated internal alkynes 4-octyne, bis(trimethylsilyl)acetylene, and diphenyl acetylene were generally unreactive, even at highly elevated temperatures (160 °C in mesitylene).
The efficiency with which stannoles undergo cycloadditions with arynes makes them desirable substrates for the synthesis of extended π-systems through multiple, successive cycloadditions. To this end, conditions were developed for the iterative addition of stannoles to bifunctional aryne precursors. Stannole 2a was treated with 1.1 equivs of a bis-benzyne precursor under standard conditions for Me3Si-OTf precursors, and this afforded the desired mono-addition product 8 in 76% isolated yield (Scheme 3). A larger excess of the aryne precursor (2 equiv.) increased the yield of 8 to 85% (see page S13†). Similarly, treatment of stannole 2a with an excess of 1,2,4,5-tetrabromobenzene (2 equiv.) and nBuLi (1.1 equiv.) afforded the mono-addition product 9 in 66% isolated yield. Both 8 and 9 participated in a second cycloaddition with stannole 2b to give unsymmetrical acene 10 in 72 and 60% yields, respectively (Scheme 4). One-pot double-addition of stannole 2a to 0.5 equivalents of 1,2,4,5-tetrabromobenzene was also successful, to generate tetrabenzopentacene 11 in 40% yield.
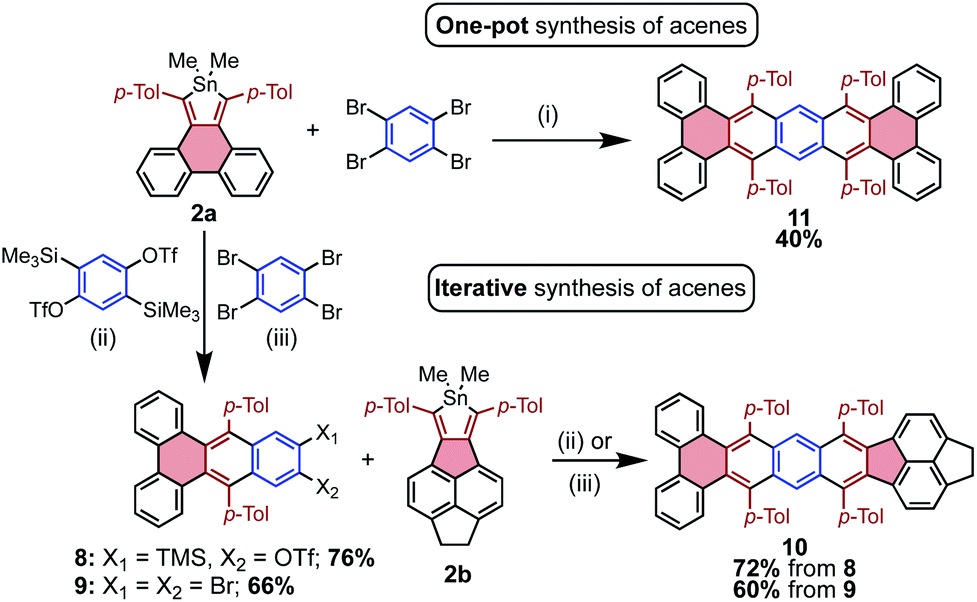 | ||
| Scheme 4 Synthesis of acenes via the multiple addition of stannoles to benzynes. Reagents and conditions: (i) aryne (0.5 equiv.), nBuLi (1.1 equiv.), toluene, 23 °C; (ii) aryne (1.1 equiv.), [nBu4N][Ph3SiF2] (3 equiv.), DMAD (1.5 equiv.), toluene, 60 °C; (iii) aryne (2 equiv.), nBuLi (1.1 equiv.), toluene, 23 °C. | ||
