
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances on drug delivery nanoplatforms for the treatment of autoimmune inflammatory diseases
Jing
Zhu†
a,
Weihong
Chen†
b,
Yuansong
Sun†
b,
Xiaoyi
Huang
c,
Ruixi
Chu
c,
Rui
Wang
c,
Deqing
Zhou
c and
Sheng
Ye
 *a
*a
aCollege of Science & State Key Laboratory of Tea Plant Biology and Utilization, Anhui Agricultural University, Hefei, Anhui 230036, China. E-mail: sye503@ahau.edu.cn
bDepartment of Emergency Surgery, the Second Hospital of Anhui Medical University, Hefei, Anhui 230001, China
cCollege of Animal Science and Technology, Anhui Agricultural University, Hefei, Anhui 230036, China
First published on 12th September 2022
Abstract
As one of the current research hotspots, drug release nanoplatforms have great potential in the treatment of autoimmune inflammatory diseases. At present, most of the drugs used in clinic do not have tissue specificity and are difficult to play a full role in the treatment of diseases. Therefore, giving full play to the real efficacy of drugs is a very challenging task. The drug treatment effect has been significantly improved by designing and developing intelligent nanoplatforms to achieve drug targeting and controlled release, which can fundamentally change the treatment strategy of autoimmune inflammatory diseases. In this review, the principles of drug delivery through different action mechanisms of nanoplatforms are reviewed, with emphasis on the specific applications of nanoplatforms in various autoimmune inflammatory diseases. Moreover, the future development of nanoplatforms to further improve the efficacy of clinical drugs is also prospected.
1. Introduction
Autoimmune inflammation is a type of chronic inflammatory disease mediated by immunity.1–3 The incidence rate of autoimmune inflammation is increasing worldwide.4 The pathogenesis of this disease is complex, and it is not completely clear at present as to what factors, such as genetic susceptibility, imbalance of the immune system, and environment are involved.5,6 The most representative diseases are rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), ulcerative colitis (UC), and autoimmune hepatitis (AIH). Due to its variable mode of onset and prolonged course, the inflammatory focus is not limited.7 An excessive immune response leads to the damage of multiple organs and tissues, accelerates the progress of the inflammatory response, and finally causes multiple organ dysfunction.8–10 Long-term illness will greatly damage the physical and mental health of patients, and thus cause a serious economic burden.11 Therefore, higher requirements are proposed for the treatment and management of autoimmune inflammatory diseases.In the past few decades, many patients have benefited from the emergence of various new therapies and the application of new effective treatment strategies.12,13 However, the overall treatment guideline followed in clinical practice still alleviates symptoms and delays the further development of the disease. The commonly used drugs include hormones, such as glucocorticoids, immunosuppressants, and biological drugs. Glucocorticoids can often quickly reduce symptoms but cannot inhibit the development of the disease.14 Drugs that can delay the progress of the disease often need to be used for a long time. From the perspective of long-term benefits, the serious side effects and high economic burden caused by sustained drug use are still urgent problems to be solved. Compared with traditional treatments, targeted therapy is highly regarded for its specificity and advanced nature, which can treat lesions to the greatest extent, while reducing side effects.15 Therefore, it is particularly important to design elaborate drug delivery systems to achieve targeted therapy.
Drug delivery systems have attracted extensive attention due to their remarkable results in the treatment of cancer and other diseases.16–18 These systems can change the pharmacokinetics of drugs, improve the therapeutic effect and reduce drug-related side effects. A nanoscale drug delivery system can realize the controlled release of drugs in a period ranging from several hours to tens of hours.19,20 Due to the unique physical and chemical properties of nanomaterials, they can transport drugs to the targeted site for sustained release, which greatly reduces the non-targeted accidental release caused by other factors.21–24 Therefore, the design and development of promising nano drug delivery systems are of great significance for the targeted treatment of autoimmune inflammatory diseases.
2. Autoimmune inflammatory diseases
Autoimmune inflammatory diseases cause a pathological autoimmune response in the body to a variety of factors and damage and destroy the body's cells and tissue components through the excessive immune response.25,26 The long-term chronic immune-inflammatory reaction will further aggravate tissue damage and organ dysfunction.27 They are generally divided into organ-specific autoimmune diseases and systemic autoimmune diseases. These diseases usually involve muscles, joints, blood, bones, soft tissues around joints, and other systems throughout the body. The organ-specific autoimmune diseases include chronic autoimmune thyroiditis, autoimmune encephalomyelitis, autoimmune orchitis, UC, and membranous glomerulonephritis. The systemic autoimmune diseases include RA, SLE, and systemic vasculitis. Due to the existence of individual differences, even for the same disease, the clinical manifestations and prognosis of different patients are very different.28At present, the specific mechanism of this type of disease is still unclear. This may be due to the influence of external factors, the change in antigens tolerated by the body, and exposure to new antigenic determinants. For example, denatured γ-globulins acquire antigenicity by exposing new antigenic determinants, thereby inducing autoantibodies (rheumatoid factors).29 Besides, the body's regulation of autoreactive B cells is also very important. It is known that in mice, their TS cells are significantly reduced with an increase in their age. Owing to the premature reduction of TS cell function, excessive autoantibodies appear, and autoimmune diseases similar to human SLE are induced.30 In addition, some viruses may avoid the tolerance of T cells by changing the determinants of their antigens, which may also act as an adjuvant of B cells (such as EBV) to promote the formation of autoantibodies and participate in the pathogenesis of SLE.31 In general, most researchers believe that it is a disease physiologic process that destroys the body's autoimmune balance and leads to autoimmune disorder under the joint action of a variety of pathogenic factors.32,33 These pathogenic factors can be summarized as genetic factors and external environmental factors. Epidemiological data showed that the occurrence of this type of disease tends to family inheritance.34,35 The external factors include physical factors, dietary factors, pathogen infection, and drug factors.36,37 In terms of treatment, given that these diseases cannot be cured, it is necessary to adopt a long-term or even lifelong treatment program. Drug therapy is still one of the most commonly used programs at present, which can be divided into non-steroidal anti-inflammatory analgesics, glucocorticoids, immunosuppressants, targeted therapy, and immunomodulatory therapy.38–40 Long-term regular medication can only alleviate symptoms or delay the progress of the disease, and it is also accompanied by many drug side effects.41 As a second-line treatment, surgical treatment is usually decided according to the patient's condition when there are complications or disabilities caused by joint injury. In addition, there are some other treatment methods, such as plasma exchange, which have some effects, but there is still not enough evidence to support their development as a mainstream treatment.
In general, the treatment and management of these diseases should be long-term and comprehensive.42 While alleviating symptoms, they can delay the progress of the disease, and actively prevent and control the occurrence of various complications to avoid repeated recurrence and greater damage to the body. Patients should have a certain understanding of the disease and pay attention to the observation of their disease in life to avoid its recurrence and get timely treatment if there is any change.43 Moreover, these patients should pay attention to adjusting their lifestyle, giving up bad hobbies, maintaining a peaceful state of mind, and avoiding infection.44 With the continuous development of medical technology, it is believed that more treatment methods that are beneficial will become available in the future, bringing more hope to patients.
3. Principle of nano-drug delivery nanoplatform
In recent years, great progress has been achieved in overcoming the shortcomings of targeted drugs in the pharmaceutical field with the help of nanotechnology.45,46 Nano-drug carriers can selectively accumulate in diseased tissues by enhancing the trafficability on the cell membrane and the retention effect in cells, changing the distribution of drugs in vivo and improving their bioavailability to achieve a therapeutic drug concentration in lesions.47–49 Meanwhile, the controlled drug release at the lesion site is also important for the size of the carrier.50 Premature or accidental release often fails to achieve good therapeutic results.51 At present, there are three main strategies to solve this problem, i.e., passive targeting, active targeting, and stimulus-responsive nanocarriers.3.1 Passive targeted release
As one of the strategies to solve the problem of rapid metabolism of drugs, passive targeting realizes the targeted treatment of drugs mainly by enhancing the penetration and retention effect to achieve the accumulation of long-cycle nanocarriers. Unfortunately, the originally designed nanocarriers have a very short circulating half-life and are quickly cleared by monocyte macrophages in the body's innate immune system after intravenous injection, and thus it is difficult to achieve an effective drug therapeutic concentration.52 Polysaccharide or polyethylene glycol residues are connected to the surface of the carrier by modifying the surface of the improved carrier, which effectively prevents the aggregation of the carrier and its interaction with blood components.53 The modified nanocarrier significantly prolongs the circulation time and effectively increases the concentration of therapeutic drugs at the lesion site. To date, various modified nanomaterials have been widely used in the treatment of autoimmune diseases. For example, Zhang et al. developed a PLGA NP delivery system.54 Polymer carriers based on PLGA and polyethylene glycol polymer (PLGA–PEG) can solve the problems of poor oral absorption and low water solubility of natural compounds Res and BA. The advantages of PLGA make it the best carrier for Res and BA, and the simple modification of polyethylene glycol (PEG) provides additional targeting ability for NPs. In vitro experiments confirmed that the nanoparticles have sustained-release properties in vitro. HPLC analysis showed that BA and Res had high encapsulation efficiency in NP. In addition, the polymer-coated co-delivery system effectively improved the anti-inflammatory effect of a single BA or Res, and its therapeutic effect on UC was significantly improved. However, the limitations of passive targeting restrict its further development, where it is difficult to ensure the release of drugs after reaching the designated site due to its nonspecific passive targeting and there is no specific relationship between the carrier and the diseased tissue;55 consequently, researchers have proposed an active targeting strategy (Fig. 1).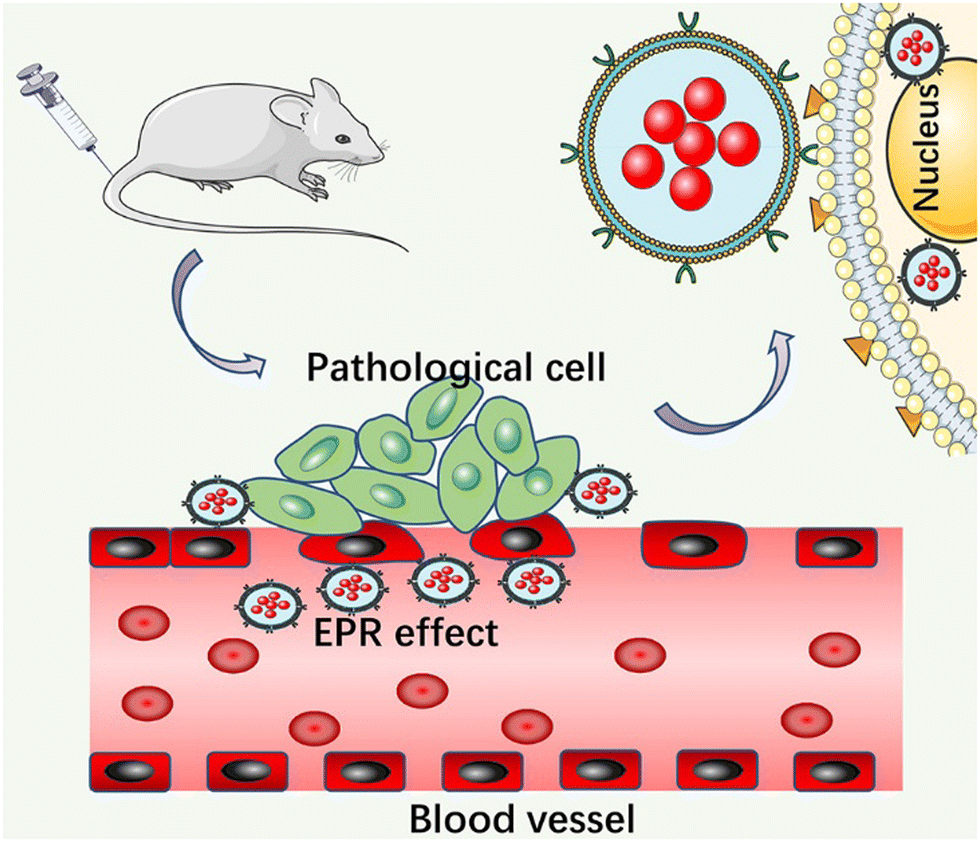 | ||
| Fig. 1 Schematic diagram of passive targeted release. | ||
3.2 Active targeted release
