
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydroxide ion-conducting metal–organic frameworks for anion-exchange membrane applications
Maria-Magdalena
Titirici
 a and
Petra Ágota
Szilágyi
*b
a and
Petra Ágota
Szilágyi
*b
aDepartment of Chemical Engineering, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK
bCenter for Materials Science and Nanotechnology (SMN), Department of Chemistry, University of Oslo, P.O. Box 1033, Blindern, N-0315 Oslo, Norway. E-mail: P.A.Szilagyi@kjemi.uio.no
First published on 5th October 2022
Abstract
With the increasing demand for green energy, there is a great urgency to develop perm-selective, economical and robust membranes for applications in fuel cells. Metal–organic frameworks are attractive materials for this application as they are typically electrically insulating, have tuneable pore size and chemistry – enabling the realisation of specific interactions with the ions -, and their processing technology has recently undergone a tremendous development, allowing for their synthesis as free-standing membranes. This critical review aims at evaluating recent advances in the field of hydroxide-ion conducting metal–organic frameworks for ion-exchange membrane applications. In addition, it will conceptualise the most promising approaches for design, synthesis and processing of the framework-based membranes.
Introduction
There is a scientific and increasingly societal consensus that the current energy paradigm largely based on fossil-fuel combustion for energy provision is unsustainable and it urgently needs to be replaced by renewables. The reason for this is manifold, including the increasingly real problem of climate change, loss of biodiversity, and health concerns, but also the constantly decreasing resources, volatile and unreliable geopolitical issues, as well as fluctuating prices. Renewable energy sources on the other hand are plentiful, and most geographical areas possess significant resources of at least some types of them and the technologies offered by them are increasingly affordable.1 Renewables are however typically fluxes and as such, they only allow for energy conversion to electricity rather than for storage. On account of their spatial and temporal distribution as well as general intermittence they are not able to provide an even load to demand and thus need to be balanced and, in peak periods, energy storage solutions need to be found.2 To this end, current technologies typically make use of electrochemical energy storage in the form of batteries. However, it is increasingly apparent that battery technologies alone are not able to afford the energy storage needed for the full transition of the energy landscape; in particular, some applications have characteristic requirements that batteries currently cannot meet, these include heavy-duty vehicles, aviation, naval transport, grid-scale storage, etc.3 For this reason, it is imperative that in conjunction with electrification alternative energy-storage technologies are also developed and marketed. In particular, the production of high energy-density fuels in peak renewable-flux periods is a realistic and desirable energy-storage solution for diverse applications, complementary to battery technologies.Hydrogen economy
Among high energy-density fuels, hydrogen is highly promising and it has been the subject of significant research interest. Its advantages include the highest energy density of all known substances by weight (120 kJ mol−1) and availability – it is the most common element in the universe, which is within the five most abundant elements on Earth and water in particular is a plentiful source of it. Furthermore, hydrogen is unrivalled in the versatility of ways it may be utilised for energy conversion and storage, combusted,4 electrochemically oxidised in a fuel cell,5 and used as a reactant to form fuels such as liquid hydrocarbons,6 which processes may take place through a variety of reaction types ranging from thermally to (photo-)electrochemically driven reactions. As a matter of fact, the hydrogen economy has been highlighted as particularly appealing as it may be included in a completely carbon-free cycle,7 provided it is generated photo/electro-chemically, i.e. green hydrogen is used. However, safe and green production is not the only challenge needing to be overcome for the hydrogen economy to be a reality, additional challenges include its low-cost, safe, efficient and effective storage, distribution, and oxidation to water.8Fuel cells
The electrochemical oxidation of hydrogen produces water as the sole product in the electrochemical device of fuel cells. This process is crucial as the combustion of hydrogen is highly exothermic, in fact, it forms an explosive mixture with air in a broad concentration range, which makes its controlled oxidation not only more efficient but also much safer and thus highly desirable.5 In fuel cells, hydrogen is stripped of electrons on the anode, while oxygen is reduced on the cathode. The two redox reactions (half reactions) are physically separated and the process is accompanied by the flow of electrons from the anode to the cathode on an external circuit. It generates electricity while ions flow between the two electrode sides of the device, which are separated by a semipermeable membrane for charge balancing. Mature fuel cell technology makes use of protons to balance charges by flowing from the anode to the cathode side across a proton conducting membrane, in the proton-exchange membrane fuel cell (PEMFC). In PEMFCs, the processes are as follows:| Anode: | H2 → 2 H+ + 2 e− |
|---|---|
| Cathode: | 1/2 O2 + 2 e− + H2O → 2 OH− |
| Electrolyte: | H+ + OH− → H2O |
| Overall: | H2 + 1/2 O2 → H2O |
Commercial PEMFCs typically feature a Nafion® proton exchange membrane, which boasts high proton conductivity, adequate chemical stability, mechanical robustness, and acceptable costs. However, the integration of polymers into devices causes increasing environmental concerns over the sustainability of polymer manufacturing as well as over their fate beyond disposal.9,10 It is important to re-iterate that hydrogen and oxygen combine to form an explosive gas mixture in a broad concentration range; therefore, the mixing of the two gases must be avoided at all times. Technically, the gases are introduced into the device via separate cell compartments, but it is also important that the dissolved gas molecules do not transfer between the two different compartments through the electrolyte (and the membrane) either. This phenomenon is called crossover, which presents a practical challenge in the development of membranes – ideally they would be perm-selective for protons. However, repulsive interactions with hydrogen molecules are difficult to achieve and in order to avoid H2 transfer into the cathode side, thicker membranes are typically applied, as a result of which a kinetic barrier to the transfer of unwanted species emerges.11 This, however, exacerbates issues regarding cost, environmental impact, etc. Consequently, viable alternatives are being sought.
Although PEMFCs equipped with Nafion® membranes are a mature commercial technology with fair efficiency, they have the drawback of, because of the ion transport process, requiring a highly acidic, corrosive environment as an electrolyte.12 Besides the obvious health and safety and environmental issues related to the use of low pH solutions in potential every-day technologies, the strongly acidic environment also requires the use of components that can withstand the corrosive environment, which is a particularly penalising limitation for the electrocatalyst, particularly for the anode, for which the costly and increasingly scarce Pt is employed.13 However, more earth-abundant, low-cost electrocatalysts, such as Fe or Ni, have been shown to display comparable performance to Pt, when used in an alkaline environment wherein they are sufficiently stable.14
Alkaline fuel cells
Alkaline electrolyte conditions are provided in alkaline anion-exchange membrane fuel cells (AAEMFCs), wherein the charge compensating ion transport is accomplished by the migration of hydroxide ions from the cathode side to the anode side. The electrode reactions in AAEMFCs are the same as in PEMFCs in the sense that hydrogen is oxidised on the anode while oxygen is reduced on the cathode to form water molecules (and generate electricity). The conceptual difference lies in the charge balancing ion nature and the direction of the ion transport. As seen before, in the case of PEMFCs, positively charged protons migrate from the anode side to the cathode side, in contrast, for AAEMFCs, negatively charged hydroxide ions are traversing the membrane from the cathode to the anode compartment (Fig. 1).15 This arrangement presents several advantages, including the possibility to employ cheaper electrocatalysts, the improved oxygen-reduction reaction kinetics in an alkaline medium, and the simpler balance of plant near ambient pressure, and, as the ion transport direction opposes that of the current, lower crossover and electrical drag are expected. Nonetheless, AAEMFC technology is not without its drawbacks, a major disadvantage for the widespread roll-out of the technology being the availability of hydroxide-ion exchange membranes with low cost, high ion conductivity, chemical and mechanical stability, easy processability, etc.16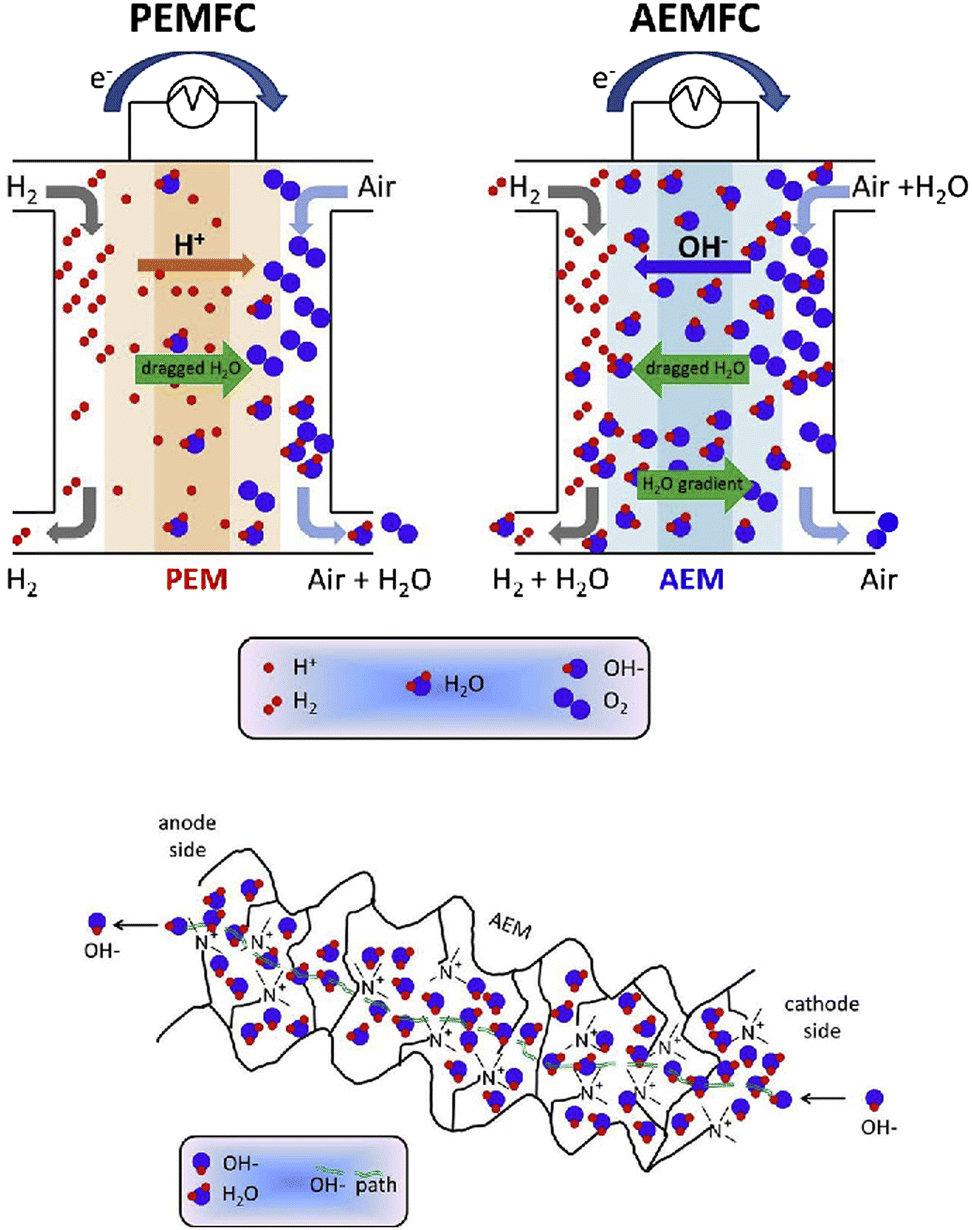 | ||
| Fig. 1 Schematic of an AEMFC as compared to a PEMFC (top scheme), and of an AEM based on a quaternary ammonium pendant functional group (bottom scheme). Reproduced with permission from ref. 15. | ||
It should be emphasised that both PEMFC and AAEFMC technologies are desirable as they enable the use of hydrogen as a fuel, contributing to a radical change in the energy landscape. Although there is a clear prospect of developing more efficient and cost-effective electrocatalysts, both technologies are limited by membranes, and to be specific by the membrane materials themselves, and thus further research efforts are needed in this direction.17,18 It is for this reason that the exploration and evaluation of other material classes is of great importance and timeliness.