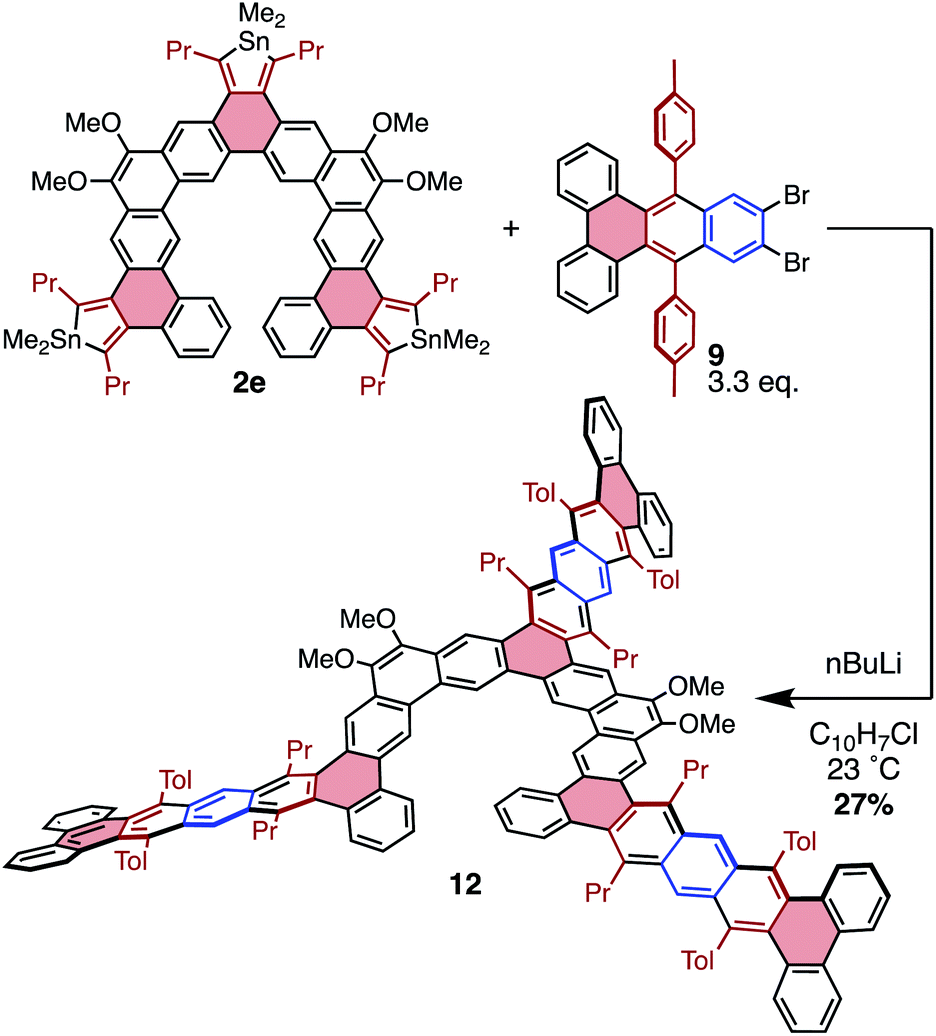
To further explore possibilities for this cycloaddition chemistry in producing unusual PAH structures, the molecular “lobster-shaped” compound 12 was chosen as a target that would be difficult to obtain using other methodologies (Scheme 5). Compound 12 was generated from stannole 2e and benzyne precursor 9 in a manner similar to that previously described; however, due to the insolubility of 9, 1-chloronaphthalene was used as a solvent to fully solubilize both components prior to benzyne formation. From these conditions, 12 was isolated in 27% yield, indicating an efficiency of approximately 65% per cycloaddition. Compound 12 displayed a propensity to aggregate in solution, producing sharp resonances in 1H NMR spectra only at concentrations below 0.4 mM. This is likely a function of the significant π-extension in this molecule, as similar compound 3e does not display this behavior in solution. This aggregation behavior complicates assessment of purity for 12, as the isolated material still displays some broad resonances in addition to sharp peaks associated with unaggregated 11 (by 1H NMR spectroscopy). The broad resonances may be due to impurities or partial aggregation. Despite this, MALDI unambiguously confirms the presence of 12, and does not display peaks corresponding to over- or under-substitution in the range of 500–3000 amu (Fig. S54†). This suggests that the broadness observed by 1H NMR spectroscopy is likely due to aggregation rather than the presence of major impurities.