Active targeting is proposed to solve the problem of the low specificity of passive targeting.56 Active targeting nanocarriers were prepared by employing active ligands for selective cell binding.57 The active ligands include nucleic acids, proteins (antibodies), peptides, and small molecules, such as polysaccharide molecules and vitamins.58–60 The corresponding receptor molecules are overexpressed or only exist on specific target cells.61 Due to the unique biological interaction between receptors and ligands, nano-carriers circulating in the blood can be directionally aggregated to the lesion site to achieve the effect of drug internalization to the target tissue.62 The receptors expressed in different autoimmune diseases have some specificity with the corresponding ligands. The special factors of the disease should be fully considered in the treatment process. For example, in the treatment of SLE, correcting the impaired clearance of apoptotic cells is considered to be an ideal choice to suppress the autoimmune response and alleviate the development of SLE. Therefore, the team of Zhu et al. rationally designed and generated a nano drug carrier based on mimicking apoptotic cells, i.e., gold nanocage (Au NC), which was delivered by the liver X receptor (LXR) agonist T0901317.63 Through in vitro experiments, PS–lipos–Au NC@T0901317 could effectively enhance the clearance of apoptotic cells by increasing the expression of Mer, one of the key phagocytosis-related receptors on macrophages, thereby reducing the production of anti dsDNA autoantibodies, reducing the inflammatory response, and alleviating the kidney damage in lupus model mice. The combination of a receptor agonist and receptor is the beginning of the active targeted release of nanomaterials. Due to the specificity of receptor binding, this process can occur accurately. In addition, PS–lipos–Au NC can improve the poor solubility of hydrophobic agonists, prolong the drug release time, and reduce related adverse reactions. Thus, this team's NP-based apoptotic analog drug delivery system for the treatment of SLE provides a promising strategy for the effective treatment of SLE. It is undeniable that the therapeutic effect is significantly improved when the drug is loaded in the active targeting nanocarriers. However, to ensure the excellent performance of active targeting nanocarriers, various factors must be considered comprehensively. Appropriate ligands and coupling methods should be selected according to the differences in the pathogenesis of different autoimmune diseases to achieve the maximum therapeutic effect (Fig. 2).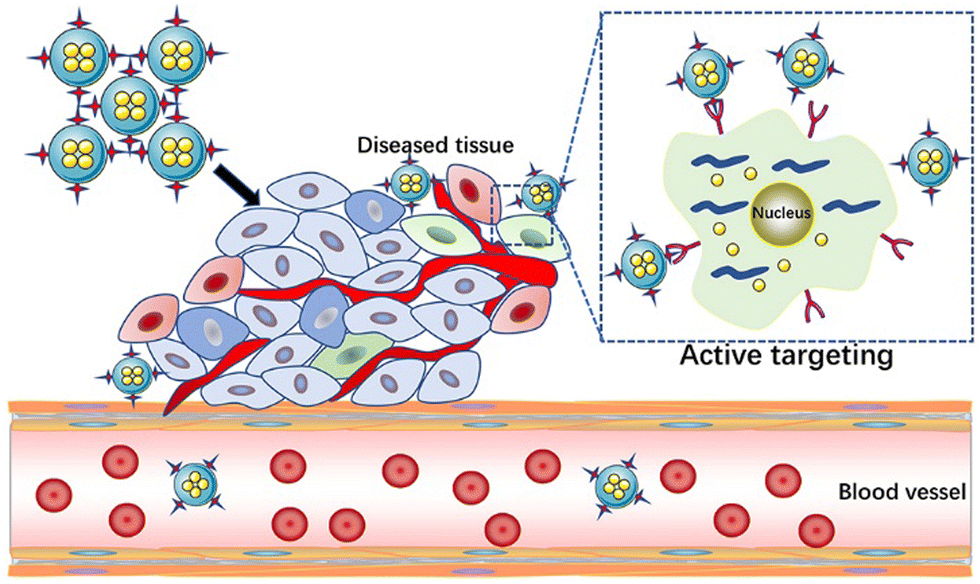 | ||
| Fig. 2 Schematic diagram of active targeted release. | ||
3.3 Stimulus-responsive release
The third strategy is to synthesize stimulus-responsive nanocarriers, including single stimulus and multiple stimulus responses. This type of nanocarrier can respond to appropriate stimuli in the surrounding environment, causing its structure change and release drugs.64,65 The common internal and external biological stimuli include biological enzymes, pH, magnetic field, redox, light, and pressure.66,67 The action mechanism of light-responsive nanocarriers is mainly carried out through a photothermal or photodynamic reaction.68,69 Near-infrared (NIR) light with a wavelength of 650–900 nm is one of the most commonly used external light sources.70 In the photothermal reaction, the nanocarrier, as a photothermal agent, absorbs the external light source and converts it into heat energy simultaneously, which induces a change in the structure of the carrier, and then stimulates the release of drugs carried in it.71,72 Due to the controllability of the external light source in time and space, the ideal controlled release of drugs can be realized after the carrier is prepared by using nanomaterials with high photothermal conversion efficiency simultaneously.73 In the photodynamic reaction, the nanocarrier acts as a photosensitizer to mediate the reaction and transfer the received light energy to the surrounding molecules through the transformation between energy states. Under the condition of sufficient oxygen, the reaction produces a large number of cytotoxic reactive oxygen species (ROS), such as singlet oxygen, hydroxyl radical, and hydrogen peroxide.74,75 While releasing the drug, the ROS molecules can cause irreversible damage to molecules such as DNA and protein, thus inducing the death of immunogenic cells and achieving the expected therapeutic effect.76,77 Compared with the previous stimulation of inducing drug release from the carrier, light-induced drug release has attracted extensive attention due to its mild reaction conditions, high biocompatibility, and easy control.There is sufficient evidence to show that multi-stimulus-response nanocarriers are more in line with the internal environmental changes of the body than single stimulus-response nanocarriers.78 The combination of pH, temperature, redox, and light stimulation induces a change in the structure, size, and chemical or physical properties of multi stimulus-responsive nano-carriers.79,80 For example, Gao et al. synthesized the metal/semiconductor composite material Au NR@CuS.81 The large octahedral void space could be used to load a large amount of drug methotrexate (MTX) for chemotherapy and modified with vasoactive intestinal peptide (VIP) and hyaluronic acid (HA) to form VIP–HA Au NR@CuS NPs targeting synovial cells in RA. Under laser irradiation, the integration of the Au NR and CuS semiconductor photocatalyst not only could generate heat but also introduce additional ˙OH into photodynamic therapy (PDT). The in vitro fluorescence signals showed that the modified nanoplatform with VIP–HA targeting moiety significantly improved the efficiency of cell internalization and improved the targeted delivery of packaged products. The synthesized nanomaterials accelerated the release of MTX under the dual stimulation of heat and ROS. Under combination therapy, VIP–HA–Au NR@CuS NPs could effectively inhibit synovial cells and the edema of CIA mice was significantly reduced. The biological metabolism of nano-carriers in vivo is also a focus of treatment. Within a few hours to days after the treatment, nanocarriers could be cleared by extravasation of the kidney and tissue, and larger ones could be phagocytized by macrophages in vivo.82,83 A drug delivery system based on nanomaterials is a novel and efficient treatment method that has a good development prospect in the treatment of autoimmune inflammatory diseases (Fig. 3).84
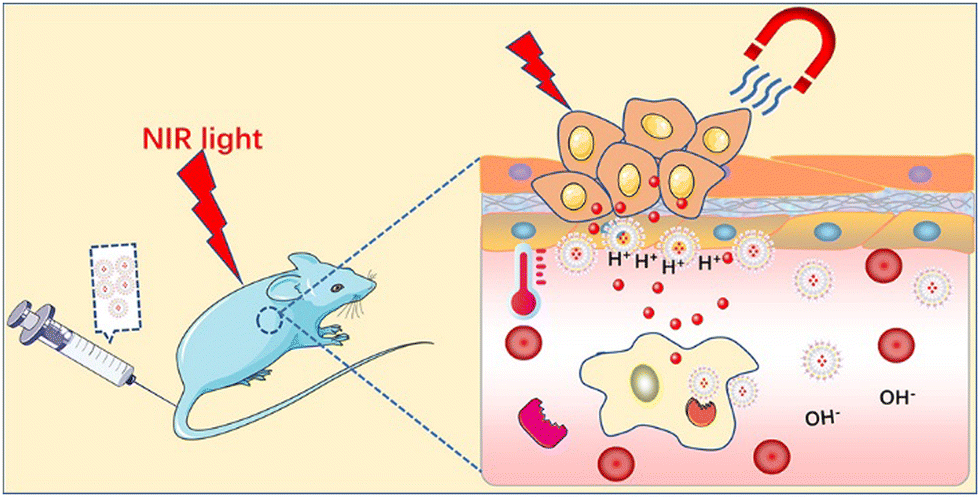 | ||
| Fig. 3 Schematic diagram of stimulus-responsive release. | ||
4. Nano-drug delivery platform for rheumatoid arthritis
RA is a common chronic autoimmune disease, which is characterized by inflammatory synovitis.85 Its feature is invasive and symmetrical arthritis of the small joints of the hands and feet, which is caused by synovial hyperplasia. Proliferative synovium secretes matrix-degrading enzymes to degrade cartilage directly, resulting in cartilage degradation and related loss of joint function.86 The etiology of RA is unknown and it is initially ignored, missing the best treatment period during its early stage of onset, which is similar to that of other inflammatory diseases. Therefore, early diagnosis is very important and can achieve effective results for the treatment of RA.87 Because of its high sensitivity and specificity, NIR-II photoacoustic (PA) molecular imaging (PMI) technology has become the latest promising method for the highly sensitive and specific diagnosis and treatment of RA. Here, Chen et al. designed an anti-rheumatism targeted drug, tocilizumab (TCZ), combined with polymer nanoparticles (PNPs), called TCZ–PNPs, to develop the first NIR-II treatment nanoplatform for the early PA-guided treatment of RA. Their work proved that TCZ–PNPs have good biocompatibility, high light stability, and a strong NIR-II extinction coefficient. The NIR-II PMI results showed that TCZ–PNPs have good targeting and could effectively and noninvasively diagnose joint tissue of RA. In addition, one month of monitoring and treatment showed that the use of TCZ PNPs could significantly inhibit RA. Thus, TCZ–PNP-assisted NIR-II PMI provides a new strategy for RA treatment and monitoring (Fig. 4).88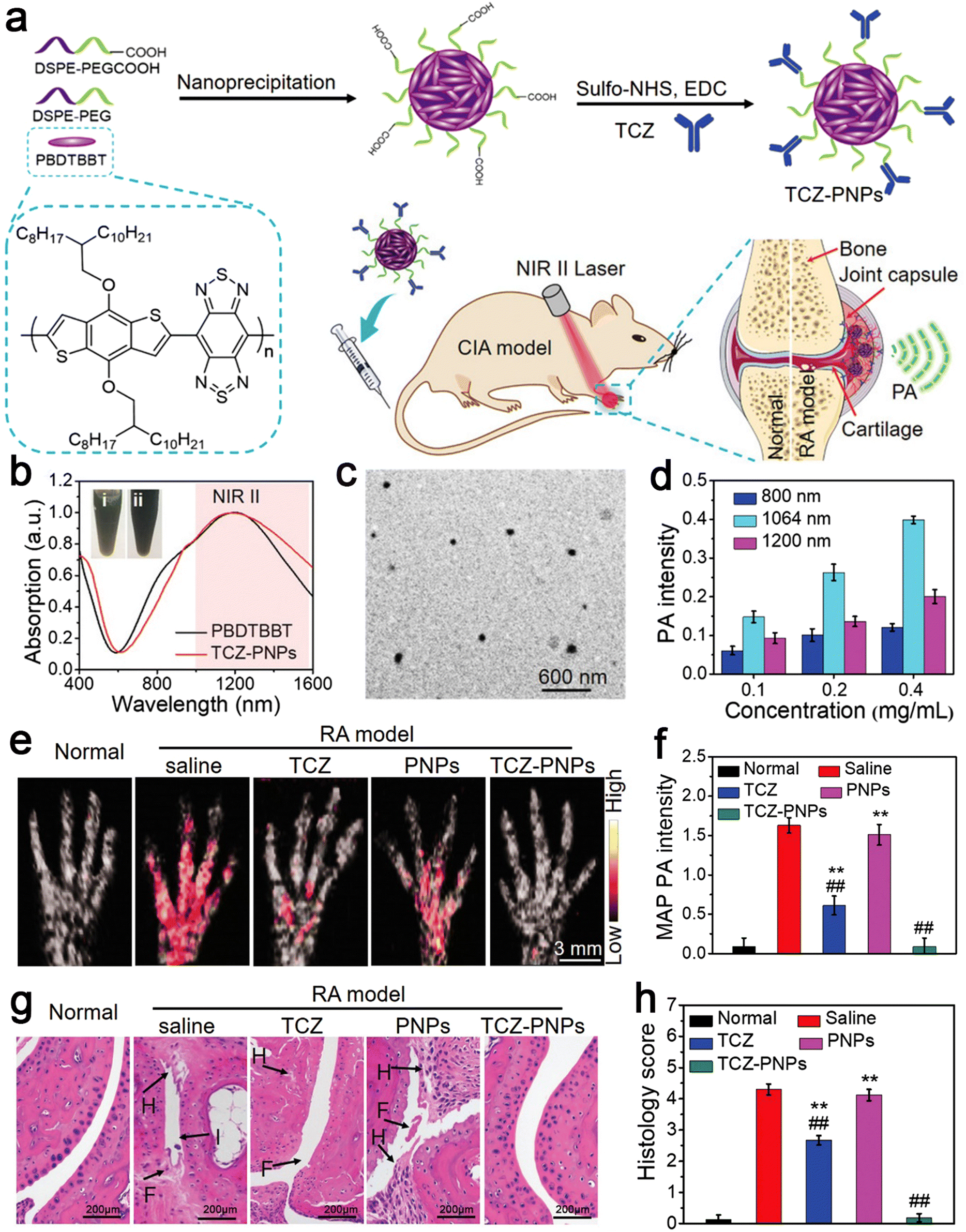 | ||
| Fig. 4 (a) Schematic illustration of TCZ–PNPs for NIR-II photoacoustic (PA) imaging and therapy of RA model mouse. (b) Normalized UV-vis-NIR absorption spectra of PBDTBBT in CHCl3 solution and the encapsulated TCZ–PNPs in water. (c) TEM images of TCZ–PNPs. (d) Statistical result of PA intensity of different concentrations of TCZ–PNPs. (e) MAP PA images. (f) Corresponding statistical data of forepaws in normal group, saline, TCZ, PNPs, and TCZ–PNPs-treated RA groups. (g) H&E staining images. (h) Histology score in the normal group, saline, TCZ, PNPs, and TCZ–PNP-treated RA groups. Adapted and reprinted with permission from ref. 85 Copyright 2020, Wiley-VCH GmbH. | ||
Besides indomethacin, non-steroidal anti-inflammatory drugs (NSAIDs) are used to treat RA and non-arthritis RA.89 However, indomethacin is known for its damaging effect on the gastrointestinal tract. A chitosan–lipid composite drug delivery system could avoid the damage to the gastrointestinal tract caused by drugs and enhance the stability of drugs. According to the current research results, microfluidic simulation seems to be the most promising method to prepare an indomethacin release system featuring the conditions of large capacity, rapid release, no serious conditions, and accurate control of the covering process, which has great potential in the clinic.90
Biological therapy, drug delivery nanoscale systems, photothermal therapy (PTT), and PDT have been used in the treatment of RA, including hydrogels, metal matrices, polymer matrices, and organic nanomaterials. However, the vast majority of nanomaterials still have various shortcomings, including low drug loading and low light conversion. It is urgent to explore new materials beyond the above-mentioned limitations. Yolk–shell nanostructures have unique morphological characteristics of a large specific surface area and low density, which have attracted much attention in the field of biomedicine.81
Huang et al. designed a metal–semiconductor composite, i.e., core–shell Au NRs@CuS nanocomposite, for the treatment of RA by PDT, PTT, and chemotherapy (Fig. 5). Au NR@CuS (100 μg mL−1) in the process of 808 nm laser irradiation exhibited an increase in temperature to about 37 °C within 5 min at the fastest rate. This experimental condition avoids the negative effects of high temperature on normal tissues. It not only could produce the heat required for treatment but also the laser irradiation without NPs will not damage normal cells. The photothermal conversion efficiency of gold calculated for Au NR@CuS under 808 nm laser irradiation was about 67.2%. ˙OH production was evaluated using methylene blue (MB). Upon irradiation at 0.5 W cm−2 for 5 min, Au NR@CuS could bleach more than 90% of MB. The semiconductor photocatalyst combines the Au NRs and CuS nanometer material with a Fenton-like reaction, which can inhibit the recombination of photogenerated electrons and holes, provide a place for photocatalytic reactions and introduce additional ˙OH photodynamic curative treatment. In addition, the big hole space in Au NR@CuS NPs can act as a nuclear power source. We can to load chemotherapeutic drugs and modify them by VIP HA Au formed by VIP and HA NR@CuS NPs for the targeted therapy of RA synovial cells. Octahedral CuS semiconductors and plasma gold nanorods were integrated in gold NR@CuS yolk–shell NPs and could effectively remove hyperplasia of synovial tissue caused by RA. Under combined treatment, VIP–HA Au NR@CuS NPs could effectively inhibit the proliferation of synovial cells in CIA mice and significantly reduce the degree of edema in CIA mice. Furthermore, in vitro and in vivo research showed that the VIP–HA Au NR@CuS NPs offer potential possibilities for the therapy of RA. MTX is one of the latest drugs for the treatment of RA. Huang et al. also confirmed that drug-loaded NPs have a perfect drug loading performance and can load the drug MTX under different and the same pH conditions to control and release the drugs.81 Guo et al. also chose metal nanomaterials as drug carriers and used a metal–organic framework (MOF) as a nanocarrier to deliver MTX with an extremely high drug loading and simple and environmental preparation method.91
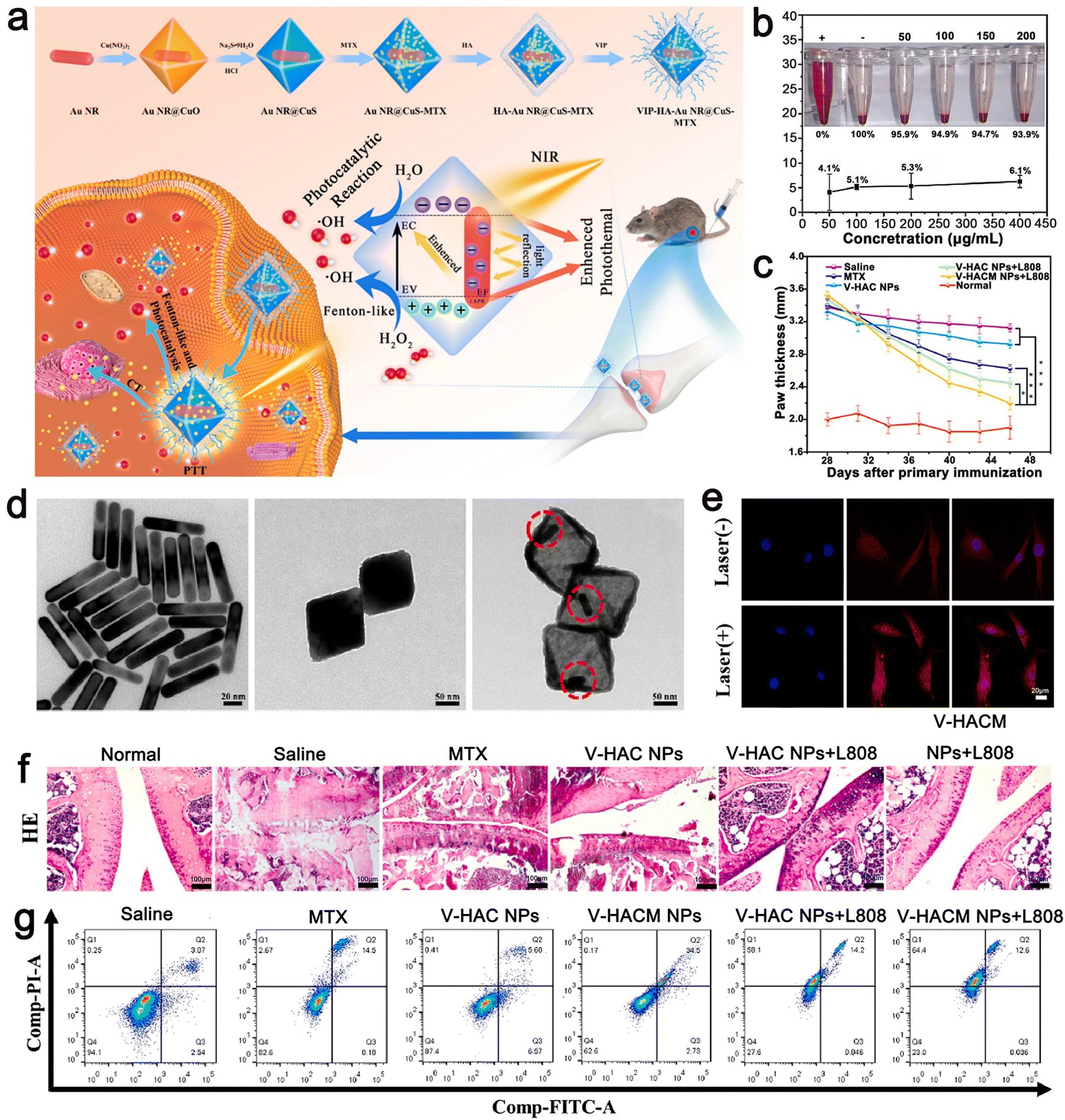 | ||
| Fig. 5 (a) Schematic illustration of the synthesis VIP–HA Au NR@CuS NPs and synergistic treatment of rheumatoid arthritis. (b) Rate of hemolysis of VIP–HA Au NR@CuS NPs in mouse red blood cells after incubation for 24 h. (c) In each group, the mean clinical index of paw thickness was determined within the times indicated. (d) TEM images of Au NR, Au NR@CuO NPs, Au NR@CuS NPs. (e) MTX release in FLS cells with or without laser irradiation. (f) H&E staining method was used to identify the histopathology of the joints in different treatment groups. (g) Flow cytometric analysis of FLS cell apoptosis/necrosis after various treatments. Adapted and reprinted with permission from ref. 81 Copyright 2021, Elsevier Ltd. | ||