Ion-exchange membranes
In order for a membrane to be viable, they should have the following characteristics: (i) high ion conductivity, protons or hydroxide ions, depending on the medium, (ii) strong chemical and mechanical robustness in the relevant medium, (iii) no crossover (or minimal), i.e. only the transfer of the desired ion occurs through the membrane and not that of any other ions or the reactant hydrogen or oxygen molecules. In particular, as previously explained, the avoidance of reactant crossover is highly important from a safety perspective, (iv) affordability and (v) low environmental footprint. When it comes to properties of the materials, these characteristics translate into (i) vacuous structure, such as porous or channelled, enabling fast ion transport, electronic insulation to avoid short-circuits, (ii) chemical inertness and morphological stability, (iii) selectivity via highly specific interactions and/or pore diameters, (iv) scalable synthesis, an abundance of precursors and ease of processing, and (v) ability to be produced under mild conditions using green chemical principles, reactants may be sourced sustainably, easily recyclable, etc. In essence, materials classes that may afford the above should be a high priority of research focus for the development of advanced membranes for fuel-cell technologies.While various material classes are being explored as ion-exchange membranes for fuel-cell applications,12,19 it is out of the scope of this review paper to evaluate the current state-of-art technology and recent progress in all classes. However, it should be noted that there are two classic material classes that are typically the focus of exploration, i.e. organic polymers and ceramics. Polymer-exchange membranes are flexible and easily processed. However, they suffer from stability issues and structural changes occurring in contact with the electrolyte, e.g. swelling, and they are typically synthesised in environmentally unsustainable approaches while they also represent significant recycling issues beyond their lifetime.20 On the other hand, with operating conditions at high temperatures, inorganic ceramic ion-exchange membranes are also investigated, which are chemically robust materials that may be produced from abundant and relatively harmless resources. However, they lack poor processability and some detrimental mechanical characteristics, such as brittleness.21
In recent years, hybrid inorganic–organic materials have emerged as potential contenders in the ion-exchange membrane field.22 Such materials are expected to intrinsically combine the desirable properties of both organic and inorganic materials, such as good mechanical flexibility and robustness, absence of swelling, chemical and structural tuneability, stability and ease of processing. This review explores advances in the design of certain hybrid materials for ion exchange purposes.
Discussion
Metal–organic frameworks
A particular class of intrinsically hybrid materials, which have been attracting tremendous research interest in applications ranging from catalysis, electrochemical energy storage, gas storage and separation, sensing, water harvesting to the removal of toxic waste is metal–organic frameworks (MOFs).23–31 These materials are composed of inorganic nodes, such as metal cations or oxidic clusters, interconnected by organic linkers, typically polycarboxylates or heterocycles, via coordination bonds. When the building blocks self-assemble, they typically form an extended crystalline porous framework (Fig. 2). Taking advantage of both organic and inorganic chemistry, the framework topologies achievable are virtually limitless, and in fact, over 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 structures have been reported in the Cambridge Crystallographic Data Centre already.32
000 structures have been reported in the Cambridge Crystallographic Data Centre already.32
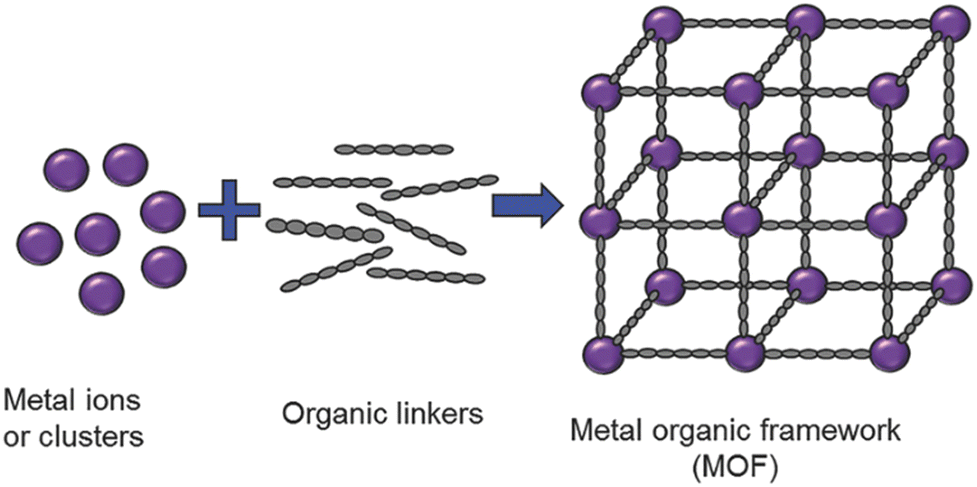 | ||
| Fig. 2 Generalized scheme for metal–organic framework preparation. Reproduced with permission from ref. 31. | ||
Importantly, MOFs have an open pore structure interlinked via channels, which are all part of the crystal structure; consequently, their geometry and chemistry may be controlled with atomic precision.33–41 This is a crucial advantage as it provides a way for engineering the interactions of loosely bonded ions with the MOF structure on the surface of the channels across the particles, which in principle should provide an unrivalled approach to control the ion transport. It is for the above reasons that metal–organic frameworks have been the subject of increasing investigations and in effect an emerging class of materials for ion-exchange membranes.42 Their ability to maximise inherent ion conductivity may be rationalised based on their structural and compositional properties: i) as a consequence of MOFs being formed on a self-assembly of inorganic and organic building blocks, in which the channels are intrinsic features of the crystal structure; they are therefore highly regular and tuneable both topologically and chemically and thus also offer a strict design-driven control over intrinsic ion mobility; ii) the intrinsically porous nature of MOFs offering open channels allows for ion conduction pathways inside the crystallite, restricting the ion transport to the43 interior to the MOF particles. Therefore, the ion transport inside macroscopic amounts of MOFs may be controlled by the careful design and crystal engineering of MOF lattices in principle. This, in combination with the unrivalled structural and compositional versatility, makes MOFs a highly desirable material class for applications relying on ion conductivity, particularly as ion-exchange membranes in fuel cells.