 | ||
| Scheme 5 Synthesis of a π-extended helicene. | ||
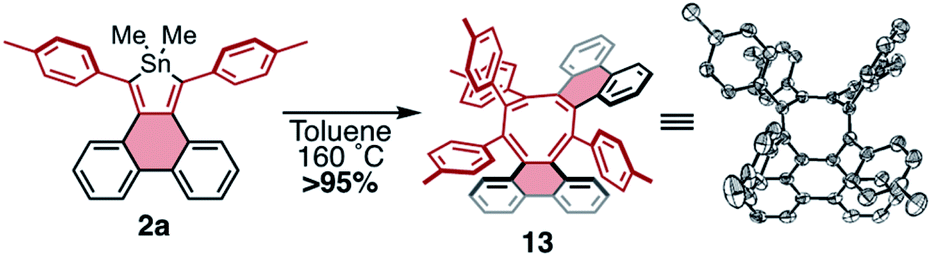
Given the facile elimination of divalent tin during the cycloadditions described above, it seemed possible that stannoles might undergo other cycloaddition–elimination processes. To explore the possibility of dimerization via cycloaddition, stannole 2a was heated to 160 °C in toluene-d8 in a sealed reaction vessel and the reaction mixture was monitored by 1H NMR spectroscopy. Over a period of 12 h a new species formed in exceptionally high yield (>95%) displaying 4 tolyl CH3 resonances of equal intensity and lacking any discernible resonances from dimethyltin. This is consistent with a product that is a highly unsymmetrical dimer, formed by elimination of Me2Sn. Single crystals of the product were grown by slow diffusion of hexanes into a concentrated sample in CDCl3, and X-ray crystallography confirmed the product as compound 13, a helically chiral 1,2,4,5-cyclooctatetraene (COT) (Scheme 6). While there is a literature report of a COT with this substitution pattern,33 it was formed as a minor product; to our knowledge, this is the first observation of a high-yield, selective synthesis of this type of substituted COT. This result highlights the potential for stannoles as versatile diene sources for a range of cycloadditions, making them valuable divergent building blocks for the synthesis of conjugated molecules.
 | ||
| Scheme 6 Formal [4 + 4] dimerization of stannoles to generate a helically chiral cyclooctatetraene. | ||
The photophysics of 3b–3e, 4, 11, and 12 were investigated by UV-vis and fluorescence spectroscopy (Fig. 1). The fine structure in the absorbance spectra displays significant sensitivity to the pattern of ring fusion, with 3c–3e displaying different relative intensities of the three main absorption bands. However, the absorption onset (λonsetabs), and consequently the optical HOMO–LUMO gap (Eopt), are fairly consistent for the series at 3.03, 2.90, and 2.90 eV respectively (Table 1). The incorporation of a five-member ring into 3b significantly alters the shape of the absorption spectrum, and imbues the structure with a notably smaller Eopt of 2.86 eV than 3a despite their similarly sized π-system. Increasing the length of the acene fragment in these systems bathochromically shifts the absorption onset and decreases the HOMO–LUMO gap, with dibenzotetracene 4 reaching 2.70 eV, and tetrabenzopentacenes 11 and 12 reaching 2.35 and 2.38 eV respectively. The fluorescence spectra of the six fully benzenoid structures display similar shapes with two broad peaks, while 3b displays three. The trends in fluorescence maximum (λmaxem) mirror those in absorption onset, with 3c–3e displaying highly similar maxima at 434, 435, and 451 nm respectively. 4, 11, and 12 are notably bathochromically shifted to 477, 551, and 538 nm respectively. These results highlight how orthogonal synthetic control over the core pattern of ring-fusion and the degree of π-extension enables tuning key aspects of the photophysical properties such as fine structure, HOMO–LUMO gap, and fluorescence.