Therefore, they combined MTX with tannic acid (TA) in the ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, after coordinating with the iron ion (Fe3+), reasonably designed MOFs, and then modified their surface with HA. The generated MOF achieved continuous drug release and an ultra-high drug loading (45%). The in vivo therapeutic evaluation showed that it could selectively recognize diseased cells to obtain anti-inflammatory effects (Fig. 6). The MOF could enhance the anti-rheumatic activity of MTX by targeting administration and minimizing its toxic effect. Khan et al. used polycaprolactone polyethylene glycol polycaprolactone (PCL–PEG–PCL) nano-carrier through ring-opening copolymerization as a carrier for loading MTX.92 This carrier enabled the drug to play an efficient utilization role in vitro and further reduced the side effects of the drug when its encapsulation efficiency was high. In lipopolysaccharide-activated macrophages, the PCL–PEG–PCL nano-carrier showed excellent properties of blood compatibility, very high absorption rate, and continuous release. In the optimized nano micelles, eucalyptus oil was used as a penetration enhancer for easy use on the skin. It significantly improved the permeability of the nano micelles through the skin. The MTX nanomicelles significantly improved the pharmacokinetics of the drug and its half-life was prolonged by 4.34 times more than that of the free MTX. The MTX nano micelle hydrogel significantly reduced the MTX-induced hepatotoxicity without activating the immune system.92
:1, after coordinating with the iron ion (Fe3+), reasonably designed MOFs, and then modified their surface with HA. The generated MOF achieved continuous drug release and an ultra-high drug loading (45%). The in vivo therapeutic evaluation showed that it could selectively recognize diseased cells to obtain anti-inflammatory effects (Fig. 6). The MOF could enhance the anti-rheumatic activity of MTX by targeting administration and minimizing its toxic effect. Khan et al. used polycaprolactone polyethylene glycol polycaprolactone (PCL–PEG–PCL) nano-carrier through ring-opening copolymerization as a carrier for loading MTX.92 This carrier enabled the drug to play an efficient utilization role in vitro and further reduced the side effects of the drug when its encapsulation efficiency was high. In lipopolysaccharide-activated macrophages, the PCL–PEG–PCL nano-carrier showed excellent properties of blood compatibility, very high absorption rate, and continuous release. In the optimized nano micelles, eucalyptus oil was used as a penetration enhancer for easy use on the skin. It significantly improved the permeability of the nano micelles through the skin. The MTX nanomicelles significantly improved the pharmacokinetics of the drug and its half-life was prolonged by 4.34 times more than that of the free MTX. The MTX nano micelle hydrogel significantly reduced the MTX-induced hepatotoxicity without activating the immune system.92
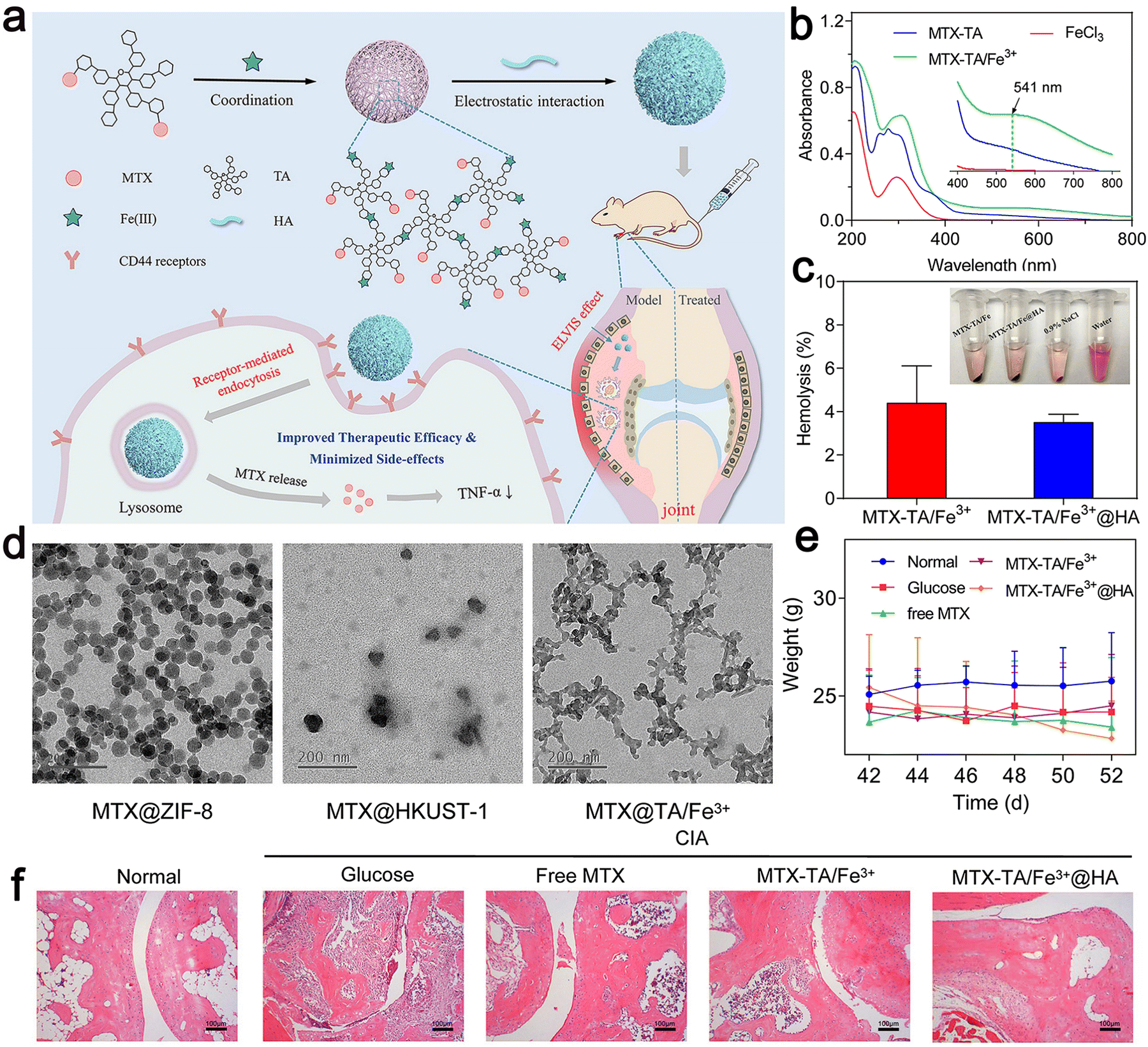 | ||
| Fig. 6 (a) Schematic illustration of the construction of MTX–TA/Fe3+@HA for targeted RA treatment. (b) UV-vis spectra of MTX–TA, FeCl3, and MTX–TA/Fe3+ MOFs. (c) Hemolysis percentage of MTX–TA/Fe3+ and MTX–TA/Fe3+@HA. (d) TEM images of ZIF-8, HKUST-1 and TA/Fe3+ MOFs. (e) Body weight of the mice after different treatments. (f) H&E staining images of the ankle joints of the mice after different treatments. Adapted and reprinted with permission from ref. 91 Copyright 2020, Elsevier B.V. | ||
Nanomaterials can avoid the hepatotoxicity caused by RA drugs. Talegonkar et al. developed HA functionalized hydroxyapatite nanoparticles (HYA-NPs) containing MTX and TEF to load and deliver MTX and TEF.93 For the % entrapment efficiency (% EE) and % drug loading (% DL), compared with HAMT NPs, it was found that the % EE and % DL of MTX and TEF were higher in HYA–HAMT NPs. The experimental results showed that the uptake and cytotoxicity of the HYA–HAMT NPs were significantly enhanced, and the radioactive percentage of HYA–HAMT NPs in the synovium area within 24 h (76.76%) significantly increased (p < 0.05). In the evaluation of hepatotoxicity, HYA–HAMT NPs reduced the hepatotoxicity to about 29.66% compared with the commercial formula.93
M1 macrophages, which secrete various inflammatory cytokines, are one of the main factors that cause RA. To alleviate synovial inflammation, it must be transformed into an anti-inflammatory M2 phenotype or directly cleared of M1 macrophages. Compared with conventional drugs acting on M1 macrophages, novel drug loading in NPs treated by this method showed less cell viability. Yang et al. developed a material that can be actively transported to M1 macrophages and synergistically induce M2 macrophage polarization and M1 macrophage reduction, which is modified by folate and silver nanoparticles (FA–Ag NPs).94 Intracellular glutathione (GSH) plays a series of key roles in anti-inflammatory, including M1 macrophage apoptosis, to promote M2 macrophage polarization. These factors contribute to the treatment of RA and the folate receptor is overexpressed on the surface of M1 macrophages. FA–Ag NPs are dissolved and sustainably release Ag+, causing GSH to exhibit a series of anti-inflammatory effects, which makes ROS clear M1 and macrophages undergo apoptosis, to promote the polarization of M2 macrophages. When activated macrophages were treated with 0.9 nm FA–Ag NPs, the percentage of apoptotic cells gradually increased with time and reached 50% after 48 h of culture. They were shown to have effective anti-inflammatory activity and play a significant therapeutic role in a high biosafety RA mouse model. The FA–Ag NPs were removed from the body mainly through feces after treatment. They did not show any obvious long-term side effects and no tissue accumulation. This experimental work used only bioactive NPs to treat RA and loaded no drugs, emphasizing the advantage of FA–Ag NPs to target the treatment of RA by simultaneous M1-to-M2 macrophage repolarization and M1 macrophage apoptosis.
The transdermal delivery of aceclofenac (ACE) hydrogel is an effective way to treat inflammatory diseases. Tyagi et al. used a nanostructured lipid carrier (NLC) based on the transdermal delivery of a high-efficiency transdermal drug delivery system (ACE–NLC) hydrogel with high stability. In addition, ACE–NLC could penetrate deeply into the skin and keep the skin completely intact.95
The multifunctional nanoplatforms designed by Li et al. are based on gold nanoparticles (Au NPs) encapsulated by 5th generation (G5) polyamine dendrimers to achieve the antioxidant co-delivery of anti-inflammatory TNF-α siRNA and tocopherol succinic acid (α-TOS) to macrophages.96 The amine-terminated G5 dendrimer macromolecule was continuously connected with 1,3-propane sulfone (1,3-PS) through a PEG spacer and PEGylated FA α-TOS was functionalized, and then encapsulated with NPs. Also, the generated Au DENPs, which were functional, showed FA-mediated targeting specificity, and the antifouling property of vitellin was compatible with double cells, enabling serum-enhanced siRNA to be transported to M1 macrophages.
5. Nano-drug delivery platform for systemic lupus erythematosus
SLE is an autoimmune connective tissue disease, which mostly occurs in young women.97 Its pathogenesis is low tolerance to autoantigens. The continuous production of pathogenic autoantibodies will cause damage to tissues and multiple organ NP-based therapies have been proven to effectively slow down the development of the SLE disease.98–100 There are some reports on nano drugs that play an important role in the pathogenesis of SLE. Here, apoptotic cells were encapsulated in a simulated Au NC by coupling phosphatidylserine (PS) to the surface of liposomes. Then, the LXR agonist T0901317 was used as the carrier. Au NC was reasonably designed and prepared, which could correct the damage of apoptotic cell clearance in SLE. Firstly, uniform Au NC with a size of 69 ± 5 nm were prepared via the current displacement process between HAuCl4 and a sacrificial template such as Ag NC. Au NC and T0901317 were incubated together and the solvent was removed from Au NC@T0901317 by vacuum freeze-drying. To improve the biocompatibility of Au NC, liposomes with high mobility and interfacial tension were coated on them based on the interaction between the liposomes and Au NC. Here, 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) was chosen to form liposomes together with 1,2-distearoyl-sn-glycero-3-[phospho-l-serine] (sodium salt) (DSPS) and 27-hydroxycholesterol because DSPS can be recognized and bind to PS receptors on macrophages, whereas 27-hydroxycholesterol constitutes an endogenous ligand for LXR. As shown in Fig. 7a, liposomes with a thickness of 4 nm were successfully coated on Au NC@T0901317 and formed relatively stable PS–lipos–Au NC@T0901317. Mer is a tyrosine kinase of phagocytosis-related receptors. It plays a key role in phagocytosis and the clearance of apoptotic cells. In SLE, it is downregulated on macrophages, leading to an autoimmune response to autoantigens and developing into chronic organ damage and inflammation. Their findings demonstrated that the PS–lipos–Au NC@T0901317 NP-based drug delivery systems that mimic apoptotic cells could specifically target macrophages and significantly enhance the phagocytic clearance of apoptotic cells by increasing Mer expression on bone marrow-derived macrophages (BMDMs) and splenic macrophages. Phagocytosis of these NPs was further assessed using confocal laser scanning microscopy (CLSM), which was consistent with the results of flow cytometry. Simultaneously, they also demonstrated that the phagocytosis of PS liposome Au NC was largely dependent on PS receptors, such as Mer. Given that BMDMs show similar biological characteristics to normal macrophages, they used BMDMs in this study to investigate whether lipos–Au NCs and PS–lipos–Au NCs were toxic to normal macrophages. After incubation with lips–Au NCs and PS–lips Au NCs for 24 and 48 h, the cell viability of BMDMs was determined by CCK8. The viable BMDMs remained at levels of 74.2% and 74.4% even at the concentration of 10 μmol L−1 in the lipos–Au NC and PS–lipos–Au NC treated groups, respectively, after co-incubation for 24 h. Therefore, it effectively slowed down the progression of SLE in mice, helped to reduce the production of anti dsDNA autoantibodies, reduced systemic inflammation, and alleviated kidney damage. Thus, drug delivery systems based on apoptotic-mimicking NP provide a promising strategy for SLE treatment by addressing the key pathogenesis of this refractory disease (Fig. 7).63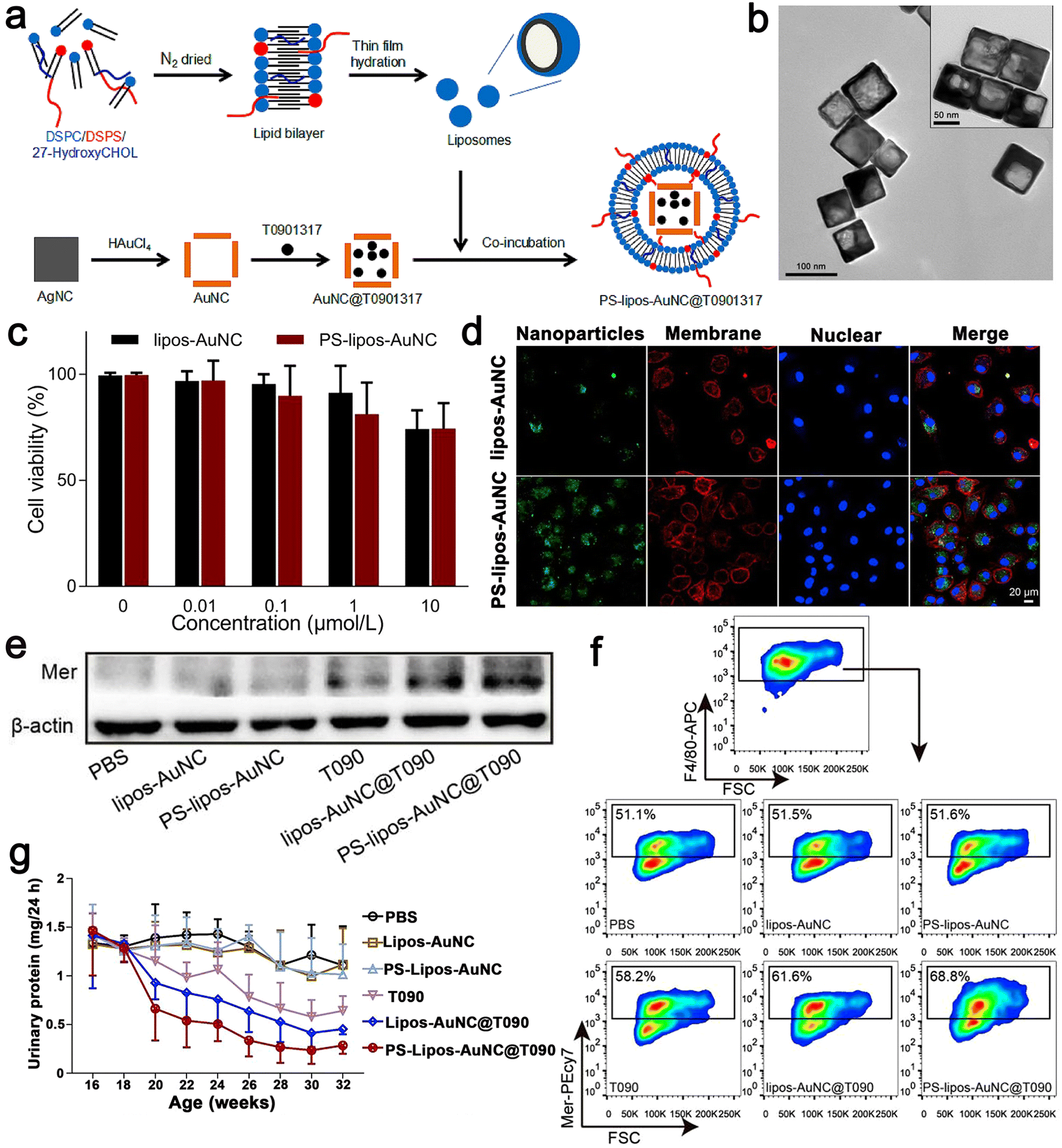 | ||
| Fig. 7 (a) Schematic illustration showing the preparation procedure of T0901317-loaded PS–lipos–Au NCs. (b) Transmission electron microscopy images of Au NCs and PS–lipos–Au NCs (upper right corner). (c) Cell viabilities of BMDMs incubated with different concentrations of lipos–Au NCs and PS–lipos–Au NCs for 24 h. (d) Confocal laser scanning microscopy images of the uptake of FITC labeled lipos–Au NCs (upper) and PS–lipos–Au NCs (lower) by BMDMs after co-incubation for 2 h. (e) Expression level of Mer protein in bone marrow-derived macrophages (BMDMs) of lupus mice. (f) Expression level of Mer on the surface of BMDMs from lupus mice. (g) 24 h urinary protein excretion levels of lupus mice injected twice weekly from 16 to 32 weeks of age. Adapted and reprinted with permission from ref. 63 Copyright 2020, Elsevier Ltd. | ||
Scientific research has presented new methods for the fabrication of nano-drug delivery systems. A monomethoxy(polyethylene glycol)–poly(D,L-lactide-co-glycolide)–poly(L-lysine) (mPEG–PLGA–PLL) nano-delivery system was constructed and used to deliver microRNA-125a (miR-125a) to splenic T cells and participate in the treatment of SLE. Their experiments proved that the miR-125a-loaded mPEG–PLGA–PLL (PEALmiR-125a) NPs could protect miR-125a from degradation and had good biocompatibility to prolong the circulation time of miRNA in vivo. Moreover, PEALmiR-125a NPs preferentially aggregated in the pathological spleen organs of SLE and effectively transported miR-125a to spleen T cells. To explore whether peal or pealmir-125a NPs have toxic effects on T cells, dexamethasone (Dex) was used as a positive control, which could directly inhibit T cell proliferation and induce T cell apoptosis (Fig. 8b). The experimental data showed that peal and PEALmiR-125a NPs had negligible effects on T cell proliferation and apoptosis. On the contrary, Dex inhibited T cell proliferation and induced apoptosis. The PEALmiR-125a NPs significantly inhibited the progress of SLE. Compared with the same dose of free miR-125a in the mouse model after four weeks of systemic medication, the PEALmiR-125a NP nano-drug delivery system wonderfully improved the treatment effect of miR-125a in vivo and had good safety.101 In addition, studies have shown that PEALmiR-125a NPs offer a treatment strategy for SLE. Thus, this formula may also be more widely used in other immune diseases (Fig. 8).
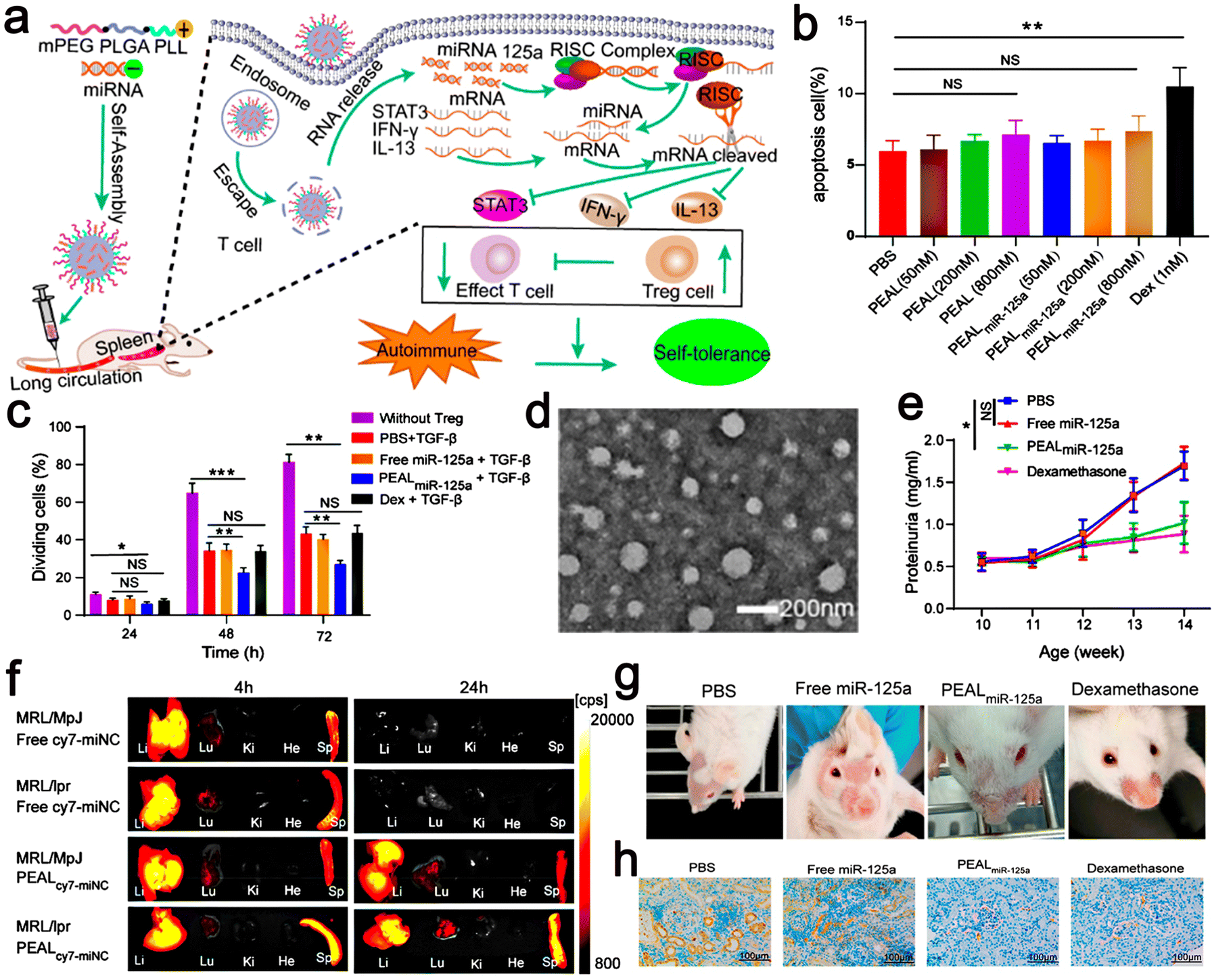 | ||
| Fig. 8 (a) Schematic illustration of microRNA-125a-loaded polymeric nanoparticles alleviate systemic lupus erythematosus. (b) Splenic T cells were incubated with Dex or different concentrations of PEAL or PEALmiR-125a for 24 h. Then, the percentage of apoptotic T cells was analyzed by flow cytometry. (c) Tregs were induced by PBS, free miR-125a, PEALmiR-125a, or Dex, plus TGF-β for 72 h. (d) TEM images of PEALmiNC NPs. (e) Proteinuria was measured at different intervals. (f) Representative fluorescence images of organs from MRL/MpJ mice and MRL/lpr mice. (g) Skin lesions from SLE mice with different treatments. (h) IHC images of kidney sections for renal deposition of anti-C3. Adapted and reprinted with permission from ref. 101 Copyright 2020, the American Chemical Society. | ||
Exosomes are extracellular vesicles (EV) encapsulated by nano lipid bilayers. Because of their inherent ability to shuttle lipids, proteins, and genes between cells and their natural affinity with target cells, they have attracted increasing attention. Their inherent characteristics, such as biocompatibility, stability, low immunogenicity, and the ability to overcome biological barriers, have prompted researchers to be interested in using exons as drug carriers. Especially in gene therapy, exons play a significant role in immune tolerance, stimulating and regulating immune signals and inflammation. Thus far, exon-based nanocarriers have been developed to treat common autoimmune diseases. Various evidence showed that the existence of EV-specific patterns and their products plays an important physiological and pathological role in SLE.102 Thus, EV have great potential for the diagnosis, prediction, prognosis, and targeted treatment of SLE.
Glucocorticoids can be used to treat SLE, but their adverse reactions offset their therapeutic advantages. A new nano hydrogel for the continuous administration of Dex was designed to ameliorate the duration and reduce the dosage of the treatment. Hydrogel is a soft material composed of water swelling cross-linked polymers. The insertion of a cyclodextrin (CD) moiety increases the location of the hydrophobic drug complex. Polyamine amine (PAA) is a biocompatible and biodegradable polymer, and it is easy to generate CD in hydrogels. In vitro experiments revealed that upon pretreatment with Dex for 24–48 h under simulated inflammatory conditions, the β-CD/PAA nanoscale gel inhibited the adhesion of Jurkat cells to human umbilical vein endothelial cells (HUVEC). The results of in vitro experiments showed that compared with the free drugs, the CD/PAA nano Dex preparation was more effective and showed an effect faster at lower doses. In addition, the β-CD/PAA nanogel inhibited COX-2 expression in Jurkat cells induced by phorbol 12-myristate 13-acetate (PMA) + A23187. The nanogel showed an effect in Jurkat cells in the concentration range of 10−8–10−5 M within 10 weeks, while Dex was only effective at 10−5 M after 48 h of treatment. Therefore, the new nanogel Dex preparation combined with faster action and a lower dose indicates that it may be easier to manage than free drugs and reduce their side effects. The morphology and physicochemical properties of the Dex gel are suitable as sustained release ophthalmic drug delivery systems. Therefore, for eye diseases caused by SLE, Dex in CD/PAA gel represents a potential new strategy for preparing eye drops, which can make up for the shortcomings of drugs (Table 1).103
| Diseases | Nanomaterials | Average size (nm) | Drugs | Ref. |
|---|---|---|---|---|
| Rheumatoid arthritis | VIP–HA–Au NR@CuS NPs | Length of 98.9 and width of 22.7 | MTX | 81 |
| Rheumatoid arthritis | TCZ–PNPs | 190 ± 5.6 | TCZ | 88 |
| Rheumatoid arthritis | MOFs | 50.0 | MTX | 91 |
| Rheumatoid arthritis | PCL–PEG–PCL | 31.0 | MTX | 92 |
| Rheumatoid arthritis | FA–Ag NPs | 0.8–0.9 | MTX | 93 |
| Rheumatoid arthritis | HYA–HAMT-NPs | 274.9 ± 64 | MTX/TEF | 94 |
| Rheumatoid arthritis | Au DENPs | 2.1 | α-TOS | 96 |
| Systemic lupus erythematosus | PS–lipos–Au NC | 69.0 ± 5.0 | T0901317 | 63 |
| Systemic lupus erythematosus | mPEG–PLGA–PLL | 118.9 ± 1.45 | miR-125a | 101 |
| Systemic lupus erythematosus | β-CD/PAA | 314 ± 3.5 | Dex | 103 |
6. Nano-drug delivery platform for ulcerative colitis
UC is a chronic nonspecific inflammatory affecting the rectum and colon.104,105 The injury of the intestinal epithelium is due to the inflammatory reaction of lumen microbiota caused by the activation of the immune system and the production of cytokines. UC can lead to a variety of complications, such as abdominal pain, diarrhea, and rectal bleeding. If the patient does not get treatment in time, UC may further lead to colorectal cancer.106ROS are a pool of chemically active oxygen-containing molecules produced by the normal metabolism of oxygen. Although low levels of endogenous ROS are required to maintain normal cellular function by regulating oxygen homeostasis and cellular signaling, their overproduction is closely related to the pathogenesis and development of inflammatory diseases. Both experimental and clinical evidence indicate that ROS overproduced by infiltrating inflammatory cells in the intestinal mucosa may amplify the inflammatory response, trigger mucosal damage, and accelerate mucosal ulceration in the pathogenesis of UC.107 The traditional and current popular treatment options for UC include amino salicylates, immunosuppressants, antibiotics corticosteroids, and biologics. Treatment with antioxidants or free radical scavengers may also reduce colitis. However, due to various reasons, only limited success has been achieved thus far. Recently, nanomedical methods have been proven to have great potential in the diagnosis, prevention, and treatment of various diseases. For the treatment of gastrointestinal diseases, NPs also show special advantages, such as protecting payloads from instability or hydrolysis, improving bioavailability, and increasing drug release/retention at disease sites.108 Thus, to overcome these limitations and to solve these problems of UC, researchers have developed a drug delivery system based on nanotechnology. Cyclooxygenase-1 (COX-1), which helps reduce inflammation, fever, and pain, and reduces side effects, is closely related to COX-1 inhibition. Celecoxib (CLX) is an NSAID, which cannot affect COX-1 and inhibit cyclooxygenase-2 (COX-2). CLX treatment may have an effective therapeutic effect on UC and reduce the side effects of traditional NSAIDs. However, although CLX has many advantages, it still has some shortcomings, such as low oral bioavailability, large distribution volume, and poor water solubility. Thus, to solve these problems, Khan et al. developed NLC to encapsulate CLX. In nano lipid formulations, a colloidal lipid carrier containing solid lipids with an average particle size of 100–1000 nm, dispersed in water or aqueous surfactant, is a solid lipid nanoparticle (SLN). SLN has the advantage of protecting active ingredients and regulating the release of drugs. However, there are only solid lipids in traditional SLN, and these solid lipids have some limitations, such as limited drug loading function, poor stability, and low release rate during storage. Alternatively, NLCs have storage capacity for numerous drugs with very few ordered lipid matrix defects. Thus, it is an effective solution to these problems including low oral bioavailability, large distribution volume, and poor water solubility because it is easy to produce and contains no organic matter. The aim of one study was the preparation of CLX-loaded Eudragit S100 (EUD S100)-coated NLC to treat colitis in Swiss albino mice induced by dextran sodium sulfate (DSS). NLC was prepared with glycerol monostearate (GMS) as a solid lipid, geraniol as a liquid lipid, and PF-127 as a water-soluble emulsifier. Besides, the main requirement of colon-targeted preparation is to resist erosion in the acidic environment of the stomach and deliver it to the colon successfully. After the CLX–NLCs were formulated, they were coated with gastric-resistant colon-specific polymer EUD S100 to ensure that the CLX is protected from the acidic stomach environment. Simultaneously, NLC loaded with CLX could be specially delivered to the colon. In this study, they successfully developed a novel celecoxib drug biocompatible nano formulation with colon-specific properties. Notably, the nanocarriers were developed using generally recognized as safe (GRAS) compounds approved by the Food and Drug Administration (FDA), which are non-toxic, available to the human gut, and cost-effective, and thus the nanocarriers are reliably safe. Their study also demonstrated that EUD S100-coated CLX-loaded NLC is a convenient nanoparticle that can adequately target colonic tissue. NLC has good properties, showing the sustained release of CLX in physiological buffer solutions, and is compatible with normal cells. EUD S100-coated CLX-loaded NLC can play an effective role in the treatment of DSS-induced inflammation, which has a good prospect in the treatment of UC.106
Mice treated with CLX alone and treated with EUD S100-coated CLX-loaded NLC resulted in significant weight gain compared with the DSS group, and the length of the colon was observably shortened with the DSS treatment. However, EUD S100-coated CLX-loaded NLC restored the length of the colon. Meanwhile, NLC cured with EUD S100 coated with CLX could protect the colon by promoting sulphomucin secretion and its conversion to sialobacilin.106 Khan et al. used EUD S100 to coat CLX to protect drugs from the harsh stomach environment, while they also used EUD S100 to coat NPs to bypass the worn stomach environment.
The aim of Khan et al. was to prepare an oral drug delivery system that bypasses the hostile gastric acid environment and can locally target the inflammatory colonic epithelium by preparing 5-aminosalicylic acid (5-ASA) gelatin NPs and wrapping these NPs on the intestinal coating polymer EUD S100. Simultaneously, 5-ASA can produce and promote an anti-inflammatory response. In this study, EUD S100 gelatin NPs loaded with 5-SAS exhibited the most ideal physical and chemical properties. The transmission electron microscopy (TEM) micrographs showed that the final NPs were nearly spherical in diameter from 225 nm to 250 nm without any aggregation. Moreover, the NPs showed a slightly positive zeta potential. A high zeta potential value is conducive to particle rejection, preventing particle aggregation and avoiding particle sedimentation, thus providing good stability for the dispersion of NPs. In addition, in the study by Khan et al., NPs showed considerable cytocompatibility both in vitro and in vivo possibly mediated by the modulation of inflammatory cell activation and accumulation and subsequent improvement in inflammatory biomarkers. Considerable cytocompatibility plays a protective role in mice. This may be through the accumulation and activation of inflammatory cells, thereby improving inflammatory symptoms. Thus, 5-ASA NP has an excellent anti-inflammatory capacity and may be another supplement to treat UC.109
In addition to using EDU S100-coated NPs, polylactic acid-glycolic acid copolymer (PLGA) can be directly used as a carrier of the agent without the need for coating EDU S100. As a Food and Drug Administration (FDA)-approved polymer, PLGA has a great ability to biodegrade, increase the solubility of hydrophobic drugs, effectually transport more drugs to the diseased part, and slowly release encapsulated drugs. Zhang et al. and Ma et al. both used PLGA-loaded drugs to synthesize NPs.
Zhang et al. developed a PLGA NP delivery system targeting P-selectors. Natural compounds are a great source of anti-inflammatory drugs. Resveratrol (Res) is a naturally occurring styrene-like substance (a polyphenol), which has been shown in animal models to improve damage in several diseases, including cardiovascular disease, inflammatory bowel disease (IBD), ischemic injury, and cancer. According to some research, Res can inhibit UC in mouse models. Meanwhile, it has also been observed that dietary triterpene birch acid (BA) can lower myeloperoxidase (MPO) in the colon and the levels of lipid peroxide, and can also restore colitis mice catalase and superoxide dismutase (SOD), drop low glutathione levels, and significantly diminish inflammation media such as matrix metalloproteinases and the expression of prostaglandin E2. Besides, BA alleviated visceral ache caused by mustard oil and acetic acid in mice. The results showed that the effect of Res on UC was complementary to Res, suggesting that the combination of BA and Res may have synergistic effects on UC. PLGA as a drug delivery carrier, has excellent biodegradability, biocompatibility, and good sustained drug release and other advantages, and simultaneously, the drug-loaded PLGA not only makes the NP PEG polymer have good stability but also helps NPs avoid removal by the reticuloendothelial system, making NPs has a longer cycle time in the body. In addition, the simple modification of PEG provides additional targeting capabilities for NPs. Importantly, PLGA-based carriers are particularly advantageous for the delivery of hydrophobic drugs. They can effectively improve the solubility of hydrophobic drugs and have high encapsulation efficiency, making them the best carrier for Res and BA. P-Selectin is expressed on the endothelial cells of most tissues, which is a membrane glycoprotein. It is worth noting that in ulcerative colon inflammation parts, higher P-selectin expression is found but is absent in healthy human and mouse tissues, showing that P-selectin is a possible nano drug to the colon conveying a specific target to treat UC. To prepare P-selectin-targeted NPs co-delivered by BA and Res to colon endothelial cells, Zhang et al. prepared surface P-selectin binding peptide (PBP)-modified PLGA NPs and bound them with two lipophilic dyes (Fig. 9).54
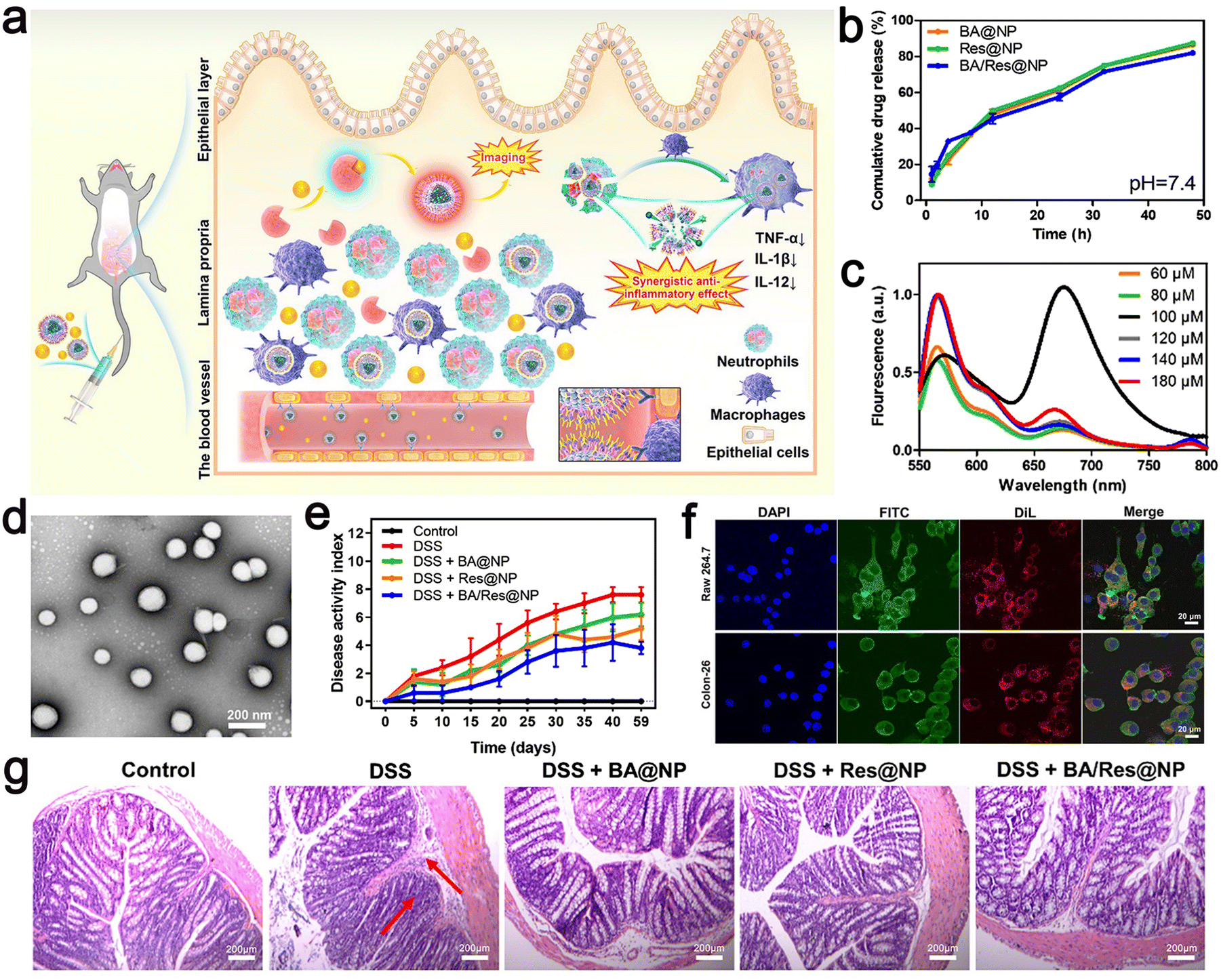 | ||
| Fig. 9 (a) Schematic illustration of targeted delivery of BA/Res@NP-PBP to the inflamed colon. (b) Cumulative release profiles of BA and Res from BA@NP, Res@NP, and BA/Res@NP in pH 7.4. (c) Fluorescence spectra of BA/Res@NP-PBP with different total concentrations. (d) TEM images of BA/Res@NP. (e) Disease activity index showing the therapeutic outcomes of BA/Res@NP against chronic UC. (f) Confocal images of cell uptake of NPs by Raw 264.7 cells and Colon-26 cells. (g) H&E staining images of the colons. Adapted and reprinted with permission from ref. 110 Copyright 2021, Elsevier Ltd. | ||
The NPs had a size of 184.3 ± 7.1 nm, a narrow poly dispersion index (PDI) of 0.051.
The total concentration of DiL and DiD was adjusted, and the measured fluorescence intensity increased from 60 μM to 80 μM. A strong fluorescence signal was observed at 670 nm, and the highest energy transfer efficiency was obtained (Fig. 9c), a negative zeta potential, and the characteristics of sustained-release in vitro. HPLC analysis showed that BA and Res had high entrapment rates in NPs. In addition, the polymer-coated co-delivery systems effectively improved the anti-inflammatory role of individual BA or Res, and p-selectin-mediated targeting increased the accumulation of NPs in the inflammatory colon. This approach provides a potential co-delivery system for the transport of natural nanomedicine to achieve UC-targeted therapies and provides a non-invasive and highly sensitive method for precisely observing inflammatory illness.
Ma et al. also used PLGA as a drug carrier. S100A9 is a potential target molecule to treat colitis, but thus far, no effective targeting method has been found. The research goal of Ma et al. is to develop an effective and secure drug transport system targeting S100A9 nanometers. Then, they developed an oral nano transport system to synthesize PLGA–TAS NPs using PLGA-loaded S100A9 inhibitor Tasquinimod. TLR4 overexpressed macrophage membranes (MMS) were used to wrap NPs to prepare MM–PLGA–TAS, which allowed the nanoparticles to accumulate specifically in the area of colitis. According to dynamic light scattering (DLS) measurements, the average diameter of MM–PLGA–TAS NPs was 207.2 nm and that of the uncoated PLGA–TAS NPs was 168.1 nm. PLGA–TAS and MM–PLGA–TAS are spherical materials with an average diameter of 162 nm and 181 nm, respectively. TEM showed that the thickness of the macrophage membrane is about 11 nm. The drug loading and entrapment efficiency of TAS in PLGA were 6.37% ± 0.26% and 67.42% ± 1.79% μg mg−1, respectively. In addition, these nanoparticles were stable under the storage condition of 4 °C, and their particle size and zeta potential remained stable in deionized water for one week. These results indicate that the synthesized nanoparticles have suitable particle size and stability and have a good cell membrane coating effect. Simultaneously, they also show that the modified nano-system is a feasible scheme for the treatment of UC.111
Ma et al. proposed a convenient oral-targeted colitis delivery system for the treatment of mice with UC. This system encouraged the drug to accumulate in the inflamed colon tissue, reducing the risk of systemic exposure, and thus is an effective treatment for UC. Ma et al. used a TLR4 overexpressed macrophage membrane (MMS) to coat NPs to prepare MM–PLGA–TAS with a certain targeting function.111 Similarly, Gao et al. developed and designed a food-derived microoral delivery system for UC, which also utilized the macrophage targeting capability of β-1,3-D dextran microcarrier yeast cell wall particles (YPs). They formed nanoparticles (EMO-NPs) by coating emodin (EMO) with lactoferrin (LF) targeting intestinal epithelial cells, and then loaded them in YPs to form the final formulation with two outer and inner targeting layers (EMO–NYPs). Notably, YPs and LF are both food-derived nanocarriers, and YPs have good ability to protect the stability of EMO–NYPs in the gastric environment and control the release of loaded EMO NP around the colon. For the inner layer targeting strategy, EMO NP loaded with LF ligand can target LF receptor (LFR) on the surface of intestinal epithelial cells, increase the uptake efficiency of EMO NPs by cells and enhance mucosal repair effect. These external and internal targeting structures may play a role in enhancing the anti-inflammatory capacity of EMO and mucosal repair, respectively. Based on cellular uptake assessment, EMO NP and EMO–NYP target the DECT-1 and LF receptors on the cell membranes of Caco-2 cells and macrophages, respectively. According to these results, EMO–NYPs are a safe and effective oral drug delivery system against UC (Fig. 10).112
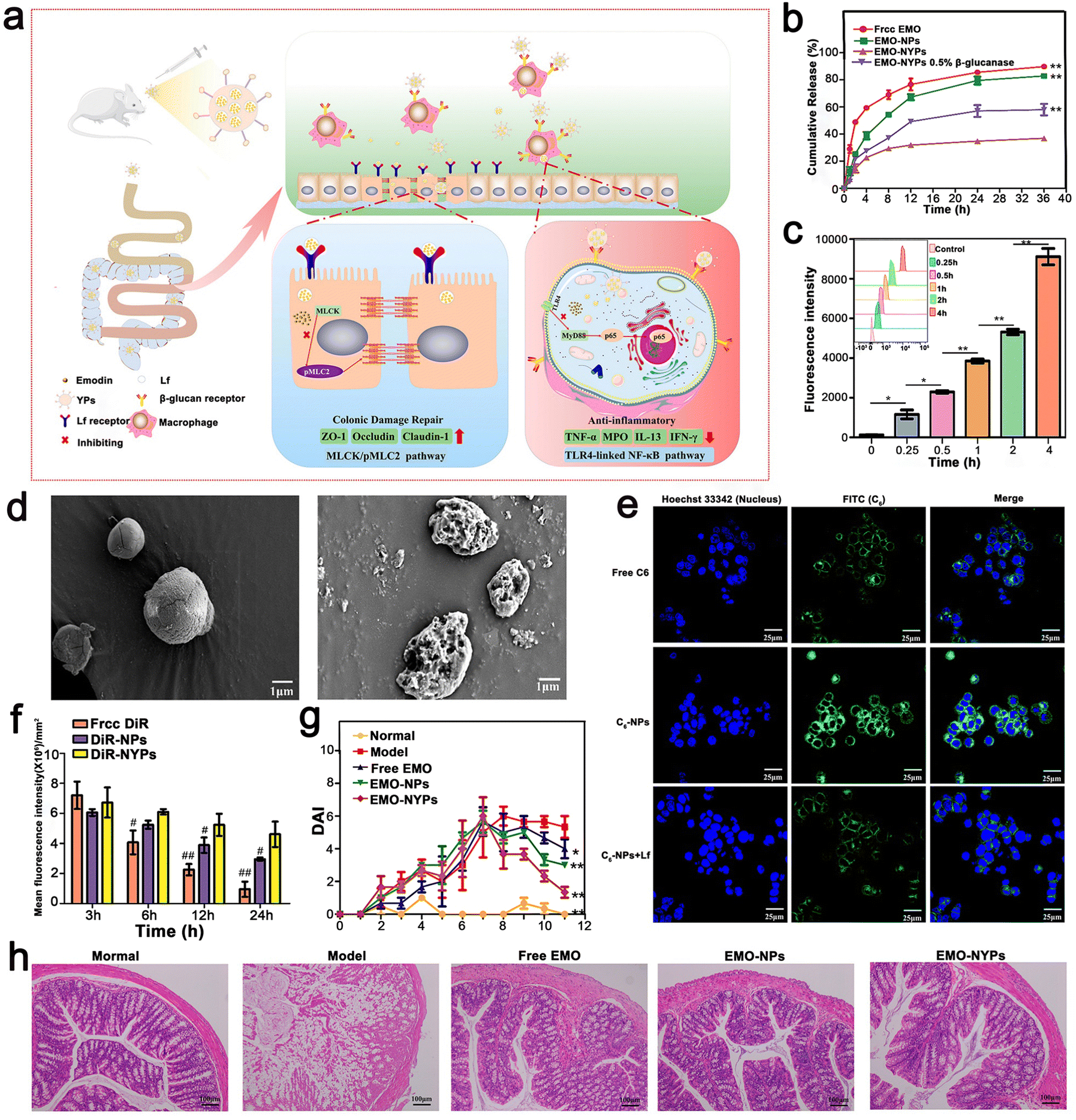 | ||
| Fig. 10 (a) Schematic illustration using EMO–NYPs in the treatment of UC in mice. (b) In vitro release profiles of free EMO, EMO-NPs, and EMO–NYPs in SGF. (c) Quantitative determination of C6–NYP uptake by RAW 264.7 cells after 4 h incubation at different times. (d) SEM images of EMO–NYPs after SGF and SCF treatment. (e) Qualitative cellular uptake evaluation of Caco-2 cells after incubation. (f) Histogram of mice at different intervals after oral administration of different preparations. (g) DAI score of different oral preparations in mice. (h) H&E staining images of the colon of mice after oral treatment with five EMO preparations. Adapted and reprinted with permission from ref. 112 Copyright 2021, Elsevier, Ltd. | ||
Gao et al. constructed EMO-NPs and EMO–NYPs to realize dual-targeting NPs. The dual biological response of Rhein (RH) in promoting the repair of colonic mucosal injury and controlling inflammatory response was combined with a dual-targeted oral nano drug delivery strategy (intestinal epithelial cells and macrophages) for the effective treatment of UC. Briefly, CP/HA/Rh NPs encapsulated in LF were modified with calcium pectin (CP) and HA carbohydrates. The CP layer made CP/HA/Rh NP steadier, which could protect them from the acidic environment of the gastrointestinal tract, and then release HA/Rh NPs to the site of colonic lesions. Because Rh had no obvious fluorescence, they used C6 as a fluorescent probe for tracking. When the concentration increased from 6.25–100 ng mL−1 and the time increased from 0 to 4, the fluorescence intensity of C6 in HA/C6 NPs increased significantly, indicating that the uptake of C6 NPs by HA/Caco-2 cells was concentration and time dependent. After Caco-2 cells were incubated with free C6, C6 NPs, or HA/C6NPs at a C6 concentration of 100 ng mL−1 for 4 h, the fluorescence intensity of C6NPs or HA/C6NPs was significantly higher than that of free C6 tested by FCM (p < 0.01). However, there was no significant difference in fluorescence intensity between C6 NP, C NP, and HA/C6 NPs. In contrast, when C6 NP or HA/C6 NP was exposed to Caco-2 cells, the intracellular fluorescence decreased significantly (p < 0.01). Caco-2 cells were pre-incubated with free LF, and LF bound to the receptor site on the surface of Caco-2 cells in advance. According to the results of the cell uptake assessment, the NPs specifically targeted with the LF and HA ligands increased uptake rates. The experimental result showed that CP/HA/Rh NPs inhibited the TLR4/MyD88/nuclear factor kappa-B (NF-κB) signaling pathway to observably reduce inflammation and accelerate colon recovery (Fig. 11). The nanostructures were specifically designed for the pharmacological effects of RH in the anti-inflammatory response and repair of colonic mucosal damage. Simultaneously, all the nanomaterials in the NP, such as CP, LF, and HA are food grade and eco-friendly. In addition, the preparation method of NPs is relatively simple, and the drugs and polymers are relatively cheap and can be used in large quantities. It is worth mentioning that most oral nanoparticle drug delivery systems only target one site or one cell. This study provides a new strategy to target two different cells simultaneously based on different mechanisms of UC treatment. Finally, they observed that RH could inhibit the expression level of proinflammatory cytokines and improve DSS-induced colitis in mice. Therefore, nanosystems are an effective, safe and relatively simple oral drug delivery system.110
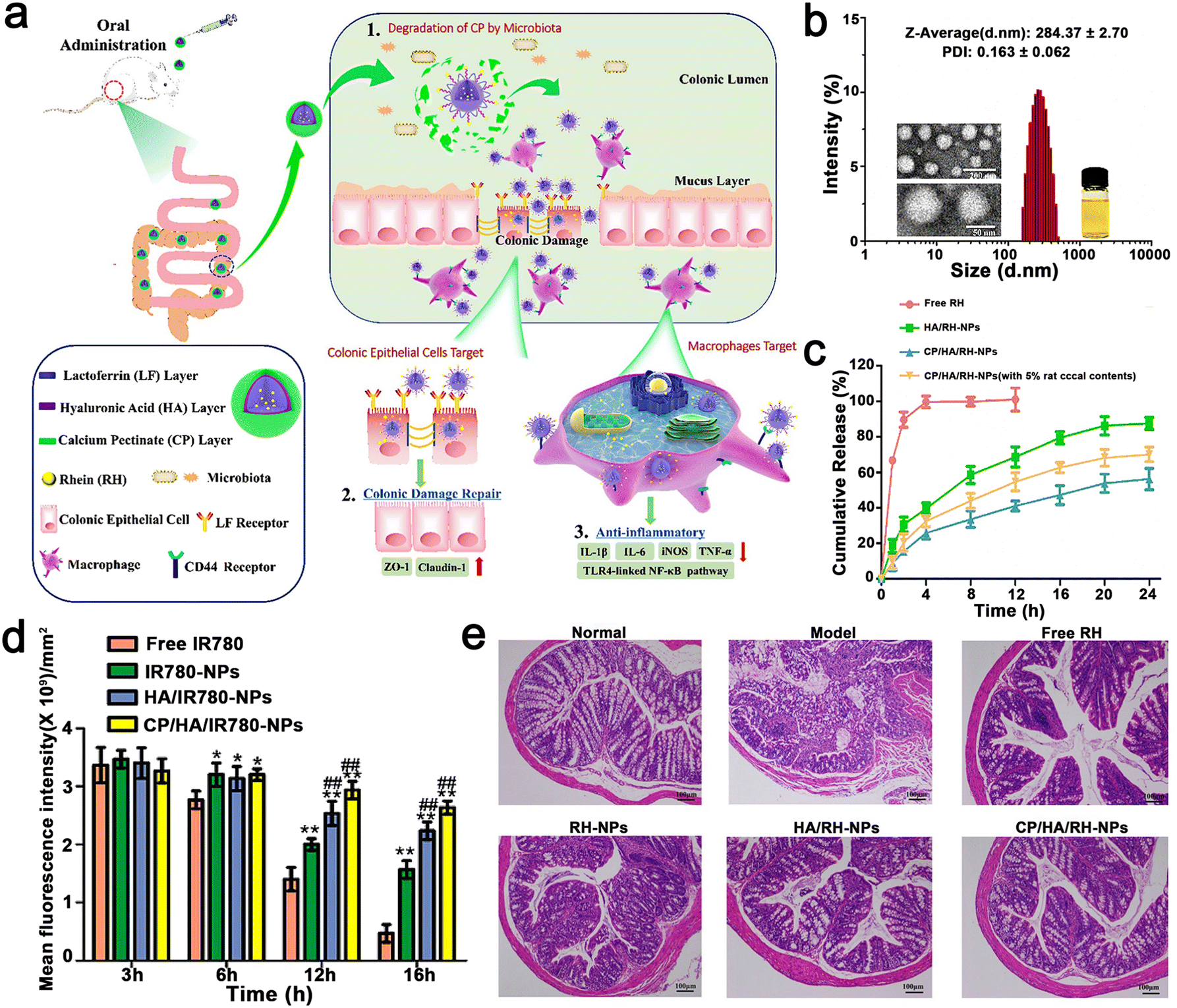 | ||
| Fig. 11 (a) Schematic illustration using CP/HA/RH NPs in the treatment of UC in mice. (b) TEM and size distribution of CP/HA/RH NPs. (c) In vitro release profiles of RH from free RH, HA/RH-NPs, and CP/HA/RH NPs in the presence and absence of rat cecal content groups in PBS. (d) Histogram of fluorescent signal analysis of UC mice after oral delivery of different formulations. (e) H&E staining images of colonic sections among the six formulations. Adapted and reprinted with permission from ref. 54 Copyright 2022, BioMed Central. | ||
The preparation of nanomaterials not only needs to overcome the wear of the gastric environment but also needs to deal with the significant degradation of therapeutic drugs caused by the lysosomal environment. pH-responsive nano bomb delivery system can significantly improve the efficiency of drugs in the cytoplasm and effectively reduce the degradation of enzymes in lysosomes, but previous pH-responsive nano bombs are often complex and expensive. Therefore, the goal of Zhang et al. was to build an L-arginine pH-activated nano bomb system loaded with PS (HA–PS@NPs) modified by HA to cure UC. The mechanism of action is L-arginine–CO2 (L-Arg–CO2) in HA–L-Arg at lysosomal acidic pH-CO2@NPs, the carrier releases carbon dioxide and produces a “nano bomb effect”, triggering the escape of PS lysosomes. Meanwhile, the inflammatory colon is effectively targeted by CD44 receptors overexpressed in colonic epithelial and macrophage cells. Oral administration can reduce inflammation and UC symptoms by down-regulating the expression of pro-inflammatory cytokines HA-NP and HA–PS@NPs. In summary, the pH-activated nano bomb delivery system has curative effects and provides an effective targeting strategy to treat UC.113
7. Nano-drug delivery platform for autoimmune hepatitis
AIH is an autoimmune liver disease caused by autoantibodies and autoreactive T cells produced by the body.114,115 It is also a type of pathology that can make use of nano agents for targeted liver treatment. Chronic progressive hepatitis may develop into cirrhosis if not treated well. Its chronic nature requires continuous and nearly chronic medical treatment to slow the progression of the disease and avoid liver transplantation if possible.116 In many circumstances, most of these diseases are treated with steroid anti-inflammatory drugs, but long-term use will lead to serious side effects.117 The wide distribution of steroids in the body easily crosses the biological barrier, causing AIH patients to abandon treatment due to severe steroid-related complications. One possible solution is to concentrate the pharmacological activity in the liver, improve the utilization efficiency of drugs and reduce the use of drugs, and thus there is a need to identify solutions that can concentrate drugs in the liver and lower the level of untargeted drugs.118,119Avidin-nucleic-acid-nano-assemblies (ANANAS) are a type of poly anti-biotin NPs based on the high affinity-driven nucleation of avidin units around non-coding plasmid DNA. ANANAS are biodegradable and highly tolerated and show a high degree of liver affinity with no need for liver targeting elements. The role of drug-loaded ANANAS depends on the binding of biotinylated drug ligands to biotinylated binding sites (BBS) that bind anti-biotin proteins on the surface of drug NPs. The distance between the biotin and the drug portion determines the interval length from ANANAS to the protein core. If the drug uses short-spaced biotin ligands, it will be located near the surface of the granular protein (inner surface layer); however, if a long-spaced ligand is used (e.g., 5 kDa PEG), it will be located near the outside of the surface layer. In ANANAS-carrying Dex (ANANAS–Dex) for AIH therapy, these drugs are attached to the vector via a long-chain (5 kDa) PEG biotin ligand using unstable hydrazone bonds, allowing these free drugs to be released after internalization by the target cell via the endosomal–lysosomal pathway.118
Morpurgo et al. investigated Dex-loaded biodegradable avidin-nucleic acid nanocomposites. They focused on nanoparticle-modified biotin-Dex alternative adhesives and assessed the properties of the junctions between biotin and drug components and their impact on the in vivo and in vitro effects of ANANAS–Dex preparations. They also studied the hydrazone bonds used in the drug formulation, i.e., hydrazone (Hz) and carbamate hydrazide–hydrazone (Cb Hz). Therefore, they developed four new biotin–Dex complexes as Dex–ANANAS tethers. All conjugated compounds were Dex–ANANAS tethers. All the conjugated compounds were evaluated for their hydrolysis stability in vitro and the most resistant conjugated compounds selected to ameliorate the second generation ANANAS–Dex.118
The results showed that the hydrolysis stability of the Dex hydrazone is significantly affected by the chemical environment.
When ANANAS–Dex was close to the avidin-biotin binding sac, it stabilized the hydrazone bond and prevented hydrolysis, especially in the Hz and Cb-Hz forms. The other momentous finding is that small changes in α-biotin-PEG in the ω end significantly affected the properties of the polymer. Especially, the introduction of six carbon spacers at the ω end of 5 kDa PEG could influence the solubility and lower the colloidal protection of the polymer. Therefore, in the formulation of the new generation ANANAS–Dex-L, considered, it contained low and high molecular weight binders and biotin methoxylated polyethylene glycol for colloidal protection. Compared to the first-generation ANANAS–Dex-A, this generation has high drug load and hydrolytic stability and similar colloidal stability. In principle, ANANAS–Dex-L should be a more outstanding choice for treatment because of these properties. Although the persistence of free drugs and their in vivo levels in different organs depends not only on the biological distribution and pharmacokinetics of the carrier but also on the hydrolytic stability of Hz–Dex, bond Dex-response genes are activated primarily in the liver after the administration of NPs, which is consistent with the hypothesis of a more goal-centered effect.118
Morpurgo et al. studied ANANAS, and their research mainly focused on the effective treatment of patients with liver inflammation using Dex-carrying nanoparticles based on ANANAS to develop a comprehensive platform to assess the efficacy of ANANAS in an AIH mouse model.119
They also prepared ANANAS–Hz–Dex by blending biotin–PEG–Hz–Dex with a core of NPS30% BBS (ANANAS–Hz–Dex30) coverage. The efficacy of ANANAS was studied mainly through in vivo and in vitro experiments using ANANAS–Hz–Dex.
Their results showed that in the animal models, ANANAS–Hz–Dex NPs were more efficacious than the free drugs in illness control; meanwhile, the NPs did not release steroids in any other body area except the liver, indicating that they are a suitable carrier to control chronic liver inflammation with great promise. Moreover, ANANAS–Hz–Dex has relatively durable free drug availability, and even low concentrations of Dex are sufficient to control disease characteristics. It should be noted that despite repeated administration, the use of ANANAS alone had no measurable negative effect on liver function. This indicates that the carrier itself has neither toxicity nor pro-inflammatory effect (Fig. 12).119
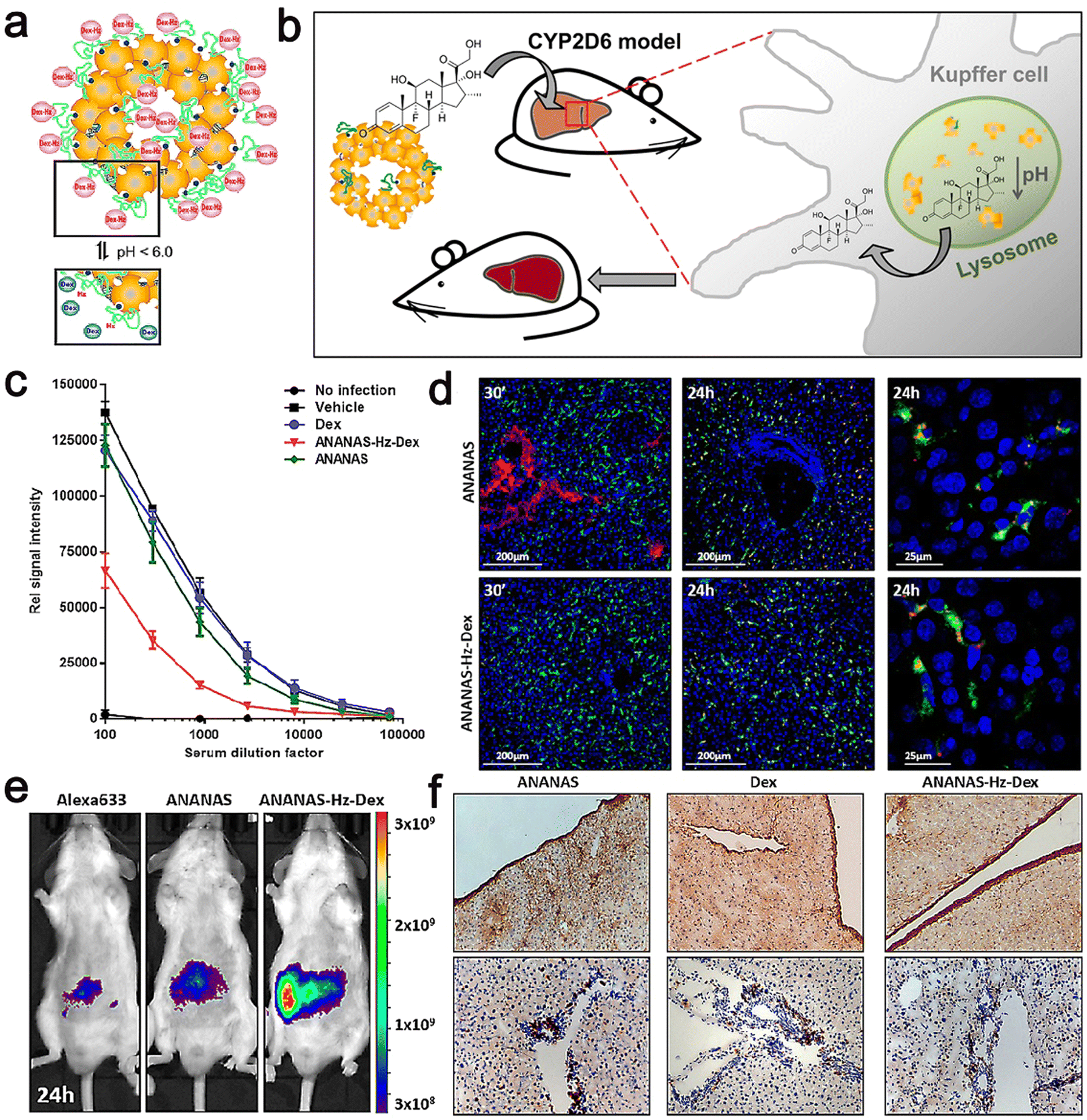 | ||
| Fig. 12 (a) Scheme of the pH-dependent release of Dex from ANANAS–Hz–Dex30. (b) Schematic illustration of biodegradable avidin-nucleic-acid-nano-assemblies in an autoimmune hepatitis murine model. (c) Anti-CYP2D6 IgG levels were analyzed in serum collected at euthanasia. (d) Confocal microscopy liver images from healthy mice sacrificed 30 min and 24 h after ANANAS and ANANAS–Hz–Dex. (e) In vivo optical imaging of mice treated with Alexa633, ANANAS, or ANANAS–Hz–Dex and scanned 24 h after treatment. (f) Collagen I and CD4 T cell staining on liver sections from mice. Adapted and reprinted with permission from ref. 119 Copyright 2019, the American Chemical Society. | ||
Selenium (Se) is an essential trace element for human beings and animals. It is a dietary nutrient that plays a significant part in lots of aspects of health, especially its antioxidant activity. It is a component of more than 25 selenium proteases and enzyme-catalyzed sites in vivo and plays a vital part in protecting tissues and cells from oxidative damage, but Se deficiency has been linked to a variety of diseases, including liver damage.120
Selenium nanoparticles (Se NPs) have aroused increasing concerns because of their excellent properties and biological activity. Se NPs are considered a potential selenium supplement because they show great potential in nutritional supplements, chemoprevention, chemotherapeutic, and nanomedicine delivery applications. The liver-protective effects of Se NPs seem promising because they can protect animals from liver damage induced by chemicals or pathogens through their antioxidant capacity.121
Bai et al. prepared chitosan-stabilized Se NPs (CS–Se NPs) by rapid ultrafiltration and evaluated their free radical scavenging and antioxidant abilities.
In addition, the protective effects of spray-dried CS–Se NP powder on autoimmune liver disease, such as reducing liver edema and improving hepatocyte necrosis, were also studied in the mouse model of liver injury induced by concanavin A (Con-A). In their study, CS-stable selenium NPs had strong free radical scavenging ability and certain antioxidant activity, which could effectively protect mice from Con-A-induced autoimmune liver injury. In conclusion, CS–Se NPs are worthy of further development as nutritional supplements or even nano drugs, which can have a good effect in the protection against liver injury induced by autoimmune diseases. Meanwhile, Se NPs may play an important role in antioxidant activity and liver protection of Se NPs.121
Nano drugs based on major histocompatibility complex (pMHC) peptides are molecules that can bind to specific receptors on immune cells and may be able to reprogram adaptive immune responses or innate responses to cure cancer and autoimmunity. NPs coated with pMHC molecules related to autoimmune diseases could effectively solve the inflammation in a variety of organ-specific autoimmune disease models and have a certain effect on AIH without damaging normal immunity.120,122,123
Santamaria et al. and Khadra et al. both carried out relevant studies on pMHC NPs. Santamaria et al. mainly studied the properties of pMHC NPs. NPs coated with major histocompatibility complexes (pMHCs) of autoimmune disease-related peptides could suppress autoimmune diseases by reprogramming homologous effector T cells into regulatory T cells, and then scaled up. To better understand the correlation between pharmacodynamics and bioavailability, they developed a new way to quantify the absolute amount of activity of the drug products. PMHC coating with protein corona orientation could protect it from protein hydrolysis and recognition by anti-drug antibodies, which is conducive to protecting its biological efficacy. Subsequently, they measured the protein corona coating pMHC using a series of characteristic quantitative local methods, and they provided clues to the structure of the ligand coating on nano drugs based on pMHC. Their series of measurements showed that the directional coupling between ligands and the NP surfaces overcomes the potential negative effects of plasma protein adsorption on the connecting properties of these compounds with their homologous receptors on the specific target cells. They developed a special and highly sensitive method to quantify intact drugs in biological samples. They used tissue ultrasound to release the compounds captured by cells for quantification and developed sensitive quantitative methods for drug products to measure the bioavailability of drugs. The measurement showed that pMHCII NPs would quickly leave the microcirculation of the liver and spleen, but would not leave the lungs, because these compounds were not observed to be captured in cells. Considering that the blood volume of mice has time to pass through the whole vascular system about 4 times in the first minute, cell scavengers in organs such as the liver and spleen have enough opportunity to capture these compounds and remove them from the circulation, and thus it can be concluded that pMHCII NPs have a short bioavailability. The experimental results proved that, surprisingly, these compounds have short bioavailability and a fast PK spectrum. In addition, the type of cell responsible for retention and organ capture was identified, and the capture of homologous T cells and the formation of T-cell receptor (TCR) micro clusters in vivo were also recorded by them.120
Meanwhile, Santamaria et al. also focused on whether the destruction of hepatocytes and/or bile duct cells in autoimmunity stimulates autoreactive T cells, which can respond to disease-related and unrelated PMHCII-based nano drugs. They realized that nano drugs showing different liver universal antigen peptides triggered the formation and expansion of T-regulatory-type-1 (Tr1)-like cells in mice experiencing all types of liver autoimmune diseases and in NSG mice humanized with PBMC from primary biliary cholangitis (PBC) patients. Thus, by suppressing liver inflammation, these nano drugs effectively slowed the incidence of AIH primary sclerosing cholangitis (PSC) and primary PBC, in different genetic contexts, even when disease severity was at its peak. Importantly, the inhibition of liver inflammation by these nano drugs does not impair immunity against viruses (vaccinia and influenza), intracellular bacteria (Listeria) or metastatic (liver) allogenic tumours. Tr1 like CD4+ T cells triggered by pMHC-based nano drugs can play a regulatory role only when they bind to homologous pMHC class II professional antigen-presenting cells (APC) carrying endogenous autoantigen. These APC must capture the autoantigens released by damaged hepatocytes, and thus they only exist in a large number in target organs or drainage lymphoid tissues, and the distal APC coordinating these immune responses is not loaded with liver-derived autoantigens. Thus, pMHC-based nano drugs will not damage the immunity to systemic infection or vaccines.122
NPs can display peptides of the major histocompatibility complex II molecules associated with autoimmune disease and trigger the formation of homologous Tr1 cells that reverse organ-specific autoimmune responses. This shows that pMHCII-NPs of the mitochondrial protein epitope can slow down the progress of AIH and can play an effective therapeutic role in various animal models affected by autoimmune diseases. Simultaneously, some studies have shown that Tr1 cells are very effective in inhibiting autoimmunity, where they mediate organ-specific immune regulation through it. However, recent proof suggests that when experimental autoimmune encephalomyelitis (EAE) and AIH occur simultaneously in animal models, pMHCII-NP treatment can produce quite confusing results, which may depend on the type of autoantigenic peptides displayed on NPs. Therefore, Khadra et al. established a zonal population model of T cells in comorbid mice to determine the mechanism. They conducted time-series simulation and bifurcation analysis and compared and analyzed different behavior patterns. The final results showed that the delayed distribution of CIR-1 cells in the central nervous system in mice with trea-1-dependent inflammation could finally be explained by the delayed distribution of CIR-1 cells in the central nervous system compared with that in mice with trea-1-dependent inflammation. The experimental result showed that local Tr1 cell retention, homologous and autoantigen expression play an important role in effectively regulating the function of cells. Therefore, these results provide new ideas for the regulation of Tr1 cell recruitment and its self-regulation function (Table 2).123
| Disease | Nanomaterials | Average size (nm) | Drugs | Ref. |
|---|---|---|---|---|
| Ulcerative colitis | CP/HA/RH-NPs | 172.0 | RH | 54 |
| Ulcerative colitis | 5-ASA NPs | 225.0–250.0 | 5-ASA | 109 |
| Ulcerative colitis | EMO–NYPs | 157.99 ± 0.13 | EMO | 110 |
| Ulcerative colitis | MM–PLGA–TAS NPs | 207.2 | Tasquinimod | 111 |
| Ulcerative colitis | PBP–PLGA–NP | 184.3 ± 7.1 | BA/Res | 112 |
| Ulcerative colitis | HA–PS@NPs | 279.9 ± 0.934 | PS | 113 |
| Autoimmune hepatitis | ANANAS | 123.0 | Dex | 118 |
| Autoimmune hepatitis | ANANAS–Hz–Dex | 125.8 ± 1.2 | Dex | 119 |
| Autoimmune hepatitis | pMHC NPs | pMHC | 122 |
8. Conclusions and perspectives
With the continuous development of modern medical technology, the treatment of autoimmune inflammatory diseases has made some progress in recent years, but its poor prognosis is still one of the factors endangering human health. Because the pathogenesis of the this disease is indeterminacy, there are no specific therapeutic drugs at present. Clinical treatment mainly aims to alleviate or reduce disease activity and minimize the damage to the body caused by the side effects of long-term medication. In recent years, with the in-depth understanding of the pathogenesis of this disease, various new targeted therapies designed for the key pathways and molecular mechanisms involved in its pathogenesis have gradually emerged and attracted extensive attention. This is intended to overcome the limitations of low bioavailability and non-specificity of traditional drugs and improve the therapeutic effect of drugs. The development of nano drug loading technology makes it possible to realize real drug-targeted therapy.According to the different interaction modes between nanocarriers and the body, they can mainly be divided into three types, i.e., active targeting, passive targeting, and stimulus response (single stimulus and multi stimulus-response). Compared with passive targeting, active targeting has higher targeting to diseased tissues through antigen-antibody binding reaction in vivo. Stimulation-responsive nanocarriers can give certain stimulation according to the change in the microenvironment in the diseased tissue or outside to realize the controlled-release treatment of drugs. After the principle of action is clear, the design and preparation of nanocarriers with excellent performance is the first problem to be solved. Secondly, the surface modification and modification of carriers are also factors affecting their performance. In addition, drug loading, stability after drug loading, and carrier metabolic clearance after treatment are also the main challenges. Therefore, how to adjust the size, morphology, and surface decoration of nanomaterials in the process of preparing carriers, and further improve the effectiveness of specific targeted therapy based on successfully obtaining qualified nanocarriers should be the research focus in the future.
Although nano-drug carrier systems and packaging technology still face many problems, with the development of modern science and technology, their unique advantages will show better prospect. To date, the research on nanodrug carriers is still in the theoretical stage, and few drugs can be used in clinical treatment, which seriously limits the wide application of nano-drug carriers. In the future, we should be committed to developing nano-drug carriers with easy synthesis, high efficiency, high drug loading, low toxicity, low cost, and clinical application value. By combining the synthesis of nanocarriers with the modification of surface groups, a new drug delivery route is established, which is conducive to the development of interdisciplinary comprehensive research and gives full play to its scientific and market value.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21902157, 51702004), Starting Fund for Scientific Research of High-Level Talents, Anhui Agricultural University (rc382108), Key Research and Development Plan of Anhui Province (2022e07020037), the Open Fund of the State Key Laboratory of Catalysis in DICP, CAS (N-21-12), and the Open Fund of the State Key Laboratory of Molecular Reaction Dynamics in DICP, CAS (SKLMRD-K202223), Innovation and Entrepreneurship Plan for College Students (ds193807, dg203801), University scientific research project of Anhui Provincial Department of Education (KJ2021A0322).References
- J. S. Smolen, D. Aletaha and I. B. McInnes, Lancet, 2016, 388, 2023–2038 CrossRef CAS
.
- E. K. Kapsogeorgou and A. G. Tzioufas, Isr. Med. Assoc. J., 2016, 18, 519–524 Search PubMed
- L. Durcan, T. O'Dwyer and M. Petri, Lancet, 2019, 393, 2332–2343 CrossRef
- A. Komori, Clin. Mol. Hepatol., 2021, 27, 58–69 CrossRef PubMed
- Y. J. Lin, M. Anzaghe and S. Schülke, Cells, 2020, 9, 880 CrossRef CAS PubMed
- B. Terziroli Beretta-Piccoli, G. Mieli-Vergani and D. Vergani, Cell. Mol. Immunol., 2022, 19, 158–176 CrossRef CAS
- R. Ungaro, S. Mehandru, P. B. Allen, L. Peyrin-Biroulet and J. F. Colombel, Lancet, 2017, 389, 1756–1770 CrossRef
- E. A. Littlejohn and S. U. Monrad, Primary Care, 2018, 45, 237–255 CrossRef PubMed
- E. Brzustewicz, I. Henc, A. Daca, M. Szarecka, M. Sochocka-Bykowska, J. Witkowski and E. Bryl, Cent. Eur. J. Immunol., 2017, 42, 259–268 CrossRef CAS
- D. Aletaha and J. S. Smolen, JAMA, 2018, 320, 1360–1372 CrossRef
- L. Du and C. Ha, Clin. Gastroenterol., 2020, 49, 643–654 CrossRef PubMed
- J. S. Smolen and D. Aletaha, Nat. Rev. Rheumatol., 2015, 11, 276–289 CrossRef PubMed
- M. A. Stoffer, M. M. Schoels, J. S. Smolen, D. Aletaha, F. C. Breedveld, G. Burmester, V. Bykerk, M. Dougados, P. Emery, B. Haraoui, J. Gomez-Reino, T. K. Kvien, P. Nash, V. Navarro-Compán, M. Scholte-Voshaar, R. van Vollenhoven, D. van der Heijde and T. A. Stamm, Ann. Rheum. Dis., 2016, 75, 16–22 CrossRef PubMed
- J. S. Smolen, D. van der Heijde, K. P. Machold, D. Aletaha and R. Landewé, Ann. Rheum. Dis., 2014, 73, 3–5 CrossRef CAS PubMed
- E. Pérez-Herrero and A. Fernández-Medarde, Eur. J. Pharm. Biopharm., 2015, 93, 52–79 CrossRef PubMed
- E. Pugliese, J. Q. Coentro and D. I. Zeugolis, Adv. Mater., 2018, 30, e1704324 CrossRef
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 1–17 Search PubMed
- J. Zhu, R. Hou, M. Liu, L. Wang, W. Chen, Y. Sun, W. Wei and S. Ye, Mater. Today Sustain., 2022, 18, 100125 CrossRef
- S. H. Chang, H. J. Lee, S. Park, Y. Kim and B. Jeong, Biomacromolecules, 2018, 19, 2302–2307 CrossRef CAS PubMed
- P. Huang, H. Song, Y. Zhang, J. Liu, J. Zhang, W. Wang, J. Liu, C. Li and D. Kong, ACS Appl. Mater. Interfaces, 2016, 8, 29323–29333 CrossRef CAS
- V. Ramalingam, K. Varunkumar, V. Ravikumar and R. Rajaram, Sci. Rep., 2018, 8, 3815 CrossRef
- Y. Dou, C. Li, L. Li, J. Guo and J. Zhang, J. Controlled Release, 2020, 327, 641–666 CrossRef CAS PubMed
- M. M. Liu, C. Y. Zhang, A. L. Han, L. Wang, Y. J. Sun, C. N. Zhu, R. Li and S. Ye, Nano Res., 2022, 15, 6862–6887 CrossRef CAS
- M. M. Liu, J. Zhu, Y. H. Liu, F. L. Gong, R. Li, H. Chen, M. Zhao, Q. K. Jiang, J. Liu and S. Ye, Chem. Eng. J., 2022, 446, 137080 CrossRef CAS
- L. Wang, F. S. Wang and M. E. Gershwin, J. Intern. Med., 2015, 278, 369–395 CrossRef CAS PubMed
- B. A. Michel, Praxis (Bern 1994), 2000, 89, 477–480 CAS
- G. Műzes and F. Sipos, Orv Hetil, 2018, 159, 908–918 CrossRef
- X. Wang, P. Wang and X. Yang, Zhejiang Daxue Xuebao, Yixueban, 2018, 47, 435–440 Search PubMed
- V. G. Dinis, V. T. Viana, E. P. Leon, C. A. Silva, C. G. Saad, J. C. Moraes, E. S. Bonfa and A. C. Medeiros-Ribeiro, Clin. Rheumatol., 2020, 39, 1747–1755 CrossRef
- A. La Cava, C. J. Fang, R. P. Singh, F. Ebling and B. H. Hahn, Autoimmun. Rev., 2005, 4, 515–519 CrossRef CAS
- M. Quaglia, G. Merlotti, M. De Andrea, C. Borgogna and V. Cantaluppi, Viruses, 2021, 13, 277 CrossRef CAS PubMed
- H. Wu, J. Liao, Q. Li, M. Yang, M. Zhao and Q. Lu, Clin. Immunol., 2018, 196, 34–39 CrossRef CAS PubMed
- G. J. Tobón, J. O. Pers, C. A. Cañas, A. Rojas-Villarraga, P. Youinou and J. M. Anaya, Autoimmun. Rev., 2012, 11, 259–266 CrossRef CAS PubMed
- S. Aslani, M. Mahmoudi, J. Karami, A. R. Jamshidi, Z. Malekshahi and M. H. Nicknam, Autoimmunity, 2016, 49, 69–83 CrossRef CAS
- J. Cárdenas-Roldán, A. Rojas-Villarraga and J. M. Anaya, BMC Med., 2013, 11, 73 CrossRef
- T. Ogura and H. Kameda, Nihon Rinsho Meneki Gakkai Kaishi, 2014, 37, 25–32 CrossRef
- H. M. Hussein and E. A. Rahal, Crit. Rev. Microbiol., 2019, 45, 394–412 CrossRef CAS PubMed
- K. H. Lee, B. S. Ahn, D. Cha, W. W. Jang, E. Choi, S. Park, J. H. Park, J. Oh, D. E. Jung, H. Park, J. H. Park, Y. Suh, D. Jin, S. Lee, Y. H. Jang, T. Yoon, M. K. Park, Y. Seong, J. Pyo, S. Yang, Y. Kwon, H. Jung, C. K. Lim, J. B. Hong, Y. Park, E. Choi, J. I. Shin and A. Kronbichler, Autoimmun. Rev., 2020, 19, 102469 CrossRef CAS PubMed
- M. Ramos-Casals, A. Roberto Perez, C. Diaz-Lagares, M. J. Cuadrado and M. A. Khamashta, Autoimmun. Rev., 2010, 9, 188–193 CrossRef CAS PubMed
- B. E. Ostrov, Immunol. Invest., 2015, 44, 777–802 CrossRef CAS PubMed
- M. Jastrzębska, M. E. Czok and P. Guzik, Cardiol. J., 2013, 20, 569–576 CrossRef PubMed
- G. S. Cooper, M. L. Bynum and E. C. Somers, J. Autoimmun., 2009, 33, 197–207 CrossRef PubMed
- R. Cervera, Scand. J. Clin. Lab. Invest., Suppl., 2001, 235, 27–30 CrossRef CAS
- F. W. Miller, Adv. Exp. Med. Biol., 2011, 711, 61–81 CrossRef CAS PubMed
- M. Naeem, U. A. Awan, F. Subhan, J. Cao, S. P. Hlaing, J. Lee, E. Im, Y. Jung and J. W. Yoo, Arch. Pharmacal Res., 2020, 43, 153–169 CrossRef CAS PubMed
- S. Naz, M. Shamoon, R. Wang, L. Zhang, J. Zhou and J. Chen, Int. J. Mol. Sci., 2019, 20, 362 CrossRef PubMed
- J. Li, Y. Ai, L. Wang, P. Bu, C. C. Sharkey, Q. Wu, B. Wun, S. Roy, X. Shen and M. R. King, Biomaterials, 2016, 76, 52–65 CrossRef CAS PubMed
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20–37 CrossRef CAS PubMed
- L. Huang, S. Zhao, F. Fang, T. Xu, M. Lan and J. Zhang, Biomaterials, 2021, 268, 120557 CrossRef CAS PubMed
- R. Goyal, L. K. Macri, H. M. Kaplan and J. Kohn, J. Controlled Release, 2016, 240, 77–92 CrossRef CAS PubMed
- A. Rodzinski, R. Guduru, P. Liang, A. Hadjikhani, T. Stewart, E. Stimphil, C. Runowicz, R. Cote, N. Altman, R. Datar and S. Khizroev, Sci. Rep., 2016, 6, 20867 CrossRef CAS PubMed
- S. M. Moghimi, A. C. Hunter and J. C. Murray, Pharmacol. Rev., 2001, 53, 283–318 CAS
- M. Wang and M. Thanou, Pharmacol. Res., 2010, 62, 90–99 CrossRef CAS PubMed
- X. Yan, C. Yang, M. Yang, Y. Ma, Y. Zhang, Y. Zhang, C. Liu, Q. Xu, K. Tu and M. Zhang, J. Nanobiotechnol., 2022, 20, 99 CrossRef CAS PubMed
- J. Xiang, R. Zhao, B. Wang, X. Sun, X. Guo, S. Tan and W. Liu, Front. Oncol., 2021, 11, 758143 CrossRef PubMed
- J. George, I. K. Yan and T. Patel, Lab. Invest., 2018, 98, 895–910 CrossRef CAS
- N. Bertrand, J. Wu, X. Xu, N. Kamaly and O. C. Farokhzad, Adv. Drug Delivery Rev., 2014, 66, 2–25 CrossRef CAS
- N. T. Huynh, E. Roger, N. Lautram, J. P. Benoît and C. Passirani, Nanomedicine, 2010, 5, 1415–1433 CrossRef CAS PubMed
- F. Danhier, O. Feron and V. Préat, J. Controlled Release, 2010, 148, 135–146 CrossRef CAS PubMed
- D. B. Kirpotin, D. C. Drummond, Y. Shao, M. R. Shalaby, K. Hong, U. B. Nielsen, J. D. Marks, C. C. Benz and J. W. Park, Cancer Res., 2006, 66, 6732–6740 CrossRef CAS PubMed
- A. Ahmad, F. Khan, R. K. Mishra and R. Khan, J. Med. Chem., 2019, 62, 10475–10496 CrossRef CAS PubMed
- T. Dai, N. Li, F. Han, H. Zhang, Y. Zhang and Q. Liu, Biomaterials, 2016, 83, 37–50 CrossRef CAS PubMed
- N. Xu, J. Li, Y. Gao, N. Zhou, Q. Ma, M. Wu, Y. Zhang, X. Sun, J. Xie, G. Shen, M. Yang, Q. Tu, X. Xu, J. Zhu and J. Tao, Biomaterials, 2019, 197, 380–392 CrossRef CAS PubMed
- S. Hajebi, N. Rabiee, M. Bagherzadeh, S. Ahmadi, M. Rabiee, H. Roghani-Mamaqani, M. Tahriri, L. Tayebi and M. R. Hamblin, Acta Biomater., 2019, 92, 1–18 CrossRef CAS PubMed
- G. Liu, J. F. Lovell, L. Zhang and Y. Zhang, Int. J. Mol. Sci., 2020, 21, 479–490 CrossRef
- A. J. Cole, V. C. Yang and A. E. David, Trends Biotechnol., 2011, 29, 323–332 CrossRef CAS PubMed
- M. Zhu, G. Nie, H. Meng, T. Xia, A. Nel and Y. Zhao, Acc. Chem. Res., 2013, 46, 622–631 CrossRef CAS PubMed
- P. Huang, X. Wang, X. Liang, J. Yang, C. Zhang, D. Kong and W. Wang, Acta Biomater., 2019, 85, 1–26 CrossRef CAS PubMed
- A. Hak, V. Ravasaheb Shinde and A. K. Rengan, Photodiagn. Photodyn. Ther., 2021, 33, 102205 CrossRef CAS PubMed
- Y. Wang, H. M. Meng and Z. Li, Nanoscale, 2021, 13, 8751–8772 RSC
- K. Yang, S. Zhang, G. Zhang, X. Sun, S. T. Lee and Z. Liu, Nano Lett., 2010, 10, 3318–3323 CrossRef CAS
- D. Zhi, T. Yang, J. O'Hagan, S. Zhang and R. F. Donnelly, J. Controlled Release, 2020, 325, 52–71 CrossRef CAS PubMed
- J. T. Robinson, S. M. Tabakman, Y. Liang, H. Wang, H. S. Casalongue, D. Vinh and H. Dai, J. Am. Chem. Soc., 2011, 133, 6825–6831 CrossRef CAS PubMed
- S. Kwiatkowski, B. Knap, D. Przystupski, J. Saczko, E. Kędzierska, K. Knap-Czop, J. Kotlińska, O. Michel, K. Kotowski and J. Kulbacka, Biomedicine, 2018, 106, 1098–1107 CAS
- A. M. Rkein and D. M. Ozog, Dermatol. Clin., 2014, 32, 415–425 CrossRef CAS PubMed
- A. P. Castano, P. Mroz and M. R. Hamblin, Nat. Rev. Cancer, 2006, 6, 535–545 CrossRef CAS PubMed
- J. Dobson, G. F. de Queiroz and J. P. Golding, Vet. J. (London, England: 1997), 2018, 233, 8–18 CrossRef CAS PubMed
- W. Xiong, W. Wang, Y. Wang, Y. Zhao, H. Chen, H. Xu and X. Yang, Colloids Surf., B, 2011, 84, 447–453 CrossRef CAS
- Q. Zheng, C. Xu, Z. Jiang, M. Zhu, C. Chen and F. Fu, Front. Chem., 2021, 9, 650358 CrossRef CAS PubMed
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed
- R. Huang, C. Zhang, Y. Bu, Z. Li, X. Zheng, S. Qiu, J. O. Machuki, L. Zhang, Y. Yang, K. Guo and F. Gao, Biomaterials, 2021, 277, 121088 CrossRef CAS PubMed
- Y. Du and B. Chen, Drug Des., Dev. Ther., 2019, 13, 1401–1408 CrossRef CAS PubMed
- S. Stanley, Curr. Opin. Biotechnol, 2014, 28, 69–74 CrossRef CAS PubMed
- M. Zhang, W. Hu, C. Cai, Y. Wu, J. Li and S. Dong, Mater. today. Bio, 2022, 14, 100223 CrossRef CAS PubMed
- A. M. Wasserman, Am. Fam. Physician, 2011, 84, 1245–1252 Search PubMed
- J. Charles, H. Britt and Y. Pan, Aust. Fam. Physician, 2013, 42, 765 Search PubMed
- J. J. Cush, Med. Clin. North Am., 2021, 105, 355–365 CrossRef
- J. Chen, J. Qi, C. Chen, J. Chen, L. Liu, R. Gao, T. Zhang, L. Song, D. Ding, P. Zhang and C. Liu, Adv. Mater., 2020, 32, e2003399 CrossRef PubMed
- A. F. Radu and S. G. Bungau, Cells, 2021, 10, 2857 CrossRef CAS
- A. Dalmoro, S. Bochicchio, S. F. Nasibullin, P. Bertoncin, G. Lamberti, A. A. Barba and R. I. Moustafine, Eur. J. Pharm. Sci., 2018, 121, 16–28 CrossRef CAS
- L. Guo, Y. Chen, T. Wang, Y. Yuan, Y. Yang, X. Luo, S. Hu, J. Ding and W. Zhou, J. Controlled Release, 2021, 330, 119–131 CrossRef CAS PubMed
- M. Qindeel, D. Khan, N. Ahmed, S. Khan and R. Asim Ur, ACS Nano, 2020, 14, 4662–4681 CrossRef CAS PubMed
- Y. Yang, L. Guo, Z. Wang, P. Liu, X. Liu, J. Ding and W. Zhou, Biomaterials, 2021, 264, 120390 CrossRef CAS PubMed
- S. Pandey, N. Rai, A. Mahtab, D. Mittal, F. J. Ahmad, N. Sandal, Y. R. Neupane, A. K. Verma and S. Talegaonkar, Int. J. Biol. Macromol., 2021, 171, 502–513 CrossRef CAS PubMed
- N. K. Garg, N. Tandel, S. K. Bhadada and R. K. Tyagi, Front. Pharmacol., 2021, 12, 713616 CrossRef CAS PubMed
- J. Li, L. Chen, X. Xu, Y. Fan, X. Xue, M. Shen and X. Shi, Small, 2020, 16, e2005661 CrossRef PubMed
- M. Kiriakidou and C. L. Ching, Ann. Intern. Med., 2020, 172, Itc81–itc96 CrossRef PubMed
- G. Fortuna and M. T. Brennan, Dent. Clin. North Am., 2013, 57, 631–655 CrossRef PubMed
- I. Gergianaki, A. Bortoluzzi and G. Bertsias, Best Pract. Res., Clin. Rheumatol., 2018, 32, 188–205 CrossRef PubMed
- E. Beccastrini, M. M. D'Elios, G. Emmi, E. Silvestri, D. Squatrito, D. Prisco and L. Emmi, Int. J. Immunopathol. Pharmacol., 2013, 26, 585–596 CrossRef CAS PubMed
- J. Zhang, C. Chen, H. Fu, J. Yu, Y. Sun, H. Huang, Y. Tang, N. Shen and Y. Duan, ACS Nano, 2020, 14, 4414–4429 CrossRef CAS PubMed
- C. Zheng, L. Xie, H. Qin, X. Liu, X. Chen, F. Lv, L. Wang, X. Zhu and J. Xu, Front. Cell Dev. Biol., 2022, 10, 835566 CrossRef PubMed
- M. Argenziano, C. Dianzani, B. Ferrara, S. Swaminathan, A. Manfredi, E. Ranucci, R. Cavalli and P. Ferruti, Gels, 2017, 3, 22 CrossRef PubMed
- J. D. Feuerstein, A. C. Moss and F. A. Farraye, Mayo Clin. Proc., 2019, 94, 1357–1373 CrossRef
- B. C. da Silva, A. C. Lyra, R. Rocha and G. O. Santana, World J. Gastroenterol., 2014, 20, 9458–9467 CrossRef PubMed
- R. K. Mishra, A. Ahmad, A. Kumar, A. Vyawahare, S. S. Raza and R. Khan, Mater. Sci. Eng., C, 2020, 116, 111103 CrossRef CAS PubMed
- C. Li, Y. Zhao, J. Cheng, J. Guo, Q. Zhang, X. Zhang, J. Ren, F. Wang, J. Huang, H. Hu, R. Wang and J. Zhang, Adv. Sci., 2019, 6, 1900610 CrossRef CAS
- Q. Zhang, H. Tao, Y. Lin, Y. Hu, H. An, D. Zhang, S. Feng, H. Hu, R. Wang, X. Li and J. Zhang, Biomaterials, 2016, 105, 206–221 CrossRef CAS PubMed
- A. Ahmad, M. M. Ansari, R. K. Mishra, A. Kumar, A. Vyawahare, R. K. Verma, S. S. Raza and R. Khan, Mater. Sci. Eng., C, 2021, 119, 111582 CrossRef CAS PubMed
- R. Luo, M. Lin, C. Fu, J. Zhang, Q. Chen, C. Zhang, J. Shi, X. Pu, L. Dong, H. Xu, N. Ye, J. Sun, D. Lin, B. Deng, A. McDowell, S. Fu and F. Gao, Carbohydr. Polym., 2021, 263, 117998 CrossRef CAS PubMed
- Z. Li, X. Zhang, C. Liu, Q. Peng, Y. Wu, Y. Wen, R. Zheng, Q. Yan and J. Ma, J. Innate Immun., 2021, 1–13, DOI:10.1159/000519363
- X. Pu, N. Ye, M. Lin, Q. Chen, L. Dong, H. Xu, R. Luo, X. Han, S. Qi, W. Nie, H. He, Y. Wang, L. Dai, D. Lin and F. Gao, Carbohydr. Polym., 2021, 273, 118612 CrossRef CAS PubMed
- W. Wei, Y. Zhang, R. Li, Y. Cao, X. Yan, Y. Ma, Y. Zhang, M. Yang and M. Zhang, Int. J. Nanomed., 2022, 17, 603–616 CrossRef PubMed
- S. Pape, C. Schramm and T. J. Gevers, United Eur. Gastroenterol. J., 2019, 7, 1156–1163 CrossRef CAS
- C. Covelli, D. Sacchi, S. Sarcognato, N. Cazzagon, F. Grillo, F. Baciorri, D. Fanni, M. Cacciatore, V. Maffeis and M. Guido, Pathologica, 2021, 113, 185–193 Search PubMed
- M. Sebode, J. Hartl, D. Vergani and A. W. Lohse, Liver Int., 2018, 38, 15–22 CrossRef PubMed
- N. K. Gatselis, K. Zachou, G. K. Koukoulis and G. N. Dalekos, World J. Gastroenterol., 2015, 21, 60–83 CrossRef CAS PubMed
- A. Ongaro, M. B. Violatto, E. Casarin, I. Pellerani, G. Marchini, G. Ribaudo, M. Salmona, M. Carbone, A. Passoni, E. Gnodi, E. Schiavon, A. Mattarei, D. Barisani, P. Invernizzi, P. Bigini and M. Morpurgo, Nanomedicine, 2022, 40, 102497 CrossRef CAS PubMed
- M. B. Violatto, E. Casarin, L. Talamini, L. Russo, S. Baldan, C. Tondello, M. Messmer, E. Hintermann, A. Rossi, A. Passoni, R. Bagnati, S. Biffi, C. Toffanin, S. Gimondi, S. Fumagalli, M. G. De Simoni, D. Barisani, M. Salmona, U. Christen, P. Invernizzi, P. Bigini and M. Morpurgo, ACS Nano, 2019, 13, 4410–4423 CrossRef CAS PubMed
- Y. Yang, K. K. Ellestad, S. Singha, M. M. Uddin, R. Clarke, D. Mondal, N. Garabatos, P. Solé, C. Fandos, P. Serra and P. Santamaria, J. Controlled Release, 2021, 338, 557–570 CrossRef CAS PubMed
- A. Carambia, C. Gottwick, D. Schwinge, S. Stein, R. Digigow, M. Şeleci, D. Mungalpara, M. Heine, F. A. Schuran, C. Corban, A. W. Lohse, C. Schramm, J. Heeren and J. Herkel, Immunology, 2021, 162, 452–463 CrossRef CAS PubMed
- C. S. Umeshappa, S. Singha, J. Blanco, K. Shao, R. H. Nanjundappa, J. Yamanouchi, A. Parés, P. Serra, Y. Yang and P. Santamaria, Nat. Commun., 2019, 10, 2150 CrossRef PubMed
- H. Jamaleddine, P. Santamaria and A. Khadra, Immunology, 2020, 161, 209–229 CrossRef CAS PubMed
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |