
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning the luminescence of transition metal complexes with acyclic diaminocarbene ligands
Mikhail A.
Kinzhalov
 *a,
Elena V.
Grachova
a and
Konstantin V.
Luzyanin
*b
*a,
Elena V.
Grachova
a and
Konstantin V.
Luzyanin
*b
aSt Petersburg University, 7/9 Universitetskaya Nab., Saint Petersburg, 199034, Russia. E-mail: m.kinzhalov@spbu.ru
bDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool L69 7ZD, UK. E-mail: konstantin.luzyanin@liverpool.ac.uk
First published on 6th December 2021
Abstract
Metal complexes featuring acyclic diaminocarbene ligands have recently emerged as powerful emitters for prospective use in OLEDs and other electroluminescent technologies. Owing to their strong σ-donating properties and broad synthetic availability, acyclic diaminocarbenes have successfully challenged the application of other ancillary ligands for fine-tuning of luminescence emission. This review systematises the data reported concerning the preparation of luminescent metal complexes bearing acyclic diaminocarbene ligands, uncovers their photophysical properties as a function of the carbene ligand structure and metal characteristics, and draws attention to the potential of ADCs and the advantages that the application of metal-(acyclic diaminocarbenes) provides to the family of transition-metal-based phosphors.
1. Introduction
Luminescent transition metal complexes underpin the development of OLEDs and other electroluminescent technologies (for the main reviews on the topic see ref. 1–10), and a broad range of applications in the fields of luminescence chemosensing,11–13 photocatalysis,14–16 sensitisation,17–19 self-assembled materials,20 and bioimaging.20–23 Although pure organic light emitters are predominantly fluorescent (triplet excitons formed are deactivated by thermal processes), the presence of strong spin–orbit coupling in transition metal complexes upholds the intersystem crossing from the singlet to the triplet state enabling phosphorescent relaxation. An electroluminescent device doped with a transition metal compound may form up to 25% singlet and 75% triplet excitons resulting in nearly quantitative luminescence efficiencies.The choice of ligands impacts the photoemissive properties of metal complexes with cyclometallated aromatic ligands dominating the field due to several reasons.1–3,24,25 The strongest bonding interaction with transition metals increases the d–d energy gap leading to a lower extent of non-emissive quenching due to the suppressed population to the higher-lying d–d excited state, while the emergence of close-lying, ligand-centred π–π* electronic transitions opens up the possibility for the facile tuning of emission wavelengths. Despite numerous reports on the photophysical properties of cyclometallated transition metal complexes, the lack of blue-phosphorescent compounds with good photoluminescence quantum yields and photostability still represents one of the main challenges. To cause blue luminescence, the energy of a triplet state of an organometallic emitter should be high enough, which may simultaneously lead to the thermal population of higher-lying triplet metal-centred (MC) ligand-field excited states. These states are nonemissive by decreasing the photoluminescence quantum yield Φ and by placing electrons into metal–ligand σ*-antibonding orbitals, causing the gradual degradation of the compound by ligand loss.1 One of the solutions that emerged is the use of a secondary set of σ-donor ligands, which would additionally destabilize unoccupied d-orbitals and their corresponding metal-centred excited states, and improve the efficiency and stability of blue phosphorescence. As a result, strong σ-donor carbene ligands, i.e., N-heterocyclic carbenes (NHCs),42 acyclic diaminocarbenes (ADCs),165 and cyclic alkylaminocarbenes,166 have been evaluated as organometallic emitters (Fig. 1).
 | ||
| Fig. 1 Representative luminescent transition metal complexes supported by cyclometallated phenylpyridines, NHCs26,27 and ADCs.28,29 | ||
The prevailing part of examples discloses the photophysical properties of metal–NHC species (for reviews on the topic see ref. 30–33; for recent manuscripts see ref. 34–38); only a few reports describing the photoluminescence properties of cyclic alkylaminocarbenes have recently emerged.39–41 At the same time, a number of experimental studies considering luminescent metal–ADC species have now reached a level, where the first survey to consider these diverse ADC-containing organometallic phosphors is timely.
The essential goals of this review are (i) to systematise the reports published until now describing the preparation and the photophysical properties of metal–ADCs; (ii) to compare, whenever possible, the photophysical properties of metal–ADCs against metal–NHC species, and (iii) to draw attention to the potential of ADCs and the advantages that the application of ADCs gives to organometallic chemistry and materials science.
2. Acyclic diaminocarbenes as ligands
NHCs and ADCs are neutral ligands featuring divalent carbon centres with six-electron valence shells.42,165 Both carbene types exhibit a singlet ground-state electronic configuration with a HOMO (Highest Occupied Molecular Orbital) and LUMO (Lowest Unoccupied Molecular Orbital) best described as a formally sp2-hybridised lone pair and an unoccupied p-orbital at the carbene carbon, respectively. The relative stability of diaminocarbene is ensured by the presence of mutually σ-electron-withdrawing and π-electron-donating nitrogens both inductively (by lowering the energy of the occupied σ-orbital) and mesomerically (by donating electron density to the empty p-orbital) (Fig. 2). The principal consequence of the ground-state electronic structure of diaminocarbenes is their propensity to act as σ-donors and bind to a wide range of metal centres. While strong σ-donor and comparatively weak π-accepting properties of aminocarbenes suggest the coordination properties closely resembling those of phosphines, the former are in general more electron-donating than the latter.43,44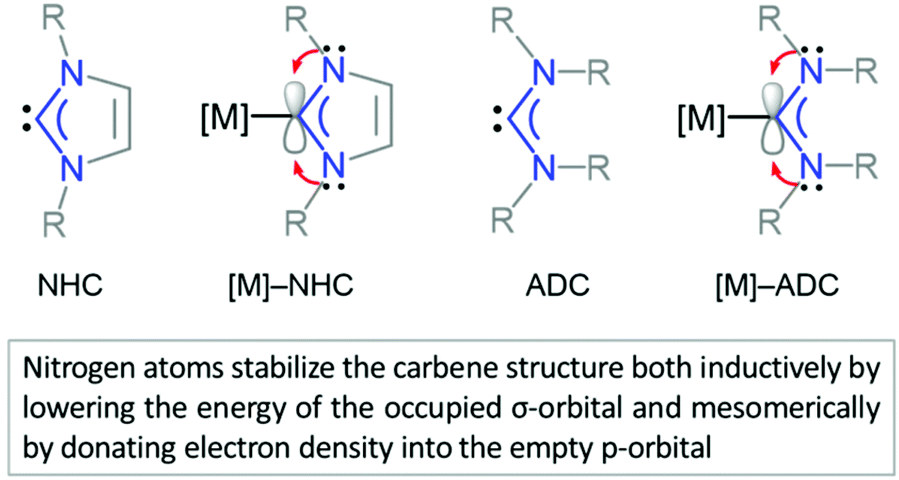 | ||
| Fig. 2 Heterocyclic (NHC)42 and acyclic (ADC)165 diaminocarbenes, and their corresponding metal complexes. | ||
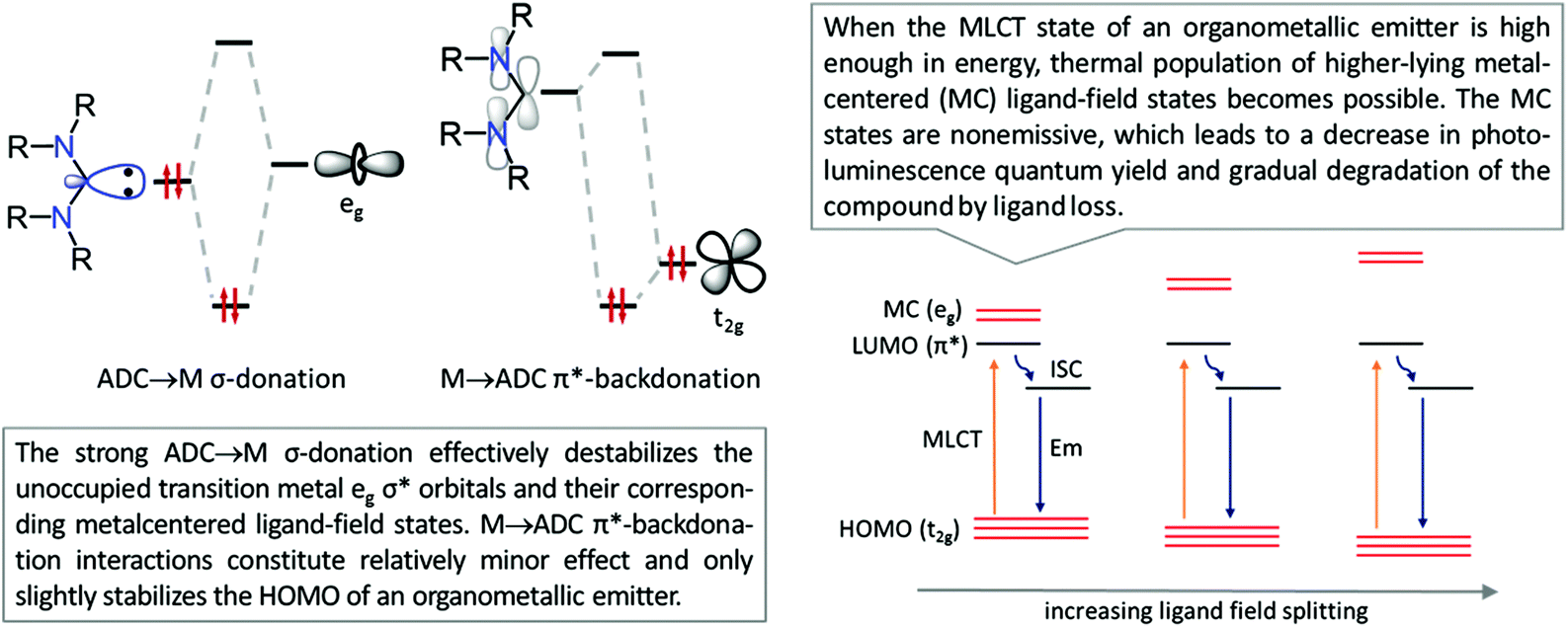
This is reflected in a stronger and shorter metal–ligand bond (as judged by its length and greater bond dissociation energies) in diaminocarbene complexes when compared to their phosphine counterparts. Due to the lower electronegativity of carbon compared to nitrogen, the in-plane lone electron pair on the former possesses significantly higher energy than that in common N-donors such as pyridyl ligands. Fig. 3 shows the bonding interactions between diaminocarbenes and transition metals; the major contribution to bonding is due to σ-donation that effectively destabilises the transition metal eg σ* orbitals, with metal-to-ligand π-backdonation interactions constituting relatively minor effects.45
 | ||
| Fig. 3 [M]-ADC bonding interactions and effect of the ADC ligand on the destabilisation of the non-emissive MC excited state of the organometallic emitter. | ||
The absence of a cyclic fragment in the ADCs causes significant changes in their steric and electronic characteristics in comparison to NHCs. Owing to the lack of electron density delocalisation (as no aromatic heterocyclic fragment is present in the structure), ADCs are generally more electron-donating than aromatic-NHCs. Moreover, ADCs are more basic and nucleophilic species than their cyclic non-aromatic counterparts insofar as a wider N–Ccarbene–N bond angle decreases the s-character of the lone pair on the carbene carbon.46–48 The better donor ability of ADCs and an increased electronic density on a metal destabilise the ligand-field states, potentially leading to higher quantum yields for pure deep blue photoluminescence and expected performance in OLED devices (Fig. 4, left).
 | ||
| Fig. 4 (Left): structurally related luminescent IrIII NHC and ADC complexes;26,28 (right): conformers of IrIII–ADC species showing distinct photophysical properties.49 | ||
Due to the acyclic structure, rotation around the Ccarbene–N bonds is enabled allowing for the acyclic diaminocarbene ligand to adopt various conformations with differing steric and electronic characteristics.50–52 For instance, in carbene species having small substituents, a rotation barrier of the Ccarbene–N bonds is lower than 13 kcal mol−1 as indicated by experimental53 and theoretical54 studies. Rotational flexibility plays a significant role in the photoemissive properties:55 different conformers of ADCs exhibit different energies of the triplet metal-to-ligand charge-transfer (MLCT) states (Fig. 4, right).49
Another important observation regarding metal–ADCs is their synthetic availability since several strategies for their preparation are established and widely used.58–62 Valuable properties of complexes of transition metals with diaminocarbene ligands associated with their photophysical applications are determined by the characteristics of the metal centre, as well as by the balance between the donor and steric properties of the diaminocarbene ligand. Each application requires complexes and, therefore, ligands with a different set of parameters, that are screened empirically, imposing stringent requirements on the method of their synthesis.
Free acyclic diaminocarbenes can be synthesized in the laboratory and used for direct coordination to metal centers to the same extent as their N-heterocyclic counterparts. At the same time, the most widespread approach is based on nucleophilic addition to metal-bound isocyanides, leading to the production of diminocarbene metal complexes without the release of free carbene.58–63 This approach is stoichiometric, atom-efficient and high-yielding as a rule allowing for an easy variation of the periphery of ADCs, thus ensuring high synthetic flexibility (Scheme 1). The elegance of this approach consisting basically of one step and leading to diverse well-defined metal–diaminocarbene species is in sharp contrast to the strategies for the generation of custom-made N-heterocyclic carbenes64 and cyclic alkylaminocarbenes,39 where only the ligand synthesis may require several consecutive steps. Note that all luminescent ADC complexes known up to date and described in this review were obtained by the isocyanide pathway.
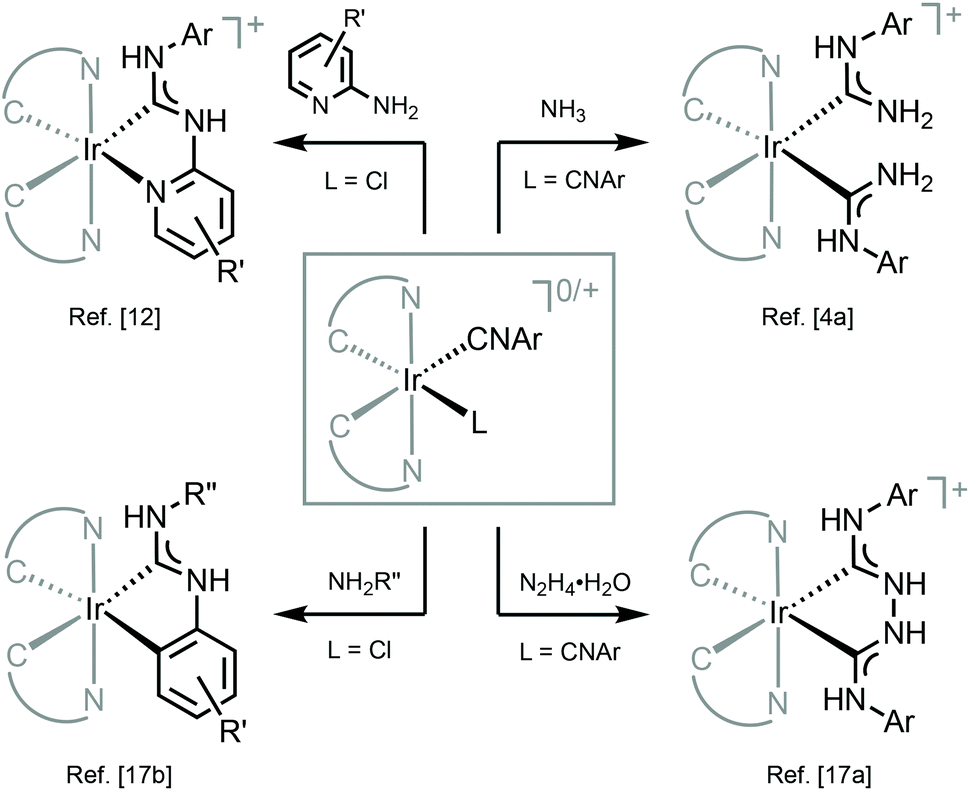 | ||
| Scheme 1 Representative examples of IrIII–ADC complexes prepared by nucleophilic addition to metal-bound isocyanides.28,49,56,57 | ||
3. Luminescent [M]-ADC complexes
Luminescent complexes of four transition metals, viz. IrIII, PtII, ReI, and AuI/III, bearing ADC ligands are considered in the current review. All the examples discussed belong to the 5d row of transition metals, and although many 4d metal–ADC complexes are known, to the best of our knowledge none of them have been described as luminescent. While the excellent donor capacity and binding properties of ADC ligands make them suitable candidates for the construction of luminescent systems based even on first-row transition metal complexes, including iron,65,66 no significant progress has been made in this area to date. Nevertheless, it has been demonstrated that the donor properties of ADCs can destabilize the metal-localized ligand-field excited states in FeII complexes, affecting their optical properties.66The first example of an emissive transition metal ADC complex was reported by Che and coworkers for PtII species in the late 90s,67 but an explosion of research in this area took place several years ago, starting with the discovery of IrIII–ADC complexes by Teets56 and Luzyanin68 groups. Of note, some of the complexes depicted below feature deprotonated forms of ADCs, that are called aminocarbenes for the sake of simplicity of the discussion; some authors suggest that they should be called formamidinyl or aminocarbene-like species from the perspective of organometallic formalism.58
3.1. Group 9: iridium(III) ADC complexes
The combination of ADCs and cyclometallating (C^N or C^C) ligands around the IrIII centre can potentially lead to bright triplet luminescence (phosphorescence) due to the strong spin–orbit coupling characteristics of IrIII complexes.2,33,69,70Bis-C^N-cyclometallated IrIII complexes bearing neutral “Chugaev-type”67,71–76 deprotonated C,C′-chelating bis(diaminocarbene) ligands 4–6 were prepared via the addition of excess hydrazine to isocyanides in 1–3 (Scheme 2).56,77 Protonation of 4–6 with one equivalent of HBF4 led to cationic bis(diaminocarbene) derivatives 7–9. Electrochemical studies were used to assess the energies of frontier orbitals; the oxidation potentials of cyclometallated IrIII complexes are linked to the HOMO energy levels consistent with the removal of an electron from an Ir-aryl-centred orbital. The first oxidation potential of 4–9 corresponds to IrIII/IrIV transition (primarily metal-centred) and is located within 0.86–1.02 V. These electrochemical potentials are somewhat similar to those for cyclometallated IrIII complexes bearing NHCs (0.97–1.10 V).78,79 When compared to starting bis-arylisocyanides species 1–3 (1.63 V, 1.68 V, and 1.44 V, respectively),56,77,80 Ir-bis-ADCs 4–9 are 0.58–0.66 V easier to oxidise, suggesting the destabilisation of the HOMO levels upon conversion of both isocyanides to a C,C′-chelating ADC ligand.
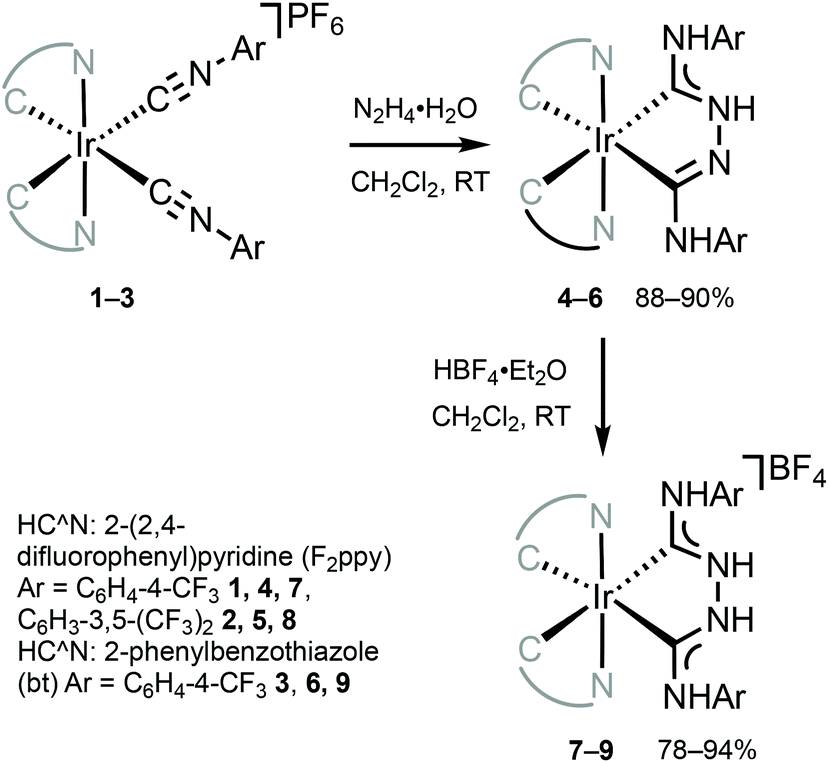 | ||
| Scheme 2 Preparation of chelating IrIII–ADCs 4–9.56,77 | ||
Metal–ADCs 4–9 display 1,3CT absorption bands tailing into the visible region (Fig. 5) in contrast to bis-isocyanides 1–3, where those occur under 400 nm. In solution, 4 and 5 are blue emitters (λmax 473–474, Φ 0.044 for 4 or 0.010 for 5) (Fig. 5). The emission spectra of 4–6 are considerably redshifted by ca. 8–12 nm (360–410 cm−1) from those of the starting bis-isocyanides 1–3, which can be attributed to the decrease of the π-accepting properties and the increase of the σ-donating properties of the ADC ligand, leading to the destabilisation of the HOMO for 4–6.59,816 emits in solution in the green to yellow regions with λmax at 519 nm and Φ of 0.19. Protonation of 4–6 results in a 2–4-fold increase in quantum yields for 7–9 (Φ 0.08 for 7, 0.02 for 8, and 0.60 for 9). Immobilisation of 4, 6, 7, and 9 in PMMA (2 wt% of the IrIII complex) brings a significant increase in Φ (Φ 0.56 for 4, 0.90 for 6, 0.68 for 7, and 0.86 for 9); both the emission energy and spectral profiles are retained. An increase in Φ can be justified as a result of decreasing the non-radiative vibrational relaxation of the excited state in the rigid environment. Even though 4–6 exhibit only moderate photoluminescence properties, this study illustrates that both electrochemical and photophysical properties are significantly impacted upon conversion of isocyanide to carbene species. As a result, this metal-mediated approach to ligand design features an alternative to a classic ligand exchange approach towards the tuning of excited states and subsequently the luminescent profiles of metal complexes.
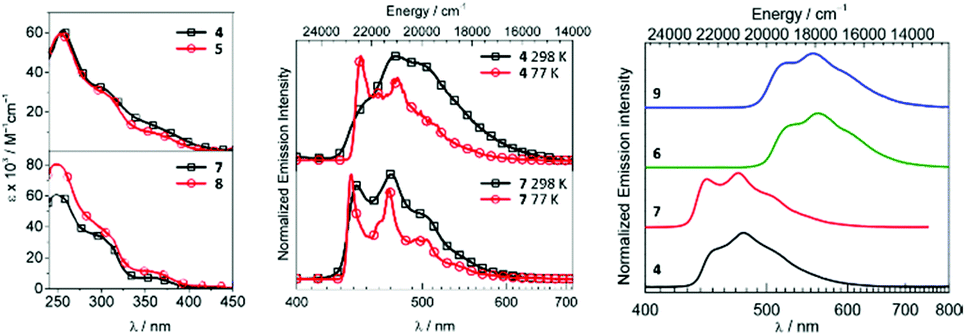 | ||
| Fig. 5 (Left): UV-vis spectra of 4, 5, 7, and 8 in CH2Cl2; (centre): normalised emission spectra of 4 and 7 in CH2Cl2 at RT and in CH2Cl2/toluene at 77 K; (right): normalised emission spectra of 4, 6, 7, and 9 in the PMMA film (2 wt%) at RT. Adapted with permission from ref. 56 and 77. Copyright (2017, 2018) American Chemical Society. | ||
The neutral tris-chelated cyclometallated IrIII complexes supported by the ADC ligands were generated via the coupling of amines with IrIII-isocyanides 10 and 11 (Scheme 3).57 Upon the nucleophilic attack by an amine, a subsequent base-promoted intramolecular activation of an aromatic C–H bond yields the final ortho-metallated IrIII–ADCs 12–15. The first oxidation potentials of 12 and 13 (0.52 and 0.54 V) are cathodically shifted by at least 0.25–0.28 V when compared to those of relevant IrIII–NHC [Ir(F2ppy)2(C,C′-NHC)] (C,C′-NHC is C,C′-cyclometallated 3-methyl-1-(4-(trifluoromethyl)phenyl)-1H-imidazol-3-ium-2-ide, 0.82 V or C,C′-cyclometallated C,C′-cyclometallated 3-methyl-1-(4-(trifluoromethyl)phenyl)-1H-benzo[d]imidazol-3-ium-2-ide, 0.78 V).79 This comparison demonstrates significantly higher-lying HOMO levels in the ADC complexes than in their cyclic counterparts, consistent with the fact that ADC is a more electron-rich donor than NHC.48
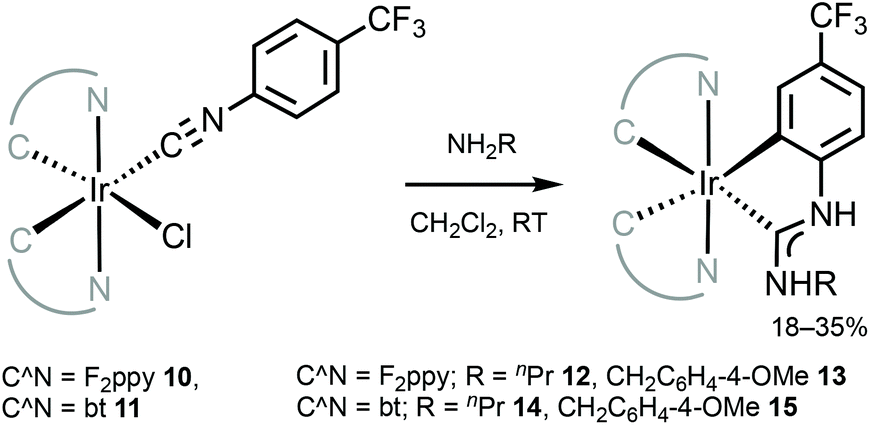 | ||
| Scheme 3 Synthesis of cyclometallated IrIII–ADC complexes 12–15.57 | ||
Iridium(III)–ADCs 12–15 exhibit reduced lowest-energy absorption bands tailing to 450 nm for F2ppy (12, 13) and 500 nm for bt (14, 15) complexes attributed to both singlet and triplet MLCT transitions (Fig. 6). The energies of the MLCT transitions are affected by varying the cyclometallating C^N ligand, while alteration of the ADC periphery induces a minor change in the extinction coefficients without affecting notably the MLCT bands. In CH2Cl2 solution at RT, complexes 12 and 13 exhibit blue phosphorescence with λmax values at 498 and 495 nm and moderate Φ values (0.22 and 0.18, respectively). In solution, the yellow-orange emission of 14 and 15 has significantly lower Φ values (0.049 for 14, 0.047 for 15). Immobilisation of metal–ADCs in the PMMA matrix increases Φ to 0.79 (for blue emitters 12 and 13) and 0.30–0.37 (for orange emitters 14 and 15) (Fig. 6). Computational studies showed that iridium-cyclometallated species possess typical characteristics of thermally activated delayed fluorescence emitters with potential to be used as novel electroluminescent devices.82
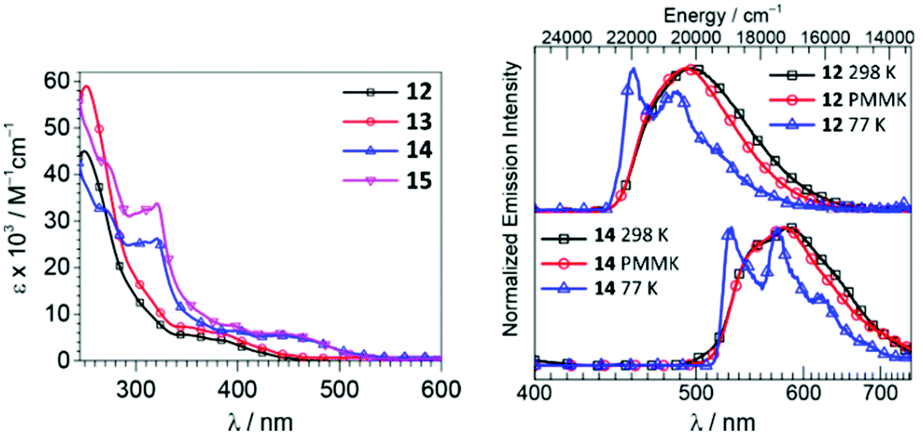 | ||
| Fig. 6 (Left): UV-vis spectra of 12–15 in CH2Cl2; (right): normalised emission spectra of 12 and 14 in CH2Cl2 solution at 298 K, in CH2Cl2/toluene glass at 77 K (λext 310 nm), and in the PMMA film (2 wt%) at 298 K (λext 365 nm). Adapted with permission from ref. 57. Copyright (2018) American Chemical Society. | ||
A combination of DFT and TD-DFT calculations were used to predict the multi-state redox switchable nonlinear optics response9 of iridium(III) ADC complexes like 7–983 and 12–15.84 The nonlinear optics response can be easily modulated by one electron redox processes and IrIII complexes like 7–15 can be applicable for redox second-order nonlinear optic switches with two redox states.
An approach aimed towards the increase of the emission energy of cyclometallated iridium complexes focused on the destabilisation of the LUMO have been reported.24,33,85 Replacing the pyridyl ring of the C^N ligands with an N-heterocyclic carbene leads to a significant increase in the LUMO energy and, consequently, increases the emission energy of complexes. The majority of the recent achievements depicting this approach involved homoleptic tris-cyclometallated Ir(C,C′-NHC)3 complexes,34,38,85–89 showing blue or near-UV phosphorescence at room temperature. A cascade reaction involving nucleophilic addition of propyl amine to a coordinated isocyanide in 16–18 with a subsequent base-assisted cyclometallation (Scheme 4) led to the heteroleptic cyclometallated derivatives 19–21 featuring two C,C′-NHC and one C,C′-ADCs.90 The first oxidation potential of 19–21 falls within the 0.16–0.25 V range, akin to those for homoleptic mer-[Ir(C,C′-NHC)3] complexes (0.14–0.23 V).34,85 Both 19–21 and the aforementioned mer-[Ir(C,C′-NHC)3] are difficult to reduce due to the high energy of the LUMO, which is being responsible for the large HOMO–LUMO gap in this type of IrIII compounds. 19 is not luminescent in solution at RT, while 20 shows a weak blue (λmax 418 nm, Φ 0.013) and 21 green (λmax 511 nm, Φ 0.39) emissions. Immobilisation in the PMMA film (2 wt%) increases Φ for 19 and 20 to 0.13 and 0.31 without affecting λmax. For 21 in the PMMA film, a hypsochromic shift for luminescence is accompanied by an increase in Φ (λmax 459 nm, Φ 0.48), Fig. 7. When compared to homoleptic mer-[Ir(C,C′-NHC)3]34,85 already used for the manufacturing of deep blue OLEDs, the mixed-carbene species 19–21 exhibit similar or superior photoluminescence characteristics, i.e. higher Φ.
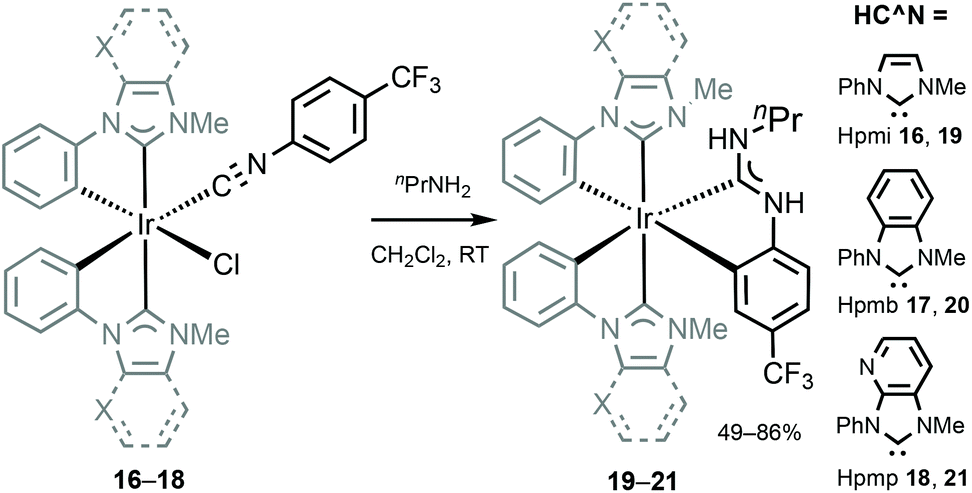 | ||
| Scheme 4 Preparation of cyclometallated IrIII–ADCs 19–21.90 | ||
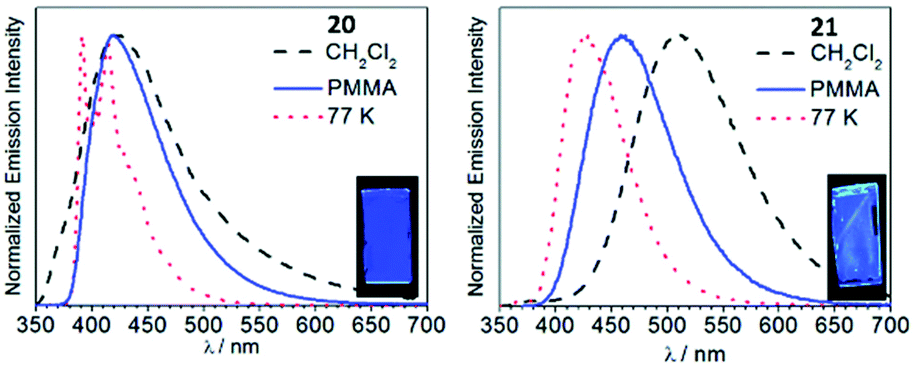 | ||
Fig. 7 Emission spectra of 20 and 21 in CH2Cl2 and in the PMMA film at RT, and in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 CH2Cl2/toluene glass at 77 K (λext 310 nm). The insets show photographs of the PMMA films under UV illumination. Adapted with permission from ref. 90. Copyright (2019) The Royal Society of Chemistry. :3 CH2Cl2/toluene glass at 77 K (λext 310 nm). The insets show photographs of the PMMA films under UV illumination. Adapted with permission from ref. 90. Copyright (2019) The Royal Society of Chemistry. | ||
Metal-mediated addition of NH3 to the coordinated isocyanides in 22–29 (Scheme 5) allows the preparation of another family of heteroleptic cyclometallated IrIII species bearing ancillary ADCs.28,68 Oxidation of the thus prepared ADCs 30–45 is manifested by an irreversible wave centred at metal (0.77–1.22 V). Conversion of one CNR in 26–29 to ADC in 34–37 brings about a decrease in the oxidation potential by ca. 0.2 V; generation of the second ADC in 38–41 induces a further drop of the oxidation potential by ca. 0.2 V.
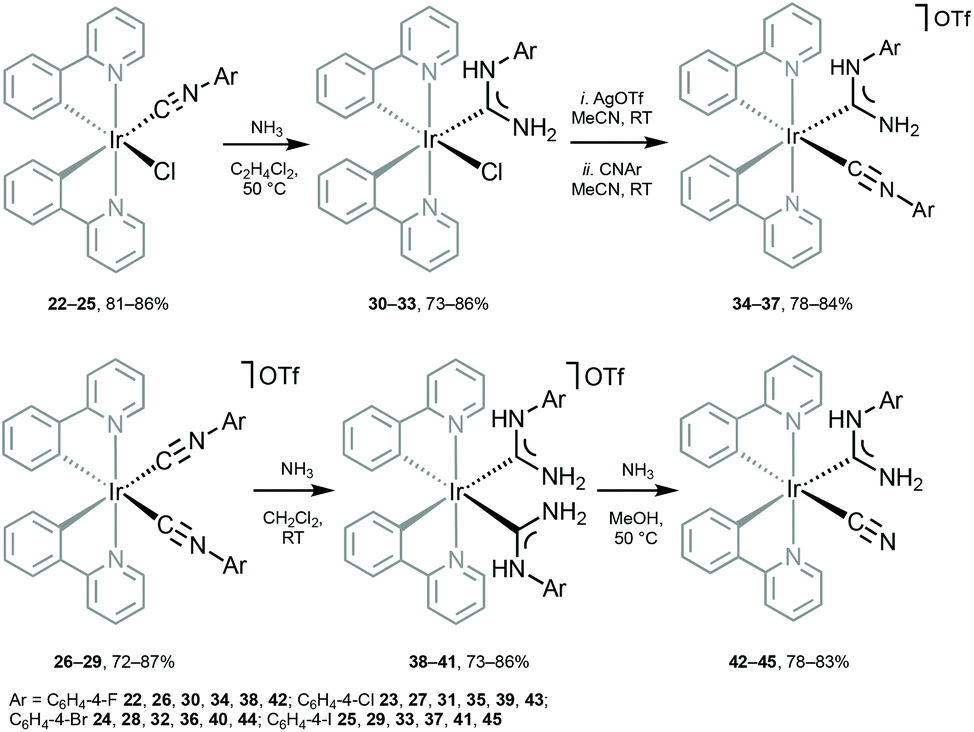 | ||
| Scheme 5 Preparation of IrIII–ADC complexes 30–45.28,68 | ||
Variation of the halogen substituent in the aryl fragment of the CNR or the corresponding ADC ligands induces no significant change in the positions of the singlet MLCT bands and only exerts a minimal influence on the extinction coefficients (Fig. 8, top). Conversion of the CNRs to ADCs impacts those transitions: a ca. 30 nm (ca. 2000 cm−1) redshift of the singlet MLCT absorption bands occurs on going from 26–29 to 38–41 (Fig. 8, bottom, for representative examples of compounds with a bromo-substituted phenyl fragment).28
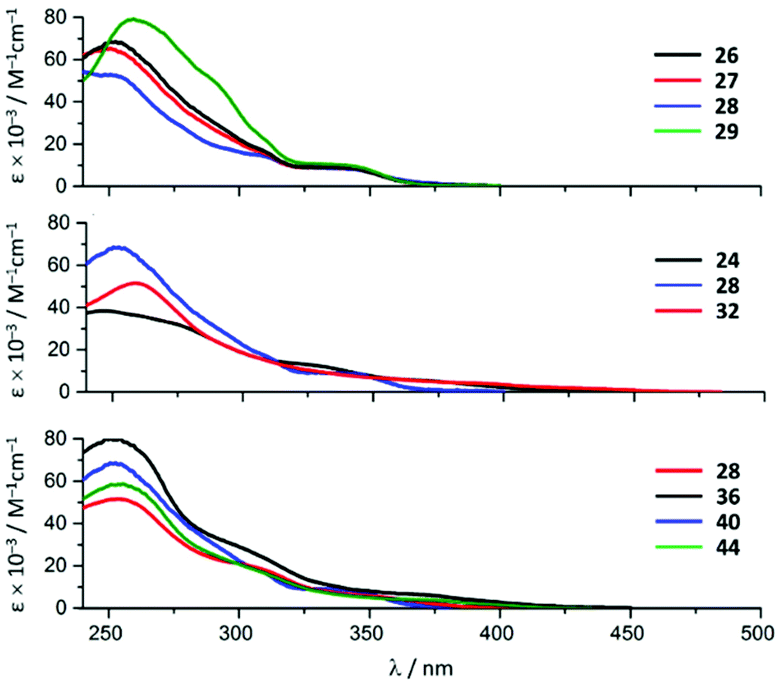 | ||
| Fig. 8 UV-vis spectra of the representative IrIII species of the 22–45 family in MeCN at RT. Adapted with permission from ref. 28. Copyright (2020) American Chemical Society. | ||
Destabilisation of the HOMO as a result of the ADC ligand having lower π-accepting and higher σ-donating properties when compared to isocyanide is suggested as a reason for the singlet MLCT transition shift. Compounds 38–41 with two ADC ligands exhibit blueshifted singlet MLCT bands relative to the complexes with ADC/Cl (30–33) or ADC/CN (42–45) ligand environments insofar as strong σ-donation from ADC increases the HOMO–LUMO gap.
In solution, upon photoexcitation by UV light complexes all 22–45 exhibit a blue-green phosphorescence (Fig. 9).28 Conversion of 22–25 (λmax 469 nm, Φ 0.10 for 22) to 30–33 (λmax 502 nm, Φ 0.04 for 26) is accompanied by a bathochromic shift of emission energy and drop in the Φ. These relatively small Φ values for 30–33 photoemission can be explained by the presence of chlorides which have a smaller ligand field splitting parameter91 and stabilise the non-emissive metal centred states.92,93 A redshift of the λmax upon the transformation of coordinated CNR to ADC could be explained by the lower π-accepting and higher σ-donating properties of ADC59 that subsequently leads to the destabilisation of the HOMO,81 as confirmed by a cathode shift of the oxidation potential by ca. 0.3 V. A similar effect was observed for 26–29 (λmax 451 nm, Φ 0.65 for 27), 34–37 (λmax 458 nm, Φ 0.40 for 35), 38–41 (λmax 472 nm, Φ 0.31 for 39) and 42–45 (λmax 473 nm, Φ 0.45 for 43). A further theoretical investigation confirmed that the transformation of the isocyanide to the ADC ligand within the 22–45 family increases the relative contribution of the iridium d orbitals to the HOMO (23 31%, 31 40%, 27 19%, 35 25%, 39 34%) inducing a drop in the HOMO–LUMO gap and enhanced mixing of the singlet MLCT and triplet LC states.28
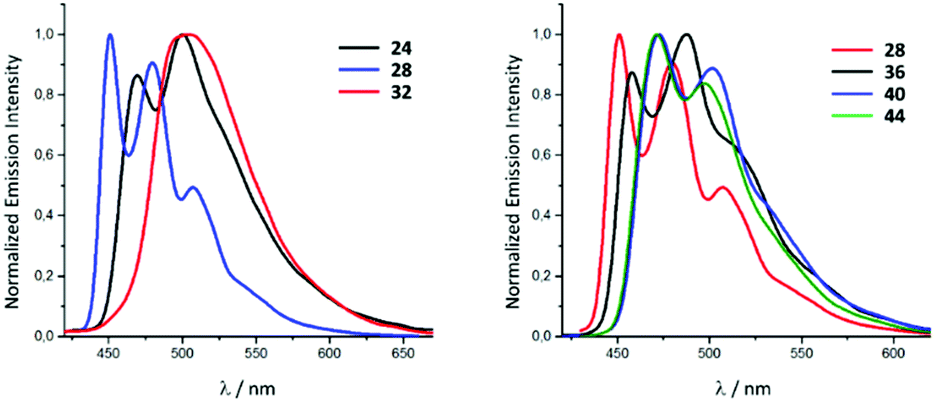 | ||
| Fig. 9 Photoluminescence spectra of the representative IrIII species of the 22–45 family recorded in MeCN at RT (λext 350–400 nm). Adapted with permission from ref. 28. Copyright (2020) American Chemical Society. | ||
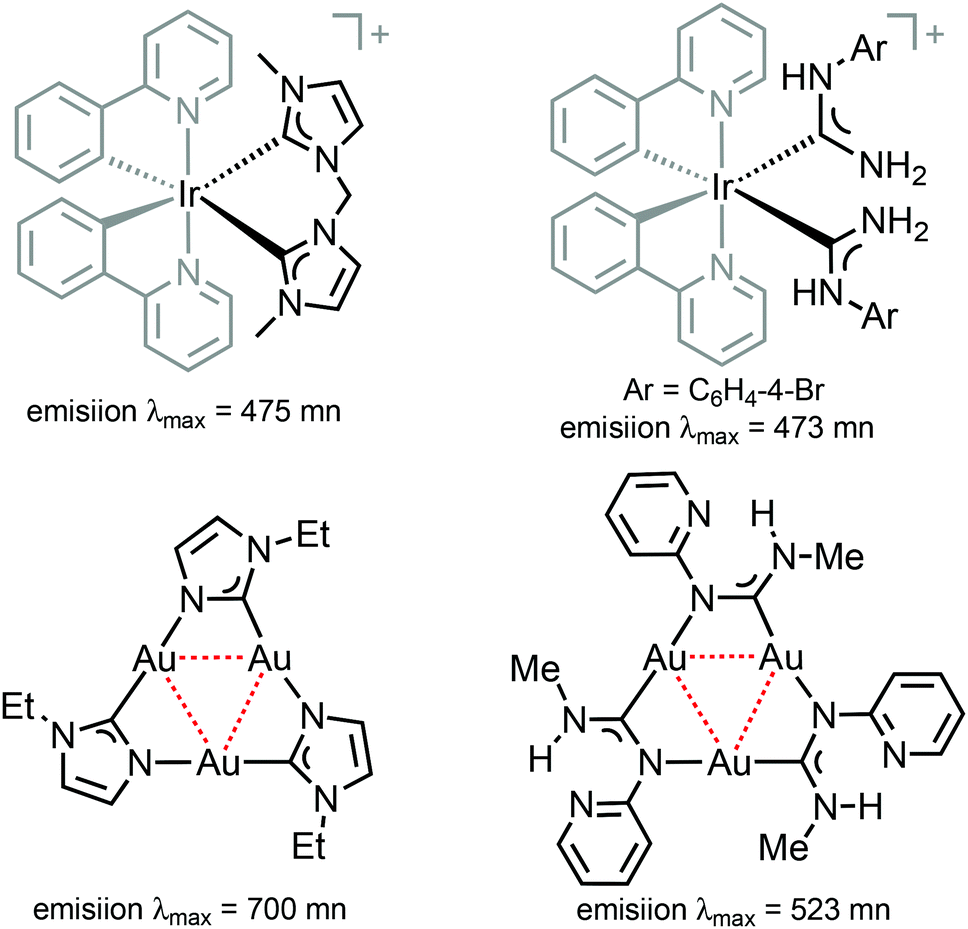
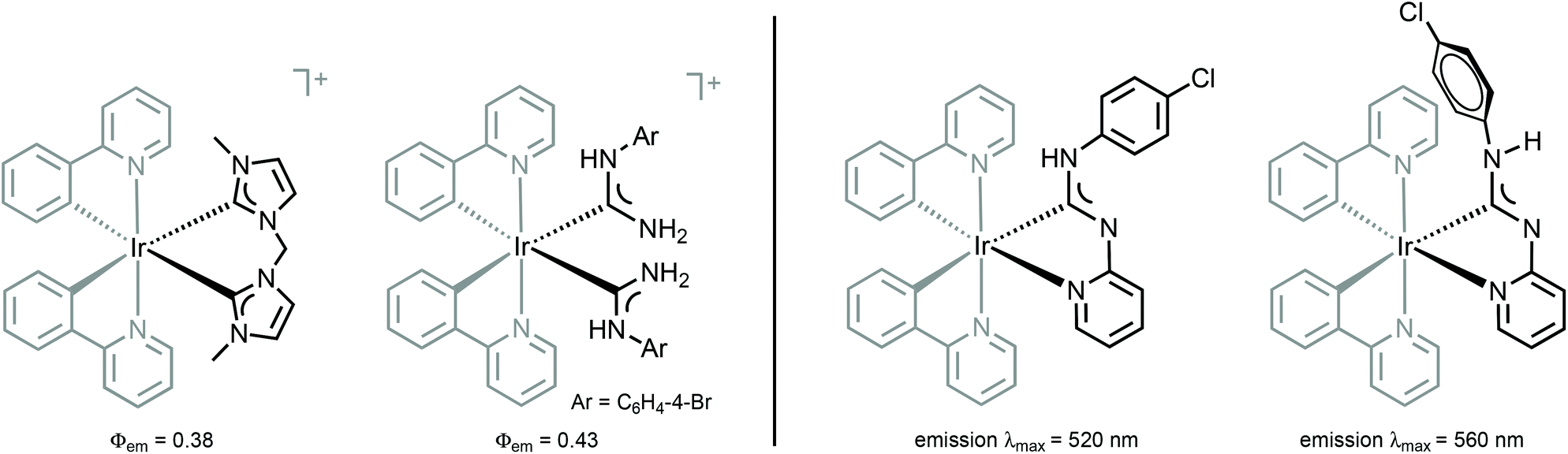
Comparison of 38–41 with the structurally related complexes with NHCs shown in Fig. 1 led to the conclusion of a rather similar λmax, as well as lifetimes of the excited states. This suggests that the emission properties of [Ir(C^N)2L2]+ complexes with carbene-based ancillary ligands are primarily controlled by the nature of C^N ligands, in contrast to the classical approach of charged complexes [Ir(C^N)2(N^N)]+ bearing chemically tunable N^N ancillary ligands with low-energy π* orbitals. The larger values of Φ for these ADC derivatives (0.43) when compared to NHC analogues (0.38) shown in Fig. 1 are explained by the less accessible higher-lying triplet MC excited states at room temperature for ADC complexes due to their superior donor properties.
Based on complexes 42–45 with ADC and cyano groups, ancillary ligands proposed a new class of organometallic chemosensors for the quantitative determination of mercury.2842–45 react selectively with Hg2+ ions, which is accompanied by a linear decrease in the luminescence intensity; limit of detection of mercury(II) ions as low as 2.63 × 10−7 M, which makes 42–45 one of the most sensitive photoluminescent chemosensors for the detection of mercury in solution.
Cyclometallated IrIII species 48–53 with deprotonated C,N-chelating ADC species represent a novel class of luminescent mechanochromic systems. Species 48–53 were prepared by the replacement of the chloride ligand of isocyano precursors with substituted 2-aminopyridines followed by an intramolecular nucleophilic attack (Scheme 6).4948–53 exhibit two moderate absorption shoulders at 340–360 and 420–440 nm, ascribed to an admixture of LC and MLCT [dπ(Ir) → π*(C,N-ADC)] transitions. In solution, complexes 48–53 display intensive triplet photoluminescence at RT with Φ ranging from 0.25 to 0.59 (Fig. 10). Solution emissions are attributed to phosphorescence derived from the predominant triplet MLCT [dπ(Ir) → π*(C,N-ADC)] state mixed with triplet LC(ppy) character as confirmed by computational studies for 48.
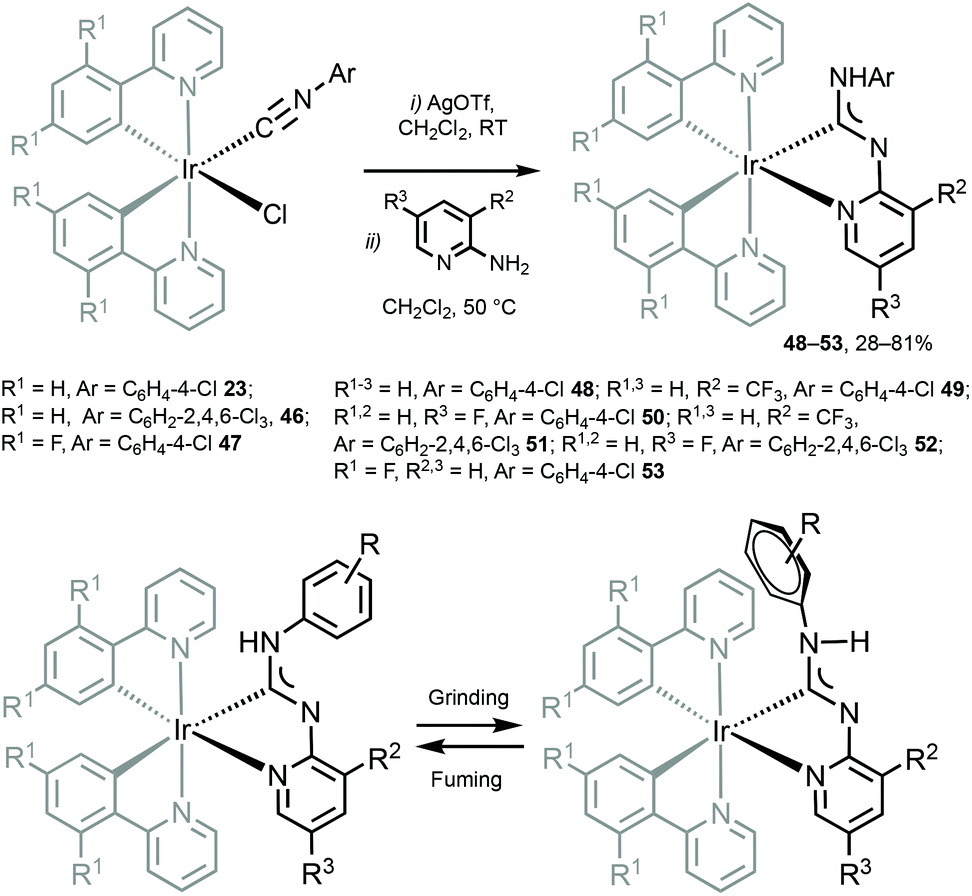 | ||
| Scheme 6 (Top): preparation of 48–53; (bottom): proposed mechanochemical change of the Ar orientation responsible for the observed change in the emission properties of diaminocarbene complexes.49 | ||
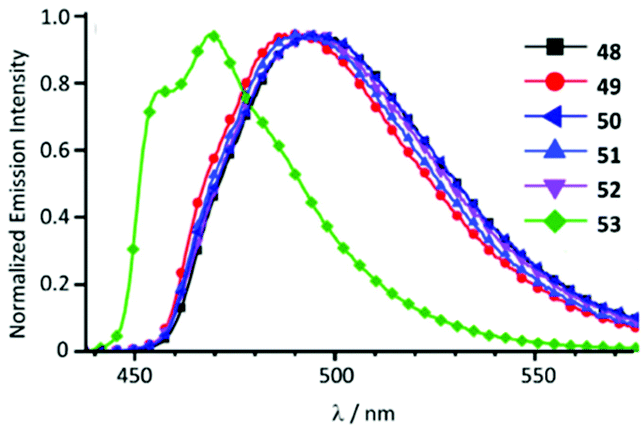 | ||
| Fig. 10 Emission spectra of 48–53 in CH2Cl2 solution at RT. Adapted with permission from ref. 49. Copyright (2020) The Royal Society of Chemistry. | ||
In the solid state 48–53 emit strong sky blue to yellow luminescence. Mechanical stimulation of 48–53 leads to a redshift of the emission tentatively associated with a conformational change of the ADC ligand (Scheme 6 and Fig. 11). Different orientations of the N-aryl moiety of the ADC ligand is reflected in the crystal packing affecting the energy of MLCT transitions and subsequently the photophysical properties of the complexes. The contribution of this effect depends on the nature of the substituents in the N-aryl moiety and indicates that the luminescent mechanochromic properties of these emitters can be tailored through systematic modification of the bidentate ADC ligands.
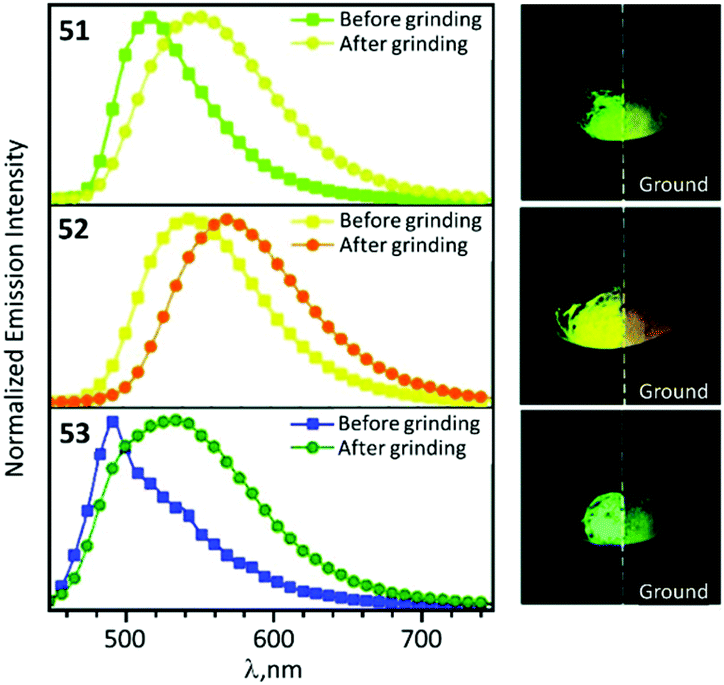 | ||
| Fig. 11 Solid-state emission spectra of 51–53 before and after grinding using λext 425 nm; photographs taken under UV lamp illumination (λext 365 nm). Adapted with permission from ref. 49. Copyright (2020) The Royal Society of Chemistry. | ||
It is worth mentioning that several different types of luminescent mechanochromic transition metal complexes have been previously reported.94–96 Luminescent mechanochromism of most systems described is associated with a reversible switching between crystalline polymorphic modifications and the amorphous state, primarily through the intermolecular or intramolecular metal–metal interactions.97–101 This makes systematic fine-tuning and modifications of the mechanochromic properties including the emission characteristics of different mechanochromic forms challenging. In the cases of 48–53, the mechanochromic properties do not depend on their supramolecular structures supported by intermolecular interactions but are controlled by steric effects of the carbene ligands. This can be used as an alternative and additional tool for tuning the mechanochromic properties of organometallic emitters. The multiple stimuli responsive properties of 48–53 can be useful for smart materials in developing multifunctional films with potential applications as security storage materials.102
3.2. Group 10: platinum(II) ADC complexes
Platinum as well as iridium has a high spin–orbit coupling constant. In contrast to d6 IrIII complexes which prefer strongly an octahedral coordination geometry, the PtII ion has an intrinsic propensity to adopt a square-planar coordination geometry, even in the presence of weak field ligands.20,103,104 This structural bias is of key importance when considering radiationless deactivation pathways of their excited states. Population of the strongly antibonding dx2−y2 orbital, e.g. by absorption of light or by thermal activation, weakens the Pt-ligand bonds and induces structural distortion towards a tetrahedral geometry. Such structural deformation decreases the ligand-field splitting, thus facilitating non-radiative deactivation via thermally excited Pt 5d states. The way to circumvent this is to use rigid multidentate ligand scaffolds and/or strong σ-donor atoms that are beneficial for the development of highly robust phosphorescent PtII complexes.PtII complexes featuring Chugaev-type67,71–76,105–107 chelating bis-ACD ligands 54 and 55 were assembled via the template reaction of tBuNC, excess hydrazine hydrate, and aqueous K2[PtCl4]; separation was achieved by fractional recrystallisation (Scheme 7, Route A).108 Through this reaction, two coordinated tBuNCs reacted with hydrazine to yield a deprotonated chelating dicarbene ligand in 54 and 55. In the case of 54, the subsequent PtII-mediated dealkylation of tBuNC gives rise to the cyanide. Reversible protonation of 54 and 55 with aqueous HBF4 generates 57 and 58 (Scheme 7, Route C).
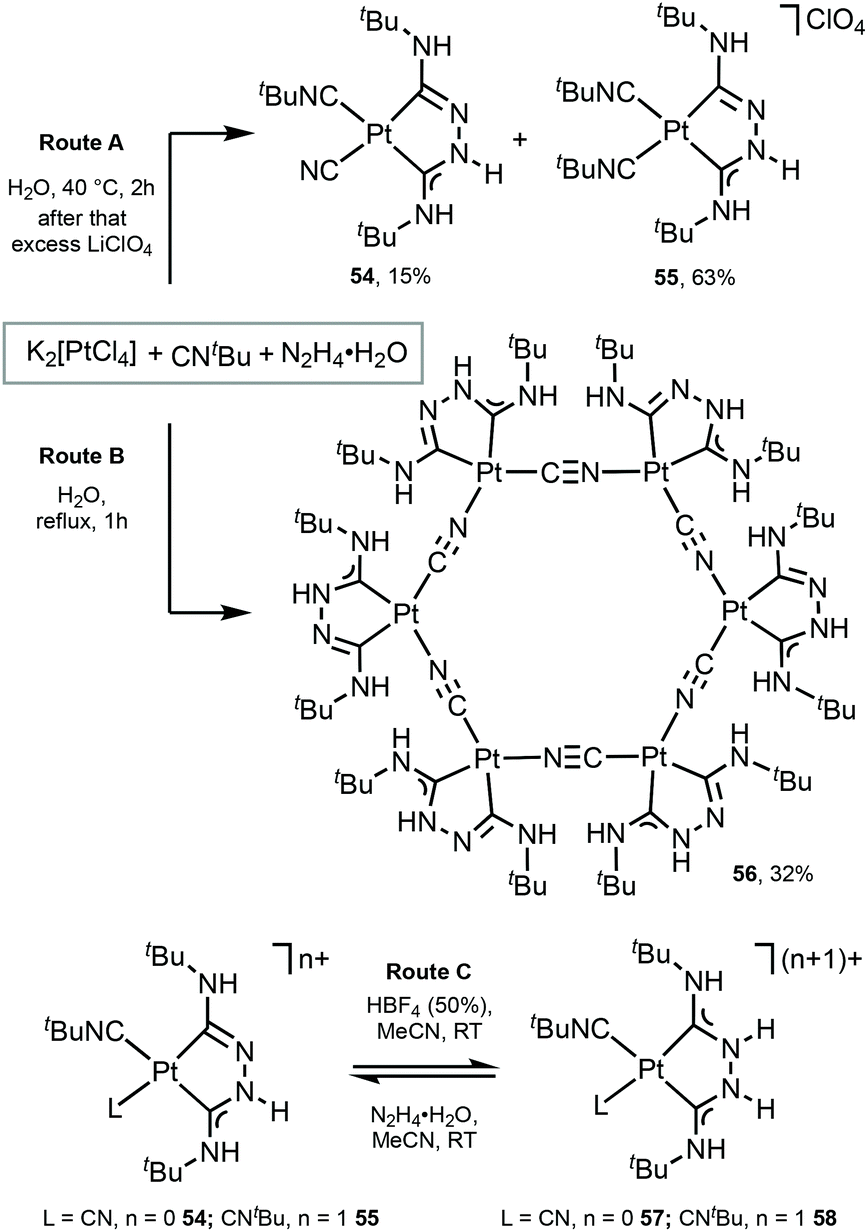 | ||
| Scheme 7 Preparation of complexes 54–58.67,108 | ||
The UV–vis spectra of 54 and 55 display a moderate absorption band at 390 nm and 406 nm, respectively, which was assigned to the MLCT transition Pt → π*(ADC); protonation results in a significant blue shift for the lowest energy absorption in the UV–vis spectra (Fig. 12). The luminescence of 54 and 55 in solution is extremely weak, however, the protonation brings about a dramatic emission enhancement (ca. 12-fold for 54) accompanied by the hypsochromic shift (Fig. 12). High-energy emissions of 57 and 58 formed upon protonation of 54 and 55 are assigned to the triplet intraligand excited state of the bis-ACD moiety.
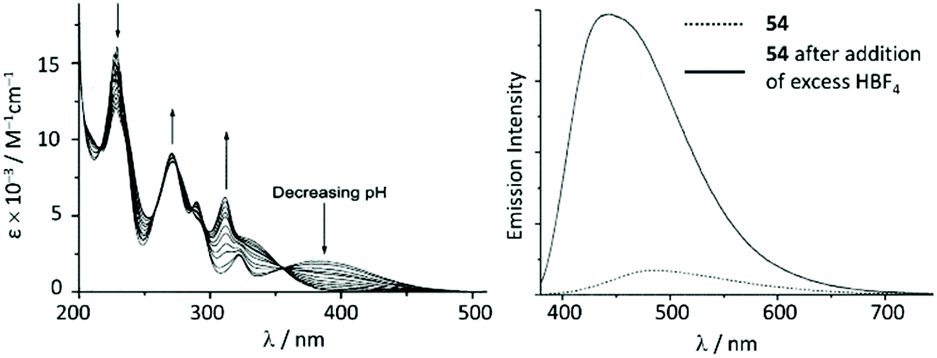 | ||
| Fig. 12 (Left): evolution of UV–vis absorption spectrum of 55 upon addition of HBF4 in 0.1 M aqueous NaCl solution at 298 K; (right): emission spectrum of 54 in MeCN at 298 K (λext 350 nm) before and after addition of excess HBF4. Adapted with permission from ref. 108. Copyright (2001) Elsevier Ltd. | ||
It is worth noting that homoleptic tetrakis(carbene) complexes with monodentate ADC [Pt{C(NHMe)2}4]Cl2109–111 and NHC [Pt{NHC}4](PF6)2 (NHC = imidazolidin-2-ylidene,112 tetrahydropyrimidine-2-ylidene,112 benzimidazolin-2-ylidene113) ligands are not luminescent, while tetra(carbene) complexes with bis (NHC) C,C-chelating ligands114–116 emit in the visible and near-UV regions of the spectra. The main difference between the luminescent and non-luminescent platinum complexes discussed above is the existence of a dπ–pπ conjugated system in the case of complexes with C,C-chelating carbene ligands and the absence of conjugation in the case of complexes with monodentate carbene ligands due to the possibility of rotation around the metal–carbon bond.
A unique Pt6 macrocycle 56 with chelating ADC and bridging CN ligands was prepared by a template reaction between K2PtCl4, tBuNC, and hydrazine hydrate in water under reflux (Scheme 7, Route B).67 Compound 56 forms through the reaction of two coordinated tBuNC ligands with hydrazine to give a chelating dicarbene ligand followed by the PtII-mediated dealkylation of tBuNC to CN species. In comparison with 54 and 55, stronger heating of the reaction mixture leads to the complete dealkylation of the isocyanide.
The UV–vis spectrum of 56 contains an intense absorption band at ca. 350 nm assigned to the Pt-π*(ADC) spin-allowed charge transfer transition (Fig. 13). Upon excitation at 350 nm in solution and in the solid state, complex 56 emits at ca. 500 nm.
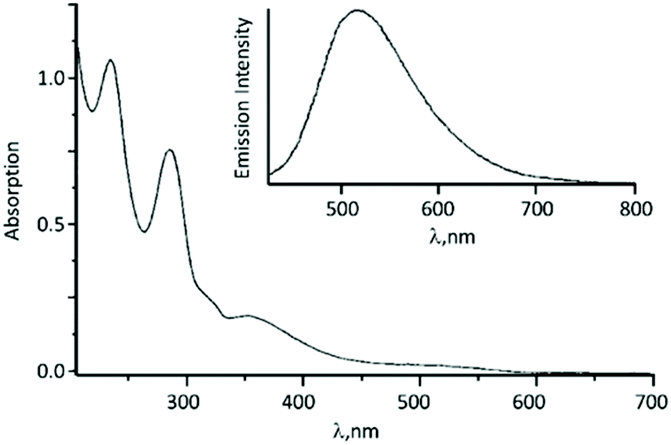 | ||
| Fig. 13 UV–vis spectrum of 56 in MeOH solution; the inset shows the emission spectrum of 56 in CH2Cl2 solution. Adapted with permission from ref. 67. Copyright (1998) John Wiley & Sons, Inc. | ||
Luminescent PtII–ADCs 61–67 were prepared by nucleophilic addition of hydrazine or amines to the coordinated isocyanide in starting from 59 and 60 (Scheme 8).117 Conversion of isocyanide ligands in 59 and 60 to the corresponding ADCs in 61–67 results in a significant bathochromic shift of emission maxima (λmaxca. 530 nm for 59 and 60), and 61–67 display emission in solution with λmax in the range 540–560 nm, that was assigned to the triplet MLCT excited states.
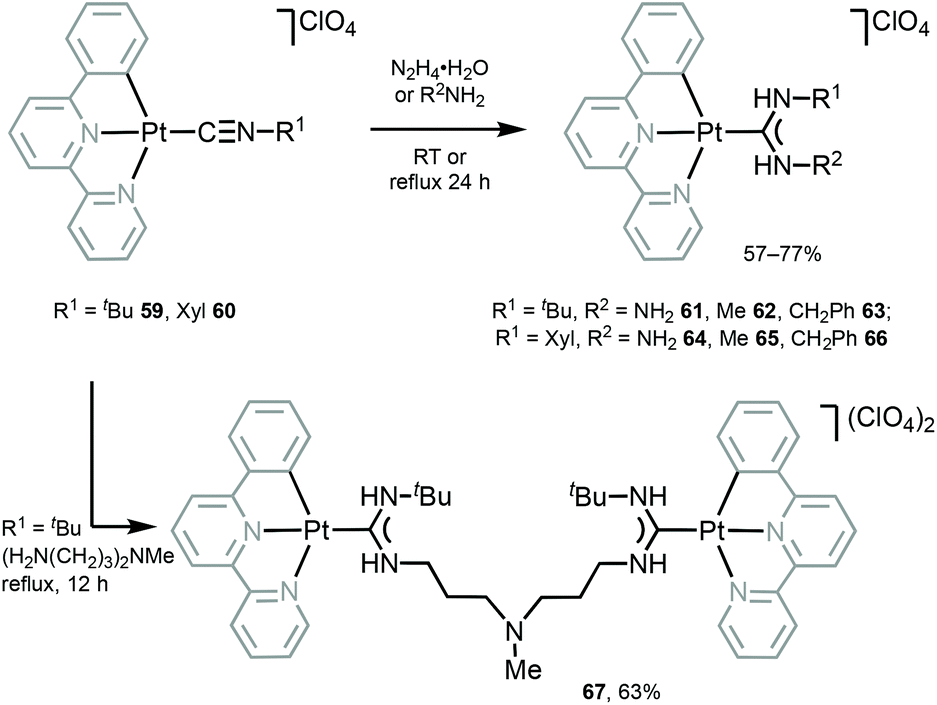 | ||
| Scheme 8 Synthesis of 61–67.117 | ||
Another family of phosphorescent cyclometallated PtII acyclic diaminocarbene complexes 70–73 was prepared by nucleophilic attack by the primary amines on the coordinated isocyanide in 68 and 69 (Scheme 9).118
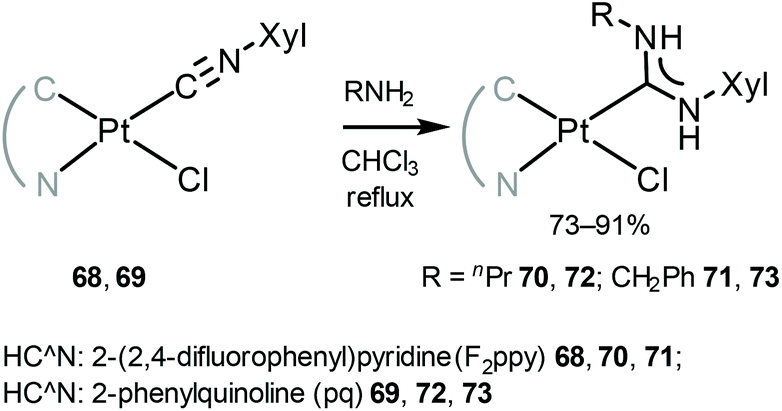 | ||
| Scheme 9 Synthesis of cyclometallated PtII ADC complexes 70–73.118 | ||
ADC derivatives 70–73 exhibit characteristic absorption bands at ca. 400 (70, 71) and 440 nm (72, 73) assigned to the transitions into singlet mixed (IL/MLCT) excited states (Fig. 14). Very weak lowest-energy bands (at ca. 430 or 460 nm) were only discernible in very concentrated solution and were attributed to the direct population of mixed triplet IL/MLCT nature favoured by the high spin–orbit coupling of the metal centre. In the solid state and immobilised in the PS matrix (5 wt% of the PtII complex) 70–73 exhibit green (70 and 71, λmax 470–475 nm) or orange (72 and 73, λmax 550–590 nm) emission with high or moderate Φ values (0.70–0.74 for 70 and 71, and 0.15–0.19 for 72 and 73). Conversion of 68 and 69 to 70–73 is therefore not accompanied by a shift of emission energy, but a notable increase of Φ values (Φ 0.25 for 68 and 0.08 for 69). This observation can be justified by a stronger donicity of the ADC causing higher ligand field splitting and reducing the thermal population of non-radiative triplet metal-centred excited states. The emission energies in THF solutions are essentially identical, although, as expected, with lower lifetimes and lower quantum yields (Φ < 0.1) than in PS films and in the solid state. Compounds 70–73 bearing ADCs are shown to be valuable organometallic emitters for OLED devices alongside commonly used [Pt(C^N)Cl(NHC)] counterparts with NHC ligands.119
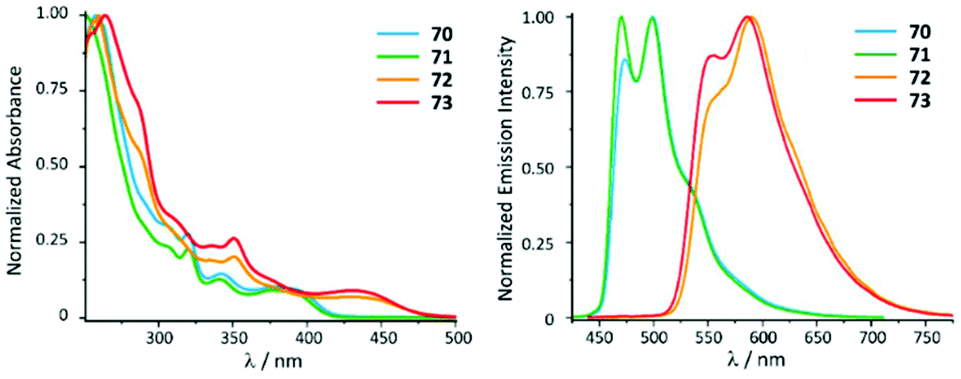 | ||
| Fig. 14 (Left): normalised UV-vis absorption spectra of 70–73 in THF; (right): normalised emission spectra of 70–73 recorded in a PS film (5 wt%) at 298 K (λext 385 for 70 and 71 and 420 nm for 72 and 73). Adapted with permission from ref. 118. Copyright (2021) The Royal Society of Chemistry. | ||
Bis-alkynyl species 76 and 77 with ADC ligands were prepared by nucleophilic addition of dimethylamine to the coordinated isocyanide in 74 and 75 (Scheme 10).120 Despite a large excess of diethylamine and a long time (3 days), addition to a solely coordinated isocyanide occurs. The authors associate the lack of reactivity of the second isocyanide to steric restrictions, which prevent the formation of bis-ADCs.
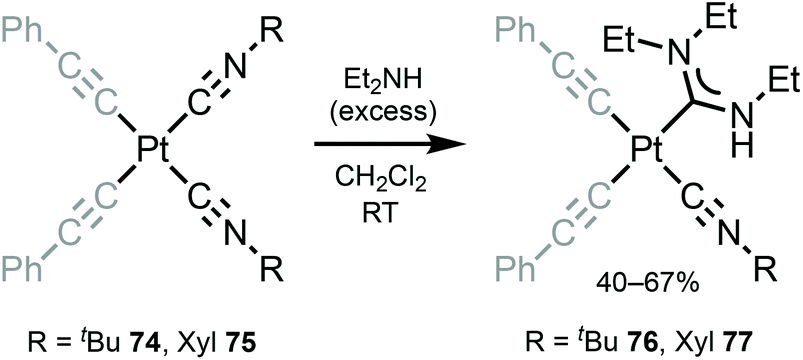 | ||
| Scheme 10 Synthesis of complexes 76 and 77.120 | ||
UV–Vis spectra of 76 and 77 contain two high-energy absorption resonances with maxima in the 252–313 region, assigned to transitions involving the intraligand π–π* excitation of the alkynyl species and charge transfer in the Pt−C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh. The degree of conjugation within the complex is reduced upon generation of ADC in 76 and 77, leading to a minor decrease in the extinction coefficient for the most intense UV band and a significant blue-shift of the lower-energy band (Fig. 15). In solution, 74–77 are weakly luminescent; a significant enhancement is observed for 74–77 as frozen solvent glass (77 K) or in PMMA films. Both isocyanide and ADC species showed identical profiles of phosphorescence with a λmax of 430 nm, indicating that both aforementioned ligands in 74–77 have a cooperative effect on the energy of the emissive triplet states.
CPh. The degree of conjugation within the complex is reduced upon generation of ADC in 76 and 77, leading to a minor decrease in the extinction coefficient for the most intense UV band and a significant blue-shift of the lower-energy band (Fig. 15). In solution, 74–77 are weakly luminescent; a significant enhancement is observed for 74–77 as frozen solvent glass (77 K) or in PMMA films. Both isocyanide and ADC species showed identical profiles of phosphorescence with a λmax of 430 nm, indicating that both aforementioned ligands in 74–77 have a cooperative effect on the energy of the emissive triplet states.
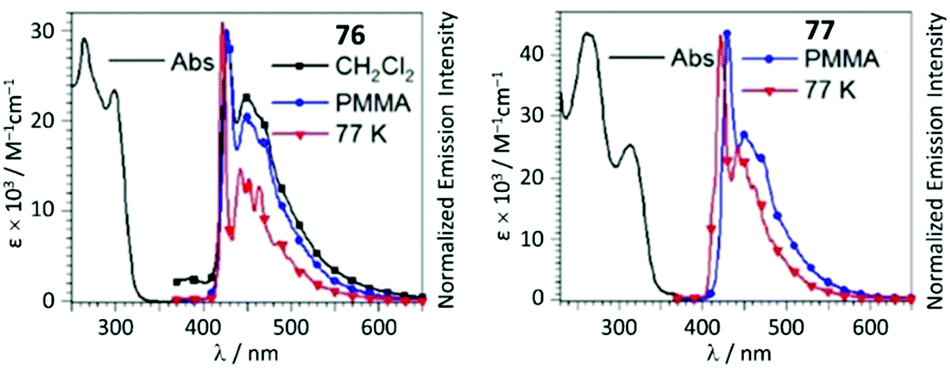 | ||
| Fig. 15 UV–Vis spectra of 76 and 77 in CH2Cl2 solution; emission spectra in different media at variable temperature, λext 310 nm. Adapted with permission from ref. 120. Copyright (2020) John Wiley & Sons, Inc. | ||
Conversion of metal-bound isocyanide to ADC results in a remarkable increase in Φ from 0.014 (for 74, PMMA) and 0.058 (for 75, PMMA) up to 0.23 (for 76, PMMA) and 0.15 (for 77, PMMA). The phosphorescence lifetime is longer for both ADC derivatives 76 and 77 than for the starting bis-isocyanides 74 and 75; the enhanced quantum efficiencies of the ADC complexes are attributed to the large destabilisation of the empty Pt-centred orbital by the strong σ-donation of ADC.
The photoluminescence properties of ADC derivatives 76 and 77 in PMMA are comparable to those of platinum(II) acetylide complexes with NHC ligands, viz. trans-[(NHC)2Pt(CCAr)2] (NHC = 1,3-substituted-1H-imidazol-2(3H)-yl or 1,3-substituted-1H-benzimidazol-2(3H)-yl), that have shown promise in the manufacturing of OLED devices.37,121
3.3. Group 7: rhenium(I) ADC complexes
Although the ADC complexes of both Mn and Re have been previously reported,122–128 a sole report of luminescent ReI ADC complexes is disclosed.122 A series of luminescent ReI complexes 81–88 with C,N-ADC was synthesised via the nucleophilic addition of amino-azaheterocycles, namely, 2-aminopyridine, 2-aminothiazole, and 2-aminobenzothiazole, to ReI isocyanides. The reaction is facilitated by the halide abstraction with silver or thallium salts, and/or thermal or photochemical CO ligand substitution (Scheme 11). The UV–vis spectra of 81–88 show intense ligand centred π–π* transitions of the isocyanide and C,N-ADC ligands in the UV region (λ < 330 nm) and the lower-energy bands (350 nm > λ > 450 nm) assigned to the MLCT transitions dπ(Re) → π*(C,N-ADC) and dπ(Re) → π*(CNR) (Fig. 16). Upon excitation at a λext of 350 nm, 81–88 exhibit strong emission with the energy spanning from blue to red regions. Using DFT calculations, emissions of 81, 82, 85, and 87 were attributed to the mixed triplet MLCT and LC excited-state origin, while for 83, 84, 86, and 88 to the triplet MLCT [dπ(Re) → π*(C,N-ADC)]. Within the 81–88 family the emission energy is redshifted in-line with a decrease of π-conjugation of the C,N-ADC ligand (λmax 467 nm (81), 489 nm (87), 499 nm (85), 570 nm (88), and 590 nm (86)). The authors justify this trend by the contribution of the empty pπ(CADC), which becomes lower-lying in the presence of a less π-conjugated N-heterocycle due to the interaction with a lower-lying lone pair electrons in the LUMO [π*(C,N-ADC)].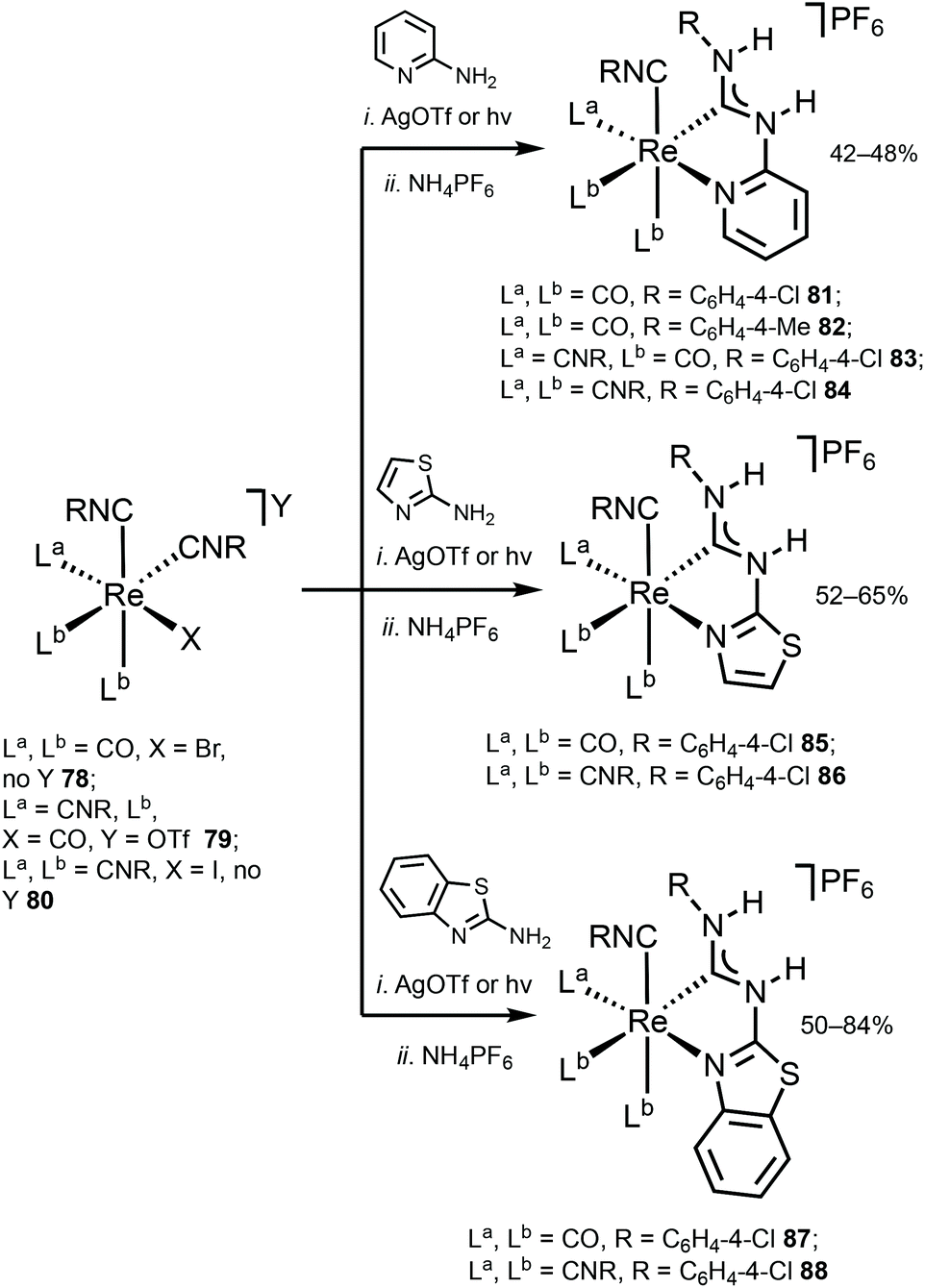 | ||
| Scheme 11 Synthesis of ADC complexes 81–88.122 | ||
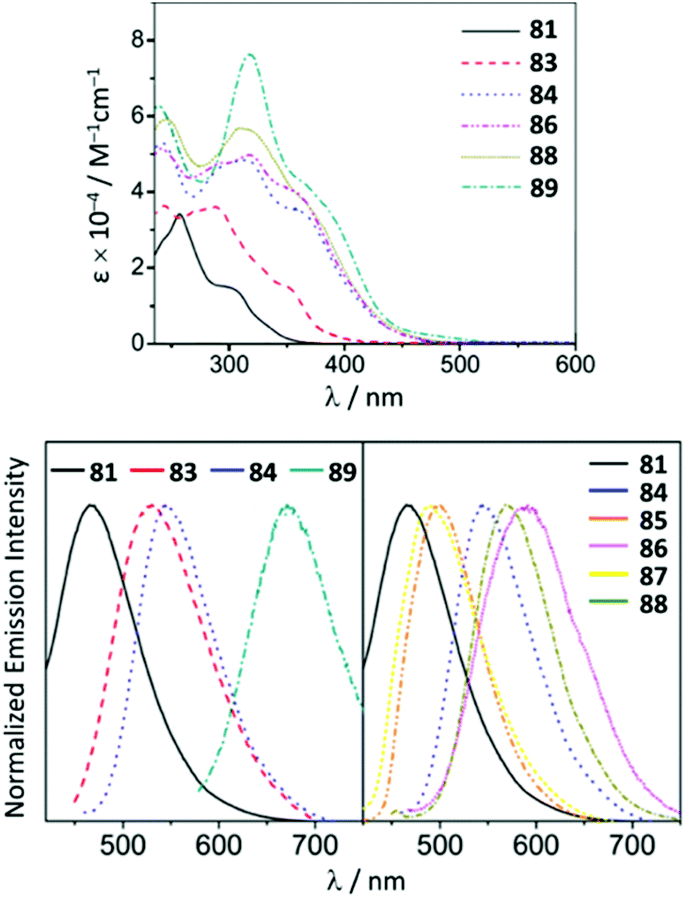 | ||
| Fig. 16 (Top): UV−vis spectra of ReI ADC complexes 81–88 in CH2Cl2 at 298 K; (bottom): emission spectra of 81–88 in CH2Cl2 at 298 K. Adapted with permission from ref. 122 Copyright (2016) American Chemical Society. | ||
Alkylation of 84 with Me2SO4 gives 89 (Scheme 12),122 showing a noticeable redshift of the emission on going from C,N-ADC 84 (λmax 547 nm) to the methylated derivative 89 (λmax 672 nm) is linked to the increased steric repulsion due to installation of methyl groups twisting the ADC moiety. It is noteworthy that while isocyano-(diimine) complexes [Re(CNR)4(N^N)](PF6) display typically orange to red phosphorescence,129 a careful design of the C,N-ADC ligands enables the MLCT phosphorescence covering the entire visible region satisfying the requirements for different applications.
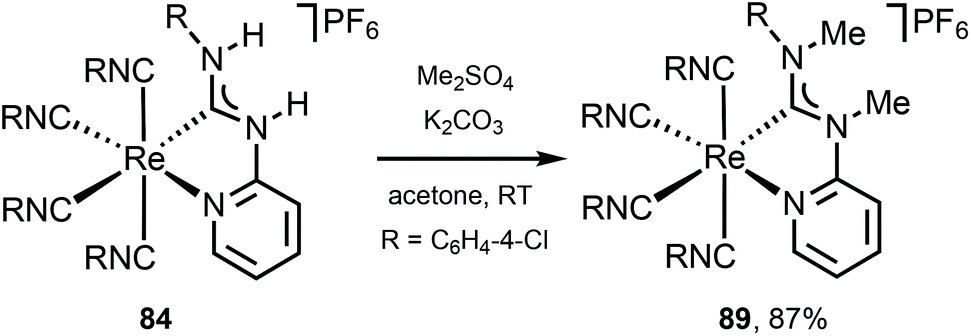 | ||
| Scheme 12 Methylation of the chelating C,N-ADC ligand in 78.122 | ||
3.4. Group 11: gold(I/III) ADC complexes
Gold(I) complexes usually require the presence of chromophores or a group promoting the formation of dimeric or polymeric species through aurophilic interactions to impart the desired luminescence, with luminescence lifetimes depending on the origin of the emissions.130–132ADC derivatives 90 and 91 were prepared by reacting H[AuCl4] or H[AuBr4] with methyl isocyanide and methylamine in an aqueous solution (Scheme 13).133,134 In solution and in the solid state, protons of two ADC species interact with X−via N–H⋯X⋯H–N (X = Cl, Br) three-centred HBs, which prevent the X− coordination to AuI (Fig. 17). Addition of ammonia salts (NBu4BF4, NH4PF6, NH4AsF6, or NH4SbF6) to a solution of 90 results in counter-ion metathesis and precipitation of colourless 92–95 (anion = BF492, PF693, AsF694, SbF695) and (93, 94) × ½(solvent) (solvent = benzene, acetone, chlorobenzene).133,135,136
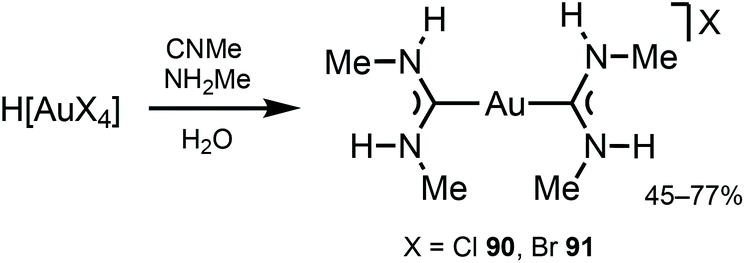 | ||
| Scheme 13 Synthesis of complexes 90 and 91.133,134 | ||
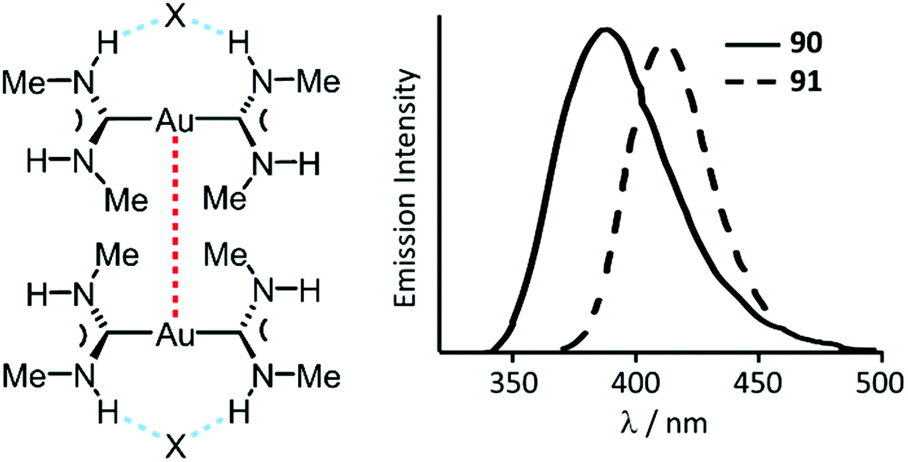 | ||
| Fig. 17 (Left): Non-covalent interactions in 90 and 91; dotted lines are the Au⋯Au aurophilic (red) and N–H⋯X HBs (blue) interactions. Au⋯Au separations are 3.1231(3) (90) and 3.1297(4) (91) Å; (right): Emission spectra of 90·H2O and 91·H2O at RT. Adapted with permission from ref. 134. Copyright (2002) American Chemical Society. | ||
Complexes 90–95 are not emissive in solution at RT.135 In frozen solutions and in the solid state, 92–95 show bright phosphorescence in the violet-green region (390–535 nm) associated with the formation of dimers (90, 91) or extended chains (92–95) supported by Au⋯Au attractions (Fig. 17 and 18).134–136 Although colourless crystals of 90 and 91 are isomorphic, they exhibit different emission maxima, λmax 391 nm (90) vs. λmax 412 nm (91), resulting from the larger distortion in the excited state for the bromide derivative due to the larger anion size. The crystal packing in both 93 and 94 is similar, and the anion interconnects with N–H groups of three ADC three organometallic cations via HBs facilitating the further self-association by aurophilic interactions (Fig. 18). The absence of luminescence in solutions of 90–95 is due to a breakdown of the supramolecular structure. The fact that the emission spectra of the frozen solutions strongly resemble the spectra of crystalline 90–95 suggest similarities in the respective supramolecular structures in both states.
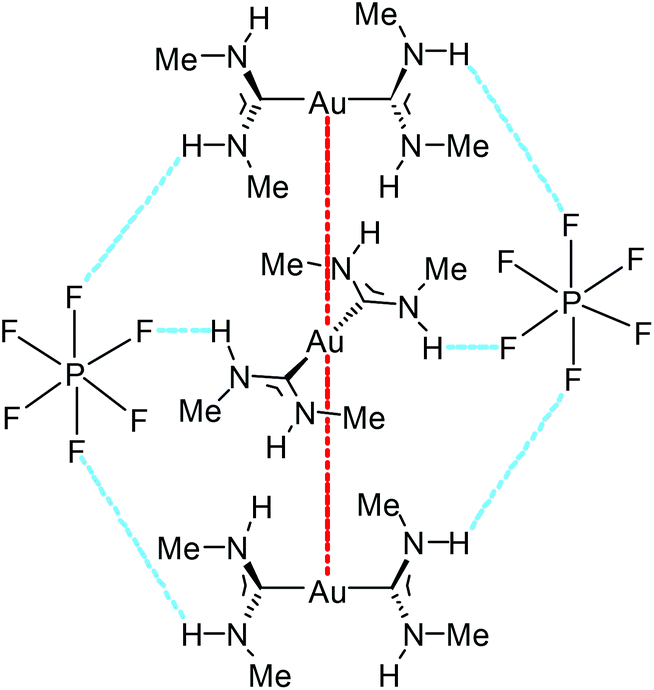 | ||
| Fig. 18 A fragment of an endless supramolecular chain in the solid structure of 93. Aurophilic interactions are indicated in red, and the N–H⋯F HBs are indicated in blue. Au⋯Au separation is 3.1882(1) Å.136 | ||
The supramolecular structure of 93 and 94 is responsive to an external stimulus; 94 demonstrates a solvatochromic luminescent effect (Fig. 19). The difference in photoluminescence energy is an outcome of bending of the columns of cations and disorder in the cation positioning within the column. Thus, the synergistic action of HBs and aurophilic attractions allows overcoming the coulombic factors which should serve to separate the cations from each other, and the supramolecular interactions (aurophilic attractions) make compounds 92–95 emissive. These results show the significant impact of the even weakly coordinating anion or solvated molecule on the aurophilic interactions. Most likely, the differences in the N–H⋯X HBs with the anions are responsible for the variations in the solid-state structures and ultimately, the photoluminescence properties.
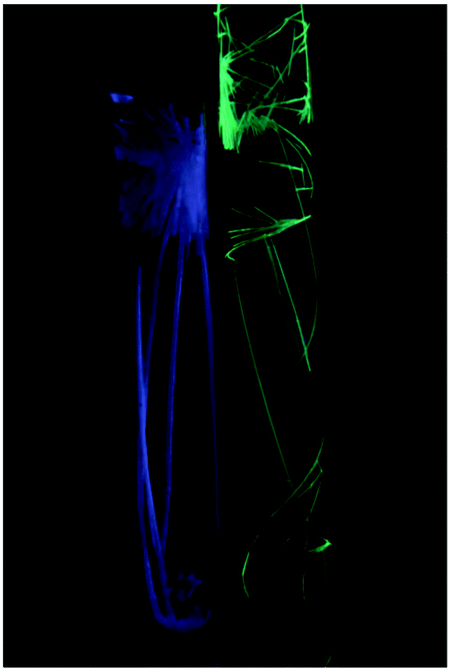 | ||
| Fig. 19 Photograph of 94 × ½(solvent) taken under irradiation with UV light in chlorobenzene λmax 490 nm (left) and in acetone λmax 468 nm (right). Reprinted with permission from ref. 136. Copyright (2008) American Chemical Society. | ||
The reaction between [Au(CNMe)2]Cl formed in situ and 2-aminoethanol or 1,2-diaminoethane led to 96137 and 97138 (Scheme 14). The shorter Au⋯Au separation in 97 relative to 90 is facilitated by the fact that in 97 the three-centred N–H⋯Cl⋯H–N HBs connect with the NH groups of two neighbouring cations (Fig. 20). In contrast, for 90 the three-centred N–H⋯Cl⋯H–N HBs bind the NH groups of the two ADC ligands from the same cation (Fig. 17). Colourless 96 and 97 show no luminescence in solution, while solid 97 is strongly emissive at RT and at 77 K with λmax 377 nm upon excitation with λext 330 nm. The luminescence observed is attributed to the aurophilic interactions present in the solid (Fig. 20).
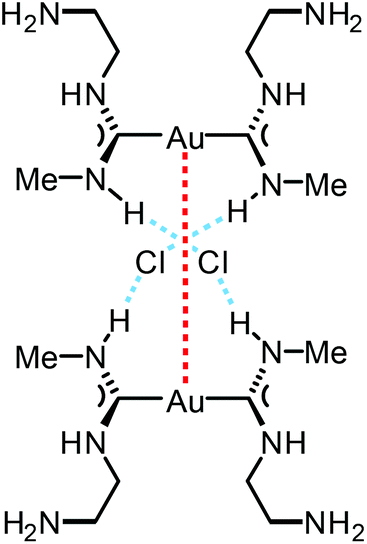 | ||
| Fig. 20 Schematic representation of the non-covalent interactions in the dimer of 97. Dotted lines indicate the Au⋯Au aurophilic interactions (red) and N–H⋯Cl HBs (blue). Au⋯Au separation is 3.0224(4) Å.138 | ||
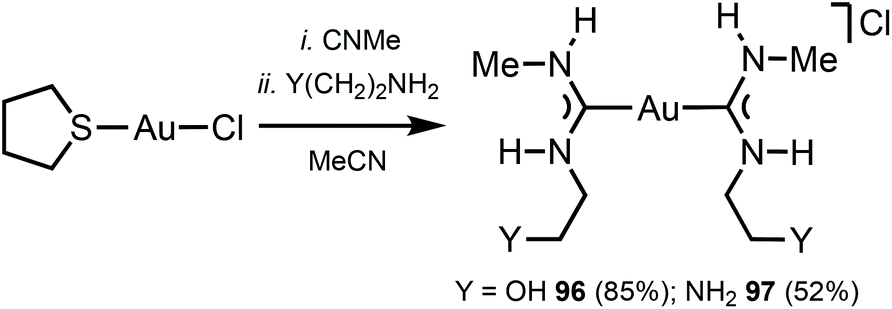 | ||
| Scheme 14 Synthesis of complexes 96 and 97.137,138 | ||
Changes in the backbone of the ADC species may induce dramatic changes in the emission associated with the change in the electron-richness of carbene species. Protic ADC ligands, i.e. those featuring one or two NH wingtips, possess Brønsted acidity, being able to act as donors of hydrogen bonding.62 For AuI–ADC complexes, aurophilic interactions arise alongside the hydrogen bonding and their synergetic interactions make these complexes capable of luminescence. As an example, complex [Au{C(NMe2)(NHMe)}2](PF6)135 bearing a sole NH proton on the ADC moiety is non-emissive in both the solution and in the solid state. Due to the absence of an effective matrix of HBs, the manifestation of Au⋯Au interactions associated with the luminescence of this class of compounds turns out to be impossible. In addition, the bis-protic gold(I)–NHC complex [Au(C3H4N2)2]Cl139 does not manifest hydrogen bonding and is therefore non-luminescent.
Switching and tuning the solid-state luminescence properties of molecular materials by modulating molecular packing through non-covalent routes is an attractive prospect.140–145 Herein, ADC ligands can uniquely play both roles enabling non-covalent binding as well as luminescence emission. Complexes 101–106 were prepared by the nucleophilic attack of primary or secondary amines or diamine on the corresponding AuI-bound 2-pyridyl isocyanides in 98–100 (Scheme 15).146 Single-crystal XRD studies of 101 and 103 indicated the formation of supramolecular 1D polymers or dimers supported by Au⋯Au aurophilic interactions and N−H⋯Cl hydrogen bonds, while 104 and 105 do not exhibit aurophilic attractions in the solid state.
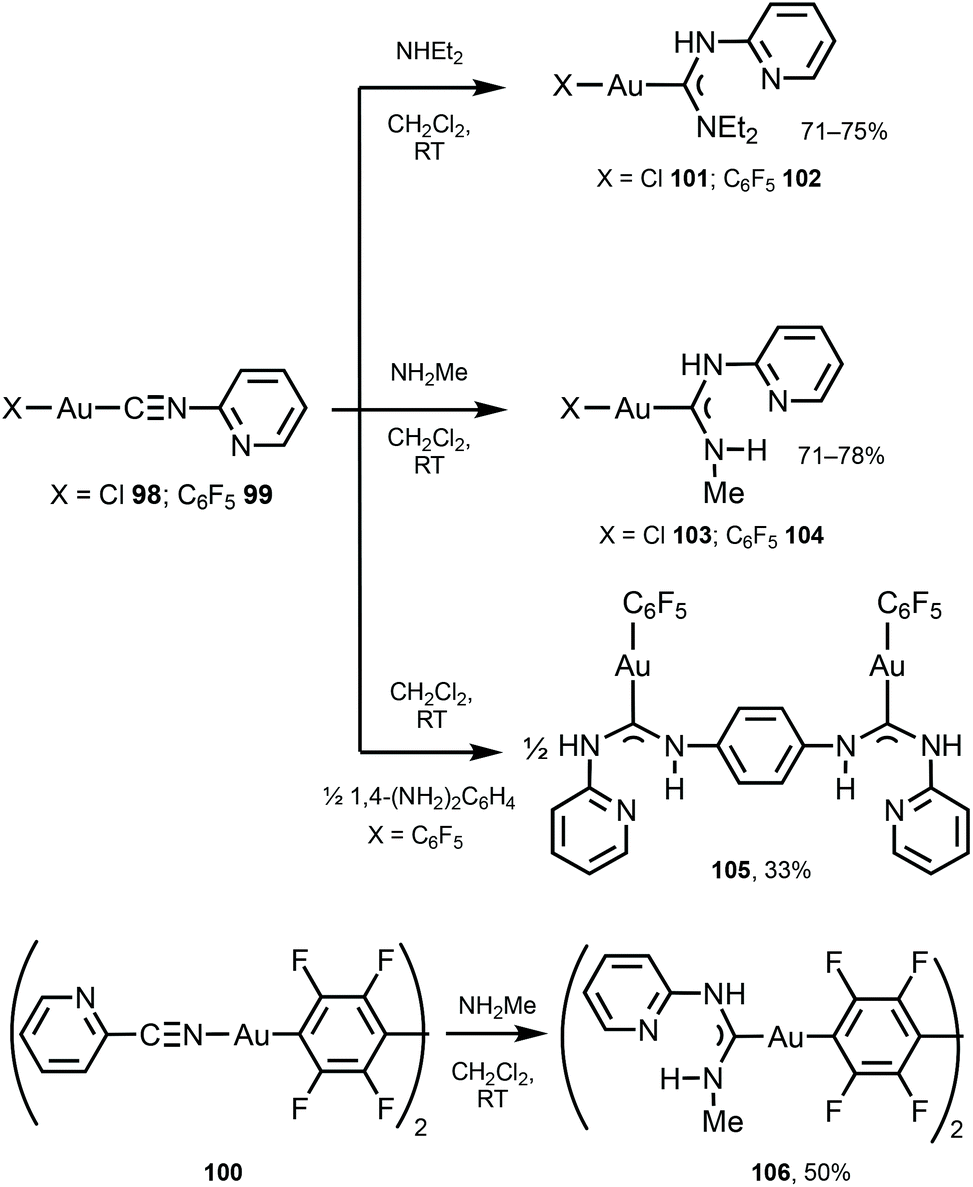 | ||
| Scheme 15 Synthesis of 101–106.146 | ||
A similar strategy enabled the preparation of 109–112 starting from 107 and 108 bearing 4-pyridyl isocyanide (Scheme 16).147109–112 lack aurophilic contacts in the solid state. The absorption spectra of 101–106 and 109–112 are very similar and contain one or two bands (at ca. 250 nm for 109–112; 250 and 280 nm for 101–106, respectively), assigned to the ligand-centred π–π* transitions associated with aromatic groups in the ligand environment. Parent AuI–CNRs 98–100, 107, and 108 are not luminescent either in the solid state or in solution, but all ADC derivatives 101–106 and 109–112 show intense yellow-green luminescence both in solution and in the solid state at RT. The emission profiles and energies of 104, 102–106, and 109–112 in solution and in the solid state are similar and are consistent with an intramolecular origin of the luminescence owing to the absence of Au⋯Au short contacts. For 101 and 103, the emission energies in the solid state (λmax 562 nm, 101 and λmax 470 nm, 103) are redshifted in comparison to those recorded in CH2Cl2 solution (λmax 457 nm, 101 and λmax 409 nm, 103), attributed to the intermolecular aurophilic interactions. A difference in the photophysical behaviour of isocyanide and ADC derivatives can be related to a higher donor capacity of ADC ligands when compared to the parent isocyanide.148,149 As a result, ADC introduces a more significant perturbation in gold(I) orbitals by enhancing the π back-bonding contribution from gold to ADC, linked to higher intersystem crossing and phosphorescence.
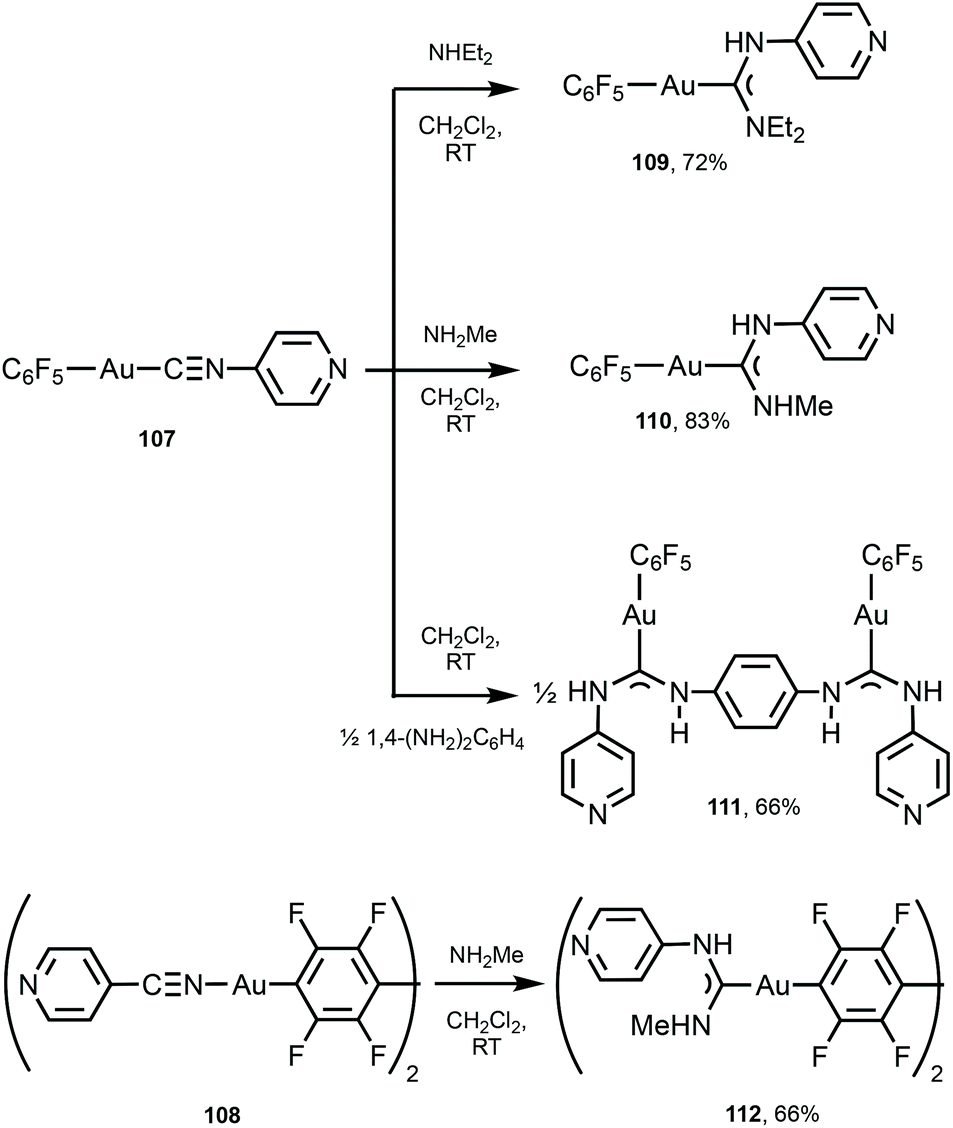 | ||
| Scheme 16 Synthesis of 109–112.147 | ||
Trinuclear gold(I) aminocarbene and pyrazolate complexes that possess intramolecular aurophilic contacts are capable of displaying interesting luminescence associated with different extents of intermolecular stacking based on aurophilic interactions;130 they can be prepared from mononuclear gold-ADCs. Thus, the deprotonation of 103 with KOH in MeOH generates formamidinyl intermediates spontaneously trimerising to cluster 113 (Scheme 17).29 Both in solution and in the solid state, 113 possesses a trigonal prismatic arrangement of six metal atoms stabilised by the Au⋯Au interactions between the two {Au3} triangles. 113 displays an intense luminescence both in the solid state with a λmax of 523 nm and in solution with a λmax of 563 nm. The long lifetime observed (84 μs) supports the phosphorescent nature of the emission that can be attributed to the presence of Au⋯Au aurophilic interactions, as reported for other trinuclear gold complexes.27,150–155
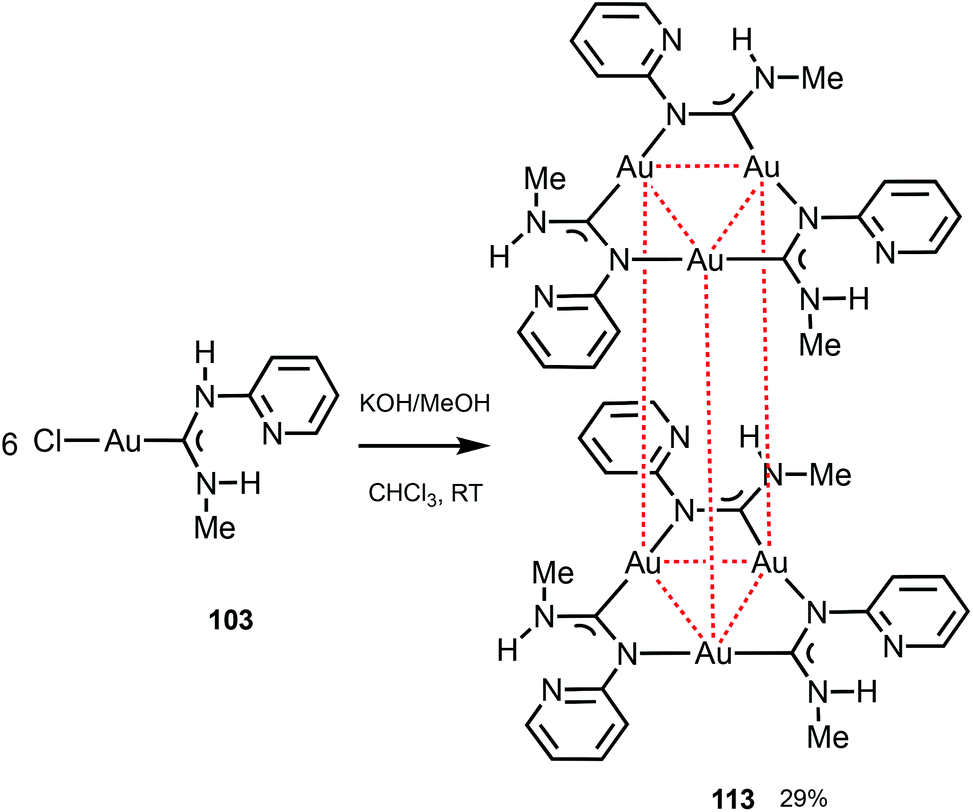 | ||
| Scheme 17 Generation of 113 and pairwise association through Au⋯Au aurophilic interactions (dotted red lines). Au⋯Au separations are 3.2532(9) Å (intramolecular contacts) and 3.2644(12) Å (intermolecular contacts).29 | ||
116 and 117 with the perylene substituents at the periphery were obtained by the nucleophilic attack of NHEt2 on the AuI-bound perylene isocyanide in 114 and 115 (Scheme 18).156 The UV-Vis absorption profiles of 114–117 are similar and dominated by the π–π* transition inside of the perylene moiety. The absorption maxima of 116 and 117 at 450 nm are blueshifted (Δ = 990 cm−1) from the parent isocyanides 114 and 115 and redshifted (Δ = 550 cm−1) from free perylene. ADCs 116 and 117, as well as the starting isocyanides 114 and 115, and free perylene isocyanide exhibit strong fluorescence in solution with a well-defined vibrational structure of the emission band (Fig. 21).
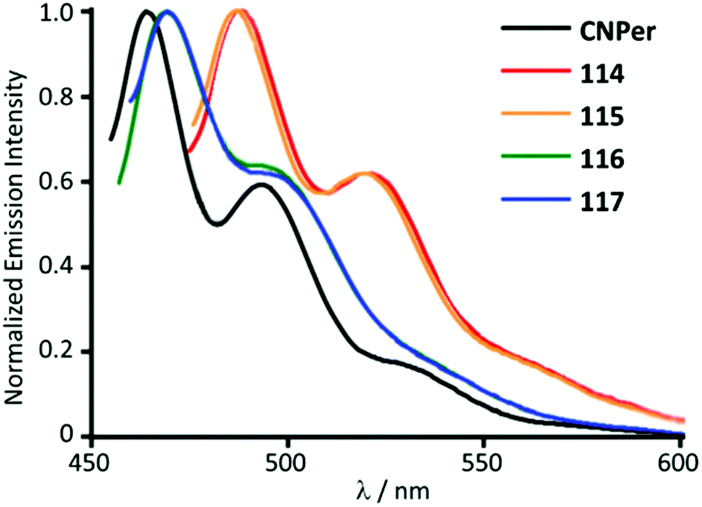 | ||
| Fig. 21 Emission spectra of 114–117 in comparison to the free perylene-isocyanide, CHCl3 solution, RT. Adapted with permission from ref. 156. Copyright (2014) The Royal Society of Chemistry. | ||
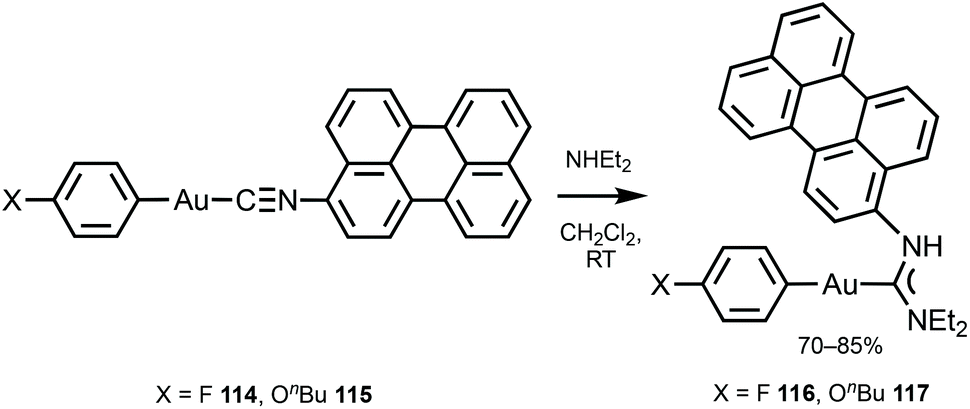 | ||
| Scheme 18 Synthesis of 116 and 117.156 | ||
The emission energies of ADCs 116 and 117 (Φ 0.69–0.93) are blueshifted (Δ = 830 cm−1) relative to the parent isocyanide species 114 and 115 (Φ 0.71–0.88) and redshifted (Δ = 1150 cm−1) relative to free perylene in-line with absorption. The resemblance of the luminescence spectra in both the vibrational structure and the spectral position suggests an intraligand π–π* transition where the ligand orbitals have been marginally modified by heavy metals.
The ADC derivatives 122–125 containing perylene diimide moieties were obtained via the nucleophilic attack of dimethylamine on the AuI-coordinated perylene diimide-isocyanide in 118–121.157 The reaction of 121 with ethylenediamine leads to the hexanuclear derivative 126 (Scheme 19). The presence of a bulky perylene diimide moiety defines the emission properties of 118–126. ADCs 122–126 demonstrate smaller quantum yields (Φ 0.10–0.21) in comparison to those of parent 118–120 (Φ 0.50–0.67), related to a photoinduced electron transfer effect involving the ADC moiety connected to perylene diimide.158 This result is consistent with a strong influence of the {AuI–ADC} fragment on the electronic density on the perylene diimide moiety, which significantly affects the photophysical properties of the complexes with charge transfer from metal to ligand (Fig. 22).
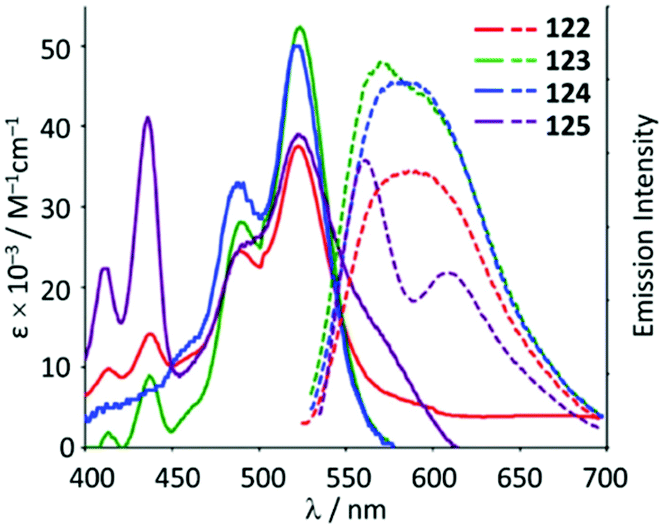 | ||
| Fig. 22 UV-Vis absorption (solid line) and emission spectra (dashed line) of 122–125 in the THF solution at RT. Adapted with permission from ref. 157 Copyright (2017) Elsevier Ltd. | ||
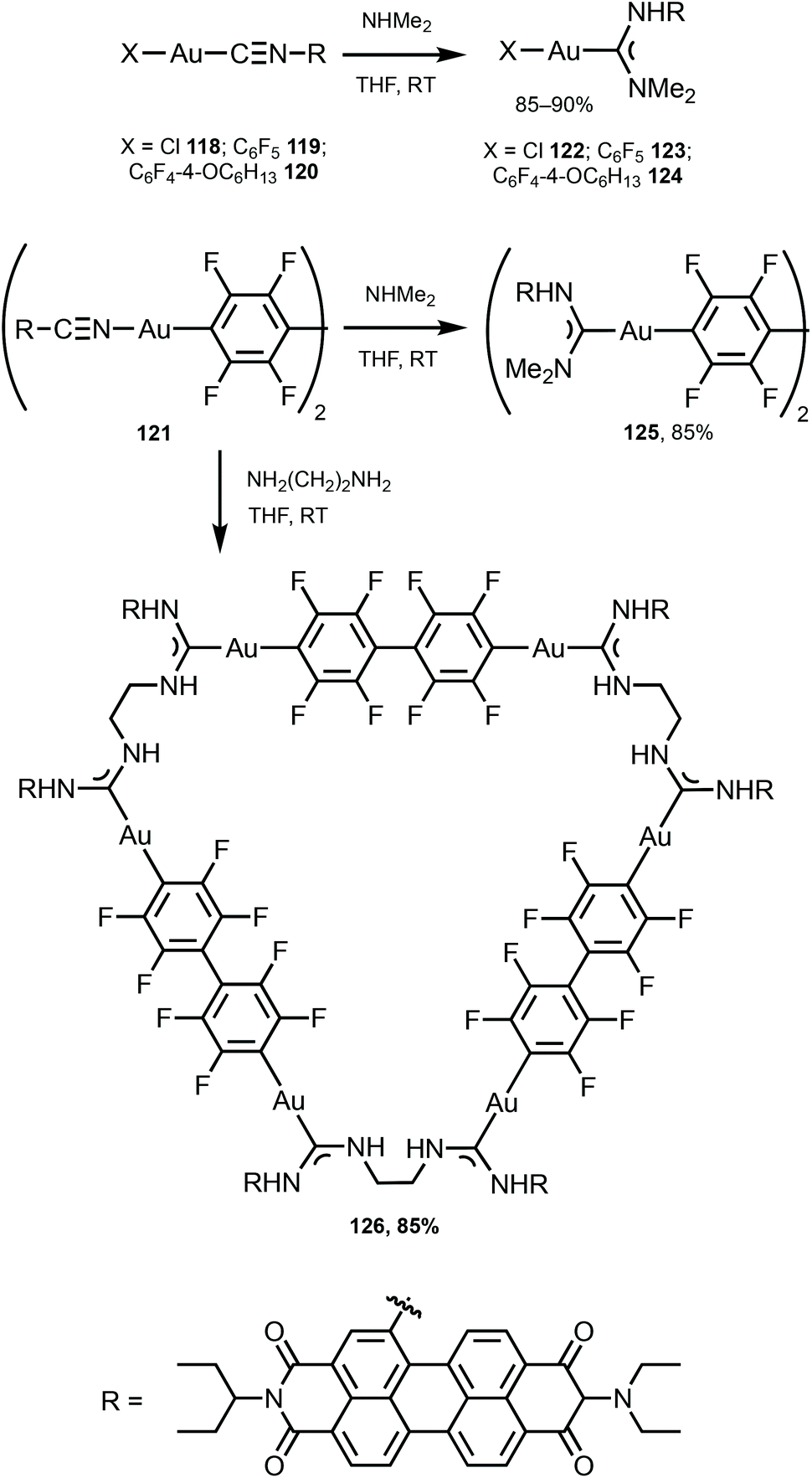 | ||
| Scheme 19 Preparation of complexes 122–126.157 | ||
In sharp contrast to AuI complexes, very few luminescent ADC complexes of AuIII have been reported. This fact also contrasts with the luminescent examples found for the isoelectronic PtII complexes.2,20,159 Low energy d–d ligand field states, which would be reached by electrons instead of the emissive excited state, and the electrophilicity of AuIII have been claimed as the possible origin of this fact. The introduction of a strong σ-donating ADC ligand increases the electron density at the metal centre and increases the energy of the d–d state.160
Gold(III)–ADC derivatives 128–130 featuring a bidentate cyclometallated C,C′-ADC ligand are obtained by reaction of [Au(C,N-pap)Cl2] (pap = 2-(2-pyridylamino)phenyl) 127 with isocyanides (Scheme 20).161
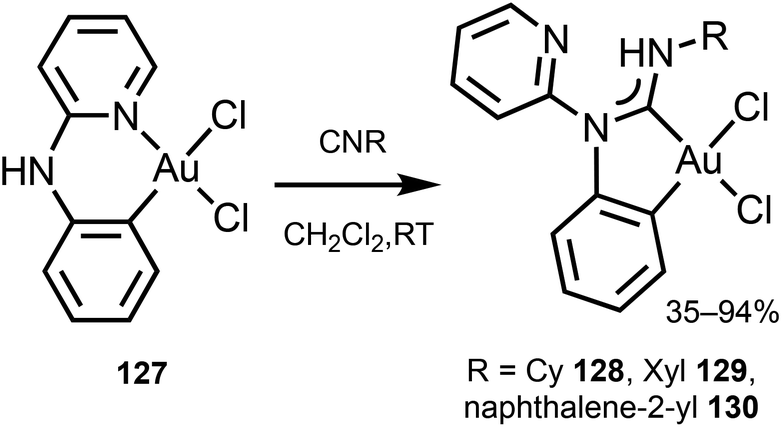 | ||
| Scheme 20 Preparation of gold(III)–ADC complexes 128–130.161 | ||
Though the starting 127 is not emissive, its ADC derivatives 128–130 are weakly luminescent (λmax 525–508 nm) in the solid state at RT. The authors suggest that the generation of ADC increases the d–d ligand field states, avoiding the non-radiative deactivation of the 3IL excited state in 128–130 as a reason for the observed difference in the properties of the starting gold-CNR and gold-ADC product.161 Lowering the temperature to 77 K induces a bathochromic shift of the emission energy of 128–130 (λmax 540–500 nm) with the lifetime in the μs range. The authors described this emission as phosphorescence, although it is better defined as thermally activated delayed fluorescence (Fig. 23).162–164
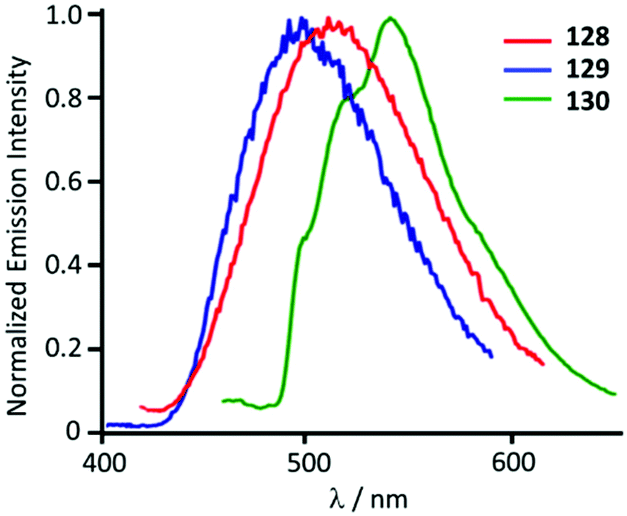 | ||
| Fig. 23 Normalised emission spectra of 128–130 in the solid state at 77 K. Adapted with permission from ref. 161. Copyright (2012) American Chemical Society. | ||
4. Conclusions and outlook
In this review, we summarised experimental studies to date disclosing the photoluminescence properties of metal complexes with acyclic diaminocarbene ligands. We have illustrated that the intrinsic σ-donating properties of acyclic diaminocarbene ligands may be connected to an efficient destabilisation of the unoccupied d orbitals and their corresponding triplet MC states, improving the efficiency and stability of phosphorescence.From this perspective, cyclometallated IrIII complexes with ancillary ADC ligands are fully coherent and support the idea that the introduction of ADC ligands leads to molecules capable of efficient luminescence. The main trend in the design of luminescent IrIII complexes is focused on the combination of properties of cyclometallating ligands with those of acyclic diaminocarbenes. The presence of cyclometallating species in the coordination sphere of the Ir-center ensures the strongest bonding interaction with transition metals, leading to a lower extent of non-emissive quenching and at the same time opening the possibility for the tuning of emission wavelengths. To overcome the problem with the thermal population of non-emissive higher-lying triplet metal-centred ligand-field states, restricting efficient blue luminescence, one of the solutions that emerged relies on the use of a secondary set of σ-donor ligands, such as acyclic diaminocarbenes. Adding ADCs to the ligands set in the coordination sphere of metal is shown to additionally destabilise the unoccupied d-orbitals and their corresponding metal-centred ligand-field states, improving the efficiency and stability of phosphorescence. At the same time, variable steric and donor properties of acyclic diaminocarbenes associated with their synthetic accessibility provided an additional tool for fine-tuning of emission profiles.
On the other hand, luminescence of gold(I) complexes is usually linked to the presence of chromophores or a group promoting the formation of dimeric or polymeric species through non-covalent interactions with luminescence lifetimes and efficiencies depending on the origin of the emissions. Herein, strong intermolecular interactions and substituents with suitable steric hindrance, such as ADCs, may be required to produce an ideal distance between two conjugated molecules, resulting in efficient luminescent excimers. In this case, ADC ligands featuring NH groups were shown to act as donors of hydrogen bonding for a synergetic combination of weak interactions, i.e., hydrogen bonding and metallophilic interactions in the solid-state providing an efficient tool for the tuning of photophysical properties. Examples, based on other metals, viz. PtII and ReI, are not yet sufficient to demonstrate general patterns, and further studies are expected to correct the status quo.
Finally, one of the advantages of the acyclic diaminocarbene ligands already evaluated for tuning the photophysical properties of organometallic emitters is the excellent synthetic availability and respective chemical stability of their metal complexes. Taking these into consideration, an expansion of the known application fields and emergence of new application fields can be expected, including but not limited to the development of chemosensing applications, and use in data protection, visible light photocatalysis, and clinical photodynamic therapy. Those may be enabled by the use of previously unexplored metal centres, and stabilisation of complexes featuring known metals in less common oxidation states, i.e. gold(III), platinum(IV), and others.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Russian Science Foundation (project 21-73-10083). KVL additionally thanks the University of Liverpool for support.References
- S. Lee and W.-S. Han, Cyclometalated Ir(III) complexes towards blue-emissive dopant for organic light-emitting diodes: fundamentals of photophysics and designing strategies, Inorg. Chem. Front., 2020, 7, 2396–2422, 10.1039/D0QI00001A.
- Y. Chi and P.-T. Chou, Transition-metal phosphors with cyclometalating ligands: fundamentals and applications, Chem. Soc. Rev., 2010, 39, 638–655, 10.1039/B916237B.
- W. P. To, Q. Y. Wan, G. S. M. Tong and C. M. Che, Recent Advances in Metal Triplet Emitters with d(6), d(8), and d(10) Electronic Configurations, Trends Chem., 2020, 2, 796–812, DOI:10.1016/j.trechm.2020.06.004.
- R. B. Bai, X. W. Meng, X. X. Wang and L. He, Blue-Emitting Iridium(III) Complexes for Light-Emitting Electrochemical Cells: Advances, Challenges, and Future Prospects, Adv. Funct. Mater., 2020, 30, 1907169, DOI:10.1002/adfm.201907169.
- Y. L. Zhuang, S. Guo, Y. J. Deng, S. J. Liu and Q. Zhao, Electroluminochromic Materials and Devices Based on Metal Complexes, Chem. – Asian J., 2019, 14, 3791–3802, DOI:10.1002/asia.201901209.
- C. E. Housecroft and E. C. Constable, Over the LEC rainbow: Colour and stability tuning of cyclometallated iridium(III) complexes in light-emitting electrochemical cells, Coord. Chem. Rev., 2017, 350, 155–177, DOI:10.1016/j.ccr.2017.06.016.
- Q.-C. Zhang, H. Xiao, X. Zhang, L.-J. Xu and Z.-N. Chen, Luminescent oligonuclear metal complexes and the use in organic light-emitting diodes, Chem. Soc. Rev., 2019, 378, 121–133, DOI:10.1016/j.ccr.2018.01.017.
- A. F. Henwood and E. Zysman-Colman, Luminescent Iridium Complexes Used in Light-Emitting Electrochemical Cells (LEECs), Top. Curr. Chem., 2016, 374, 1–41, DOI:10.1007/s41061-016-0036-0.
- A. Colombo, C. Dragonetti, V. Guerchais, C. Hierlinger, E. Zysman-Colman and D. Roberto, A trip in the nonlinear optical properties of iridium complexes, Coord. Chem. Rev., 2020, 414, 213293, DOI:10.1016/j.ccr.2020.213293.
- X. Li, Y. Xie and Z. Li, Diversity of Luminescent Metal Complexes in OLEDs: Beyond Traditional Precious Metals, Chem. – Asian J., 2021, 16, 2817–2829, DOI:10.1002/asia.202100784.
- D.-L. Ma, S.-Y. Wong, T.-S. Kang, H.-P. Ng, Q.-B. Han and C.-H. Leung, Iridium(III)-based chemosensors for the detection of metal ions, Methods, 2019, 168, 3–17, DOI:10.1016/j.ymeth.2019.02.013.
- D.-L. Ma, S. Lin, W. Wang, C. Yang and C.-H. Leung, Luminescent chemosensors by using cyclometalated iridium(III) complexes and their applications, Chem. Sci., 2017, 8, 878–889, 10.1039/C6SC04175B.
- V. Sathish, A. Ramdass, M. Velayudham, K. L. Lu, P. Thanasekaran and S. Rajagopal, Development of luminescent sensors based on transition metal complexes for the detection of nitroexplosives, Dalton Trans., 2017, 46, 16738–16769, 10.1039/c7dt02790g.
- F. Glaser and O. S. Wenger, Recent progress in the development of transition-metal based photoredox catalysts, Coord. Chem. Rev., 2020, 405, 213129, DOI:10.1016/j.ccr.2019.213129.
- J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, The merger of transition metal and photocatalysis, Nat. Rev. Chem., 2017, 1, 0052, DOI:10.1038/s41570-017-0052.
- M. Parasram and V. Gevorgyan, Visible light-induced transition metal-catalyzed transformations: beyond conventional photosensitizers, Chem. Soc. Rev., 2017, 46, 6227–6240, 10.1039/C7CS00226B.
- J.-H. Shon and T. S. Teets, Molecular Photosensitizers in Energy Research and Catalysis: Design Principles and Recent Developments, ACS Energy Lett., 2019, 4, 558–566, DOI:10.1021/acsenergylett.8b02388.
- B. T. Luppi, D. Majak, M. Gupta, E. Rivard and K. Shankar, Triplet excitons: improving exciton diffusion length for enhanced organic photovoltaics, J. Mater. Chem. A, 2019, 7, 2445–2463, 10.1039/C8TA10037C.
- X. Guo, Y. Liu, Q. Chen, D. Zhao and Y. Ma, New Bichromophoric Triplet Photosensitizer Designs and Their Application in Triplet–Triplet Annihilation Upconversion, Adv. Opt. Mater., 2018, 6, 1700981, DOI:10.1002/adom.201700981.
- K. Li, G. S. M. Tong, Q. Wan, G. Cheng, W.-Y. Tong, W.-H. Ang, W.-L. Kwong and C.-M. Che, Highly phosphorescent platinum(ii) emitters: photophysics, materials and biological applications, Chem. Sci., 2016, 7, 1653–1673, 10.1039/C5SC03766B.
- L. Holden, C. S. Burke, D. Cullinane and T. E. Keyes, Strategies to promote permeation and vectorization, and reduce cytotoxicity of metal complex luminophores for bioimaging and intracellular sensing, RSC Chem. Biol., 2021, 2, 1021–1049, 10.1039/d1cb00049g.
- X. Zhen, R. Qu, W. Z. Chen, W. Wu and X. Q. Jiang, The development of phosphorescent probes for in vitro and in vivo bioimaging, Biomater. Sci., 2021, 9, 285–300, 10.1039/d0bm00819b.
- S. Shaikh, Y. H. Wang, F. U. Rehman, H. Jiang and X. M. Wang, Phosphorescent Ir (III) complexes as cellular staining agents for biomedical molecular imaging, Coord. Chem. Rev., 2020, 416, 213344, DOI:10.1016/j.ccr.2020.213344.
- A. F. Henwood and E. Zysman-Colman, Lessons learned in tuning the optoelectronic properties of phosphorescent iridium (III) complexes, Chem. Commun., 2017, 53, 807–826, 10.1039/c6cc06729h.
- K. Li, Y. Chen, J. Wang and C. Yang, Diverse emission properties of transition metal complexes beyond exclusive single phosphorescence and their wide applications, Coord. Chem. Rev., 2021, 433, 213755, DOI:10.1016/j.ccr.2020.213755.
- F. Monti, F. Kessler, M. Delgado, J. Frey, F. Bazzanini, G. Accorsi, N. Armaroli, H. J. Bolink, E. Ortí, R. Scopelliti, M. K. Nazeeruddin and E. Baranoff, Charged Bis-Cyclometalated Iridium (III) Complexes with Carbene-Based Ancillary Ligands, Inorg. Chem., 2013, 52, 10292–10305, DOI:10.1021/ic400600d.
- M. M. Ghimire, V. N. Nesterov and M. A. Omary, Remarkable Aurophilicity and Photoluminescence Thermochromism in a Homoleptic Cyclic Trinuclear Gold(I) Imidazolate Complex, Inorg. Chem., 2017, 56, 12086–12089, DOI:10.1021/acs.inorgchem.7b01679.
- A. A. Eremina, M. A. Kinzhalov, E. A. Katlenok, A. S. Smirnov, E. V. Andrusenko, E. A. Pidko, V. V. Suslonov and K. V. Luzyanin, Phosphorescent Iridium(III) Complexes with Acyclic Diaminocarbene Ligands as Chemosensors for Mercury, Inorg. Chem., 2020, 59, 2209–2222, DOI:10.1021/acs.inorgchem.9b02833.
- C. Bartolomé, M. Carrasco-Rando, S. Coco, C. Cordovilla, P. Espinet and J. M. Martín-Alvarez, Structural Switching in Luminescent Polynuclear Gold Imidoyl Complexes by Intramolecular Hydrogen Bonding, Organometallics, 2006, 25, 2700–2703, DOI:10.1021/om0601753.
- L. Mercs and M. Albrecht, Beyond catalysis: N-heterocyclic carbene complexes as components for medicinal, luminescent, and functional materials applications, Chem. Soc. Rev., 2010, 39, 1903–1912, 10.1039/B902238B.
- R. Visbal and M. C. Gimeno, N-heterocyclic carbene metal complexes: photoluminescence and applications, Chem. Soc. Rev., 2014, 43, 3551–3574, 10.1039/C3CS60466G.
- M. Elie, J. L. Renaud and S. Gaillard, N-Heterocyclic carbene transition metal complexes in light emitting devices, Polyhedron, 2018, 140, 158–168, DOI:10.1016/j.poly.2017.11.045.
- A. Bonfiglio and M. Mauro, Phosphorescent Tris-Bidentate IrIII Complexes with N-Heterocyclic Carbene Scaffolds: Structural Diversity and Optical Properties, Eur. J. Inorg. Chem., 2020, 2020, 3427–3442, DOI:10.1002/ejic.202000509.
- J. Lee, H.-F. Chen, T. Batagoda, C. Coburn, P. I. Djurovich, M. E. Thompson and S. R. Forrest, Deep blue phosphorescent organic light-emitting diodes with very high brightness and efficiency, Nat. Mater., 2015, 15, 92, DOI:10.1038/nmat4446.
- C.-F. Chang, Y.-M. Cheng, Y. Chi, Y.-C. Chiu, C.-C. Lin, G.-H. Lee, P.-T. Chou, C.-C. Chen, C.-H. Chang and C.-C. Wu, Highly efficient blue-emitting iridium(III) carbene complexes and phosphorescent OLEDs, Angew. Chem., Int. Ed., 2008, 47, 4542–4545, DOI:10.1002/anie.200800748.
- Z. Chen, S. Suramitr, N. Y. Zhu, C. L. Ho, S. Hannongbua, S. M. Chen and W. Y. Wong, Tetrafluorinated phenylpyridine based heteroleptic iridium(iii) complexes for efficient sky blue phosphorescent organic light-emitting diodes, J. Mater. Chem. C, 2020, 8, 2551–2557, 10.1039/c9tc05779j.
- J. D. Bullock, Z. Xu, S. Valandro, M. Younus, J. Xue and K. S. Schanze, trans-N-(Heterocyclic Carbene) Platinum(II) Acetylide Chromophores as Phosphors for OLED Applications, ACS Appl. Electron. Mater., 2020, 2, 1026–1034, DOI:10.1021/acsaelm.0c00064.
- A. K. Pal, S. Krotkus, M. Fontani, C. F. R. Mackenzie, D. B. Cordes, A. M. Z. Slawin, I. D. W. Samuel and E. Zysman-Colman, High–Efficiency Deep–Blue–Emitting Organic Light–Emitting Diodes Based on Iridium(III) Carbene Complexes, Adv. Mater., 2018, 30, 1804231, DOI:10.1002/adma.201804231.
- G. D. Frey, R. D. Dewhurst, S. Kousar, B. Donnadieu and G. Bertrand, Cyclic (alkyl)(amino)carbene gold(I) complexes: A synthetic and structural investigation, J. Organomet. Chem., 2008, 693, 1674–1682, DOI:10.1016/j.jorganchem.2008.01.026.
- D. Di, A. S. Romanov, L. Yang, J. M. Richter, J. P. H. Rivett, S. Jones, T. H. Thomas, M. Abdi-Jalebi, R. H. Friend, M. Linnolahti, M. Bochmann and D. Credgington, High-performance light-emitting diodes based on carbene-metal-amides, Science, 2017, 356, 159–163, DOI:10.1126/science.aah4345.
- A. S. Romanov, S. T. E. Jones, Q. Gu, P. J. Conaghan, B. H. Drummond, J. Feng, F. Chotard, L. Buizza, M. Foley, M. Linnolahti, D. Credgington and M. Bochmann, Carbene metal amide photoemitters: tailoring conformationally flexible amides for full color range emissions including white-emitting OLED, Chem. Sci., 2020, 11, 435–446, 10.1039/C9SC04589A.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, An overview of N-heterocyclic carbenes, Nature, 2014, 510, 485, DOI:10.1038/nature13384.
- R. H. Crabtree, NHC ligands versus cyclopentadienyls and phosphines as spectator ligands in organometallic catalysis, J. Organomet. Chem., 2005, 690, 5451–5457, DOI:10.1016/j.jorganchem.2005.07.099.
- N. S. Antonova, J. J. Carbo and J. M. Poblet, Quantifying the Donor-Acceptor Properties of Phosphine and N-Heterocyclic Carbene Ligands in Grubbs’ Catalysts Using a Modified EDA Procedure Based on Orbital Deletion, Organometallics, 2009, 28, 4283–4287, DOI:10.1021/om900180m.
- H. Jacobsen, A. Correa, A. Poater, C. Costabile and L. Cavallo, Understanding the M(NHC) (NHC=N-heterocyclic carbene) bond, Coord. Chem. Rev., 2009, 253, 687–703, DOI:10.1016/j.ccr.2008.06.006.
- P. Frémont, N. Marion and S. P. Nolan, Carbenes: Synthesis, Properties, and Organometallic chemistry, Coord. Chem. Rev., 2009, 253, 862–892, DOI:10.1016/j.ccr.2008.05.018.
- R. W. Alder, P. R. Allen, M. Murray and A. G. Orpen, Bis(diisopropylamino)carbene, Angew. Chem., Int. Ed. Engl., 1996, 35, 1121–1123, DOI:10.1002/anie.199611211.
- D. Martin, Y. Canac, V. Lavallo and G. Bertrand, Comparative Reactivity of Different Types of Stable Cyclic and Acyclic Mono- and Diamino Carbenes with Simple Organic Substrates, J. Am. Chem. Soc., 2014, 136, 5023–5030, DOI:10.1021/ja412981x.
- J. Han, K.-M. Tang, S.-C. Cheng, C.-O. Ng, Y.-K. Chun, S.-L. Chan, S.-M. Yiu, M.-K. Tse, V. A. L. Roy and C.-C. Ko, Mechanochemical changes on cyclometalated Ir(III) acyclic carbene complexes – design and tuning of luminescent mechanochromic transition metal complexes, Inorg. Chem. Front., 2020, 7, 786–794, 10.1039/C9QI01278H.
- D. R. Snead, S. Inagaki, K. A. Abboud and S. Hong, Bis(2-alkylpyrrolidin-1-yl)methylidenes as Chiral Acyclic Diaminocarbene Ligands, Organometallics, 2010, 29, 1729–1739, DOI:10.1021/om901112n.
- E. L. Rosen, D. H. Sung, Z. Chen, V. M. Lynch and C. W. Bielawski, Olefin Metathesis Catalysts Containing Acyclic Diaminocarbenes, Organometallics, 2010, 29, 250–256, DOI:10.1021/om9008718.
- E. L. Rosen, M. D. Sanderson, S. Saravanakumar and C. W. Bielawski, Synthesis and study of the first N-aryl acyclic diaminocarbene and its transition-metal complexes, Organometallics, 2007, 26, 5774–5777, DOI:10.1021/om7007925.
- B. V. Johnson and J. E. Shade, Trisubstituted diaminocarbene complexes of iron, J. Organomet. Chem., 1979, 179, 357–366, DOI:10.1016/S0022-328X(00)91751-0.
- R. W. Alder, M. E. Blake and J. M. Oliva, Diaminocarbenes; Calculation of Barriers to Rotation about Ccarbene−N Bonds, Barriers to Dimerization, Proton Affinities, and 13C NMR Shifts, J. Phys. Chem. A, 1999, 103, 11200–11211, DOI:10.1021/jp9934228.
- M. S. Collins, E. L. Rosen, V. M. Lynch and C. W. Bielawski, Differentially Substituted Acyclic Diaminocarbene Ligands Display Conformation-Dependent Donicities, Organometallics, 2010, 29, 3047–3053, DOI:10.1021/om1004226.
- H. Na, A. Maity, R. Morshed and T. S. Teets, Bis-Cyclometalated Iridium Complexes with Chelating Dicarbene Ancillary Ligands, Organometallics, 2017, 36, 2965–2972, DOI:10.1021/acs.organomet.7b00428.
- H. Na and T. S. Teets, Highly Luminescent Cyclometalated Iridium Complexes Generated by Nucleophilic Addition to Coordinated Isocyanides, J. Am. Chem. Soc., 2018, 140, 6353–6360, DOI:10.1021/jacs.8b02416.
- J. Vignolle, X. Catton and D. Bourissou, Stable noncyclic singlet carbenes, Chem. Rev., 2009, 109, 3333–3384, DOI:10.1021/cr800549j.
- V. P. Boyarskiy, K. V. Luzyanin and V. Y. Kukushkin, Acyclic diaminocarbenes (ADCs) as a promising alternative to N-heterocyclic carbenes (NHCs) in transition metal catalyzed organic transformations, Coord. Chem. Rev., 2012, 256, 2029–2056, DOI:10.1016/j.ccr.2012.04.022.
- L. M. Slaughter, Acyclic Aminocarbenes in Catalysis, ACS Catal., 2012, 2, 1802–1816, DOI:10.1021/cs300300y.
- V. P. Boyarskiy, N. A. Bokach, K. V. Luzyanin and V. Y. Kukushkin, Metal-Mediated and Metal-Catalyzed Reactions of Isocyanides, Chem. Rev., 2015, 115, 2698–2779, DOI:10.1021/cr500380d.
- M. A. Kinzhalov and K. V. Luzyanin, Reactivity of acyclic diaminocarbene ligands, Coord. Chem. Rev., 2019, 399, 213014, DOI:10.1016/j.ccr.2019.213014.
- M. A. Kinzhalov and V. P. Boyarskii, Structure of isocyanide palladium(II) complexes and their reactivity toward nitrogen nucleophiles, Russ. J. Gen. Chem., 2015, 85, 2313–2333, DOI:10.1134/S1070363215100175.
- L. Benhamou, E. Chardon, G. Lavigne, S. Bellemin-Laponnaz and V. César, Synthetic Routes to N-Heterocyclic Carbene Precursors, Chem. Rev., 2011, 111, 2705–2733, DOI:10.1021/cr100328e.
- J. K. McCusker, Electronic structure in the transition metal block and its implications for light harvesting, Science, 2019, 363, 484–488, DOI:10.1126/science.aav9104.
- B. C. Paulus, K. C. Nielsen, C. R. Tichnell, M. C. Carey and J. K. McCusker, A Modular Approach to Light Capture and Synthetic Tuning of the Excited-State Properties of Fe(II)-Based Chromophores, J. Am. Chem. Soc., 2021, 143, 8086–8098, DOI:10.1021/jacs.1c02451.
- S. W. Lai, K. K. Cheung, M. C. W. Chan and C. M. Che, [{Pt(CN)(C10H21N4)}6]: A Luminescent Hexanuclear Platinum(II) Macrocycle Containing Chelating Dicarbene and Bridging Cyanide Ligands, Angew. Chem., Int. Ed., 1998, 37, 182–184, DOI:10.1002/(SICI)1521-3773(19980202)37:1/2<182::AID-ANIE182>3.0.CO;2-X.
- M. A. Kinzhalov, A. A. Eremina, A. S. Smirnov, V. V. Suslonov, V. Y. Kukushkin and K. V. Luzyanin, Cleavage of acyclic diaminocarbene ligands at an iridium(III) center. Recognition of a new reactivity mode for carbene ligands, Dalton Trans., 2019, 48, 7571–7582, 10.1039/C9DT01138B.
- T.-Y. Li, J. Wu, Z.-G. Wu, Y.-X. Zheng, J.-L. Zuo and Y. Pan, Rational design of phosphorescent iridium(III) complexes for emission color tunability and their applications in OLEDs, Coord. Chem. Rev., 2018, 374, 55–92, DOI:10.1016/j.ccr.2018.06.014.
- H.-T. Mao, G.-F. Li, G.-G. Shan, X.-L. Wang and Z.-M. Su, Recent progress in phosphorescent Ir(III) complexes for nondoped organic light-emitting diodes, Coord. Chem. Rev., 2020, 413, 213283, DOI:10.1016/j.ccr.2020.213283.
- A. L. Balch and J. E. Parks, Platinum and Palladium Complexes Formed by Chelative Addition of Amines to Isocyanides, J. Am. Chem. Soc., 1974, 96, 4114–4121, DOI:10.1021/ja00820a009.
- W. M. Butler and J. H. Enemark, Chelative addition of hydrazine to coordinated isocyanides. Structure of 1,1′-dichloropallado-2,5-di(methylamino)-3,4-diazacyclopentadiene, [Me2C2N4H4]PdCl2, Inorg. Chem., 1971, 10, 2416–2419, DOI:10.1021/ic50105a010.
- A. Burke, A. L. Balch and J. H. Enemark, Palladium and platinum complexes resulting from the addition of hydrazine to coordinated isocyanide, J. Am. Chem. Soc., 1970, 92, 2555–2557, DOI:10.1021/ja00711a063.
- R. Usón, A. Laguna, M. D. Villacampa, P. G. Jones and G. M. Sheldrick, Reactions of cis-di-isocyanidebis(perfluorophenyl)gold(III) complexes with hydrazines. Crystal and molecular structure of cis-[Au{C(NHC6H4Me-p)=N–N(Ph)C(NHC6H4Me-p)}(C6F5)2], Dalton Trans., 1984, 1984, 2035–2038, 10.1039/DT9840002035.
- J. R. Stork, M. M. Olmstead and A. L. Balch, Polymorphs with Varying Platinum(II)−Thallium(I) Interactions, J. Am. Chem. Soc., 2005, 127, 6512–6513, DOI:10.1021/ja050014a.
- A. I. Moncada, S. Manne, J. M. Tanski and L. M. Slaughter, Modular Chelated Palladium Diaminocarbene Complexes: Synthesis, Characterization, and Optimization of Catalytic Suzuki-Miyaura Cross-Coupling Activity by Ligand Modification, Organometallics, 2006, 25, 491–505, DOI:10.1021/om050786f.
- H. Na, P. N. Lai, L. M. Cañada and T. S. Teets, Photoluminescence of Cyclometalated Iridium Complexes in Poly(methyl methacrylate) Films, Organometallics, 2018, 37, 3269–3277, DOI:10.1021/acs.organomet.8b00446.
- C.-H. Yang, J. Beltran, V. Lemaur, J. Cornil, D. Hartmann, W. Sarfert, R. Fröhlich, C. Bizzarri and L. De Cola, Iridium Metal Complexes Containing N-Heterocyclic Carbene Ligands for Blue-Light-Emitting Electrochemical Cells, Inorg. Chem., 2010, 49, 9891–9901, DOI:10.1021/ic1009253.
- T.-Y. Li, X. Liang, L. Zhou, C. Wu, S. Zhang, X. Liu, G.-Z. Lu, L.-S. Xue, Y.-X. Zheng and J.-L. Zuo, N-Heterocyclic Carbenes: Versatile Second Cyclometalated Ligands for Neutral Iridium(III) Heteroleptic Complexes, Inorg. Chem., 2015, 54, 161–173, DOI:10.1021/ic501949h.
- H. Na, A. Maity and T. S. Teets, Bis-cyclometalated iridium complexes with electronically modified aryl isocyanide ancillary ligands, Dalton Trans., 2017, 46, 5008–5016, 10.1039/C7DT00694B.
- J. Li, P. I. Djurovich, B. D. Alleyne, M. Yousufuddin, N. N. Ho, J. C. Thomas, J. C. Peters, R. Bau and M. E. Thompson, Synthetic Control of Excited-State Properties in Cyclometalated Ir(III) Complexes Using Ancillary Ligands, Inorg. Chem., 2005, 44, 1713–1727, DOI:10.1021/ic048599h.
- Y.-J. Gao, T.-T. Zhang and W.-K. Chen, Predicting Excited-State and Luminescence Properties of a Cyclometalated Iridium(III) Complex: Quantum Mechanics/Molecular Mechanics Study, J. Phys. Chem. C, 2021, 125, 5670–5677, DOI:10.1021/acs.jpcc.0c09464.
- Y. Shen, X. Li, J. Ye and Y. Qiu, A DFT study on second-order NLO properties of bis-cyclometalated Iridium(III) complexes with chelating dicarbene auxiliary ligands, Comput. Theor. Chem., 2019, 1163, 112535, DOI:10.1016/j.comptc.2019.112535.
- X. Li, H.-Q. Wang, J.-T. Ye, Y. Zhang and Y.-Q. Qiu, Second-order NLO properties of bis-cyclometalated iridium(III) complexes: Substituent effect and redox switch, J. Mol. Graphics Modell., 2019, 89, 131–138, DOI:10.1016/j.jmgm.2019.03.005.
- T. Sajoto, P. I. Djurovich, A. Tamayo, M. Yousufuddin, R. Bau, M. E. Thompson, R. J. Holmes and S. R. Forrest, Blue and Near-UV Phosphorescence from Iridium Complexes with Cyclometalated Pyrazolyl or N-Heterocyclic Carbene Ligands, Inorg. Chem., 2005, 44, 7992–8003, DOI:10.1021/ic051296i.
- J.-H. Kim, S.-Y. Kim, S. Choi, H.-J. Son and S. O. Kang, Peripheral Ligand Effect on the Photophysical Property of Octahedral Iridium Complex: o-Aryl Substitution on the Phenyl Units of Homoleptic IrIII(C^C)3 Complexes (C^C=1-Phenyl-3-methylimidazolin-2-ylidene-C,C2′) for Deep Blue Phosphorescence, Inorg. Chem., 2021, 60, 246–262, DOI:10.1021/acs.inorgchem.0c02882.
- R. J. Holmes, S. R. Forrest, T. Sajoto, A. Tamayo, P. I. Djurovich, M. E. Thompson, J. Brooks, Y. J. Tung, B. W. D’Andrade, M. S. Weaver, R. C. Kwong and J. J. Brown, Saturated deep blue organic electrophosphorescence using a fluorine-free emitter, Appl. Phys. Lett., 2005, 87, 243507, DOI:10.1063/1.2143128.
- Z. Chen, L. Wang, S. Su, X. Zheng, N. Zhu, C.-L. Ho, S. Chen and W.-Y. Wong, Cyclometalated Iridium(III) Carbene Phosphors for Highly Efficient Blue Organic Light-Emitting Diodes, ACS Appl. Mater. Interfaces, 2017, 9, 40497–40502, DOI:10.1021/acsami.7b09172.
- H. Sasabe, J.-i. Takamatsu, T. Motoyama, S. Watanabe, G. Wagenblast, N. Langer, O. Molt, E. Fuchs, C. Lennartz and J. Kido, High-Efficiency Blue and White Organic Light-Emitting Devices Incorporating a Blue Iridium Carbene Complex, Adv. Mater., 2010, 22, 5003–5007, DOI:10.1002/adma.201002254.
- H. Na, L. M. Cañada, Z. Wen, J. I. Wu and T. S. Teets, Mixed-carbene cyclometalated iridium complexes with saturated blue luminescence, Chem. Sci., 2019, 10, 6254–6260, 10.1039/C9SC01386E.
- D. Reinen, M. Atanasov, P. Köhler and D. Babel, Jahn–Teller coupling and the influence of strain in Tg and Eg ground and excited states – A ligand field and DFT study on halide MIIIX6 model complexes [M=TiIII–CuIII; X=F−, Cl−], Coord. Chem. Rev., 2010, 254, 2703–2754, DOI:10.1016/j.ccr.2010.04.015.
- K. Dedeian, J. Shi, E. Forsythe, D. C. Morton and P. Y. Zavalij, Blue phosphorescence from mixed cyano-isocyanide cyclometalated iridium(III) complexes, Inorg. Chem., 2007, 46, 1603–1611, DOI:10.1021/ic061513v.
- Y.-J. Cho, S.-Y. Kim, C. M. Choi, N. J. Kim, C. H. Kim, D. W. Cho, H.-J. Son, C. Pac and S. O. Kang, Photophysics and Excited-State Properties of Cyclometalated Iridium(III)–Platinum(II) and Iridium(III)–Iridium(III) Bimetallic Complexes Bridged by Dipyridylpyrazine, Inorg. Chem., 2017, 56, 5305–5315, DOI:10.1021/acs.inorgchem.7b00384.
- P. C. Xue, J. P. Ding, P. P. Wang and R. Lu, Recent progress in the mechanochromism of phosphorescent organic molecules and metal complexes, J. Mater. Chem. C, 2016, 4, 6688–6706, 10.1039/c6tc01503d.
- S. Ito, Recent Advances in Mechanochromic Luminescence of Organic Crystalline Compounds, Chem. Lett., 2021, 50, 649–660, DOI:10.1246/cl.200874.
- B. H. Di and Y. L. Chen, Recent progress in organic mechanoluminescent materials, Chin. Chem. Lett., 2018, 29, 245–251, DOI:10.1016/j.cclet.2017.08.043.
- H. Ito, T. Saito, N. Oshima, N. Kitamura, S. Ishizaka, Y. Hinatsu, M. Wakeshima, M. Kato, K. Tsuge and M. Sawamura, Reversible Mechanochromic Luminescence of [(C6F5Au)2(μ-1,4-Diisocyanobenzene)], J. Am. Chem. Soc., 2008, 130, 10044–10045, DOI:10.1021/ja8019356.
- T. Seki, T. Ozaki, T. Okura, K. Asakura, A. Sakon, H. Uekusa and H. Ito, Interconvertible multiple photoluminescence color of a gold(i) isocyanide complex in the solid state: solvent-induced blue-shifted and mechano-responsive red-shifted photoluminescence, Chem. Sci., 2015, 6, 2187–2195, 10.1039/C4SC03960B.
- S. Yagai, T. Seki, H. Aonuma, K. Kawaguchi, T. Karatsu, T. Okura, A. Sakon, H. Uekusa and H. Ito, Mechanochromic Luminescence Based on Crystal-to-Crystal Transformation Mediated by a Transient Amorphous State, Chem. Mater., 2016, 28, 234–241, DOI:10.1021/acs.chemmater.5b03932.
- N. M.-W. Wu, M. Ng and V. W.-W. Yam, Photochromic Benzo[b]phosphole Alkynylgold(I) Complexes with Mechanochromic Property to Serve as Multistimuli-Responsive Materials, Angew. Chem., Int. Ed., 2019, 58, 3027–3031, DOI:10.1002/anie.201806272.
- K. Chen, M. M. Nenzel, T. M. Brown and V. J. Catalano, Luminescent Mechanochromism in a Gold(I)–Copper(I) N-Heterocyclic Carbene Complex, Inorg. Chem., 2015, 54, 6900–6909, DOI:10.1021/acs.inorgchem.5b00821.
- J. Han, Y.-K. Chun, S.-L. Chan, S.-C. Cheng, S.-M. Yiu and C.-C. Ko, Development of Dual Phosphorescent Materials Based on Multiple Stimuli-Responsive Ir(III) Acyclic Carbene Complexes, CCS Chem., 2021, 3, 2345–2359, DOI:10.31635/ccschem.021.202101016.
- J. Herberger and R. F. Winter, Platinum emitters with dye-based σ-aryl ligands, Coord. Chem. Rev., 2019, 400, 213048, DOI:10.1016/j.ccr.2019.213048.
- M. A. Soto, R. Kandel and M. J. MacLachlan, Chromic Platinum Complexes Containing Multidentate Ligands, Eur. J. Inorg. Chem., 2021, 2021, 894–906, DOI:10.1002/ejic.202001117.
- G. Rouschias and B. L. Shaw, The chemistry and structure of Chugaev's salt and related compounds containing a cyclic carbene ligand, J. Chem. Soc. A, 1971, 2097–2104, 10.1039/J19710002097.
- A. I. Moncada, M. A. Khan and L. M. Slaughter, A palladium Chugaev carbene complex as a modular, air-stable catalyst for Suzuki–Miyaura cross-coupling reactions, Tetrahedron Lett., 2005, 46, 1399–1403, DOI:10.1016/j.tetlet.2005.01.033.
- J. R. Stork, M. M. Olmstead and A. L. Balch, Effects of Pt⋯Pt Bonding, Anions, Solvate Molecules, and Hydrogen Bonding on the Self-Association of Chugaev's Cation, a Platinum Complex with a Chelating Carbene Ligand, Inorg. Chem., 2004, 43, 7508–7515, DOI:10.1021/ic049226j.
- S.-W. Lai, M. C. W. Chan, Y. Wang, H.-W. Lam, S.-M. Peng and C.-M. Che, Luminescent metal complexes derived from carbene and related ligands: tuning excited-state properties with metal–carbon multiple bonds, J. Organomet. Chem., 2001, 617–618, 133–140, DOI:10.1016/S0022-328X(00)00723-3.
- S. Z. Goldberg, R. Eisenberg and J. S. Miller, Crystal and molecular structure of tetrakis(bis(methylamino)carbene)platinum(II) hexafluorophosphate, [Pt(C(NHMe)2)4](PF6)2, Inorg. Chem., 1977, 16, 1502–1507, DOI:10.1021/ic50172a053.
- J. S. Miller and A. L. Balch, Preparation and reactions of tetrakis(methyl isocyanide) complexes of divalent nickel, palladium, and platinum, Inorg. Chem., 1972, 11, 2069–2074, DOI:10.1021/ic50115a017.
- J. R. Stork, D. Rios, D. Pham, V. Bicocca, M. M. Olmstead and A. L. Balch, Metal−Metal Interactions in Platinum(II)/Gold(I) or Platinum(II)/Silver(I) Salts Containing Planar Cations and Linear Anions, Inorg. Chem., 2005, 44, 3466–3472, DOI:10.1021/ic048333a.
- V. Blase, A. Flores-Figueroa, C. Schulte to Brinke and F. E. Hahn, Template Synthesis of Tetracarbene Platinum(II) Complexes with Five- and Six-Membered-Ring N-Heterocyclic Carbenes, Organometallics, 2014, 33, 4471–4478, DOI:10.1021/om5006947.
- F. E. Hahn, V. Langenhahn, T. Lügger, T. Pape and D. Le Van, Template Synthesis of a Coordinated Tetracarbene Ligand with Crown Ether Topology, Angew. Chem., Int. Ed., 2005, 44, 3759–3763, DOI:10.1002/anie.200462690.
- Y. Unger, A. Zeller, S. Ahrens and T. Strassner, Blue phosphorescent emitters: new N-heterocyclic platinum(ii) tetracarbene complexes, Chem. Commun., 2008, 2008, 3263–3265, 10.1039/B804019B.
- Y. Unger, A. Zeller, M. A. Taige and T. Strassner, Near-UV phosphorescent emitters: N-heterocyclic platinum(ii) tetracarbene complexes, Dalton Trans., 2009, 24, 4786–4794, 10.1039/B900655A.
- Y. Unger, D. Meyer and T. Strassner, Blue phosphorescent platinum(ii) tetracarbene complexes with bis(triazoline-5-ylidene) ligands, Dalton Trans., 2010, 39, 4295–4301, 10.1039/B927099J.
- S.-W. Lai, M. C.-W. Chan, K.-K. Cheung and C.-M. Che, Carbene and Isocyanide Ligation at Luminescent Cyclometalated 6-Phenyl-2,2′-bipyridyl Platinum(II) Complexes: Structural and Spectroscopic Studies, Organometallics, 1999, 18, 3327–3336, DOI:10.1021/om990256h.
- M. T. Moreno, E. Lalinde, M. Martínez-Junquera, E. Alfaro-Arnedo, I. López, I. Larráyoz and J. G. G. Pichel, Luminescent Cyclometalated Platinum(II) Complexes with Acyclic Diaminocarbene Ligands: Structural, Photophysical and Biological Properties, Dalton Trans., 2021, 50, 4539–4554, 10.1039/D1DT00480H.
- A. I. Solomatina, D. V. Krupenya, V. V. Gurzhiy, I. Zlatkin, A. P. Pushkarev, M. N. Bochkarev, N. A. Besley, E. Bichoutskaia and S. P. Tunik, Cyclometallated platinum(ii) complexes containing NHC ligands: synthesis, characterization, photophysics and their application as emitters in OLEDs, Dalton Trans., 2015, 44, 7152–7162, 10.1039/C4DT03106G.
- Y. Wu, Z. Wen, J. I.-C. Wu and T. S. Teets, Efficient Deep Blue Platinum Acetylide Phosphors with Acyclic Diaminocarbene Ligands, Chem. – Eur. J., 2020, 26, 16028–16035, DOI:10.1002/chem.202002775.
- J. D. Bullock, A. Salehi, C. J. Zeman, K. A. Abboud, F. So and K. S. Schanze, In Search of Deeper Blues: Trans-N-Heterocyclic Carbene Platinum Phenylacetylide as a Dopant for Phosphorescent OLEDs, ACS Appl. Mater. Interfaces, 2017, 9, 41111–41114, DOI:10.1021/acsami.7b12107.
- C.-O. Ng, S.-C. Cheng, W.-K. Chu, K.-M. Tang, S.-M. Yiu and C.-C. Ko, Luminescent Rhenium(I) Pyridyldiaminocarbene Complexes: Photophysics, Anion-Binding, and CO2-Capturing Properties, Inorg. Chem., 2016, 55, 7969–7979, DOI:10.1021/acs.inorgchem.6b01017.
- C. Lih-Chiou, C. Min-Yuan, C. Jia-Hwa, W. Yuh-Sheng and L. Kuang-Lieh, Synthesis and reactivity of rhenium(I) isocyanide complexes. Crystal and molecular structures of ReBr(CO) 4{C(NHPh)(NHCHMe2)} and ReBr(CO)3(PPh3)IC(NHPh)(NHCHMe2), J. Organomet. Chem., 1992, 425, 99–111, DOI:10.1016/0022-328X(92)80025-S.
- K.-L. Lu, C.-M. Wang, H.-H. Lee, L.-C. Chen and Y.-S. Wen, Facile and successive orthometallation in rhenium carbene complex, J. Chem. Soc., Chem. Commun., 1993, 2008, 706–707, 10.1039/C39930000706.
- K.-L. Lu, H.-H. Lee, C.-M. Wang and Y.-S. Wen, Synthesis and reactivity of cyclometalated rhenium diaminocarbene complexes, Organometallics, 1994, 13, 593–599, DOI:10.1021/om00014a034.
- J.-S. Fan, J.-T. Lin, C.-C. Chang, S.-J. Chou and K.-L. Lu, Successive Reactions of Rhenium Isocyanide Complexes with Nitrogen-Containing Ligands. One-Flask Conversion of ReBr(CO)4(CNPh) to [Re(CO)3(NH2R)2{C(NHPh)(NHR)}]+Br, Organometallics, 1995, 14, 925–932, DOI:10.1021/om00002a045.
- J.-S. Fan, F.-Y. Lee, C.-C. Chiang, H.-C. Chen, S.-H. Liu, Y.-S. Wen, C.-C. Chang, S.-Y. Li, K.-M. Chi and K.-L. Lu, Synthesis, structure, and reactivity of rhenium N-isocyanide complexes ReBr(CO)3(CNR)(CNNPPh3), J. Organomet. Chem., 1999, 580, 82–89, DOI:10.1016/S0022-328X(98)01084-5.
- X. Schoultz, T. I. A. Gerber and E. C. Hosten, Formation of a methine carbon-to-rhenium σ bond in an oxorhenium(V)-benzothiazole complex, Inorg. Chem. Commun., 2016, 68, 13–16, DOI:10.1016/j.inoche.2016.03.020.
- C.-C. Ko, J. W.-K. Siu, A. W.-Y. Cheung and S.-M. Yiu, Synthesis, Characterization, and Photophysical and Emission Solvatochromic Study of Rhenium(I) Tetra(isocyano) Diimine Complexes, Organometallics, 2011, 30, 2701–2711, DOI:10.1021/om200074t.
- V. W.-W. Yam and E. C.-C. Cheng, Highlights on the recent advances in gold chemistry—a photophysical perspective, Chem. Soc. Rev., 2008, 37, 1806–1813, 10.1039/B708615F.
- C.-M. Che and S.-W. Lai, Structural and spectroscopic evidence for weak metal–metal interactions and metal–substrate exciplex formations in d10 metal complexes, Coord. Chem. Rev., 2005, 249, 1296–1309, DOI:10.1016/j.ccr.2004.11.026.
- I. Kondrasenko, K.-y. Chung, Y.-T. Chen, J. Koivistoinen, E. V. Grachova, A. J. Karttunen, P.-T. Chou and I. O. Koshevoy, Harnessing Fluorescence versus Phosphorescence Ratio via Ancillary Ligand Fine-Tuned MLCT Contribution, J. Phys. Chem. C, 2016, 120, 12196–12206, DOI:10.1021/acs.jpcc.6b03064.
- J. E. Parks and A. L. Balch, Gold carbene complexes: preparation, oxidation, and ligand displacement, J. Organomet. Chem., 1974, 71, 453–463 CrossRef CAS.
- R. L. White-Morris, M. M. Olmstead, F. Jiang and A. L. Balch, New Insights into the Effects of Self-Association of the Cation [Au{C(NHMe)2}2]+on Its Solid State Structure and Luminescence, Inorg. Chem., 2002, 41, 2313–2315, DOI:10.1021/ic020030y.
- R. L. White-Morris, M. M. Olmstead, F. Jiang, D. S. Tinti and A. L. Balch, Remarkable Variations in the Luminescence of Frozen Solutions of [Au{C(NHMe)2}2](PF6)·0.5(Acetone). Structural and Spectroscopic Studies of the Effects of Anions and Solvents on Gold(I) Carbene Complexes, J. Am. Chem. Soc., 2002, 124, 2327–2336, DOI:10.1021/ja012397s.
- D. Rios, D. M. Pham, J. C. Fettinger, M. M. Olmstead and A. L. Balch, Blue or Green Glowing Crystals of the Cation [Au{C(NHMe)2}2]+. Structural Effects of Anions, Hydrogen Bonding, and Solvate Molecules on the Luminescence of a Two-Coordinate Gold(I) Carbene Complex, Inorg. Chem., 2008, 47, 3442–3451, DOI:10.1021/ic702481v.
- D. Rios, M. M. Olmstead and A. L. Balch, Colorless, non-luminescent; colorless, luminescent; and yellow, luminescent crystals of the cation [Au{C(NHCH3)(NHCH2CH2OH)}2]+. The roles of anions and hydrogen bonding in determining the aggregation of two-coordinate gold(i) cations, Dalton Trans., 2008, 31, 4157–4164, 10.1039/B806878J.
- D. Rios, M. M. Olmstead and A. L. Balch, Blue-, Green- and Non-luminescent Crystals from One Reaction. Isolation and Structural Characterization of a Series of Gold(I)-Siver(I) Heterometallic Complexes Utilizing a Gold Carbene Metalloligand Containing Free Amino Groups, Inorg. Chem., 2009, 48, 5279–5287, DOI:10.1021/ic900243v.
- P. C. Kunz, C. Wetzel, S. Kögel, M. U. Kassack and B. Spingler, [(C3H4N2)2Au]Cl—a bis protic gold(i)-NHC, Dalton Trans., 2011, 40, 35–37, 10.1039/C0DT01089H.
- B. Lu, S. Liu and D. Yan, Recent advances in photofunctional polymorphs of molecular materials, Chin. Chem. Lett., 2019, 30, 1908–1922, DOI:10.1016/j.cclet.2019.09.012.
- C. Wang and Z. Li, Molecular conformation and packing: their critical roles in the emission performance of mechanochromic fluorescence materials, Mater. Chem. Front., 2017, 1, 2174–2194, 10.1039/C7QM00201G.
- W. Wang, Y. Zhang and W. J. Jin, Halogen bonding in room-temperature phosphorescent materials, Coord. Chem. Rev., 2020, 404, 213107, DOI:10.1016/j.ccr.2019.213107.
- I. O. Koshevoy, M. Krause and A. Klein, Non-covalent intramolecular interactions through ligand-design promoting efficient photoluminescence from transition metal complexes, Coord. Chem. Rev., 2020, 405, 213094, DOI:10.1016/j.ccr.2019.213094.
- E. R. T. Tiekink, Supramolecular assembly based on “emerging” intermolecular interactions of particular interest to coordination chemists, Coord. Chem. Rev., 2017, 345, 209–228, DOI:10.1016/j.ccr.2017.01.009.
- S. Varughese, Non-covalent routes to tune the optical properties of molecular materials, J. Mater. Chem. C, 2014, 2, 3499–3516, 10.1039/C3TC32414A.
- C. Bartolome, M. Carrasco-Rando, S. Coco, C. Cordovilla, J. M. Martin-Alvarez and P. Espinet, Luminescent gold(I) carbenes from 2-pyridylisocyanide complexes: Structural consequences of intramolecular versus intermolecular hydrogen-bonding interactions, Inorg. Chem., 2008, 47, 1616–1624, DOI:10.1021/ic702201e.
- C. Bartolome, M. Carrasco-Rando, S. Coco, C. Cordovilla, P. Espinet and J. M. Martin-Alvarez, Gold(i)-carbenes derived from 4-pyridylisocyanide complexes: supramolecular macrocycles supported by hydrogen bonds, and luminescent behavior, Dalton Trans., 2007, 45, 5339–5345, 10.1039/b711430c.
- F. M. C. Menezes, M. L. Kuznetsov and A. J. L. Pombeiro, Isocyanide Complexes with Platinum and Palladium and Their Reactivity toward Cycloadditions with Nitrones to Form Aminooxycarbenes: A Theoretical Study, Organometallics, 2009, 28, 6593–6602, DOI:10.1021/om900513b.
- C. Singh, A. Kumar and H. V. Huynh, Stereoelectronic Profiling of Acyclic Diamino Carbenes (ADCs), Inorg. Chem., 2020, 59, 8451–8460, DOI:10.1021/acs.inorgchem.0c00886.
- O. Elbjeirami, M. D. Rashdan, V. Nesterov and M. A. Rawashdeh-Omary, Structure and luminescence properties of a well-known macrometallocyclic trinuclear Au(i) complex and its adduct with a perfluorinated fluorophore showing cooperative anisotropic supramolecular interactions, Dalton Trans., 2010, 39, 9465–9468, 10.1039/C0DT00736F.
- T. Osuga, T. Murase, K. Ono, Y. Yamauchi and M. Fujita, [m × n] Metal Ion Arrays Templated by Coordination Cages, J. Am. Chem. Soc., 2010, 132, 15553–15555, DOI:10.1021/ja108367j.
- T. Osuga, T. Murase, M. Hoshino and M. Fujita, A Tray-Shaped, PdII-Clipped Au3 Complex as a Scaffold for the Modular Assembly of [3×n] Au Ion Clusters, Angew. Chem., Int. Ed., 2014, 53, 11186–11189, DOI:10.1002/anie.201404892.
- J. Ruiz, D. Sol, M. A. Mateo, M. Vivanco and R. Badia-Laino, A new approach to the synthesis of trinuclear gold(I) imidazolate complexes and their silver(I)-induced photoluminescence behavior, Dalton Trans., 2020, 49, 6561–6565, 10.1039/d0dt01246g.
- M. Kiguchi, J. Inatomi, Y. Takahashi, R. Tanaka, T. Osuga, T. Murase, M. Fujita, T. Tada and S. Watanabe, Highly Conductive [3×n] Gold-Ion Clusters Enclosed within Self-Assembled Cages, Angew. Chem., Int. Ed., 2013, 52, 6202–6205, DOI:10.1002/anie.201301665.
- R. Hahn, F. Bohle, W. Fang, A. Walther, S. Grimme and B. Esser, Raising the Bar in Aromatic Donor–Acceptor Interactions with Cyclic Trinuclear Gold(I) Complexes as Strong π-Donors, J. Am. Chem. Soc., 2018, 140, 17932–17944, DOI:10.1021/jacs.8b08823.
- S. Lentijo, J. E. Expósito, G. Aullón, J. A. Miguel and P. Espinet, Highly fluorescent complexes with 3-isocyanoperylene and N-(2,5-di-tert-butylphenyl)-9-isocyano-perylene-3,4-dicarboximide, Dalton Trans., 2014, 43, 10885–10897, 10.1039/C4DT01016G.
- C. Domínguez, M. J. Baena, S. Coco and P. Espinet, Perylenecarboxydiimide-gold(I) organometallic dyes. Optical properties and Langmuir films, Dyes Pigm., 2017, 140, 375–383, DOI:10.1016/j.dyepig.2017.01.057.
- R. Martínez-Máñez and F. Sancenón, Fluorogenic and Chromogenic Chemosensors and Reagents for Anions, Chem. Rev., 2003, 103, 4419–4476, DOI:10.1021/cr010421e.
- V. W. W. Yam and A. S. Y. Law, Luminescent d(8) metal complexes of platinum(II) and gold(III): From photophysics to photofunctional materials and probes, Coord. Chem. Rev., 2020, 414, 213298, DOI:10.1016/j.ccr.2020.213298.
- V. W.-W. Yam, S. W.-K. Choi, T.-F. Lai and W.-K. Lee, Syntheses, crystal structures and photophysics of organogold(III) diimine complexes, Dalton Trans., 1993, 1993, 1001–1002, 10.1039/DT9930001001.
- O. Crespo, M. C. Gimeno, A. Laguna, S. Montanel-Pérez and M. D. Villacampa, Facile Synthesis of Gold(III) Aryl–Carbene Metallacycles, Organometallics, 2012, 31, 5520–5526, DOI:10.1021/om300495u.
- W.-P. To, G. Cheng, G. S. M. Tong, D. Zhou and C.-M. Che, Recent Advances in Metal-TADF Emitters and Their Application in Organic Light-Emitting Diodes, Front. Chem., 2020, 8, 653, DOI:10.3389/fchem.2020.00653.
- J. C. Lima and L. Rodríguez, Highlights on Gold TADF Complexes, Inorganics, 2019, 7, 124 CrossRef CAS.
- M.-C. Tang, M.-Y. Leung, S.-L. Lai, M. Ng, M.-Y. Chan and V. W. W. Yam, Realization of Thermally Stimulated Delayed Phosphorescence in Arylgold(III) Complexes and Efficient Gold(III) Based Blue-Emitting Organic Light-Emitting Devices, J. Am. Chem. Soc., 2018, 140, 13115–13124, DOI:10.1021/jacs.8b09205.
- M. A. Kinzhalov and K. V. Luzyanin, Synthesis and Contemporary Applications of Platinum Group Metals Complexes with Acyclic Diaminocarbene Ligands (Review), Russ. J. Inorg. Chem., 2022, 67(1), 48–90, DOI:10.1134/S0036023622010065.
- M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Cyclic (Alkyl)(amino)carbenes (CAACs): Recent Developments, Angew. Chem., Int. Ed., 2017, 56(34), 10046–10068, DOI:10.1002/anie.201702148.
| This journal is © the Partner Organisations 2022 |