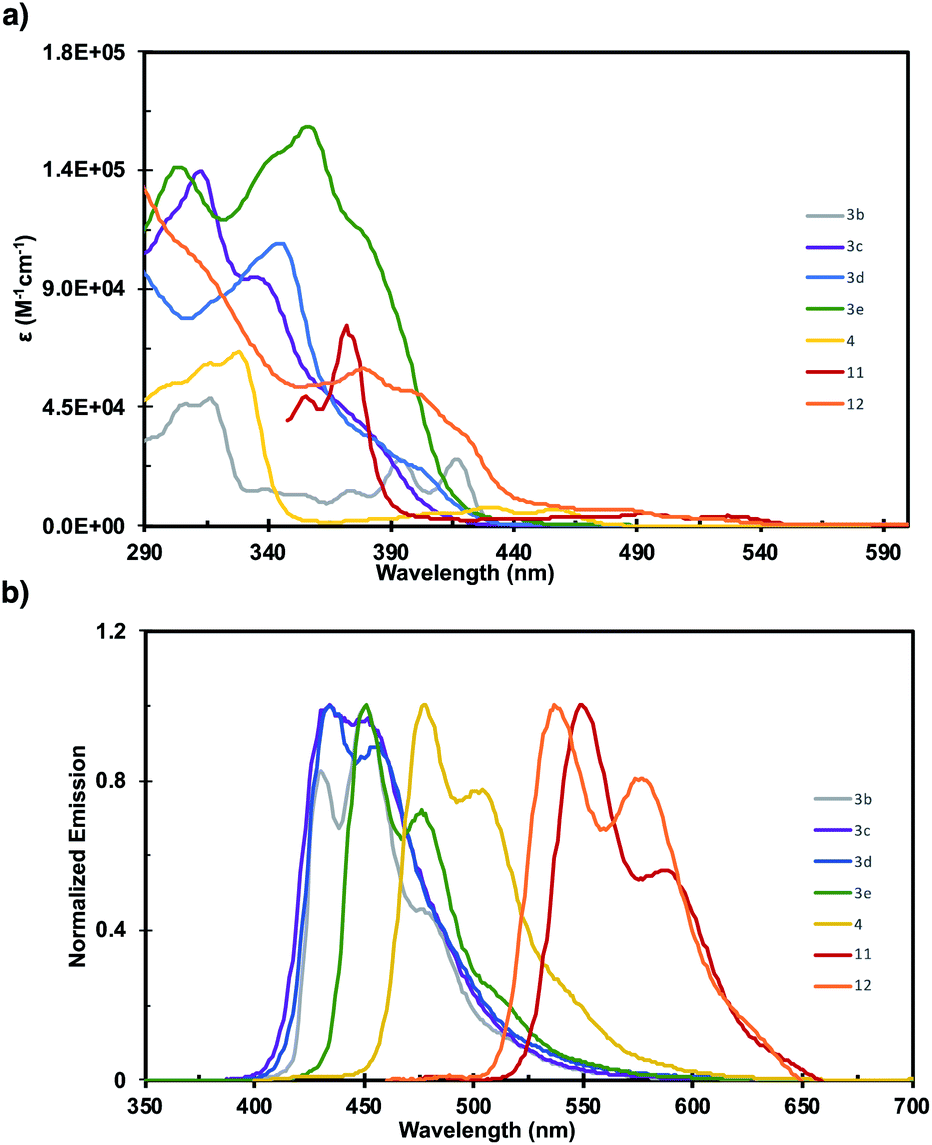 | ||
| Fig. 1 Steady-state photophysics of 3b (gray), 3c (purple), 3d (blue), 3e (green), 4 (yellow), 11 (red), and 12 (orange). (a) UV-vis and (b) fluorescence spectroscopy acquired at 5 × 10−6 M in THF (3b, 3c, 3d, 3e, 4, 12) and 1-chloronaphthalene (11). All fluorescence spectra were normalized to their respective λmax. | ||
| Compound | λ maxabs (nm) | λ onsetabs (nm) | λ maxem (nm) | E opt (eV) |
|---|---|---|---|---|
| 3b | 316 | 434 | 451 | 2.86 |
| 3c | 312 | 409 | 434 | 3.03 |
| 3d | 344 | 428 | 435 | 2.90 |
| 3e | 357 | 428 | 451 | 2.90 |
| 4 | 327 | 459 | 477 | 2.70 |
| 11 | 372 | 527 | 551 | 2.35 |
| 12 | 308 | 521 | 538 | 2.38 |
Conclusions
In conclusion, stannoles represent a versatile diene source in [4 + 2] cycloadditions for the synthesis of π-extended aromatic systems. The high yielding cycloaddition–aromatization cascade and the broad compatibility with arynes suggest that stannoles can be superior to commonly employed cyclopentadienones for many applications in PAH synthesis, and their generation via [2 + 2 + 1] cycloadditions provides a complimentary approach to established methods for use of reactive dienes in the expansion of diverse PAH cores. Additionally, the high-yield formation of COT derivative 13 suggests that stannoles may be useful synthons for cycloadditions beyond [4 + 2], where their spontaneous aromatization is highly desirable for the synthesis of unusual conjugated ring structures.Data availability
Crystallographic data for 13 has been deposited at the CCDC under 2121655.Author contributions
H. M. B. and T. D. T. conceived of the idea. H. M. B. developed the methodology. D. D. B. expanded the methodology and substrate scope. H. M. B., D. D. B., and G. R. K. acquired the data for synthesis and characterization. R. C. H. acquired and analysed the crystallographic data for compound 13. H. M. B., D. D. B., Y. L., and T. D. T. wrote and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the National Science Foundation under Grant No. CHE-2103696. Work performed at the Molecular Foundry was supported by the Office of Science, Office of Basic Energy Sciences, of the US Department of Energy under Contract No. DE-AC02-05CH11231. The authors thank Dr Hasan Celik for assistance with NMR spectroscopy and Justin Jurczyk for useful discussions. Crystallographic analysis was performed at UC Berkeley CheXray facility, which is supported by the NIH Shared Instrumentation Grant S10-RR027172.Notes and references
- S. Günes, H. Neugebauer and N. S. Sariciftci, Conjugated Polymer-Based Organic Solar Cells, Chem. Rev., 2007, 107, 1324–1338, DOI:10.1021/cr050149z.
- T. M. Figueira-Duarte and K. Mullen, Pyrene-Based Materials for Organic Electronics, Chem. Rev., 2011, 111, 7260–7314, DOI:10.1021/cr100428a.
- J. Wu, W. Pisula and K. Müllen, Graphenes as Potential Material for Electronics, Chem. Rev., 2007, 107, 718–747, DOI:10.1021/cr068010r.
- N. Panwar, A. M. Soehartono, K. K. Chan, S. Zeng, G. Xu, J. Qu, P. Coquet, K. T. Yong and X. Chen, Nanocarbons for Biology and Medicine: Sensing, Imaging, and Drug Delivery, Chem. Rev., 2019, 119, 9559–9656, DOI:10.1021/acs.chemrev.9b00099.
- Z. Sun and J. Wu, Open-Shell Polycyclic Aromatic Hydrocarbons, J. Mater. Chem., 2012, 22, 4151–4160, 10.1039/c1jm14786b.
- J. Wu and K. Müllen, All-Benzenoid Polycyclic Aromatic Hydrocarbons: Synthesis, Self-Assembly and Applications in Organic Electronics, Carbon-Rich Compounds: From Molecules to Materials, John Wiley and Sons, 2006, pp. 90–139, DOI:10.1002/3527607994.ch3.
- A. Narita, X. Y. Wang, X. Feng and K. Müllen, New Advances in Nanographene Chemistry, Chem. Soc. Rev., 2015, 44, 6616–6643, 10.1039/c5cs00183h.
- M. B. Goldfinger, K. B. Crawford and T. M. Swager, Directed Electrophilic Cyclizations: Efficient Methodology for the Synthesis of Fused Polycyclic Aromatics, J. Am. Chem. Soc., 1997, 119, 4578–4593, DOI:10.1021/ja9642673.
- H. Ito, Y. Segawa, K. Murakami and K. Itami, Polycyclic Arene Synthesis by Annulative π-Extension, J. Am. Chem. Soc., 2019, 141, 3–10, DOI:10.1021/jacs.8b09232.
- K. P. C. Vollhardt, Cobalt-Mediated [2 + 2 + 2]-Cycloadditions: A Maturing Synthetic Strategy [New Synthetic Methods (43)], Angew. Chem., Int. Ed., 1984, 23, 539–556, DOI:10.1002/anie.198405393.
- S. Han, A. D. Bond, R. L. Disch, D. Holmes, J. M. Schulman, S. J. Teat, K. P. C. Vollhardt and G. D. Whitener, Total Syntheses and Structures of Angular [6]-and [7]Phenylene: The First Helical Phenylenes (Heliphenes), Angew. Chem., Int. Ed., 2002, 41, 3223–3227, DOI:10.1002/1521-3773(20020902)41:17<3223::AID-ANIE3223>3.0.CO;2-G.
- T. Jin, J. Zhao, N. Asao and Y. Yamamoto, Metal-Catalyzed Annulation Reactions for π-Conjugated Polycycles, Chem.–Eur. J., 2014, 20, 3554–3576, DOI:10.1002/chem.201304640.
- F. Teplý, I. G. Stará, I. Starý, A. Kollárovič, D. Šaman, L. Rulíšek and P. Fiedler, Synthesis of [5]-, [6]-, and [7]Helicene via Ni(0)- or Co(I)-Catalyzed Isomerization of Aromatic Cis,Cis-Dienetriynes, J. Am. Chem. Soc., 2002, 124, 9175–9180, DOI:10.1021/ja0259584.
- D. Pérez, D. Peña and E. Guitián, Aryne Cycloaddition Reactions in the Synthesis of Large Polycyclic Aromatic Compounds, Eur. J. Org. Chem., 2013, 2013, 5981–6013, DOI:10.1002/ejoc.201300470.
- O. T. Dyan, G. I. Borodkin and P. A. Zaikin, The Diels–Alder Reaction for the Synthesis of Polycyclic Aromatic Compounds, Eur. J. Org. Chem., 2019, 7271–7306, DOI:10.1002/ejoc.201901254.
- Y. Xiao, J. T. Mague, R. H. Schmehl, F. M. Haque and R. A. Pascal, Dodecaphenyltetracene, Angew. Chem., Int. Ed., 2019, 58, 2831–2833, DOI:10.1002/anie.201812418.
- R. G. Clevenger, B. Kumar, E. M. Menuey and K. V. Kilway, Synthesis and Structure of a Longitudinally Twisted Hexacene, Chem.–Eur. J., 2018, 24, 3113–3116, DOI:10.1002/chem.201705676.
- H. M. Duong, M. Bendikov, D. Steiger, Q. Zhang, G. Sonmez, J. Yamada and F. Wudl, Efficient Synthesis of a Novel, Twisted and Stable, Electroluminescent “Twistacene”, Org. Lett., 2003, 5, 4433–4436, DOI:10.1021/OL035751V.
- X. Qiao, M. A. Padula, D. M. Ho, N. J. Vogelaar, C. E. Schutt and R. A. Pascal, Octaphenylnaphthalene and Decaphenylanthracene, J. Am. Chem. Soc., 1996, 118, 741–745, DOI:10.1021/ja953669s.
- J. W. Barton, M. K. Shepherd and R. J. Willis, Polycyclic Biphenylenes. Part 6. Direct Routes to Benzo[b]Biphenylene and Related Systems via Cycloaddition Reactions, J. Chem. Soc., Perkin Trans. 1, 1986, 967–971, 10.1039/p19860000967.
- J. Nakayama, S. Yamaoka, T. Nakanishi and M. Hoshino, 3,4-Di-Tert-Butylthiophene 1,1-Dioxide, a Convenient Precursor of o-Di-Tert-Butylbenzene and Its Derivatives, J. Am. Chem. Soc., 1988, 110, 6598–6599, DOI:10.1021/ja00227a069.
- Q. Miao, X. Chi, S. Xiao, R. Zeis, M. Lefenfeld, T. Siegrist, M. L. Steigerwald and C. Nuckolls, Organization of Acenes with a Cruciform Assembly Motif, J. Am. Chem. Soc., 2006, 128, 1340–1345, DOI:10.1021/ja0570786.
- W. Steglich, E. Bochmann, G. Gansin and L. Wilschitz, Herstellung Und Reaktionen von 2,5-DiphenyI-6-Oxo-1,3,4-Oxadiazin, Synthesis, 1977, 04, 252–253, DOI:10.1055/s-1977-24339.
- M. Ramirez, E. Darzi, J. Donaldson, K. N. Houk and N. K. Garg, Cycloaddition Cascades of Strained Alkynes and Oxadiazinones, Angew. Chem., Int. Ed., 2021, 60, 18201, DOI:10.1002/anie.202105244.
- G. R. Kiel, M. S. Ziegler and T. D. Tilley, Zirconacyclopentadiene-Annulated Polycyclic Aromatic Hydrocarbons, Angew. Chem., Int. Ed., 2017, 56, 4839–4844, DOI:10.1002/anie.201700818.
- K. Kunô, K. Kobayashi, M. Kawanisi, S. Kozima and T. Hitomi, Diorganostannylenes. III. Formation of Dimethylstannylene from 1,1-Dimethyl-2,3,4,5-Tetraphenyl-1-Stannacyclopentadiene and Acetylenes, J. Organomet. Chem., 1977, 137, 349–354, DOI:10.1016/S0022-328X(00)81441-2.
- C. Grugel, W. P. Neumann and M. Schriewer, The First Stable Derivative of 7-Stannanorbornane and Its Thermolysis, Angew. Chem., Int. Ed., 1979, 18, 543–544, DOI:10.1002/anie.197905431.
- B. Wrackmeyer and U. Klaus, New Phosphabenzenes by [4 + 2] Cycloaddition of Stannoles to 1-phospha-1-alkynes – Determination of Signs of Coupling Constants nJ(31P, 13C), nJ(31P, 1H), 2J(31P, 29Si), 2J(119Sn, 31P), J. Organomet. Chem., 1996, 520, 211–226, DOI:10.1016/0022-328X(96)06316-4.
- K. Yoshio, S. Akio, K. Shintaro, C. Kazumi and A. Wataru, Zirconocene Coupling Route to 1,2-disilacyclobutanes, Chem. Lett., 2000, 29, 1082–1083, DOI:10.1246/cl.2000.1082.
- W. Yue, J. Gao, Y. Li, W. Jiang, S. Di Motta, F. Negri and Z. Wang, One-Pot Synthesis of Stable NIR Tetracene Diimides via Double Cross-Coupling, J. Am. Chem. Soc., 2011, 133, 18054–18057, DOI:10.1021/ja207630a.
- I. Nagao, M. Shimizu and T. Hiyama, 9-Stannafluorenes: 1,4-Dimetal Equivalents for Aromatic Annulation by Double Cross-Coupling, Angew. Chem., Int. Ed., 2009, 48, 7573–7576, DOI:10.1002/anie.200903779.
- Y. Ura, Y. Li, Z. Xi and T. Takahashi, Cu(I) Catalyzed or Promoted Metallacycle Transfer of Zirconacycles to Stannacycles, Tetrahedron Lett., 1998, 39, 2787–2790, DOI:10.1016/S0040-4039(98)00430-4.
- J. Kubitschke, H. Hopf, P. G. Jones, I. Dix and L. Ernst, A Cyclobutadiene Intermediate in the Intramolecular Cycloaddition of 4,15-Bis(Phenylethynyl)[2.2]Paracyclophane, Eur. J. Org. Chem., 2008, 548–554, DOI:10.1002/EJOC.200700702.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2121655. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2sc00397j |
| This journal is © The Royal Society of Chemistry 2022 |