MOFs for ion-exchange membrane applications
In particular, for MOFs as proton-exchange membranes, there has been a tremendous advance both in terms of fundamental understanding of the underlying processes, experimental characterisation, processing, and development of materials, resulting in recorded proton conductivities exceeding 10−2 S cm−1 in operational conditions, with viable stabilities and manufacturing approaches, while a deep understanding in the conductivity mechanisms is also being developed, putting MOFs in the vanguard of cutting-edge proton conductor materials. It should be noted that recently there have been a number of excellent authoritative reviews in this field and the readers are encouraged to consult them.35,36,39,44–51 However, hydroxide-ion conductive MOFs and their membranes are a much less explored area, likely the consequence of the maturity of the PEM-FC technology over that of the AAEM-FC; therefore, it is the latter, which is the focus of the present work.Challenges
As seen, MOFs are a highly promising material class for ion-exchange membrane applications; nevertheless, some of the intrinsic challenges arising from their chemical composition and physical characteristics need to be considered and applied in the evaluation of their performance. In particular, the stability of MOFs in highly alkaline media is a limiting factor; most metal–organic frameworks feature hard acids in their nodes and therefore combine easily with hydroxide ions, resulting in framework decomposition.52 Even MOFs considered as highly stable chemically, such as UiO-66(Zr), ZIF-8 or MIL-53(Al), show signs of degradation in alkaline media.53,54 In order to construct a reliable fuel-cell device, it is indispensable that the MOF-based membrane withstand the harsh conditions, potentially amounting to 6 M KOH,55 in its pure form, or an alternative processing technique must be developedto improve the frameworks’ chemical resistance. Although ion-exchange membranes when integrated into a fuel cell are immersed in a liquid electrolyte, in order to achieve the very high ion conductivity approaching that afforded by the current proton-exchange membrane technology, high intrinsic hydroxide-ion conductivities must be achieved in the frameworks, as it will be seen, current materials offer significantly lower ion conductivities for hydroxide ions than for protons. MOFs are typically synthesised as powders, which form is not useful for membrane production. Recent advances in developing processing and fabrication technologies, such as monolith, thin film, or fibre-mat production give confidence that such challenges regarding the processing of MOFs are easily amenable to membrane formation with controlled porosity and thickness will be overcome.56–64 It should be noted that with the advent of industrial production of certain MOFs, it has been shown that their production costs can be reduced significantly, as highlighted by recent reports.65,66 However, to date there is only a handful of commercially available MOFs and their price is currently not competitive with well-established productions such as ceramics or common polymers.Sustainability issues associated with polymer materials are well known, but it is a less explored area for more recent classes of materials, such as metal–organic frameworks. As a general rule, MOFs are formed of coordinative bonds, which are easier to break than covalent ones and thus the recyclability of MOFs is not such a grave problem. In addition, recent advances in applying green chemical principles for MOF syntheses have paved the way for the emergence of sustainable MOFs, making them a more environmentally benign option so long as their component metals are captured and re-used after their lifespan.67–70
Ion transport
In addition to practical considerations, the mechanism through which hydroxide ions are transported within the ion-exchange membrane, and in particular inside the MOF channels needs to be evaluated and quantified. In a general sense, mass transport is described through the self-diffusion coefficient; in porous materials, there are three main mechanisms to mass transport the relative contribution of which depend on the conditions and the host-guest interaction strength: (i) the diffusion of molecules/ions, say when immersed in a solvent or exposed to gas pressure the molecules/ions physically located inside the pores may collide and an effective motion thus takes place down on a concentration gradient; (ii) the Knudsen diffusion, which occurs on the collision of guest molecules/ions with the pore and channel surfaces; and (iii) the surface diffusion, which consists of the adsorption and subsequent desorption of guest particles on a surface site, resulting in effective surface-hopping motions.71,72More to the point of applications in ion-exchange membranes, the transport of ions in the medium of electrolytes needs to be regarded. In the case of proton transport in aqueous solutions, the mobility of protons is exceptionally high, ca. and order of magnitude higher than that of Li+ or Na+.73 This is a direct consequence of the specific interaction of protons with water molecules. In addition, considering its practical importance, the quantitative description of the proton transport mechanism has received significant interest, and it is conventionally described using two models: (i) the vehicle mechanism, or the direct diffusion of the proton carriers (e.g. H3O+ in water), is essentially a generic diffusion model valid to any solute, while (ii) the Grotthus mechanism takes into consideration the specific interactions that exist between protons and water molecules. In particular, according to the Grotthus mechanism, the protons are allowed to jump from one molecule capable of H-bonding (H2O in this case) to a neighbouring one without the need for the significantly heavier O atom to move, resulting in an effective acceleration of the proton mobility. Thus, it is the favoured mechanism when fast ion transport is needed (Fig. 3).74,75 In fact, Nafion®, the benchmark and commercial proton-exchange membrane integrated into PEM-FCs features proton conductivity pathways according to the Grotthus mechanism, in which, similar to the mechanism in aqueous solutions, protons are exchanged on clusters of H2O and H3O+ that are supported by the membrane's sulphonic acid groups.76 Such insights paved the way to developing MOFs with high proton conductivity. While initial studies were aimed at accelerating ion carriers; today, the focus has shifted to exploring channel spaces, capable of accommodating both the proton carriers and the conduction medium, i.e. H3O+ and H2O. This approach effectively engineers proton conduction pathways from a crystallographic viewpoint.50,77
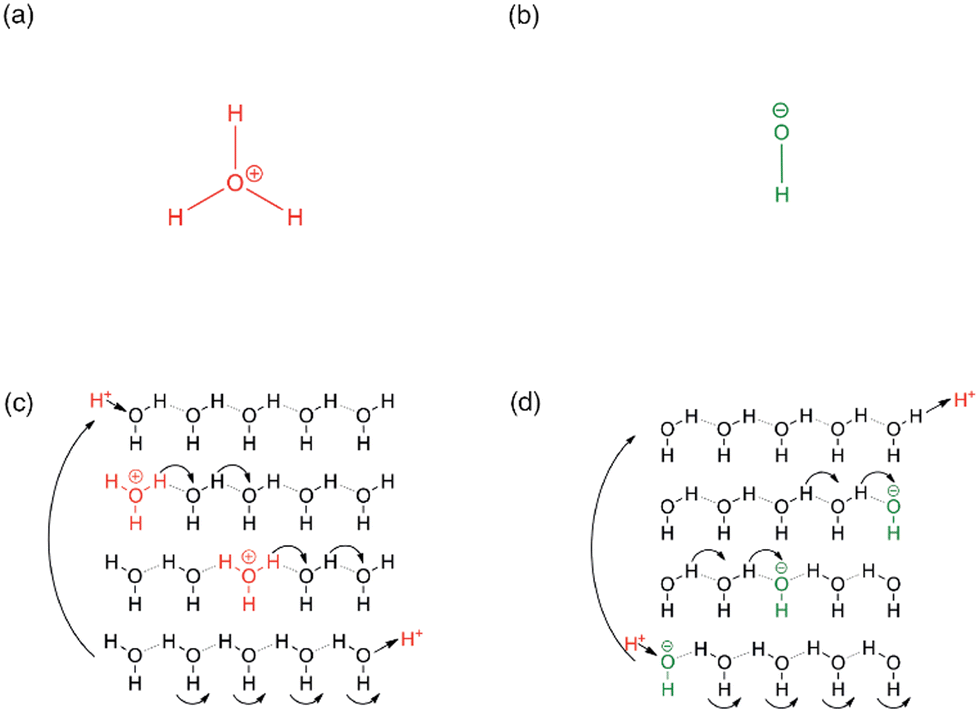 | ||
| Fig. 3 (a) Hydronium ion (b) hydroxide ion (c) hop and turn Grotthus mechanism for the conductivity of H+ as hydronium ion along a proton wire (d) Equivalent mechanism for OH− conductivity as proton holes along a proton wire. Reproduced with permission from ref. 74. | ||
The mobility of hydroxide ions remains lower than that of protons, although the mobility of OH− is significantly higher in aqueous solutions than that of other ionic solutes. It stands to reason that the exploitation of Grotthus- or Grotthus-like mechanisms should result in significantly improved hydroxide-ion conductivities in porous media, similarly to what has been observed for proton conductors (Fig. 3). In fact, theoretical calculations suggest the existence of a distinct transport mechanism, which may be viewed as an inverse proton transfer between the charge carrier OH− and the H2O conduction medium.38
Measuring hydroxide-ion transport
In the most general terms, mass transport in a particular medium is expressed through the self-diffusion coefficient. As such, mass transport in MOFs can be measured using a range of techniques; considering the large interaction cross-section of hydrogen with neutrons, neutron-based techniques in general, quasi-elastic neutron scattering (QENS) in particular is one of the most accurate and sensitive tools in determining the diffusion coefficient of hydroxide ions in a given material;78 additionally, nuclear magnetic spin resonance (NMR) techniques are well suited in directly measuring dynamic effects in proton-bearing species such as OH−, and pulsed-field gradient NMR spectroscopy is a powerful method in quantifying them.79 Furthermore, other techniques, such as interference microscopy, infrared microscopy, luminescence quenching, quartz-crystal microbalance, and confocal laser scanning microscopy, have also been shown to yield diffusion coefficients in solids.80–84Ion conductivity measurements at the same time are most typically carried out using electrochemical impedance spectroscopy, wherein a pellet, thin film, or membrane of the specimen is compressed to a known thickness and held in place between two electrodes under given conditions (temperature, electrolyte/relative humidity, and RH) under a set voltage within a particular frequency range under open circuit conditions. Effectively, the charge transference or the amount of charges passing through the specimen in the course of a certain duration is measured. Care must be taken for the piece of equipment to be in a controlled atmosphere, not only to ensure constant temperature and RH value throughout the experiment, both of which influence the OH− conductivity, but also to avoid the 2 OH− + CO2 → CO32− + H2O reaction, as the technique normally relies on measuring ‘any’ ion transport. Thus, conductivity from CO32− would also contribute to the measured value thereby burdening the experimental data with an additional yet unknown degree of error, in effect poisoning the fuel cell. Often, the Bode and Nyquist impedance plots are also acquired to apply the Nyquist semicircle method to calculate the charge transfer resistance and subsequently the conductivity.85 It should be noted that experimental challenges arise from the MOF forms, as mentioned before, MOFs are typically synthesised as powders but for conductivity measurements, a free-standing form of them must be obtained. While significant progress has been made in MOF thin-film syntheses, this approach is not always available, practical or indeed convenient to determine bulk properties for instance on account of orientation effects;61 at the same time, the precise measurement of the specimen width is crucial for accurate conductivity measurement, while the disintegration of the specimens also must be avoided, both of which are challenging with any of the MOF-forming methods.
MOF topologies in alkaline environments
As seen before, MOFs are promising materials for hydroxide ion-exchange membrane applications on account of their intrinsic electric insulating properties, inherent and tuneable ion-conductivity pathways, ability to be processed, etc.; however, major challenges include their stability in alkaline aqueous environments. In fact, electrolytes in AAEMFCs typically are made up of an aqueous KOH solution of 6–9 M in concentration, which evidently results in challenges to the stability of the exchange membrane regardless of the material classes used. In the case of metal–organic frameworks, there is limited information available as to which MOFs are stable in alkaline environments. Jiang et al. have assessed the current status and improvement strategies of the chemical stability of MOFs in a review paper, which may serve as a future inspiration and help identifying new directions in design tactics.86 Although there is only very scarce data available on the stability of MOFs in the required pH range, some frameworks have been reported at pH > 10, which may already be useful for evaluating the suitability of certain MOFs as hydroxide-ion exchange membranes for fuel cells.It should be noted that the MOFs stable in alkaline conditions are very diverse both in terms of composition and topology, comprising O- and N-donor linkers, various transition metal nodes (both, single cations as well as oxidic clusters), as well as featuring 1D channels or 3D intersected channels. Most representatives of extremely high hydroxide-ion tolerance, i.e. stability at pH ≥ 14 are however high-pKa azolate derivatives and relatively soft metals.87 These most promising frameworks include PCN-601 (Ni2TPP, H4TPP – 5,10,15,20-tetra(1H-pyrazol-4-yl)porphyrin)88 whose stability in saturated NaOH solution has been reported, ZIF-8 (ZnMeIM, MeIM – 2-methylimidazole) reportedly stable in 8 M NaOH solution at 100 °C,89 as well as ZrPP-1 (PP = pyrogallic porphyrin), whose ability to withstand 20 M NaOH for a week87 is outstanding and a highly promising prospect. In addition, PCN-602(Ni) (Ni2TPPP, H4TPPP - 5,10,15,20-tetrakis(4-(1H-pyrazol-4-yl)phenyl)porphyrin)90 and La(BTB)H2O (BTB = 1,3,5-tris(4-carboxyphenyl)benzene)91 have also been shown to be stable at pH = 14, underlining their potential for exploitation in applications at highly basic conditions. Additionally, relatively common MOF topologies of MIL-101, MIL-53, and UiO-6652 have demonstrated a degree of stability in alkaline media (pH = 12) and thus raising hopes for potential applications at high pH values.
As a more general design strategy, once again the HSAB (Hard and Soft Acid and Base Theory) principle92 may be used as a guidance, high-valent metal cations combined with hard base oxidic donor ligands lead to strong coordination bonds extending framework stability, while low-valent metals combined with soft base N-heterocycles with high pKa value favour the stability of the resultant coordination bonds, particularly in alkaline media. In addition, Jiang and co-workers have identified additional strategies to enhance the stability of MOFs in aqueous alkaline media, which include the use of several metals as a node, i.e. mixed-metal MOFs, introduction of azole-moieties in the carboxylic linkers, increasing hydrophobicity either via linker decoration with hydrophobic ligands or hydrophobic surface treatment, integration of (typically pyridinic) pillar ligands within the MOF structure, interpenetration, and compositing e.g. with carbonaceous materials or polymers.86
The above strategies fine-tune, in essence, weaken, the interactions between the framework and the medium, particularly those of hydroxide ions. Such approaches, alongside topological tuning of the framework, and, as seen, facilitating a Grotthus-like ion transport mechanism, are at the heart of controlling hydroxide-ion conductivity in a MOF and, perhaps not surprisingly, some of the very same strategies are being considered to maximise OH− transport through MOF channels.
Considering that recent advances in (i) approaches for improving MOF stability in alkaline environments;86 (ii) shaping and forming MOFs for more suitable mechanical characteristics56–64 and new ways of improving static robustness;93 (iii) more sustainable approaches to production;67–70 and (iv) lowered costs65,94 – all of which have been explored in excellent recent review papers. We identify the hydroxide-ion conductivity as the principal remaining bottleneck and therefore our continued discussion will be focussed on that.
Effecting OH− transport in MOFs
Fundamentally, all strategies of tuning hydroxide-ion mobility in MOFs rely on the engineering of the interactions between the ions and the framework itself. Kitagawa et al. have put forward a three-level classification:44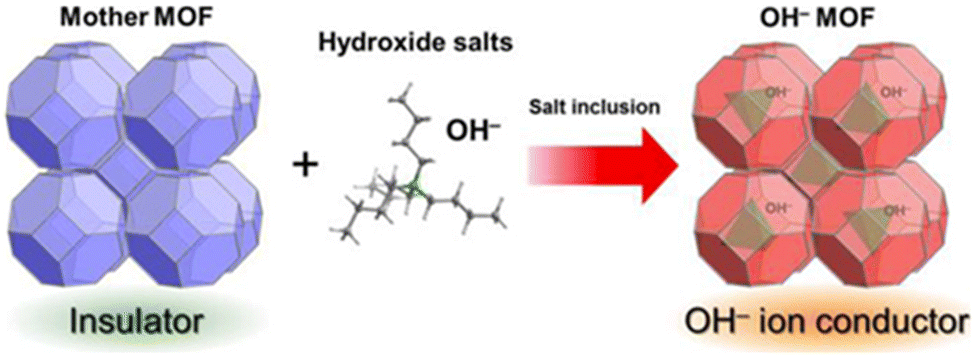 | ||
| Fig. 4 Schematic representation of the inclusion of an OH− containing salt in a MOF's pores. Reproduced with permission from ref. 38. | ||
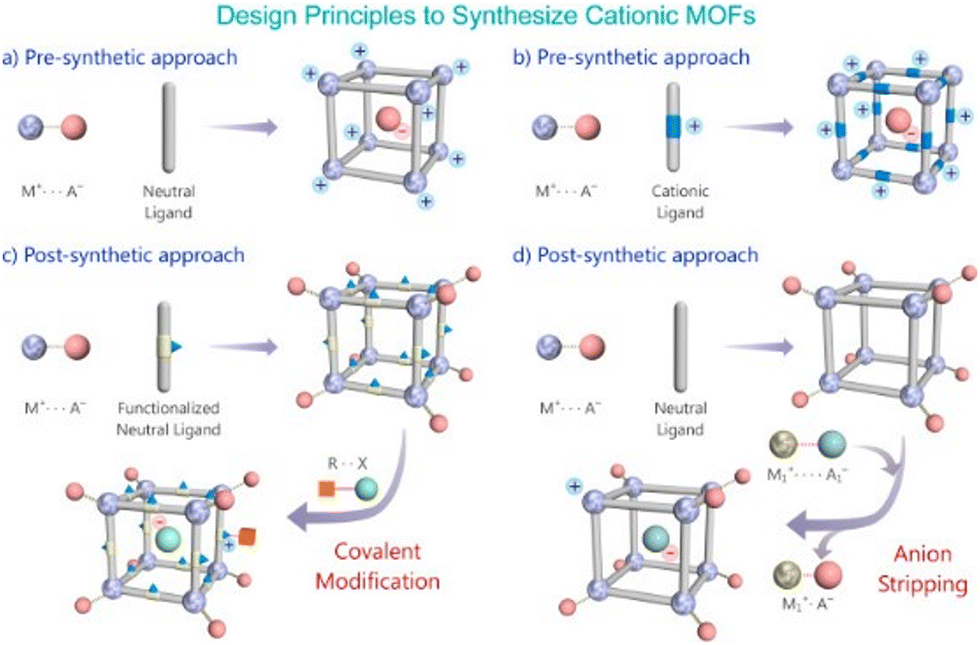 | ||
| Fig. 5 Schematic representation of routes to build a cationic MOF via pre- and post-synthetic approach. Reproduced with permission from ref. 95. | ||
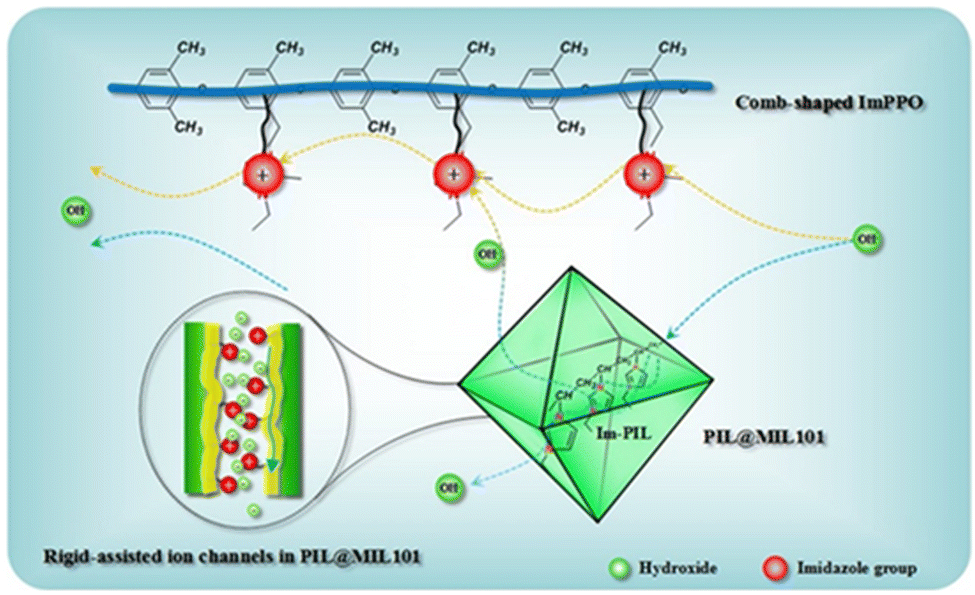 | ||
| Fig. 6 Schematic illustration of the insertion of PILs into MOF channels and of hydroxide transport along the ion nanochannels. Reproduced with permission from ref. 96. | ||
In addition, the above approaches can also be combined with each other to increase both the hydroxide-ion concentration and mobility, and therefore effectively its transference number.
Analysis of OH− conductivity data in MOFs
Table 1 summarises the hydroxide-ion conductivities in MOF-based materials measured to date at given conditions. Furthermore, it highlights which, if any, of the 3 approaches discussed above were used and whether additional specimen processing has been carried out. In addition to the main approaches to enhance ion conductivity, it should be recalled that for achieving high proton conductivity, the facilitations of the Grotthus mechanism had been successfully implemented, while there is some evidence that such a technique could also be employed for hydroxide-ion conductivity. It stands to reason to relate methodologies aimed at increasing MOF stability in alkaline media with those aimed at improving hydroxide ion conductivity on account of (i) the necessity for the MOF to be stable under the operating conditions and (ii) the fundamental analogy of engineering MOF-ion interactions.| MOF | Additive | Approach | Processing | σ (mS cm−1) | RH (%) | T (°C) | Ref. |
|---|---|---|---|---|---|---|---|
| a Samples were further processed as membranes.IL-Ionic liquid; PIL-poly-ionic liquid; PSM-post-synthetic modification; Py-2,5-pyridinedicarboxylate; Pymca-pyrimidine-2-carboxylato; BDP_X-1,4-bis(pyrazol-4-yl)benzene-4-X (X = H, OH, NH2); dcbpy-4,4′-dicarboxy-2,2′-bipyridine; Cho-choline; BMIM-1-butyl-3-methylimidazolium hydroxide; Bu4N-tetrabutylammonium; BPPO-bromomethylated poly(2,6-dimethyl-1,4-phenylene oxide); QA-N,N,N′,N′-tetramethyl-1,6-hexanediamine and allyl bromide; EVIm-1-ethyl-3-vinylimidazolium; ImPPO-imidazolated poly (2,6-dimethyl-1,4-phenylene oxide); ImPEEK-imidazolated poly (ether ketone); PVA-polyvinyl alcohol; PVP-polyvinylpyrrolidone; PVDF-polyvinylidene fluoride; PEI-polyetherimide; SEBS-hydr. styrene-ethylene/butylene-styrene triblock copolymer | |||||||
| ZIF-8 (Zn) | Cho-OH | 1-IL | n/a | 4 × 10−4 | 98 | 25 | 97 |
| 1-ILa | PVA membrane matrix | 8.4 × 10−2 | 98 | 60 | |||
| ZIF-8 (Zn) | Cho-OH | 1-ILa | IL-templated synthesis + PVA membrane | 2.6 × 10−1 | 98 | 60 | 98 |
| ZIF-8 (Zn) | BMIM-OH | 1-IL | n/a | 6.8 × 10−1 | 98 | 80 | 99 |
| BMIM-OH | 1-ILa | PVP/PVDF membrane blend | 1 | 98 | 80 | ||
| ZIF-8 (Zn) | n/a | n/a | n/a | 2.3 × 10−9 | >96 | 25 | 38 |
| Bu4N-OH | 1-IL | 2.8 × 10−5 | >96 | ||||
| Bu4N-OH + NaOH | 1-IL + salt | 6.2 × 10−4 | >96 | ||||
| ZIF-8 (Zn) | Bu4N-OH | 1-ILa | PEI membrane | 1 | 100 | 25 | 100 |
| 1.46 | 100 | 55 | |||||
| ZIF-8 (Zn) + ZIF-67 (Co) mixture | Bu4N-OH | 1-ILa | PEI membrane | 1.5 | 100 | 25 | |
| 2.7 | 100 | 55 | |||||
| ZIF-67 (Co) | Bu4N-OH | 1-ILa | PEI membrane | 1 | 100 | 25 | |
| 1.5 | 100 | 55 | |||||
| MIL-100 (Cr)–OH | n/a | 2-Anion stripping from node | n/a | 2.1 × 10−2 | 99.9 | 50 | 101 |
| NH2-MIL-101 (Cr) | n/a | 3 | N-Vinylimidazole: N-vinyl-2-pyrrolidone/divinylbenzene | 120 | — | 80 | 102 |
| Semi-interpenetrating membranes | |||||||
| MIL-101 (Cr) | Poly-N-vinylimidazolium | 3a-PIL | SEBS binder used to prepare membrane of ImPEEK | 36.6 | — | 20 | 103 |
| MIL-101 (Cr) | Poly-1-vinyl-3-ethylimidzolium | 3a-PIL | Comb-shaped ImPPO membrane | 138 | 100 | 80 | 96 |
| MIL-101 (Cr) | n/a | 2a-linker modification | (Both polymer and MOF) ImPEEK membrane matrix | 36 | 100 | 50 | 104 |
| 47 | 100 | 60 | |||||
| NH2-MIL-101 (Fe)-F | BPPO | 2a-anion stripping | PVA coating covalently linked on the two sides | 145 | 100 | 80 | 105 |
| [NH2-UiO-66 (Zr)]+Cl− | QA (PIL) | 2 & 3a-Anion stripping then PIL | Membrane formation through casting with BPPO | 123 | 100 | 80 | 106 |
| NH2-UiO-66 (Zr) w 10 eq. acetic acid of different ratios of an acetic acid modulator | n/a | n/a | Light OFF | 2 × 10−×3 | 95 | 55 | 107 |
| Light ON | ca. 2 × 10−5 | 95 | |||||
| NH2-UiO-66 (Zr)/Py-UiO-66(Zr) | n/a | 2a-Linker modification | Im-PEEK | 73 | — | 80 | 108 |
| FJU-66 (Cu) | n/a | n/a | n/a | 2.1 × 10−5 | 95 | 30 | 109 |
| [EVIm]OH | 1-IL | 57 | 95 | 30 | |||
| 91 | 95 | 85 | |||||
| 3 eq. KOH | 1-Salt | 6 | 95 | 30 | |||
| 59 | 95 | 85 | |||||
| 0.9 eq. Bu4N-OH | 1-IL | 1.99 × 10−4 | 95 | 30 | |||
| 3.44 × 10−3 | 95 | 85 | |||||
| [Ni2(μ-pymca)3]OH·nH2O | n/a | 2 | n/a | 8 × 10−1 | 99 | 27 | 110 |
| 2.5 × 10−2 | 95 | 30 | |||||
| 5.6 × 10−2 | 95 | 90 | |||||
| [Ni8(OH)3(H2O)(BDP_H)5] | n/a | n/a | n/a | 7.64 × 10−5 | 22 | 40 | 111 |
| [Ni8(OH)3(H2O)(BDP_OH)5] | n/a | n/a | n/a | 5.86 × 10−6 | 22 | 40 | |
| [Ni8(OH)3(H2O)(BDP_NH2)5] | n/a | n/a | n/a | 7.32 × 10−5 | 22 | 40 | |
| K[Ni8(OH)5(EtO)(BDP_H)5.5] | KOH | 1 & 2 | PSM-missing linker | 3.91 × 10−3 | 22 | 40 | |
| K3[Ni8(OH)3(EtO)(BDP_O)5 | KOH | 1 & 2 | PSM-missing linker | 1.82 × 10−3 | 0 | 40 | |
| 2.75 × 10−2 | 22 | 40 | |||||
| 11.6 | 100 | 40 | |||||
| K[Ni8(OH)5(EtO)(BDP_NH2)5.5] | KOH | 1 & 2 | PSM-missing linker | 3.73 × 10−5 | 0 | 40 | |
| 2.78 × 10−2 | 22 | 40 | |||||
| 1.47 | 100 | 40 | |||||
| Nd7(OH)5[Ru(dcbpy)3]4·4nH2O | n/a | 2 | n/a | 10−3–10−4 | 90 | 25 | 112 |
| Ce7(OH)5[Ru(dcbpy)3]4·4nH2O | n/a | 2 | 90 | 25 | |||
| La7(OH)5[Ru(dcbpy)3]4·4nH2O | n/a | 2 | 90 | 25 | |||
| Ln7(OH)5[Ru(dcbpy)3]4·4nH2O | n/a | 2 | ca. 10−6 | 40 | 25 | ||
First, it should be noted that the measurement conditions and sample production and processing methods are very different in most cases, and therefore, a straight-forward comparison of the results and subsequent drawing of conclusions are virtually impossible. Nevertheless, some considerations can be made, which can help identify further pathways to improving performance. In particular, the success of the various approaches and their combination applied to increasing hydroxide ion conductivity will be discussed below.
Topological and compositional remarks
From a compositional and topological perspective, one can notice a relative lack of diversity in the frameworks explored for hydroxide-ion conductivity so far. On account of the small number of MOF structures with demonstrated ability to withstand highly alkaline media, it should come as little surprise that most notable MOF topologies reported as hydroxide-ion conductive ones fall into the few more stable topologies and chemistries. Particularly, ZIF-8,89 ZIF-67,113 MIL-101,114 and UiO-66115 have been among the most studied frameworks. Interestingly, these frameworks that are not of these well-known and investigated families have compositional similarities, in the sense that they are all built up of (relatively) soft acids of Cu+,109 Ni2+110 or Ru2+112 combined with high pKa heterocycles of 2,7-bis(3,5-dimethyl) dipyrazol-1,4,5,8-naphthalene-tetracarboxydiimide, 1,4-bis(pyrazol-4-yl)benzene-4-X (X = H, OH or NH2), and 4,4′-dicarboxy-2,2′-bipyridine, respectively, which is in line with the HSAB theory.92Approaches to induce OH− conductivity and their combination
Among the hydroxide-ion conductors reported to date, the data on the conductivity of pure, i.e. non-modified or as-synthesised, MOFs are very scarce. However, some examples exist, including the conductivity of ZIF-8, NH2-UiO-66(Zr), FJU-66 and a series of Ni-bis(pyrazol)benzene MOFs. Although these values are generally low, e.g. 2.3 × 10−9 S cm−1 in the case of the fairly hydrophobic ZIF-8,38 other representatives have higher conductivities, including NH2-UiO-66(Zr) with a remarkable performance of 2 × 10−3 S cm−1 in dark conditions,107 when the formation of a hydrogen-bonded network of water molecules facilitated by the amino groups is unhindered. An interesting and somewhat contradictory effect was observed for the [Ni8(OH)4(H2O)2(BDP_X)6] (H2BDP_X = 1,4-bis(pyrazol-4-yl)benzene-4-X, X = H, OH, NH2) metal–organic frameworks.111 For these FCC MOFs, Navarro et al. observed a slight but measurable conductivity change as a function of the X ligand of the linker, intriguingly, the most hydrophobic ligand, i.e. H, displayed the highest conductivity and the worst performance was met by the most hydrophilic ligand, i.e. OH. While this observation was not explained in the manuscript, an attempt to interpret the results will be given later.The rest of the conductivities reported so far have been measured on a specimen, which has been modified according to the approaches discussed previously. Generally speaking, MOFs built up of soft acids and heterocyclic linkers have been modified with Approach 1, some of which were combined with Approach 2, or with Approach 2 only. However, MOFs built up of hard acids and carboxylate linkers have either been modified with Approach 2 or Approach 3.
The other framework topology explored for enhancing hydroxide-ion conductivity through the encapsulation of ionic species is the FJU-66. Here, Xiang and co-workers encapsulated two types of ILs, 1-ethyl-3-vinylimidazolium ([EVIm]+) hydroxide and Bu4N-OH, as well as KOH.109 They found the addition of 1 eq. [EVIm]-OH yielded the highest ion conductivity of 91 mS cm−1 at 85 °C – a stunning, >5 orders of magnitude improvement over the empty framework, whereas the addition of 3 eq. of KOH resulted in somewhat lower but still high conductivity of 59 mS cm−1. Interestingly, the addition of 0.9 eq. Bu4N-OH resulted in comparatively low performance of 3.44 × 10−3 mS cm−1 under the same conditions. The authors attempted to rationalise these differences based on IL+-MOF interactions being the strongest for [EVIm]+ on account of its sp2 C and N atoms as possible binding sites to the naphthalene diimide groups on FJU-66 via hydrogen bonding or lone pair-π interactions, whereas K+ are expected to be hydrated and thus offering a high concentration of localised H2O molecules facilitating hydroxide-ion transport.109
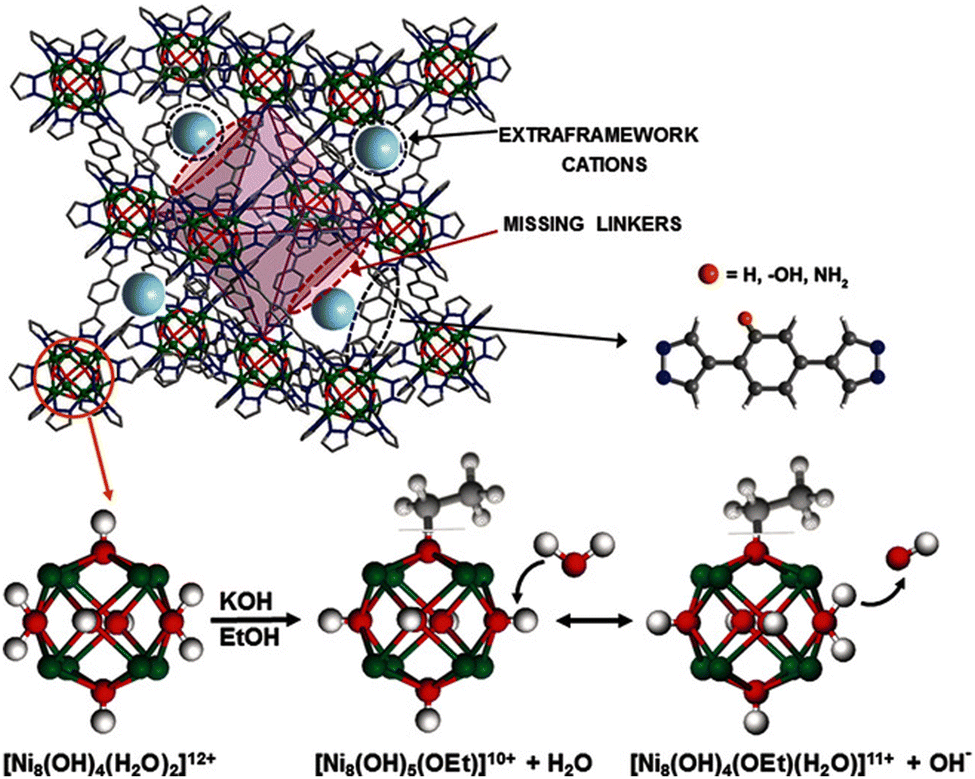 | ||
| Fig. 7 Top: Schematic representation of the structure of the K[Ni8(OH)5(EtO)(BDP_X)5.5] systems. Bottom: Cluster deprotonation taking place during the conversion into K[Ni8(OH)5(EtO)(BDP_X)5.5] materials and proposed proton transfer mechanism to explain the ion conductivity of the hydrated materials. Figure reproduced with permission from [111]. | ||
Approaches 2 and 3 have also been successfully combined by Tang et al. who prepared cationic NH2-UiO-66(Zr)-OH via anion stripping, the channels of which were subsequently impregnated with the PIL precursors allyl bromide and N,N,N′,N′-tetramethyl-1,6-hexanediamine to form the PIL materials ‘QA’ in situ, inside the pores of MOFs.106 The so-obtained QA@MOF was then further processed in the form of a membrane using brominated polyphenylene oxide. Similarly to other examples in which PILs have been introduced in the pores of MOFs, the measured OH− conductivity was very high 123 mS cm−1 at 80 °C and 100% RH, similar to PIL performance, as seen before. Once again, it appears that the MOF plays the role more of a structural element than an active contributor to ion conductivity.
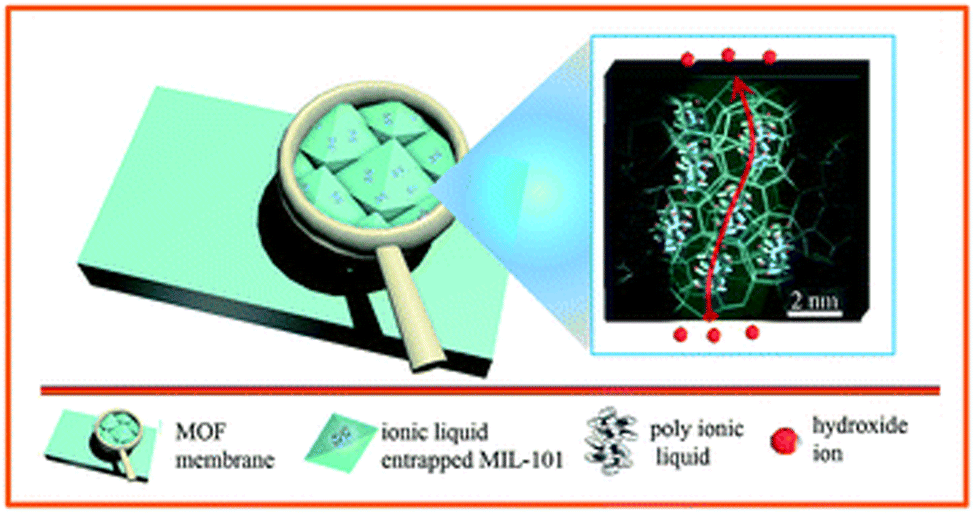 | ||
| Fig. 8 Schematic illustration of in situ assembly of a poly(ionic liquid) in metal–organic framework. Reproduced with permission from ref. 103. | ||
Conclusions and perspectives
To conclude, very high OH− conductivities have been measured for MOF-based materials prepared in various approaches. To put the conductivity values into perspective, the proton conductivity of Nafion®, which exceeds 10 mS cm−1 up to 80 °C, can be used.76 Considering that on account of its size, the transport of hydroxide ions compared with protons would be expected to be more sluggish. Therefore, one might consider σ > 10 mS cm−1 values determined under similar conditions (RH close to 100% and temperatures up to 80 °C) to be remarkable and promising ones. In fact, all three approaches for enabling hydroxide-ion conductivity, i.e. filling pores with hydroxide-ion conducting salts, formation of cationic frameworks with hydroxide counter-anions, and infiltration of hydroxide-ion conducting polymers into MOF pores, seen earlier have resulted in frameworks with high ion mobilities. Particularly spectacular results have been achieved for MOFs incorporating ion-conducting polymers in their channels; however, it appears that their conductivities are determined by the polymers themselves116 and not the MOFs, whose role is likely more one of the structural agents. While this is of course highly interesting, it should be nevertheless considered whether MOFs are the best choice of materials for use as a mechanical agent alone. This role may be fulfilled by other materials, such as porous ceramics, which may be cheaper and more robust chemically and thermally. Currently, the other two approaches, both of which increase hydroxide-ion concentrations in the MOF pores, underperform the one relying on conducting polymers. However, there are some highly promising examples, wherein the ion conductivity of these MOFs approaches that of the polymers themselves. The most promising one involved the addition of IL [EVIm]OH into the pores of FJU-66, where [EVIm]+ ions are strongly anchored in the MOF pores, presumably weakening the electrostatic interactions between the cations and the hydroxide ions, which renders them more mobility in the framework at high RH%.109 The combination of engineering cationic MOFs through missing linker effects and concurrent KOH inclusion into MOFs also led to promising hydroxide-ion conductivities, specifically at high RH%. The significance of high RH% for OH− conducting MOFs without PILs indicates that water molecules adsorbed on the MOF channels have a substantial importance. Such an effect suggests that the ion conductivity may be governed by the Grotthus mechanism, similarly to the proton conductivity. This is highly desirable because, as seen earlier, the Grotthus mechanism affords much lower activation energy (typically 2–3 kcal mol−1)75 compared with the vehicular or en masse diffusion mechanism, in which the ions hop from one binding site to another.117 What is surprising is that while for the development of proton-exchange membranes based on MOFs, significant effort has been put into nanochannel engineering for ensuring ion conductivity via the Grotthus mechanism,51 such kind of rational MOF selection and/or engineering has not yet gained a foothold in the development of hydroxide-ion conducting MOFs. Perhaps the reason for this lies in the relatively poorer stability of most MOFs in alkaline conditions when compared with acidic media. On the other hand, there is some evidence that the structural processing of MOFs, e.g. as membranes, can also accelerate hydroxide-ion motions down a potential or concentration gradient. Interestingly, this is also one of the key suggestions for enhancing the MOF stability towards harsher alkaline conditions. Once again, this approach has not been knowingly explored, and consequently significantly verified and exploited to date. It is therefore hereby suggested that this could be an important future perspective in developing MOF-based hydroxide-ion exchange membranes. Additional strategies for improving the alkaline stability of MOFs include the use of mixed-metal MOFs, the introduction of azole moieties, and affecting framework interpenetration, all of which may have the potential in accelerating hydroxide ion conductivity as they modulate OH−-MOF interaction strengths. However, it should be noted that the ability to induce ion transport via the Grotthus mechanism appears to have by far the largest effect. It is for this reason that increasing hydrophobicity, one of the most promising methods to improve MOF stability towards alkaline conditions, will likely be counterproductive, as it would result in a lowered intrapore concentration of the ion-carrier water molecules.It should be noted that the current status of hydroxide-ion conductivity in MOFs is relatively underdeveloped when compared with proton conductivity, for instance. This very well may be a direct consequence of PEMFC technology being more established than AAEMFCs, however, this relative lack of advance also comes with a less standardised way of data reporting and measurement conditions. As the ion conductivity performances depend strongly on variables such as temperature and relative humidity (or water concentration), reported results and thus performances would be more straightforward to compare if there were a standard set of conditions to carry out the experiments in.
Furthermore, in order to integrate MOF-based membranes into operational fuel cells, the crossover of gaseous molecules should be avoided. Although the polymer community regularly reports crossover data on ion-conducting polymer membranes,118 such evaluations are completely absent for MOF ion conductors. It is hereby encouraged that techniques are adopted and/or developed to enable this important assessment of MOF materials for applications as ion-exchange membranes in fuel cells.
As a final thought, metal–organic frameworks still face a number of challenges to overcome before they can be commercialised as integral parts of AAEMFCs. The three main challenges include the stability of MOFs in harsh alkaline media, their facile and scalable processing as membranes, as well as their affordability. Interestingly, one of the main conclusions of this critical review, namely the potential of the processing of MOFs via their compositing into polymer membranes could potentially simultaneously address all these challenges, therefore bringing the application of MOF-based OH−-exchange membranes closer to a commercial technology.
Author contributions
P. Á. Szilágyi conceptualised the remit of the manuscript, analysed the available literature data, wrote, reviewed, and edited the manuscript. M-M. Titirici contributed towards the conceptualisation through discussions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge funding from the Centre for Advanced Materials for Renewable Energy Generation (CAMREG #EP/P007805/1). PAS would like to acknowledge the financial support of the University of Oslo.References
- D. Bogdanov, M. Ram, A. Aghahosseini, A. Gulagi, A. S. Oyewo, M. Child, U. Caldera, K. Sadovskaia, J. Farfan, L. De Souza Noel Simas Barbosa, M. Fasihi, S. Khalili, T. Traber and C. Breyer, Energy, 2021, 227, 120467 CrossRef.
- H. Zsiborács, N. H. Baranyai, A. Vincze, L. Zentkó, Z. Birkner, K. Máté and G. Pintér, Electronics, 2019, 8, 729 CrossRef.
- C. E. Thomas, Int. J. Hydrogen Energy, 2009, 34, 6005–6020 CrossRef CAS.
- A. Onorati, R. Payri, B. Vaglieco, A. Agarwal, C. Bae, G. Bruneaux, M. Canakci, M. Gavaises, M. Günthner, C. Hasse, S. Kokjohn, S.-C. Kong, Y. Moriyoshi, R. Novella, A. Pesyridis, R. Reitz, T. Ryan, R. Wagner and H. Zhao, Int. J. Engine Res., 2022, 23, 529–540 CrossRef CAS.
- L. Fan, Z. Tu and S. H. Chan, Energy Reports, 2021, 7, 8421–8446 CrossRef.
- Z. He, M. Cui, Q. Qian, J. Zhang, H. Liu and B. Han, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 12654–12659 CrossRef CAS PubMed.
- A. Züttel, A. Remhof, A. Borgschulte and O. Friedrichs, Philos. Trans. R. Soc., A, 2010, 368, 3329–3342 CrossRef PubMed.
- M. Yue, H. Lambert, E. Pahon, R. Roche, S. Jemei and D. Hissel, Renewable Sustainable Energy Rev., 2021, 146, 111180 CrossRef.
- R. C. Thompson, C. J. Moore, F. S. vom Saal and S. H. Swan, Philos. Trans. R. Soc., B, 2009, 364, 2153–2166 CrossRef CAS PubMed.
- J.-P. Lange, ACS Sustainable Chem. Eng., 2021, 9, 15722–15738 CrossRef CAS.
- M. Schoemaker, U. Misz, P. Beckhaus and A. Heinzel, Fuel Cells, 2014, 14, 412–415 CrossRef CAS.
- S. Ahmad, T. Nawaz, A. Ali, M. F. Orhan, A. Samreen and A. M. Kannan, Int. J. Hydrogen Energy, 2022, 47, 19086–19131 CrossRef CAS.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- L. Yang, J. Shui, L. Du, Y. Shao, J. Liu, L. Dai and Z. Hu, Adv. Mater., 2019, 31, 1804799 CrossRef PubMed.
- D. R. Dekel, J. Power Sources, 2018, 375, 158–169 CrossRef CAS.
- Z. F. Pan, L. An, T. S. Zhao and Z. K. Tang, Prog. Energy Combust. Sci., 2018, 66, 141–175 CrossRef.
- V. Vijayakumar and S. Y. Nam, J. Ind. Eng. Chem., 2019, 70, 70–86 CrossRef CAS.
- A. Alaswad, A. Omran, J. R. Sodre, T. Wilberforce, G. Pignatelli, M. Dassisti, A. Baroutaji and A. G. Olabi, Energies, 2021, 14, 144 CrossRef CAS.
- M. Shabani, H. Younesi, M. Pontié, A. Rahimpour, M. Rahimnejad and A. A. Zinatizadeh, J. Cleaner Prod., 2020, 264, 121446 CrossRef CAS.
- Y. Xing, H. Li and G. Avgouropoulos, Materials, 2021, 14, 2591 CrossRef CAS PubMed.
- G. Kaur, V. Kumar, F. Baino, J. C. Mauro, G. Pickrell, I. Evans and O. Bretcanu, Mater. Sci. Eng., C, 2019, 104, 109895 CrossRef CAS PubMed.
- R. K. Nagarale, G. S. Gohil, V. K. Shahi and R. Rangarajan, Macromolecules, 2004, 37, 10023–10030 CrossRef CAS.
- C. Xu, R. Fang, R. Luque, L. Chen and Y. Li, Coord. Chem. Rev., 2019, 388, 268–292 CrossRef CAS.
- T. Jia, Y. Gu and F. Li, J. Environ. Chem. Eng., 2022, 10, 108300 CrossRef CAS.
- Ü. Anik, S. Timur and Z. Dursun, Microchim. Acta, 2019, 186, 196 CrossRef PubMed.
- J. F. Olorunyomi, S. T. Geh, R. A. Caruso and C. M. Doherty, Mater. Horiz., 2021, 8, 2387–2419 RSC.
- L. G. Gordeeva, Y. D. Tu, Q. Pan, M. L. Palash, B. B. Saha, Y. I. Aristov and R. Z. Wang, Nano Energy, 2021, 84, 105946 CrossRef CAS.
- M. J. Kalmutzki, C. S. Diercks and O. M. Yaghi, Adv. Mater., 2018, 30, 1704304 CrossRef PubMed.
- P. Á. Szilágyi and A. J. Sobrido, J. Energy Chem., 2022, 73, 348–353 CrossRef.
- S. Zhang, J. Wang, Y. Zhang, J. Ma, L. Huang, S. Yu, L. Chen, G. Song, M. Qiu and X. Wang, Environ. Pollut., 2021, 291, 118076 CrossRef CAS PubMed.
- A. S. Varela, W. Ju and P. Strasser, Adv. Energy Mater., 2018, 8, 1703614 CrossRef.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- J. L. C. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3–14 CrossRef CAS.
- Y. Ye, W. Guo, L. Wang, Z. Li, Z. Song, J. Chen, Z. Zhang, S. Xiang and B. Chen, J. Am. Chem. Soc., 2017, 139, 15604–15607 CrossRef CAS PubMed.
- A.-L. Li, Q. Gao, J. Xu and X.-H. Bu, Coord. Chem. Rev., 2017, 344, 54–82 CrossRef CAS.
- J. Li, M. Yi, L. Zhang, Z. You, X. Liu and B. Li, Nano Sel, 2022, 3, 261–279 CrossRef CAS.
- M. Sadakiyo, T. Yamada, K. Honda, H. Matsui and H. Kitagawa, J. Am. Chem. Soc., 2014, 136, 7701–7707 CrossRef CAS PubMed.
- M. Sadakiyo, H. Kasai, K. Kato, M. Takata and M. Yamauchi, J. Am. Chem. Soc., 2014, 136, 1702–1705 CrossRef CAS PubMed.
- M. Sadakiyo and H. Kitagawa, Dalton Trans., 2021, 50, 5385–5397 RSC.
- M. Sadakiyo, T. Yamada and H. Kitagawa, J. Am. Chem. Soc., 2009, 131, 9906–9907 CrossRef CAS PubMed.
- D. N. Dybtsev, V. G. Ponomareva, S. B. Aliev, A. P. Chupakhin, M. R. Gallyamov, N. K. Moroz, B. A. Kolesov, K. A. Kovalenko, E. S. Shutova and V. P. Fedin, ACS Appl. Mater. Interfaces, 2014, 6, 5161–5167 CrossRef CAS PubMed.
- G. Alberti and M. Casciola, Solid State Ionics, 2001, 145, 3–16 CrossRef CAS.
- X. Li, L. Dong, S. Li, G. Xu, J. Liu, F. Zhang, L. Lu and Y. Lan, J. Am. Chem. Soc., 2012, 134, 51–54 CrossRef PubMed.
- J.-S. M. Lee, K.-i Otake and S. Kitagawa, Coord. Chem. Rev., 2020, 421, 213447 CrossRef CAS.
- D.-W. Lim and H. Kitagawa, Chem. Rev., 2020, 120, 8416–8467 CrossRef CAS PubMed.
- K. Biradha, A. Goswami, R. Moi and S. Saha, Dalton Trans., 2021, 50, 10655–10673 RSC.
- X.-X. Xie, Y.-C. Yang, B.-H. Dou, Z.-F. Li and G. Li, Coord. Chem. Rev., 2020, 403, 213100 CrossRef CAS.
- Y. Ye, L. Gong, S. Xiang, Z. Zhang and B. Chen, Adv. Mater., 2020, 32, 1907090 CrossRef CAS PubMed.
- M. Sadakiyo, T. Yamada and H. Kitagawa, ChemPlusChem, 2016, 81, 691–701 CrossRef CAS PubMed.
- D. I. Kolokolov, D.-W. Lim and H. Kitagawa, Chem. Rec., 2020, 20, 1297–1313 CrossRef CAS PubMed.
- D.-W. Lim, M. Sadakiyo and H. Kitagawa, Chem. Sci., 2019, 10, 16–33 RSC.
- K. Leus, T. Bogaerts, J. De Decker, H. Depauw, K. Hendrickx, H. Vrielinck, V. Van Speybroeck and P. Van Der Voort, Microporous Mesoporous Mater., 2016, 226, 110–116 CrossRef CAS.
- D. Bůžek, S. Adamec, K. Lang and J. Demel, Inorg. Chem. Front., 2021, 8, 720–734 RSC.
- J. Wei, D. Zhang, L. Zhang, H. Ouyang and Z. Fu, ACS Appl. Mater. Interfaces, 2019, 11, 35597–35603 CrossRef CAS PubMed.
- J. O. Jensen, D. Aili, M. K. Hansen, Q. Li, N. J. Bjerrum and E. Christensen, ECS Trans., 2014, 64, 1175–1184 CrossRef CAS.
- Y. Dou, W. Zhang and A. Kaiser, Adv. Sci., 2020, 7, 1902590 CrossRef CAS PubMed.
- R. Ostermann, J. Cravillon, C. Weidmann, M. Wiebcke and B. M. Smarsly, Chem. Commun., 2011, 47, 442–444 RSC.
- D. Zacher, O. Shekhah, C. Wöll and R. A. Fischer, Chem. Soc. Rev., 2009, 38, 1418–1429 RSC.
- C. Crivello, S. Sevim, O. Graniel, C. Franco, S. Pané, J. Puigmartí-Luis and D. Muñoz-Rojas, Mater. Horiz., 2021, 8, 168–178 RSC.
- S. Han and C. B. Mullins, ChemSusChem, 2020, 13, 5433–5442 CrossRef CAS PubMed.
- Z.-G. Gu and J. Zhang, Coord. Chem. Rev., 2019, 378, 513–532 CrossRef CAS.
- J. Fonseca and T. Gong, Coord. Chem. Rev., 2022, 462, 214520 CrossRef CAS.
- D. G. Madden, D. O’Nolan, N. Rampal, R. Babu, C. Çamur, A. N. Al Shakhs, S.-Y. Zhang, G. A. Rance, J. Perez, N. P. Maria Casati, C. Cuadrado-Collados, D. O’Sullivan, N. P. Rice, T. Gennett, P. Parilla, S. Shulda, K. E. Hurst, V. Stavila, M. D. Allendorf, J. Silvestre-Albero, A. C. Forse, N. R. Champness, K. W. Chapman and D. Fairen-Jimenez, J. Am. Chem. Soc., 2022, 144, 13729–13739 CrossRef CAS PubMed.
- X.-M. Liu, L.-H. Xie and Y. Wu, Inorg. Chem. Front., 2020, 7, 2840–2866 RSC.
- D. DeSantis, J. A. Mason, B. D. James, C. Houchins, J. R. Long and M. Veenstra, Energy Fuels, 2017, 31, 2024–2032 CrossRef CAS.
- J. Ren, X. Dyosiba, N. M. Musyoka, H. W. Langmi, B. C. North, M. Mathe and M. S. Onyango, Int. J. Hydrogen Energy, 2016, 41, 18141–18146 CrossRef CAS.
- G. Hoyez, J. Rousseau, C. Rousseau, S. Saitzek, J. King, P. Á. Szilágyi, C. Volkringer, T. Loiseau, F. Hapiot, E. Monflier and A. Ponchel, CrystEngComm, 2021, 23, 2764–2772 RSC.
- I. Pakamorė, J. Rousseau, C. Rousseau, E. Monflier and P. Á. Szilágyi, Green Chem., 2018, 20, 5292–5298 RSC.
- M. C. Ribadeneyra, J. King, M. M. Titirici and P. Á. Szilágyi, Chem. Commun., 2022, 58, 1330–1333 RSC.
- M. Titirici, S. G. Baird, T. D. Sparks, S. M. Yang, A. Brandt-Talbot, O. Hosseinaei, D. P. Harper, R. M. Parker, S. Vignolini, L. A. Berglund, Y. Li, H.-L. Gao, L.-B. Mao, S.-H. Yu, N. Díez, G. A. Ferrero, M. Sevilla, P. Á. Szilágyi, C. J. Stubbs, J. C. Worch, Y. Huang, C. K. Luscombe, K.-Y. Lee, H. Luo, M. J. Platts, D. Tiwari, D. Kovalevskiy, D. J. Fermin, H. Au, H. Alptekin, M. Crespo-Ribadeneyra, V. P. Ting, T.-P. Fellinger, J. Barrio, O. Westhead, C. Roy, I. E. L. Stephens, S. A. Nicolae, S. C. Sarma, R. P. Oates, C.-G. Wang, Z. Li, X. J. Loh, R. J. Myers, N. Heeren, A. Grégoire, C. Périssé, X. Zhao, Y. Vodovotz, B. Earley, G. Finnveden, A. Björklund, G. D. J. Harper, A. Walton and P. A. Anderson, J. Phys.: Mater., 2022, 5, 032001 Search PubMed.
- E. G. Schlosser, Chem. Ing. Tech., 1978, 50, 244 CrossRef.
- S. Qiu, M. Xue and G. Zhu, Chem. Soc. Rev., 2014, 43, 6116–6140 RSC.
- P. Atkins, P. W. Atkins and J. de Paula, Atkins' physical chemistry, Oxford university press, 2014 Search PubMed.
- T. Miyake and M. Rolandi, J. Phys.: Condens. Matter, 2015, 28, 023001 CrossRef PubMed.
- N. Agmon, Chem. Phys. Lett., 1995, 244, 456–462 CrossRef CAS.
- H. G. Haubold, T. Vad, H. Jungbluth and P. Hiller, Electrochim. Acta, 2001, 46, 1559–1563 CrossRef CAS.
- D.-W. Lim and H. Kitagawa, Chem. Soc. Rev., 2021, 50, 6349–6368 RSC.
- N. Rosenbach Jr., H. Jobic, A. Ghoufi, F. Salles, G. Maurin, S. Bourrelly, P. L. Llewellyn, T. Devic, C. Serre and G. Férey, Angew. Chem., Int. Ed., 2008, 47, 6611–6615 CrossRef PubMed.
- F. Stallmach, S. Gröger, V. Künzel, J. Kärger, O. M. Yaghi, M. Hesse and U. Müller, Angew. Chem., Int. Ed., 2006, 45, 2123–2126 CrossRef CAS PubMed.
- J. Kärger, T. Binder, C. Chmelik, F. Hibbe, H. Krautscheid, R. Krishna and J. Weitkamp, Nat. Mater., 2014, 13, 333–343 CrossRef PubMed.
- C. Wang and W. Lin, J. Am. Chem. Soc., 2011, 133, 4232–4235 CrossRef CAS PubMed.
- O. Zybaylo, O. Shekhah, H. Wang, M. Tafipolsky, R. Schmid, D. Johannsmann and C. Wöll, Phys. Chem. Chem. Phys., 2010, 12, 8093–8098 RSC.
- W. Zhou, C. Wöll and L. Heinke, Materials, 2015, 8, 3767–3775 CrossRef CAS.
- S. Han, T. M. Hermans, P. E. Fuller, Y. Wei and B. A. Grzybowski, Angew. Chem., Int. Ed., 2012, 51, 2662–2666 CrossRef CAS PubMed.
- P. Vadhva, J. Hu, M. J. Johnson, R. Stocker, M. Braglia, D. J. L. Brett and A. J. E. Rettie, ChemElectroChem, 2021, 8, 1930–1947 CrossRef CAS.
- M. Ding, X. Cai and H.-L. Jiang, Chem. Sci., 2019, 10, 10209–10230 RSC.
- E.-X. Chen, M. Qiu, Y.-F. Zhang, Y.-S. Zhu, L.-Y. Liu, Y.-Y. Sun, X. Bu, J. Zhang and Q. Lin, Adv. Mater., 2018, 30, 1704388 CrossRef PubMed.
- K. Wang, X.-L. Lv, D. Feng, J. Li, S. Chen, J. Sun, L. Song, Y. Xie, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 914–919 CrossRef CAS PubMed.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O’Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed.
- X.-L. Lv, K. Wang, B. Wang, J. Su, X. Zou, Y. Xie, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2017, 139, 211–217 CrossRef CAS PubMed.
- J. Duan, M. Higuchi, S. Horike, M. L. Foo, K. P. Rao, Y. Inubushi, T. Fukushima and S. Kitagawa, Adv. Funct. Mater., 2013, 23, 3525–3530 CrossRef CAS.
- R. G. Pearson, J. Chem. Educ., 1968, 45, 581 CrossRef CAS.
- P. Z. Moghadam, S. M. J. Rogge, A. Li, C.-M. Chow, J. Wieme, N. Moharrami, M. Aragones-Anglada, G. Conduit, D. A. Gomez-Gualdron, V. Van Speybroeck and D. Fairen-Jimenez, Matter, 2019, 1, 219–234 CrossRef.
- X. Dyosiba, J. Ren, N. M. Musyoka, H. W. Langmi, M. Mathe and M. S. Onyango, Sustainable Mater. Technol., 2016, 10, 10–13 CrossRef CAS.
- A. Karmakar, A. V. Desai and S. K. Ghosh, Coord. Chem. Rev., 2016, 307, 313–341 CrossRef CAS.
- J. Chen, M. Guan, K. Li and S. Tang, J. Ind. Eng. Chem., 2021, 94, 465–471 CrossRef CAS.
- C. Liu, S. Feng, Z. Zhuang, D. Qi, G. Li, C. Zhao, X. Li and H. Na, Chem. Commun., 2015, 51, 12629–12632 RSC.
- C. Liu, G. Zhang, C. Zhao, X. Li, M. Li and H. Na, Chem. Commun., 2014, 50, 14121–14124 RSC.
- T. Zhang, G. Yu, X. Liang, N. Zhao, F. Zhang and F. Qu, Int. J. Hydrogen Energy, 2019, 44, 14481–14492 CrossRef CAS.
- J. Vega, A. Andrio, A. A. Lemus, L. F. del Castillo and V. Compañ, Electrochim. Acta, 2017, 258, 153–166 CrossRef CAS.
- C. Mao, R. A. Kudla, F. Zuo, X. Zhao, L. J. Mueller, X. Bu and P. Feng, J. Am. Chem. Soc., 2014, 136, 7579–7582 CrossRef CAS PubMed.
- N. Anahidzade, M. Dinari, A. Abdolmaleki, K. F. Tadavani and M. Zhiani, Energy Fuels, 2019, 33, 5749–5760 CrossRef CAS.
- Z. Li, W. Wang, Y. Chen, C. Xiong, G. He, Y. Cao, H. Wu, M. D. Guiver and Z. Jiang, J. Mater. Chem. A, 2016, 4, 2340–2348 RSC.
- X. He, M. Gang, Z. Li, G. He, Y. Yin, L. Cao, B. Zhang, H. Wu and Z. Jiang, Sci. Bull., 2017, 62, 266–276 CrossRef CAS.
- B. Wu, L. Ge, D. Yu, L. Hou, Q. Li, Z. Yang and T. Xu, J. Mater. Chem. A, 2016, 4, 14545–14549 RSC.
- K. Li, J. Chen, M. Guan and S. Tang, Int. J. Hydrogen Energy, 2020, 45, 17813–17823 CrossRef CAS.
- S. Wang, S. Fan, Z. Fang, Y. Hu, M. Dong and X. Peng, ACS Appl. Nano Mater., 2021, 4, 8352–8359 CrossRef CAS.
- J. Ren, J. Xu, M. Ju, X. Chen, P. Zhao, L. Meng, J. Lei and Z. Wang, Advanced Powder Materials, 2022, 1, 100017 CrossRef.
- Z. Li, Z. Zhang, Y. Ye, K. Cai, F. Du, H. Zeng, J. Tao, Q. Lin, Y. Zheng and S. Xiang, J. Mater. Chem. A, 2017, 5, 7816–7824 RSC.
- S. S. Nagarkar, B. Anothumakkool, A. V. Desai, M. M. Shirolkar, S. Kurungot and S. K. Ghosh, Chem. Commun., 2016, 52, 8459–8462 RSC.
- C. Montoro, P. Ocón, F. Zamora and J. A. R. Navarro, Chem. – Eur. J., 2016, 22, 1646–1651 CrossRef CAS PubMed.
- A. Watanabe, A. Kobayashi, E. Saitoh, Y. Nagao, S. Omagari, T. Nakanishi, Y. Hasegawa, W. M. Sameera, M. Yoshida and M. Kato, Inorg. Chem., 2017, 56, 3005–3013 CrossRef CAS PubMed.
- R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Science, 2008, 319, 939–943 CrossRef CAS PubMed.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- E. Abouzari-Lotf, M. V. Jacob, H. Ghassemi, M. Zakeri, M. M. Nasef, Y. Abdolahi, A. Abbasi and A. Ahmad, Sci. Rep., 2021, 11, 3764 CrossRef CAS PubMed.
- P. Ramaswamy, N. E. Wong and G. K. H. Shimizu, Chem. Soc. Rev., 2014, 43, 5913–5932 RSC.
- Q. Tang, B. Li, D. Yang, P. Ming, C. Zhang and Y. Wang, Int. J. Hydrogen Energy, 2021, 46, 22040–22061 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |